
Abstract
Organic nitrates, such as nitroglycerin (GTN), isosorbide-5-mononitrate and isosorbide dinitrate, and pentaerithrityl tetranitrate (PETN), when given acutely, have potent vasodilator effects improving symptoms in patients with acute and chronic congestive heart failure, stable coronary artery disease, acute coronary syndromes, or arterial hypertension. The mechanisms underlying vasodilation include the release of •NO or a related compound in response to intracellular bioactivation (for GTN, the mitochondrial aldehyde dehydrogenase [ALDH-2]) and activation of the enzyme, soluble guanylyl cyclase. Increasing cyclic guanosine-3′,-5′-monophosphate (cGMP) levels lead to an activation of the cGMP-dependent kinase I, thereby causing the relaxation of the vascular smooth muscle by decreasing intracellular calcium concentrations. The hemodynamic and anti-ischemic effects of organic nitrates are rapidly lost upon long-term (low-dose) administration due to the rapid development of tolerance and endothelial dysfunction, which is in most cases linked to increased intracellular oxidative stress. Enzymatic sources of reactive oxygen species under nitrate therapy include mitochondria, NADPH oxidases, and an uncoupled •NO synthase. Acute high-dose challenges with organic nitrates cause a similar loss of potency (tachyphylaxis), but with distinct pathomechanism. The differences among organic nitrates are highlighted regarding their potency to induce oxidative stress and subsequent tolerance and endothelial dysfunction. We also address pleiotropic effects of organic nitrates, for example, their capacity to stimulate antioxidant pathways like those demonstrated for PETN, all of which may prevent adverse effects in response to long-term therapy. Based on these considerations, we will discuss and present some preclinical data on how the nitrate of the future should be designed. Antioxid. Redox Signal. 23, 899–942.
II. Organic Nitrate Bioactivation
A. High-potency (affinity) pathway involving the ALDH-2 in GTN bioactivation
2. Impact of gene polymorphism on GTN bioactivation by ALDH-2
4. Dihydrolipoic acid and thioredoxin as physiological reducing factors of ALDH-2
5. Molecular mechanism of ALDH-2 bioactivation of organic nitrates via its reductase activity
6. Impact of organic nitrate-induced ROS formation on ALDH-2 activity
7. Purified ALDH-2 as a peroxynitrite synthase in the presence of GTN
VI. Development of New Organic Nitrates Devoid of Side Effects
VII. Summary and Future Perspectives for the Class of •NO Donors
I. Introduction
A. Historical view
T
In early 1847, he announced his discovery to his old master, Theophile Pelouse, a French doctor and chemist. Part of this letter was printed in an 1847 Forum of Comptes Rendus (292). In it, he described the method of production of nitroglycerin and he also described nitroglycerin as looking like olive oil with a slight yellow color, as being insoluble in water, but soluble in ether and alcohol, and as having no odor, but a sweet, piquant aromatic flavor. Sobrero is indeed today universally accepted as the founder of nitroglycerin (293).
Shortly after, Alfred Nobel stepped into the picture (1833–1896). He had heard about nitroglycerin from his tutor, N.N. Zinin. By using porous silica to absorb the unstable nitroglycerin, Alfred Nobel created an easily handled explosive called dynamite. With this, Nobel became increasingly successful in the business of dynamite manufacture all over Europe.
Due to the commercial success, Nobel was one of the richest people of his era. Reportedly, when he realized how much damage was caused by military use of his invention, he decided to found the Nobel Prize for the benefit of humankind to pay back his dues. Interestingly, when Nobel was taken ill with heart disease, his doctor prescribed him nitroglycerin. Nobel refused to take it, knowing that it caused headache and ruling out that it could eliminate chest pain. In a letter, Nobel wrote, “It is ironical that I am now ordered by my physician to eat nitroglycerin.”
Ten years before the commercial success of GTN as an explosive in 1857, patients with angina pectoris symptoms were treated with inhaled amyl nitrite by the Scottish physician, T. Lauder Brunton (1844–1916), providing the rationale for commercial use of nitrovasodilators in the clinical setting (28).
Following industrial production of GTN and dynamite, it was also noticed by the medical community that workers with heart disease in the GTN and dynamite factories showed less anginal symptoms during work. Interestingly, they suffered from increased frequency of angina pectoris at the weekend, identifying GTN as an antianginal drug for clinical use. Today, we know that this so-called rebound ischemia phenomenon is responsible for more frequent angina attacks over the weekend upon withdrawal of the drug. For those workers in GTN manufacturing facilities, the effects of withdrawal at the weekend also include a Monday morning headache in those experiencing regular GTN exposure in the workplace. Over the weekend, the workers lose their tolerance, and when they are re-exposed on Monday, they develop a headache again (283).
Mostly due to the inadequate dosing, GTN prescription has replaced amyl nitrite in clinical therapy since the 1870s. From a pharmacological point of view, both compounds belong to the class of nitrovasodilators and are thought to produce nitric oxide or a related species in the organism upon bioactivation (7).
In 1888, the American physician, D.D. Stewart, made an important discovery. Following chronic treatment of a patient with GTN, he required a 20-fold higher dose of GTN compared with the initial dose to induce comparable antianginal effects (297). Stewart's report describes for the first time the phenomenon of nitrate tolerance.
During the last two decades, much effort has been put into the characterization of the mechanisms of the pharmacological action of organic nitrates (221 –223, 238). The key findings were the identification of the GTN bioactivation process by Jonathan Stamler's group (41), the demonstration of involvement of reactive oxygen species (ROS) causing tolerance and cross-tolerance (236), and also the findings that not all nitrates are the same, rather representing a very heterogeneous family of compounds with differing vasodilator principles, tolerance mechanisms, and capacities to induce antioxidant enzymes (110, 238).
B. Hemodynamic effects of nitrates in patients with coronary artery disease and acute and chronic congestive heart failure
Organic nitrates dilate venous capacitance vessels, large- and medium-sized coronary arteries, collaterals (4, 5) (Fig. 1), and also the aorta, while coronary and peripheral arterioles with a diameter <100 μm have been demonstrated to be nitrate resistant (287, 288). Thus, in the setting of stable angina, the preferential venodilation induced by anti-ischemic doses of nitrates results in venous pooling and therefore preload reduction, leading to a reduction of left ventricular end-diastolic filling pressure and wall tension. The consequences are a reduction in myocardial workload and oxygen demand (4, 5).
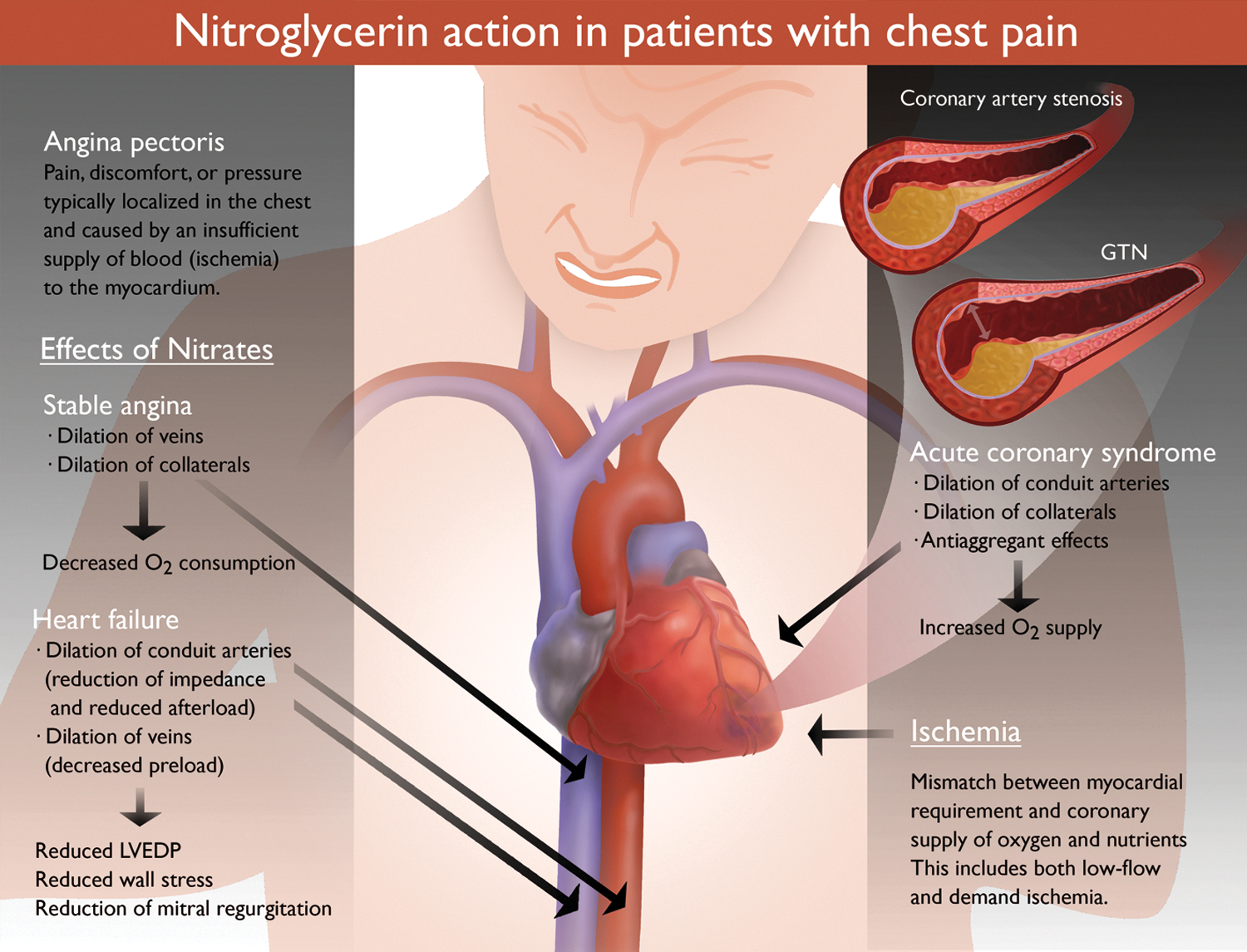
Beyond these effects, in patients with acute coronary syndrome, nitrates have potent anti-ischemic effects that depend on the dilation of large epicardial coronary arteries and coronary collaterals. This leads to an improved blood perfusion and therefore oxygen delivery to subendocardial regions, mainly by increasing total coronary conductance. In addition, since arteriolar tone is largely unaffected by nitrates (287), coronary steal phenomena as well as reflex tachycardia are in general avoided. Taken together, the mismatch between oxygen demand and oxygen supply in ischemic regions is rapidly relieved on short-term administration of organic nitrates.
Through similar hemodynamic effects, nitrates also markedly improve left ventricular function in patients with acute and chronic congestive heart failure (CHF). Nitrates decrease the right atrial pressure with a redistribution of blood from the central circulation into larger capacitance veins. Nitrates also cause an unloading of the failing afterload-dependent ventricle by reducing the impedance to the left ventricular ejection, mainly via dilation of large capacitance arteries such as the aorta. The increase in compliance of the arterial vasculature in turn leads to a reduction in the magnitude, frequency, and velocity of reflected waves in the arterial circulation (349).
Thus, these hemodynamic afterload effects of nitrates cause an increase in cardiac output, a reduction in left ventricular filling pressure and wall tension, and also a reduction in mitral regurgitation, thereby shifting the stroke volume/left ventricular end-diastolic pressure relationship from a negative to a positive slope.
In general, the potent anti-ischemic effects of organic nitrates such as GTN might be explained by their improved vasodilatory potency in ischemic vascular tissues as shown for isolated porcine coronary arteries under hypoxic conditions (97, 269). It remains to be established whether reduced ROS formation during hypoxia improves the redox-sensitive bioactivation of GTN by mitochondrial aldehyde dehydrogenase (ALDH-2) or if the potential product of this bioactivation process, inorganic nitrite, is reduced more easily to a vasodilator under hypoxic conditions (see section on Organic Nitrate Bioactivation).
C. Cellular mechanisms of vasodilation by organic nitrates
The activation of the enzyme, soluble guanylyl cyclase (sGC), by nitrate-derived nitric oxide (•NO) was identified as the principal mechanism of action of these drugs [for review, see Munzel et al. (225)]. Activation of sGC leads to increased bioavailability of cyclic guanosine-3′,-5′-monophosphate (cGMP) and activation of cGMP-dependent protein kinases, such as the cGMP-dependent protein kinase I (cGK-I). The relaxation downstream to these processes requires Ca2+-dependent and/or -independent mechanisms. cGK-I inhibits the inositol-1,4,5-trisphosphate [IP3]-dependent calcium release mediated by phosphorylation of the IP3 receptor-associated cGMP kinase substrate (IRAG) and activates the big calcium-activated potassium channel (BKCa) through phosphorylation, leading to hyperpolarization and reduced calcium influx.
The inhibitory effect of the •NO-cGMP-cGK-I signaling pathway on voltage-gated Ca2+ channels was meanwhile confirmed by three independent studies (10, 128, 162). Then, cGK-I activates the Ca2+-ATPase pump and thereby the efflux of calcium to the extracellular space. Ca2+-independent relaxation by cGK-I involves phosphorylation of the myosin-binding subunit (e.g., myosin phosphatase targeting subunit 1 [MYPT1]) (Fig. 2).
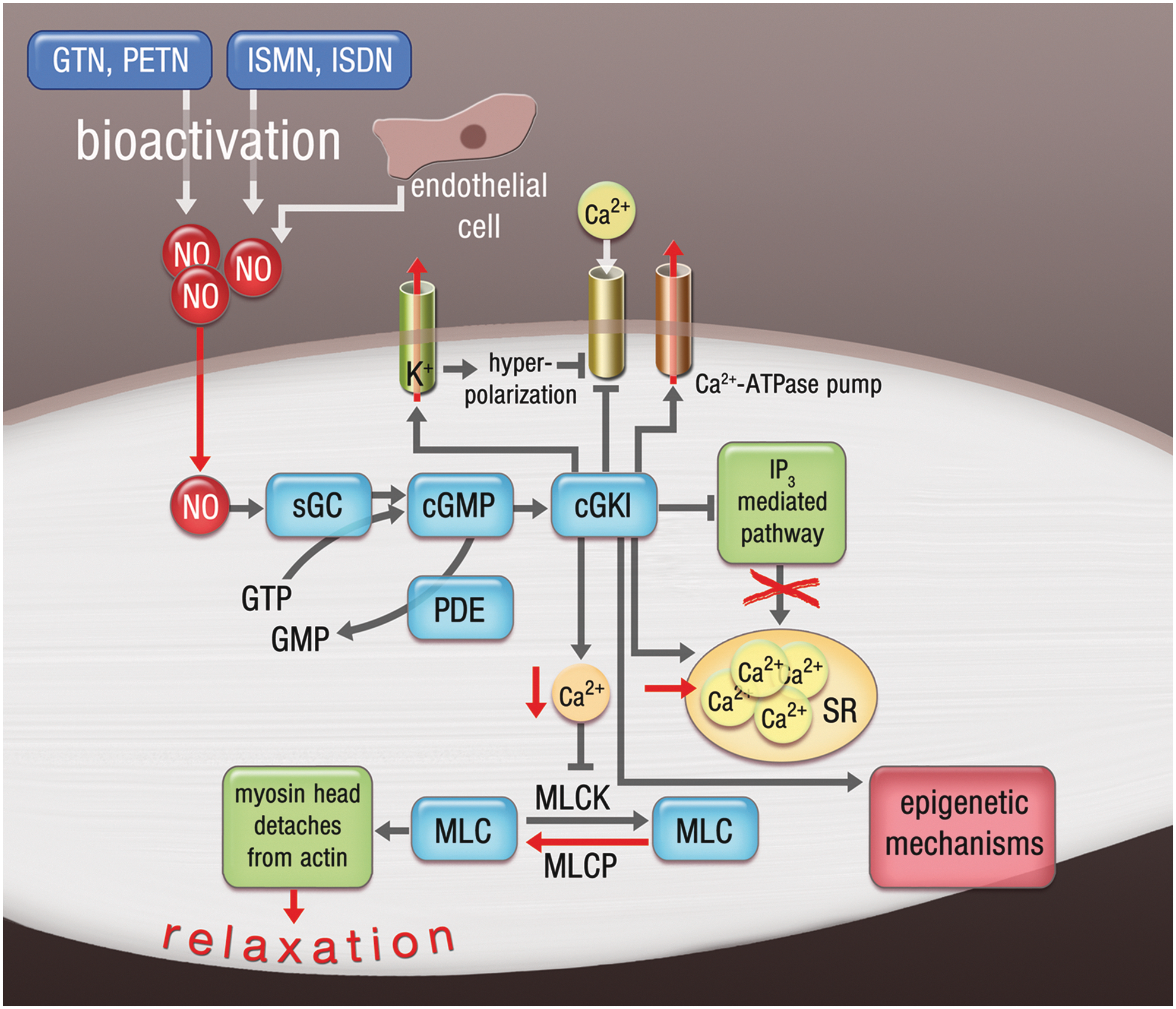
Furthermore, cGK-I might phosphorylate and thereby inhibit the small GTP-binding protein, RhoA, leading to decreased Rho kinase (ROK) activity and conserved activity of myosin light chain phosphatase (MLCP), all of which are vasodilatory. ROK can also directly phosphorylate and increase the contractility of the myosin light chain (MLC). cGK-I also induces a feedback mechanism (which lowers the intracellular cGMP concentration) by the phosphorylation and activation of phosphodiesterases (PDEs). More recent studies also put emphasis on epigenetic regulation of nitrate-induced smooth muscle relaxation by demonstrating that GTN increases histone acetylase activity and Nɛ-lysine acetylation of contractile proteins influences GTN-dependent vascular responses (47).
D. No •NO from nitroglycerin?
Interestingly, the concept that the group of organic nitrates causes vasodilation in general by •NO release was recently challenged by two independent studies demonstrating an almost 100-fold discrepancy between GTN-evoked •NO formation and vasodilation, whereas, for example, a direct correlation between these parameters was shown (158). In addition, Nunez et al. (249) reported a similar discrepancy between hemodynamic effects in response to GTN administration and •NO release, suggesting that the action of GTN is unrelated to its bioconversion to •NO. Thus, it appears that at least GTN-induced vasodilation might be mediated by an •NO-related species, but not by •NO itself. A detailed discussion on the identity of the vasodilating species, which might be formed by organic nitrates, was provided in previous review articles, but could comprise, for example, iron–nitrosyl or S-nitroso species (67, 223). A significant increase in iron–nitrosyl or S-nitroso species can be seen some minutes after oral intake of GTN in human volunteers or animals (147, 248, 330).
Further evidence is provided by recent studies showing that so-called •NO donors such as pentaerithrityl tetranitrate (PETN) and GTN have substantially different effects on gene expression (65, 264). Treatment with GTN resulted in a larger expression of cardiotoxic genes and inhibition of the expression of cardioprotective proteins, whereas PETN treatment enhances the expression of genes in the opposite direction (Fig. 3). A possible explanation for this differential gene expression profile in response to both •NO donors may be the release of different vasoactive molecules upon bioactivation (264).

E. Clinical uses/usefulness of organic nitrates
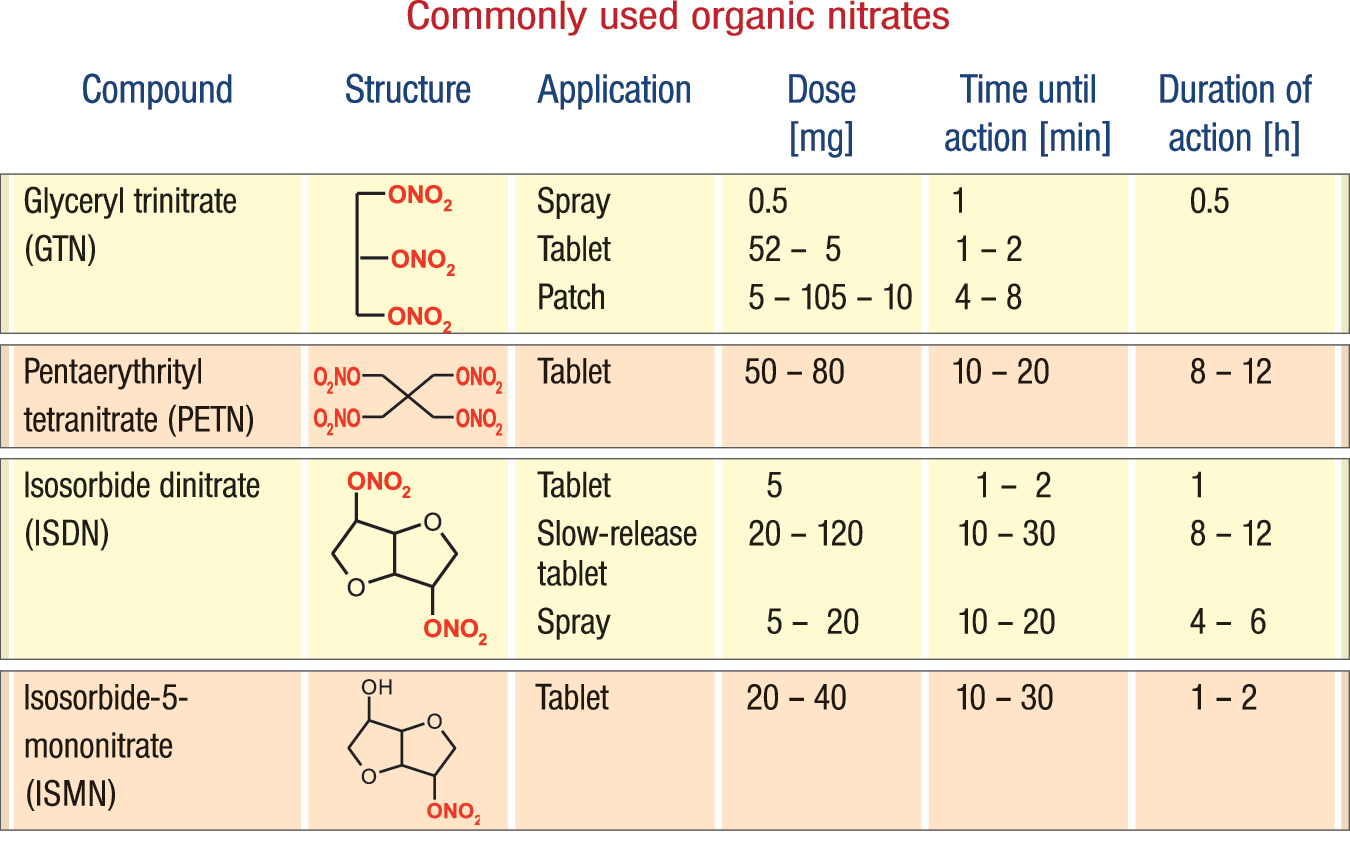
In general, treatment of patients with coronary artery disease (CAD), chronic CHF, or arterial hypertension with nitrates comprises organic nitrates such as GTN (mainly spray, capsule, patch, and infusion), PETN (tablet), isosorbide-5-mononitrate (ISMN) (tablet), and isosorbide dinitrate (ISDN) (tablet) (Fig. 4). Although nitrates in general failed to improve prognosis in patients with CAD and CHF (except for the combination ISDN and hydralazine in patients with CHF), they can be considered for acute and long-term treatment in these patient groups. The current guidelines for stable angina (310), acute coronary syndromes (126), acute and chronic CHF (nitrates alone for patients with CHF and angina and ISDN+hydralazine for patients with CHF alone) (203), and arterial hypertension (197) still more or less recommend the use of oral or intravenous organic nitrates.
An interesting observation was recently published by Ambrosio et al. (9). The authors found that in a large multinational registry, treatment of patients with CAD with organic nitrates was associated with a shift away from ST-segment elevation myocardial infarction (MI) to the non-ST-segment elevation infarction and that this shift was associated with a less pronounced release of cardiac markers. The authors speculated that this beneficial effect may be mediated, at least in part, by the positive effects of nitrates on ischemic preconditioning (9).
F. Do nitrates beneficially influence prognosis in patients with CAD?
It is also important to note that most of the studies with organic nitrates in the setting of heart failure and CAD demonstrated improvement in symptoms, but mostly failed to demonstrate an improvement in prognosis. Large trials such as ISIS-4 (2) or GISSI-3 (1) did not provide any prognostic benefit of prolonged nitrate therapy in patients with acute MI. On the contrary, several retrospective studies (meta-analysis) performed in postinfarct patients (143, 153, 240), patients following percutaneous coronary intervention and diabetes (345), or patients with vasospastic angina revealed that the nitrate use is associated with an unfavorable prognosis. In a study by Nakamura et al., the use of oral nitrates such as ISDN and ISMN was associated with an increased risk for cardiac death (240). Similarly, Ishikawa et al. and Kanamasa et al. described that long-term continuous use of nitrates increased cardiac events in patients with healed MI (143, 153).
More recent studies in patients with diabetes mellitus revealed that long-term therapy with ISMN leads to more cardiovascular events consisting of cardiovascular revascularization, nonfatal MI, and cardiovascular death (143, 153). Moreover, in patients with vasospastic angina, a multicenter registry study from Japan just recently revealed that in patients treated with organic nitrates, GTN use (patches) was associated with a significant increase in the incidence of major adverse cardiovascular events, including cardiac death, nonfatal MI, hospitalization due to unstable angina and heart failure, and appropriate implantable cardioverter defibrillator shocks (308).
Thus, these results may indicate that the use of GTN and ISMN may cause more cardiovascular events in patients with acute and chronic CAD or with vasospastic angina, all of which may be linked to the well-described side effects of these nitrates in causing oxidative stress, nitrate tolerance, endothelial dysfunction, or an increase in sensitivity of the vasculature to vasoconstrictors, a phenomenon which is likely linked to the stimulation of autocrine endothelin production within the vessel wall.
With respect to ISDN, there is also clear evidence that long-term administration without a sufficient nitrate-free interval causes tolerance in patients with CAD (262) and also CHF (82). Interestingly, after washout of ISDN due to a sufficiently long nitrate-free interval, the anti-ischemic and hemodynamic effects in patients with CHF are preserved with the disadvantage that in the nitrate-free interval, the patients are not covered with nitrate therapy. It is important to note that like ISMN and GTN, ISDN causes tolerance and also endothelial dysfunction (286).
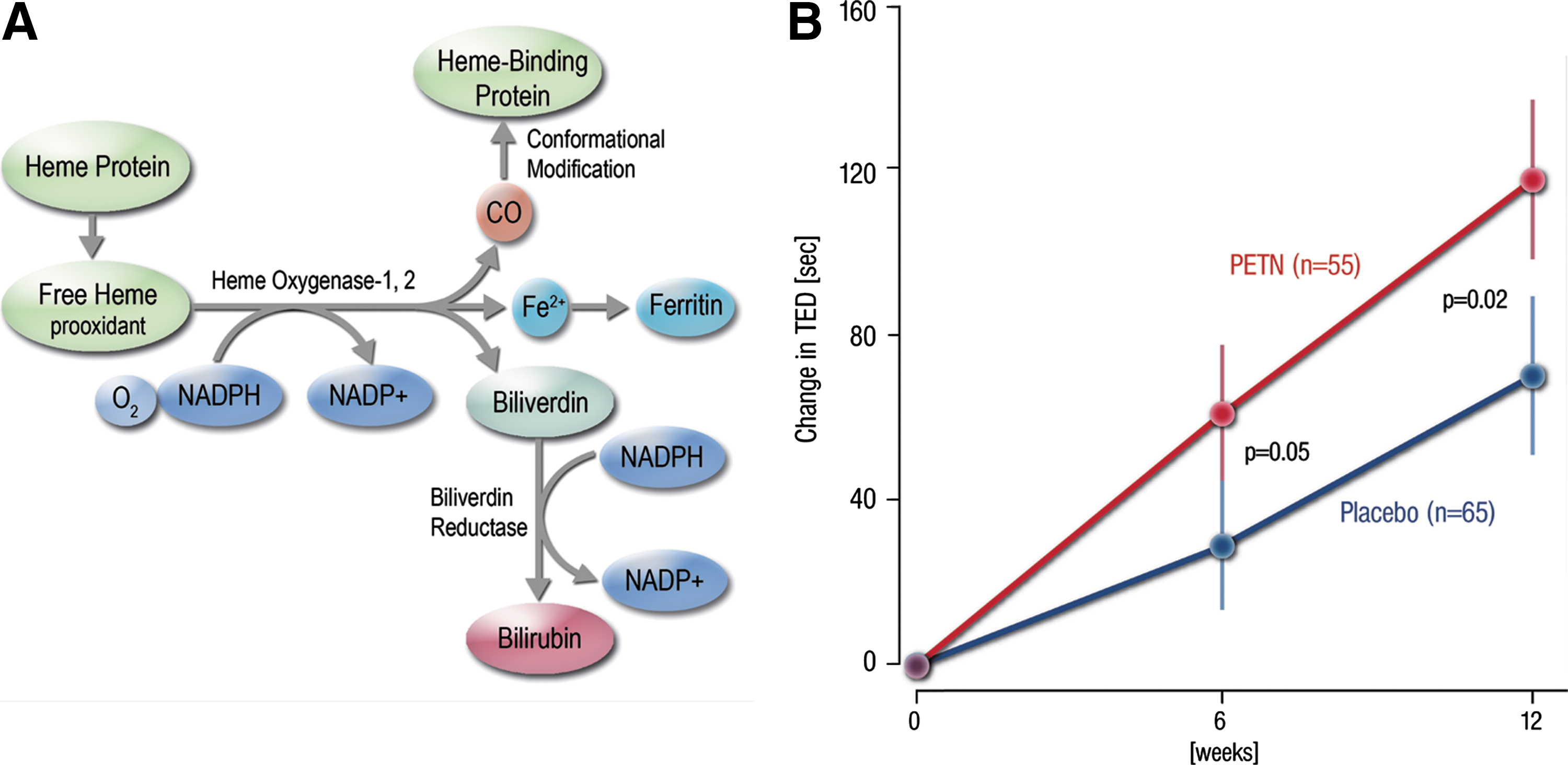
PETN is an organic nitrate with completely different effects on the vasculature. PETN does not cause tolerance, it does not increase oxidative stress within the vasculature, and it does not cause endothelial dysfunction when treating patients with CAD (275). The reason for this is that this nitrate stimulates the expression of heme oxygenase-1 (HO-1) in endothelial and smooth muscle cells, which in turn leads to enhanced production of one of the most potent antioxidants in our body, bilirubin (251, 252, 327). It also induces the expression of ferritin, which in turn diminishes the levels of free iron and therefore the production of hydroxyl radicals (•OH) via Fenton chemistry (251, 252). Moreover, it also causes an increase in the production of carbon monoxide (CO), which in turn stimulates the sGC and therefore lowers vascular tone (327).
We recently performed a large randomized, double-blind, placebo-controlled multicenter trial (CLEOPATRA study) to investigate the anti-ischemic efficacy of 80 mg of PETN (b.i.d., given in the morning and at midday) over placebo in patients with stable angina pectoris. A total of 655 patients, evaluated with the intention to improve anti-ischemic therapy, were randomized to PETN or placebo and completed the study. After 6 and 12 weeks of treatment, patients underwent treadmill exercising tests as a functional readout of exercise capacity (233). Treatment with PETN for 12 weeks did not modify the primary endpoint of total exercise duration. In a prespecified subgroup analysis of patients with reduced exercise capacity, however, PETN appeared more effective than placebo treatment (Fig. 5). Superiority over placebo treatment was evident, particularly in patients who were symptomatic in low exercise levels. PETN 80 b.i.d. was well tolerated and the overall safety profile was comparable with placebo (233).
Despite the fact that PETN therapy alone did not provide any additional benefit in unselected patients with known CAD, its administration in combination with modern anti-ischemic drugs could increase exercise tolerance in symptomatic patients with reduced exercise capacity.
G. Organic nitrate chemistry and pharmacokinetics
Organic nitrates represent a group of nitric acid esters and are typically formed from nitric acid and alcohol by a condensation reaction (R-OH + HNO3 → R-ONO2 + H2O). The powerful explosive properties of organic nitrates are based on the fact that thermal decomposition of nitrate esters mainly yields the gases, molecular nitrogen (N2) and carbon dioxide (4 CH2ONO2-CHONO2-CH2ONO2 → 12 CO2 + 10 H2O + 5 N2 + 2 •NO). GTN (nitroglycerin) was used for decades and is still in use as an industrial explosive in the form of dynamite. In addition, PETN is frequently used as an explosive for military and mining industry purposes. PETN, also known as PENT, PENTA, TEN, corpent, penthrite, or nitropenta, can be mixed with a plasticizer yielding a plastic explosive.
Due to these properties, besides the clinical or mining use, a large scientific interest is directed toward the detection of organic nitrates (for prevention of bombing) as well as the biological degradation of aliphatic organic nitrates (for biodegradation in contaminated soil). Minor biochemical research is dedicated to the atmospheric chemistry of organic nitrates (e.g., formation, removal, interaction with ozone). Figure 4 displays the structures of the most frequently clinically used organic nitrates. ISMN and ISDN are mono- and dinitrates, in which the nitrate esters are covalently bound to the sugar derivative, isosorbide. The covalent bond to the sugar backbone largely reduces the explosiveness of these organic nitrates.
To become a vasodilator, all organic nitrates need to undergo a 3-electron reduction, leading to nitric oxide and the respective alcohol (R-ONO2 + 3 e− + 3 H+ → R-OH +•NO + H2O). It is still elusive whether or not under biological conditions this reduction involves inorganic or organic nitrite as an intermediate.
The chemical structure of the organic nitrates currently in clinical use differs markedly (Fig. 4). ISMN and ISDN have one or two −ONO2 groups linked to the isosorbide sugar ring, whereas GTN has three −ONO2 groups linked to the glycerol backbone; PETN has a more spherical shape due to linkage of four methylene −ONO2 groups to the central carbon atom. These differences in their chemical structure result in quite distinct pharmacokinetic profiles, including different transporters for cellular uptake [for review, see Daiber et al. (67)].
Of note, after oral application of PETN to humans, only the dinitrate and mononitrate metabolites (pentaerithrityl dinitrate [PEDN] and pentaerithrityl mononitrate [PEMN]) can be found in the blood (246), while PETN itself has never been quantified and pentaerithrityl trinitrate (PETriN) has only been detected occasionally in very low concentrations. This suggests that despite its high stability in aqueous media, PETN is rapidly bioconverted by intestinal microorganisms yielding the less effective mono- and dinitrate metabolites, which are bioabsorbed and produce their therapeutic effects (130, 285, 322). This also reflects in different pharmacokinetic properties: in contrast to GTN (whose absorption and metabolism upon administration show a peak at 30 min) (302, 337), PETN metabolites reach their maximum plasma concentration 2–3 h after oral administration (322).
II. Organic Nitrate Bioactivation
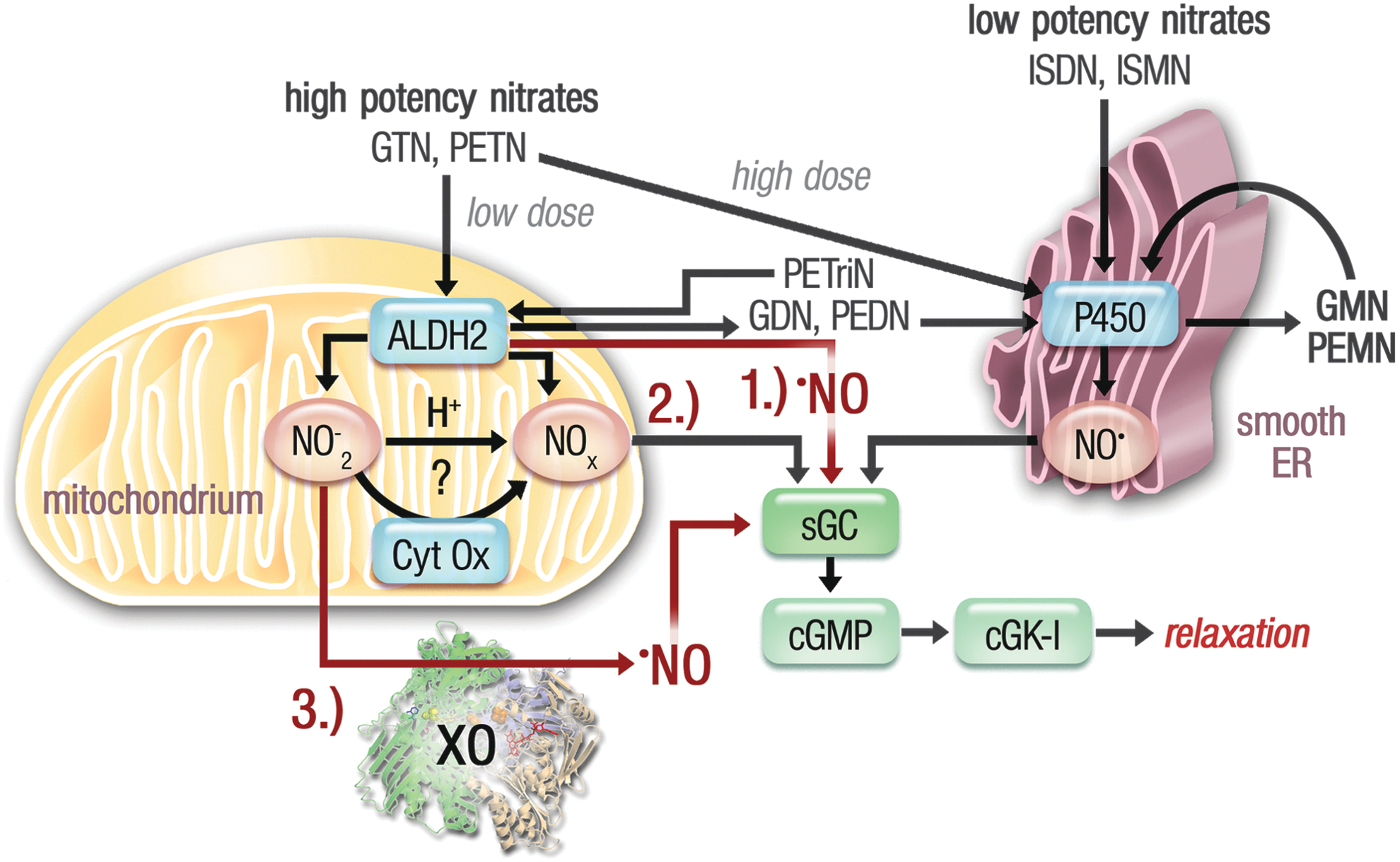
Organic nitrates are supposed to get biotransformed to release a vasoactive molecule. In particular, GTN has been intensively studied and it has been demonstrated that the nitrate is biotransformed by two different pathways, a high- and a low-affinity pathway (Fig. 6). GTN concentration–relaxation curves are biphasic, suggesting a high-potency pathway (operative at clinically relevant GTN concentrations <1 μM) and a low-potency pathway (operative at suprapharmacological GTN concentrations >1 μM).
A. High-potency (affinity) pathway involving the ALDH-2 in GTN bioactivation
In 2002, Chen et al. identified the mitochondrial isoform of aldehyde dehydrogenase (ALDH-2) as a key enzyme in a clinically relevant (high affinity) bioactivation process of GTN (41) and therefore provided new important information for nitrate pharmacology. The ALDH-2 is well known from the alcoholism research field and accounts for the removal of toxic acetaldehyde upon ethanol consumption by conversion to acetic acid. Individuals with the East Asian inactive variant of the enzyme show the so-called flushing syndrome in response to alcohol intake (340). More recently, ALDH-2 was implicated in the prevention of cocaine addiction and relapse in experimental models (344). Besides the new nitrate reductase activity, the enzyme displays two physiological enzymatic activities (dehydrogenase and esterase) (67).
The isolated enzyme generates nitrite (NO2 −) and 1,2-glyceryl dinitrate (1,2-GDN) from GTN and the reaction is accelerated by NAD+, probably by an allosteric action. Nonspecific inhibitors of this enzyme (disulfiram, cyanamide, chloral hydrate) and high substrate concentrations (acetaldehyde) attenuated the vasodilating, cGMP-eliciting, and blood pressure-lowering activity of GTN in rats and inhibited the organic nitrate reductase activity of ALDH-2 (41, 168, 304, 347).
GTN-dependent vasodilation of isolated aortic rings and GTN bioactivation to 1,2-GDN in cultured cells were acutely prevented by daidzin, a highly specific inhibitor of ALDH-2 (61, 304). Using isolated rat aortic rings, a marked attenuation of the GTN vasodilator potency following incubation with acetaldehyde and choral hydrate was observed (304), as previously observed in rabbit aortic rings (347), as well as with benomyl and daidzin. In addition, the activation of cGK-I (as assessed by P-VASP) and vasodilation by GTN were markedly inhibited by the ALDH-2 inhibitor, benomyl (10 μM), whereas benomyl did not modify SNP- or ACh-induced phosphorylation of VASP and vasorelaxation (304).
We also showed that treatment of RAW 264.7 macrophages with GTN or exposure to benomyl or daidzin reduced GTN bioactivation (1,2-GDN formation). These results confirmed the observations by Chen et al. and pointed to a specific role of ALDH-2 in the cGMP-mediated GTN-induced vasorelaxation (41). Additionally, by depleting endothelial cells of functional mitochondria (so-called ρ0 cells), we could also show that GTN-stimulated increases in cGMP were markedly attenuated (304).
Interestingly, inhibition of ALDH-2 did not completely abolish the vasodilator and cGK-I-stimulating activity of GTN. The concentration–response curve was shifted to the right, and with higher concentrations of GTN, a maximal relaxant response was still achieved (304). This finding supports our concept of two independent pathways accounting for bioactivation of GTN, where only the high-potency pathway is dependent on ALDH-2 activity; other enzymatic (e.g., cytochrome P450 enzymes) and nonenzymatic (e.g., low-molecular-weight thiols) pathways may account for the low-potency pathway (Fig. 6).
The reduction of GTN by thiol groups in the active site of ALDH-2 has been proposed as a mechanism to explain the nitrate reductase activity of ALDH-2 (41). During this reaction, a hypothetic thionitrate intermediate will be formed from GTN in the active center of ALDH-2 (99), with concomitant release of 1,2-GDN. The intermediate then reacts by nucleophilic attack of an adjacent second cysteine thiol group under formation of a disulfide bridge releasing nitrite as the leaving group (see the Redox Regulation of the ALDH-2 section for more details). The inactive thiol-oxidized enzyme can be reduced and reactivated by thiol donors such as dithiothreitol (DTT) and 2-mercaptoethanol.
Our observations demonstrated that mitochondrial lipoic acid may function as the natural reducing agent (323), which was later supported by others (80). Chen et al. suggested that •NO, an S-nitrosothiol or a nitroso–metal complex may be formed from nitrite (NO2 −). This could involve intermediate formation of nitrous acid (HNO2), catalysis by components of the mitochondrial respiratory chain (e.g., cytochrome c oxidase), or conversion by the xanthine oxidoreductase (Fig. 6) (41). Feelisch and coworkers have demonstrated extensive S-nitros(yl)ation of blood and tissue proteins in response to 0.1–100 mg/kg GTN treatment of rats (147).
The potential involvement of cytochrome c oxidase in the mitochondrial bioactivation of inorganic nitrite to •NO, especially under hypoxic conditions, was recently characterized in all mechanistic details (134, 135). The role of cytochrome c oxidase as a nitrite reductase in plant metabolism was also recently reviewed (140). However, others described cytochrome c oxidase as a sink of •NO and attribute the hypoxic vasodilation by inorganic nitrite to its bioactivation by hemoglobin and formation of bioactive •NO metabolites rather than free •NO (316).
However, a direct •NO formation by therapeutic concentrations of GTN (<1 μM) and xanthine oxidoreductase or isolated mitochondria has not yet been demonstrated. In principle, this concept is a revival of the Needleman thiol theory, which already suggested an interaction of organic nitrates with the mitochondria (swelling and increased oxygen uptake) as well as a depletion of mitochondrial thiol pools in response to chronic GTN treatment (242). Involvement of NO2 − in GTN-induced relaxation is supported by recent reports on nitrite-mediated protection from ischemic damage (32). In addition, nitrite-driven ATP release from erythrocytes (a vasodilatory pathway) (104) provides an attractive explanation for the contribution of nitrite to the potent anti-ischemic properties of GTN. The concept of inorganic nitrite as an essential intermediate during nitrovasodilator bioactivation was previously put forward by Feelisch and coworkers (248).
The concept of GTN-induced mitochondrial nitrite formation with subsequent reduction to •NO was recently challenged by Mayer and coworkers, who demonstrated that sGC activation was triggered by bioactivation of nitrite with mitochondrial cytochrome c oxidase and GTN in the presence of mitochondria, but these triggers differed markedly with respect to effects of respiratory chain substrates and inhibitors (167). Based on these observations, the authors negated a role of nitrite in the GTN-induced vasodilation process, although no direct comparison was provided on the potency of GTN versus nitrite-triggered sGC activation in the presence of mitochondrial homogenate.
In 2005, involvement of ALDH-2 in the GTN bioactivation process was proven at the molecular level using ALDH-2-deficient (ALDH-2−/− ) mice that demonstrated impaired relaxation in response to GTN, but not SNP or ISDN (39). In the same year, there was the first report on a role of ALDH-2 in the bioactivation process in humans (195). These authors treated healthy volunteers with the ALDH inhibitor, disulfiram, a drug used for treatment of alcoholism (e.g., antabuse), and observed significantly impaired GTN, but not SNP, -induced blood flow increases in the forearm. They also observed similar effects in East Asian volunteers with ALDH-2 Glu504Lys mutation polymorphism (ALDH2*2) (195). Bioactivation of GTN by ALDH-2 is also of high epidemiological interest since a large part of the East Asian population carries the ALDH2*2 polymorphism and therefore demonstrates impaired responsiveness to GTN (184).
1. ALDH-2 is also involved in PETN bioactivation
In 2007, we demonstrated the bioactivation of PETN and its trinitrate metabolite, PETriN, by ALDH-2 using ALDH-2−/− mice (324). In contrast, the more denitrated metabolites of PETN, PEDN and PEMN, as well as the GTN metabolite, 1,3-GDN, and the mononitrate, ISMN, were not bioactivated by ALDH-2. These observations exclude that differences between GTN and ISDN are not only simply due to structural differences but also due to differences in the reactivity toward thiol groups since the latter parameter determines the affinity for bioactivation by the ALDH-2 (324). Others later confirmed the bioactivation of PETN by ALDH-2 (121).
2. Impact of gene polymorphism on GTN bioactivation by ALDH-2
The widely distributed East Asian variant of ALDH-2 (ALDH2*2) with the point mutation, E487K, displays a similar decrease in NAD+ binding affinity compared with E268Q mutant (177). Accordingly, the ALDH2*2 variant displayed not only a significantly reduced dehydrogenase and esterase activity (177) but also impaired GTN reductase activity (184). Thus, the vasodilatory potency of GTN is blunted in East Asian individuals with the ALDH2*2 polymorphism as well as in humans under therapy with the ALDH inhibitor, disulfiram (195). In a recently published work, these findings were supported by data obtained with purified ALDH2*2 variant displaying a reduced dehydrogenase, esterase, and nitrate reductase activity compared with the wild-type enzyme (ALDH2*1) (18). Accordingly, the ALDH2*2 variant yielded decreased levels of 1,2-GDN and •NO in the presence of GTN and finally evoked less pronounced activation of sGC.
Interestingly, the previously described ALDH-2 activator, Alda-1 (37), induced only minor activation of the dehydrogenase activity of wild-type ALDH-2, but a 4-fold increase in ALDH2*2 variant dehydrogenase activity (18). The effect of Alda-1 on esterase activity of the ALDH2*1 enzyme was moderate, but caused an 8-fold increase in the ALDH2*2 variant. It was a disappointment to see that Alda-1 neither increased the GTN bioactivation nor the sGC activity (18) and obviously cannot be clinically employed to increase the vasodilatory potency of GTN in (nitrate-tolerant) patients under chronic treatment or to improve the anti-ischemic effects of GTN in East Asian individuals with ALDH2*2 polymorphism. It remains elusive whether individuals with the East Asian variant of ALDH-2 (ALDH2*2) show reduced hemodynamic effects for PETN therapy.
3. Redox regulation of the ALDH-2
The crystal structure of bovine ALDH-2 revealed an active site with three neighboring cysteine residues (Cys301–303), making the enzyme prone to inactivation by various oxidants. Besides the already discussed inactivation by reactive oxygen and nitrogen species (RONS) (superoxide and peroxynitrite) (254, 323) and organic nitrates (GTN and PETN/ISDN at higher concentrations) (20, 61, 99, 254), a number of redox-sensitive pathways were described for the regulation of ALDH-2 activity (Table 1). Lipid peroxidation products such as 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA) form covalent Michael adducts with the ALDH-2, leading to deactivation of the enzyme (254, 294). In addition, nitro fatty acids (as generated by peroxynitrite) were highly potent inhibitors of purified ALDH-2 (254).
4-HNE, 4-hydroxynonenal; ALDH-2, mitochondrial aldehyde dehydrogenase.
Oxidative modification of ALDH-2 involves the nitration of tyrosine residues in response to GTN or ISMN treatment, resulting in the loss of nitrate-induced antiaggregatory effects, increase in cGMP levels, and decrease in blood pressure, all of which were prevented with the catalytic antioxidant, MnTBAP (214). Reversible inhibition of ALDH-2 activity and detection of S-nitros(yl)ated protein were observed in response to GSNO (215). In addition, reversible regulation of ALDH-2 activity by S-glutathionylation was described in the presence of GTN and peroxynitrite (323).
Likewise, the expression level of ALDH-2 is controlled in a redox-sensitive manner by GTN (likely by increased superoxide/peroxynitrite formation) (306, 327). The decrease in ALDH-2 protein was more pronounced in GTN-tolerant veins compared with arteries (54). As a possible explanation, Wölkart et al. demonstrated increased 26S proteasomal degradation of ALDH-2 protein with no requirement of alterations of transcriptional control of ALDH-2 expression (335). It is noteworthy that the 26S proteasome was demonstrated to be activated upon peroxynitrite-dependent nitration of a tyrosine residue (341, 342).
Another post-translational inhibitory mechanism for ALDH-2 is its hyperacetylation due to inhibition of the redox-sensitive deacetylase, sirtuin 3 (294), for example, by 4-HNE (95). Different phosphorylation sites were reported for ALDH-2, among them were activating and deactivating ones. The most interesting phosphorylation modifications may be those by redox-sensitive JNK- (inactivating) and PKCɛ- (activating) dependent ones (294). A cross talk between AMPK and ALDH-2 was also discussed (194). A human study revealed that phosphatidylinositol 3-kinase (PI3K)-mediated phosphorylation of ALDH-2 confers the cardioprotective phenotype observed in females (175).
Additionally, in 2008, the group of Mochly-Rosen identified another important therapeutic potential of ALDH-2 consisting of the anti-ischemic protection in a model of MI (37), which was later confirmed by others (193, 301). These authors observed that infarct size in mice was reduced by ALDH-2 activation, whereas ischemic damage was increased in response to ALDH-2 inhibition (e.g., by GTN treatment). More recently, it was shown that pharmacological activation of ALDH-2 by the activator, Alda-1, can reduce infarct size in a model of MI (303) and improve cardiac function in heart failure (108), whereas inhibition/deficiency of ALDH-2 in experimental diabetes (317) or pressure overload (289) was associated with cardiac dysfunction. Likewise, ALDH-2 transgenic mice were protected from autophagy and cardiac dysfunction in a type 1 diabetes mellitus model (123), and ALDH-1×ALDH-2 double knockout mice displayed a Parkinson's phenotype with neurodegeneration and motor dysfunction (333).
4. Dihydrolipoic acid and thioredoxin as physiological reducing factors of ALDH-2
Addition of dihydrolipoic acid only partially restored ALDH-2 activity in mitochondria upon challenges with authentic peroxynitrite or GTN (323). Likewise, treatment of mitochondria with antimycin A, a complex III inhibitor, caused mitochondrial superoxide and hydrogen peroxide formation-triggered partial irreversible inhibition of ALDH-2, which was not completely reversible by adding dihydrolipoic acid. The concept of dihydrolipoic acid-mediated protection was also confirmed in a human study (330). Based on these observations, we postulated a redox regulation of ALDH-2 activity by GTN and RONS (mainly peroxynitrite) as well as irreversible inhibition by formation of sulfonic acid by oxidizing species (58, 66, 323). In 2011, we also characterized the role of the thioredoxin/thioredoxin reductase system for the repair/reduction of ALDH-2 (254).
5. Molecular mechanism of ALDH-2 bioactivation of organic nitrates via its reductase activity
In a first step, ALDH-2 catalyzes the formation of a hypothetical thionitrate intermediate (99) from the reaction of GTN, PETN, and its trinitrate, PETriN, with a reactive thiol group at the active site of the enzyme generating the denitrated metabolite (1,2-GDN, PETriN, or PEDN) (Fig. 7) (67, 324). The thionitrate is stabilized by spontaneous nucleophilic attack of a second neighboring cysteine thiol group by formation of a disulfide bridge and release of nitrite. Another possibility could be the direct generation of •NO by the thionitrate (99, 176), but it is unclear to this date whether this process requires transition metals or not (19).
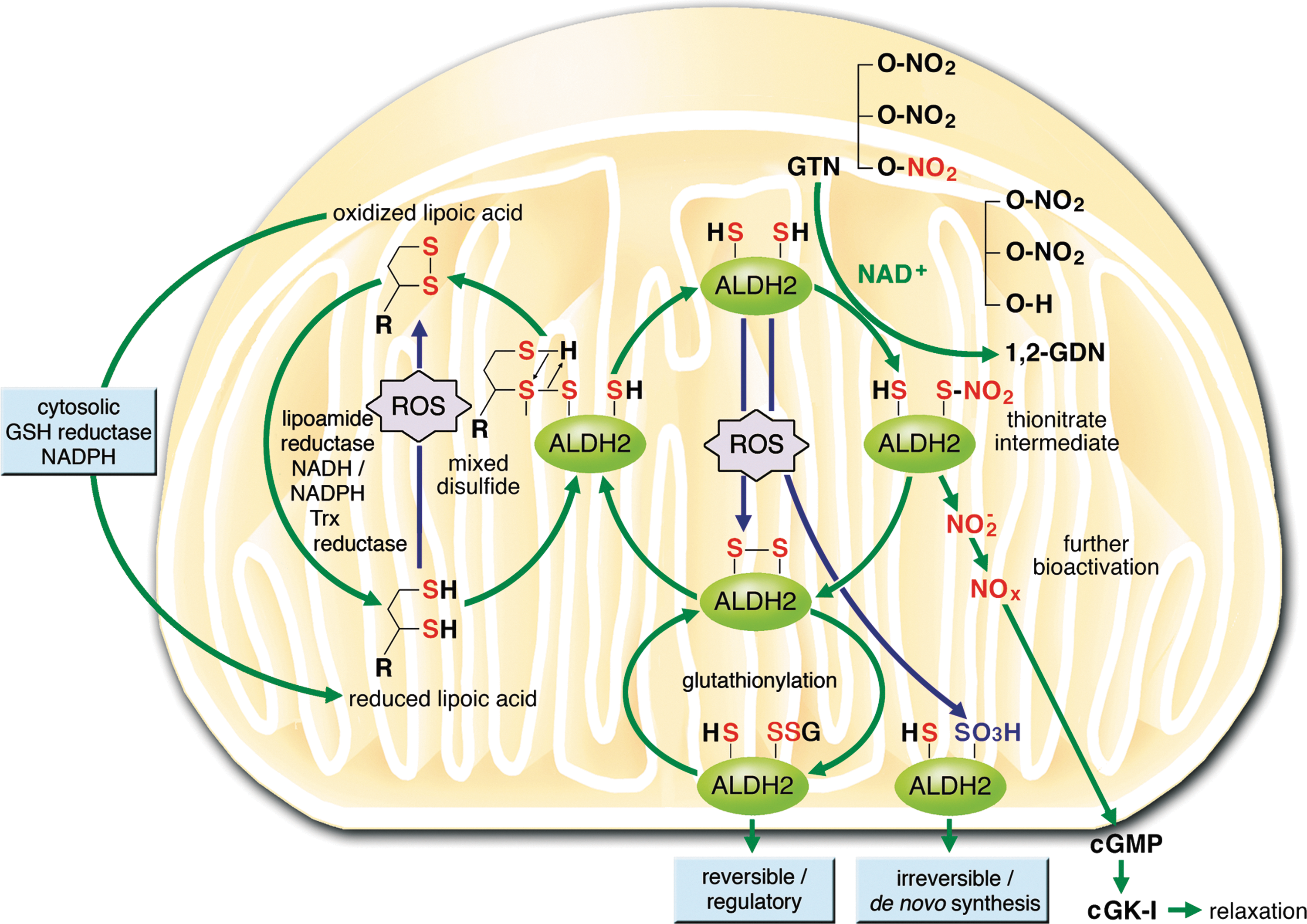
Meanwhile, the thionitrate intermediate was confirmed at the molecular level by resolving the X-ray structure of the protein with the thionitrate group at the active site (Fig. 7) (176). The enzymatic activity can be further regulated by reaction of the disulfide bridge with glutathione, which forms quite a stable adduct that can be detected with a specific antibody against GSH. This regulatory process is called S-glutathionylation and is probably due to a narrow active site of the enzyme prohibiting the attack of a second GSH molecule, which would result in GSSG formation and the reactivated enzyme.
6. Impact of organic nitrate-induced ROS formation on ALDH-2 activity
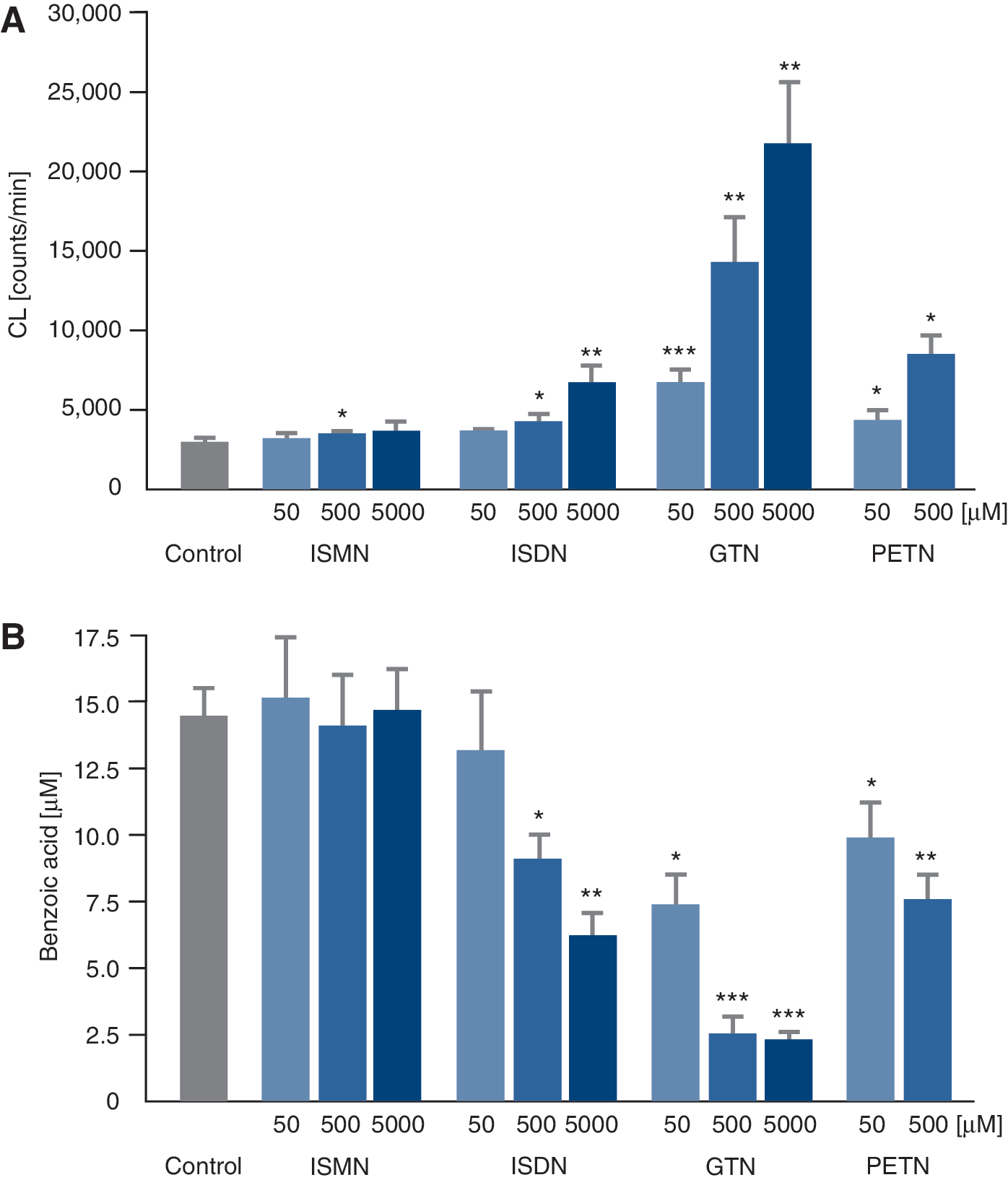
There is also irreversible inhibition of the enzyme, probably via formation of sulfonic acid groups by oxidants such as superoxide or peroxynitrite (323), which requires de novo synthesis of the ALDH-2. In cells/mitochondria with intact redox state, the disulfide bridge (or sulfenic acid −SOH upon reaction with peroxynitrite) is reduced by dithiol compounds such as the mitochondrial dihydrolipoic acid or synthetic compounds such as DTT (40, 323). In isolated mitochondria, there is a direct correlation between the induction of ROS formation by different organic nitrates and their ability to inhibit ALDH-2 in the same samples (Fig. 8) (61). Similar inhibition of the enzyme was observed upon treatment of the isolated ALDH-2 enzyme or isolated mitochondria with peroxynitrite or •NO/superoxide-generating systems and, to a lesser extent, with hydrogen peroxide (61, 254, 323). It was striking that ISMN induced no mitochondrial ROS formation, ISDN some, PETN some more, and GTN the most (Fig. 8).
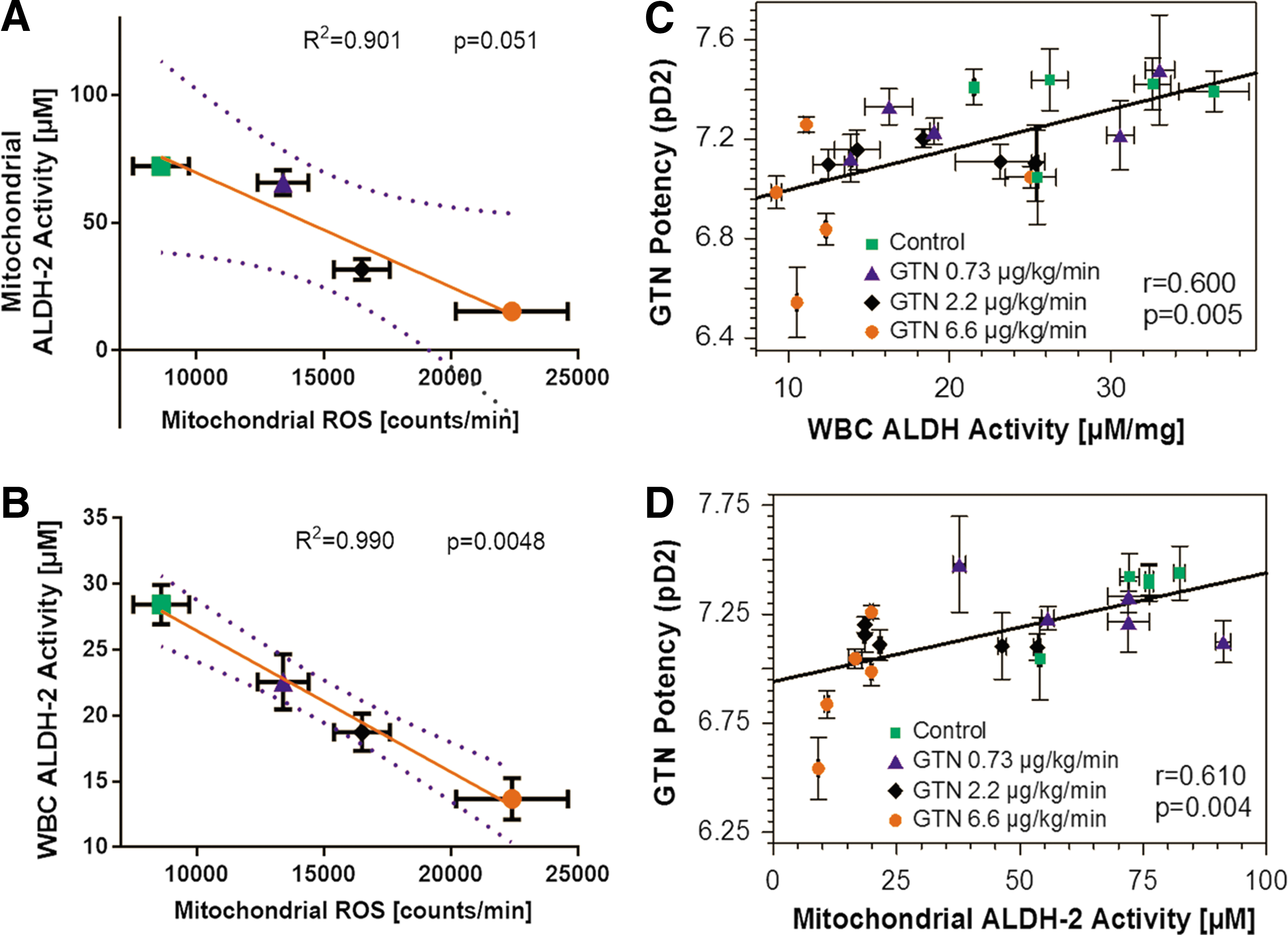
Likewise, the induction of mitochondrial ROS formation and inhibition of ALDH-2 depended on the GTN dose that was applied in vivo (Fig. 9) (330). mtROS formation correlated well with the ALDH-2 activity in isolated mitochondria as well as white blood cells (WBCs). Vice versa, ALDH-2 activity in isolated mitochondria as well as WBCs showed a clear correlation with the vasodilator potency of GTN, all of which show a GTN dose-dependent pattern (Fig. 9) (330).
7. Purified ALDH-2 as a peroxynitrite synthase in the presence of GTN
The purified ALDH-2 enzyme itself produces RONS in the presence of GTN (331). As an explanation for this RONS formation, the conformational change was proposed as taking place upon the binding of NAD+ to the enzyme. Accordingly, the E268Q mutation reduced the binding affinity of NAD+ due to the absence of glutamic acid and thereby suppressed RONS formation by the enzyme in the presence of GTN (331). Interestingly, the nitrate reductase activity of ALDH-2 was significantly affected by E268Q mutation.
Nitric oxide formation from wild-type ALDH-2 and GTN could only be observed in the presence of superoxide dismutase (SOD), providing evidence for involvement of superoxide in the breakdown of •NO. In contrast, the E268Q variant displayed significant •NO formation in the absence of SOD, indicating that superoxide formation in the variant was less pronounced. It should be noted that high GTN concentrations (>10 μM) were used for the detection of •NO. The dehydrogenase and esterase activities are almost absent in the E268Q mutant, whereas basal nitrate reductase activity is similar to the one in wild-type ALDH-2. However, nitrate reductase activity of the wild-type enzyme was increased 7-fold upon addition of NAD+ (by a structural change) and this increase was completely lost in the E268Q (18).
Replacement of a cysteine residue at the active site by a serine group (C302S) resulted in complete loss of all three enzymatic activities (dehydrogenase, esterase, and reductase) and a complete suppression of the 1,2-GDN, as well as •NO formation in the presence of GTN (331).
Previously, we could demonstrate that purified human ALDH-2 was significantly deactivated by GTN, but to a minor extent by PETN (Fig. 10). In addition, the RONS formation by ALDH-2 was significantly less pronounced in the presence of PETN compared with GTN (Fig. 10).
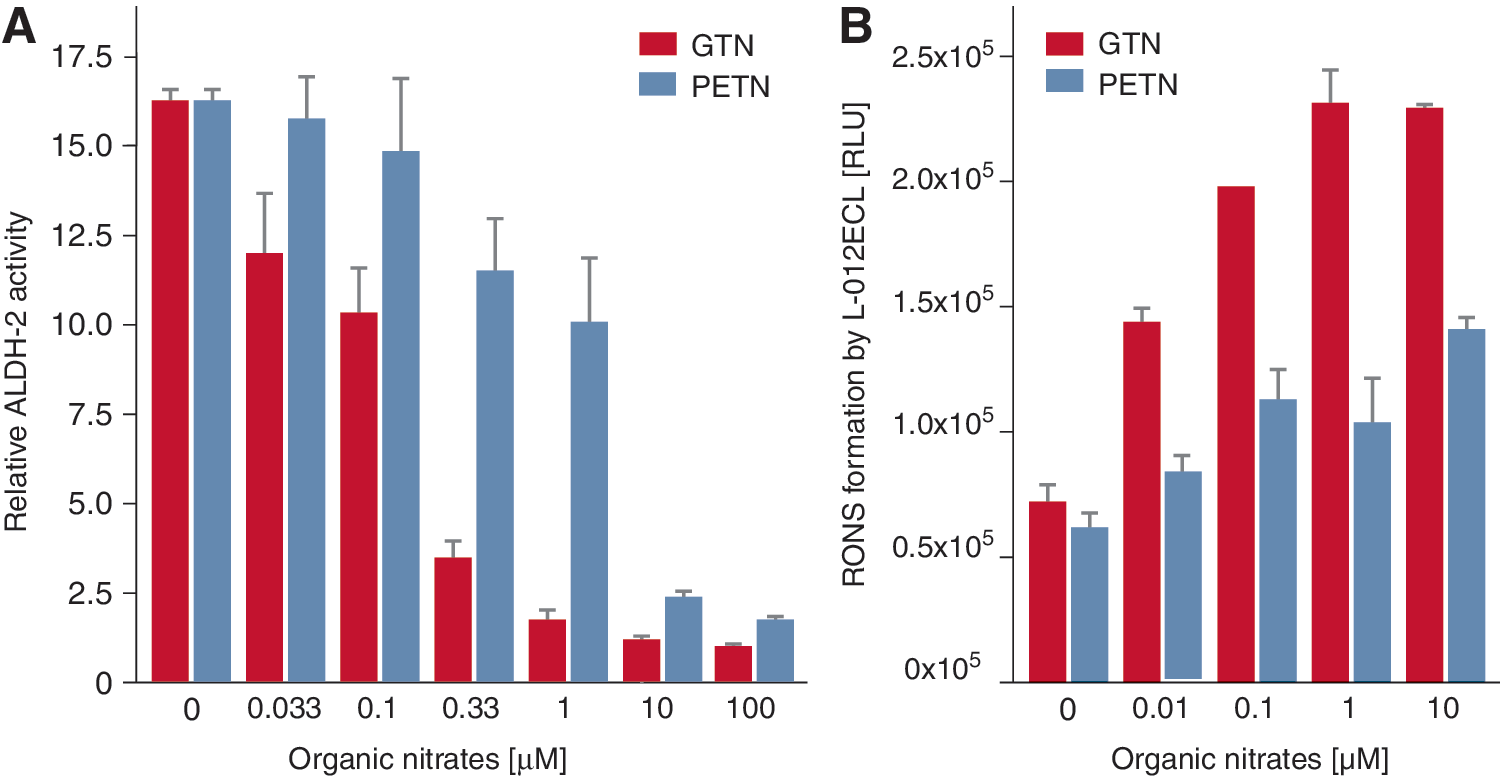
These observations provide an attractive additional explanation for the tolerance-devoid and beneficial effects of PETN under chronic therapy, which were previously defined to rely solely on induction of HO-1 and ferritin (250, 252, 324), as well as on the controlled and slow uptake of the drug upon oral treatment (164, 330). If PETN really prevents inactivation of its own bioactivating enzyme, ALDH-2, then this provides an additional explanation for the lack of tolerance development by this particular organic nitrate.
The observation that the purified ALDH-2 enzyme generates RONS in the presence of GTN (and to a minor extent also PETN) suggests that ALDH-2 itself could represent a source of GTN-dependent RONS formation. As proposed by Mayer and coworkers, ALDH-2 could represent a relevant peroxynitrite synthase under in vivo treatment with GTN (331). However, this conclusion is at variance with previous data from our group and others, indicating that ALDH is rather an RONS scavenger and prevents degradation of •NO since knockdown of ALDH-2 in cultured cells increased RONS (306). In a subsequent study, it was demonstrated in ALDH-2 knockout mice that deficiency in ALDH-2 results in increased RONS levels under chronic GTN therapy (326).
GTN-triggered RONS formation from purified ALDH-2 rather seems to be an artificial side effect as frequently observed with simplified, purified enzymatic systems (e.g., due to lack of essential cofactors or antioxidants). The ALDH-2 independent formation of RONS by GTN, mainly by direct superoxide/peroxynitrite formation by the mitochondrial respiratory chain and RONS-triggered activation of secondary sources of ROS, is discussed in detail in the Increased NADPH oxidase activity and Oxidative stress impairs GTN biotransformation sections. The differential effects of GTN and PETN on ALDH-2 activity are of great interest for the research field of nitrate tolerance (221, 303) and deserve further investigation. Due to the reported cardiac protection by ALDH-2, this research might also have significant impact for the field of heart disease (31, 37, 108, 163, 193).
B. The low-potency (affinity) pathway for bioactivation of GTN—low-molecular-weight systems and enzymatic systems of nitrate bioactivation besides ALDH-2
The low-potency pathway leads to formation of measurable amounts of •NO in vascular tissues in vivo (218) and in vitro (158). Therefore, •NO is a vasoactive principle of GTN applied at higher concentrations. Previous studies that focused on the identification of enzymes and/or low-molecular-mass factors, which could generate •NO from GTN, identified cysteine, N-acetylcysteine, and thiosalicylic acid (85, 277), as well as deoxyhemoglobin, deoxymyoglobin (16), cytochrome P450 (CYP) (200), and xanthine oxidase (XO) (76). While •NO formation by deoxyhemoproteins and XO will be confined to tissues of low oxygen tension (hypoxia), the CYP pathway is likely to account for •NO formation from GTN accumulating in high concentrations in the liver, lung, and kidney (218).
Since the nonenzymatic reaction of GTN with thiols requires high concentrations of these thiols (mM range) as well as GTN (μM range), this reaction may lack physiological significance. Optimized systems observed activation of purified sGC by cysteine (1 mM) and GTN in the low micromolar range, a quite low concentration, but still 100- to 1000-fold less potent than vasodilation of isolated vessels by GTN (117).
Another low-molecular-weight (but also low affinity) pathway was postulated for the ascorbate system, although activation of purified sGC required at least 10 μM GTN in the presence of ascorbate (10 mM) (166). It should be noted that inorganic nitrite was even more effective in this system and evoked more sensitive activation of sGC in the presence of ascorbate, which could be of relevance for the involvement of intermediary formation of inorganic nitrite during GTN bioactivation (see section II.A.).
When used in vivo, ascorbate and thiol compounds often significantly improve organic nitrate action or efficiently prevent development of tolerance. However, it is unclear whether their action is based on direct bioactivation of the organic nitrates or on secondary antioxidant effects on nitrate-induced RONS formation since all of them are potent antioxidants (see below for further details).
1. Cytochrome P450 enzymes
Induction of hepatic CYP isoforms by glucocorticoids and other agents increases •NO and cGMP formation from GTN in the liver, lung, and kidney (17), whereas a 3-day infusion of GTN decreases hepatic CYP expression (210, 211). A similar effect was not observed with sodium nitroprusside infusion, indicating that the decrease of CYP expression may specifically depend on GTN metabolism by CYP. In addition to its function in hepatic metabolism of GTN, CYP is also a favorable candidate for catalyzing •NO formation from GTN in vascular tissues exposed to high concentrations of GTN (Fig. 6).
Different isoforms of CYP have been shown to account for •NO release from 10 μM GTN in isolated blood vessels of human (208) and animal origin, the most active CYP isoform in rats being CYP1A2 (209). Induction of vascular CYP1A2 by a 5-day treatment of rats with i.p. acetone strongly increased •NO release from GTN by isolated veins, abdominal arteries, and thoracic aorta, whereas downregulation of CYP1A2 by 48 h of GTN infusion decreased vascular •NO formation (209).
In cultured rat lung fibroblasts (RFL-6 cells), the inhibitor of cytochrome P450, proadifen (0.1 mM), decreased cGMP stimulation by GTN (1–100 μM) by up to 81% (276). cGMP stimulation by ISDN was inhibited to a similar degree under these conditions. However, proadifen did not affect cGMP stimulation by SNP that spontaneously releases nitric oxide. The bioactivation pathways for ISMN and ISDN (as well as other di- and mononitrate metabolites from GTN and PETN) are less carefully defined, but it is assumed that cytochrome P450 enzymes contribute to vasodilation by bioactivation of those less potent nitrates when reaching high enough concentrations in tissues (Fig. 6) (208, 210). However, another group reported differences in P450-mediated organic nitrate bioactivation, identifying GTN, but not ISDN, as a substrate (218).
According to mechanistic studies of the Zweier group, organic nitrite is the initial product in the process of cytochrome P450-mediated organic nitrate activation and is the precursor of •NO and nitrosothiols, serving as the link between organic nitrate and sGC activation (183). These findings on the organic nitrate bioactivation process show similarities to recent reports on inorganic nitrite-mediated protection from ischemic damage (32) as well as formation of •NO from inorganic nitrite (32).
2. Xanthine oxidoreductase
XO was identified as an ISDN and ISMN-metabolizing enzyme with higher turnover with xanthine instead of NADH as the source of electrons (76). However, most of the investigations on nitrate bioactivation by purified enzymes or enriched cellular compartments were performed using high suprapharmacological concentrations of the organic nitrates. Therefore, it is strongly recommended to repeat these studies using clinically relevant concentrations of the drugs, even in vivo animal or at least cellular models. It was suggested that inorganic nitrite is the initial product in the process of XO-mediated organic nitrate biotransformation and serves as the link between organic nitrate and sGC activation to provide the precursor of •NO and nitrosothiols (182). Inorganic nitrite formation was also suggested as an intermediate during ALDH-2-mediated bioactivation of GTN (41).
It is well established that microorganisms such as bacteria and fungi display enzymatic activities, reducing inorganic nitrate. Therefore, bioactivation of inorganic nitrate by the microorganisms in the mouth and the gastrointestinal tract makes them likely candidates for the bioactivation of dietary inorganic nitrate (192). For mammalian tissues or homogenates, a nitrate-reducing activity has been postulated for several years. In 2008, xanthine oxidoreductase was identified to display nitrate-reducing activity in vitro, ex vivo, and in vivo (148). The exact extent of contribution of the bacterial and mammalian pathways to overall conversion of inorganic nitrate to nitrite, and subsequently bioactivation of nitrite to the vasodilator nitric oxide, is still a matter of debate.
3. Glutathione-S-transferase
Early observations with isolated glutathione-S-transferase (GST) isozymes (or at least enriched subcellular fractions) suggested that different GTN metabolic pathways are linked to these isozymes (178). Based on results obtained with pharmacological inhibitors for the GSTs, these enzymes mediate GTN bioactivation, leading to guanylyl cyclase activation and relaxation of vascular smooth muscle (247, 291). More recent studies suggest that both GST and ALDH-2 are involved in GTN action, while ALDH-2 plays a major role, and the change of calcitonin gene-related peptide contents closely correlates with the bioactivation of GTN (348). Overexpression of GST isozymes can protect endothelial cells and smooth muscle cells against oxidative stress associated with GTN (and markedly alter cellular responses to repeated doses) or tolerance (318). By manipulating GSTs, physiological tolerance to GTN may be diminished or eliminated (149).
4. Glyceraldehyde-3-phosphate dehydrogenase
The reaction of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) with GTN yielded inorganic nitrite and glyceryl-1,2-dinitrate (284). The reaction was inhibited by the thiol-modifying compound, disulfiram, whereas reducing agents such as DTT or tris(2-carboxyethyl) phosphine promoted the reduction of GTN. In the absence of reducing equivalents, the enzyme was inhibited in the presence of GTN, in accordance with previous reports that GAPDH is easily inhibited by S-nitrosation (213) or oxidation (295).
All of these observations suggest a thiol-dependent bioactivation process of GTN by GAPDH. Although the GTN concentrations used in this study were again suprapharmacological (in the upper μM range), the high abundance of GAPDH in cells and tissues may confer some clinical relevance to this enzymatic reaction of GTN. This would also be in accordance with the previously described observation that exposure of intact human erythrocytes to GTN or its metabolite, nitrite, causes an immediate spike in ATP release (104), leading to a powerful vasodilatory response by binding to purinergic receptors on the endothelium, which stimulates •NO production by endothelial nitric oxide synthase (eNOS).
5. Other ALDH isoforms
Besides ALDH-2, cytosolic aldehyde dehydrogenase (ALDH-1) in the presence of GTN also evoked activation of purified sGC (measured by cGMP formation), although the potency was 30-fold lower compared with ALDH-2 (19) and therefore is likely to contribute to actions of GTN in tissue or cells at suprapharmacological concentrations. For purified cytosolic isoforms, ALDH1a1 and ALDH3A1, it was even shown that they not only bioactivate GTN but also ISMN and ISDN, again at suprapharmacological concentrations (186, 315).
III. Side Effects of Chronic Nitroglycerin Therapy
A. Nitrate tolerance
The clinical introduction of organic nitrates at the end of the 19th century was soon followed by the observation that the hemodynamic and clinical effects of GTN, ISMN, and ISDN invariably wane upon continuous therapy. In the setting of CAD, nitrate tolerance has been demonstrated as the loss of effects on treadmill walking time and time of onset of angina (262). In CHF, it has been described as the loss of hemodynamic effect of the administered nitrate (81), and in hypertension, it is evident as the rapid loss of the hypotensive effects of these drugs.
Rather controversial data have been reported for the antiplatelet effects of GTN. Preclinical studies demonstrated that tolerance is associated with a paradoxical activation of platelets (87), and another report showed that prior exposure to GTN, even in very low doses, induces tolerance to the antiaggregatory effects of the drug (87). In contrast, other studies in both rats and humans have shown that platelet responsiveness is preserved, despite hemodynamic tolerance (24, 138).
Another issue is the so-called nitrate resistance defined by the reduced effectiveness of organic nitrates in the setting of cardiovascular disease (and therefore increased oxidative stress). Nitrate resistance shares the same features with nitrate tolerance, but impaired nitrate effectiveness is not induced by prior nitrate therapy. For instance, McVeigh et al. reported reduced hemodynamic effects in diabetic patients and (as mentioned above) that GTN-induced inhibition of platelet aggregability is blunted in patients with CAD or diabetes (204). To date, it remains unclear whether these different forms of reduced responsiveness to nitrates share common mechanisms (e.g., dysfunction of downstream •NO signaling pathways) or should rather be considered as two distinct entities. The mechanism of nitrate bioactivation is a highly complex process.
However, the mechanism of the development of nitrate tolerance is even more complex since it involves neurohormonal counter-regulation, expansion of plasma volume (collectively classified as pseudotolerance), and intrinsic vascular processes defined as true tolerance (Table 2).
GTN, glyceryl trinitrate.
1. Pseudotolerance
The vasodilation evoked by intravenous, oral, and transdermal nitrate therapy causes the release of catecholamines (260), an increase in catecholamine rates (298) and plasma vasopressin levels (228, 260,) and increases in plasma renin activity (228, 260) and aldosterone levels (228, 260). Such activation of neurohormonal vasoconstrictor forces has been demonstrated in patients with CAD, patients with heart failure (257), and healthy subjects (260). In line with these data, long-term continuous transdermal GTN therapy has been associated with altered autonomic neural function, including impaired baroreflex activity and prevalence of sympathetic to parasympathetic tone in the regulation of heart rate (112).
Additionally, in both animal and human studies, long-term therapy with organic nitrates was associated with increased sensitivity to a receptor-dependent vasoconstrictor such as serotonin, phenylephrine, angiotensin II, and thromboxane A (133, 226). A marked increase in intravascular volume, secondary to the transvascular shift of fluid and/or to aldosterone-mediated salt and water retention (228, 260), has also been observed in patients treated with GTN. Although these changes could attenuate the preload effect of GTN, evidence suggests that these mechanisms are not sufficient to fully explain the loss of nitrate effectiveness. For instance, there is a difference in the time frame of neurohormonal activation, plasma expansion, and development of tolerance (228); furthermore, studies testing the effects of diuretics, beta-blockers, or angiotensin II converting enzyme (ACE) inhibitors did not invariably reverse or prevent tolerance. Thus, although the possible prognostic implications of these changes need to be acknowledged, other mechanisms of tolerance and a hypothesis that explained all these changes had to be found.
2. True vascular tolerance mechanisms
True vascular tolerance is thought to be due to an inability of the vascular tissue to respond to organic nitrates in the absence of the neurohormonal environment. Thus, vessels from animals pretreated with nitrates demonstrated a blunted reaction in response to, for example, GTN. In the mid 80s, true vascular tolerance mechanisms comprised impaired GTN biotransformation, intracellular SH-group depletion, a desensitization of the •NO target enzyme, sGC, as well as an increase in PDE activity.
The concept of nitroglycerin-induced depletion of thiol stores is mainly based on the observations by Needleman on the inhibition of thiol-dependent mitochondrial proteins in response to different organic nitrates (242). This group also reported the importance of sulfhydryl groups for the enzymatic function of essential proteins and linked GTN-induced inhibition of these enzymes and loss of vasodilation to the depletion of thiol groups (146, 243). Later, the loss of cellular thiol groups in response to nitroglycerin and the excessive formation of S-nitrosothiols as a potential mechanism of nitrate tolerance (impaired bioactivation) were put forward by Feelisch et al. and Fung et al. (85, 101).
Subsequent studies provided support of this concept by the prevention of nitrate tolerance and associated side effects by sulfhydryl donors such as N-acetylcysteine (see also the Sulfhydryl group donors section) (23, 106). However, it will be almost impossible to figure out whether direct antioxidant effects of the thiol compounds, improved enzymatic bioactivation or direct reaction of the thiols with GTN to generate •NO, account for the prevention of nitrate tolerance (100).
First evidence for a contribution of PDE enzymes to nitroglycerin-induced tolerance was based on the observation that cGMP turnover was changed in nitrate-tolerant vessels (12). In addition, the unspecific PDE inhibitor, dipyridamole, showed synergistic effects on vasodilation and other hemodynamic effects (6). The first direct proof for GTN-mediated induction of the expression of PDE (isoform 1A1) contributing to vascular nitrate tolerance and cross-tolerance against endogenous vasodilators dates back to the year 2001 (156). In this study, •NO/cGMP-mediated vasodilation was desensitized and Ca2+-mediated vasoconstriction was supersensitized, all of which were ameliorated by the PDE1-specific inhibitor, vinpocetine. For more details on PDE inhibition in nitrate tolerance, see the PDE inhibitors section.
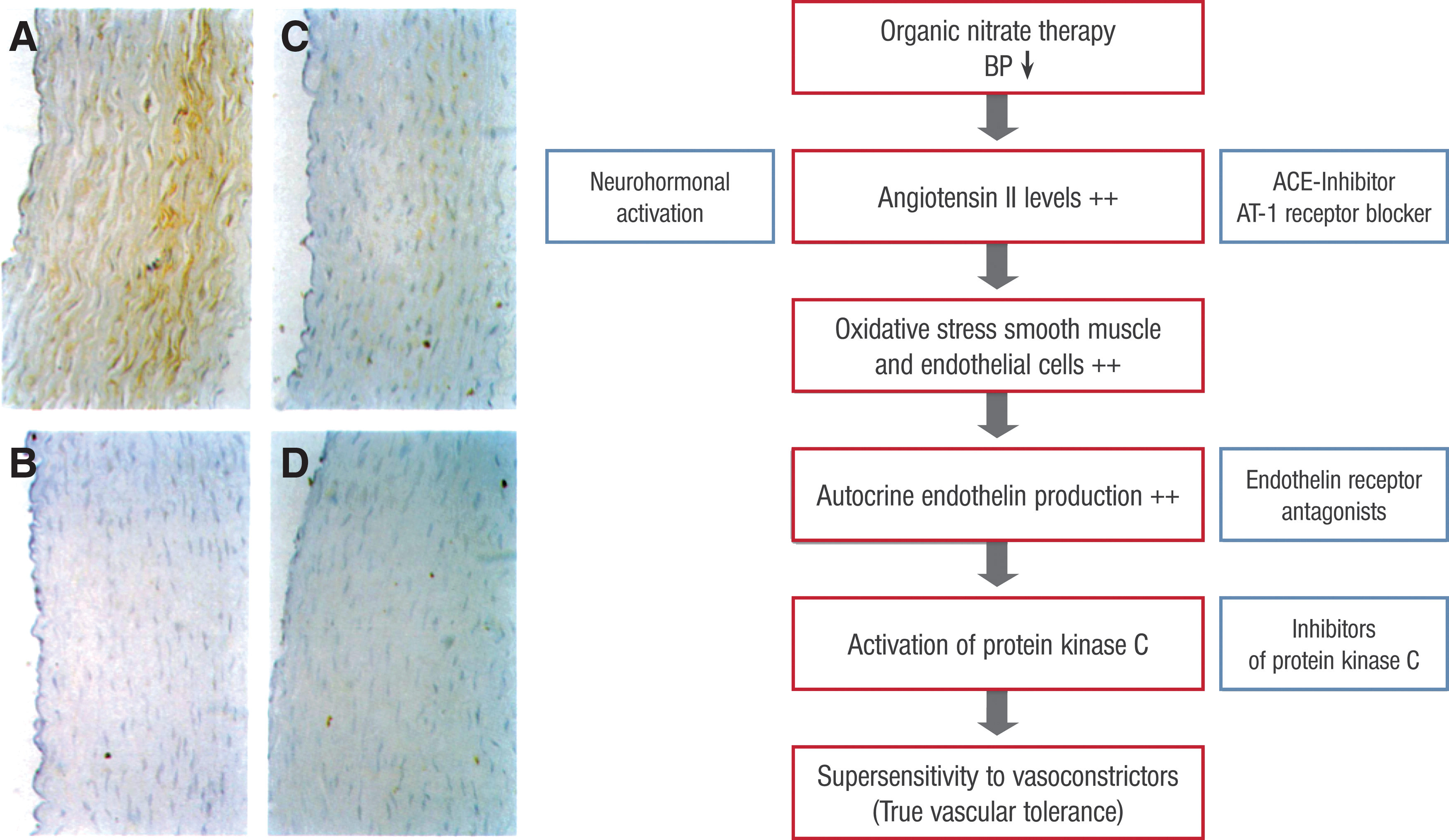
As depicted in Table 2, within the last 30 years, several additional mechanisms have come into play, being responsible for the true vascular tolerance phenomenon. These include an increase in oxidative stress in the tolerant vasculature as well as an increase in sensitivity to vasoconstrictors such as serotonin, phenylephrine, angiotensin II, and thromboxane A (133, 226), mainly due to increased autocrine endothelin-1 (ET-1) production (172, 221, 226, 253).
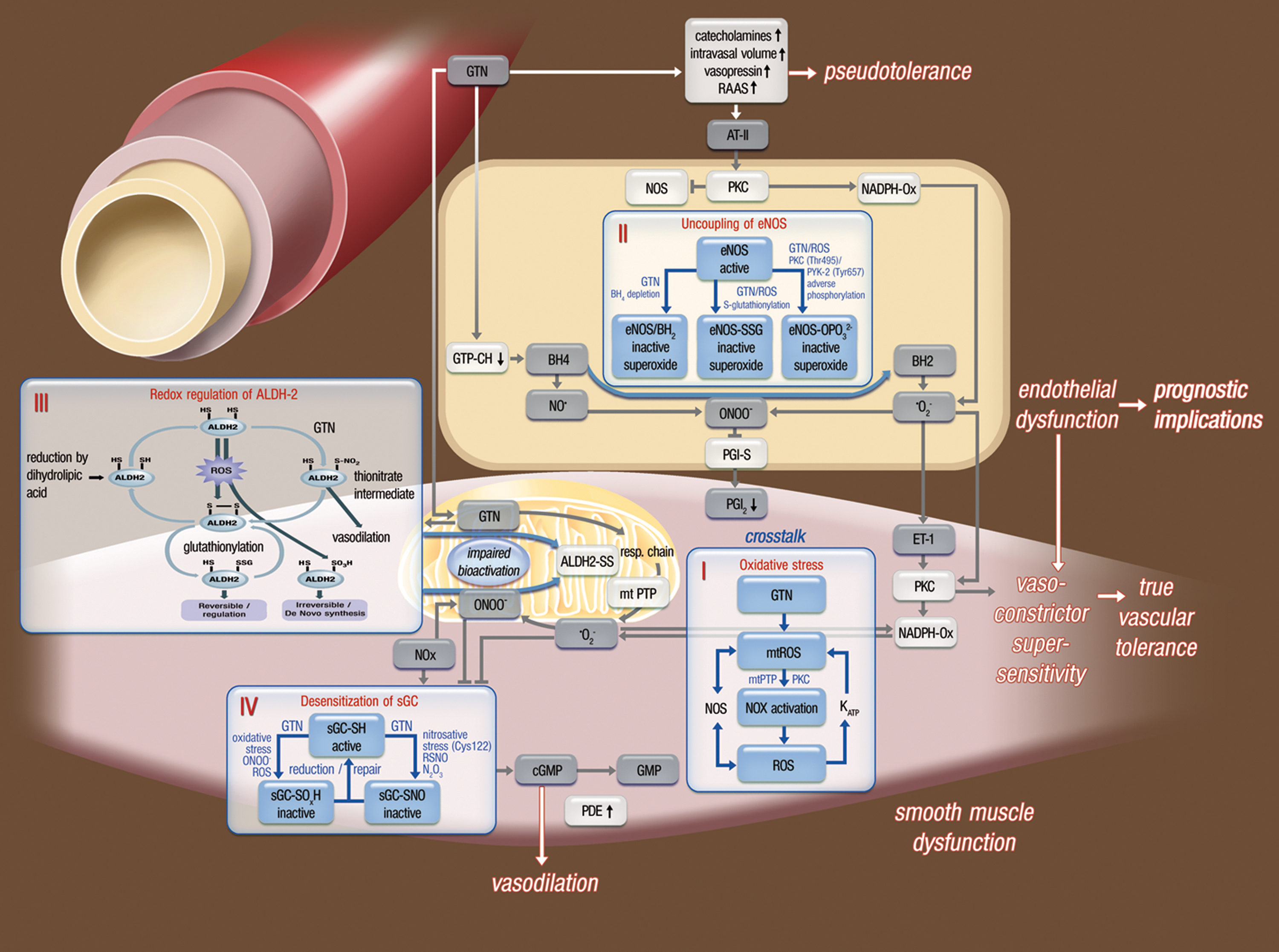
There is a growing body of evidence to suggest that increased RONS (superoxide and peroxynitrite) production in response to GTN therapy may be actually responsible for almost all true tolerance phenomena, such as decreased sGC responsiveness and impaired GTN biotransformation by the ALDH-2 simply due to oxidation of critical SH-groups of these enzymes (67, 222). There is also strong evidence that RONS production contributes to the induction of ET-1 (151, 152). It is also clear that the group of organic nitrates is quite heterogeneous. Nitrates are considerably different in regard to their mechanism of vasodilation, tolerance development, increases in oxidative stress, and the development of endothelial dysfunction (110, 238).
a. GTN increases oxidative stress and causes tolerance
In 1995, we proposed a new molecular mechanism for GTN tolerance and cross-tolerance (236). Critical to this concept was the evidence that the bioavailability of ROS (superoxide) in tolerant vessels mounted to about twice that in controls and that this abnormality was corrected by the addition of liposomal SOD, which dismutates O2 •− to H2O2 and oxygen (236). Subsequently, we demonstrated that GTN treatment stimulates the vascular (and particularly endothelial) production of peroxynitrite (137), a highly reactive intermediate generated from the rapid diffusion-limited reaction of •NO with O2 •−. Evidence of GTN-induced increased ROS production in humans was then obtained ex vivo in arterial segments and in blood or platelets taken from patients rendered tolerant to GTN (42, 204, 268, 281).
GTN tolerance was also associated with increased markers of free radical-induced lipid peroxidation such as cytotoxic aldehydes and isoprostanes (150), esterified 8-epi-prostaglandin F2α (PGF2α) (201), and with a mild reduction in the responsiveness to the •NO donor SNP in healthy volunteers (115), which might also be compatible with ROS-mediated interference with •NO signaling. From these findings, we proposed the existence of a unifying hypothesis, which, founded on the concept of GTN-induced increased oxidative stress, could be compatible with the multiple different observations associated with long-term nitrate therapy (115, 223, 237). Several sources of oxidative stress have been discussed to significantly contribute to nitrate tolerance such as NADPH oxidases (96, 172, 226) and the mitochondria respiratory chain (56, 64, 304).
There is an ongoing discussion on the question whether low-dose GTN (in the clinical setting) induces vascular oxidative stress and endothelial dysfunction. Several groups observed these side effects at lower doses of GTN in animal studies (2.1–2.7 [rabbit], 9.5, 10.5, and 14.4 [rat] mg/kg/day) (141, 231, 232, 236, 304, 319, 327) and even in clinical trials using excess vascular tissues from bypass surgery upon GTN treatment (0.72 mg/kg/day for 24–48 h) (136, 281). In contrast, only a trend of impaired endothelium-dependent relaxation (calcium ionophore A23187) was observed in patients when treated with a 3.5-fold lower dose and shorter exposure of GTN (0.21 mg/kg/day for 24 h), but the authors still reported on GTN-induced vascular oxidative stress (268).
In addition, numerous clinical studies reported on impaired acetylcholine-dependent vasoreactivity, or flow-mediated dilation (FMD), in the forearm and coronaries of GTN (0.2 mg/kg/day)-treated individuals and improvement of these adverse effects by the antioxidants, vitamin C and the eNOS cofactor BH4 (34, 109, 114, 189), also supporting GTN-induced cross-tolerance to endothelium-dependent vasodilators and a role for oxidative stress. Very early studies of Murad and coworkers reported on GTN-induced endothelial dysfunction in rats (200 mg/kg/day) (212), whereas Bassenge and colleagues did not observe changes in acetylcholine- or FMD-mediated intra-arterial dilations in dogs (2.2 mg/kg/day) (299). Likewise, the contribution of oxidative stress to the development of GTN-induced nitrate tolerance was questioned by studies in ascorbate-deficient animals (332, 336).
In summary, these data indicate that the effect of chronic GTN therapy on endothelial function and vascular oxidative stress shows appreciable variations with respect to different species, vascular beds, and dose and duration of GTN treatment and therefore requires cautious interpretation. Higher doses in rodents might be justified by the different metabolism and resulting accelerated pharmacokinetics.
b. Oxidative stress impairs GTN biotransformation
The recognition of the role of a mitochondrial enzyme in the biotransformation of organic nitrates and of a role of mitochondrial oxidative stress in the development of tolerance provided a link between only two apparently separate hypotheses (reduced bioactivation vs. ROS-mediated •NO scavenging or ROS-mediated inactivation of •NO signaling). This hypothesis is essentially based on the concept that the oxidation of thiol groups may cause inhibition of several enzymes [including ALDH-2 (61, 323) and sGC (271)] and therefore both reduced GTN biotransformation and inhibited •NO signal transduction (222). In line with this, treatment of tolerant animals with mitochondria-targeted antioxidants completely prevented or reversed GTN tolerance (83, 103), and heterozygous knockout of manganese superoxide dismutase (MnSOD+/− mice) markedly aggravated tolerance development in response to GTN (64).
These data reconcile the bioactivation and oxidative stress hypotheses and provide an interesting clinical corollary of the original observations published by Needleman and Hunter, showing that incubation with high concentrations of nitrates induced swelling of isolated cardiac mitochondria, stimulated oxygen consumption, and uncoupled oxidative phosphorylation (146, 242, 243), all data that are consistent with a mitochondrial source of nitrate-elicited ROS. In support of this concept, we were able to demonstrate that in vivo and in vitro GTN treatment increases RONS (most likely peroxynitrite) formation in isolated mitochondria and decreases bioactivation of GTN to the 1,2-GDN metabolite. Furthermore, the GTN-dependent activation of sGC was lost upon chemical depletion of mitochondrial proteins (Fig. 11) (304).
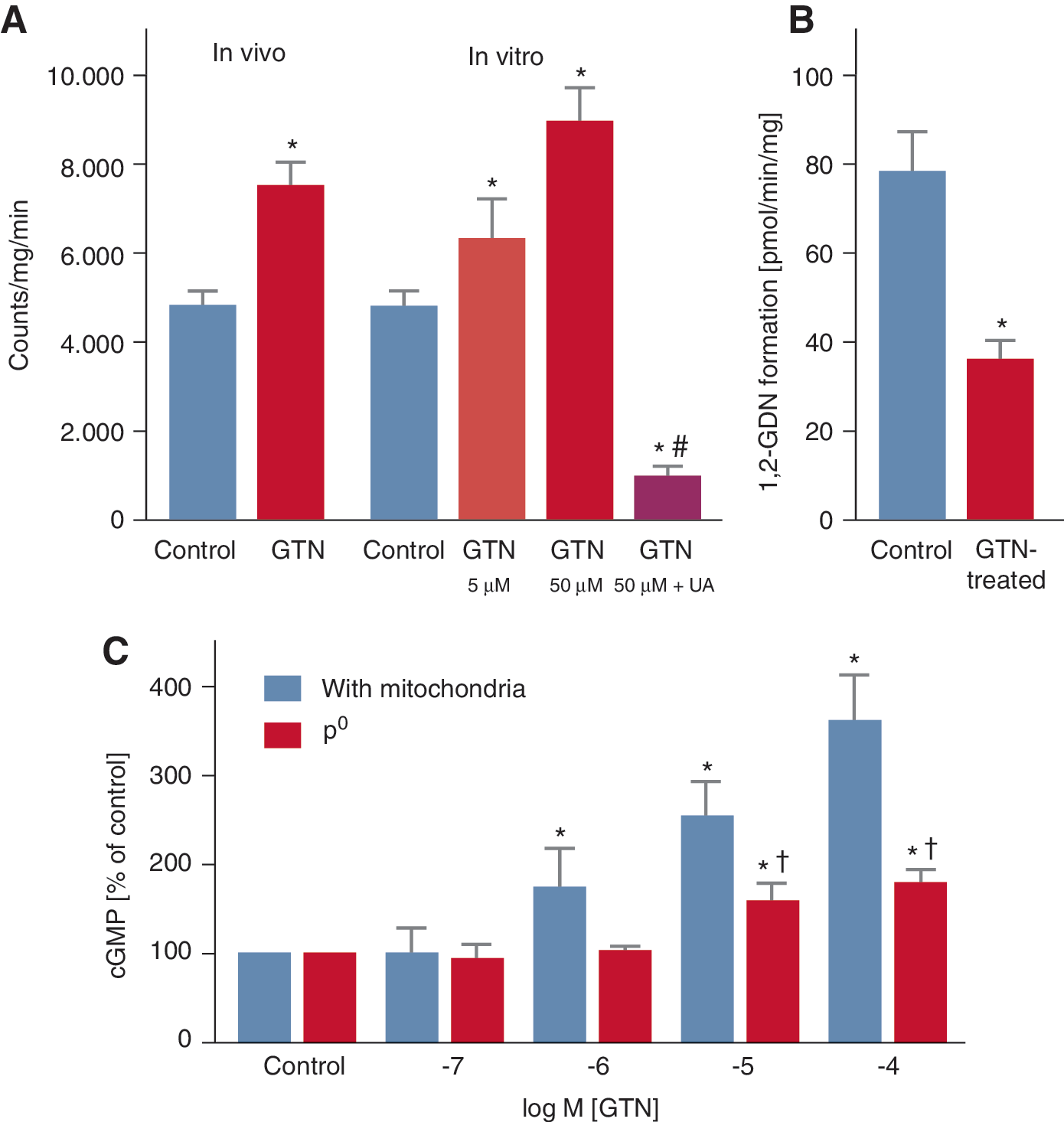
Importantly, these considerations apply to GTN tolerance, however, most likely not to ISMN or ISDN tolerance because these drugs do not undergo mitochondrial metabolism (61). Regardless of the exact mechanism by which GTN stimulates mitochondrial ROS production (e.g., premature release of partially reduced oxygen from mitochondrial complex I or III, initiation of lipid peroxidation, depolarization of mitochondrial membrane potential, mitochondrial swelling) (67), these observations support the idea that oxidative stress may directly impair GTN biotransformation either by oxidative inhibition of ALDH-2 or by depletion of essential repair cofactors such as lipoic acid (323). Recent data obtained with purified ALDH-2 provide evidence that ALDH-2 could be even a source of GTN-triggered ROS formation (254). The pathways leading to pseudo and true vascular tolerance in response to GTN are summarized in Figure 12.
A critical role for ALDH-2 in causing nitrate (GTN) tolerance was also provided in human studies. To address this issue, patients undergoing bypass surgery were treated for 48 h with the organic nitrate GTN intravenously, with a dosage recommended to treat patients with an acute coronary syndrome. In excised vessels, we observed not only a marked degree of tolerance to GTN but also cross-tolerance to the endothelium-dependent vasodilator, acetylcholine. Tolerance was associated with an inhibition in the activity of the ALDH-2 in veins and the mammary artery and also with a decreased expression of ALDH-2 (136) (Fig. 13).
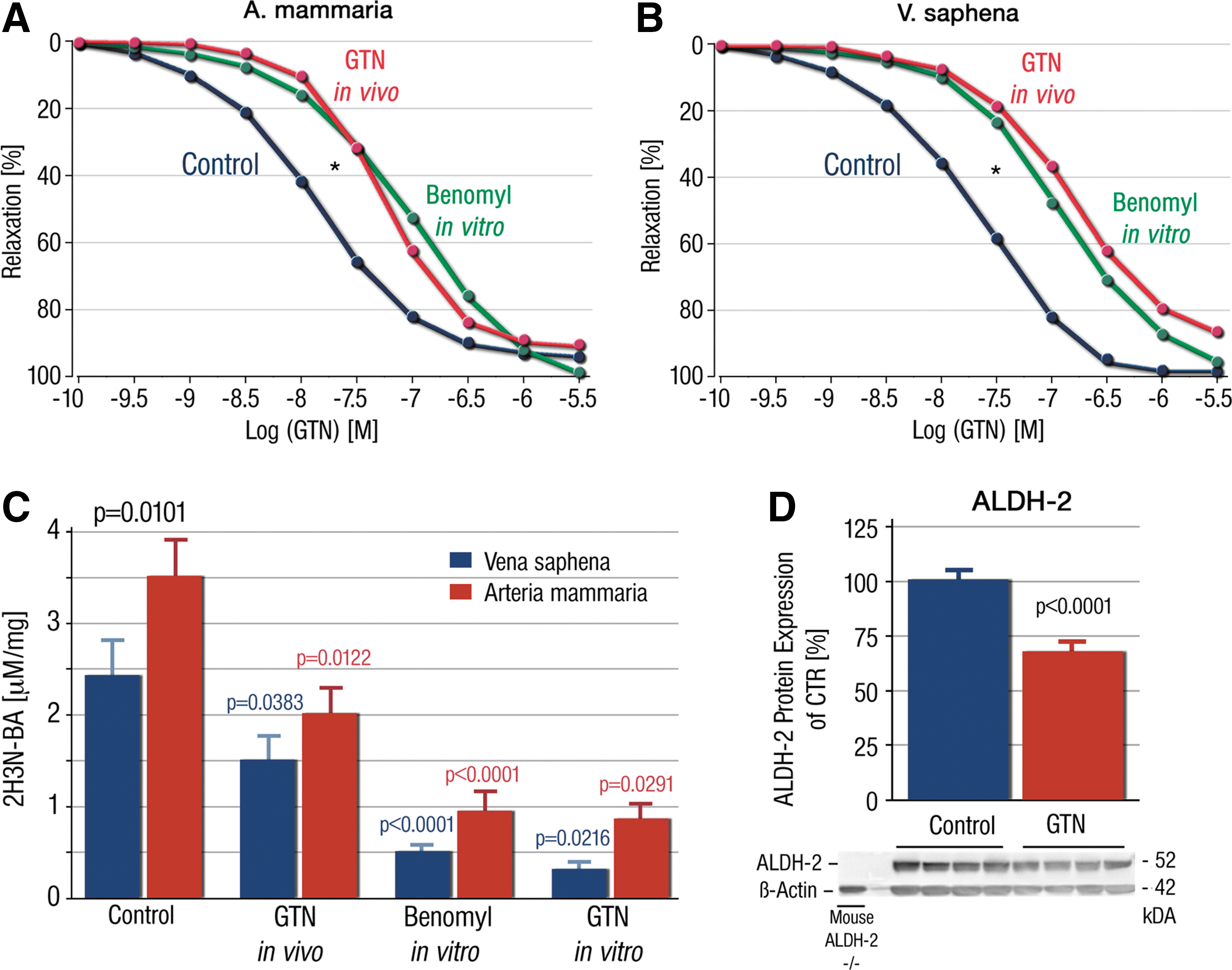
These results were later confirmed by Eschenhagen and coworkers (139). These authors also confirmed the mainly mitochondrial localization of ALDH-2 in smooth muscle cells in human veins. The latter observation is, however, at variance with a more recently proposed concept that ALDH-2 (the mitochondrial ALDH isoform) is mainly located in the cytosol of smooth muscle cells (in rat and human vascular tissue) and mitochondria-located ALDH-2 does not contribute significantly to the GTN bioactivation (21). This highly interesting hypothesis has not been confirmed by other groups so far and is at variance with the reported mitochondrial localization of ALDH-2 in human smooth muscle cells (139), loss of GTN bioactivation in mitochondria-depleted endothelial (ρ0) cells (304), mitochondrial localization of ALDH-2 in cardiac tissue (105), and the commonly observed mitochondrial localization of ALDH-2 in liver tissue (21, 105). However, there are indications that ALDH-2 may play a less pronounced role for GTN bioactivation in other species (e.g., bovine or porcine aorta) (245).
Since we and others have repeatedly shown that ALDH-2 activity was decreased in response to GTN in vivo and in vitro treatment in animals and humans, we speculated whether this decrease could be used not only as a reliable marker for nitrate tachyphylaxis (acute overload of the GTN bioactivating system) but also clinical tolerance (330). For this purpose, we isolated WBCs from the buffy coat by the dextran sedimentation method and size exclusion centrifugation by Ficoll, as previously described (60). According to our data, monocytes/lymphocytes have higher ALDH-2 activity compared with granulocytes (mainly PMN) (69). Therefore, WBCs seem to be the most reliable markers to monitor ALDH-2 activity in human whole blood and were used for subsequent clinical studies in human volunteers.
There, we showed that ALDH activity of WBCs is a reliable marker for organic nitrate tachyphylaxis and may be a useful marker for clinical tolerance as well since a single sublingual administration of GTN already significantly impaired ALDH-2 activity in human and rat WBC preparations isolated from these subjects (66, 330). Chronic GTN infusion resulted in decreased ALDH-2 activity in rat WBC preparations. Importantly, there was a highly significant linear correlation of mitochondrial and WBC ALDH-2 activity with GTN potency and all parameters dose-dependently decreased in parallel with increasing doses of i.v. GTN (see also Fig. 9).
We also showed that lipoic acid cotherapy was able to eliminate all negative side effects of GTN administration in humans (such as the decrease in WBC ALDH activity) (330). This is indicative, as shown before in the experimental setting, of lipoic acid being able to prevent nitrate tolerance in GTN-treated rats (324).
These results were further supported in a murine genetic model of heterozygous manganese superoxide dismutase (MnSOD, SOD2, the mitochondrial SOD isoform), where the 50% deficiency strikingly increased the susceptibility of the mice for development of not only nitrate tolerance but also endothelial dysfunction (64). Likewise, deficiency of another antioxidant enzyme, glutathione peroxidase-1 (GPx-1), favored the development of endothelial dysfunction even in response to low-dose GTN (69).
c. Desensitization of the sGC
In the late 80s, the desensitization of the sGC was suggested as a mechanism of tolerance (212, 219). This desensitization is compatible with the evidence that patients treated with one nitrate also show reduced sensitivity to other •NO-dependent vasodilators (so-called cross-tolerance). Importantly, it has been shown that S-nitrosylation of sGC results in decreased responsiveness to •NO characterized by loss of •NO-stimulated sGC and cGK-I activity (270). Desensitization of sGC was concentration- and time-dependent on exposure to S-nitrosocysteine, and it was proposed that S-nitrosylation of sGC is a means by which memory of •NO exposure is kept in smooth muscle cells and could be a mechanism of •NO tolerance.
The authors extended this in vitro evidence to in vivo observations by demonstrating that development of nitrate tolerance and cross-tolerance by a 3-day chronic GTN treatment correlates with S-nitrosylation and desensitization of sGC in tolerant tissues and that tolerance was reversed by concomitant treatment with the sulfhydryl donor, N-acetylcysteine (271). In line with this, our group just established that GTN-induced tolerance is partially prevented in rats by therapy with an sGC activator, but not with an sGC stimulator (145), suggesting that oxidation of sGC is a critical event in causing desensitization of the enzyme (see also the sGC stimulators and activators section).
Transnitrosation by S-nitrosocysteine (267) or reductive nitros(yl)ation of the cysteine residues, Cys78 and Cys122, in sGC was also shown to cause desensitization of the enzyme by a conformational change of the enzymatic structure (86). In addition, other transnitrosating agents or •NO donors, such as S-nitrosoglutathione and sodium nitroprusside, caused concentration-dependent deactivation of the purified sGC enzyme. This was associated with S-nitros(yl)ation of two cysteine residues in the sGC heterodimers that was prevented in the presence of glutathione (199). Likewise, in angiotensin II-induced hypertension and nitrosative stress (due to inflammation and inducible nitric oxide synthase [iNOS] induction), sGC deactivation was correlated with Cys516 nitros(yl)ation and cells overexpressing Cys516A mutant were protected from angiotensin II-dependent desensitization of the •NO/cGMP pathway (51).
Due to the fact that at least four cysteine residues were reported for S-nitros(yl)ation-dependent sGC desensitization (Cys78, 122, 243, 516) and are distributed between the heterodimers of the enzyme, the enzyme is highly susceptible to all kinds of oxidative and nitrosative stress. Oxidative inactivation and nitrosative desensitization of sGC were previously shown for superoxide (27), peroxynitrite (321), and iNOS induction under inflammatory conditions (155). Inflammation is a new confirmed risk factor for cardiovascular disease (154, 202) and inactivation of sGC under these conditions is very likely.
d. Increased sensitivity to vasoconstrictors in response to GTN therapy
GTN has also been shown to trigger a supersensitivity of the vasculature to vasoconstrictors, such as angiotensin II, phenylephrine, and serotonine, a phenomenon that may compromise the vasodilatory effects of nitrates (172). Interestingly, it appears that increased autocrine levels of endothelin within the vasculature, with subsequent activation of phospholipase C (PLC) and protein kinase C (PKC), are responsible for this true tolerance mechanism (Fig. 14) (226).
These pathways (PLC and PKC) depend on increased intracellular calcium levels and activate the myosin light chain kinase, leading not only to increased contractility of the myosin–actin filaments (Fig. 2) but also provide the link to cytosolic oxidative stress. Similarly, agonist-driven calcium-independent activation of the RhoA/ROK pathway contributes to vasoconstriction via inhibition of MLCP.
Importantly, this phenomenon of increased vascular sensitivity to vasoconstrictors is likely due to increased oxidative stress within endothelial and smooth muscle cells since ROS have been shown to increase the expression of endothelin within endothelial and smooth muscle cells, which in turn activates PKC (151, 152) (Fig. 14). The increased sensitivity to vasoconstriction was shown for norepinephrine, KCl, serotonin, angiotensin II, and PKC activators (Fig. 15) and was normalized following inhibition of PKC (226).
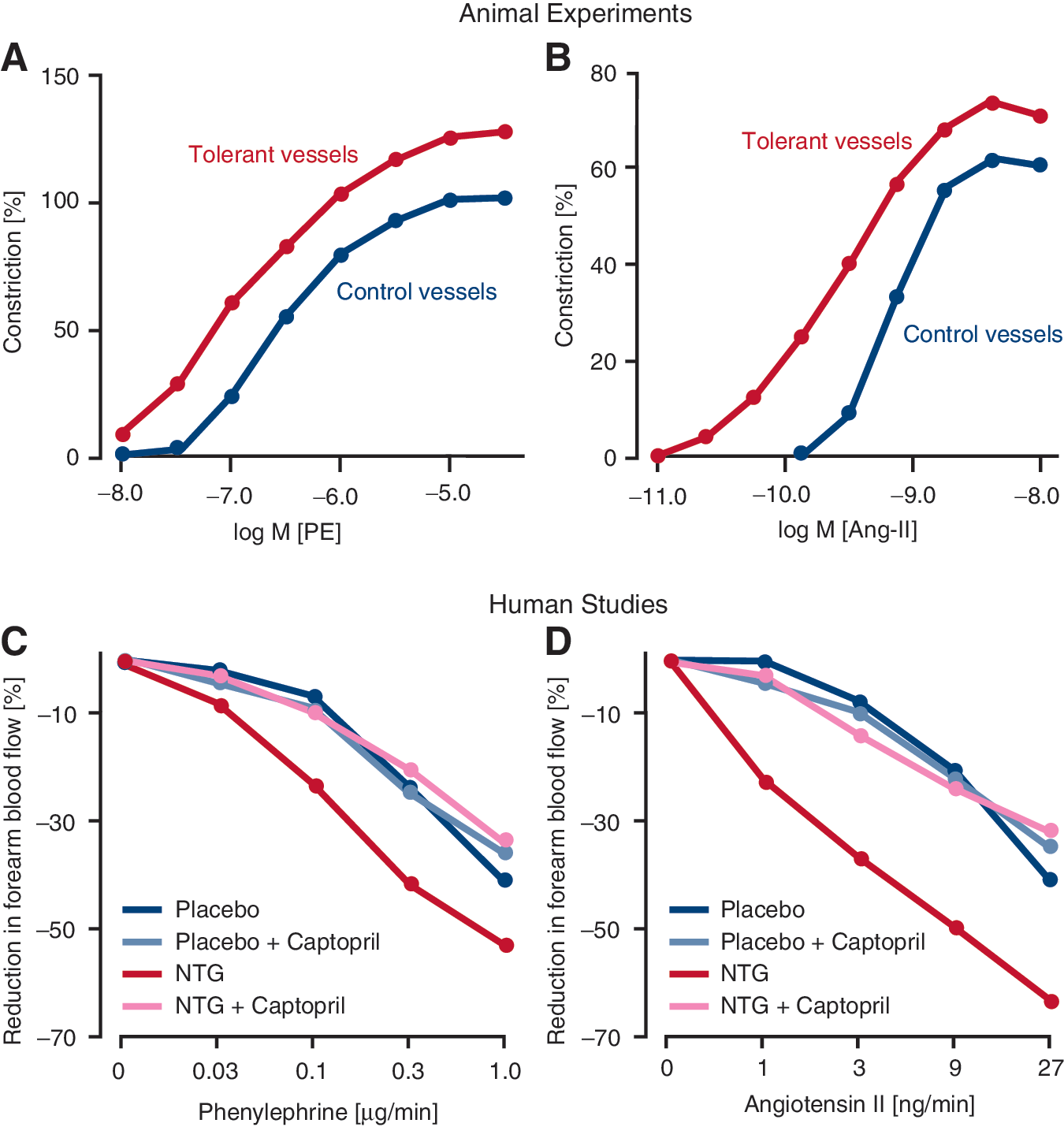
Studies in patients with CAD also demonstrated that long-term infusion of GTN for 48 h in a clinically relevant concentration of 0.5 μg/kg/min causes a supersensitivity of forearm arterioles to vasoconstrictors, such as angiotensin II and noradrenaline, all of which were completely corrected by concomitant treatment with the ACE inhibitor, captopril (Fig. 15) (132). Thus, it is tempting to speculate that enhanced vasoconstriction in GTN-tolerant patients may quite substantially contribute to the attenuation of the GTN vasodilatory effects.
e. Increased sensitivity to vasoconstrictors in response to ISMN therapy
Even more importantly, ISMN treatment, the most frequently used oral organic nitrate worldwide, was also associated with a strong increase in the expression of ET-1, mainly within the endothelial cell layer and the adventitia, and by increased sensitivity of the vasculature to vasoconstricting agents, such as phenylephrine and angiotensin II, as demonstrated before in GTN-tolerant vessels (Fig. 16) (253). Incubation of inflammatory cells with ISMN activated the phagocytic NADPH oxidase and caused an oxidative burst, all of which were blocked in vitro by the endothelin receptor blocker, bosentan, and normalized in vivo by gp91phox deficiency (253). In aortic tissue, ISMN induced a similar increase in ROS (superoxide) formation, most likely by Nox2, which was prevented by the unspecific flavin-dependent oxidoreductase inhibitor apocynin by gp91phox deficiency, as well as by the ET receptor antagonist bosentan (Fig. 16).
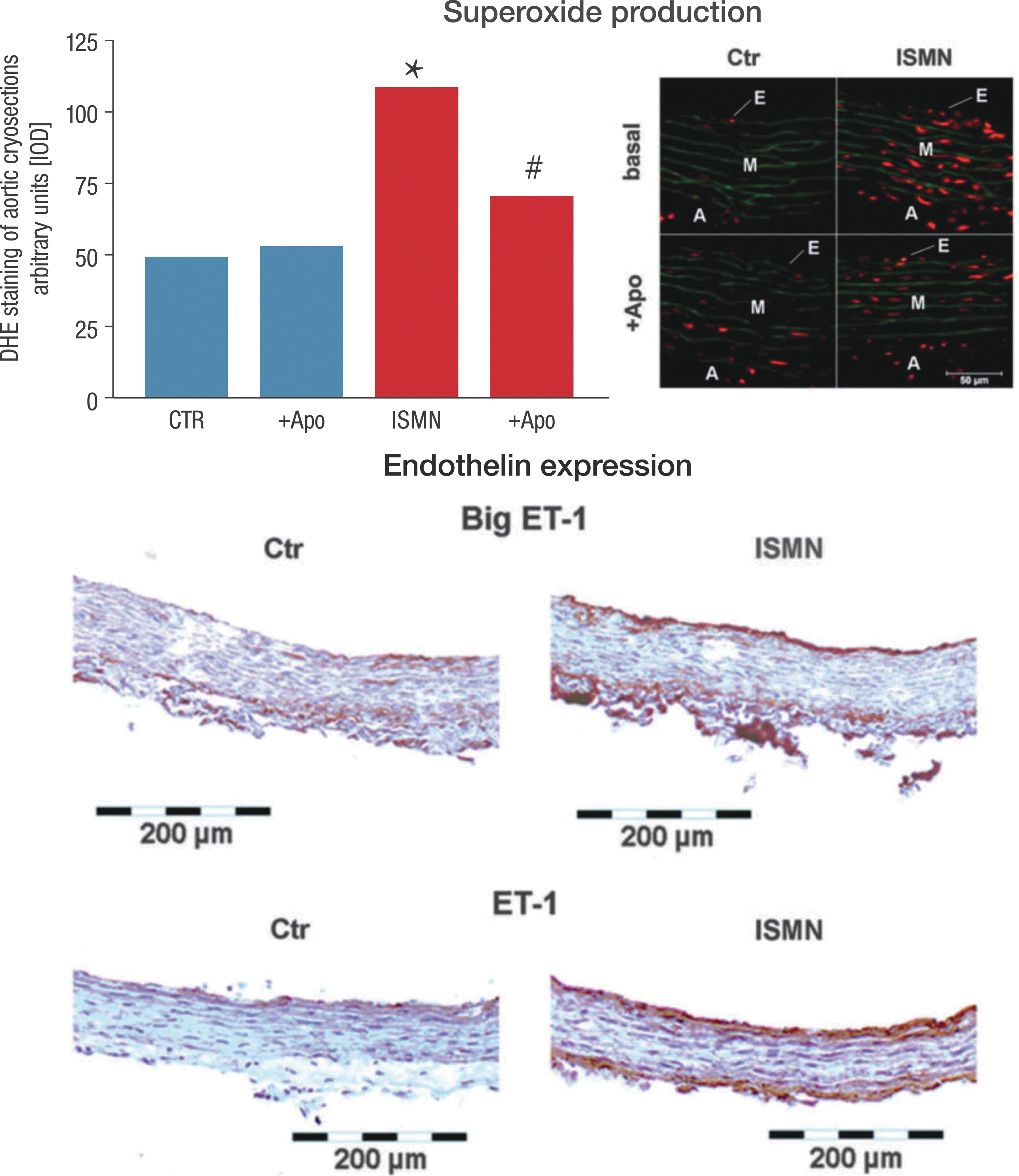
Although these adverse effects of ISMN look similar to the previous observations in response to GTN treatment, there are several fundamental differences. First, in contrast to GTN, ISMN is not bioactivated by mitochondrial ALDH-2, a process that leads to a marked increase in mitochondrial ROS production, and second, NADPH oxidase activation in response to ISMN is not dependent on the cross talk between ROS-producing mitochondria and NADPH oxidase and is an independent phenomenon triggered by unclear mechanisms. Thus, these findings may explain, at least in part, why therapy of postinfarct patients with ISMN leads to an increased rate of coronary events (240).
f. Epigenetic mechanisms of GTN-induced tolerance
More recent studies have demonstrated for the first time the involvement of epigenetic mechanisms in the development of nitrate tolerance. Colussi et al. observed that GTN administration increased not only cGMP production but also protein Nɛ lysine acetylation in cultured rat smooth muscle cells, including MLC phosphorylation and actomyosin formation (47). These effects were abolished by pretreatment with GTN and restored by treatment with trichostatin. Ex vivo experiments performed on aortic rings where tolerance was induced in vivo with subcutaneous injections of GTN revealed that all proacetylation drugs studied caused a reversal of nitrate tolerance.
In addition, the vasodilator response of GTN was abolished by the histone acetylase inhibitor, anacardic acid. These findings suggest that GTN therapy increases histone acetylase activity, all of which is lost upon chronic treatment with GTN. These findings may also indicate that a combination therapy of epigenetic drugs with GTN may be a novel approach to prevent GTN tolerance development. In addition, epigenetic drugs may also affect the progression of atherosclerosis in general since more recent evidence linked DNA methylation to the progression of atherosclerosis (49).
B. Organic nitrates cause endothelial dysfunction in the clinical setting
A number of lines of evidence show that therapy with most organic nitrates in clinically used doses impairs responsiveness to stimuli for the release of endothelium-derived •NO (Fig. 17). This phenomenon, also known as endothelial dysfunction, has been observed in animal studies and in humans during prolonged GTN (34, 115), ISMN (312), and ISDN therapy (286). In large coronary arteries, continuous treatment (5 days) with transdermal GTN leads to enhanced acetylcholine-induced paradoxical constriction instead of endothelium-dependent vasodilation (34). Evidence of impaired responses to acetylcholine was found in arteries removed from patients undergoing nitrate therapy at the time of bypass surgery, 48 h (281), and continuous treatment with transdermal GTN resulted in a marked reduction of acetylcholine-induced increases in forearm blood flow in healthy volunteers treated with GTN for 6 days (115).
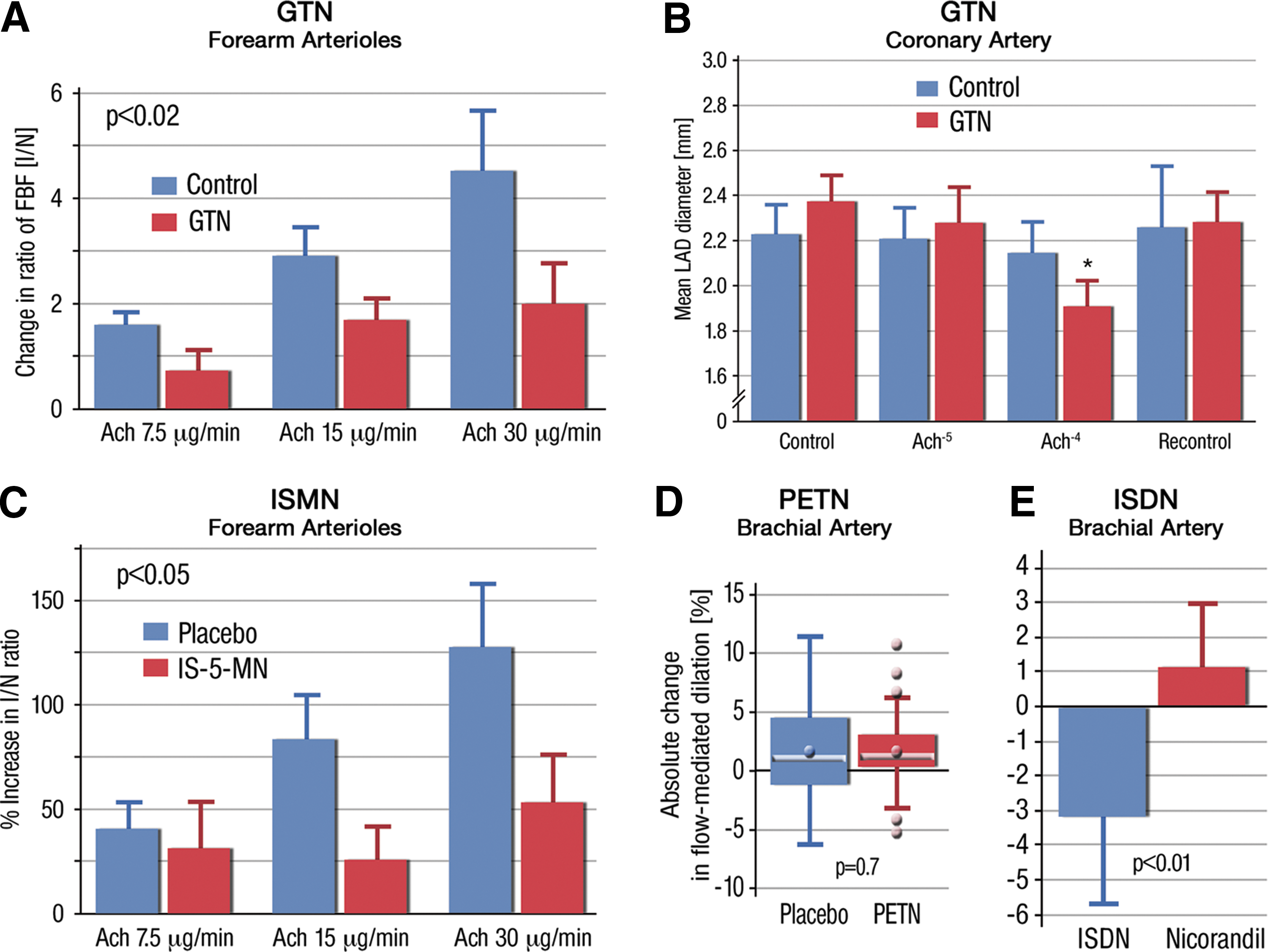
Emphasizing the existence of specific abnormalities at the level of the eNOS, the vasoconstriction elicited in control subjects by the infusion of a specific inhibitor of eNOS, L-NMMA, was significantly blunted after GTN therapy. Finally, at the lowest concentration, the eNOS inhibitor even caused a paradoxical dilation, which was interpreted as human in vivo evidence of GTN-induced eNOS uncoupling (and resulting in paradoxical production of a vasoconstrictor) (115). In line with this finding, GTN-induced endothelial dysfunction was prevented by folic acid, a compound that facilitates recoupling of eNOS (109).
Most importantly, endothelial dysfunction is not limited to treatment with GTN in tolerance-inducing doses; it was also demonstrated after treatment even with intermittent dosing of ISMN (312). Evidence of endothelial dysfunction has been shown on withdrawal of nitrate therapy, an observation that might suggest a role of impaired endothelial homeostasis in the rebound phenomenon (34). So far, the mechanisms (and the enzymatic superoxide source) involved in the increased oxidative stress in response to long-term ISMN treatment remain obscure. No endothelial dysfunction was observed in response to long-term PETN treatment (Fig. 17) (275), which is likely due to the previously mentioned upregulation of the antioxidant enzyme HO-1 (327). Compared with equipotent dosages of nicorandil, ISDN (40 mg/day) treatment for 3 months was associated with a reduction in endothelium-dependent FMD, an effect similar to that observed with GTN (Fig. 17) (286).
In 2002, we reported on the effects of chronic GTN treatment on the vasculature of patients undergoing coronary artery bypass surgery with and without prolonged GTN treatment before surgery (281). With these studies, we demonstrated that GTN pretreatment caused not only a marked degree of tolerance but also endothelial dysfunction in the mammary and radial artery and also in veins (Fig. 18). Tolerance and endothelial dysfunction were not only associated with increased vascular superoxide production as assessed with the superoxide-specific dye, dihydroethidine (DHE), but also with decreased phosphorylation of the vasodilator-stimulated phosphoprotein at 239 (P-VASP), reflecting decreased activity of the cGMP-dependent kinase I. In summary, evidence that chronic nitrate treatment causes endothelial dysfunction is now substantial and there is also growing evidence that increased oxidative stress within the vasculature is a major player in causing this phenomenon (221, 222, 238), especially mitochondrial RONS formation (66, 69).
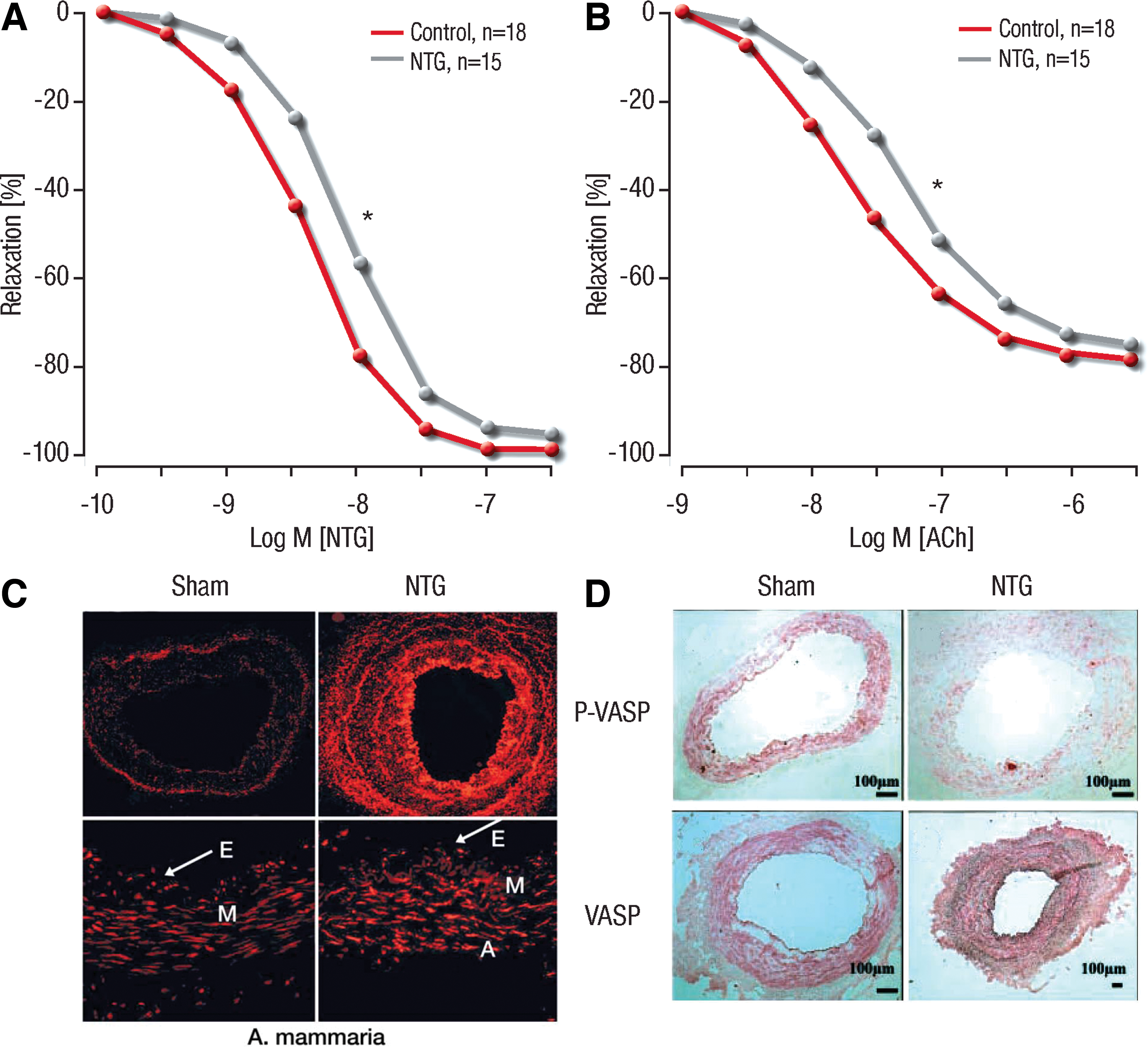
1. Molecular mechanisms underlying nitrate-induced endothelial dysfunction
Endothelial dysfunction in response to organic nitrates may be caused by different mechanisms, including decreased expression and/or activity of the endothelial NOS or even uncoupling of the enzyme (224, 280). A summary of all relevant redox-regulated processes (redox switches) that might lead to the dysfunction or uncoupling of eNOS in general or in the setting of nitrate tolerance is provided in Figure 19. Besides GTN-triggered oxidative depletion of the eNOS cofactor BH4, S-glutathionylation of cysteines in the reductase domain and adverse phosphorylation might contribute to eNOS uncoupling in the setting of tolerance (all explained in detail in the subsequent sections and Figure 20).
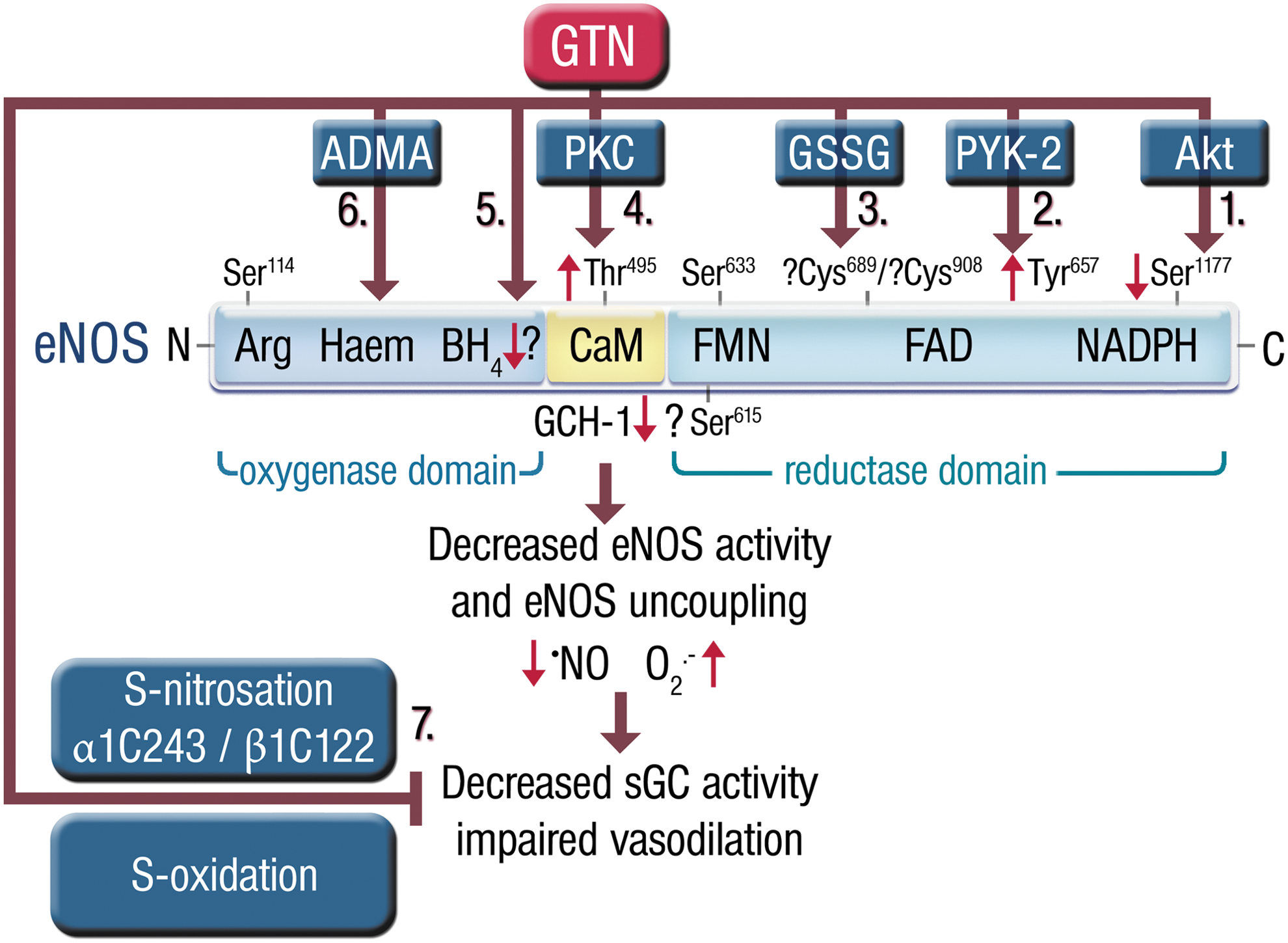
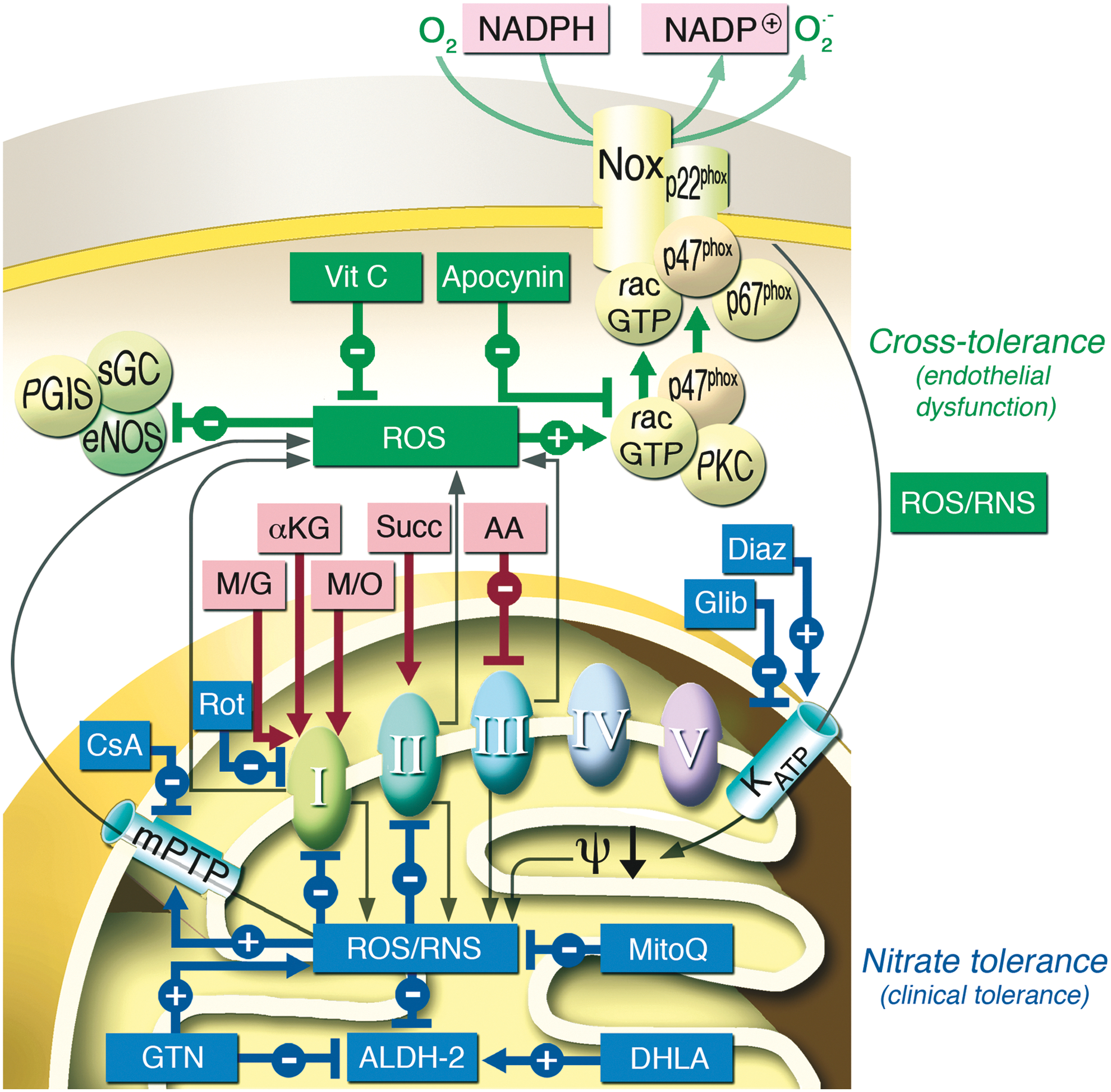
Furthermore, eNOS might be redox regulated by oxidative disruption of the zinc–sulfur complex in the dimer-binding interface (351), although evidence for this pathway was not yet provided for GTN-induced tolerance. Likewise, GTN treatment of cultured endothelial cells increased ROS formation and decreased ALDH-2 activity, all of which were associated with accumulation of the eNOS inhibitor, asymmetric dimethylarginine (ADMA) (Fig. 19) (346). Moreover, addition of exogenous ADMA significantly enhanced ROS production and MDA concentration and led to ALDH-2 inhibition, whereas overexpression of the ADMA-degrading enzyme, dimethylarginine dimethylaminohydrolase (DDAH), normalized GTN-induced oxidative stress and impaired ALDH-2 activity. Increased ADMA synthesis and decreased degradation by DDAH enzymes are well established under oxidative stress conditions and can therefore be regarded as redox switches for eNOS activity (282, 305).
Another redox switch in the •NO/cGMP signaling cascade is sGC, which has multiple reactive thiol groups that are prone to GTN-induced oxidation (220, 272) and S-nitrosation (271). There might also be increased vascular superoxide production of scavenging •NO produced by eNOS and a decreased activity and/or expression of prostacyclin synthase, which acts in concert with the NOS synthase to continuously lower vascular tone (63). Previously, we have demonstrated that in the setting of nitrate tolerance, the expression of the •NO synthase is rather up- than downregulated, suggesting that higher enzyme expression was a compensation for a dysfunctional, for example, uncoupled eNOS enzyme (232).
To address the potential role of an uncoupled eNOS, we studied vessels from control and GTN-treated animals and exposed them to the •NO synthase inhibitor, L-NNA. While L-NNA increased superoxide levels in control vessels, it inhibited the per se elevated superoxide levels in GTN-tolerant vessels, pointing to an uncoupled eNOS (232). The mechanism leading to decreased eNOS-mediated •NO production or even eNOS uncoupling is summarized in the next paragraphs.
a. BH4 deficiency
Further studies revealed that the administration of the eNOS cofactor, tetrahydrobiopterin (BH4) (122), as well as folic acid (116) was able to improve GTN-induced endothelial dysfunction, suggesting that a depletion of BH4 may be responsible for the uncoupling of the enzyme. The concentration of intracellular BH4 clearly depends on its de novo synthesis and its oxidation/degradation. Since endothelial nitrotyrosine concentrations increase in response to chronic GTN treatment (319), it is tempting to speculate that high levels of peroxynitrite may be able to oxidize BH4 to the BH3 • or BH4 •+ radical (Fig. 19) (173), which would in turn cause a functional depletion of BH4 and therefore eNOS uncoupling. Indeed, BH4 levels decreased under GTN therapy and were improved by pioglitazone cotherapy (141).
Additional evidence for intracellular BH4 depletion as one of the mechanisms of eNOS uncoupling was provided by studies demonstrating that GTN treatment causes a decrease in the expression of the BH4-synthesizing enzyme, GTP cyclohydrolase 1, and a counter-regulatory increase in the expression of the BH2-recycling enzyme, dihydrofolate reductase (159). A similar picture was found in ISMN-treated animals where endothelial dysfunction was associated with decreased GTP cyclohydrolase 1 expression (253). Opposite observations were made in models of arterial hypertension and type 1 diabetes mellitus, where PETN therapy increased the expression levels of GTP cyclohydrolase 1 and also rescued endothelial dysfunction in the setting of hypertension and diabetes (278, 279). The role of BH4 for GTN-induced nitrate tolerance was questioned by Mayer and coworkers, who did not observe decreased BH4 levels in GTN or peroxynitrite-treated vascular tissue or cultured cells (273). Instead, these authors suggested that improvement of GTN vasodilator potency by BH4 in tolerant tissue is based on a direct protective (antioxidant) effect of BH4 on sGC activity (272).
b. Changes in eNOS phosphorylation and S-glutathionylation
Further pieces of evidence for a dysfunctional uncoupled eNOS in response to nitrate treatment were demonstrated by changes in the eNOS phosphorylation pattern. GTN therapy markedly inhibited eNOS phosphorylation at Ser1177, which in turn leads to a decreased basal and stimulated •NO release of the enzyme (159). GTN treatment also increases phosphorylation of eNOS at Thr495 (Fig. 19) (159). It is known that PKC, which becomes activated during GTN treatment (226), also causes a phosphorylation of the eNOS enzyme at Thr495 and this phosphorylation has been shown to cause dysfunction (and maybe uncoupling) of the enzyme (90, 185). It is noteworthy that PKC is activated under oxidative stress conditions (282). Likewise, eNOS activity is regulated by protein tyrosine kinase-2-dependent phosphorylation at Tyr657 (Fig. 19) (190), which, however, was not observed in the setting of nitrate tolerance so far.
In addition, not only GTN therapy but also therapy with ISMN leads to an enhanced S-glutathionylation at one or more cysteine residues of the reductase domain contributing to uncoupling and therefore endothelial dysfunction (Fig. 19) (159, 253). eNOS S-glutathionylation was first reported by Zweier and coworkers as a trigger of eNOS uncoupling (36), which is meanwhile well established as a marker of eNOS uncoupling in models of hypertension (36, 102, 170), diabetes (278), acute lung injury (338), hypoxia/reoxygenation (71), and aging (255). eNOS S-glutathionylation can be regarded as a real redox switch for eNOS activity since it is easily reversible and even enzymatically regulated by removal of GSH by glutaredoxin-1 (35). Finally, experimental data suggest that BH4 levels and the eNOS S-glutathionylation state show a direct correlation and influence each other (50).
c. Increased NADPH oxidase activity
NADH/NADPH oxidases have been implicated in GTN-induced oxidative stress since 1995 (236). NADPH oxidase (Nox1, Nox2, Nox3, Nox4, Nox5, and Duox1/2) is an enzyme family with the sole job of producing superoxide and hydrogen peroxide (26, 334). NADPH oxidases were not only identified as important contributors to and triggers of cardiovascular disease (33, 120) but also confer important functions for vascular homeostasis (216, 307). Therefore, Nox family enzymes represent interesting therapeutical targets (8). For all other enzymatic systems, ROS production has to be considered as an undesired side effect. In general, it remains to be established whether increased vascular ROS production in response to chronic GTN treatment originates from NADPH oxidase (230, 236) or the mitochondria accounting for tolerance and/or nitrate-induced endothelial dysfunction (61, 304).
With recent studies, we could demonstrate that mitochondrial superoxide production was increased in the presence of substrates for mitochondrial complex I and II, pointing to an important role of this cellular compartment for tolerance (Fig. 20) (325). In 2008, we reported for the first time on a cross talk between mitochondrial and cytosolic RONS in a model of increased mitochondrial GTN-induced oxidative stress (325). In this animal model, endothelial dysfunction and nitrate tolerance were dependent on the activation of both superoxide sources (Fig. 20) (325). This established cross talk between mitochondria and the NADPH oxidase was blocked by in vivo and ex vivo administration of the mitochondrial permeability pore inhibitor, cyclosporine A, which interestingly selectively decreased endothelial dysfunction, whereas nitrite tolerance was not affected. In contrast, the respiratory complex I inhibitor rotenone reduced endothelial dysfunction and tolerance.
Conversely, in vivo or ex vivo treatment with the KATP opener, diazoxide, caused a nitrate tolerance-like phenomenon in control animals, whereas the KATP inhibitor, glibenclamide, decreased tolerance in nitroglycerin-treated animals. gp91phox−/− and p47phox−/− mice developed tolerance, but no endothelial dysfunction, in response to nitroglycerin treatment. The findings of this study are summarized and discussed in view of similar observations in different animal models in recent review articles (55, 282) (Fig. 20).
Very similar effects of rotenone (Rot), cyclosporine A (CsA), diazoxide (Diaz), and glibenclamide (Glib) have been recently demonstrated by another group in an experimental model of angiotensin II-induced hypertension (77), which were confirmed by subsequent studies in this arterial hypertension model (74, 75, 77, 170). A role of KATP channels for NADPH oxidase-driven activation of mitochondrial ROS formation via changes in the membrane potential was previously proposed (25, 157). In addition, this hyperactivated mitochondrial ROS formation in hypertensive animals caused downstream activation of Nox2 (Nox1), resulting in a vicious cycle of angiotensin II-triggered ROS formation (170, 241). The mechanism underlying this concept is based on mtROS-driven PKC and tyrosine kinase (cSrc) activation, which in turn activates NADPH oxidases. The NADPH oxidase-dependent cytosolic RONS formation will then uncouple eNOS and nitrate prostacyclin synthase and desensitize sGC.
Previous experimental studies have shown that increased oxidative stress in cellular tissue per se is able to activate the oxidase in a positive feedback manner (98), and mitochondrial ROS were demonstrated to trigger vascular NADPH oxidase activity and dysfunction in models of aging (255, 328). Thus, nitroglycerin-induced mitochondrial superoxide production may cause a secondary activation of Nox (Fig. 20).
d. Nitration of prostacyclin synthase
Protein tyrosine nitration is regarded as a footprint of peroxynitrite in vivo (53, 118). Evidence for peroxynitrite formation in the setting of nitrate tolerance (again most pronounced for GTN) comes not only from increased levels of 3-nitrotyrosine-positive proteins in vessels from control rabbits but also hyperlipidemic Watanabe rabbits (319). We established a nitrated prostacyclin synthase within the endothelium and the subendothelial space in vessels from tolerant rats (137), a specific marker for increased peroxynitrite formation in vivo (Fig. 21) (274, 350). Nitration and inhibition of prostacyclin synthase in GTN-treated rats were confirmed by immunohistochemistry as well as immunoprecipitation.
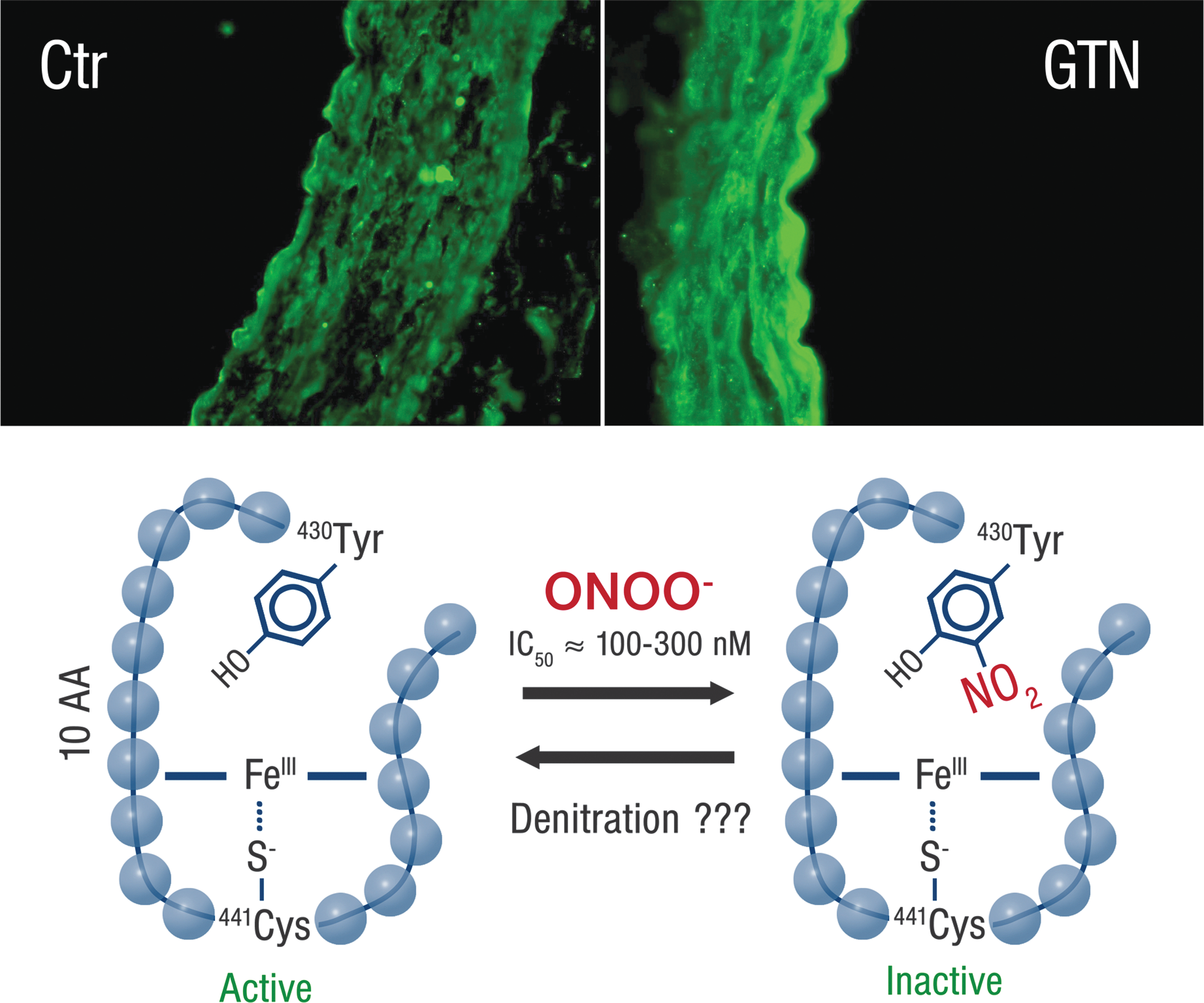
More evidence for peroxynitrite formation in GTN-treated animals was established by demonstration of increased L-012 chemiluminescence levels, a dye with a very high specificity for peroxynitrite (56, 60), giving remarkable signals in nitrate-treated tissues (60, 62, 328). Protein tyrosine nitration was blocked by preincubation with the peroxynitrite scavenger uric acid, SOD, or the inhibitor of PKC chelerythrine (3). Finally, the peroxynitrite scavenger, hydralazine, reduced GTN-induced nitration and improved the phenotype of nitrate-tolerant animals (62).
IV. Strategies to Prevent Development of Nitrate Tolerance and Nitrate-Induced Endothelial Dysfunction
A. Nitrate-free interval
Tolerance toward the hemodynamic effects of GTN can be avoided by treatment regimens with nitrate-free intervals of at least 12 h that allow regeneration of the GTN bioactivating enzymes as well as antioxidant defense systems (257). Although this strategy is intrinsically flawed by the fact that patients cannot receive a 24-h treatment (and typically do not receive nitrate therapy in the early morning hours when the incidence of acute coronary syndromes is highest), it is effective in maintaining the hemodynamic effects of the nitrate. In addition, withdrawal of nitrate therapy results in the development of rebound ischemia, likely associated with the unrestrained effect of endothelial dysfunction and hypersensitivity to vasoconstrictors. The clinical correlate of this phenomenon [reviewed elsewhere Parker (259)] is that during the nitrate-free interval, the frequency of ischemic episodes is significantly increased, compensating for the benefit of nitrates.
In patients with stable angina, Freedman et al. (94) have shown an increase in the duration of silent ischemia compared with patients treated with placebo, and other authors reported a decreased angina threshold after patch removal in both small (261) and larger multicenter trials (265). In the catheterization laboratory, acute nitrate withdrawal increased the coronary vasoconstrictor responses to acetylcholine, suggesting that the rebound phenomenon is secondary to an increased sensitivity to vasoconstrictors (13); similar findings were reported in animal studies (234). Although it is unclear whether the rebound phenomenon occurs only at the vascular level (131), a nitrate-free interval cannot be taken as an acceptable solution. Interestingly, clinical trials in patients failed to demonstrate the rebound phenomenon in patients with CAD treated with nitrates and concomitant treatment with ACE inhibitors (263) or beta-receptor blockers (144). This latter observation emphasizes how exposure to other therapies may alter the pharmacodynamic effects of nitrates and encourages the search for more novel strategies to prevent the development of nitrate side effects.
B. Sulfhydryl group donors
Following the Needleman and Johnson (244) hypothesis that nitrate tolerance could be induced by depletion of the thiol groups necessary for the biotransformation of GTN, a number of animal and human studies have tested the effect of coadministration of sulfhydryl donors such as N-acetylcysteine (229, 257) and
C. Antioxidants
A corollary of the oxidative stress hypothesis of nitrate tolerance (236) is that treatment with antioxidants may be successful in preventing this phenomenon. Studies published by Bassenge et al. (15) demonstrated that concomitant treatment with a variety of antioxidants preserves the sensitivity of the vasculature to organic nitrates in different animal experimental models. The translation of these results to clinical practice is limited by the fact that oral administration of an effective antioxidant has proved to be more complicated than initially thought (207). This failure appears to mirror the lack of efficacy of oral antioxidants in improving cardiovascular prognosis (227).
Although intra-arterial administration of high-dose vitamin C consistently appears to reverse, for example, nitrate-induced endothelial dysfunction (312), a positive effect of oral formulations appears to be much less reproducible (207). The development of more potent, more targeted, and more bioavailable antioxidants, particularly targeted at the mitochondria, will address these issues (83) in future.
D. ACE inhibitors and angiotensin II type 1 receptor blockers
The counter-regulatory mechanisms triggered by nitrate therapy may offset the direct vasodilatory effects of GTN and, together with sodium and water retention, may coincide to counterbalance the hemodynamic benefit of this drug. Animal studies revealed that during chronic therapy with GTN within 3 days of treatment, angiotensin II type 1 (AT1) receptor expression as well as vascular ACE activity was significantly increased (172). This was associated with a marked increase in sensitivity of the vasculature to the vasoconstricting action of angiotensin I and II. Likewise, an increase in vascular superoxide production was observed. Concomitant treatment with the angiotensin II type 1 (AT-1) receptor blocker, losartan or telmisartan, normalized superoxide levels and prevented tolerance and cross-tolerance to the endothelium-dependent vasodilator, acetylcholine (159, 172). Finally, a hybrid nitrate containing valsartan as a backbone showed no signs of tolerance and other side effects in vitro (161).
Importantly, the critical role of the renin–angiotensin axis is further confirmed by the evidence that ACE inhibitors prevent the development of tolerance in animal (22) and human studies (205). In line with the role of ACE inhibitors, nitrate tolerance and nitrate-induced oxidative stress in animal models are markedly attenuated at the level of both conductance and resistance vessels during concomitant administration of angiotensin II type 1 receptor blockers (93). Although it is unclear whether this also applies to humans (206), given the systematic use of ACE inhibitors (e.g., in the setting of CHF), these studies reinforce the concept that nitrate therapy in the modern clinical setting might have totally different implications than what is observed in experimental models (and in former clinical practice).
E. Hydralazine
A combination therapy of ISDN with hydralazine was able to significantly improve the prognosis in patients with CHF in the Vasodilator Heart Failure Trial (V-HeFT) I trial compared with vasodilator therapy with the alpha blocker prazosin (45) and significantly improved exercise capacity compared with enalapril in the V-HeFT II trial (46).
In addition, the double-blind, randomized African American Heart Failure Trial (A-HeFT) demonstrated that the combination of ISDN and hydralazine was markedly effective in improving the composite endpoint of the trial, which included death from any cause, a first hospitalization for heart failure, and quality of life measures (311). Interestingly, next to its effects on arteriolar tone, hydralazine has also been shown to be a highly potent scavenger of peroxynitrite (Fig. 22) (62). Peroxynitrite has been accused of being responsible not only for tolerance but also for endothelial dysfunction development in response to long-term nitrate therapy. Thus, the peroxynitrite scavenging effects of hydralazine may explain, at least in part, why this combination therapy is devoid of tolerance and why a combination therapy improves prognosis in patients with chronic CHF (Fig. 22) (127).
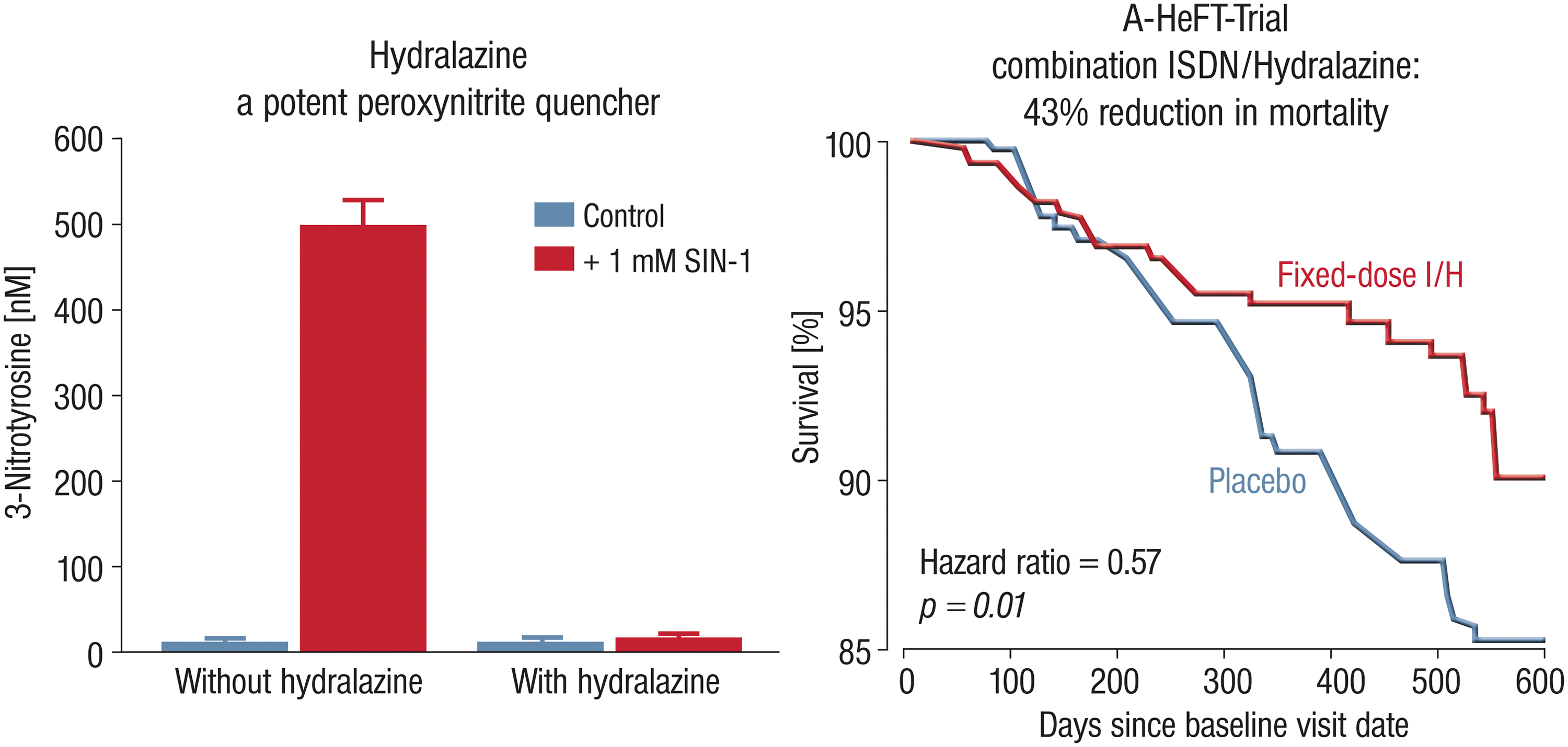
In addition to the effects on pre- and afterload of this combination therapy, hydralazine, as an effective peroxynitrite inhibitor, may prevent the development of tolerance by correcting the disrupted nitroso-redox balance in the setting of chronic CHF (127).
F. Carvedilol
Third-generation beta blockers, such as carvedilol, possess additional pleiotropic properties that might modify the development of tolerance. In animal models, the effect of carvedilol in preventing tolerance was similar to that of intravenous vitamin C infusions (320) and was probably based on the previously reported indirect antioxidant properties of this third-generation beta blocker (11). In studies in patients with arterial hypertension or CHF, carvedilol, but not atenolol, metoprolol, or doxazosin preserved the vasodilatory effect of GTN in resistance arteries, despite the administration of transdermal GTN in tolerance-inducing doses (320). Carvedilol also suppressed RONS formation in chronically GTN-treated dogs (88).
G. Statins
Therapy with statins has been associated with a number of benefits that are independent of their effect on lipid levels. In animal studies, both pravastatin and atorvastatin prevented nitrate tolerance and vascular superoxide formation induced by subcutaneous GTN injections (92), an effect that was associated with increased basal cGMP levels and was abolished when the rats received an inhibitor of the eNOS concomitantly with GTN. These animal data were confirmed by a recent human study showing that administration of atorvastatin prevents both GTN-induced endothelial dysfunction and tolerance in healthy volunteers (189). Finally, therapy with statins also appears to improve platelet reactivity to GTN in patients with stable or unstable coronary syndromes (43).
H. Endothelin receptor blockers
The demonstration that ISMN and GTN therapy stimulates autocrine ET-1 production within the vascular wall may indicate that concomitant therapy with endothelin receptor blockade, for example, with bosentan (nonselective endothelin receptor blocker), may beneficially influence the tolerance and cross-tolerance phenomena. Indeed, bosentan added to GTN-treated animals markedly reduced vascular superoxide production and largely prevented the development of nitrate tolerance (172). Likewise, bosentan improved endothelial dysfunction and increased vascular superoxide production in ISMN-treated animals (253). Thus, nonselective ET-1 receptor blockade may represent a promising tool to prevent the development of vascular side effects in response to chronic therapy with organic nitrates, in particular GTN and ISMN.
I. sGC stimulators and activators
Just recently, we were able to test the effects of sGC activation and stimulation on GTN-induced tolerance, endothelial dysfunction, and increased oxidative stress. Using the sGC activator, BAY 60-2770, and the stimulator, BAY 41-8543, we found that sGC activation, but not sGC stimulation, was able to restore vascular •NO levels and therefore the activity of the cGMP-dependent kinase in tolerant tissue (Fig. 23). Furthermore, BAY 60-2770 displayed potent inhibitory effects on oxidative stress and partially reversed GTN-induced endothelial dysfunction and vascular tolerance (Fig. 23) (145). It is important to note that by using the electron spin resonance (EPR) technique, we could also demonstrate for the first time that in vivo treatment with GTN decreases significantly ambient vascular •NO levels (Fig. 23).
Importantly, the sGC activators are compounds that activate the sGC •NO independently, even when the enzyme is oxidized or heme depleted. In contrast, sGC stimulators are substances that enhance the activation of sGC by its endogenous activator •NO. Thus, the demonstration of exclusively positive effects by sGC activation in our tolerance model suggests that the oxidation of the sGC may, at least in part, contribute to GTN-induced vascular tolerance and endothelial dysfunction (145), which would be in accordance with a previous report on GTN-induced S-nitrosation and inactivation of sGC (Fig. 19) (271).
So far, it was reported that the sGC activator, BAY 60-2770, binds to Cys122 S-nitros(yl)ated sGC. Cys122 is located in the regulatory heme domain and can affect •NO-binding and allosteric effects on the catalytic subunit upon S-nitros(yl)ation, thereby causing desensitization of the enzymatic activity (171). Overall, these data are in accordance with previously published results on a structurally related sGC activator (BAY 58-2667), the vasodilatory potency of which was not affected ex vivo by GTN-induced nitrate tolerance (296).
J. PDE inhibitors
Already in 1985, Ahlner et al. reported on synergistic hemodynamic effects of GTN and the unspecific PDE inhibitor, dipyridamole (6). These observations were later confirmed by the PDE inhibitor, zaprinast (258, 290). In the clinical setting, a reversal of GTN-induced tolerance in healthy volunteers was not found for dipyridamole, but for zaprinast cotherapy (70, 314). The beneficial effect of PDE inhibition on nitrate tolerance was also later found for the PDE1-specific inhibitor, vinpocetine (156), and selective PDE type 5 inhibitors (188, 196). Finally, a hybrid nitrate containing cilostazol, a PDE5-specific inhibitor, as a backbone showed no signs of tolerance and other side effects in vitro (161).
V. Pleiotropic Effects of Organic Nitrates
Organic nitrates are not just vasodilators but also have several pleiotropic effects. In particular, the effects of GTN on ischemic conditioning or the antioxidant properties of PETN by stimulating the activity and expression of the HO-1 will be discussed in the next paragraphs.
A. Ischemic preconditioning
The term ischemic preconditioning identifies a condition whereby a given tissue shows a reduced sensitivity to ischemia and reperfusion (IR) injury. This protective phenotype can be induced by prior exposure to a brief ischemia (239); however, the possibility that such a protective state could also be induced pharmacologically has important implications in the prevention of ischemic events (174, 343). A number of studies have shown that pharmacologic preconditioning with nicorandil, adenosine, bradykinin, and several other drugs reduces myocardial injury in the setting of cardiac surgery, coronary angioplasty, physical exercise, and pacing (266). Similarly, short-term administration of GTN has been shown to reduce electrocardiographic evidence of myocardial ischemia during exercise stress and during angioplasty in patients with cardiovascular disease (52, 180).
One hypothesis is that the mechanism of this (nonhemodynamic) protective effect depends on the sudden release of ROS during acute GTN administration (56, 304): this could trigger opening of the mitochondrial permeability transition pore and a variety of downstream effects, mostly not understood (111, 113, 129, 179). Therefore, evidence now exists that the transient ROS production associated with GTN administration (56) has two types of effects: (i) upon short-term GTN administration, it would cause preconditioning-based protection against IR (187), which, however, can be blocked by antioxidant supplementation (contributing the phenomenon of the antioxidant paradox) (79); (ii) upon prolonged administration, via oxidative damage, accumulation of ROS would lead to the toxic effects of GTN, such as endothelial and autonomic dysfunction, as described above. PETN also conferred preconditioning effects under chronic conditions (187), whereas GTN even induced a tolerance to the preconditioning effects, probably based on oxidative damage by GTN-induced RONS formation (110).
An alternative hypothesis for the preconditioning, as well as for the other nonhemodynamic effects of organic nitrates, could be that these effects are mediated by the release of inorganic nitrite (107). Interestingly, there is consistent evidence that this compound has potent cytoprotective properties, especially in IR injury (29, 72, 148), potentially mediated by nitrosative inhibition of mitochondrial respiratory chain I, which causes a reduced ROS production at reperfusion. The loss of anti-ischemic protection by chronic GTN therapy was associated with inhibition of ALDH-2 (187). Of note, ALDH-2 inhibition per se suppressed remote preconditioning in experimental models and humans (48).
B. Antioxidant properties of PETN: Nrf2 and HO-1
Another important observation was (as discussed above) that PETN and its trinitrate metabolite, PETriN, increase, in contrast to GTN, the activity and expression of the very potent antioxidant enzyme HO-1 in vitro (59, 250 –252) and in vivo (57, 279, 324). Via breakdown of porphyrins, HO-1 produces the antioxidant molecule, bilirubin (which is formed from biliverdin by biliverdin reductase) (91), and the vasodilator, CO (250, 324). HO-1 in turn stimulates the expression of a second antioxidant protein, ferritin, via the HO-1-dependent release of free iron from endogenous heme sources (252).
According to work by Kleinert and coworkers, the induction of HO-1 by PETN is probably based on a direct activation of the HO-1 promoter region via Nrf2 (either directly via Keap1 or indirectly via •NO) as well as a stabilization of the HO-1 mRNA by •NO-triggered HuR binding, effects that are not shared at all by other nitrates (Fig. 24) (65, 253, 278). The induction of extracellular SOD and GPx-1 (78) may represent another important intrinsic antioxidant pathway activated by PETN in vivo treatment (256) and may explain its beneficial effects in hyperlipidemia (124, 165) and arterial hypertension (279).
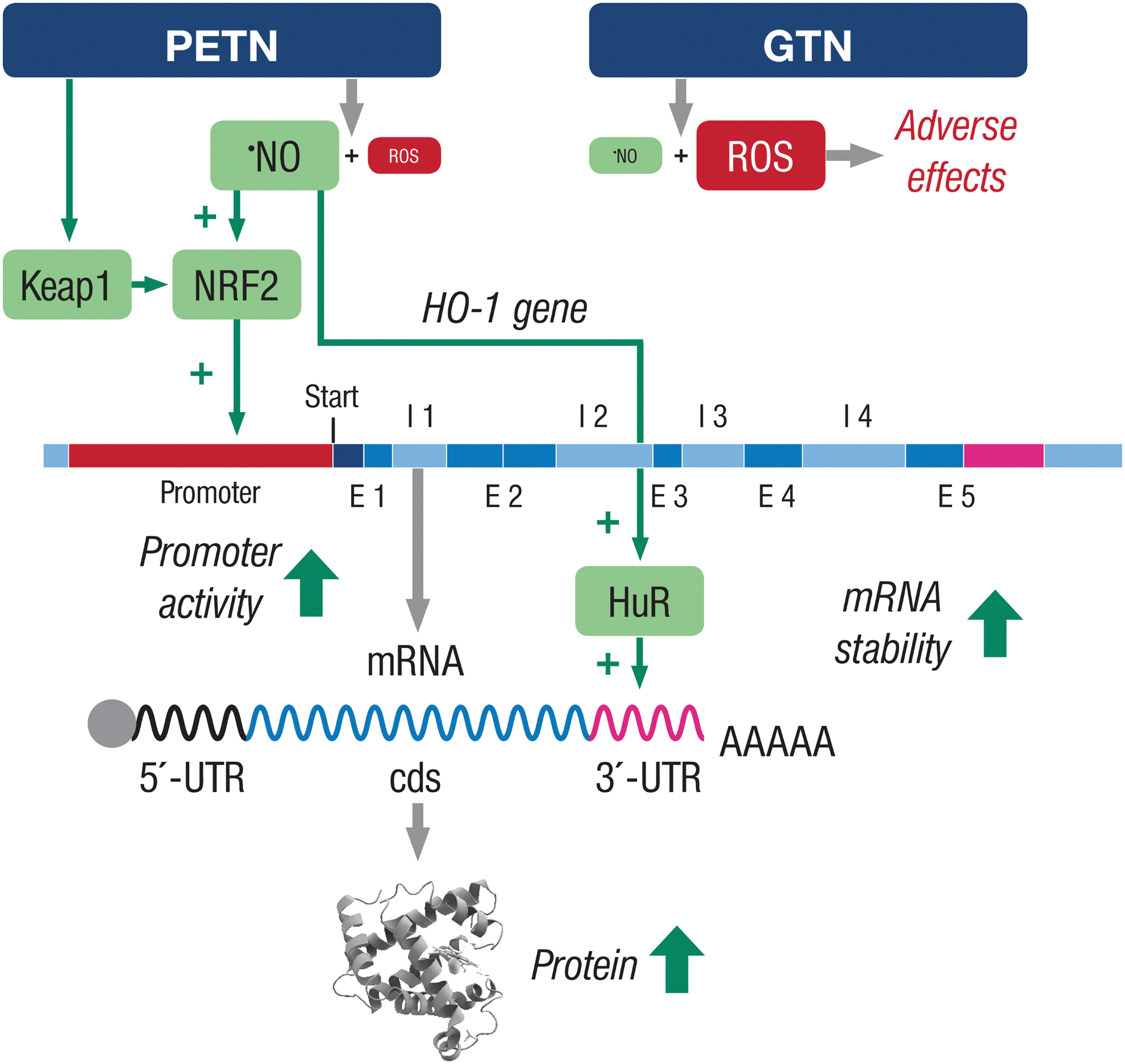
These protective properties of HO-1 upregulation may explain why PETN mimics ischemic preconditioning (79). The beneficial effect of HO-1 induction on the development of nitrate tolerance was documented by the fact that pharmacological modulation of HO-1 activity significantly affected the degree of tolerance and all associated side effects. For example, HO-1 activation by hemin (a potent HO-1 inducer) completely prevented tolerance in GTN-treated animals, while an inhibitor of HO-1, apigenin (an HO-1 suppressor), was able to induce tolerance in animals treated with PETN, a nitrate usually devoid of tolerance (323). Likewise, PETN treatment induces nitrate tolerance in HO-1+/− mice and low-dose nitroglycerin treatment induces severe loss of nitroglycerin potency in these mice with partial deficiency in HO-1 (279).
C. Epigenetic pathways, microRNA, and gene regulation
More recently, PETN was shown to induce heritable epigenetic changes that cause blood pressure reduction in female offspring of PETN-treated hypertensive rats via enhanced histone 3 lysine 27 acetylation and histone 3 lysine 4 trimethylation with subsequent induction of aortic eNOS, mitochondrial SOD, GPx-1, and HO-1 (339). More evidence for regulation of epigenetic pathways by organic nitrates comes, as already mentioned, from a report by Colussi et al. The authors observed that GTN administration increased not only cGMP production but also protein Nɛ lysine acetylation in cultured rat smooth muscle cells, including MLC phosphorylation and actomyosin formation (47). GTN-induced tolerance was reversed by all proacetylation drugs, including the normalization of p300/CREB-binding protein-associated factor activator, pentadecylidenemalonate 1b (SPV106), whereas the vasodilator potency of GTN was abolished by the histone acetylase inhibitor, anacardic acid. Previous observations suggest that chronic oral GTN treatment contributes to carcinogenesis in rats via genetic and/or epigenetic factors (309).
D. The effect of organic nitrates on endothelial progenitor cells
Administration of organic nitrates also has profound effects on the number and function of circulating endothelial progenitor cells (EPCs). Both PETN and ISDN increase the level of circulating EPCs not only in control animals but also in animals with decreased EPC levels with ischemic cardiomyopathy (313). Interestingly, ISDN, but not PETriN, increases intracellular ROS formation, which was associated with a 50% decrease in EPC migration and incorporation. In humans, GTN treatment increases apoptosis, decreases phenotypic differentiation, migration, and mitochondrial dehydrogenase activity of EPCs (73). The effect of PETN remains to be studied.
VI. Development of New Organic Nitrates Devoid of Side Effects
The development of new organic nitrates may represent an attractive strategy to find new clinical applications for this class of drugs by overcoming their clinical side effects. Based on results from a recent study from Lehmann and Daiber, aminoethyl nitrate (AEN) showed an almost similar potency compared with GTN, although being only a mononitrate (66). In contrast to triethanolamine trinitrate (TEAN) and GTN, AEN bioactivation did not depend on ALDH-2 and caused no in vitro tolerance. In vivo treatment with TEAN and GTN, but not with AEN, induced cross-tolerance to acetylcholine-dependent or GTN-dependent relaxation. Although all nitrates tested induced tolerance, only TEAN and GTN significantly increased mitochondrial oxidative stress in vitro and in vivo. The results of this study demonstrate that not all high-potency nitrates are bioactivated by ALDH-2 and that high potency of a given nitrate is not necessarily associated with induction of oxidative stress or nitrate tolerance. Obviously, there are distinct pathways for bioactivation of organic nitrates with high potency.
AEN may represent a new class of organic nitrates, which is devoid of induction of in vitro tachyphylaxis as well as oxidative stress, and showed an impressively high potency compared with not only all tested mononitrates so far but also with di- and trinitrates. Despite these beneficial and pharmacological-relevant properties, AEN induced severe in vivo tolerance to itself, requiring further mechanistic studies to reveal the bioactivation pathway as well as its mechanism of action. This may be of clinical interest since AEN is part of the structure of the potassium channel opener, nicorandil, which consists of a fused organic nitrate moiety. Nicorandil is devoid of clinical tolerance, releases •NO, and also has hyperpolarizing properties (potassium channel opener) (286), which has been demonstrated to prevent the activation of the NADPH oxidase. Therefore, future strategies to develop organic nitrate-based vasodilators and hybrid molecules may include the insertion of AEN-like structures.
The development of new nitrate hybrid molecules and the introduction of nitrate functions into established cardiovascular drugs represent another interesting field of nitrate development (198). There are several reports in the literature on nitrate hybrid molecules: Next to nicorandil (142), there may be a candidate 2NTX-99, a thromboxane synthase inhibitor or thromboxane receptor antagonist (30); GT-094, a nonsteroidal anti-inflammatory drug (NSAID) with •NO-releasing function (organic nitrate group) (125); the antifungal drug ketoconazole with a diazen-1-ium-1,2-diolate or an organic nitrate moiety (169); •NO-donor-tacrine hybrids as hepatoprotective anti-Alzheimer drug candidates (84); the NicOx compound nitroaspirin, reviewed in Gresele and Momi (119); •NO-releasing celecoxib analogs, inhibitors of inducible cyclooxygenase (44); and a vitamin E analog with •NO donor function (191).
At present, numerous established pharmaceutical enterprises and start-up companies are interested in the development of new hybrid molecules with •NO-releasing properties. The idea behind this research is not only to develop new beneficial functions of established drugs or to improve known principles of action but also to suppress side effects of these drugs. We ourselves have synthesized and tested a number of organic nitrate hybrids that are based on the known cardiovascular drugs, cilostazol (a PDE inhibitor) and pioglitazone (peroxisome proliferator-activated receptor-γ agonist), with potent vasodilator properties and beneficial profile regarding the side effects (oxidative stress, endothelial dysfunction, and nitrate tolerance) (160, 161).
VII. Summary and Future Perspectives for the Class of •NO Donors
Nowadays, organic nitrates should not be considered as a homogenous exchangeable class of •NO-releasing vasodilating compounds since they display a considerable diversity (110). Especially, GTN induces clinical tolerance, oxidative stress, and endothelial dysfunction, side effects, which are, to an extent, more or less shared with the other nitrates, ISMN and ISDN. ISMN is more likely to induce endothelial dysfunction and increases oxidative stress via activation of the vascular/phagocytic NADPH oxidase. In contrast, PETN is obviously devoid of these adverse side effects via acting on the powerful antioxidant enzyme HO-1.
Much less is known about ISDN. GTN and PETN are both bioactivated by the ALDH-2, and GTN, but not PETN, therapy causes simultaneously a deactivation of its own bioactivating pathway. Oxidative stress plays an important role in the development of nitrate tolerance and endothelial dysfunction in response to organic nitrates, although the extent and the cellular source of oxidative stress differ markedly between the ISMN, ISDN, and GTN. Oxidative stress induction represents a major limitation for the clinical use of organic nitrates since RONS formation per se is a hallmark of cardiovascular disease and actively contributes to their progression (38, 300). Organic nitrate therapy could therefore even elicit additional harmful effects in concert with RONS-associated cardiovascular disease (319).
Thus, cotreatment with direct and indirect antioxidants (e.g., vitamin C, ACE inhibitors, statins, hydralazine, or lipoic acid) has been shown to prevent these side effects, which clearly may limit their long-term clinical efficacy. A new group of nitrates, the so-called aminoalkyl nitrates, provides a good compromise between induction of nitrate tolerance and vasodilatory potency. Another strategy to avoid tolerance and endothelial dysfunction may be based on the development of hybrid molecules (e.g., combination of •NO-releasing function and NSAIDs in one molecule, see nitroaspirin). Thus, there are multiple examples for the development of these hybrid molecules, which could provide the basis for the nitrate of the future with intrinsic antioxidant properties guaranteeing continued therapeutic effects.
Footnotes
Acknowledgments
The authors thank Margot Neuser and Thilo Weckmüller for graphical support as well as Abby Russel for professional English editing service. This study was supported by the Center for Translational Vascular Biology (CTVB) and the Center for Thrombosis and Hemostasis (CTH, funded by the Federal Ministry of Education and Research, BMBF 01EO1003) of Johannes Gutenberg University Medical Center, Mainz, Germany, and the “Stiftung Mainzer Herz,” as well as Actavis Deutschland GmbH (A.D. and T.M.) with long-lasting support of our research on organic nitrates. T.M. is PI of the DZHK (German Center for Cardiovascular Research), Partner Site Rhein-Main, Mainz, Germany. A.D. and T.M. received honoraries and research support from Actavis Deutschland GmbH (Langenfeld, Germany), the manufacturer of PETN.