
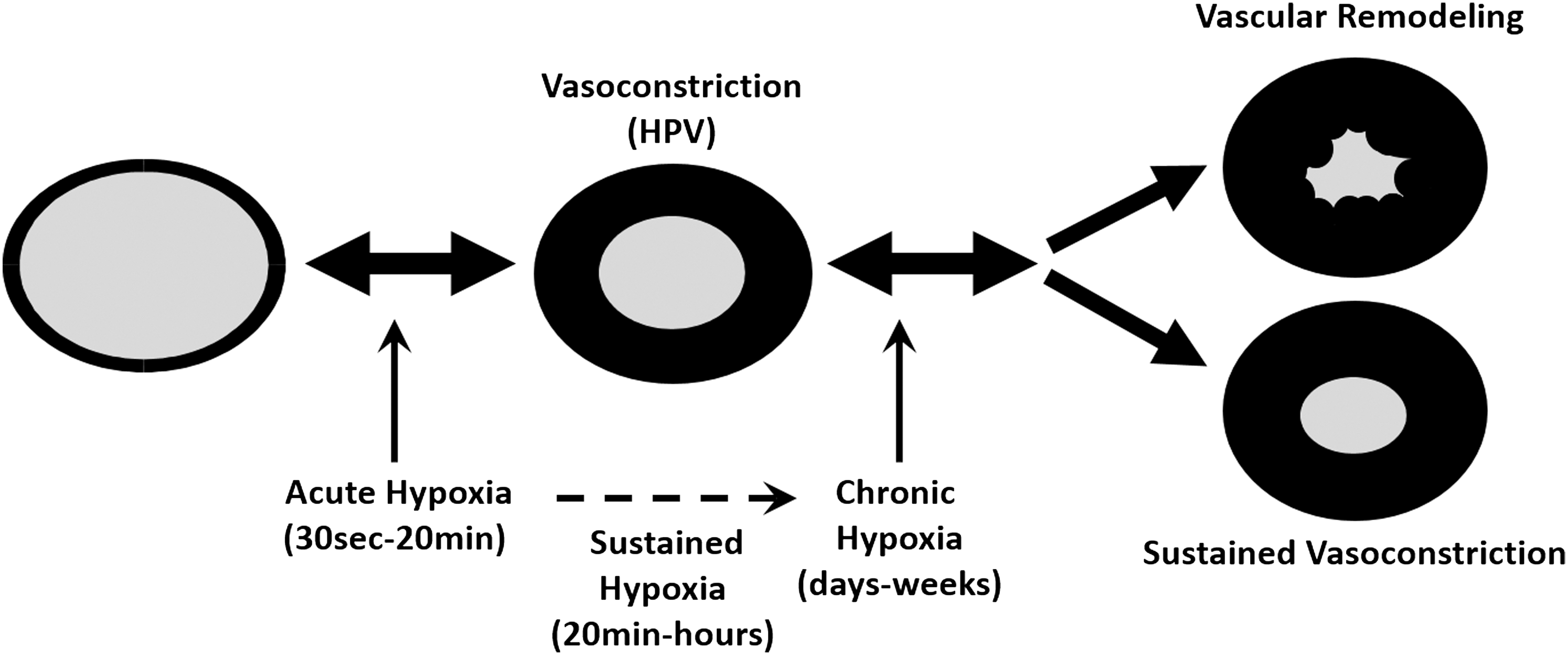
Abstract
Introduction
T
The recognition of the involvement of ion channels in hypoxic events of the pulmonary vasculature uncovered L-type calcium (Ca2+) channels and potassium (K+) channels as important players (8, 146). Several other types of potassium and calcium channels were discovered, which also contribute to HPV and chronic hypoxia-induced PH (138, 160). These ion channels are highly sensitive to redox changes (109). In this regard, two antithetical models are currently discussed. The first model favors the closure of Kv-channels mediated by a reduced mitochondrial ROS release, which activates vasoconstrictive, pro-proliferative, and anti-apoptotic signaling cascades (6). This proposal is based on the opposing observations in terms of increased or decreased ROS production in the pulmonary circulation during hypoxia. There is also a debate about the potential sources of ROS production (i.e., mitochondria or NADPH oxidases) and their downstream targets (Fig. 2).
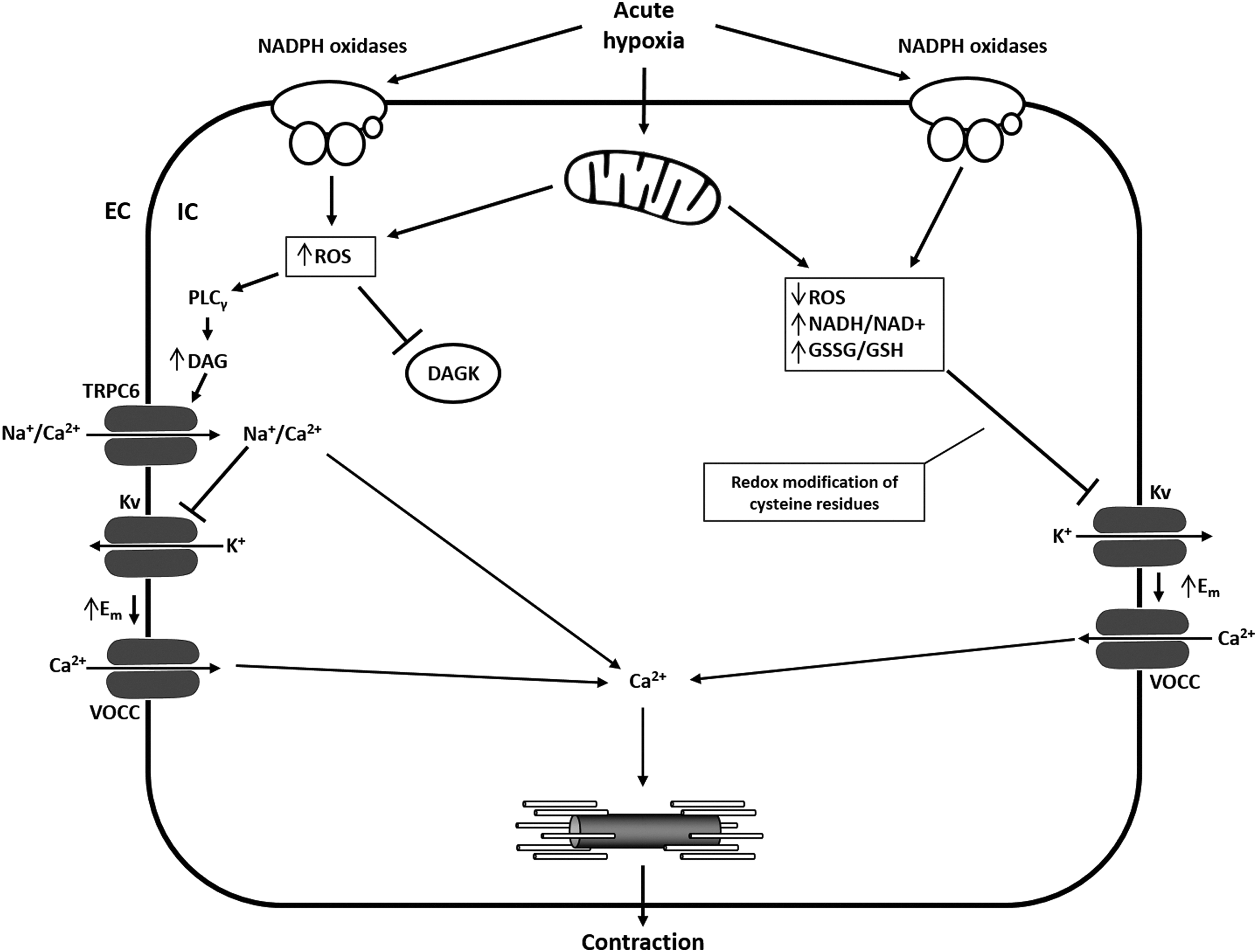
The second model proposes that an increase of ROS from mitochondria or NADPH oxidases triggers such events via different membrane channels, including Kv-, transient receptor potential (TRP)-, and L-type Ca2+ channels and/or intracellular Ca2+ release (52) (Fig. 2). The discrepancies between the two models might be explained by studies that revealed differences in the basal oxidation state among the subcellular compartments. During acute hypoxia, the cytosol and intermembrane space showed increased ROS generation, whereas ROS production in the mitochondrial matrix was decreased (133). Under normoxic conditions, the mitochondrial complexes I and III produce small basal amounts of superoxide radicals (proportional to alveolar PaO2) (19). Superoxide from complex I and the Qi side of complex III enter the matrix, while superoxide formed on the Qo side of complex III goes to the intermembrane space and is then converted to hydrogen peroxide (H2O2), which is able to pass the outer mitochondrial membrane and to enter the cytosol (133). Besides the mitochondrial respiratory chain, the ROS locally produced by the Nox family of NADPH oxidases during normoxia elicit a plethora of cellular responses required for physiological growth factor signaling (21, 22, 86). However, under hypoxic conditions, increased (52, 133, 152) and decreased (5, 6, 156) ROS production during acute and chronic hypoxia has been described.
The determination of the pulmonary vascular tone by ion channels in response to hypoxia is widely accepted. Whether due to an increase of decrease of ROS, Ca2+ entry through L-type channels, and release of calcium from the sarcoplasmic reticulum (SR) causes an increase in intracellular Ca2+, and initiates and maintains contraction of pulmonary vascular smooth muscle cells (VSMC) in response to hypoxia (151, 158).
Against this background, we, in this review, will focus on the role of redox regulation of ion channels during acute and chronic hypoxia. We are aware that we are not able to refer to all investigations which have been performed in this context. In addition, in a variety of instances, we refer also to nonpulmonary investigation if lung-specific literature is missing.
Potassium Channels
Potassium channels conduct K+ ions across the cell membrane and are crucial for the pattern of action potentials (electric impulse formation), epithelial function, cell volume regulation, hormone secretion, and adjusting plasma membrane potential (59). The conduction follows the electrochemical gradient for K+, and these channels are extremely selective for K+ (63). All known K+ channels share the same structure of this very selective pore region. K+ channels can be divided into five subcategories and differ mainly in the ways in which K+ channels are gated open: (i) inward rectifiers (Kir), including classical Kir, G-protein-gated channels, ATP-sensitive K+ channels (KATP), and K+-transport channels; (ii) four transmembrane segments-2 pores (K2P), including pH, temperature, fatty acid, voltage, and membrane stretch-regulated channels; (iii) voltage-gated (Kv); (iv) the Slo family (KCa), having a very large conductance and including the “big” K+ channels (BK); and (v) Ca2+-activated SK family (SKCa), having a small conductance (SK channels) (59).
Four major types of K+ channels have been identified in the pulmonary vasculature and the pulmonary arterial smooth muscle: (i) Kv channels, (ii) KCa channels, (iii) KIR channels, and (iv) K2P channels (25, 102). In PASMC, efflux of K+ after activation of K+ channels leads to membrane hyperpolarization and, subsequently, vasodilation. In contrast, inhibition of K+ channels causes depolarization and vasoconstriction (32). Inhibition of K+ channels and influx of K+ (rather the reduction in K+ conductance) was shown to initiate membrane depolarization, activation of voltage-operated Ca2+ channels (VOCCs), and vasoconstriction. In PASMC, this role has mainly been assigned to Kv and K2P channels (105), while KCa channels may be important in fetal or newborn animals (34, 120, 126) and both KCa and ATP-sensitive potassium channels (KATP) may modulate hypoxic depolarization in coronary arterial SMC (38, 39).
Voltage-gated K+ channels
Although various types of K+ channels are expressed in the pulmonary vasculature, much interest has been placed on the role of Kv channels, regarding the membrane potential (107, 171, 173), changes in pulmonary vascular tone (42, 119), and PASMC proliferation (80, 81, 118). Kv channels represent the largest and most diverse family of K+ channels. The family is composed by 40 genes: 36 genes of six transmembrane K+ channels (KCNA [Kv1 family], KCNB [Kv2], KCNC [Kv3], and KCND [Kv4], KCNQ [Kv7], KCNH [Kv10, Kv11, and Kv12]), and a nonconducting group of four gating modulators (KCNF [Kv5], KCNG [Kv6], KCNV [Kv8], and KCNS [Kv9]). The functional channel is formed by four α-subunits (tetrameric organization), with the pore lying in the axis. Each subunit has six transmembrane domains. Kv channels arrange as complexes of homo-tetramers or hetero-tetramers with many possible combinations (59, 127). Under acute hypoxia, the hypoxic stimulus triggers an inhibition of Kv channel activity in PASMC (122, 172). This effect has been described not only for PASMC but also for hypoxic-induced contraction in pulmonary vein smooth muscle cells (45).
In contrast, hypoxia neither inhibits Kv channel activity nor changes expression of Kv channels in systemic SMC (119, 147, 149, 172). Thus, Kv channels appear to be a hypoxic effector in the pulmonary circulation, but not in systemic SMC, conducting vasoconstriction.
Inhibition of Kv channels has also been suggested to be mediated by both decreased (6, 124, 156, 161) and increased (31, 101) ROS production from mitochondria and/or NADPH oxidases. In general, redox modification of cysteine residues is important for Kv activity. Sahoo et al. suggest that physiological levels of ROS trigger a positive feedback mechanism, which reduces Kv channel activity (131). Mittal et al. described a mechanism by which an Nox4-derived increase in ROS production induces Kv channel current inhibition (101). Furthermore, Cogolludo et al. showed that activation of NADPH oxidase and the subsequent production of H2O2 are involved in the Kv channel inhibition and the contractile response induced by thromboxane receptor activation in rat pulmonary arteries (31).
In contrast, the complex I (NADH oxidoreductase) inhibitor rotenone and the flavoprotein inhibitor diphenyleneiodonium (DPI) were shown to inhibit HPV and Kv channel currents (124, 161). However, the effect of rotenone on mitochondrial respiration strongly depends on the concentration and was shown to trigger HPV, as well as to inhibit pulmonary vasoconstrictor responses (138). Rotenone-induced pulmonary vasoconstriction (using rotenone concentrations >350 nM), which was similar to the degree of HPV, has been attributed to nonselective effects rather than to altered ROS generation (138). Other inhibitors of the proximal and distal mitochondrial respiratory chain have also shown to elicit opposing affects regarding HPV (132).
While acute hypoxia acts via inhibition of Kv channel activity, during chronic hypoxia K+ channel density and Kv channel protein expression (Kv1.5 and Kv2.1) is decreased, although a Kv current is still detectable (124). However, both induction and repression of Kv channel subunit expression under chronic hypoxia has been described (44, 61, 91, 147). Patch clamp studies showed that the hypoxic inhibition of the Kv current in PASMC is unchanged even after 2 days of ambient hypoxia, when HPV is already lost (169). However, after 3 weeks of chronic hypoxia, PASMC membrane potential was depolarized, Kv1.5 and Kv2.1 channel protein was decreased, and acute hypoxic inhibition of whole cell K+ current was lost (124). Other studies suggest Kv channel upregulation due to chronic impairment of the thioredoxin system under oxidative stress (pathophysiological ROS levels) (142). Further, interaction with pyridine nucleotides (76) and S-nitrosylation of Cys445 has been described (10).
Ca2+-activated K+ channels
Ca2+-activated K+ (KCa) channels are subcategorized according to their conductance: large (BK), intermediate (IK), and small (SK). Here, we will focus on BKCa channels. In contrast to the BKCa channel, the role of SKCa and IKCa channels in VSMC is not well understood (168). BKCa channels are ubiquitously expressed in VSMC and can be activated by changes in both membrane potential and intracellular Ca2+ concentration (79). These channels were shown to act as a negative-feedback mechanism in response to depolarization and increased cytosolic Ca2+ concentration during vasoconstriction. An increasing intracellular Ca2+ concentration was shown to decrease the BKCa current and increase the Kv current (35). Thus, cytosolic Ca2+ levels not only play a major role regulating these channels but are also sensitive to voltage changes (58).
BKCa channels are present in PASMC, but their role in whole cell K+ currents depends on the species and varies in different pulmonary artery tree regions (94). Proximal segments contain a larger proportion of KCa-enriched PASMC, whereas distal segments contain more Kv-enriched PASMC (7, 96). In addition, it has been proposed that the hypoxic response due to K+ channels changes as PASMC mature from fetal to neonatal and adult PASMC (33). It has been suggested that KCa channel activity is prominent in hypoxia-induced fetal pulmonary vasodilation (33, 125). The contribution to the hypoxic response by BKCa is still unclear. During acute hypoxia, BKCa channel activity was attenuated in PASMC (84, 113, 121), while Ca2+ release from SR increased BKCa channel activity (18).
Since Ca2+ release in the SR is linked to the activation of large-conductance KCa channels and membrane hyperpolarization (28), it remains unsolved whether acute hypoxia-mediated Ca2+ release triggers membrane depolarization in PASMC. Not much is known about the redox regulation of KCa channels in PASMC or the pulmonary vasculature of adults. At least for mouse lungs, it was shown that knockout of the functional essential BK channel alpha-subunit alters neither acute and sustained HPV nor chronic hypoxia-induced PH (130). KCa channels were shown to be important in mediation of HPV in fetal lungs, but this was due to stimulation of a cyclic nucleotide-dependent kinase, resulting in KCa-channel activation, membrane hyperpolarization, and vasodilation (33).
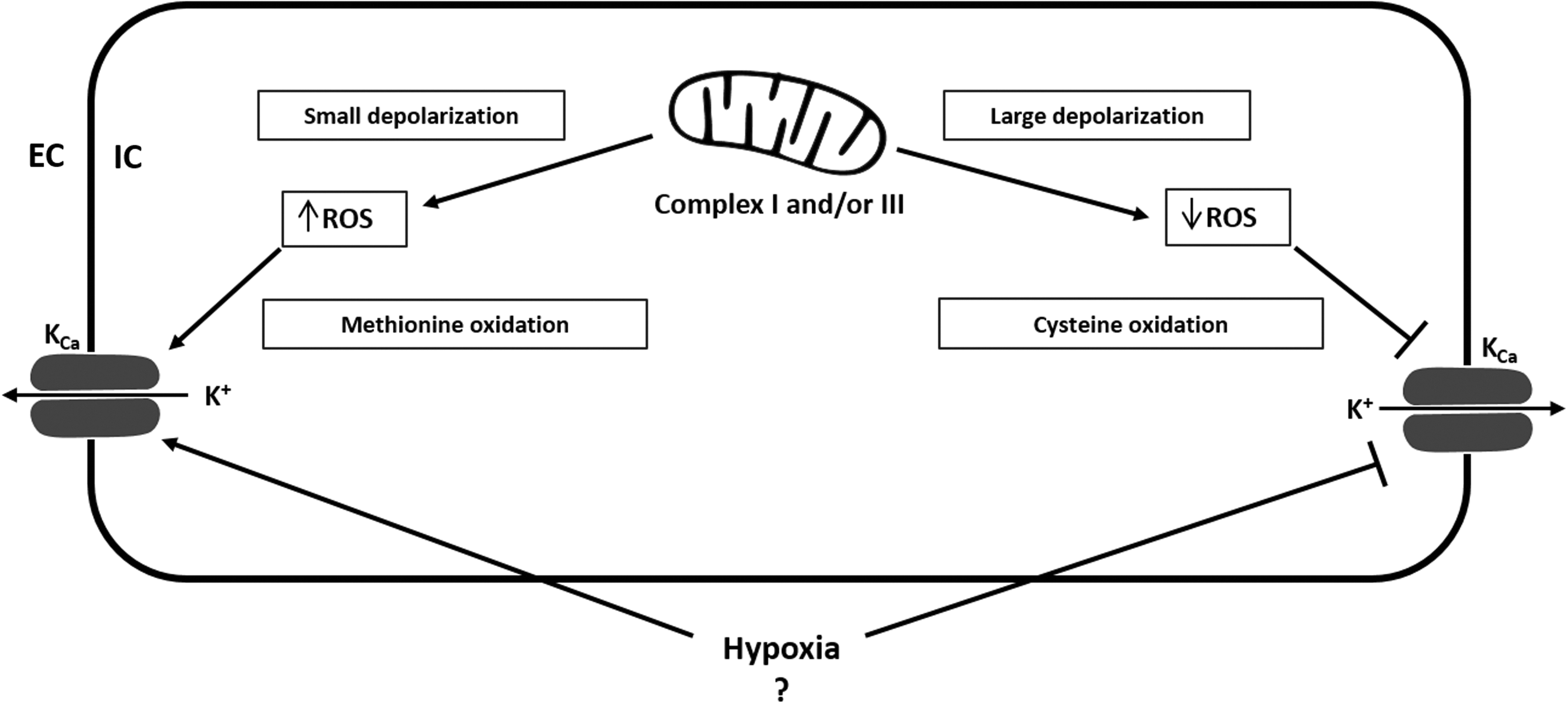
The effect of ROS on BKCa activity is, from our point of view, not conclusive and is mainly derived from nonpulmonary investigations. Figure 3 summarizes the proposed redox-regulation of KCa channels. While the selective BKCa channel inhibitor tetraethylammonium (TEA) (104) was shown to inhibit superoxide-induced vasodilation (154), it had no significant effect on open state probability of BKCa channels (88). H2O2 has been reported to induce both activation and inhibition of BKCa channel activity, depending on the experimental conditions (135, 143, 144). While cysteine oxidation decreased the currents of large-conductance Ca2+-activated K+ channels, methionine oxidation increased currents (143). Furthermore, H2O2 decreased activity of BKCa channels by shifting the voltage sensitivity to a more positive direction (40). In VSMC, peroxynitrite was shown to inhibit BKCa channel activity (24) by suppression of whole-cell KCa current and reduction of open-state probability of single KCa channels (88). In contrast, endothelial BKCa channels did not react to peroxynitrite. The authors suggest that this behavior is due to an insensitivity of endothelial BKCa channels to the interaction between superoxide and nitric oxide (NO) (43), which arises from the different β-subunit of BKCa channels expressed in SMC and endothelial cells (168).
ATP-sensitive K+ channels
KATP are a subclass of inwardly rectifying K+ channels (138), show little or no voltage dependence, and have low open probability under basal conditions. The channels consist of an octameric complex of four pore-forming inward rectifier K+ channel subunits (Kir 6.1 or 6.2) and four sulfonylurea receptors (SURs) (3). Coexpression of these subunits produces two distinct channels, nucleotide diphosphate-sensitive K+ channels (KNDP) and KATP (15). Coexpression of Kir 6.1 with SUR2B has been detected in human PASMC (37), and contribution to resting membrane potential in PASMC has been suggested. In contrast, inhibitors of KATP did not increase normoxic pulmonary vascular resistance in adult mammals, suggesting that these channel types do not control basal pulmonary arterial tone. To date, there is no proposed role for KATP channels in HPV and they have been suggested to be closed in the pulmonary arteries and not to be activated by the levels of hypoxia that cause a constriction (128).
As for KCa channels, the redox regulation of KATP channels in PASMC is not well described. In general, KATP channels are gated by intracellular nucleotides, linking energy metabolism to membrane excitability. In tissues other than the lung, there is increasing evidence the KATP channel activity is likely regulated by redox state (12, 13, 82). Unfortunately, almost all redox dependent regulatory mechanisms were described in other tissues than the lung. Thus, we will not describe these mechanisms here.
Two-pore-domain K+ channels
Two-pore-domain or K2P channels contain four transmembrane domains and two pore domains. A dimer of two subunits forms a single pore and thus the functional channel (two pores in total). N- and C-Terminus is located in the cytosol. K2P channels are selective to K+ and are important for the regulation of the resting membrane potential (background K+ channels), thereby regulating cellular excitability and K+ permeability (27, 95, 140). The regulation of K2P channels is quite complex, as these channels respond to many stimuli, including pH, stretch, temperature, fatty acids, O2 tension, sumoylation, phosphorylation, dephosphorylation, and osmolarity (117, 140). Due to the K+ selectivity and the voltage-independent gating [TREK-1 is voltage gated when S348 is phosphorylated (92), and TASK-1 was shown to be voltage dependent in rabbit PASMC (62)], K2P channels are well suited for mediating background K+ currents.
K2P comprise six subfamilies: TWIK, TREK, TASK, TASK-2, THIK, and TRESK channels (132). In the pulmonary vasculature expression of TASK-1, TASK-2, TREK-2, THIK-1, and TWIK-2 has been demonstrated (53). However, only for TASK-1 and TASK-2 (mainly TASK-1), involvement in a noninactivating background K+ conductance has been shown, where hypoxia-induced inhibition of TASK-1 contributed to PASMC depolarization (47, 108) and HPV (53, 57).
Since TASK-1 cannot sense O2 itself, the NADPH oxidase NOX4 has been suggested to be the O2-sensing partner modulating the O2 sensitivity of TASK-1. In HEK293 cells, hypoxia-induced activation of NOX4 inhibited TASK-1 activity (85). In this process, the heme moiety and FAD-binding domain were proposed to be responsible for the NOX4 regulation of TASK-1 (114). Further, in HeLa cells, TASK-1, TASK-3, and a TASK-1/3 heteromer were shown to be activated by H2O2. This effect was independent of the oxidation of–SH groups, suggesting that H2O2 acts directly on the channel protein.
In contrast, TREK-1, TREK-2, TALK-1, TASK-2, and TRESK did not respond to H2O2 treatment. Superoxide derived from a xanthine/xanthine oxidase mixture only affected TASK-2 activity in HeLa cells (112). It should be noted that H2O2 had no significant effect at concentrations till 16.3 mM, which is a rather unphysiological concentration (137) and might cause unspecific effects. Earlier, Kim et al. published conflictive results showing that H2O2 did not affect TASK-1, TASK-3, and TRAAK currents when the channels were expressed in CHO cells. However, TREK-2 was activated by H2O2, presumably as a response to H2O2-induced myosin light chain kinase (MLCK) activation (77). The different findings by Kim et al. might be explained by the much lower H2O2 concentration (5 mM) and the different cell type used in their study and are partially supported by research published by Turner and Buckler (145). Their results show that hypoxia (and thus ROS) inhibits single channel activity of TASK-1 and TASK-2 in type-1 cells isolated from the carotid body.
Calcium Channels
Intracellular Ca2+ concentration is central for the regulation of vessel tone. During homeostasis, intracellular Ca2+ is ∼100 nM intracellular and 1.6 mM extracellular (67). This huge concentration gradient between the intracellular and the extracellular Ca2+ concentration shows the importance of tightly controlled cellular Ca2+ homeostasis. Ca2+-permeable channels allow Ca2+ to enter into the cell through the membrane due to its electrochemical gradient. Ca2+ pumps transport Ca2+ against its concentration gradient, and the Ca2+ exchangers that can transport Ca2+ to the intra- or extracellular milieu, depending on the mode of action (109). The following channels coordinate cytosolic Ca2+ concentration in PASMC: (i) extracellular Ca2+ entry via VOCCs, (ii) receptor-operated cation channels (ROCs), and (iii) store-operated channels (SOCs) activated by depletion of the SR (109). Alterations of the intracellular Ca2+ concentration play an important role in muscle contraction (skeletal, cardiac, and smooth muscle) and cell motility, neurotransmitter release, neuronal excitability, learning and memory, fertilization and development, cell proliferation, differentiation, apoptosis, and gene transcription. The Ca2+ influx is crucial for hypoxic constriction of the precapillary pulmonary arteries (136). The best characterized pathways of Ca2+ entry into PASMC are through VOCCs (regulated by the resting membrane potential) and TRP channels (TRPC; voltage-independent nonselective cation channels, SOCs and ROCs) (160). An increase of intracellular Ca2+ concentration in PASMC has been widely accepted to be a critical event for HPV (151). In the pulmonary vasculature, the acute hypoxic Ca2+ release in PASMC is dependent to a lesser degree on VOCCs (inhibition attenuated hypoxic Ca2+ release by 30%) and to a greater degree on other transmembrane channels such as TRPC (inhibition attenuated hypoxic Ca2+ release by 60%) (141). These channels are mainly responsible for the sustained PASMC contraction associated with HPV and modulation of the pulmonary hypoxic response.
Voltage-operated Ca2+ channels
The cellular membrane potential of PASMC is largely regulated by K+ channels. Inhibition of K+ efflux (e.g., during hypoxia) causes depolarization of the cell. VOCCs are activated when the depolarization reaches a certain threshold, a mechanism that is also known as excitation–contraction coupling (23). Moreover, the discovery that inhibitors of L-type channels suppress HPV led to the long-standing hypothesis that HPV is primarily caused by redox-mediated inhibition of delayed-rectifier K+ channels, depolarization, and voltage-dependent Ca2+ entry (97, 157).
VOCCs are ubiquitously expressed in VSMC. Their subunits include α1, β1–β4, γ1–γ8, and α2δ1–α2δ3, each containing six transmembrane spanning domains (S1–S6) enclosed by N- and C-termini. Many of these subunits can co-assemble, causing the heterogeneity of VOCCs. The α1 subunit is the major subunit, containing the Ca2+-selective pore (loop between S5 and S6) and voltage sensor (S4), is essential for channel function, and contains sites for channel regulation via intracellular second messengers, toxins, and drugs. The combination of α1 subunits with different accessory subunits forms six functionally distinct subfamilies: the L-, N-, P/Q-, R-, and T-type channels (26). These major subgroups of the VOCC family are expressed in many cell types and are responsible for various cellular functions, including muscle contraction, control of action potential, secretion, and gene expression (17). The dihydropyridine-sensitive, high-voltage-activated and slowly inactivating L-type and the low-voltage-activated, rapidly inactivating T-type channels were most extensively studied in VSMC.
L-type channels have been extensively studied in PASMC and play an important role in increasing cellular Ca2+ concentration during hypoxia (48). L-type channels are high-voltage-activated (167) and regulate excitation-contraction coupling (56). L-type channels were shown to be upregulated in chronic hypoxia-induced PH and associated with a Ca2+-dependent resistance (68). Compared with conduit arteries, the density of L-type calcium channels is two-fold higher in PASMC of the resistance arteries (48).
The other common Ca2+ channel in the pulmonary vasculature is the low-voltage-activated T-type channel (174). These channels are insensitive to common L-type channel blockers, and their physiological relevance is poorly characterized (83). In the pulmonary vasculature, expression and function of T-type channels is not well described and their electrophysiological properties need to be fully characterized. Recent studies have also suggested that T-type channels are important in the proliferation of human PASMC (129). R-type currents have been shown to be activated by endothelin-1 (16) and cause enhanced cerebral artery constriction during subarachnoid hemorrhage (71, 87).
While influx of Ca2+ into the PASMC of resistance arteries is enhanced by hypoxia, the influx into SMC of the conduit arteries is inhibited (like systemic arteries) (48, 49). As previously mentioned, the acute hypoxic Ca2+ release in SMC of the pulmonary resistance arteries is dependent to a lesser degree on VOCCs (inhibition attenuated hypoxic Ca2+ release by 30%) and to a greater degree on other transmembrane channels such as TRPC (inhibition attenuated hypoxic Ca2+ release by 60%) (141). Nevertheless, VOCCs were one of the first Ca2+ channels to be identified as redox sensitive. Oxidants affect VOCC activity, expression, trafficking, open time, and open probability. Cysteine residues in the pore-forming α1-subunit are the molecular targets for ROS (98), and both activation and inhibition of channel activity by oxidation have been described (64, 69, 175) (Fig. 4).
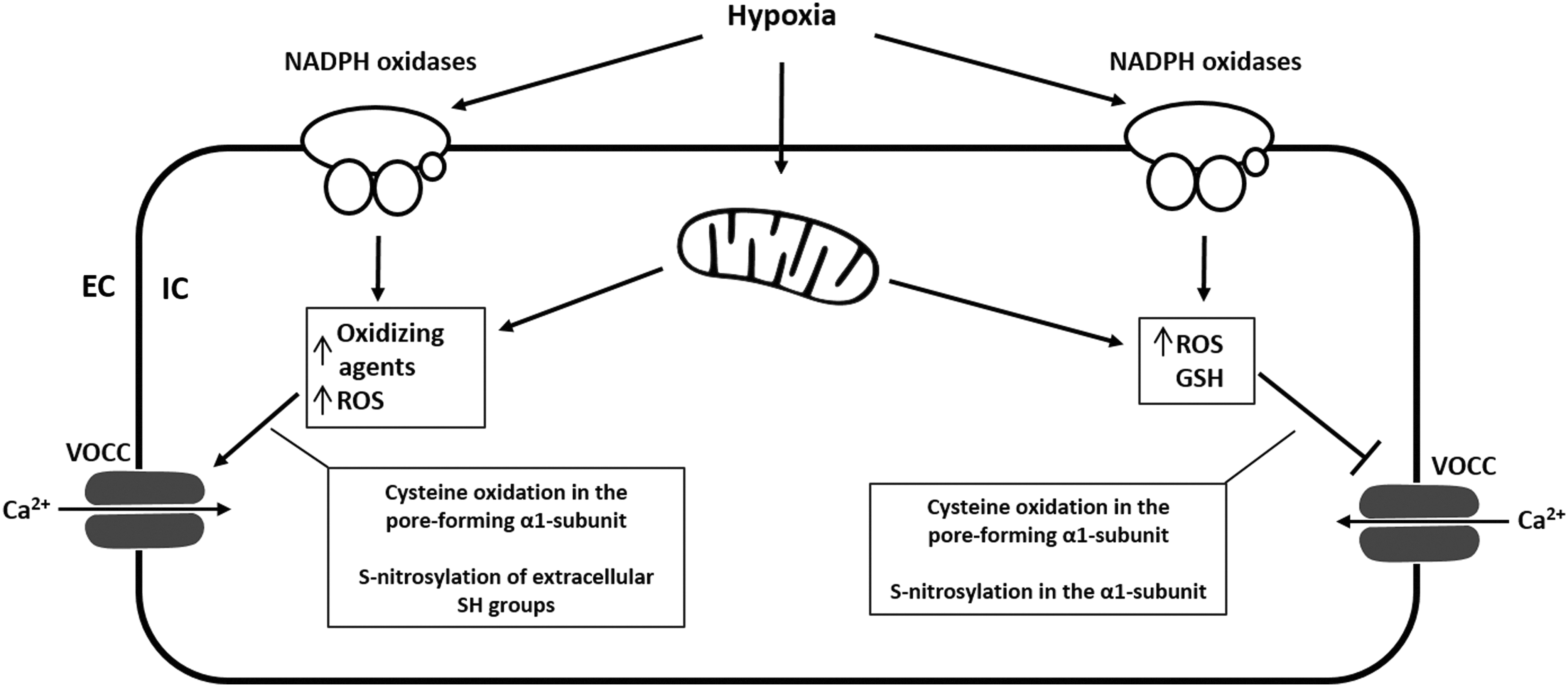
Although almost all studies were done on myocytes or cardiac L-type channels, it has been suggested that the mode of action of the redox regulation from other tissues and cells can probably be translated into VOCCs of PASMC (109). However, this assumption is challenged by observations describing different effects of hypoxia on Ca2+ influx into the PASMC of resistance arteries (enhanced by hypoxia), and on the influx of Ca2+ into SMC of the conduit arteries (inhibited by hypoxia) (48, 49). Furthermore, acute hypoxic inhibition of the pore-forming α1-subunit is known to mediate hypoxic arterial vasodilatation, but hypoxia was also shown to selectively increase the L-type Ca2+ channels in PC12 cells and cerebellar granule neurons. Thus, it remains questionable whether the redox regulation of VOCCs can be translated from one tissue to another.
Hudasek et al. reported that human cardiac L-type α1-subunits expressed in HEK 293 cells showed increased currents after application of 100 μM H2O2 in a voltage-dependent manner. Catalase treatment reduced these currents. In contrast, the NADPH oxidase inhibitors diphenylene iodonium and phenylarsine oxide had no effect on either basal Ca2+ currents or responses to hypoxia. The authors concluded that endogenous production of H2O2 regulates the α1-subunit, but neither suppression of H2O2 levels nor inhibition of NADPH oxidase was involved in O2-dependent regulation of the Ca2+ channel (70). The hypoxia-induced increase in functional L-type Ca2+ channel expression has been verified in a recombinant expression system (HEK-293 cell line stably expressing the human L-type α1-subunit). However, increased functional expression was attributed to hypoxia-induced alterations of α1-subunit trafficking (116). In general, generation of ROS is altered during acute and chronic hypoxia, and oxidation of SH groups by ROS decreased cardiac L-type Ca2+-currents (54, 55), whereas oxidation of SH groups by other oxidizing agents (DTNB) caused stimulation of Ca2+-currents in ventricular myocytes. Thus, the effect of oxidizing agents on L-type channels seems to depend on the species and the mode of action.
Store- and receptor-operated Ca2+ channels
Influx of Ca2+ across the plasma membrane can also be triggered by depletion of Ca2+ from the endoplasmic reticulum (ER) and the SR. This so-called store-operated Ca2+ entry is mediated by SOCs. Besides the inhibition of K+ channels due to oxidation of channel residues, specifically on Kv1.5 (9, 102), depolarization of PASMC in response to hypoxia has been shown to be initiated by a number of other mechanisms by which hypoxia could lead to inhibition of these and other Kv channels. These mechanisms include elevation of cytosolic Ca2+ concentration owing to Ca2+ release from stores (73, 121), and there is mounting evidence that hypoxia-induced depolarization of PASMC is at least partially due to activation of SOCs (1, 148, 155). Activation of membrane receptors by ligand binding, diacylglycerol (DAG), and protein kinase C (PKC) can stimulate ROCs, causing Ca2+ influx and Na+ efflux. Activation of Na+ influx via nonselective cation channels as a result of either Ca2+ store depletion by SOCs or activation of a receptor–G protein–second messenger pathway by ROCs has been shown to be an important primary cause of depolarization and subsequent voltage-gated Ca2+ entry in PASMC (134, 148, 155). In the vasculature in general, TRPC are capable of forming functional ROCs and SOCs (150).
Similar to VOCC, TRPC are a part of the superfamily of six transmembrane spanning cation channels, but lack the voltage sensitivity (voltage independent). TRPC are nonselective cation channels, but carry predominantly Ca2+ ions (110). In the pulmonary vasculature, several subtypes of have been identified according to their mechanism of activation and presence of regulatory domains in the N- and C-termini. These subtypes include the classical or canonical TRP (TRPC1–TRPC7), vanilloid-receptor-related TRP (TRPV1–TRPV4), and melastatin-related TRP (TRPM1–TRPM8) channels (115). In VSMC, more than 10 TRP isoforms have been detected. However, TRPC1, TRPC4, and TRPC6 protein expression was shown to be higher in the distal pulmonary artery than in the proximal pulmonary artery, correlating with changes in cytosolic Ca2+ concentration occurring during HPV (89), and TRPC1 was further shown to play an important role in pulmonary vascular remodeling underlying the development of hypoxia-induced PH (93). TRPC6 is highly expressed in lung tissue as well as in pulmonary and VSMC and endothelial cells (41). In TRPC6−/− mice, the acute phase of HPV is completely absent, while the sustained phase is not significantly affected (162) (Fig. 5). TRPC6 and TRPC3 were the first ion channels shown to be activated by DAG (Hofmann 1998). Under normoxia, DAG is localized in the cytoplasm. Under hypoxic conditions, DAG translocates to the plasma membrane gating TRPC6 (162).
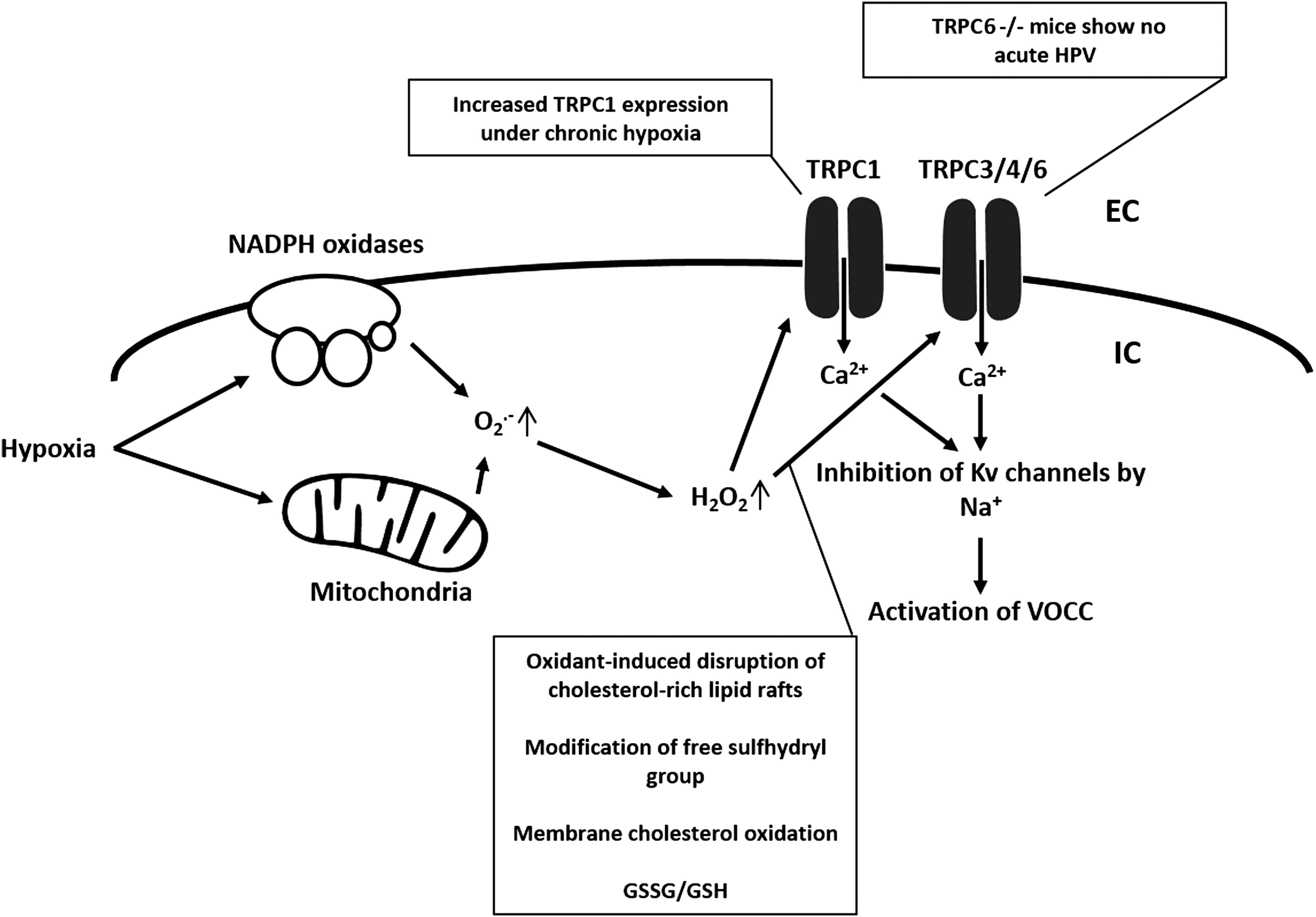
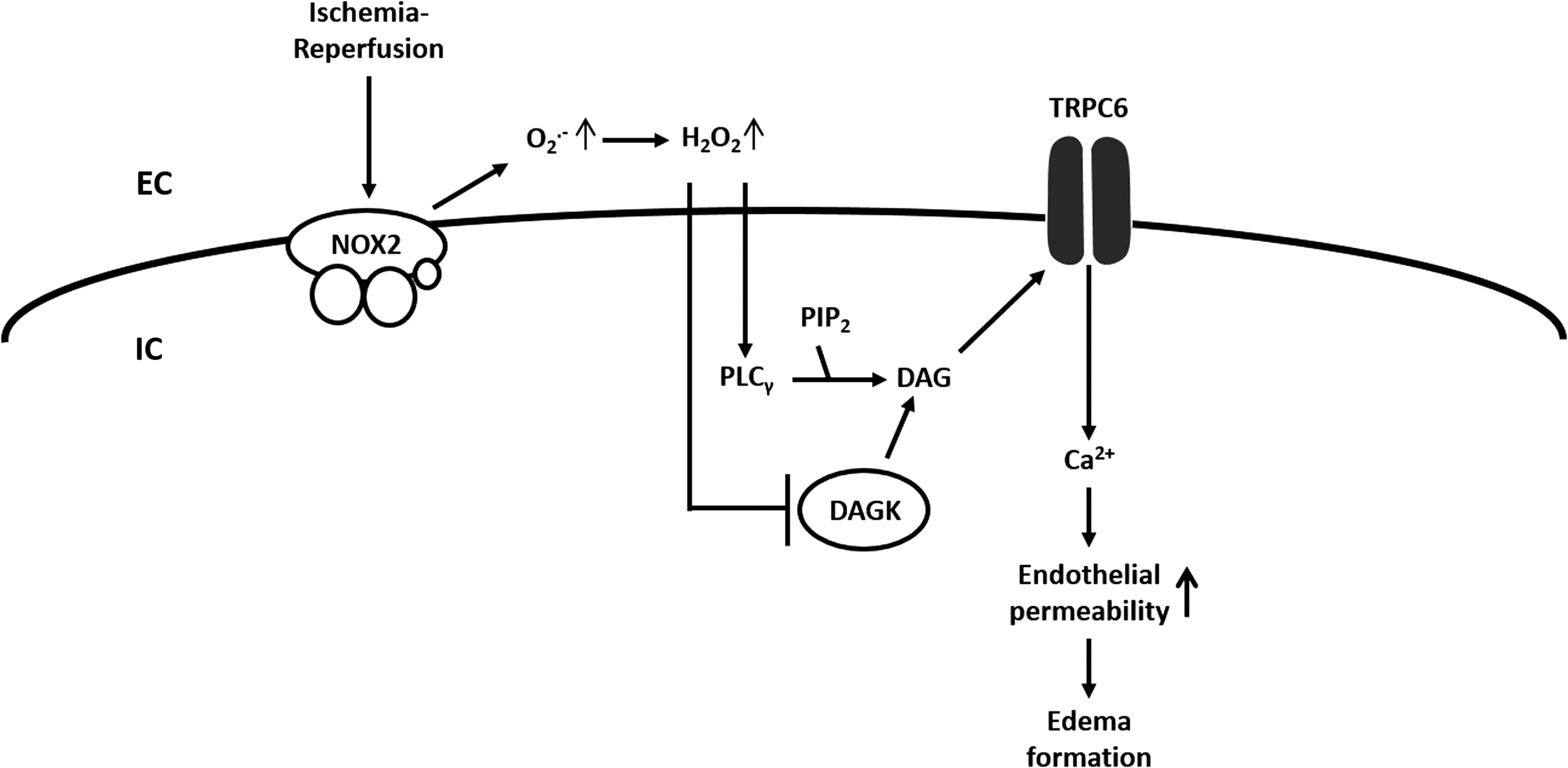
Possibly, the DAG translocation is triggerd by increased ROS as an investigation in an animal model of lung ischemia–reperfusion (I/R)-induced edema showed that TRPC6 in endothelial cells can be activated by endothelial Nox2-derived production of superoxide during the hypoxic phase of I/R with subsequent activation of phospholipase C-γ, and inhibition of DAG kinase, (164). According to this concept, superoxide is converted to H2O2, which, via an extracellular loop, triggers the TRPC6 response (164) (Fig. 6). As previously mentioned, TRPC are nonselective cation channels. Thus, we further speculate that sodium entry through TRPC6 and increasing sub-sarcolemmal Na+-concentrations inhibits Kv-channels and activates L-type Ca2+ channels (50) (Fig. 2).
It is well known that increased ROS generation can lead to Ca2+ release from intracellular stores as well as to Ca2+ entry across the plasma membrane (103). Especially in nonexcitable cells (e.g., endothelial cells) (106), although pulmonary microvascular endothelial cells express a functional voltage-gated T-type calcium channel (174), the major Ca2+ entry pathways are through SOCs and ROCs. TRPC7 (TRPM2) and TRPC3/TRPC4 have been shown to be regulated by ROS in endothelium (30). TRPC7 is no longer considered a TRPC family member (30). TRPC3/4 containing channels were shown to be activated and contributing to endothelial cell depolarization in the presence of oxidants (14) and to be regulated by oxidative stress (60) by oxidant-induced disruption of cholesterol-rich lipid rafts.
Similar to TRPC6, TRPC3 activation is regulated by C-type phospholipase (PLC) and by PLC-mediated hydrolysis of membrane-bound PIP. However, the mechanism underlying oxidative stress-mediated TRPC3 activation does not involve PIP hydrolysis. It has been speculated that membrane cholesterol oxidation by ROS might be the signaling event that activates TRPC3 (60). Poteser et al. concluded that TRPC3 and TRPC4 contribute subunits to the redox-sensitive channel. The identity of the other two subunits is unclear (123). The role of PLC in TRPC1 activation has not yet been addressed. SOCs were also shown to be indirectly regulated by cellular redox status through a nonselective cation channel that is covalently modified by glutathione disulfide (GSSG), an antioxidant molecule. It has also been suggested that SOCs may itself be a direct target of GSSG (thus of the cellular redox state) or some other ROS/reactive nitrogen species (30). In general, not much is known about redox regulation of SOC entry channels in the lung. Acute hypoxia (i.e., more ROS) was shown to enhance capacitative Ca2+ entry through SOCs in distal PASMC with subsequent depolarization and activation of VOCCs, suggesting a role for SOCs in HPV (148). Further, SOC and VOCC antagonists inhibited PASMC contraction during hypoxia as well as SOC-dependent activation of VOCCs (155). A hypothesis about ROS-dependent TRPC1 and TRPC3/4 regulation is given in Figure 5.
It should be noted that TRPM2, TRPM7, TRPC5, and TRPV1 are activated by ROS (2, 72, 170). In the case of TRPC5, TRPV1, and TRPA1, activation was triggered via oxidation of a free cysteine sulfhydryl group (139, 170). It remains to be determined whether these findings are consistent with other TRPC or TRPC in the lung.
Conclusion
Redox regulation of ion channel expression and gating under hypoxia and hypoxia-associated conditions, as well as under other conditions with an impact on the cellular redox state is a widely accepted mechanism. Direct reversible effects of ROS on ion channels include, but are not restricted to, oxidation of thiol groups, oxidation of arginine and lysine residues, and oxidation of methionine. Indirect reversible effects comprise, for example, alterations of GSH levels, activation of PLC (DAG), activation of PKC, and alterations of cytosolic Ca2+ levels.
Although our understanding of ion channel redox regulation is quite detailed for some ion channels (Kv channels, TRPC6), it remains generally scant. In addition to the the debate of increased or decreased ROS production during hypoxia, we need to understand why different oxidants under different conditions can cause both activation and inhibition of channel activity. According to the available literature, the ROS from an identical source can act on different amino-acid residues of an ion channel, thereby mediating opening or closing of the channel.
At the moment, most of the described redox regulatory mechanisms of ion channels are based on speculations and extrapolation of a few known redox regulatory mechanisms. While the upstream pathways affecting channel gating are often well described, the effects of ROS on the individual proteins of the pathways remain mostly unknown. One issue is to get a better understanding of redox protein modifications to be able to determine the complexity of ion channel redox regulation. It will be crucial to decipher how ROS are orchestrated and what role the spatial distribution of ROS plays in this regard. It has become obvious that physiological redox signaling is confined both spatially and temporally in subcellular compartments and microdomains. The redox status of a cell is not necessarily a global imbalance of oxidants and reducing molecules, but rather the net status of the redox status in different cellular compartments.
As the most redox-active compartment in the cell, mitochondria are a prominent site for ROS production (20, 75). Although mitochondria have a very high antioxidative capacity, excessive ROS release can cause a variety of disorders (74). The cytoplasm can also represent a subcellular compartment. Stimulation of the plasma membrane can trigger oxidation of specific proteins in the cytosol without affecting other organelles (100). Other redox active compartments are the nucleus, the ER lumen (36), peroxisomes (46), endosomes, and lysosomes (11). Within these compartments, ROS in microdomain generation can also be very diverse (90). Against this background, NADPH oxidases, another prominent cellular ROS source, have been shown to be expressed in such ROS microdomains [e.g., in caveolae (65) and endosomes (99)]. It has even been suggested that single Nox isoforms (Nox1) can have multiple signaling effects by occupying different microdomains within the cell (4, 29).