
Abstract
Introduction
H
Several reports have demonstrated that umbilical cord blood-derived mesenchymal stromal cells (UCB-MSCs) during long-term expansion become susceptible to senescence paralleled with an altered secretory phenotype, termed the senescence-associated secretory phenotype (SASP). However, to what extent and by what means the SASP contributes to the senescence of adult tissue stem cells remain unknown. We showed that monocyte chemoattractant protein-1 (MCP-1) is a crucial component of the SASP in human UCB-MSC senescence and determines their responses to allergic asthma treatment in vivo. MCP-1 induction is epigenetically regulated by a reduction in BMI1 in senescent UCB-MSCs. The interplay between BMI1 and MCP-1 consolidates the senescence program in UCB-MSCs, possibly determining their therapeutic potency.
Cellular senescence is triggered by the gradual accumulation of DNA damage and epigenetic alterations that can directly affect the expression of senescence-associated genes (48). Due to incomplete and erratic DNA replication, senescent cells create an intrinsic biological clock for cellular aging and converge on the so-called Hayflick limit of cell division at the molecular level (7, 19, 37, 52). The nuclei of senescent cells are characterized by irreversible changes, resulting in an aberrant nuclear shape (30), loss of heterochromatin-associated protein (e.g., heterochromatin protein-1) structures (22), altered patterns of histone modification that are frequently observed in aged cells (46), and transcriptionally repressive heterochromatin structure called senescence-associated heterochromatin foci, which induces the stable repression of E2F-target genes and represses some growth-promoting genes through the recruitment of the retinoblastoma (Rb) tumor suppressor (36). Furthermore, the conventional cell culture protocol that uses 21% of ambient oxygen could generate reactive oxygen species (ROS) (26), which induce aging-associated cellular responses by activating various signaling pathways, such as tumor protein p53 (p53), nuclear factor of kappa light polypeptide gene enhancer in B-cells (NFκB), phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/v-akt murine thymoma viral oncogene homolog (AKT), and extracellular signal-regulated kinase (ERK)/c-Jun kinase (JNK)/p38 mitogen-activated protein kinase (p38-MAPK) (15). Although it is suggested that the culture conditions and intrinsic nuclear alterations could cross talk during the senescence process, the detailed mechanism remains to be determined.
Recently, it has been reported that cellular senescence is paralleled by a striking increase in the secretion of many factors that participate in intercellular signaling, termed the senescence-associated secretory phenotype (SASP) (16, 29, 44). Although several groups have reported that the execution of cellular senescence involves, and often requires, the secretion of a plethora of factors, it is not immediately clear which soluble factors are major contributors to the SASP and what exact role it plays in cellular senescence (4). In the present study, we show for the first time that senescent UCB-MSCs secrete monocyte chemoattractant protein-1 (MCP-1) as a dominant secreted chemokine that positively relays senescence signaling via its cognate receptor chemokine (c-c motif) receptor 2 (CCR2) and reinforces senescence by increasing the protein levels of p53 and p21 via ROS or p38-MAPK signaling. Accordingly, the knockdown (KD) of CCR2 significantly improves the therapeutic outcome of UCB-MSCs by decreasing both cellular and inflammatory mediators in a severe asthma animal model. Moreover, BMI1 protein, a member of the polycomb repressor complex-1 (PRC1), binds to regulatory elements of the MCP-1 locus. Accordingly, a decrease in BMI1 during UCB-MSC senescence derepresses the transcription of MCP-1. Our results thus provide the first evidence supporting the existence of the SASP in senescent UCB-MSCs and contribute novel insights into the role of SASP signaling, which is a causative contributor to UCB-MSC senescence and positively connects the intrinsic and extrinsic circuitry of the process.
Results
Long-term expansion of UCB-MSCs induces cellular senescence
During long-term expansion of UCB-MSCs (more than 11 passages), the cells reached a senescent phase and stopped proliferating in vitro, although the growth rate depended on the donor and passage number (Supplementary Fig. S1A; Supplementary Data are available online at
Senescent UCB-MSCs possess a secretion profile
The senescent cells showed their characteristic protein secretion profile (secretome), referred to as the SASP (16). MSCs secrete a variety of cytokines and growth factors with paracrine and autocrine activities that can determine their therapeutic outcomes (20, 42). Thus, we hypothesized that senescent UCB-MSCs would also use a similar SASP phenomenon to actively regulate the senescence process. To address this hypothesis, we performed a cytokine array containing 200 proteins (interleukins, 35; chemokines, 11; other cytokines, 154; Supplementary Fig. S3A) using conditioned medium (CdM) from early- and late-phase UCB-MSCs. The levels of five proteins—MCP-1, interleukin-6 (IL-6), neurotrophin 4 (NT-4), chemokine cxc motif ligand 16 (CXCL16), and Fas-ligand (FASLG)—were markedly increased in the CdM of late-phase cells. Of these, the increase in IL-6 and MCP-1 was the highest (Fig. 1A). When we measured their mRNA expression levels, only the MCP-1 transcript was significantly increased in the late phase of UCB-MSCs compared with early-phase cells (Fig. 1B).
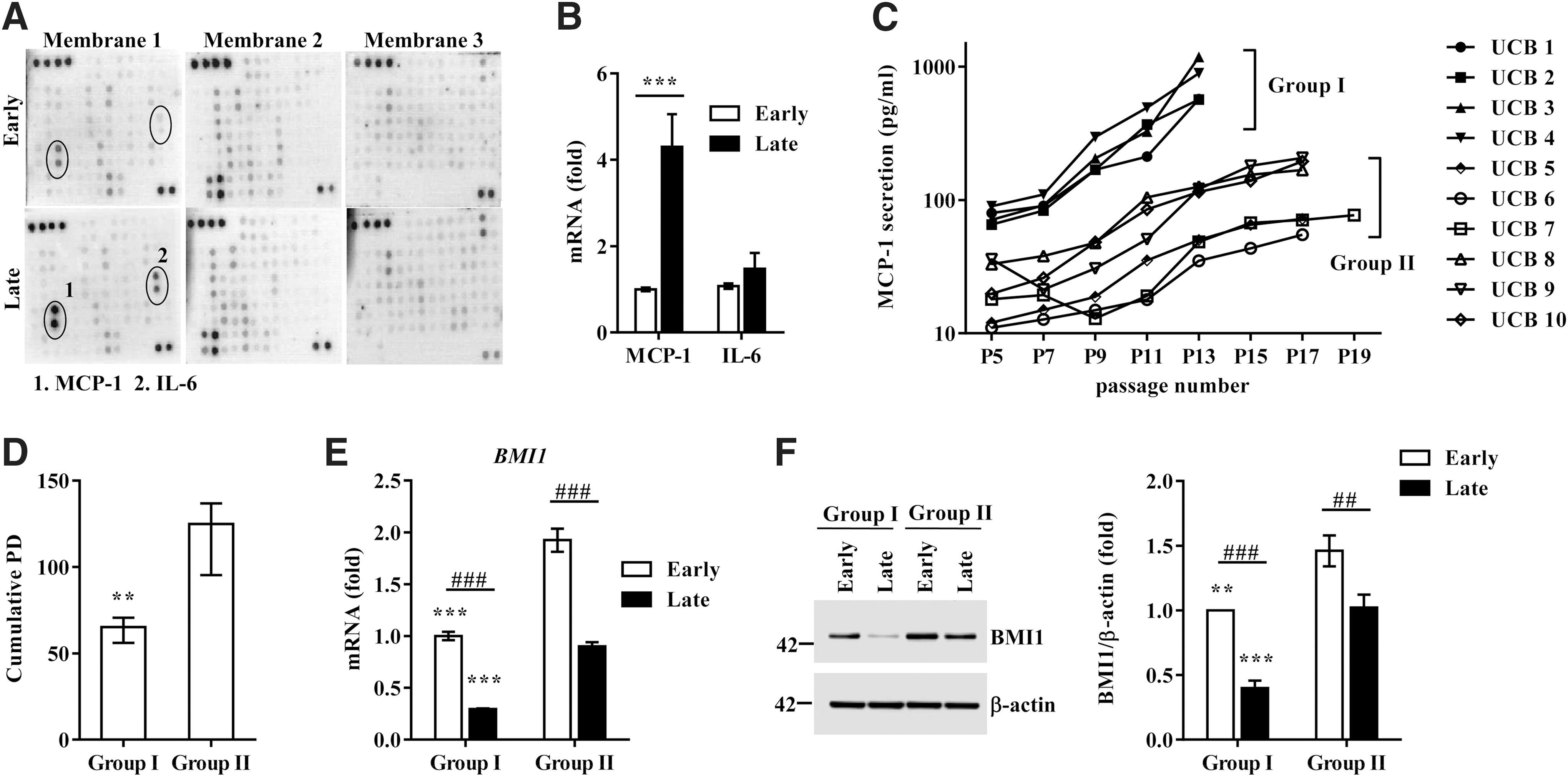
To further validate the association between MCP-1 secretion and UCB-MSC senescence, we quantified the amount of MCP-1 protein in CdM of UCB-MSCs derived from 10 individual donors. As shown in Figure 1C, the secretion of MCP-1 gradually increased as the passage number increased in all of the UCB-MSCs tested. Notably, according to the amount and rate of the increase in MCP-1 secretion with each passage, UCB-MSCs could be classified into two groups. These groups showed significant differences in basal MCP-1 secretion at the initial passage (group I vs. group II: 76.7 ± 10.8 pg/ml vs. 21.6 ± 11.0 pg/ml; p < 0.05) and the increase from the initial passage to the final passage (group I vs. group II: 10.6 ± 4.1-fold vs. 5.7 ± 2.3-fold; p < 0.01). Particularly, the cells in group I ceased to proliferate sooner and characteristically showed a faster senescence pattern with a lower cumulative PD and shorter time to doubling cessation than the cells in group II (group I vs. group II: 63.9 ± 6.7 PD vs. 119.7 ± 20.2 PD; p < 0.01) (Fig. 1D). In addition, the levels of both BMI1 transcript and protein were significantly higher in group II than in group I (Fig. 1E, F). We also observed significantly increased secretion of IL-6 protein in the late-phase UCB-MSCs (Supplementary Fig. S3B). However, this tendency varied considerably depending on the donor, which could be the reason for the minor change in the IL-6 transcript (Fig. 1B). Taken together, these data demonstrate that long-term cultivation of UCB-MSCs stimulates the SASP, which releases MCP-1 protein and determines the heterogeneity regarding the senescence process in UCB-MSCs.
MCP-1 increases senescence in UCB-MSCs
To examine the causative role of MCP-1 in UCB-MSC senescence, 100 ng/ml of recombinant MCP-1 protein (54, 56) was added for 48 h to UCB-MSCs at each passage from the intermediate phase (average P9–11), in which cells showed prominent growth arrest and MCP-1 secretion, up to the late phase. Compared with the untreated control, MCP-1 treatments significantly increased SA-β-gal activity with passaging by up to eight-fold (Fig. 2A and Supplementary Fig. S4A), concomitantly with a considerable delay in cell growth (Fig. 2B). MCP-1 treatment activated the p53-p21 signaling cascade, increasing the protein level of p53 phosphorylated at Ser392 and total p21 (Fig. 2C). In contrast, the pRb pathway was largely unaffected by MCP-1 treatment.
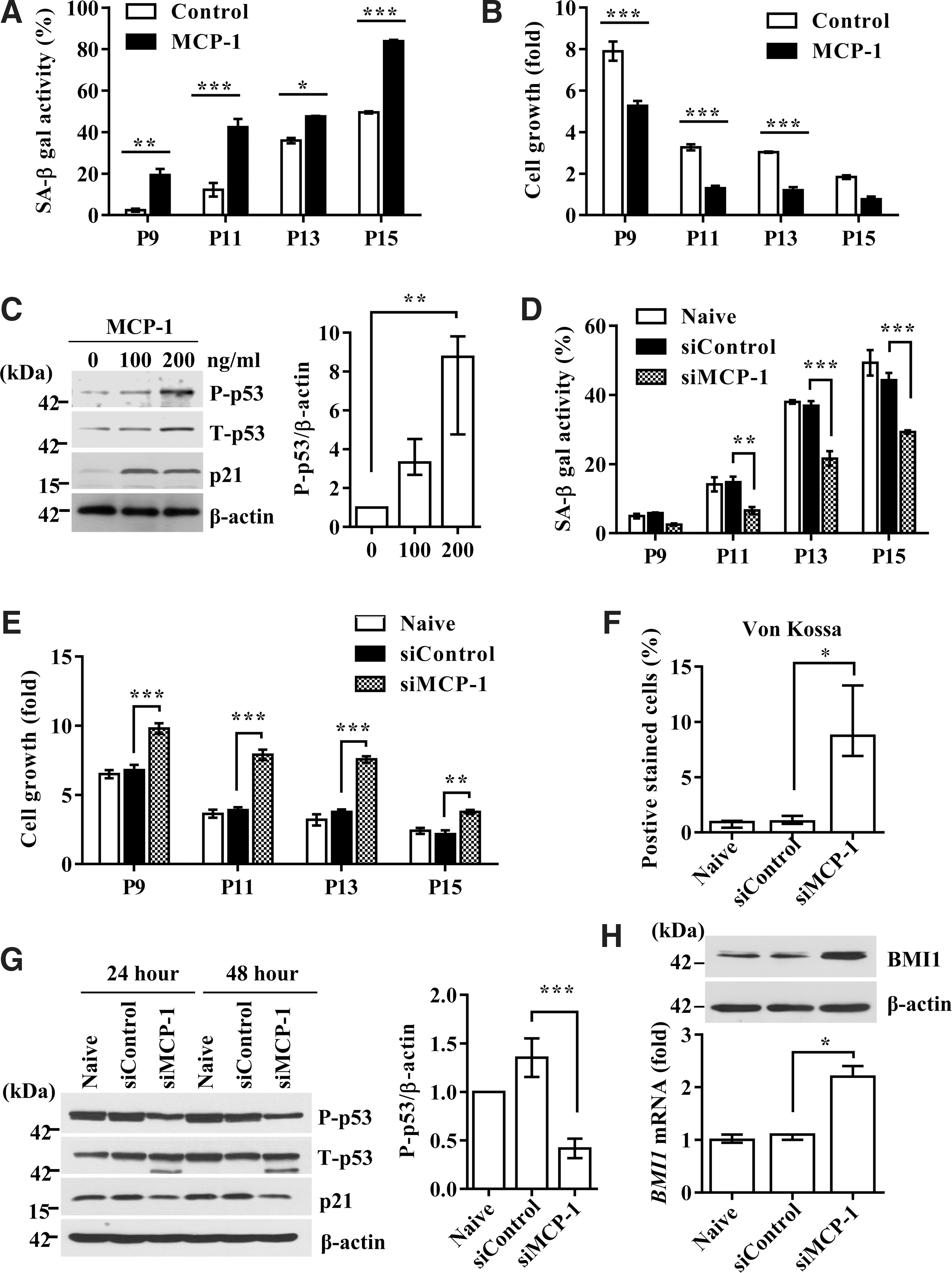
The use of siRNA to silence MCP-1 in UCB-MSCs (Supplementary Fig. 4B) significantly blocked both SA-β-gal activity induction and delayed cell growth (Fig. 2D, E, and Supplementary Fig. S4C). Moreover, MCP-1 KD UCB-MSCs showed restored osteogenic differentiation with passaging (Fig. 2F and Supplementary Fig. S4D). At the molecular level, MCP-1 KD cells showed attenuated activation of the p53-p21 pathway (Fig. 2G) and, simultaneously, a 2.2-fold increased expression of BMI1 transcript, which increased the level of BMI1 protein (Fig. 2H). Taken together, these data demonstrate that MCP-1 protein secreted as a component of the SASP plays a causative role in inducing the senescence of UCB-MSCs.
MCP-1-induced UCB-MSC senescence is mediated by its canonical receptor, CCR2
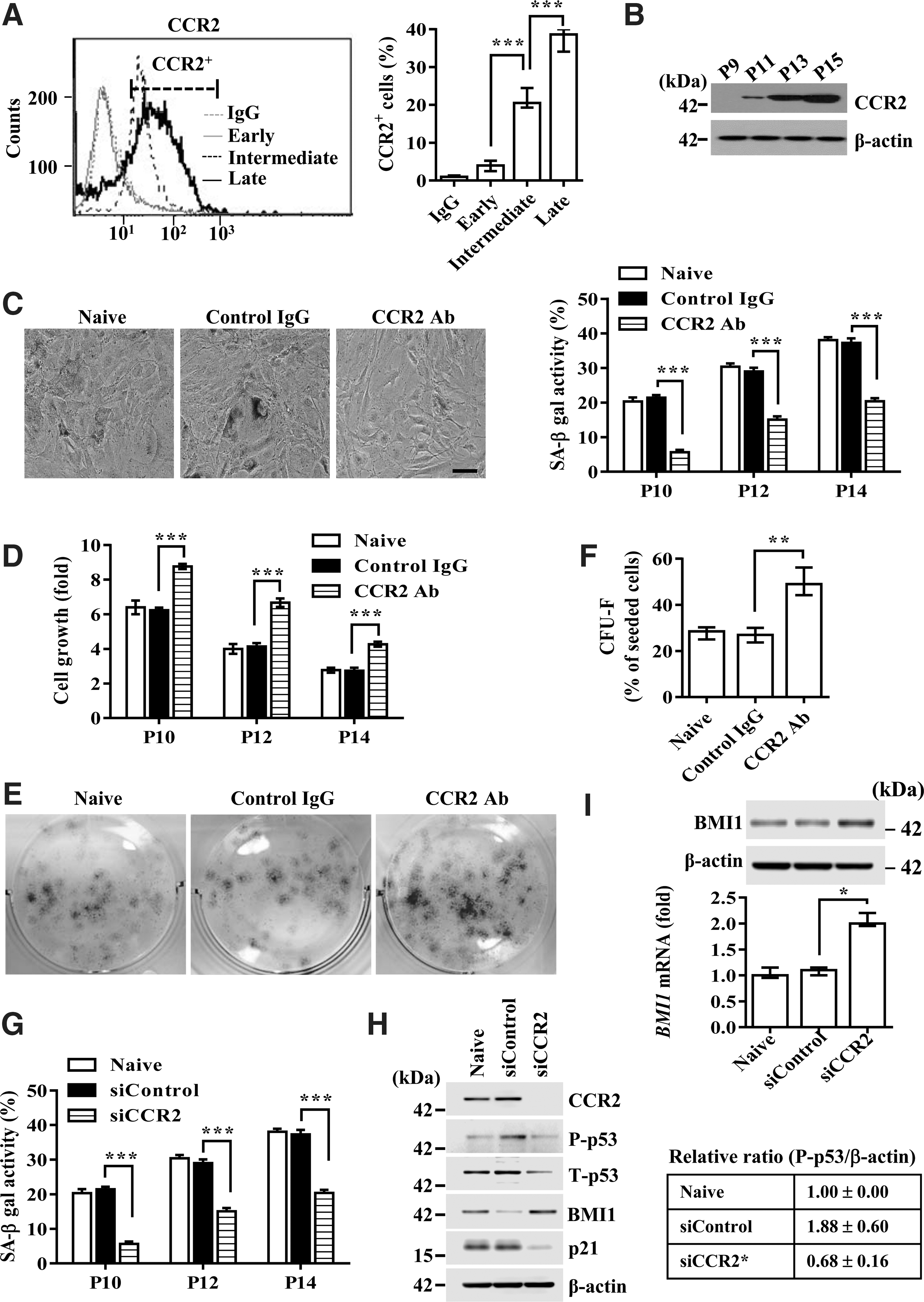
MCP-1 is a proinflammatory cytokine that exerts its biological effects through its cognate receptor, CCR2 (5, 45). The expression of CCR2 in UCB-MSCs was lower in the early phase, but it was considerably upregulated in the intermediate phase and further increased in the terminal senescent phase (Fig. 3A, B). Importantly, treatment with CCR2 blocking antibody significantly reduced SA-β-gal expression (Fig. 3C) and enhanced the growth rate (Fig. 3D) and colony-forming capacity (Fig. 3E, F) of UCB-MSCs in the intermediate phase. The significance of CCR2 was further validated by the fact that CCR2 KD cells had decreased SA-β-gal activity (Fig. 3G) and p53-p21 pathway activation (Fig. 3H). However, they had a 1.9-fold increased expression of BMI1 transcript, which simultaneously increased the level of BMI protein (Fig. 3I). Furthermore, overexpression of CCR2 in early-phase UCB-MSCs remarkably increased cellular senescence, as indicated by induction of SA-β-gal activity (Supplementary Fig. S5A), activation of the p53-p21 signaling cascade, and downregulation of BMI1 expression (Supplementary Fig. S5B). Notably, silencing of MCP-1 less affected the senescence triggered by overexpression of CCR2. These results collectively suggest that CCR2 is a key signaling factor for the secreted MCP-1 that triggers the cellular senescence of UCB-MSCs.
Autocrine/paracrine-positive loop of MCP-1 secretion through CCR2
Cellular senescence can be stably sustained by the autocrine/paracrine-positive regulatory loop of secreted SASP factors (28, 43). To test whether the MCP-1 and CCR2 signaling cascade can use a similar mechanism, we first examined whether MCP-1 and CCR2 could regulate each other's expression. Exposure of intermediate-phase UCB-MSCs to MCP-1 protein increased the expression of CCR2 in a time-dependent manner (Fig. 4A, B). Similarly, the repression of CCR2 by either siRNA or a blocking antibody decreased the secretion of MCP-1 by 1.5-fold (Fig. 4C), suggesting positive feedback between MCP-1 and CCR2.
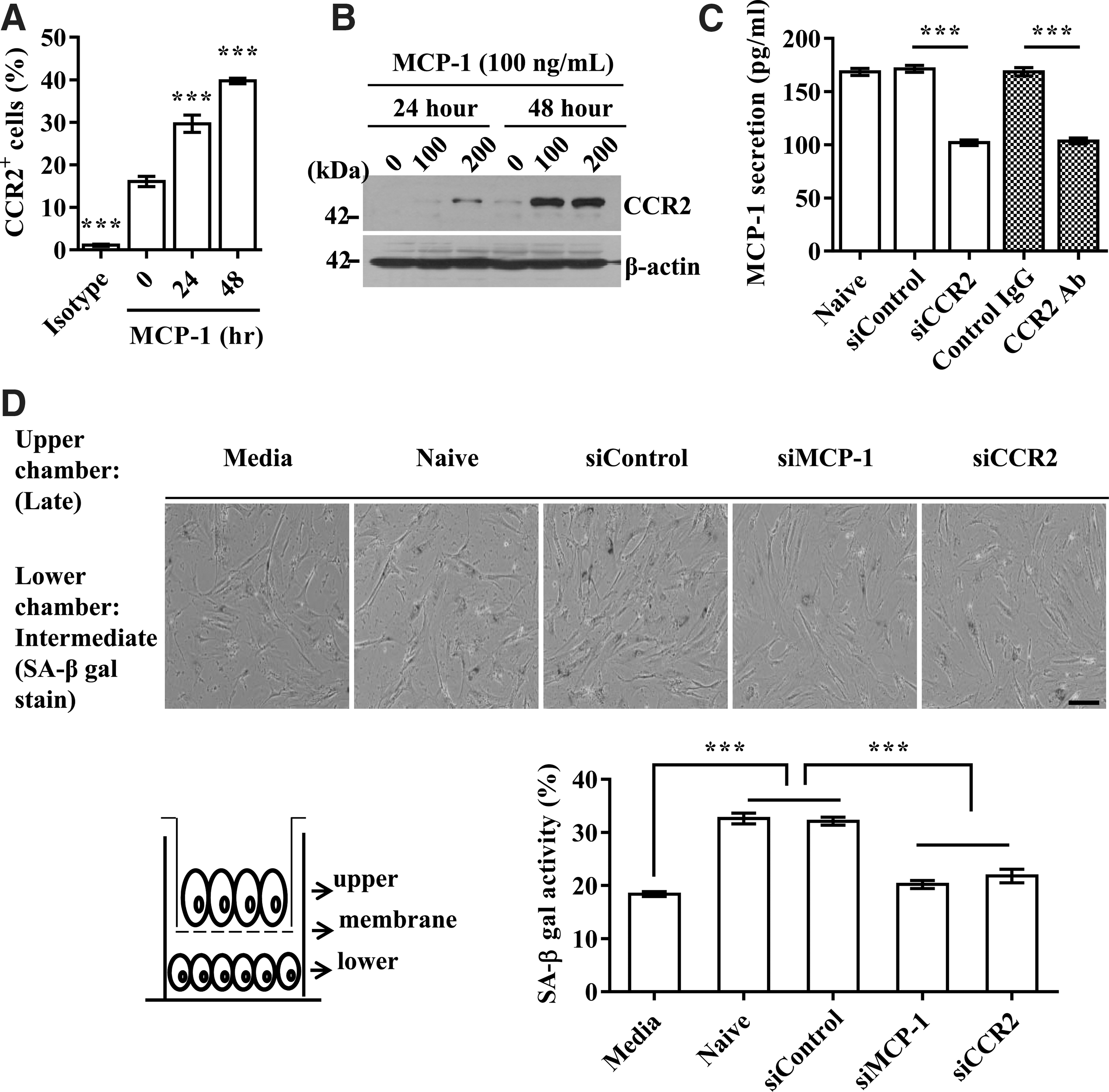
Next, to determine whether the MCP-1 and CCR2 cascade can trigger UCB-MSC senescence as a paracrine action, we cocultured intermediate- and late-phase UCB-MSCs in a Transwell chamber that prevents direct cell–cell contact due to its small pore size (1 μm). The SA-β-gal activity in intermediate-phase cells was increased by coculture with late-phase senescent cells. However, SA-β-gal activity was significantly reduced when the late-phase cells in the upper chamber were treated with MCP-1 or CCR2 siRNA (Fig. 4D). Together, these results demonstrate that the autocrine/paracrine amplification feedback between MCP-1 and CCR2 is essential to the progression of UCB-MSC senescence.
Role of oxidative stress and p38-MAPK in MCP-1-mediated UCB-MSC senescence
Oxidative stress is a central contributor to senescence-like cell arrest (12,18) and is induced by the MCP-1 and CCR2 cascade in immune cells (17). Accordingly, the level of ROS was significantly increased by long-term expansion of UCB-MSCs (Fig. 5A) and treatment with MCP-1 recombinant protein. However, this increase was prevented by CCR2 blocking antibody (Fig. 5B). Moreover, p38-MAPK, a crucial mediator of ROS-induced senescence, was activated from 8 h and peaked at 18 h after MCP-1 stimulation (Fig. 5C). Importantly, treatment with SB203580, a chemical inhibitor of p38-MAPK, significantly abrogated the increase in phosphorylated p53 and p21 proteins induced by MCP-1 stimulation (Fig. 5D, E). Addition of N-acetyl-L-cysteine (NAC), an ROS scavenger, also markedly suppressed the activation of both p38-MAPK and p53-p21 cascades (Fig. 5D, F). Accordingly, SA-β-gal activity (Fig. 5H) as well as the decrease in the levels of BMI1 transcript and protein (Fig. 5G) in the MCP-1-treated UCB-MSCs was significantly abrogated by the interference of p38-MAPK or ROS. Thus, these results suggest a significant role of ROS in MCP-1-mediated UCB-MSC senescence.
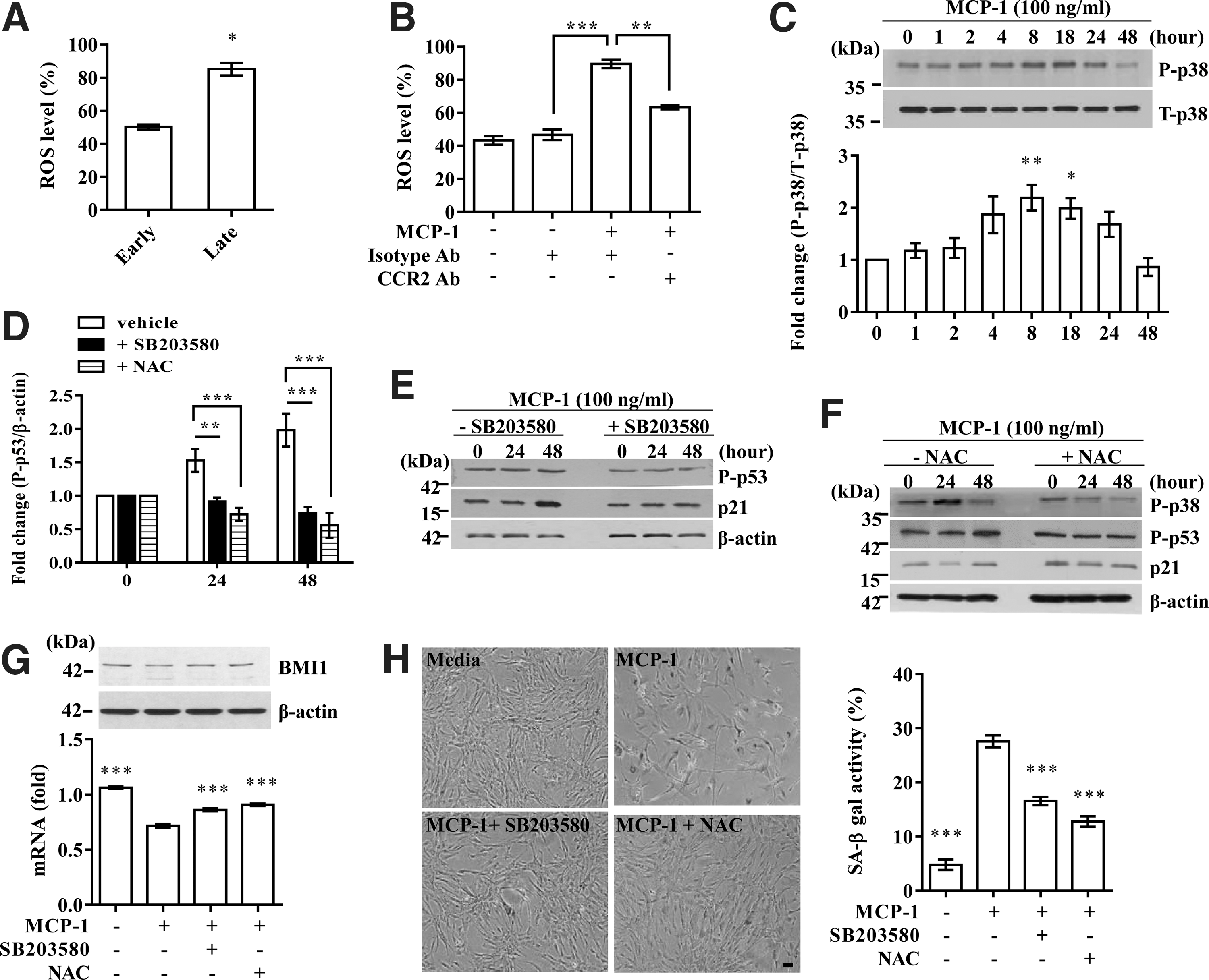
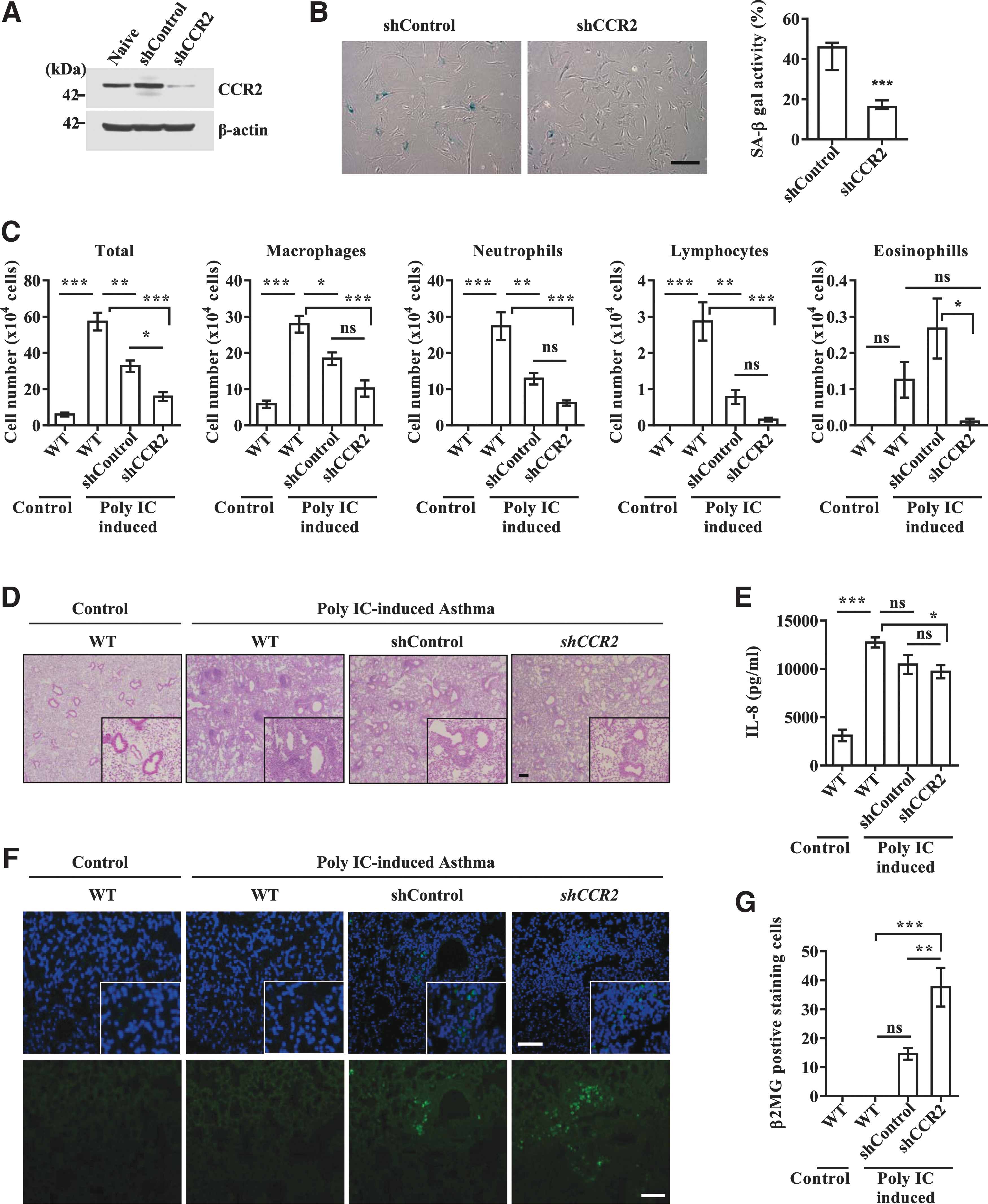
Blocking the MCP-1-CCR2 cascade enhances the therapeutic capacity of UCB-MSCs in an asthma animal model
Next, we investigated the significance of MCP-1-mediated UCB-MSC senescence under in vivo conditions. To address this issue, we compared the therapeutic outcome of UCB-MSCs stably expressing shRNA targeted against CCR2 and scrambled shRNA using a severe murine asthma model. Before injection of the cells, we confirmed the decreased level of CCR2 protein and SA-β-gal activity in CCR2 KD UCB-MSCs (Fig. 6A, B). Furthermore, silencing of CCR2 cells significantly prevented the MCP-1-induced senescent phenotypes, including activation of SA-β-gal and p53-p21 cascades (Supplementary Fig. S5C, D). Similar to previous reports (3, 31), the OVA- and polyIC-sensitized mice had significantly increased levels of cellularity and inflammatory cells, including macrophages, neutrophils, and lymphocytes, in bronchoalveolar lavage fluid (BALF) (Fig. 6C) and around the bronchial and vascular area in lung tissues (Fig. 6D). These parameters were reduced by administration of both control and CCR2 KD UCB-MSCs. However, CCR2 KD cells more effectively attenuated the infiltration of inflammatory cells in BALF and lung tissues than control UCB-MSCs (Fig. 6C, D). When inflammatory cytokines were measured, injection of UCB-MSCs reduced the level of IL-8 proteins in BALF regardless of the expression level of CCR2, with a greater reduction in CCR2 KD UCB-MSC-injected mice (Fig. 6E). The expression levels of IL-5, IFN-γ, and IL-17 were largely unaffected in the BALF of all of the groups tested (data not shown). When we compared the number of engrafted cells by staining the lung tissue with antibody specific to human β2 microglobulin, CCR2 KD UCB-MSCs yielded greater engraftment capacity in the lung than control cells (Fig. 6F, G). Thus, these results demonstrate that repression of the MCP-1 and CCR2 cascade reinforces the ability of UCB-MSCs to alleviate the inflammatory response in the experimental allergic asthma model.
Decline in BMI1 during UCB-MSC senescence derepresses the transcription of MCP-1
Next, we investigated whether the cell-intrinsic senescent program could be linked to the MCP-1-mediated SASP. According to our molecular analysis, p53, p21, and BMI1 were consistently implicated in the MCP-1-mediated SASP observed in senescent UCB-MSCs. The significance of p53 was further confirmed by the finding that ectopic expression of p53 or treatment with nutilin-3, a p53 activator, remarkably increased the secretion of MCP-1 in early-phase UCB-MSCs (Supplementary Fig. S6).
BMI1, a transcriptional repressor, prevents the premature activation of senescence-associated genes, including INK4a and ARF, in a variety of adult stem cells by binding to its target loci (8, 34, 39). Thus, we tested whether MCP-1 expression could be epigenetically regulated by BMI1 protein during UCB-MSC senescence. According to DNase I hypersensitivity and the ChIP-seq database (histone modifications and two transcription repressors, EZH2 and HDAC1) of the entire MCP-1 locus, which is available via the ENCODE project with the UCSC genome browser (
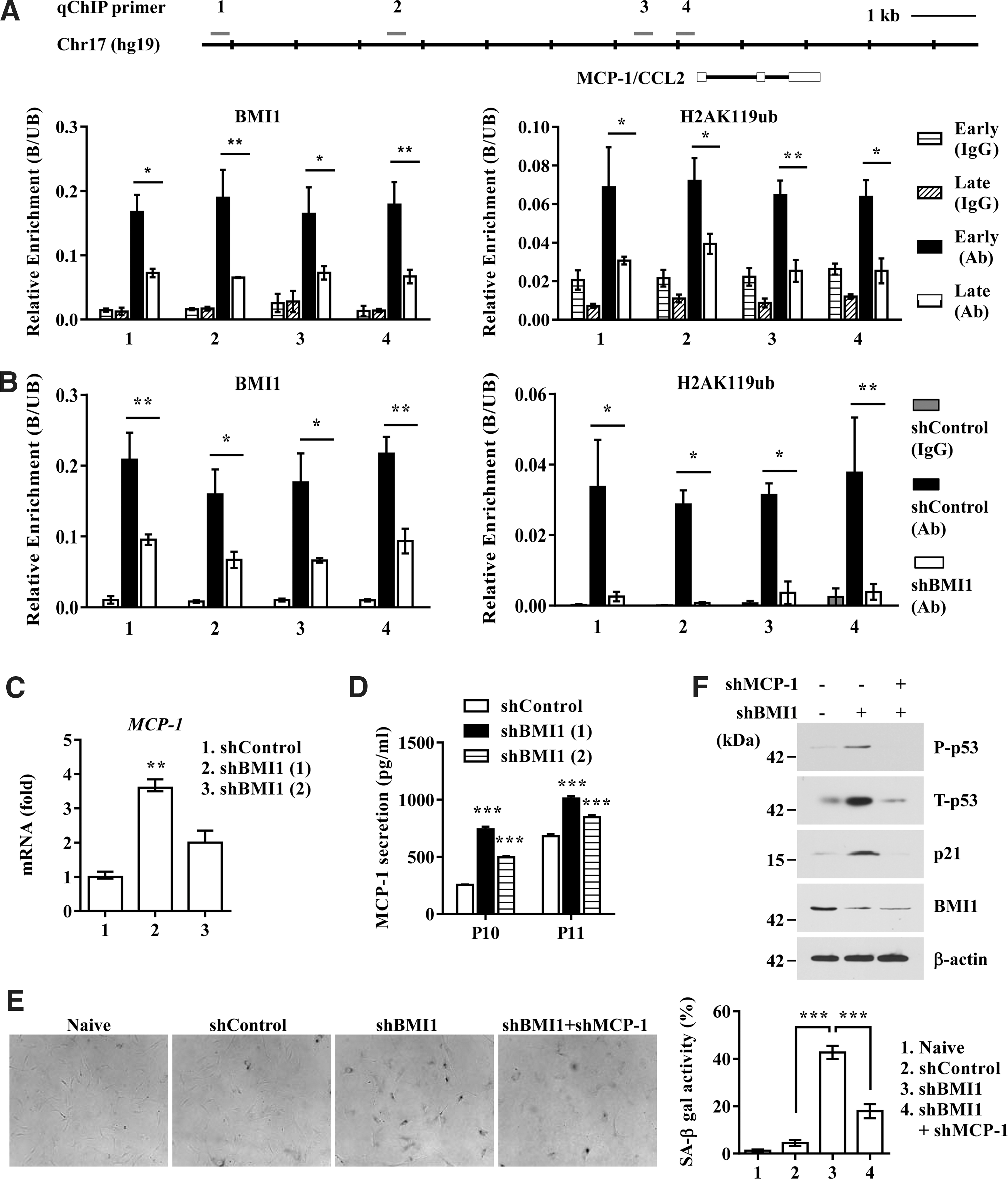
Finally, to better support a role for BMI1 protein in maintaining the repressive epigenetic status of the MCP-1 locus in early-phase UCB-MSCs, we established stable cells expressing BMI1 shRNA (Supplementary Fig. S8A). As previously reported, BMI1 KD UCB-MSCs exhibited the typical senescent phenotype, such as an enlarged and flattened morphology and increased SA-β-gal activity (Supplementary Fig. S8B, C). As expected, the promoters of MCP-1 in BMI1 KD cells lost the recruitment of BMI1 and the concomitant H2AK119Ub histone modification (Fig. 7B). The epigenetic change in the MCP-1 locus in BMI1 KD cells coincided with derepression of MCP-1 transcription (Fig. 7C), which resulted in increased secretion of MCP-1 protein (Fig. 7D). The enhanced secretion of MCP-1 protein in BMI1 KD cells triggered cellular senescence, as judged by the increase in SA-β-gal activity (Fig. 7E) and the activation of the p53-p21 signaling pathway (Fig. 7F). Most importantly, the senescent phenotypes shown in BMI1 KD cells were significantly rescued after silencing of MCP-1 (Fig. 7E, F). Taken together, these results demonstrate that BMI1, using a similar mechanism to that seen in the INK4 locus, is responsible for sustaining the repressive epigenetic stability in the promoter of MCP-1, which plays a key role in SASP-mediated stimulation of senescence of UCB-MSCs.
Discussion
Extensive ex vivo expansion by long-term cultivation is required to acquire sufficient numbers of cells for MSC-based therapies. Our present study demonstrated that senescent features of UCB-MSCs that develop through expansion in culture were stimulated by MCP-1, a main mediator of the SASP in UCB-MSCs whose expression was epigenetically regulated by the BMI1 polycomb group protein. In addition, depending on basal MCP-1 secretion, UCB-MSCs exhibited heterogeneous susceptibility to senescence during sustained culture (Fig. 1C), and the expression level of CCR2 was strongly associated with the senescent phenotype of UCB-MSCs (Fig. 3). These results suggest that the status of MCP-1 and CCR2 signaling activity could be adopted as an index for assessing the commitment of the cell to senescence. The MCP-1 and CCR2 signaling activity could also be a good predictor of susceptibility to senescence in response to environmental stresses, which is critical for their therapeutic outcomes. Indeed, in an allergic asthma animal model, CCR2 KD in UCB-MSCs led to greater beneficial effects by not only improving the engraftment capacity of infused stem cells but also reducing the airway inflammation response (Fig. 6). Moreover, since senescence-associated MCP-1 secretion was also seen in bone marrow-derived MSCs (data not shown), the roles of the MCP-1-centered SASP in other adult stem cells, such as hematopoietic stem cells (HSCs) and neural stem cells (NSCs), should be investigated to verify whether the SASP could be exploited to regulate the senescence progress of adult stem cells. Interestingly, extracellular vesicles released from bone marrow-derived MSCs play a beneficial paracrine role in reversing injury to renal (9, 10) and lung (41) tissues. The secreted extracellular vesicles, enclosed in a lipid bilayer vesicle with a diameter of ∼50–100 nm, can alter the phenotype of neighboring cells by delivering proteins, mRNA, microRNA, and long noncoding RNA contained in the extracellular vesicle to target cells (41). Thus, elucidation of a potential role of extracellular vesicles in the SASP in UCB-MSCs could be required for further studies.
As a general cause of senescence, oxidative stress and activation of its associated signaling cascades (e.g., p38-MAPK, NF-κB, C/EBPβ, p53, p21) could be generated by chronic exposure to extrinsic stresses. The secreted MCP-1 protein, acting through the CCR2 receptor, increased ROS and in turn activated p38-MAPK and p53/p21 signaling (Fig. 5A–F). Notably, p53, activated by either ectopic expression or its chemical activator, significantly increased the secretion of MCP-1 (Supplementary Fig. S6). Thus, oxidative stress acts as a pivotal second messenger in consolidating MCP-1-mediated stem cell senescence through positive feedback amplification. Indeed, treatment with NAC effectively prevented the senescence of UCB-MSCs after exposure to recombinant MCP-1 protein (Fig. 5G, H). It is well recognized that stem cells in the mesenchymal niches in the body have a relatively low concentration of oxygen at 1%–8% (6, 32). Thus, 21% ambient oxygen would produce oxidized biological macromolecules and result in the generation of ROS (26). Indeed, UCB-MSCs maintained under hypoxic (5% oxygen) conditions showed improved cell growth capacity (Supplementary Fig. S9A) and expression of BMI1 protein (Supplementary Fig. S9B), but reduced induction of the p53-p21 signaling cascade (Supplementary Fig. S9B) and MCP-1 protein secretion (Supplementary Fig. S9C) compared with cells under normoxic (21% oxygen) conditions. These results suggest that the culture conditions relevant to the cellular level of ROS could be a practical strategy to retard the progression of cellular senescence during ex vivo expansion of UCB-MSCs.
In addition, we suggest BMI1 as another possible candidate that mediates this positive feedback because its expression in UCB-MSCs was decreased by MCP-1 and because BMI1 stably maintained the repressive epigenetic status of the MCP-1 locus in early-phase UCB-MSCs (Fig. 7). Since BMI1 is downregulated in replicative senescent fibroblasts, both cell-intrinsic and -extrinsic stimuli converged on reduced BMI1 activity, which could stably reprogram the stressed cells to adopt a senescent phenotype by an epigenetic mechanism.
BMI1 is highly expressed in numerous adult stem cells to protect them against premature aging by epigenetically repressing aging-associated gene expression (38). Indeed, a deficiency of Bmi-1 in HSCs and NSCs results in premature aging and a decreased regenerative capacity (34, 39). The best-characterized target of Bmi-1 is the Ink4 locus, which, by alternative splicing, encodes two important tumor suppressors, p16Ink4a and p19Arf (8). As Bmi-1 tends to be repressed during aging, the Ink4 locus genes become progressively derepressed in several tissues (27) and stem cells (47) from older individuals. Notably, deletion of Ink4a or Arf from Bmi-1 −/− mice partially rescued the self-renewal capability of HSCs and NSCs (33,51), suggesting that an additional pathway must also function downstream of Bmi-1 in regulating stem cell aging. The present study provides experimental evidence that MCP-1 is a novel target gene for BMI1 protein to orchestrate stem cell senescence (Fig. 7).
Chronic inflammation has long been regarded as a causative contributor to age-associated degenerative ailments, including diabetes mellitus, atherosclerosis, osteoarthritis, neurodegenerative diseases, and cancers, which are major potential targets of MSC-based cell therapeutics (40). Chronic inflammatory responses redirect the protective inflammatory response to danger signals in the acute phase of inflammation into a self-destructive, damaging, and deregulated proliferation-prone environment. In this regard, chronic inflammation might be the origin site of cancer, and inflammatory microenvironments could be more common in aged tissues. Given that cell therapies are being clinically administered to treat chronic degenerative diseases with persistent inflammation, such an inflammation-prone micromilieu might affect the final outcome of cell therapies by affecting the senescence program of MSCs in a cell-autonomous or noncell-autonomous manner (1, 28, 43). Several studies have reported that a long-lasting deviant profile of inflammatory molecules, including MCP-1, was causatively involved in the initiation and maintenance of aging-related diseases and aging processes (21, 35, 50).
With this in mind, several new angles on the preparation of next-generation MSC-based cellular therapies should be considered. First, ready-to-use MSC-based cell therapies should be cautiously monitored to remove contamination from senescent cells, which could be conducted via a surrogate marker such as MCP-1 and CCR2 that can reveal how far the cells have committed to senescence. Even a small population of senescent cells in the therapy may trigger a stress response inside the other young healthy cells that reinforces itself through a vicious loop. Accordingly, the fewer the cells undergoing senescence in the therapy, the greater the benefit to patients. Once the senescence program in MSCs begins, functional compromise of their multipotency and consecutive loss of therapeutic competence inevitably result. Therefore, screening and depletion of the CCR2-expressing population could be of tremendous value when monitoring the quality of the expanded UCB-MSCs. Notably, these important concepts for MSC therapy were experimentally proven in vivo using the severe murine asthma model (Fig. 6). Thus, cell therapies should be furnished as interfered MCP-1/CCR2 signaling acting on the senescence program, although the preparation of such customized cell therapies would be costly and challenging.
In the present study, we provide the first evidence supporting the existence of the SASP as a causative contributor to UCB-MSC senescence and positive feedback between the intrinsic and extrinsic circuitry of the process. Therefore, our current results provide important insights into an enhanced and integrative understanding of both the functional aspects and regulation of the SASP, which may open up new therapeutic modalities for the preparation of next-generation MSC-based cell therapies.
Materials and Methods
Cell culture
This study was approved by the Institutional Review Board of MEDIPOST Co., Ltd. UCB was collected from umbilical veins after neonatal delivery with maternal informed consent (Supplementary Table S1). UCB harvests were processed within 24 h of collection. UCB was separated by isolating mononuclear cells with Ficoll-Hypaque solution (density, 1.077 g/cm3; Sigma, St. Louis, MO). The separated mononuclear cells were washed, suspended in Minimum Essential Medium α modification (MEM α; Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Gibco), and seeded at a concentration of 5 × 105 cells/cm2. Cultures were maintained at 37°C in a humidified atmosphere (21% O2) containing 5% CO2 with a change of culture medium twice a week (55). To maintain cells under hypoxic conditions, the oxygen concentration in the chamber was maintained at 5% with a residual gas mixture consisting of 5% carbon dioxide and balanced nitrogen. Primary bone marrow MSCs (five lots) were purchased from Lonza (PT-2501; Basel, Switzerland).
Reagents
Human recombinant MCP-1 protein (279-MC) was purchased from R&D Systems (Minneapolis, MN). The p38-MAPK inhibitor, SB203580, was obtained from Sigma. Anti-CCR2 antibody (ab32144; Abcam, Cambridge, MA) was used to block CCR2 stimulation. NAC, used to assess antioxidant effects, was purchased from Sigma. Detailed information on the reagents is provided in Supplementary Table S2.
Growth kinetics
Expansion during time in culture was measured using the trypan blue exclusion method. Each passage was cultured for 5 days and cells were obtained with trypsin-EDTA (Gibco), counted, and reseeded at a density of 2000 cells/cm2. Culture medium was replaced twice weekly. The PD was calculated for each passage by dividing the logarithm of the fold increase value obtained at the end of the passage by the logarithm of 2 (24). The PD and cumulative PD were continuously monitored until cells ceased to proliferate between 11 and 19 passages (Supplementary Fig. S1A). Cell growth in the experimental conditions was depicted as the fold increase in the cell number for each passage. A colony-forming unit–fibroblast (CFU-F) assay was performed by seeding 100 cells in a 35-mm culture dish (BD Biosciences, San Jose, CA) and incubating them in humidified 5% CO2 at 37°C; culture medium was exchanged every 3 days. After 2 weeks, the dishes were washed twice with phosphate-buffered saline (PBS; Gibco), fixed with 100% methanol, and stained with 3% crystal violet (Sigma). The number of colonies was counted.
Detection of the intracellular ROS level
ROS stress was assessed using a fluorescent probe (carboxy-H2DCFDA; C400; Invitrogen, La Jolla, CA) as previously described (53). Briefly, cells seeded in a T75 tissue culture flask at a density of 2000 cells/cm2 were maintained for 5 days before incubating with 10 μM C400 for 1 h. The ROS fluorescence intensity was analyzed using an FACSCalibur flow cytometer, and the ROS level was represented as the percentage of C400-stained cells.
Evaluation of the multilineage differentiation potential
Multilineage potential was assessed by incubating cells under specific conditions to induce differentiation into osteocytes, chondrocytes, and adipocytes (25). The procedures used are detailed in the Supplementary Materials and Methods.
Cytokine antibody array and enzyme-linked immunosorbent assay
To obtain CdM, cells were serum starved for 24 h, washed, and then cultivated for a further 24 h with fresh serum-free medium. CdM was collected and clarified by centrifugation at 200 g for 5 min. The presence of cytokines within the harvested CdM was detected using a commercially available proteome profiler array membrane, the Human Inflammatory Cytokine Array C2000 and Analysis Tool (AAH-CYT-2000) (RayBiotech, Norcross, GA). The detail of array maps in each membrane is described in Supplementary Figure S3A. Human MCP-1 (DCP00) and IL-6 (D6050) protein were quantified by an enzyme-linked immunosorbent assay (ELISA; R&D Systems) according to the manufacturer's protocol. Results were obtained by measuring absorbance at 450 nm and were normalized per 1 × 104 cell number.
Real-time polymerase chain reaction
Total RNA was isolated using an RNeasy Mini Kit (Qiagen, Crawley, UK) according to the manufacturer's instructions. One microgram of total RNA was reverse transcribed to cDNA using oligo-dT primers (Invitrogen) and superscript II polymerase (Invitrogen). Amplification was performed with the LightCycler real-time polymerase chain reaction (PCR) (Roche, Mannheim, Germany) system using LightCycler TaqMan Master Mixtures (Roche), sequence-specific probes (Roche), and specific primers (Supplementary Table S3). Relative quantities of the mRNA of interest were calculated by the comparative threshold cycle (2−ΔΔCt) method and normalized to those of β-actin. Values are expressed as fold increases relative to the levels of uninduced cells, defined as 1.
RNA interference
siRNAs for MCP-1, CCR2, and control were purchased from Dharmacon (Chicago, IL). The siRNA pool consisted of four duplexes (Supplementary Table S3). Cells were treated with control siRNA (50 nM), MCP-1 siRNA (50 nM), or CCR2 siRNA (50 nM) for 24 h using DharmaFECT reagent (Dharmacon). When cells were examined at multiple passages, de novo transfection of siRNAs was performed at each passage. To establish UCB-MSCs stably expressing MCP-1, CCR2, or BMI1 shRNA, the shRNA was cloned into pSicoR lentiviral vector (Addgene plasmid 12084). Lentivirus was produced by a four-plasmid transfection system according to the manufacturer's instructions (Invitrogen) and concentrated by precipitation using polyethylene glycol 6000. The concentrated virus was infected into UCB-MSCs with 10 μg/ml polybrene (Invitrogen). The cells were further maintained for at least 3 weeks in the presence of 1 μg/ml puromycin (Invitrogen) for selection, and the effect of ectopic expression was examined by quantitative PCR (qPCR) and Western blotting. The target sequences for each shRNA are indicated in Supplementary Table S3.
Overexpression and activation of p53 and CCR2
To overexpress the p53 protein, the coding sequence of full-length human p53 was cloned into the PCDNA3 expression vector. Cells were then transfected using transfection reagent (Invitrogen). For p53 activation, cells were treated with 10 μM nutilin-3 for 24 h (Sigma). Human CCR2 cDNA amplified by PCR using a human CCR2 open-reading frame clone (BC074751; Dharmacon) as a template was cloned into the pLenti7.5/V5 TOPO lentiviral vector (Invitrogen). Five days after infection of lentivirus containing the CCR2-expressing cassette, the effects of ectopic expression of CCR2 were examined using Western blot analysis.
SA-β-gal staining
SA-β-gal staining was used as a biomarker of senescence in UCB-MSCs. SA-β-gal activity was qualitatively assessed with a histochemical staining kit (CS0030; Sigma) according to the manufacturer's instructions, followed by inverted microscopy. The percentage of senescent cells was represented by the number of stained cells per the total number of cells.
Telomerase activity assay
Telomerase activity was measured by a telomeric repeat amplification protocol (TRAP) assay with a telomerase PCR ELISA kit (TeloTAGGG Telomerase PCR ELISA, 11854666910; Roche) according to a published protocol (14). Approximately 2 × 105 cells were harvested for each reaction and centrifuged at 3000 g for 10 min at 4°C. The absorbance for the final product was acquired by measuring absorbance at 450 nm.
Western blotting
Cell extracts were prepared in buffer containing 9.8 M urea, 4% CHAPS, 130 mM dithiothreitol, 40 mM Tris-HCl, and 0.1% sodium dodecyl sulfate. Protein concentrations were measured by a bicinchoninic acid assay (Sigma). Protein extracts (15 μg) were separated by SDS-PAGE and the resolved proteins were transferred to nitrocellulose membranes. Each membrane was incubated with anti-phospho-p53 (Ser392) (#9281), anti-p53 (#9282), anti-CCR2 (ab32144; Abcam), anti-p21 (#3688), anti-phospho-Rb (Ser780) (#9307), anti-Rb (#9309), anti-phospho-p38 (Thr180/Tyr182) (#9211), anti-p27 (#3688) (Cell Signaling, Danvers, MA), anti-BMI1 (39993; Active Motif, Carlsbad, CA), or anti-β-actin (A5441; Sigma). The density of signals for the indicated proteins was measured and quantified using NIH ImageJ software.
Asthma animal model
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Ulsan College of Medicine. To generate a murine model of severe asthma, 6-week-old BALB/c mice (OrientBio, Gapyong, Gyeonggi-do, Korea) were sensitized and challenged by intranasal administration with 75 μg OVA (Sigma) and 10 μg polyI:C (Calbiochem) at days 0, 1, 2, 3, 7, 14, 21, 22, and 23. Mice were injected with 3 × 105 UCB-MSCs by intravascular injection at day 15. BALF, lymph nodes, and lung tissues were obtained from the mice 24 h after the last immunization. The number of monocytes, eosinophils, neutrophils, and lymphocytes and the concentrations of IL-10 and IL-8 in BALF were measured as previously described (3, 31). For histopathological evaluation, the lungs were perfused with 5 ml PBS through the right ventricle and inflated with 1 ml PBS through the trachea. The inflated lungs were fixed by immersing them in 10% neutral buffered formalin solution for 24 h. Fixed lung tissues were embedded in paraffin and sectioned to 4 μm thickness and the magnitude of inflammation around the bronchial and vascular area was examined by hematoxylin and eosin staining. The engraftment of the infused UCB-MSCs was determined by immunofluorescence analysis of human B2-microglobulin (ab15976; Abcam) visualized using an FITC-labeled secondary antibody. Nuclei were counterstained using 4′,6-diamino-2-phenylindole (DAPI; Sigma).
ChIP assay
ChIP analysis was performed using a Magna ChIP™ G kit (Upstate-Millipore, Billerica, MA) according to the manufacturer's instructions. The procedures used are detailed in the Supplementary Materials and Methods section.
Statistical analyses
Data are reported as mean ± standard error of the mean (SEM) if the total sample size was more than 20 or as median ± interquartile range if the total sample size was smaller than 20 and were analyzed by GraphPad Prism 6.0 software (GraphPad Software, La Jolla, CA). Differences and significance were verified by one-way or two-way ANOVA, followed by Bonferroni post hoc tests when the total sample size was more than 20. Otherwise, we performed a nonparametric Kruskal–Wallis and Mann–Whitney U test for comparing three and two groups, respectively. All p-values less than 0.05 were considered statistically significant.
Supplementary Data
Supplementary data include Supplementary Materials and Methods, nine figures, and three tables and can be found with this article online.
Footnotes
Acknowledgments
The authors thank Dr. Mariusz Ratajczak for critical comments. This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) (2010-0029521 and 2015R1A2A2A01003235), the Korea Korean Health Technology R&D Project, Ministry of Health and Welfare of the Republic of Korea (A120216 and HI14C3339), and the Asan Institute for Life Science (2014-526).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.