
Abstract
Introduction
T
A reduction in brain GCase activity has been linked to sporadic PD and normal aging, and may contribute to the susceptibility of vulnerable neurons to degeneration. This report demonstrates that systemic reduction of GCase activity using chemical inhibition, leads to neuropathological changes in the brain reminiscent of α-synucleinopathy.
A homozygous loss-of-function mutation in GBA1 causes the well-characterized lysosomal storage disorder, Gaucher disease (22). Gaucher disease patients typically express less than 15% of functional glucocerebrosidase (GCase) (25, 62), which causes accumulation of glucosylceramide (GluCer) and glucosylsphingosine (GluSph) (22). The initial association between GBA1 mutations and the α-synucleinopathies occurred in the clinic when a subset of Gaucher disease patients developed parkinsonian symptoms (32, 42, 58, 59). Moreover, the prevalence of PD was higher in relatives of Gaucher disease patients in comparison with sporadic PD patients who do not have relatives with Gaucher disease (21, 23).
To date, ∼300 different GBA1 mutations have been identified, including missense, nonsense, and frameshift mutations, as well as insertions, deletions, and complex alleles (26, 54). The majority of these mutations result in a significant loss of lysosomal GCase activity (39, 54). Individuals who are heterozygous carriers of a GBA1 mutation that results in a 30–50% reduction in GCase activity are at an increased risk for developing PD at a frequency of 4–7% (3, 31, 43, 61); 6–7% of early-onset PD patients are GBA1 mutation carriers (GBA1-PD), which is not surprising, given that GBA1-PD are associated with more severe cognitive symptoms and increased α-synuclein accumulation relative to PD patients who are not GBA1 mutation (nonGBA1-PD) carriers (40, 43, 53, 54). In addition to this genetic association between GBA1 and PD, nonGBA1-PD patients also show a significant reduction in lysosomal GCase, suggesting that GCase levels may be important to the pathophysiology of PD (19, 47).
Age-dependent lysosomal dysfunction likely contributes to the onset and progression of the α-synucleinopathies by promoting accumulation of oligomeric α-synuclein. Aging lysosomes undergo dramatic changes, including impaired volume regulation, accumulation of indigestible materials, and impaired regulation of intralysosomal pH (35). GCase activity gradually declines with age in the substantia nigra and putamen, eventually becoming comparable with nonGBA1-PD patients (47). This reduction in GCase activity is accompanied by an accumulation of GluSph (47). These age-related changes may be early indicators of PD.
Deficits in autophagy–lysosomal degradation are implicated in the pathophysiology of PD (14, 40). The number of lysosomal-associated membrane protein (LAMP)-1-positive lysosomes is reduced and the macroautophagy-related protein, microtubule-associated protein 1A/1B-light chain 3 (LC3)-II, is elevated in the substantia nigra of PD patients in comparison with age-matched control brains (14). Macroautophagy is the major pathway by which cytoplasmic contents are degraded in the lysosome and this process relies on vesicular trafficking rather than direct import of substrates into lysosomes. Macroautophagy is initiated by the conversion of LC3-I into its lipidated form, LC3-II, initiating the formation of double-layered autophagosomes (27). Mature autophagosomes will engulf cytoplasmic material tagged with the ubiquitin-like protein, p62/SQSTM1, transport its contents to the lysosome, fuse with the outer lysosomal membrane, and release its contents for degradation (29). Mice defective in macroautophagy die soon after birth, and conditional knockdown of a macroautophagy-related protein (Atg7) in dopamine neurons causes neurodegeneration and inclusion formation (1, 30). GCase null neurons are deficient in autophagy, which correlate with accumulation of p62/SQSTM1 ubiquitinated proteins, and insoluble α-synuclein (45).
Interventions that increase lysosomal GCase may preserve lysosomal function by enhancing autophagy and reducing the progression of the disease. To date, the only known regulator of GCase expression is the transcription factor EB (TFEB), which also regulates autophagy and lysosomal biogenesis (50, 52). Overexpression of TFEB or GBA can reduce α-synucleinopathy and prevent neurodegeneration in rodent models of α-synucleinopathy (13, 48, 49). As accumulation of toxic α-synuclein oligomers is considered to be pathogenic in PD (38), deficits in lysosomal degradation of α-synuclein could have a substantial impact on the disease process.
The current article critically tests the hypothesis whether long-term inhibition of GCase can disrupt autophagy–lysosomal degradation and promote accumulation of α-synuclein aggregation in mice. Our data demonstrate that chronic pharmacological inhibition of GCase using the selective inhibitor for lysosomal GCase, conduritol-β-epoxide (CBE), promotes the accumulation of α-synuclein aggregates and neuronal cell death by disrupting lysosomal function and inducing widespread neuroinflammation.
Results
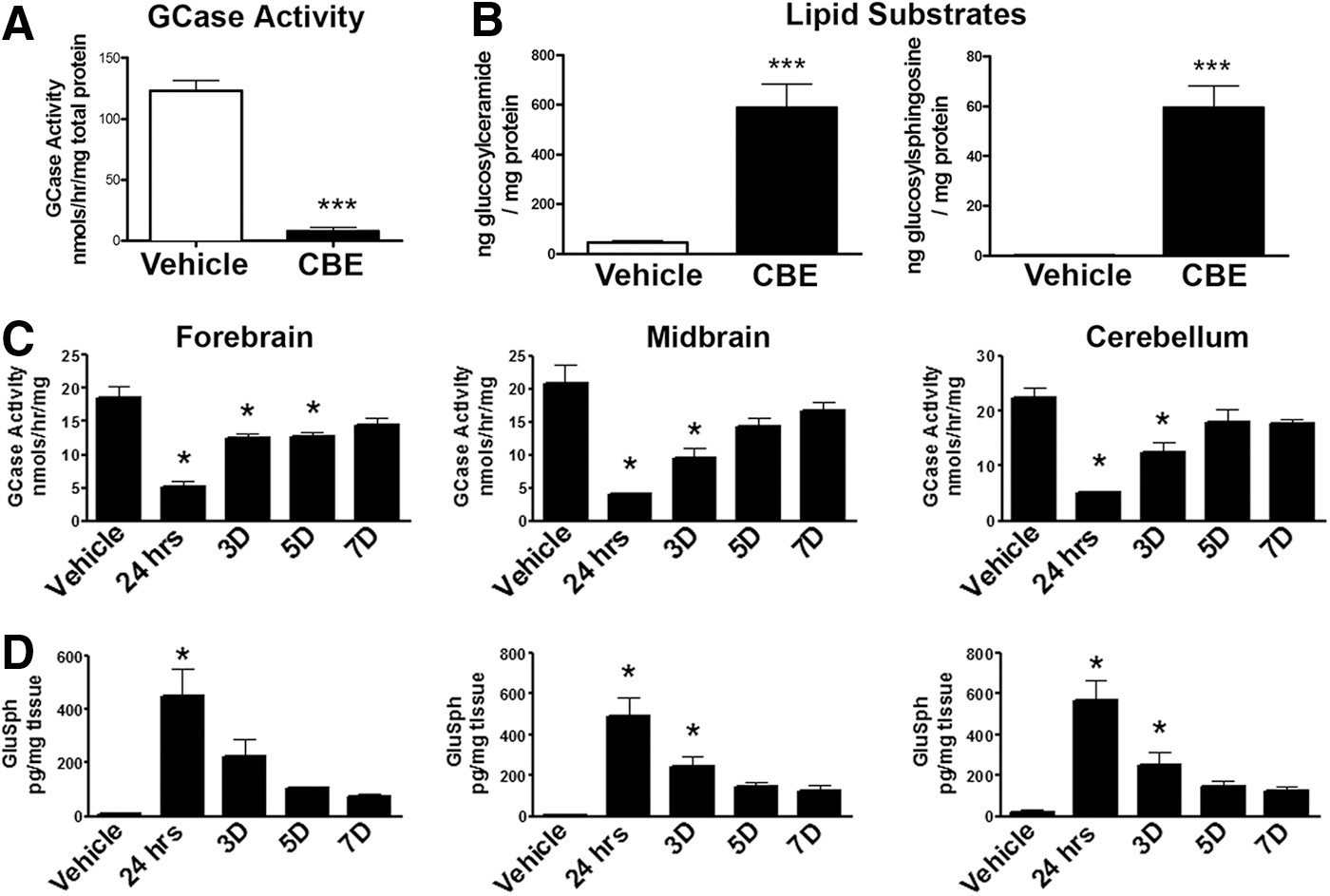
Chronic CBE treatment inhibited GCase activity and promoted the accumulation of lipid substrates
Pharmacological inhibition of GCase in mice was achieved using a selective and irreversible competitive inhibitor of GCase, CBE. Mice at 3–4 months of age were injected systemically with 100 mg/kg of CBE for 28 consecutive days and euthanized 24 h later. Chronic CBE treatment significantly blocked GCase activity in the forebrain of mice compared with vehicle treatment (T1,3=14.09, p<0.05; Fig. 1A). Inhibition of GCase was paralleled by increased levels of GluCer (T1,8=5.756, p<0.05) and GluSph (T1,8=4.027, p<0.05; Fig. 1B). To determine whether these outcomes were transient, a separate cohort of mice was injected with 100 mg/kg of CBE for 28 consecutive days and euthanized 24 h, 3 days, 5 days, or 7 days after the final injection (Fig. 1C). Data indicate that GCase activity remained significantly decreased in the forebrain at 24 h, 3 days, and 5 days (F5,14=21.8, p<0.05) and gradually returned to baseline levels by 7 days (Fig. 1C). Similarly, GCase activity remained significantly decreased in the midbrain and cerebellum at 24 h and 3 days (F5,14=21.8, and 18.32, p<0.05, respectively) following the last CBE injection and gradually returned to baseline levels by 5 days (Fig. 1C). In parallel, we measured GluSph levels in these same homogenates and found that GluSph levels were significantly increased in the forebrain at 24 h (F5,14=10.1, p<0.05) and gradually returned to baseline levels by 3 days following the last CBE injection (Fig. 1D). Similarly, GluSph levels remained significantly elevated in the midbrain and cerebellum at 24 h and 3 days (F5,14=10.1, and 15.3, p<0.05, respectively) following the last CBE injection and returned to baseline levels by 5 days (Fig. 1D).
GCase inhibition induced widespread neuroinflammatory response as well as synaptic, axonal transport, and cytoskeletal changes in mice
We have previously shown that alterations in proteins involved in neuroinflammation, synaptic transmission, axonal transport, and the cytoskeleton precede degeneration of dopamine neurons in the substantia nigra and striatum using an animal model of α-synucleinopathy (9). Therefore, we sought to determine whether prolonged inhibition of GCase caused neuroinflammatory, synaptic, cytoskeletal, and axonal transport changes.
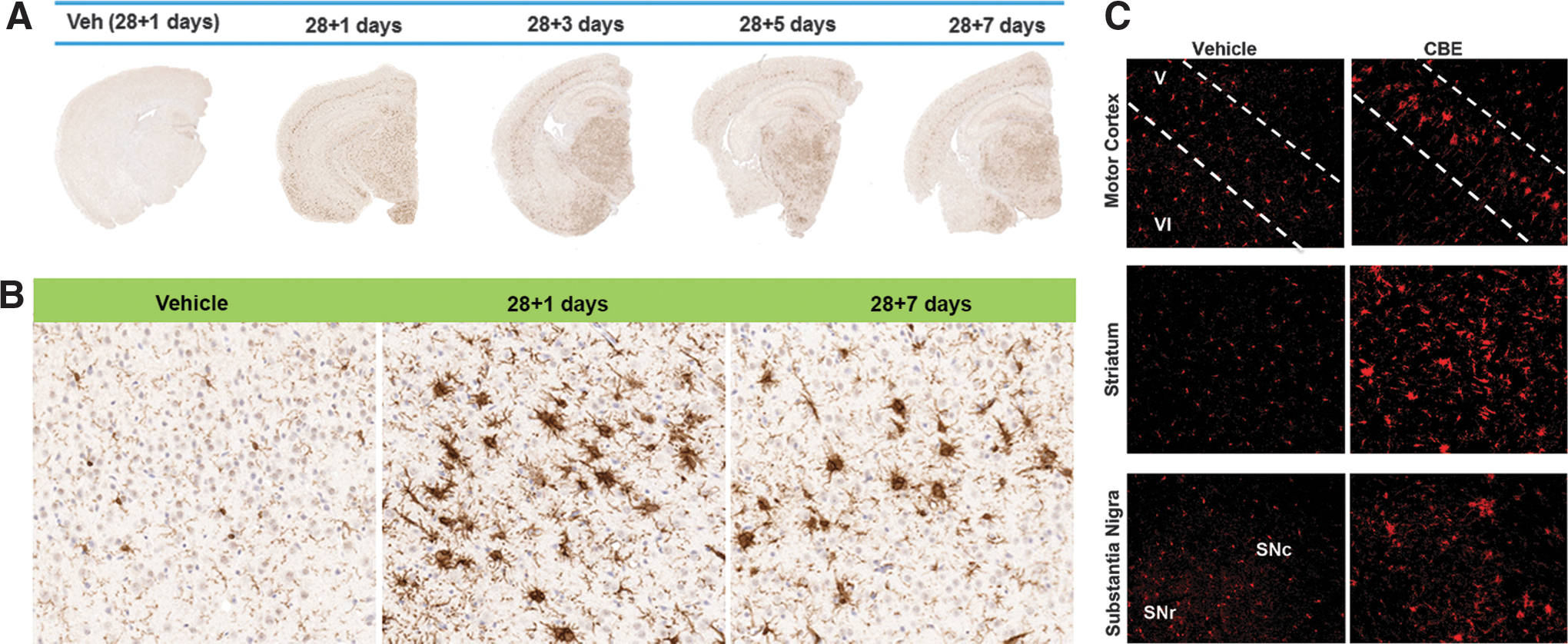
Inhibition of GCase for 28 days caused widespread microglial activation that remained elevated for 7 days following cessation of CBE (Fig. 2A, B). Widespread Iba-1-positive reactive microglia were evident throughout the brain, particularly in layer V of the cortex, as seen with higher magnification (Fig. 2B). Fluorescent staining using Iba-1 clearly illustrated a heightened microglial response in the motor cortex, striatum, and substantia nigra in CBE-treated mice in comparison with vehicle-treated mice (Fig. 2C). It has been suggested that the classical complement pathway, which is activated during developmental synaptic pruning, is reactivated during neurodegenerative diseases and is involved with removal of weak synapses (57). Our data show that chronic inhibition of GCase induced C1q expression, the first subcomponent of the C1 complex of the classical complement pathway, in the striatum (T1,6=2.416, p<0.05), substantia nigra (T1,8=6.072, p<0.05), and motor cortex (T1,6=3.278, p<0.05; Fig. 3A).
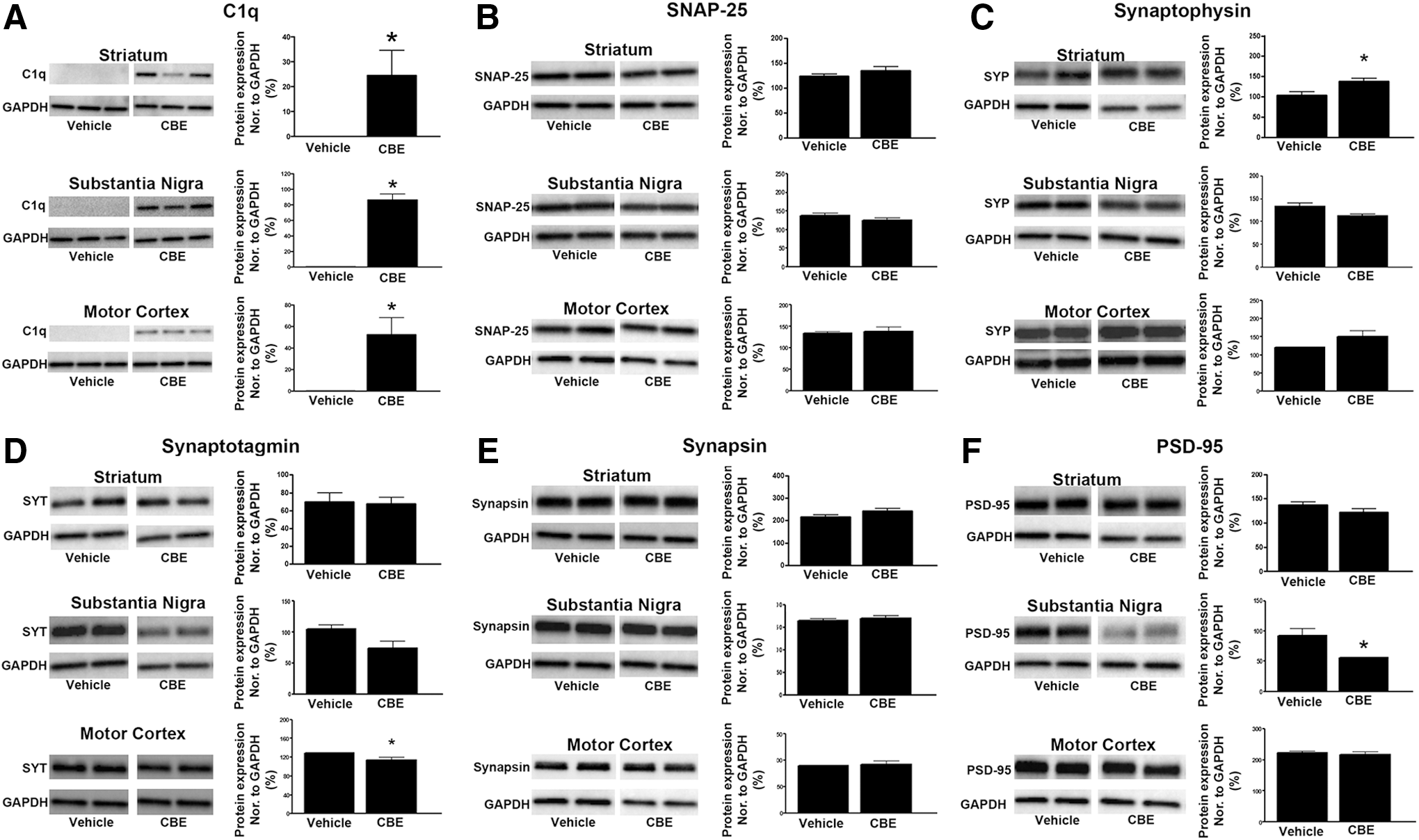
A large battery of synaptic, axonal transport, and cytoskeletal proteins was investigated in the substantia nigra, striatum, and motor cortex of mice treated with CBE for 28 consecutive days (Figs. 3 –5). Chronic inhibition of GCase did not induce changes in protein levels of the SNARE complex-associated protein, SNAP-25, in the striatum, substantia nigra, or motor cortex (Fig. 3B). Levels of synaptophysin were increased in the striatum (T1,8=2.481, p<0.05) and did not change in the substantia nigra and motor cortex (Fig. 3C). However, levels of synaptotagmin were significantly decreased in the motor cortex (T1,6=2.647, p<0.05), but remained unchanged in the striatum and substantia nigra of mice treated with CBE (Fig. 3D). Levels of the presynaptic protein, synapsin, were not altered in the striatum, substantia nigra, and motor cortex (Fig. 3E). Lastly, levels of postsynaptic density protein 95 (PSD-95) were decreased in the substantia nigra (T1,7=2.89, p<0.05) following CBE treatment (Fig. 3E).
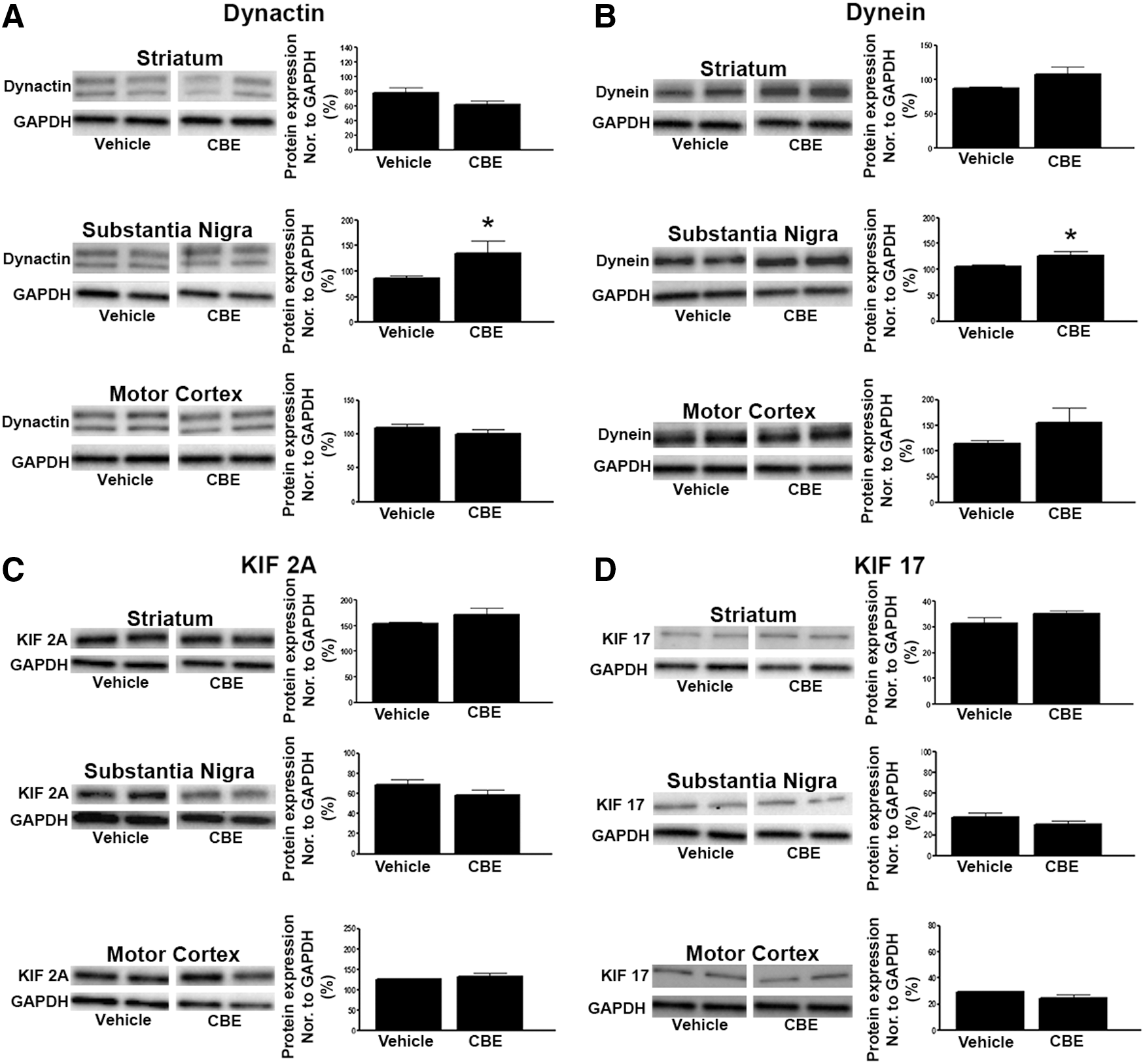
Several retrograde and anterograde transport proteins were also measured to determine if GCase inhibition can disrupt axonal transport. CBE caused an increase in the levels of the retrograde transport proteins, dynactin (T1,6=2.52, p<0.05) and dynein (T1,6=2.532, p<0.05), in the substantia nigra (Fig. 4A, B). However, levels of dynactin and dynein remained unchanged in striatum and motor cortex. Levels of the anterograde transport proteins, KIF 2A and KIF 17, remained unchanged in these three brain regions (Fig. 4C, D).
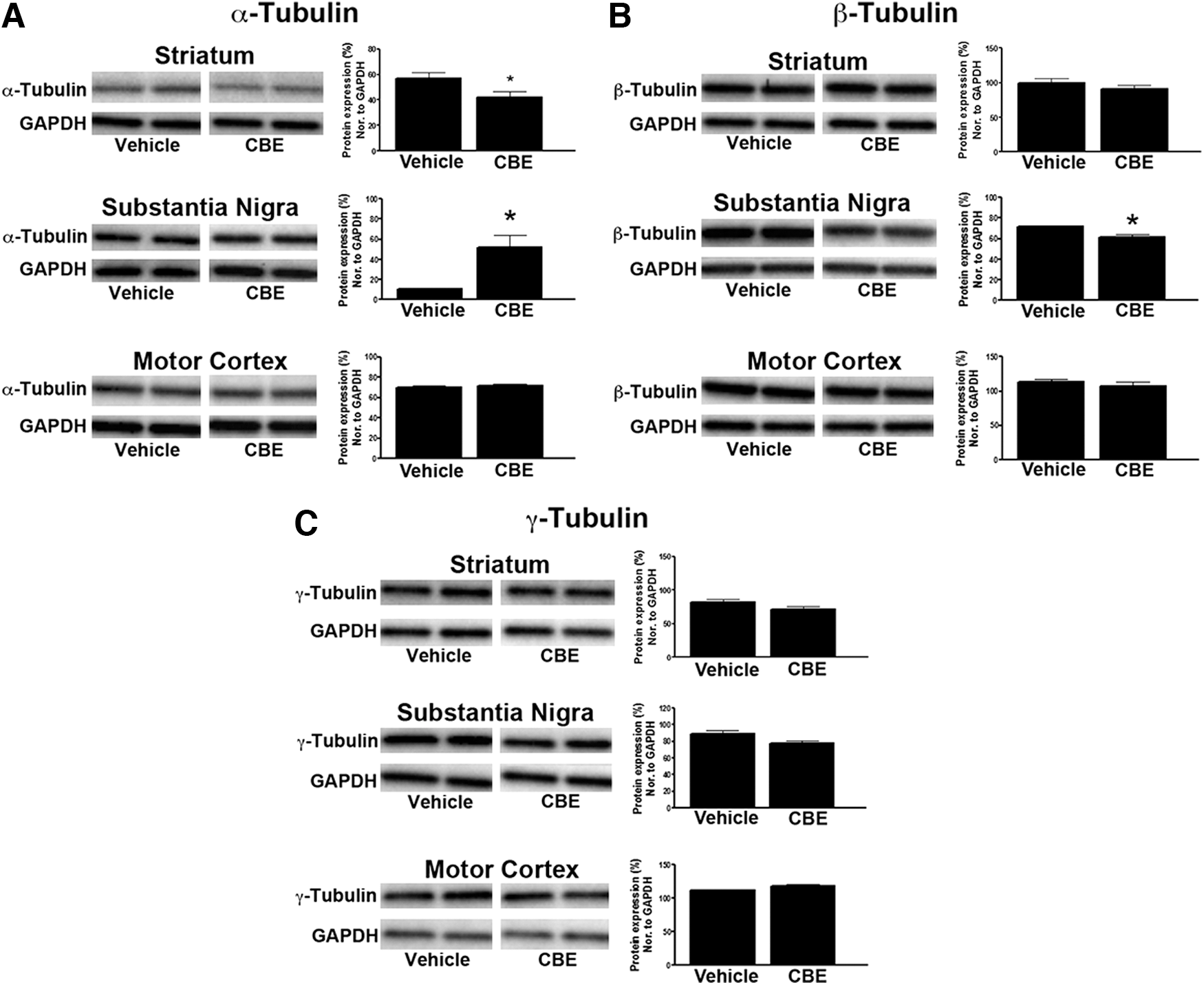
Three cytoskeletal proteins were also measured to determine if chronic GCase inhibition disrupted the microtubules. The cytoskeletal protein, α-tubulin, was significantly decreased in the striatum (T1,6=2.527, p<0.05) and increased in the substantia nigra (T1,6=3.525, p<0.05) of mice treated with CBE (Fig. 5A). Levels of α-tubulin were not altered in the motor cortex following CBE treatment (Fig. 5A). Protein levels of β-tubulin were decreased in the substantia nigra (T1,6=5.665, p<0.05) of mice treated with CBE and remained unchanged in the striatum and motor cortex (Fig. 5B). Finally, levels of γ-tubulin were not altered in mice treated with CBE in any region studied (Fig. 5C).
GCase inhibition caused accumulation of proteinase-K-resistant insoluble α-synuclein aggregates and induced autophagy–lysosomal proteins
Immunohistochemistry was used to determine if chronic inhibition of GCase could cause the accumulation of insoluble α-synuclein aggregates. The number and size of the α-synuclein aggregates were quantified in the substantia nigra of mice 24 h after the final treatment of CBE (Fig. 6A). There was a significant increase, ∼25-fold, in the number of proteinase-K resistant α-synuclein aggregates between 1 and 10 μm2 (T1,5=3.034, p<0.05), a 6-fold increase in the number of aggregates between 11 and 20 μm2 (T1,5=2.75, p<0.05), a 10-fold increase in the number of aggregates between 21 and 50 μm2 (T1,5=2.73, p<0.05), a 3-fold increase in the number of aggregates between 51 and 150 μm2 (T1,5=3.076, p<0.05), and finally a 2-fold increase in the number of aggregates over>151 μm2 (T1,5=8.6, p<0.05; Fig. 6A).
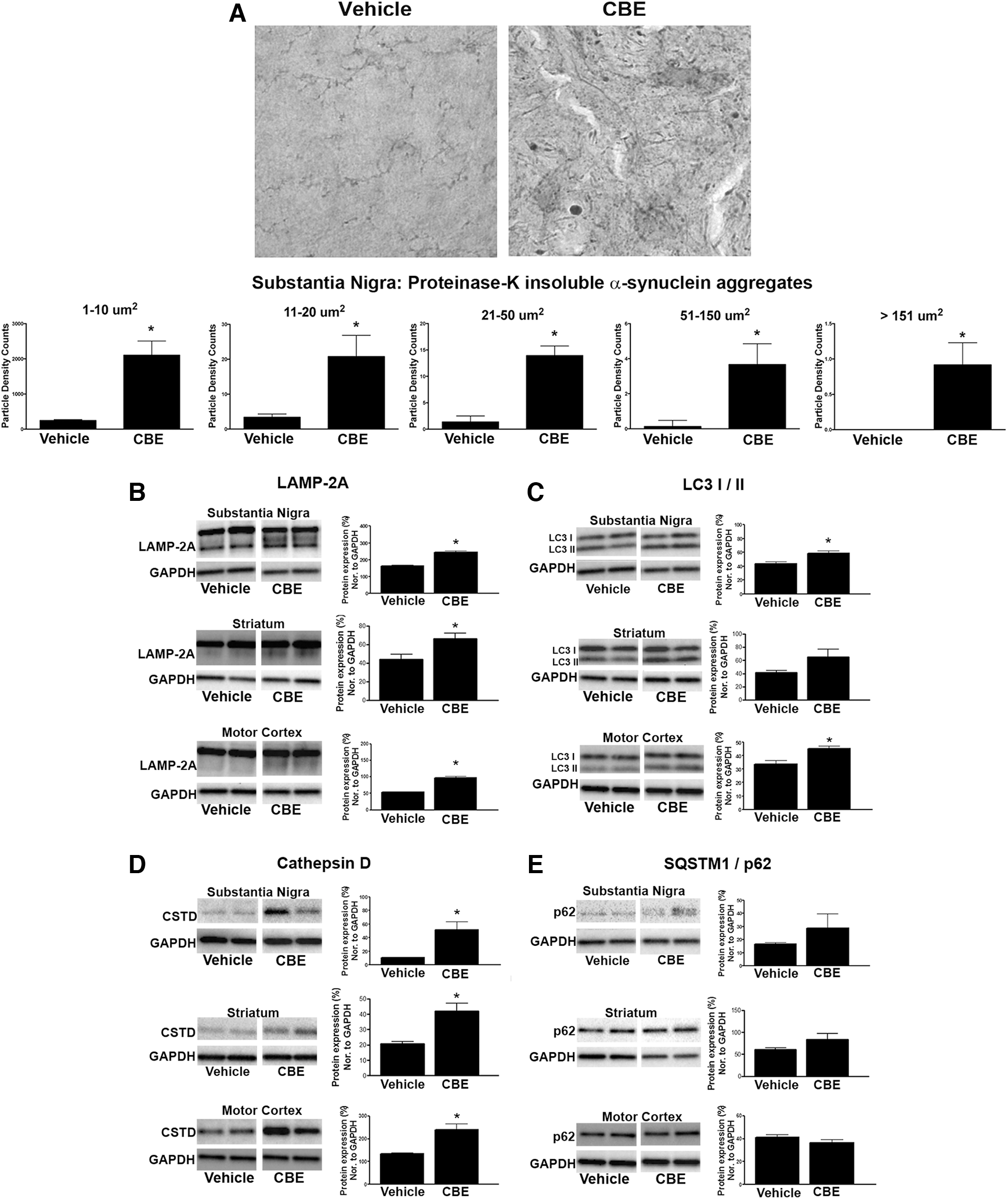
We chose to measure specific proteins involved in chaperone-mediated autophagy (CMA; LAMP-2A) and macroautophagy (LC3-I/II and SQSTM1/p62) following GCase inhibition. The lysosomal protease, cathepsin D, has also been reported to be necessary for α-synuclein degradation (12); therefore, we measured whether inhibition of GCase by CBE altered levels of cathepsin D. Chronic 28-day dosing of CBE increased the expression of LAMP-2A in the substantia nigra (T1,6=6.313, p<0.05), striatum (T1,6=2.723, p<0.05), and motor cortex (T1,6=7.805, p<0.05) compared with vehicle treatment (Fig. 6B). CBE also increased the expression of the lipidated form of LC3 (LC3-II) in the substantia nigra (T1,6=3.182, p<0.05), striatum (T1,6=2.73, p<0.05), and motor cortex (T1,6=3.580, p<0.05) of CBE-treated mice compared with vehicle treatment (Fig. 6C). Similarly, levels of cathepsin D were increased in the substantia nigra (T1,6=3.525, p<0.05), striatum (T1,6=3.56, p<0.05), and motor cortex (T1,6=3.921, p<0.05) of mice treated with CBE compared with vehicle treatment (Fig. 6D). Lastly, we examined levels of the ubiquitin-like protein, p62/SQSTM1, and despite the modest increase in SQSTM1/p62 within the substantia nigra and striatum, it did not reach statistical significance compared with vehicle treatment (Fig. 6E).
Inhibition of GCase induced cortical cell loss in mice
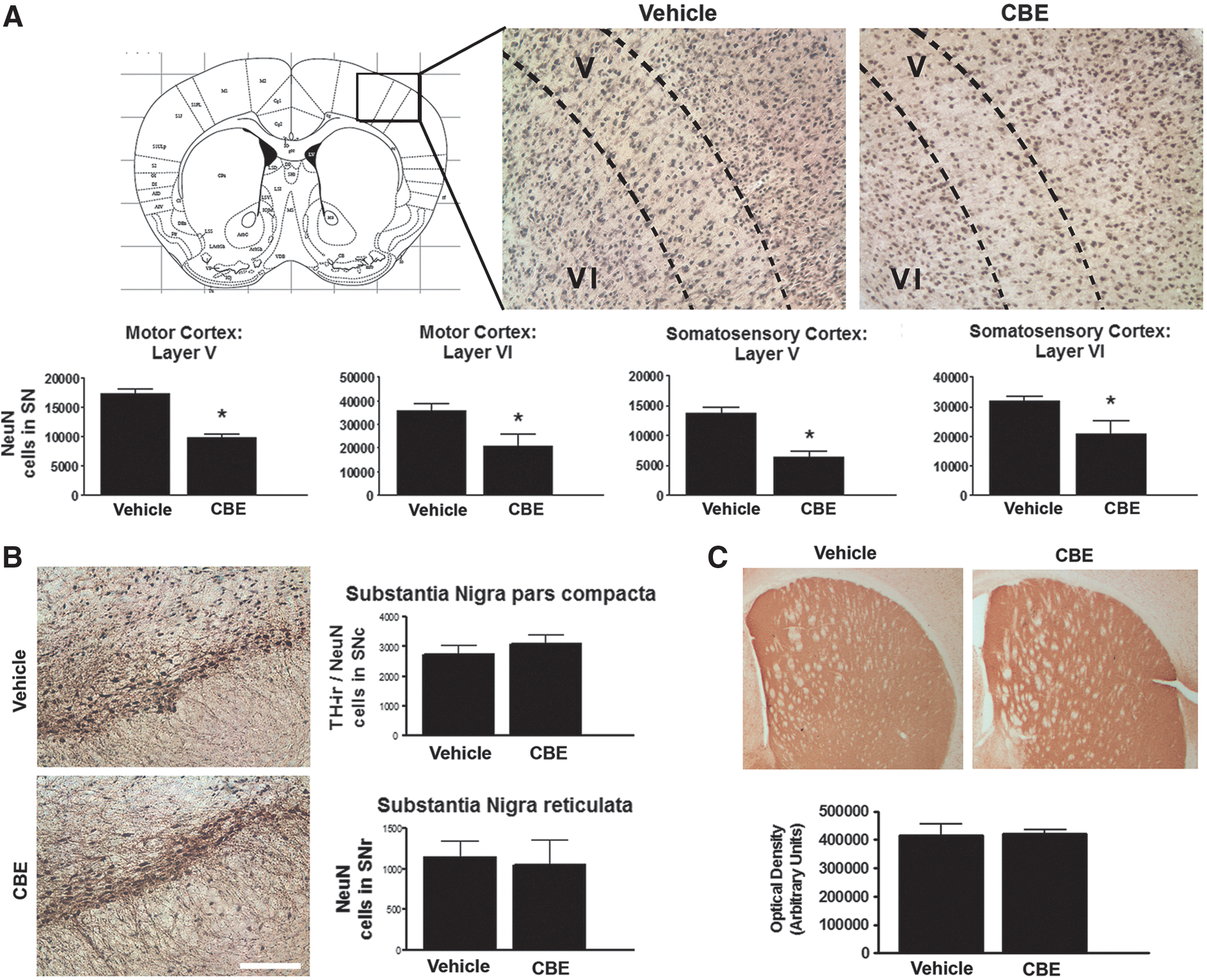
Stereological cell counts were conducted in the cortex of mice treated with CBE and revealed that chronic inhibition of GCase induced neuronal cell loss in the motor and somatosensory cortices. CBE induced a significant reduction in the number of NeuN-positive neurons in layer V (T1,5=7.315, p<0.05) and layer VI (T1,5=4.951, p<0.05) of the motor cortex in comparison with vehicle-treated mice (Fig. 7A). Similarly, CBE induced a significant reduction in the number of NeuN-positive neurons in layer V (T1,5=2.845, p<0.05) and layer VI (T1,5=3.038, p<0.05) of the somatosensory cortex in comparison with vehicle-treated mice (Fig. 7A). The number of NeuN-positive cell bodies in the substantia nigra pars reticulata and tyrosine hydrolase (TH)/NeuN-positive neurons in the substantia nigra pars compacta were not altered following chronic CBE treatment (Fig. 7B). Similarly, optical density counts in the striatum revealed that chronic inhibition of GCase for 28 days did not affect the number of TH fibers in the striatum (Fig. 7C).
Discussion
A current challenge in α-synucleinopathy research is determining what pathological events initiate the accumulation of toxic α-synuclein oligomers and cause neurodegeneration. Our data provide clear evidence that systemic inhibition of lysosomal GCase causes several cellular changes throughout the brain that eventually lead to accumulation of α-synuclein aggregates and region-specific predegenerative changes and neurodegeneration. CBE-induced inhibition of GCase caused accumulation of glycosphingolipids, widespread neuroinflammation, and altered levels of proteins involved in synaptic transmission and axonal transport. These changes were accompanied by deficits in lysosomal–autophagy degradation pathways, accumulation of α-synuclein, and neurodegeneration. Our data indicate that massive inhibition of GCase by CBE causes disturbances in brain regions that are vulnerable in PD and related α-synucleinopathies and provide insight into how age-related or genetic deficits in autophagy–lysosomal degradation systems may contribute to neurodegenerative diseases.
GCase inhibition caused deficits in lysosomal degradation and accumulation of α-synuclein
High levels of glycosphingolipids in neurons can cause pathological accumulation of α-synuclein oligomers (36), which may promote the onset and progression of PD. GCase is responsible for hydrolyzing GluCer in the lysosome, and under physiological conditions, GluCer maintains lysosomal function by modulating endolysosomal pH (55); therefore, changes in GluCer levels may disrupt normal lysosomal function and promote α-synuclein aggregation. Our data clearly show that inhibition of GCase by CBE causes α-synucleinopathy and neurodegeneration. The high levels of GluCer and GluSph either alone or in combination with the observed deficits in autophagy–lysosomal degradation may have caused the observed α-synucleinopathy in the CBE model. High levels of GluCer can promote fibril formation of α-synuclein and stabilize soluble oligomeric α-synuclein (36). Similarly, inhibition of macroautophagy and CMA can also increase α-synuclein levels (44, 64). Further studies will be required to determine whether reduced lysosomal GCase contributes to the pathophysiology of PD by promoting accumulation of α-synuclein oligomers, either through a direct interaction between glycosphingolipids and α-synuclein (36) or by disrupting autophagy–lysosomal degradation.
Both CMA and macroautophagy participate in degradation of α-synuclein (8, 11, 60, 63). CMA is an inducible form of autophagy that uses heat shock cognate protein 70 (Hsc70) to transport proteins containing a specific consensus peptide sequence to LAMP-2A for lysosome degradation (10). We show that inhibition of GCase by CBE caused an increase in LAMP-2A, which may be an attempt to increase degradation of α-synuclein and prevent cellular pathologies. A recent report demonstrated that LAMP-2A protein is reduced in the anterior cortex of early-stage PD patients, which is associated with a reduction in cholesterol and increased α-synuclein (40). Our observed increase in LAMP-2A may therefore illustrate changes that occur during the prodromal stage of PD. Similar to other cellular systems, it is possible that CMA initially increases in an attempt to compensate for the accumulation of α-synuclein. As the disease progresses with dopaminergic neuron degeneration and manifestation of clinical symptoms of PD, CMA may eventually become dysfunctional causing a decrease in LAMP-2A levels. Indeed, CMA can influence the survival of DA neurons, and overexpression of LAMP-2A prevents α-synuclein-induced neurodegeneration (66). Moreover, cholesterol can regulate CMA by modulating LAMP-2A levels on the lysosomal membrane (10, 28), and normal cholesterol regulation is disrupted under conditions promoting high GluCer levels (56). Therefore, it is also possible that CBE-induced accumulation of GluCer and GluSph may cause the observed disruption of LAMP-2A levels.
LC3-II protein levels are elevated in the substantia nigra of PD patients and are likely caused by deficits in macroautophagy, given that LC3-II colocalizes with α-synuclein within Lewy neurites and Lewy bodies (14). This increase in LC3-II has been replicated in transgenic mice that overexpress α-synuclein and in neurons exposed to the mitochondrial complex 1 inhibitor, MPP+ (14, 34). Our data showed that inhibition of GCase by CBE caused a significant increase in LC3-II protein levels in the striatum and substantia nigra. Elevated LC3-II protein levels occur upon induction of macroautophagy, where LC3-II initiates the formation of autophagosomes. However, high levels of LC3-II may also suggest a deficit in autophagosome–lysosome fusion (27). Measuring protein levels of the Ub-like protein, SQSTM1/p62, is the most informative way to distinguish induction from deficits in macroautophagy (27) and our data indicated a trend toward increased levels of the Ub-like protein, SQSTM1/p62, following GCase inhibition by CBE. It is possible that SQSTM1/p62 levels may take longer to accumulate in comparison with LC3-II, and if SQSTM1/p62 levels had been measured at a later time point following CBE cessation, then the increase may have become significant. Nonetheless, our data suggest that inhibition of GCase disrupts macroautophagy, which may contribute to the accumulation of insoluble α-synuclein aggregates.
In addition to the CMA and macroautophagy, our data also show that inhibition of GCase caused widespread elevation in cathepsin D levels. Increased cathepsin D levels may be upregulated in response to high levels of α-synuclein in the lysosomes. Cathepsin D is a major lysosomal aspartyl protease and has been shown to be essential for the degradation of α-synuclein (12, 24). Cathepsin D-deficient mice exhibit a progressive neurodegeneration that is accompanied by an accumulation of α-synuclein oligomers (12).
Collectively, our data demonstrate that inhibition of lysosomal GCase resulted in intracellular changes that are reminiscent of PD. The observed accumulation of insoluble α-synuclein may be caused by accumulation of GluCer and GluSph and/or a breakdown in autophagy–lysosomal degradation systems. Further studies will be required to elucidate the exact mechanism that is responsible for the CBE-induced accumulation of insoluble α-synuclein aggregates.
GCase inhibition induced widespread neuroinflammation and cortical degeneration
We previously demonstrated that overexpression of mutant α-synuclein causes progressive degeneration of the nigrostriatal dopamine neurons, which is preceded by microglial activation (9). Our current study shows that inhibition of GCase caused widespread microglia activation throughout the brain, which was accompanied by neurodegeneration in the motor and the somatosensory cortices, but not in the nigrostriatal pathway. It is possible that cortical neurons, are more sensitive to glycolipid accumulation following chronic CBE dosing in comparison with dopaminergic neurons' and thus degenerate faster. The systemic CBE dosing paradigm used in our study could be considered to be more of a model of neuronopathic Gaucher disease rather than sporadic PD because it causes a complete inhibition of GCase (16, 65). In comparison, nonGBA1-PD and Lewy body with dementia are clinically associated with a ∼30–50% reduction in brain GCase activity, and the clinical symptoms associated with the neurodegenerative changes manifest at a later age and are more gradual in comparison with those observed with neuronopathic Gaucher disease patients. Nonetheless, several of the pathological changes observed in the substantia nigra and striatum in mice treated with CBE in our current study are reminiscent of predegenerative changes that we have previously reported in a model of viral-induced α-synucleinopathy (9). In that model of α-synucleinopathy, altered levels of proteins related to axonal transport, the cytoskeleton, and synaptic function preceded degeneration of nigral dopaminergic neurons by at least 8 weeks and were accompanied by altered striatal dopamine homeostasis (15). It is possible that examination of CBE-treated mice at a later time point may have revealed nigrostriatal degeneration. This is further supported by recent data demonstrating that mice treated with a different, subchronic dosing regimen of CBE resulted in α-synuclein accumulation and reduced striatal dopamine release (20), indicative of dysfunction in the nigrostriatal dopamine system. Transgenic Gaucher disease mice and wild-type (WT) mice treated with CBE caused progressive neuronal loss in the cerebrum and spinal cord 2 and 5 months after the final CBE injection, despite recovery of GCase levels (67). These data suggest that lysosomal deficits caused by inhibiting GCase may be sufficient to cause progressive degeneration in the nigrostriatal pathway reminiscent of PD pathology.
GCase inhibition induced synaptic, axonal transport, and cytoskeletal protein changes in mice
Efficient axonal transport is required to transport newly synthesized proteins and organelles to the synapse and, conversely, to remove damaged proteins and organelles destined for degradation. Our data showed that chronic treatment with CBE increased levels of the retrograde axonal transport proteins, dynein and dynactin, in the substantia nigra. Neurons predominantly rely on macroautophagy for protein and organelle turnover. Autophagosome biogenesis typically occurs distally at the axon tip and travels to the cell body using retrograde dynein–dynactin transport (33). Based on these data, we hypothesized that inhibition of lysosomal degradation by CBE may impair retrograde axonal transport and promote accumulation of α-synuclein by preventing autophagosome transport from the synapse to the cell body. Indeed, mutations in dynactin can disrupt this heavily regulated axonal transport process and lead to neurodegeneration (17, 46). Mutations in DCTN1 impair dynactin-1 function and cause an atypical early-onset parkinsonism referred to as Perry Syndrome. These patients exhibit impaired microtubule binding, which is accompanied by severe dopaminergic neuron loss (2). Moreover, transgenic mice with a point mutation in the dynein heavy chain gene exhibit impaired retrograde axonal transport and motor and behavioral abnormalities, including hind limb clasping, loss of muscle tone, and incoordination, which is accompanied by striatal atrophy and dystrophic neurites (5). These studies support a role for dynactin and dynein in the pathogenesis of neurodegenerative disorders.
A disruption in α- and β-tubulin cytoskeletal protein levels in both the striatum and substantia nigra accompanied the observed changes in retrograde transport proteins. Impaired retrograde transport can prevent maturation of autophagosomes, which can be further impaired by high levels of synaptic α-synuclein (7,9). Oligomeric α-synuclein can inhibit tubulin polymerization, disrupting proper axonal transport (7). Therefore, impaired transport mechanisms in the distal axon may lead to dystrophic neurites and accumulation of protein inclusions. Based on these data, CBE-induced abnormalities in α-tubulin levels may promote α-synuclein inclusion accumulation by disrupting proper retrograde and anterograde axonal transport along the cytoskeleton. Further studies will be required to determine whether the axonal transport and cytoskeletal changes are the cause or a consequence of the autophagy deficits and α-synuclein inclusion accumulation.
In addition to the axonal transport and cytosketal changes, our data indicate that CBE caused changes to various proteins involved with synaptic function, including C1q, synaptotagmin, synaptophysin, and PSD-95. Evidence shows that synaptic dysfunction and loss precede degeneration of the cell soma and may be an early pathological event in several neurodegenerative diseases, including PD (9). α-Synuclein is abundantly expressed in presynaptic terminals of dopamine neurons in the striatum and is necessary to maintain conformation and function of SNARE proteins (6). Impaired autophagy–lysosomal degradation of α-synuclein that results in an accumulation of synaptic α-synuclein will likely disrupt synaptic functioning. This is supported by data demonstrating that overexpression of α-synuclein can disrupt neurotransmitter release by disrupting synaptic vesicle recycling before detectable neuropathology changes (41). Moreover, daily dosing of CBE to mice can disrupt dopamine neurotransmission by impairing striatal dopamine release and causing a reduction in postsynaptic density size (20). Synaptic accumulation of α-synuclein is associated with redistribution of the synaptic SNARE proteins, SNAP-25, syntaxin-1, and synaptobrevin-2 (18). Collectively, these data provide new insights into the mechanisms underlying synaptic dysfunction, accumulation of α-synuclein inclusions, and neuroinflammation in the pathophysiology of PD and related α-synucleinopathies.
It is known that synapses are tagged during normal postnatal development by the classical complement pathway for normal synaptic pruning (51, 57). Microglial recruitment participates in the removal of unwanted and redundant synapses by upregulating the complement pathway (4, 57). C1q is the initiating protein of the complement pathway and is required for the phagocytosis of dead cells, debris, and pathogens. Recently, C1q and the complement pathway have also been implicated in synaptic removal during neurodegeneration (57). Our data clearly demonstrate that GCase inhibition induces widespread C1q expression, neuroinflammation, predegenerative changes in the nigrostriatal pathway, and neuronal loss in the cortex. Therefore, synapses and axons may be early targets of inflammation-induced neurodegeneration in the α-synucleinopathies via the complement pathway.
Conclusion
The current article demonstrates that inhibition of GCase using a pharmacological inhibitor can induce the aggregation of α-synuclein and disrupt autophagy–lysosomal degradation. Inhibition of GCase also resulted in widespread neuroinflammation, which was accompanied by complement activation and changes in synaptic and transport proteins. These changes were paralleled by neurodegeneration in the motor and somatosensory cortices. Collectively, these data validate the GBA and lysosomal pathways as therapeutic targets to reduce levels of aggregated α-synuclein in PD.
Materials and Methods
Animals
Male BDF1 mice, 3–4 months of age, were used for all experimental procedures. Animals were housed in standard conditions in a 12-h dark/12-h light cycle, with ad libitum access to food and water. All animal procedures were performed in accordance with National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee (IACUC) at McLean Hospital, Harvard Medical School.
Pharmacological inhibition of lysosomal GCase
CBE is a selective irreversible inhibitor of lysosomal GCase activity and was purchased from EMD Millipore (Cat No. 234599). Mice received daily intraperitoneal (i.p.) injections, either CBE at 100 mg/kg or vehicle for 28 days, as previously described (59). Mice were euthanized at 24 h, 3 days, 5 days, or 7 days following the final i.p. injection.
GCase activity
GCase activities were determined as previously described with slight modifications (50); mouse brain tissues (∼5 mg) were homogenized in 300 μl of water. Samples were diluted in a 2 mg/ml bovine serum albumin, citric acid sodium phosphate buffer (pH 5). Ten microliters of sample was added to 75 μl of 10 mM 4-methylumbelliferyl-β-D-glucopyranoside (Sigma) substrate. After incubation with the substrate for 60 min at 37°C, the reaction was terminated using 200 μl of stop solution (0.3 M glycine/0.2 M sodium carbonate, pH 10.7). Plates were read (Ex 360/Em 460) in a Molecular Devices SPECTRAmax plate reader using Softmax Pro software. Enzymatic activity was assessed from a 4-methylumbelliferyl (Sigma) standard curve and normalized to protein content in each sample as determined using a bicinchoninic acid (BCA) assay (Thermo Scientific Pierce).
Liquid chromatography–tandem mass spectrometry analysis of GlcCer and GluSph
A 50 μl aliquot of homogenate was used to prepare samples for the quantitation of GlcCer and GluSph. Eight hundred microliters of acetonitrile and methanol 1:1 solvent was used to extract GluCer and GluSph from the tissue. The samples were sonicated, vortexed, and centrifuged before the supernatant was transferred to an injection plate for liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis. For quantification of GlcCer, a 2 μl sample was injected onto a Supelco 2.1 mm i.d.×25 cm LC-Si HPLC column (Sigma) connected to a Shimadzu LC Prominence UFLC solvent delivery system. The elution was performed using isocratic elution from 6:10:920 (water:acetic acid:acetonitrile) that contains 6 mM ammonium formate at a flow rate of 500 μl/min for 17 min. The column temperature was 45°C. The eluent from the column was introduced into an Applied Biosystems QTrap 5500 triple quadrupole mass spectrometer operated in positive ion electrospray mode. GlcCer species (C16:0, C18:0, C20:0, C22:0, C24:0, C24:1, C24:0-OH) were identified by precursor ion scan for m/z 264, and d3-C16 was used as the internal standard for GlcCer analysis. Quantification of GlcCer was performed using the selective reaction monitoring (SRM). The GlcCer concentration was calculated from the combined peak area ratios of each of the seven molecular species. For quantitation of GluSph, the same LC-MS/MS system was used. The isocratic solvent system consisted of 0.08 ml/min mobile phase A (water) and 1.0 ml/min mobile phase B (CH3CN/CH3OH/CH3COOH 97:2:1 with 5 mM of ammonium formate). The SRM transitions monitored for GluSph and the internal standard plant GluSph were m/z 462 to m/z 282 and m/z 460 to m/z 280, respectively.
Immunohistochemistry and antibodies
Animals were terminally anesthetized with sodium pentobarbital and perfused transcardially with 25 ml phosphate-buffered 0.9% saline (PBS), followed by 100 ml of 4% paraformaldehyde in phosphate buffer. Brains were removed and postfixed in 4% paraformaldehyde for 4 h before placing them in 25% sucrose. Coronal sections were then cut, 40 μm thick, on a freezing microtome and stored in antifreeze at −20°C until use. For fluorescent microscopy, sections were blocked with 10% normal serum and incubated with anti-rabbit Iba1 (Wako; 1:200) at 4°C overnight, visualized with Alexa Fluor 568 (Invitrogen; 1:500), and mounted using Vecatshield (Vector Laboratories). Images were taken using a Zeiss LSM 510.
Staining and quantification of insoluble α-synuclein aggregates
To visualize insoluble α-synuclein aggregates, tissue sections were premounted on gelatin-coated slides and incubated with proteinase-K solution (10 μg/ml; Promega) for 20–30 min at 37°C. Endogenous peroxidases were quenched in 3% hydrogen peroxide for 7 min and placed in a blocking solution (Vectashield MOM kit) for 1 h at room temperature. Tissue sections were incubated with anti-α-synuclein (1:1000; BD Transduction) overnight at 4°C using primary antibody diluent (Vectashield MOM kit). Sections were incubated with anti-mouse biotinylated secondary antibody (1:200) for 1 h at room temperature and visualized using a standard peroxidase-based method (Vectastain Elite, ABC kit; Vector Laboratories) and the chromogen, 3,3′-diaminobenzidine (Sigma). Insoluble α-synuclein aggregates were quantified using the threshold function in ImageJ.
Stereology
To quantify the number of neurons in the substantia nigra and cortex, every 6th brain section was collected. Free-floating tissue sections were rinsed in PBS before endogenous peroxidases were quenched in 3% hydrogen peroxide for 7 min. After rinsing, the sections were incubated in 0.1% Triton X-100 in PBS containing 10% normal serum. Tissue sections were then incubated with the primary antibodies, anti-rabbit TH (Pelfreeze; 1:300) and anti-mouse NeuN (Millipore; 1:1000), overnight at 4°C. After washing in PBS, sections were incubated in biotinylated secondary antibody (1:200) and visualized using a standard peroxidase-based method (Vectastain Elite, ABC kit; Vector Laboratories), and the chromogen, 3,3′-diaminobenzidine (Sigma) and the substantia nigra sections were counterstained with Nissl. Unbiased stereology (StereoInvestigator 7; MFB Bioscience) was used to quantify neurons in the substantia nigra (compacta and reticulata) and cerebral cortex using the optical fractionator method at 20× magnification, using the following parameters: a 150×150 μm counting frame and 200×200 μm grid. All counts were performed blinded. One-way ANOVA statistical analyses were performed, followed by the Bonferroni post hoc test. All analyses were conducted using GraphPad Prism (Version 5.0) (GraphPad Software, Inc.). Statistical significance was determined at the alpha level of 0.05.
Optical density analysis of striatal TH-stained tissue sections was performed using ImageJ software (Version 1.46r) using images taken with a Zeiss Axioskop microscope and using the Spot RT color camera (Diagnostic Instruments, Inc.). Striatal images were taken at 2.5× magnification, ∼+0.8 (rostral) mm and −1.0 (caudal) relative to bregma in the anterior–posterior direction. Optical density values of inverted grayscale photomicrographs were then obtained for each image by drawing a contour around the rostral and caudal striatum (excluding the nucleus accumbens). Optical density values were first normalized to background levels by subtracting values of the corpus callosum. The level of TH fiber optical density was expressed as arbitrary units and presented as % of levels measured on the contralateral side.
All quantification was carried out in blind. Statistical analysis was performed using Prism software (version 5.0b). Either one-way ANOVA tests, with Dunnett's post hoc analysis, or T-tests were used.
Western blotting
Mice were terminally anesthetized and perfused transcardially with heparinized saline (0.1% heparin in 0.9% saline), and brains were sliced at 750 μm with the use of a tissue chopper. A previously published homogenization and Western blot protocol was followed (57). Membranes were incubated overnight at 4°C with the following primary antibodies: α-tubulin (Upstate; 1:5000), β-tubulin (Chemicon; 1:5000), γ-tubulin (Sigma; 1:5000), kinesin-like protein (KIF)-2A (Abcam; 1:5000), KIF 17 (Abcam; 1:500), dynein (Santa Cruz; 1:250), dynactin (Santa Cruz; 1:1000), C1q (Abcam; 1:1000), synapsin (Thermo; 1:1000), synaptotagmin (BD Trans; 1:5000), synaptophysin (Santa Cruz; 1:500), SNAP-25 (Chemicon; 1:4000), PSD-95 (NeuroMab; 1:250), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Millipore, 1:5000). Horseradish peroxidase-conjugated (HRP) goat anti-chicken, anti-rabbit, and anti-mouse secondary antibodies (all Jackson ImmunoResearch; 1:10,000) were used for Western blot detection. After washing in Tris-buffered saline-Tween, HRP-conjugated secondary antibodies were then applied for 1 h at room temperature. The blots were treated with ECL-Plus (Amersham Biosciences) and exposed using ChemiDoc™ XRS with image Lab™ software. Optical density analysis (NIH image) was used to determine the relative abundance of each protein of interest. Bands were normalized to GAPDH for that same sample and gel.
Footnotes
Acknowledgments
This work was supported by the Consolidated Anti-Aging Foundation, the Harvard Stem Cell Institute Translational Neuroscience Fund, the Poul Hansen family, the Harold and Ronna Cooper family, a fellowship from the Canadian Institute of Health Research (E.R.), and a grant from Shire Pharmaceuticals (O.I.).
Author Disclosure Statement
No competing financial interests exist.