
Abstract
Introduction
P
YchF/Ola1 represent largely uncharacterized ATP-hydrolyzing members of the universally conserved GTPase family. Our data demonstrate that Escherichia coli YchF undergoes a redox-dependent dimerization. This inhibits its ATPase activity and prevents the inhibition of the antioxidant response. The monomer–dimer equilibrium of YchF as well as its ATPase activity is regulated by thioredoxin 1, which was identified by site-directed cross-linking as a major contact partner of YchF in vivo. Our data reveal a new regulatory mechanism for controlling the oxidative stress response and identify the YchF/Ola1 subfamily of A(G)TPases as first redox-regulated members of the universally conserved GTPase family.
The available crystal structures of Haemophilus influenzae YchF (47) and its human homologue hOla1 (24), show three distinct domains: the N-terminal domain displays a typical TRAFAC (translation factor)-type fold with a six-stranded mostly parallel β-sheet surrounded by α-helices (Fig. 1A). The second domain is characterized by two α-helices, which often serve as recognition motifs for protein and nucleic acid interaction (35). This YchF domain is similar to the coiled-coil domain in transcription elongation factor GreA (45), pointing to possible RNA interaction. The third domain is located at the C-terminus and shows similarity to TGS domains (ThrRS-GTPases-SpoT), which are associated with nucleotide-dependent regulatory functions and nucleic acid binding (2).
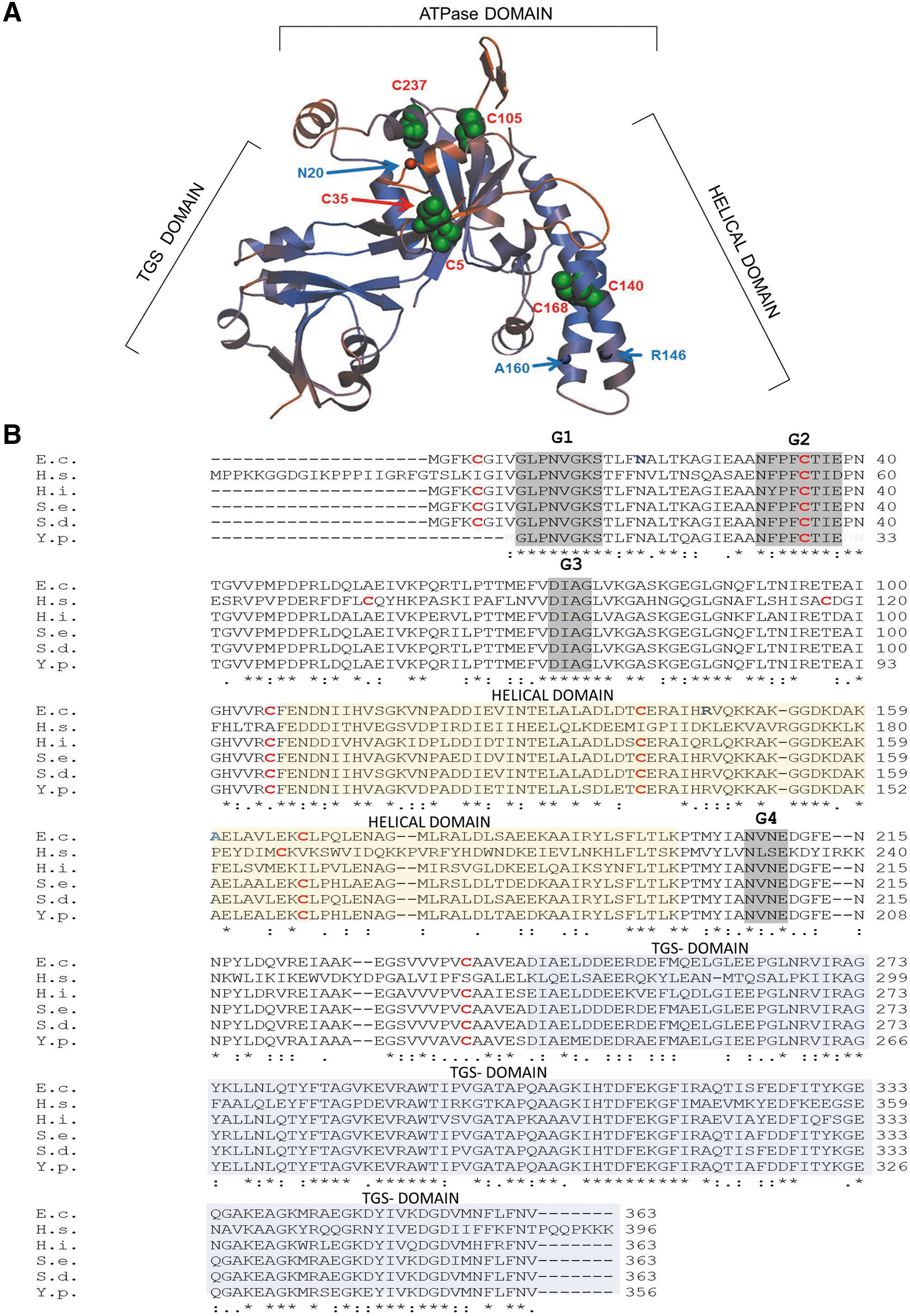
Despite its universal conservation, the physiological function of YchF/Ola1 is still unknown. Many Obg-like G/ATPases are involved in ribosome biogenesis and ribosome binding has been observed for Escherichia coli (6, 49) and Trypanosoma cruzi (16) YchF. The exact impact of YchF on ribosome biogenesis or translation is unknown, but yeast Ola1 possibly influences translational fidelity (39). Whether YchF/Ola1 interact with ribosomal proteins or rRNA is unknown, but the cleft formed by the helical domain and the TGS domain would be wide enough to accommodate nucleic acids (Fig. 1A). A possible role in DNA repair was also postulated because human hOla1 is downregulated upon DNA damage and upregulated in many tumors (46). This was validated by the observation that hOla1 interacts with BRCA1 (breast cancer-associated gene 1) at the centrosome and that depletion of hOla1 induced centrosome fragmentation (29). Other functions of Ola1/YchF include a possible role in iron uptake in pathogens (8, 15) and salinity stress in plants (9, 10).
The postulated functions of YchF/Ola1 in different species are difficult to reconcile with a single mode of action. However, in both E. coli and humans, overexpression of YchF/Ola1 resulted in H2O2 hypersensitivity (53, 56), suggesting a role of YchF/Ola1 in oxidative stress response. This is so far the only predicted function of YchF/Ola1 that appears to be evolutionarily conserved from bacteria to humans. In E. coli, YchF was shown to interact with catalase KatG, the major H2O2-detoxifying enzyme, and overexpression of YchF decreased catalase activity in E. coli cell extracts (53). YchF/Ola1 probably act via a transcription- and translation-independent mechanism (56) because overexpression of YchF in E. coli had no significant influence on the OxyR-controlled expression of KatG (53). OxyR is a LysR-like transcription factor, which serves as master regulator of the H2O2 response in E. coli (11). OxyR is activated by H2O2, resulting in increased expression of at least 30 proteins that either detoxify H2O2 or protect macromolecules against oxidative damage (19). A few genes are also repressed by oxidized OxyR; this includes oxyR itself and, intriguingly, ychF (19, 53). This demonstrates that YchF is part of the E. coli OxyR regulon and probably acts as a negative regulator of the oxidative stress response. In addition, YchF appears to be regulated by complex post-translational modifications, including dephosphorylation of serine 16 (28) and so far unknown modifications, which are required for full ATPase activity in response to H2O2 (53).
In the current study, we have analyzed YchF in the model organism E. coli. Based on our data, we propose a model for the redox regulation of YchF and its inhibitory effect on the antioxidant response.
Results
YchF forms redox-sensitive dimers
The activity of E. coli YchF is regulated by an H2O2-dependent dephosphorylation and additional, so far unknown, stress-dependent modifications (53). Redox-sensitive pathways are commonly regulated by either thiol modifications (3) or histidine oxidation (26). E. coli YchF contains six cysteine residues, which are highly conserved within enterobacteria and also weakly in other YchF/Ola1 homologues (Fig. 1B). In the structural model of E. coli YchF, which was generated using the H. influenzae YchF structure (85% sequence identity to E. coli YchF), the six cysteine residues are surface exposed (Fig. 1A), making them suitable for regulation via thiol oxidation.
To analyze this, YchF was purified from E. coli and separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) under reducing conditions (+25 mM DTT). Western blotting using α-YchF antibodies detected YchF as a strong band at ∼40 kDa. In addition, a weak band at ∼90 kDa was also detected, which could reflect a YchF dimer (Fig. 2A). Under nonreducing conditions (−DTT), the putative YchF dimer at ∼90 kDa was much stronger and less of the monomer was detected (Fig. 2A). In addition, two weaker bands (*) of ∼50 kDa and 100 kDa were detected. The band migrating at 90 kDa could, in principle, also correspond to a complex between YchF and additional proteins. To exclude this possibility, the 90 kDa band was cut out of the gel and separated on SDS-PAGE under reducing conditions. Coomassie staining revealed a single band at ∼40 kDa (Fig. 2B), demonstrating that the 90 kDa band indeed corresponded to the YchF dimer.
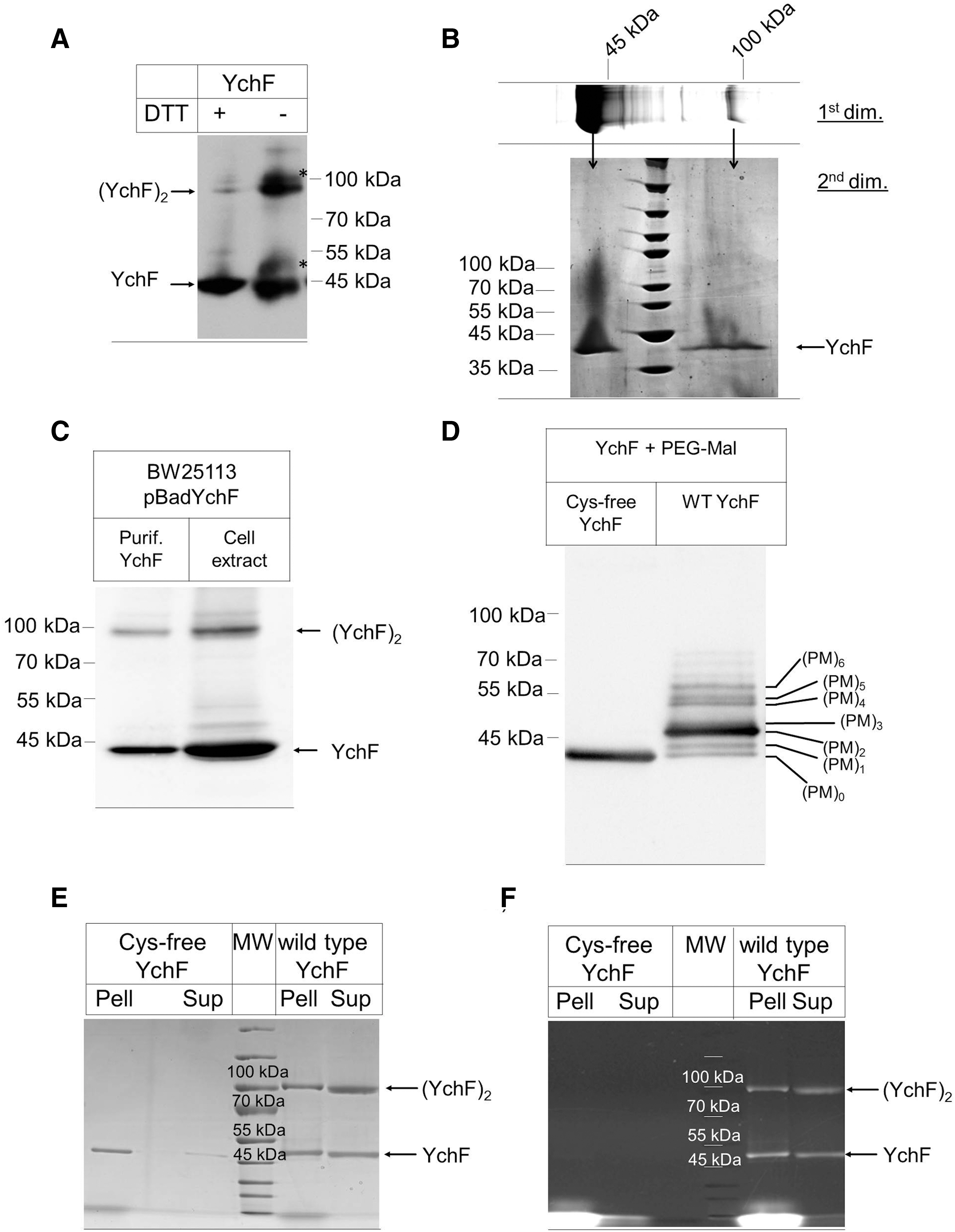
Dimerization of purified YchF could be an artifact of the in vitro conditions and we therefore analyzed dimerization in cell extracts of E. coli cells expressing YchF under an arabinose-inducible promoter. Western blotting identified the YchF monomer and dimer in these crude cell extracts (Fig. 2C), demonstrating that dimerization is not only observed with purified proteins. We also tried to detect YchF and its dimeric form in wild-type E. coli cells, but the low cellular concentration of YchF and the sensitivity of our antibody did not allow for an unambiguous detection of the dimer in whole cells.
The dithiothreitol (DTT)-sensitive YchF dimerization supports the involvement of cysteine residues in dimerization and this was further analyzed by testing their surface accessibility using the thiol-modifying agent, methoxypolyethylene glycol maleimide (PEG-Mal). PEG-Mal binding to cysteines induces a mass shift that can be monitored by SDS-PAGE (7). When YchF was reduced by tris(2-carboxyethyl)phosphine (TCEP), incubated with PEG-Mal, and subsequently analyzed by SDS-PAGE under reducing conditions, we observed seven prominent bands of increasing mass (Fig. 2D). As a control, we constructed and purified a cysteine-free YchF mutant. The Cys-free YchF did not show any mass shift upon PEG-Mal incubation (Fig. 2D). These data indicate that all six cysteine residues are accessible to PEG-Mal modification and thus are surface exposed as predicted from the homology model (Fig. 1A).
The contribution of the cysteine residues to YchF dimerization was further analyzed by comparing wild-type YchF and the Cys-free YchF mutant by SDS-PAGE under nonreducing conditions. Cys-free YchF showed a tendency to aggregate and purified proteins were therefore subjected to a centrifugation step before SDS-PAGE. For wild-type YchF, we observed both monomer and dimer by Coomassie staining (Fig. 2E). Both forms were almost equally distributed between the supernatant and pellet fraction after centrifugation. For the Cys-free mutant, we observed only the monomeric form, but no dimer (Fig. 2E), which demonstrates that YchF dimerization is cysteine dependent. The Cys-free YchF monomer was predominantly recovered from the pellet fraction after centrifugation, indicating that the cysteine residues are important for folding or solubility of YchF. Dimerization was also analyzed by the cysteine-specific fluorescent dye fluorescein-5-maleimide, which stained both monomer and dimer of wild-type YchF, but not the Cys-free mutant (Fig. 2F). This verifies the absence of cysteine residues in the Cys-free YchF derivative and indicates that not all cysteine residues of wild-type YchF are involved in dimerization. Cys-free YchF was purified under denaturing conditions and subsequently refolded. For excluding the possibility that the absence of the dimer observed for the Cys-free YchF is the result of the purification procedure, we also purified wild-type YchF under denaturing conditions and found both the monomeric and dimeric forms (Supplementary Fig. S1; Supplementary Data are available online at
The conserved cysteine residue 35 is required for YchF dimerization
For identifying the cysteine residues involved in YchF dimerization, purified YchF was treated with iodoacetamide for blocking free cysteine residues and separated by SDS-PAGE. After colloidal Coomassie staining, the monomer and dimer bands were excised and analyzed by high-resolution mass spectrometry (MS). This approach identified two uniquely cysteine-linked peptides (Table 1 and Fig. 3). In the YchF monomer, peptides containing Cys5 and Cys35 formed a prominent intramolecular disulfide bridge, which was virtually absent in the YchF dimer (Fig. 3A). Instead, Cys35-Cys35-linked peptides were present in the dimer and not in the monomer (Fig. 3B, C). This demonstrates that YchF dimerizes via disulfide bridge formation between the Cys35 residues of each monomer, whereas in the monomeric YchF, Cys5 forms an intramolecular disulfide bond to Cys35.
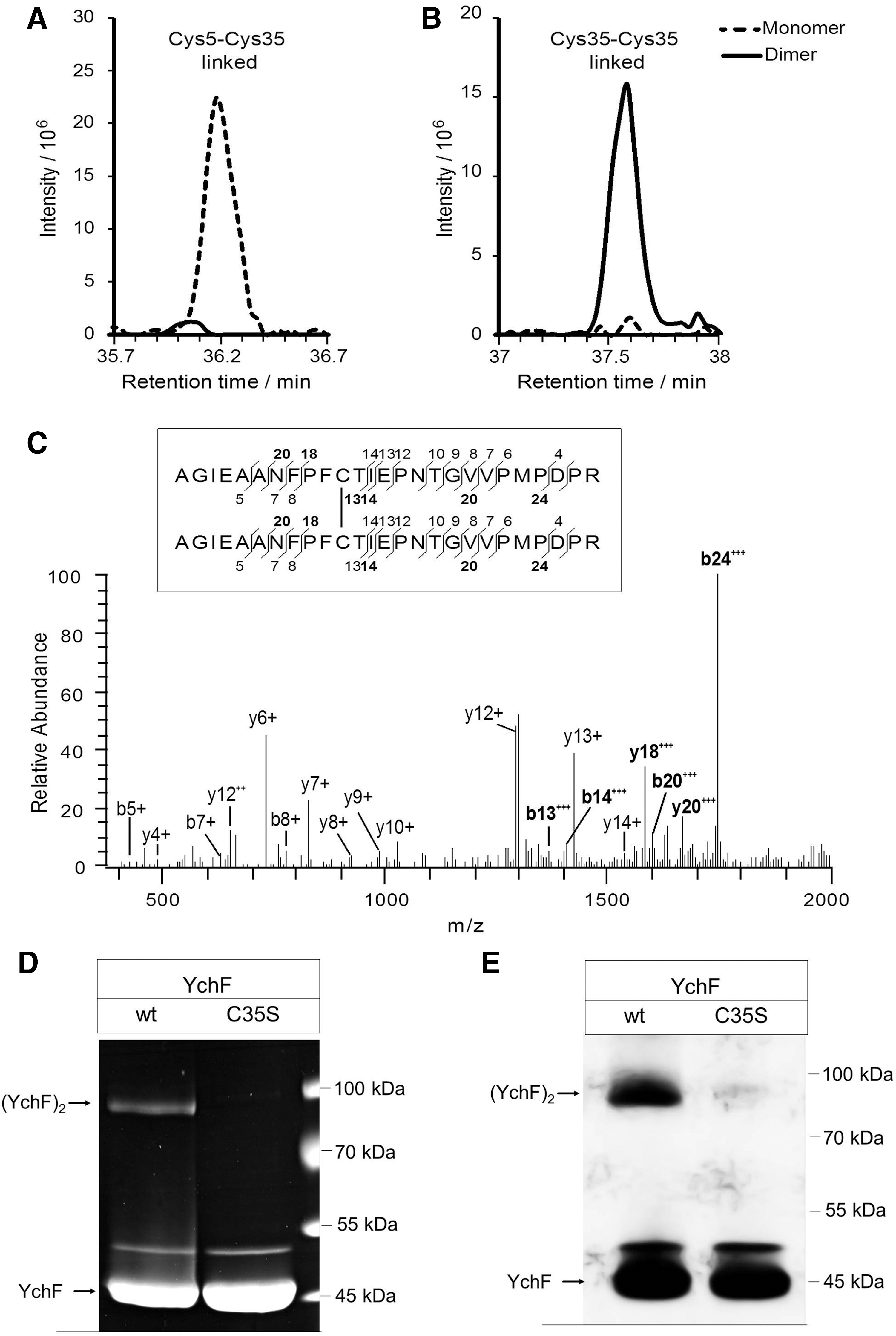
Monomer and dimer bands were excised from the polyacrylamide gel and subjected to in-gel digestion using trypsin, followed by LC-MS/MS analysis. For the identification of disulfide-linked peptides, mass spectrometric data were analyzed using the xQuest/xProphet software suite. Quantitative data analysis was performed with the Skyline software.
chromatographic peak area summed for molecular ion MS1 isotope peaks.
M*, oxidized methionine; n.d., not detected.
The involvement of Cys35 in YchF dimerization was further verified by analyzing dimer formation in a YchF mutant that contained a cysteine 35-to-serine replacement. In this mutant, almost no YchF dimer was detectable by Coomassie staining or Western blotting (Fig. 3D, E). This confirms that Cys35 is essential for dimer formation. For wild-type YchF and the YchF(C35S) mutant, an additional band at ∼50 kDa was detected, suggesting a further Cys35-independent form of YchF. Using MS, we confirmed that YchF is the main component (99.5%) in this band. As minor contamination (0.5%), we only detected 3-dehydroquinate synthase (AroB), which has a molecular mass of 39 kDa and is probably copurifying with YchF. Thus, further analysis is needed to reveal the identity of this Cys35-independent 50-kDa YchF form.
Intriguingly, among the cysteine residues present in different YchF/Ola1 species, Cys35 shows the highest conservation (Fig. 1B). This may indicate that Cys35-linked dimer formation is a general feature of the YchF/Ola1 protein family.
YchF displays redox-activated ATPase activity
To determine the physiological significance of the redox-dependent dimerization, we monitored the ATPase activity of YchF. We first measured the ATPase activity of purified wild-type YchF under reducing (+DTT) and oxidizing conditions (+tetrathionate). The addition of DTT stimulated the ATPase activity of YchF about two-fold, while the addition of tetrathionate significantly reduced the ATPase activity (Fig. 4A).
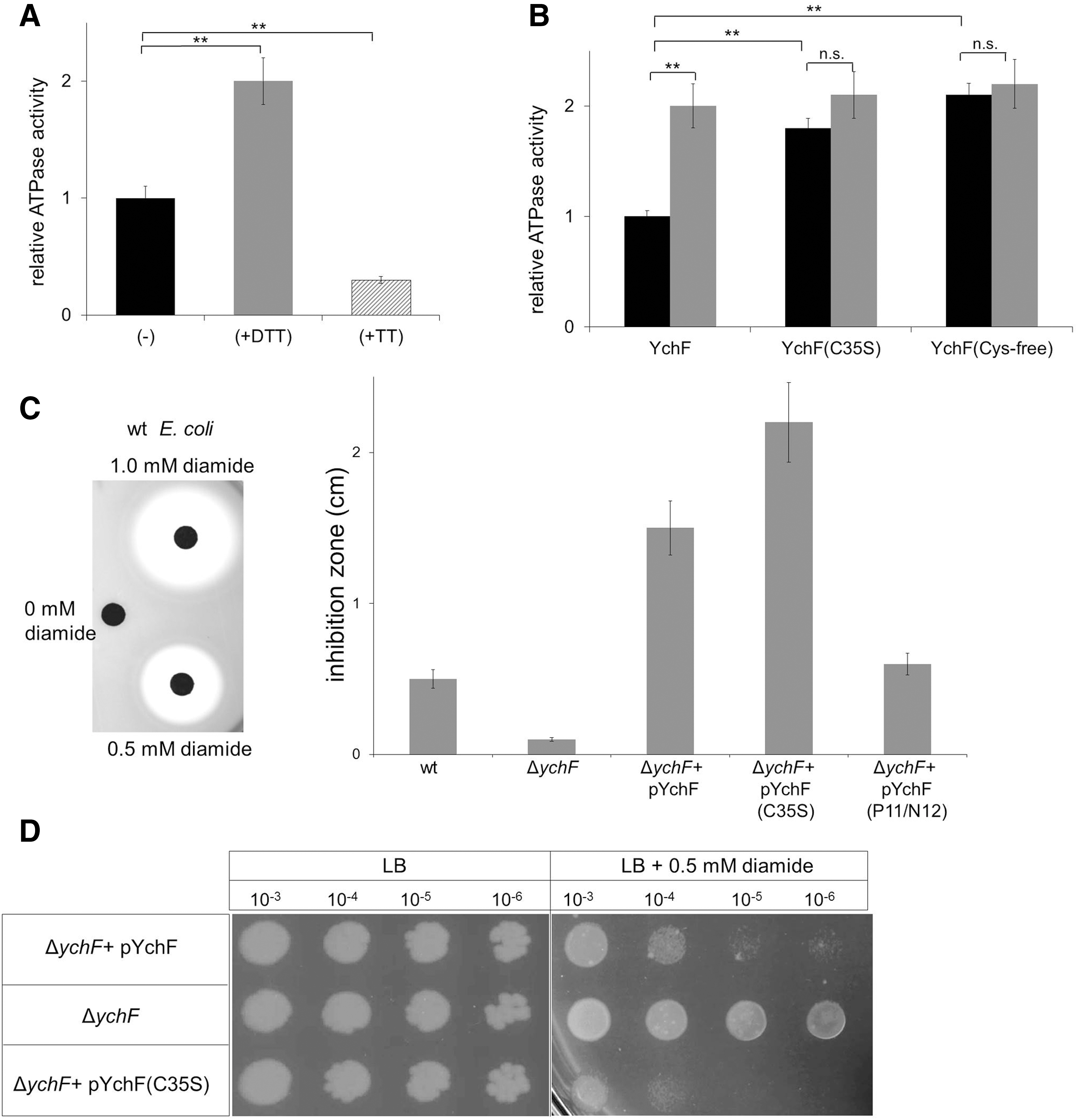
We then compared the ATPase activity of wild-type YchF with the activity of the YchF(C35S) mutant and the Cys-free YchF. In both the C35S and the Cys-free mutant, we detected an approximately two-fold higher ATPase activity than in the wild type. The addition of DTT had no significant effect on the Cys-free or the C35S YchF mutants (Fig. 4B). In summary, these data demonstrate that the ATPase activity of YchF is determined by its redox-sensitive dimerization. The YchF dimer exhibits only low activity, but the dissociation into the monomer strongly stimulates its ATPase activity.
The overexpression of YchF/Ola1 in E. coli or humans increases the sensitivity toward oxidative stress (53, 56). This phenotype depends on the ATPase activity of YchF (53) and we therefore reasoned that expressing the YchF(C35S) mutant should result in enhanced oxidative stress sensitivity, which was induced by the thiol-modifying agent, diamide. Diamide induces reversible non-native disulfide bonds in cytoplasmic proteins (19), thereby reducing the free thiol concentration. Diamide sensitivity was analyzed by agar diffusion assays (Fig. 4C) and by spotting serially diluted cells on diamide-containing plates (Fig. 4D). Both assays demonstrated increased sensitivity of the YchF(C35S)-expressing ΔychF strain compared with the ΔychF strain expressing wild-type YchF or the plasmid-free ΔychF strain (Fig. 4C, D). Expressing the ATPase-deficient YchF(P11A/N12A) derivative, which shows reduced ATPase activity (53), had only a minor effect on diamide sensitivity (Fig. 4C). These data indicate that the high ATPase activity of the YchF(C35S) mutant inhibits the ability of E. coli to cope with oxidative stress.
YchF interacts with thioredoxin 1 in vivo
The redox-sensitive monomer–dimer transition of YchF and its effect on the ATPase activity raised the question about the physiological redox partner proteins of YchF. This was addressed by using a site-directed in vivo photo-cross-linking approach with para-benzoyl-L-phenylalanine (pBpa). pBpa can be incorporated specifically at amber stop codon positions in the presence of a plasmid-borne orthogonal aminoacyl-tRNA synthetase/tRNACUA pair (37). pBpa was first incorporated into position 20 of YchF. Position 20 is located between cysteine residues, Cys5 and Cys35, and had been cross-linked to catalase KatG (53). E. coli cells expressing pBad-YchF(N20pBpa) were UV exposed to induce the cross-link reaction in vivo and YchF and its cross-linked partner proteins were then purified. As a control, we analyzed samples without UV exposure and samples from cells expressing pBad-YchF without pBpa. Following SDS-PAGE and MS analysis of cross-linked samples and the respective controls, we analyzed the data specifically for cross-links between YchF and proteins that are either associated with the oxidative stress response or involved in maintaining redox homeostasis. In support of previous data (53), we found the E. coli catalases, KatG and KatE, as well as the large subunit of the alkyl hydroperoxide reductase AhpF cross-linked to YchF at position 20 (Table 2). KatG, KatE, and AhpF were also identified in the purified YchF sample without UV exposure (Table 2), supporting the previously observed copurification of these proteins together with YchF. When searching the MS data for potential YchF cross-links to proteins controlling redox homeostasis, we found strong cross-links to thioredoxin 1 (TrxA) and weaker cross-links to thioredoxin 2 (TrxC), peroxiredoxin, and glutaredoxin-4 (Table 2).
Cells expressing YchF with the UV-activated cross-linker pBpa at position 20 of YchF were grown on LB medium and one half was UV exposed (+UV), while the other half served as control (−UV). YchF was purified from both samples and separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Equal gel slices were cut out from both lanes, followed by in-gel digestion using trypsin and mass spectrometric analysis. Shown are data obtained for Escherichia coli proteins involved in antioxidant response.
Protein identified.
Calculated molecular mass.
Molecular mass of cross-linking product determined by extrapolation.
Relative intensity observed in gel slices from the control lane (−UV) compared with the +UV lane.
Sequence coverage of total sequence by detected peptides.
Number of unique peptides detected.
The strong cross-link between YchF and TrxA was further verified by immune detection. When YchF was purified from UV-exposed E. coli pBad-YchF(N20pBpa) cells, we observed a strong UV-dependent band at ∼50 kDa, which was recognized by α-TrxA antibodies (Fig. 5A). This band was absent when pBad-YchF(N20pBpa) was expressed in a ΔtrxA knockout strain. These data demonstrate that YchF interacts with TrxA in vivo. We also noticed that TrxA copurified with YchF because it was detected as an ∼10 kDa band in the purified YchF sample (Fig. 5A). Upon UV exposure, the copurifying TrxA amount became significantly weaker, presumably because most of TrxA was cross-linked to YchF (Fig. 5A). To analyze the contribution of the cysteine residues of YchF to the TrxA interaction, we reduced the concentration of available thiol groups by treating the cells before cross-linking with a low concentration of diamide. At concentrations below 100 μM, diamide does not induce the OxyR response (58) and, in agreement with this, the diamide-treated sample did not show significantly changed YchF or TrxA levels (Fig. 5B). However, upon diamide treatment, we observed a significant reduction in the YchF-TrxA cross-linking efficiency (Fig. 5C). We also analyzed the cross-link between YchF and peroxiredoxin and found reduced amounts of the cross-linked product upon diamide treatment (Fig. 5D). Thus, decreasing the number of available thiol groups impairs the interaction of YchF with TrxA and peroxiredoxin, which suggests that the cysteine residues of YchF are important for these interactions.
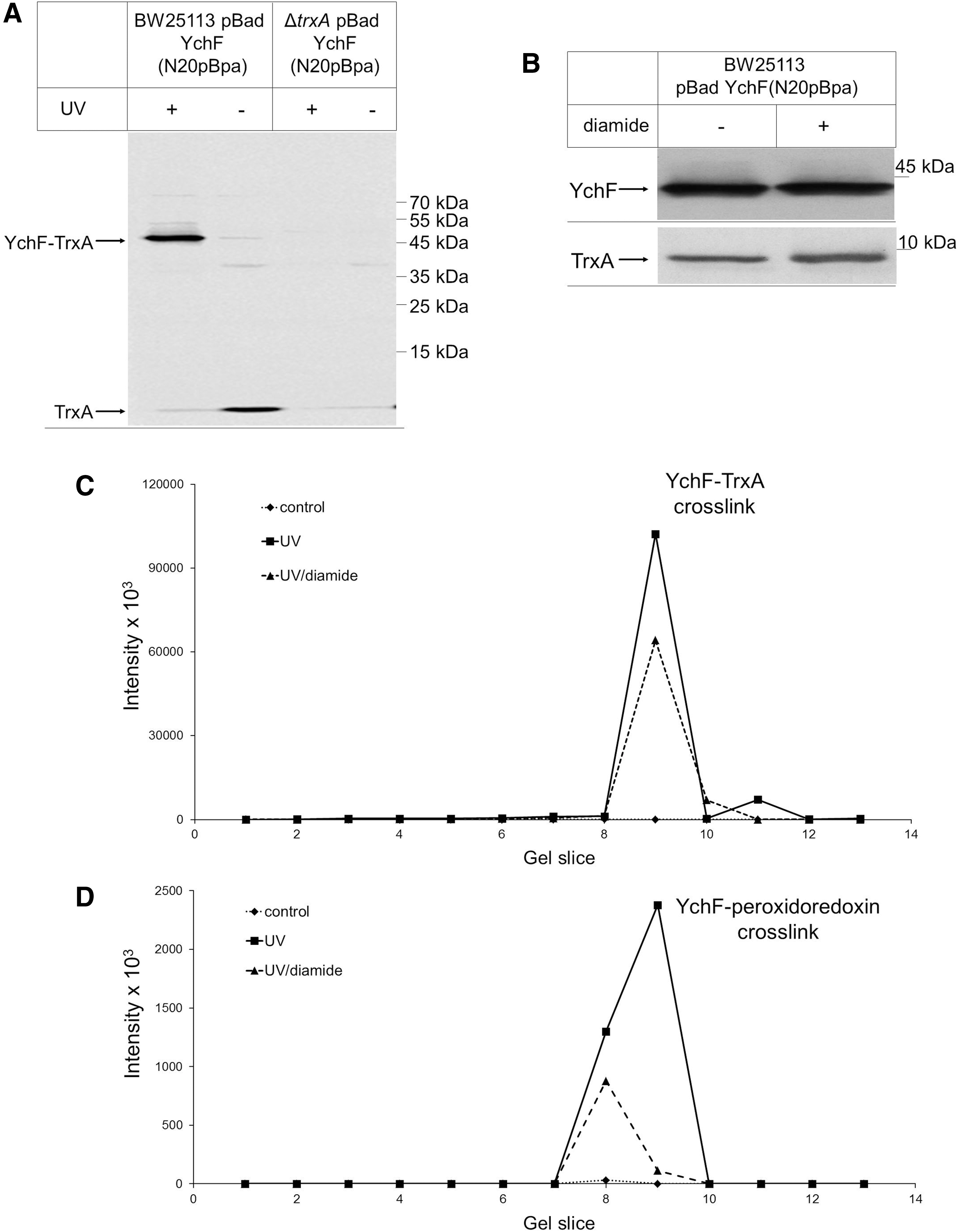
The specificity of the YchF-TrxA interaction was further analyzed by incorporating pBpa into positions 146 and 160 of YchF. These residues are close to Cys140 and Cys168 and located within a predicted coiled-coil structure of YchF. Western blotting showed YchF-TrxA cross-links for both positions (Fig. 6), which were confirmed by MS. However, the cross-linked products from positions 146 and 160 appeared weaker than the cross-link from the N20 residue. Furthermore, both cross-linked products migrated as a double band; a weaker band migrating at ∼50 kDa, and a stronger band migrating at ∼55 kDa. Position-dependent mobility of pBpa cross-linked products on SDS-PAGE is frequently observed (25, 31, 38) and probably the result of differences in the three-dimensional structures of the cross-linking products. We also noticed less TrxA copurifying with YchF that had pBpa inserted into positions 140 and 160, which could indicate that pBpa insertion at these residues reduces the YchF-TrxA interaction.
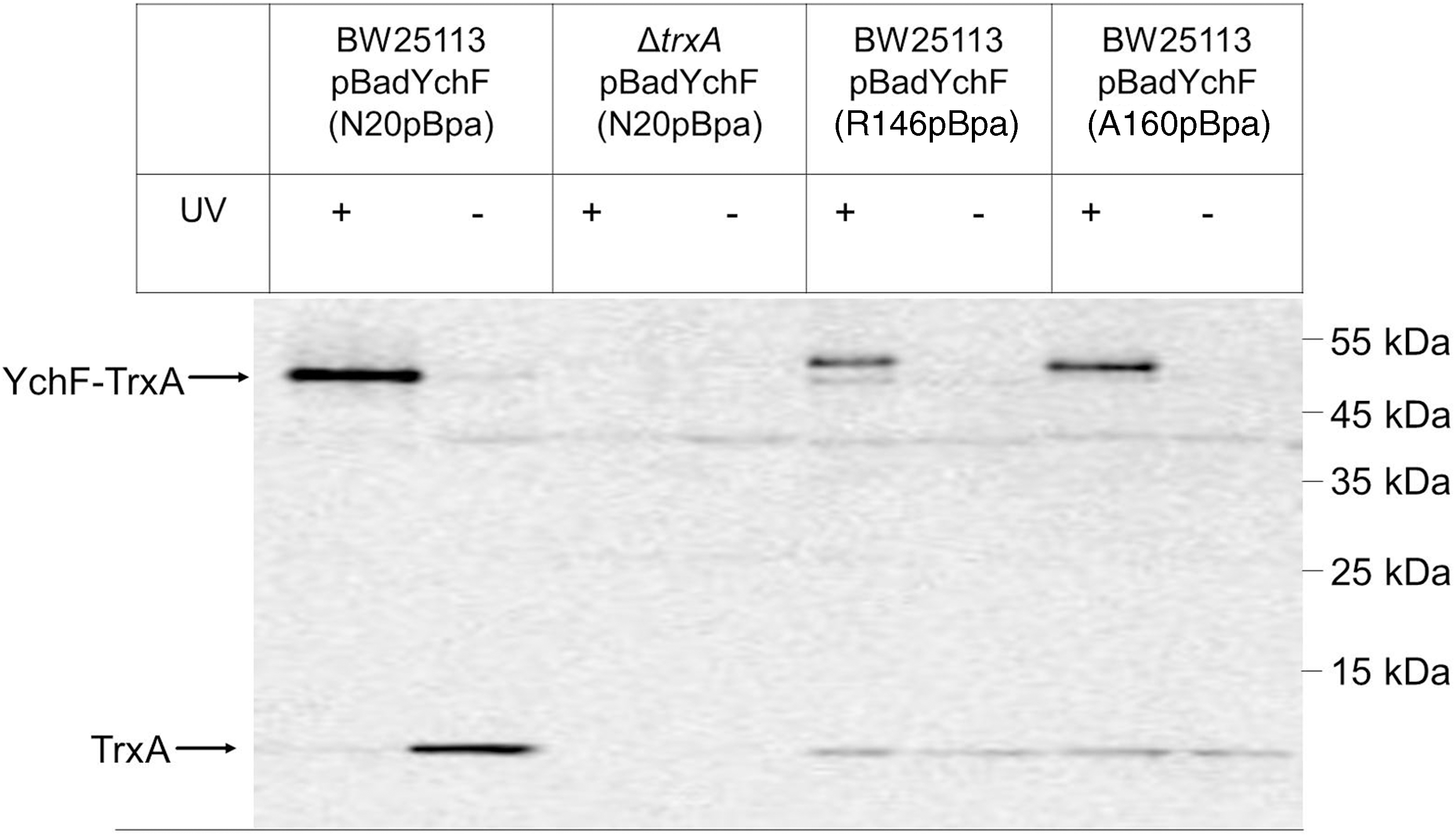
In summary, our data show that the N-terminus of YchF and the predicted coiled-coil domain are cross-linked to TrxA. Reducing the concentration of free thiol groups by diamide treatment reduces the YchF-TrxA cross-linking efficiency, suggesting that the YchF-TrxA interaction probably involves mixed disulfides.
Thioredoxin 1 activates the ATPase activity of YchF by dimer dissociation
The physiological significance of the YchF-TrxA interaction was determined by analyzing the ATPase activity of YchF in the presence or absence of TrxA. TrxA was added to purified YchF and the ATPase activity was measured. In the presence of equimolar amounts of TrxA, we observed about two-fold stimulation of the YchF ATPase activity, which was comparable with the stimulatory effect of DTT (Fig. 7A). The stepwise increase of the TrxA concentration to a YchF:TrxA ratio of 1:4 did not significantly stimulate the ATPase activity further (Fig. 7A). These data demonstrate that the YchF-TrxA interaction accelerates the ATPase activity of YchF and establish the physiological significance of the YchF-TrxA interaction observed via in vivo cross-linking.
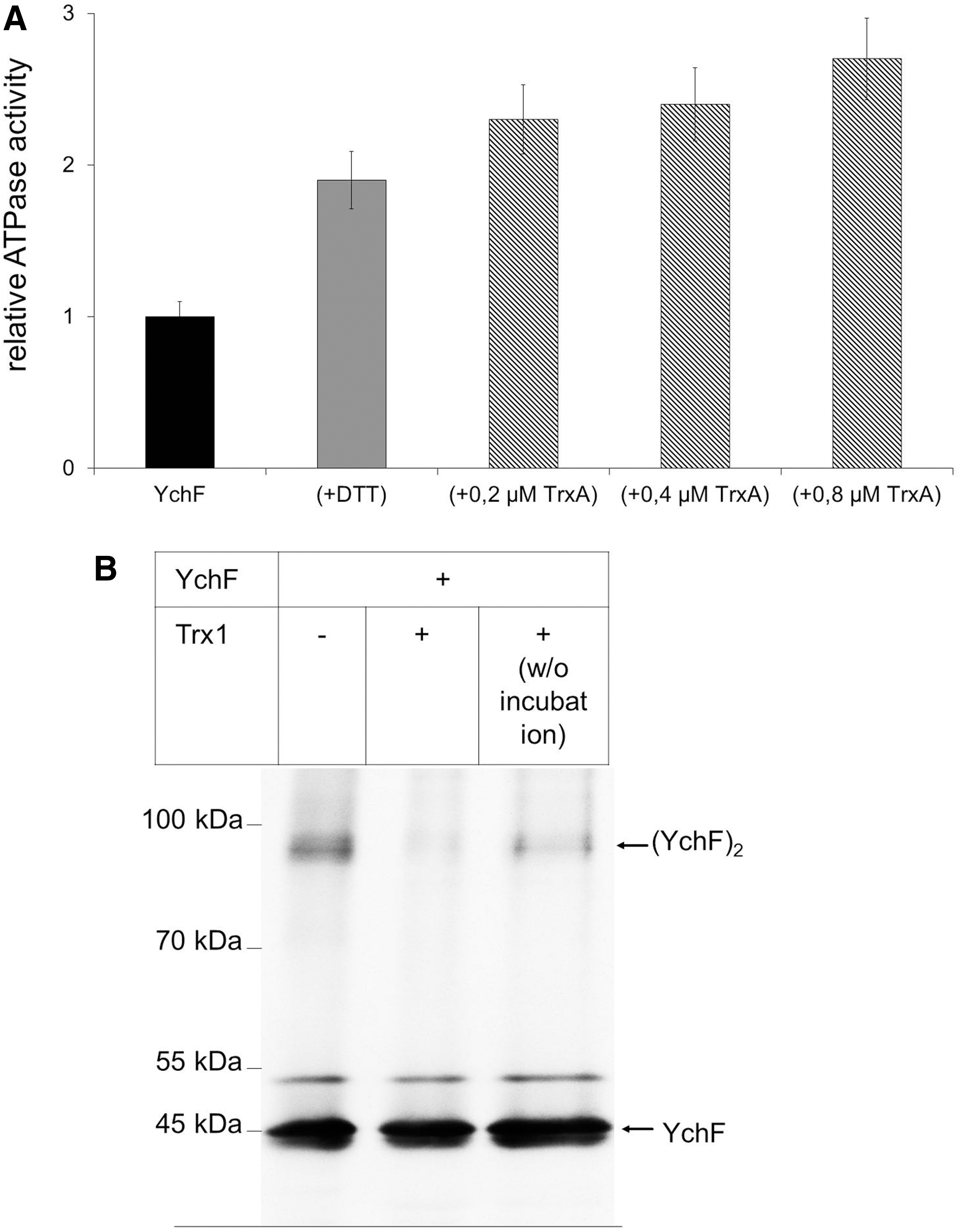
We also tested whether TrxA would dissociate the YchF dimer. When purified YchF was separated under nonreducing conditions by SDS-PAGE, we observed both the YchF monomer and dimer by Western blotting (Fig. 7B). However, when YchF was incubated with TrxA before SDS-PAGE and Western blotting, the YchF dimer was almost undetectable. As a control, we added TrxA to YchF without further incubation and separated it directly on SDS-PAGE. Even without additional incubation, TrxA significantly reduced the YchF dimer. Thus, TrxA dissociates the YchF dimer, which concomitantly stimulates the ATPase activity of YchF.
Discussion
The YchF/Ola1 subfamily constitutes one of the most conserved members of the P-Loop GTPase family and is characterized by its unique preference for ATP (24, 52). Sequence comparison between E. coli YchF and human hOla1 revealed 45% sequence identity and 62% sequence similarity, but there is so far no clear consensus about the possible role of this protein family. Disclosing cellular targets of YchF/Ola1 is of significant importance because human Ola1 appears to play a major role in pathogenicity and disease development. Ola1 expression is regulated by DNA damage (32, 46) and is upregulated in many human tumors (46). On the other hand, a downregulation of Ola1 is observed in interferon β-treated multiple sclerosis patients (14). Furthermore, cell proliferation and apoptosis seem to be influenced by Ola1 (23). In both bacteria and humans, overexpression of YchF/Ola1 increases the sensitivity toward oxidative stress (53, 56, 57) and an imbalance of the oxidative stress response could potentially provide the link between DNA damage repair (40), the pathogenesis of multiple sclerosis (30), and apoptosis (43).
The YchF/Ola1 mode of action is unknown, but the available data support a role as inhibitor of the oxidative stress response by a transcription- and translation-independent mechanism (53, 56). E. coli cells expressing ATPase-deficient YchF derivatives do not display H2O2 hypersensitivity (53). This suggests that the ATPase activity of YchF is required for inhibition of the oxidative stress response, probably by regulating the interaction of YchF with proteins such as KatG or the iron scavenger Dps (19, 53). The ATPase activity is subject to a complex regulatory regime (53) and our data now demonstrate that YchF undergoes a reversible redox-sensitive dimerization, which inactivates its ATPase activity. Dimer formation depends on Cys35, which is conserved from E. coli to humans and located next to the conserved threonine residue within the G2 motif of the ATPase domain (Fig. 1). This provides the explanation for the ATPase-inhibiting effect of YchF dimerization. The G2 motif serves as the effector region of GTPases, and hydrogen bonds between the threonine residue and the γ-phosphate are required for catalysis and the subsequent conformational changes (54). This so-called loaded spring mechanism is most likely impaired upon disulfide bond formation of the nearby cysteine residue.
TrxA or reducing agents such as DTT dissociate the YchF dimer and activate its full ATPase activity. Thus, the redox-regulated monomer–dimer equilibrium of YchF appears to be a key event in the functional cycle of YchF. Wild-type E. coli cells produce ∼15 μM H2O2/s (41), which is too low for significant thiol oxidation (19). Still, we observed YchF dimer formation in E. coli cells without H2O2 treatment. This could indicate that the active site thiols of YchF are unusually reactive, which has also been observed for OxyR and AhpCF (4). In addition, the detection of YchF in E. coli cells by Western blotting required moderate overexpression. Assuming that YchF inhibits the antioxidant response, the endogenous H2O2 concentration in YchF-overexpressing cells might actually be higher than in wild-type E. coli cells, which would promote the formation and stability of the YchF dimer even in the absence of exogenous H2O2. Our current model suggests that under native conditions, YchF exists predominantly as a monomer due to the reducing conditions in the bacterial cytosol. This would allow for the full ATPase activity, which is required for inactivation of antioxidant enzymes. In the presence of oxidative stress, YchF dimerization would prevent ATP hydrolysis and the inhibitory effect of YchF on antioxidant enzymes diminishes (Fig. 8). Redox-dependent dimerization is a well-known effect in biology and has been observed in response to oxidative stress for peroxiredoxins (5), mitofusins (42), or the redox chaperone, Hsp33 (18). Dimer dissociation of peroxiredoxin and Hsp33 is facilitated by the thioredoxin and glutaredoxin systems (18, 27), similar to what we observe for YchF. Intriguingly, our data identified TrxA as the predominant redox partner of YchF. In contrast to TrxC, TrxA in E. coli is not under OxyR control (59, 60) and thus not upregulated in the presence of oxidative stress. This probably reduces YchF monomerization under oxidative stress conditions.
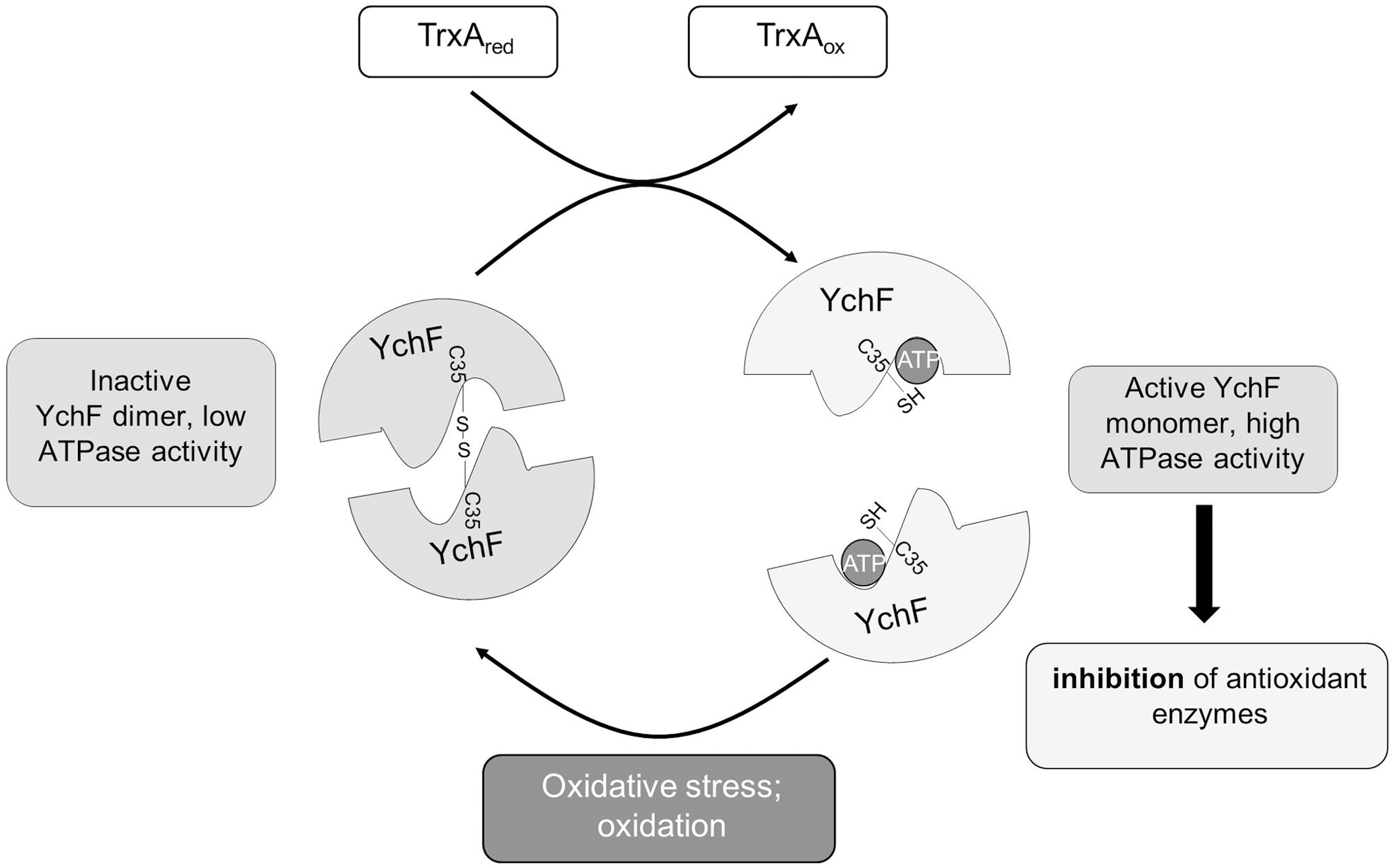
In YchF-overexpressing cells, catalase activity is reduced (53) and the interaction of YchF with the three main H2O2-detoxifying enzymes, KatG, KatE, AhpCF, in E. coli has been demonstrated (Table 2) (53). Still, the exact mechanism of catalase inhibition remains to be identified. YchF could serve as a trap for these enzymes under nonstress conditions, allowing their immediate reactivation upon oxidative stress. This strategy could be part of the adaptive response against oxidative stress that has been observed in both bacteria and eukaryotes (20). The regulation of H2O2-degrading enzymes is also important for H2O2-dependent signaling processes, which stimulate cell proliferation and migration in eukaryotic cells (51). A role of Ola1/YchF in regulating the endogenous H2O2 concentration is supported by several observations. Knockdown of Ola1 has been shown to influence the migration of cancer cells, probably by modulation of the intracellular ROS levels (57). In addition, focal adhesion kinase is downregulated in H2O2-treated mesenchymal stem cells (44), but upregulated upon Ola1 knockdown (22), which is expected if Ola1 inhibits H2O2-degrading enzymes. The existence of H2O2-dependent signaling processes in bacteria has not been clearly demonstrated, but the catalytic mechanism of catalases might be a further reason for their inhibition in the absence of oxidative stress. At low H2O2 concentrations, the two-step catalytic cycle of catalases can stall with the heme cofactor in its ferryl intermediate state (36), which will then abstract electrons from the surrounding polypeptide and thus function as a damaging oxidant. In addition, catalase peroxidases also efficiently oxidize NAD(P)H, which is linked to superoxide radical formation (19). Thus, the post-translational inhibition of antioxidant enzymes in the absence of oxidative stress is probably important for both unicellular and multicellular organisms.
The interaction between YchF and TrxA probably adds to the multiple effects that are associated with YchF/Ola1 overexpression because this will likely reduce the concentration of thioredoxin available for other cellular processes. In eukaryotic cells, the thioredoxin-interacting protein, Txnip, has been shown to be a key element of cellular redox regulation because it inhibits thioredoxin activity (34). Txnip promotes apoptosis, increases ROS production, and influences metastasis (55), phenotypes which have also been associated with increased Ola1 concentrations (23, 46, 56).
Finally, the involvement of YchF/Ola1 in regulating oxidative stress could also explain why these proteins hydrolyze ATP rather than GTP. Guanine is particularly sensitive to oxidation due to its low redox potential (17), and the product of guanine oxidation, 7,8-dihydro-8-oxoguanine, is routinely determined for monitoring oxidative DNA damage (21). ATP has a significantly higher redox potential compared with GTP (48) and is less sensitive toward oxidation. Furthermore, upon oxidative stress, the ATP level transiently increases, probably due to the inhibition of energy-consuming processes (1). Using ATP rather than GTP could therefore reflect the functional adaptation of the Ola1/YchF subfamily to regulation of the oxidative stress response.
Materials and Methods
Bacterial strains and growth conditions
E. coli strains, BW25113, MRE600, BL21, and BL21(DE3), were used as wild-type strains and were grown in LB medium at 37°C; 25 μg/ml kanamycin was added for the growth of JW1194 (ΔychF) and MY1013 (ΔtrxA); 50 μg/ml ampicillin was added for strains carrying pBad-ychF or its derivatives; and 35 μg/ml chloramphenicol for strains carrying pSup-BpaRS-6TRN (37). Plasmid construction and mutagenesis are described in Supplementary Data. Spot assays and inhibition assays in the presence of diamide were performed as described for H2O2 (53).
Protein purification and ATPase assay
To purify N-terminally His-tagged thioredoxin 1, BL21-carrying pET19b-trxA was grown to an optical density (OD) of 0.5, and then induced by 1 mM IPTG. After 3 h, cells were harvested, resuspended in EPTX buffer (25 mM HEPES KOH, pH 7.5, 1 M NH4-acetate, 0.4 mM Mg-acetate), and stored at −20°C. After thawing, Complete protease inhibitor mixture (Roche) and phenylmethylsulfonyl fluoride (PMSF, final concentration 0.5 mM) were added. Cells were lysed thrice by a French pressure cell (Thermo Scientific). Cell debris was removed by centrifugation (30 min, 15,500 rpm, Sorvall SS-34 rotor). The supernatant was centrifuged for 1 h at 50,000 rpm (Beckmann Ti50 rotor). The supernatant was mixed with EPTX (+5 mM imidazole)-equilibrated Talon beads (Clontech) and incubated at 4°C for 2 h. The Talon beads were washed five times with 3 ml 5 mM imidazole in EPTX buffer and proteins were eluted with an imidazole gradient (100–300 mM) in EPTX buffer. Eluted proteins were buffer exchanged to 1× HT buffer (50 mM Hepes-KOH, pH 7.6; 100 mM K-acetate, 10 mM Mg-acetate) using PD10 columns (GE Healthcare).
Purification of YchF carrying a C-terminal His-tag followed the same protocol, but cells were induced with 0.001% arabinose. Cysteine-free YchF was purified from inclusion bodies after the first centrifugation step. The pellet was resuspended in 6 M urea in EPTX buffer at 4°C for 5 h. After centrifugation (15,500 rpm, 30 min, Sorvall SS-34 rotor) the supernatant was incubated with EPTX-equilibrated Talon beads and further purified as described above. ATPase activity of YchF was determined by measuring hydrolysis of γ-33P-labeled ATP (53).
Cell disruption with lysozyme
E. coli cells were grown to an OD of 0.5 and YchF expression was induced by 0.001% arabinose. Two hours after induction, cells were harvested and washed twice with Tris-HCl (pH 8.0). After centrifugation, the cells were resuspended in Tris-HCl (pH 8.0), 1 mM EDTA, 25 mM NaCl containing lysozyme (Sigma) (10 mg/ml), Complete protease inhibitor (Roche), and 0.5 mM PMSF. The suspension was incubated for 30 min at 37°C. Subsequently, the cells were frozen five times in liquid nitrogen and thawed in water. The suspension was centrifuged (14,000 rpm, 10 min) and the supernatant was loaded onto an SDS-PAGE.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis
Samples were denatured at 37°C for 10 min. Samples for nonreducing SDS-PAGE were resuspended in DTT-free 4x Laemmli loading buffer (278 mM Tris-HCl, pH 6.8, 44.4% glycerol, 4.4% SDS, 0.02% bromophenol blue). Reducing loading buffer contained fresh DTT at a final concentration of 25 mM.
For two-dimensional SDS-PAGE, purified YchF was separated by nonreducing SDS-PAGE as the first dimension. The gel was stained with colloidal Coomassie and the YchF monomer and dimer bands were excised and incubated in 200 μl of denaturing buffer (50 mM Tris-HCl pH 6.8, 30% glycerol, 2% SDS, 6 M urea,1 M DTT) for 30 min at 54°C. The sample was separated by reducing SDS-PAGE as the second dimension and stained with colloidal Coomassie.
Western blot analyses
For Western blot analyses, the proteins were blotted onto nitrocellulose membranes (GE Healthcare). α-YchF antibodies were raised in rabbits against the peptide VNEDGFENNPYLDQC. α-TrxA antibodies were obtained from Sigma. A horseradish peroxidase-coupled secondary antibody was used for detection; blots were incubated for 1 min with homemade enhanced chemiluminescence reagent and signals were detected by a CCD camera.
In vivo site-directed cross-linking
E. coli cells carrying pSUP-BpaRS-6TRN and pBad-YchFN20, pBad-YchFR146, or pBad-YchFA160 were grown overnight and used for the inoculation of 400 ml LB; 0.5 mM pBpA in 1 M NaOH was added and cells were grown at 37°C to an OD of 0.3 before they were induced by 0.001% arabinose. Cells were harvested after 3 h of growth and resuspended in 8 ml PBS buffer. Half of the sample was transferred into a six-well microtiter plate and treated with UV light (0.12 J/cm2) (UV Transilluminator Vilber Lourmat BLX-365) for 20 min. The other half was protected against UV light. YchF was then purified from UV-exposed and control cells following the protocol described above. To analyze the effect of thiol depletion on in vivo cross-linking, cells were treated for 30 min with 50 μM diamide (Sigma) at 37°C before harvesting.
Protein identification and quantification by mass spectrometry
Lanes of an SDS gel of purified samples were cut into equal slices and subjected to in-gel digestion using trypsin, followed by nano-UHPLC/MS analysis essentially as described (38). The LTQ-FT-Ultra (Thermo Fisher Scientific) was operated with settings as described (33), except that survey full MS spectra were collected at a resolution of 25,000 with an automatic gain control target value of 2 × 106 ions. Proteins were identified by database searches using the MaxQuant program version 1.3.0.5 (12, 13) as described (33), selecting carbamidomethylation of cysteine as a fixed modification. Protein sequences were taken from the UniProt database (50) for E. coli (version 2013_07, taxonomy: 83333, keywords: 181 and 1185). Intensities per protein (representing the sum of unique peptide intensities) and per gel slice were used to generate profiles for individual gel lanes and to estimate the molecular mass of cross-link products by extrapolation. Relative quantification was done by dividing the total intensity per protein observed in the control lane (−UV) by that observed in the +UV lane.
Identification of disulfide bonds by mass spectrometry
To block free cysteine residues, purified YchF (3.5 mg) was treated with 100 mM iodoacetamide for 15 min at 37°C before separation by SDS-PAGE. After colloidal Coomassie staining, the monomer and the dimer bands were excised. Gel slices were washed and destained by five successive alternate 10-min incubations with 10 mM NH4HCO3, followed by 50% ethanol in 10 mM NH4HCO3. To minimize disulfide scrambling, the pH was adjusted to 6.5 with acetic acid. Furthermore, 2 mM N-ethylmaleimide (NEM) was included in both solutions to prevent the formation of non-native disulfide bonds. In-gel protein digestion using trypsin was carried out at pH 6.5 in 10 mM NH4HCO3 supplemented with 2 mM NEM. Samples were incubated with 66 ng trypsin (sequencing grade modified, Promega) per gel slice for 15 h at 37°C. Peptides were eluted in 0.05% trifluoroacetic acid (TFA)/50% acetonitrile (ACN), dried in vacuum, and redissolved in 15 μl 0.1% TFA. Samples were analyzed by nano-HPLC/ESI-MS/MS as described (33), except that peptides were separated on a 50 cm × 70 μm C18 column (Acclaim PepMap RSLC column, 2 μm particle size, 100 Å pore size; Thermo Scientific) at a flow rate of 250 nl/min with a linear gradient consisting of 1% to 65% solvent B (0.1% formic acid, 4% dimethylsulfoxide, 30% methanol, 48% ACN) for 30 min. The identification of disulfide-linked peptides is described in Supplementary Data.
Cysteine labeling with PEG-Mal
Purified YchF in 50 mM Tris-HCl, pH 7.5, 5 mM MgCl2, and 200 mM NaCl was reduced by incubating it for 1 h at 4°C with 150 μl Thermo Scientific™ Immobilized TCEP Disulfide Reducing Gel and subsequently incubated with 0.75 mM PEG-MAL (in DMSO) for 15 min at 25°C. Loading buffer (−DTT) was added and the samples were denatured at 37°C for 10 min and loaded onto SDS-PAGE.
Cysteine labeling with fluorescein-5-maleimide
Purified YchF (5 mg) was reduced by incubation with TCEP Reducing Gel for 2 h at 4°C and continuous shaking (750 rpm). After centrifugation (21, 000 g, 3 min), ∼50 μg of fluorescein-5-maleimide (in HT buffer) was added to the supernatant, followed by a 2-h incubation at room temperature. Precipitates were removed by centrifugation (21, 000 g, 1 min) and both supernatant and pellet were denatured for 10 min at 54°C in Laemmli loading buffer (−DTT). After SDS-PAGE, fluorescence was detected using a CCD camera.
Footnotes
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft to B.W. and H.G.K., the Excellence Initiative of the German Federal and State Governments (Grant EXC 294 BIOSS Centre for Biological Signalling Studies) to B.W., the Else-Kröner-Fresenius Stiftung and the Motivate MD College of the University Freiburg Medical School to L.H., and the Chinese Scholarship Council to Q.B. The authors thank the National Institute of Genetics, Japan, for E. coli strains, JW1194 (ΔychF) and MY1013 (ΔtrxA).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.