
Abstract
Introduction
N
GSK2795039 directly inhibits NADPH oxidase 2 (NOX2) as demonstrated by its inhibition of reactive oxygen species (ROS) production and NADPH consumption in a recombinant NOX2 system. It is selective for NOX2 as shown in NOX-specific cell-based assays addressing oxygen consumption and horseradish peroxidase-independent ROS detection in vitro. GSK2795039 shows in vivo target engagement in the periphery as well as protective effects in a model of acute pancreatitis. The present study rigorously demonstrates that GSK2795039 is a novel small molecule NOX2 inhibitor that can be used to explore both NOX2 biology in vivo and the therapeutic potential of NOX2 inhibition in disease.
In addition to their role in normal physiology, several lines of evidence suggest that chronic activation or overproduction of cytotoxic ROS by NOX enzymes leads to oxidative stress, a key pathological mechanism thought to underlie various disease states (29, 36, 46), such as atherosclerosis (5, 32, 56), diabetic nephropathy (25, 28), acute lung injury (29), pulmonary fibrosis (3, 10), and amyotrophic lateral sclerosis (66). Numerous animal studies using genetic ablation of gp91phox and/or p47phox support the hypothesis that pharmacological inhibition of NOX2 may be a therapeutic strategy (15). However, complete abrogation of NOX2 is known to lead to increased infections and increased sensitivity to autoimmune disorders and should be carefully considered for such treatments.
Throughout the last decade, efforts to validate the relative contribution of specific NOX enzymes to either normal physiological processes or to pathological states have been made primarily via the characterization of genetic knockout animals or with the use of peptide-based inhibitors (12, 37). However, both approaches have different caveats such as the potential for compensatory mechanisms in constitutive knockout mice and the limited biodistribution and efficacy of peptides. As such, the development of small-molecule NOX inhibitors is not only needed to characterize NOX biology but also to validate their potential as therapeutic agents (3).
To date, a number of small molecules have been shown to block ROS production in primary neutrophils or related cell lines and hence reported as NOX inhibitors (3, 31). However, for most of these inhibitors, a full understanding of their mode of action, selectivity, in vivo pharmacokinetics, and pharmacodynamics is lacking; therefore, their utility in characterizing NOX2 biology in complex biological systems is limited. Hence, we undertook a screening campaign to identify novel small molecule NOX2 inhibitors with the aim of identifying a potent inhibitor that could specifically inhibit the NOX2 enzyme following systemic administration in rodents.
To truly validate a small molecule as an NOX inhibitor, extensive characterization of its pharmacology is required to prove that apparent activity in enzyme and cell-based assays is specific to the particular NOX enzyme and is not due to free radical scavenging, interference with the ROS detection reagents, or inhibition of upstream pathways. The commonly used inhibitor, diphenyleneiodonium (DPI), blocks ROS production in phagocytes via accepting an electron from flavin. This enables DPI to interfere not only with NOX but also with other flavin-dependent enzymes, such as nitric oxidase synthase, xanthine oxidase, mitochondrial complex I, and cytochrome P-450 reductase (49). In addition to its broad selectivity, the utility of DPI as an in vivo tool to evaluate NOX biology is limited by its irreversible binding to flavin, low solubility, and high toxicity in rodents (LD50<10 mg/kg) (20, 49).
Another small molecule, apocynin, has also been commonly used as a tool inhibitor to evaluate the function of NOX2 in biological systems. As a natural methoxy-substituted catechol isolated from Picrorhiza kurroa (59), apocynin showed efficacy in a wide range of rodent disease models, including hypertension (47, 52), diabetic nephropathy (24), cardiac hypertrophy (4, 41), retinal vascular inflammation (1), and stroke (22, 62). While apocynin has been shown to inhibit the phagocyte respiratory burst in some cellular assays (57), its mechanism of action is still a matter of debate because it also has radical scavenging properties and does not inhibit NOX2 in a cell-free enzyme system (27, 31).
The pharmacology of other small molecule NOX2 inhibitors in vivo has not been well defined. As such, there is a clear need for the development of directly acting NOX enzyme inhibitors that are suitable for testing in vivo to accurately define the biological functions of NOX enzymes and to assess their potential as therapeutic drug targets.
In this study, we report the pharmacological characterization of GSK2795039, a novel small molecule that is a potent NOX2 enzyme inhibitor in various in vitro and in vivo systems. In parallel, we have characterized other reported NOX inhibitors and highlighted apparent differences in their pharmacology. Our study highlights the potential flaws inherent to the use of ROS detection systems and emphasizes the need for several unrelated assays to measure NOX activity. Finally, we demonstrate that GSK2795039 inhibits the NOX2 enzyme following systemic dosing in rodents and is therefore a suitable tool molecule to further evaluate the pathophysiological role of NOX2.
Results
GSK2795039 is a potent inhibitor of NOX2 in cell-free and whole-cell assays
GSK2795039 (Fig. 1), N-(1-Isopropyl-3-(1-methylindolin-6-yl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-1-methyl-1H-pyrazole-3-sulfonamide, was discovered following a high-throughput screen and subsequent lead optimization campaign to identify inhibitors of the NOX2 enzyme. GSK2795039 is a novel 7-azaindole structure wherein sulfonamide functionality is critical for its NOX2 activity. This feature is unique to GSK2795039 and was an early indication of its differentiated mode of action from other reported NOX inhibitors.
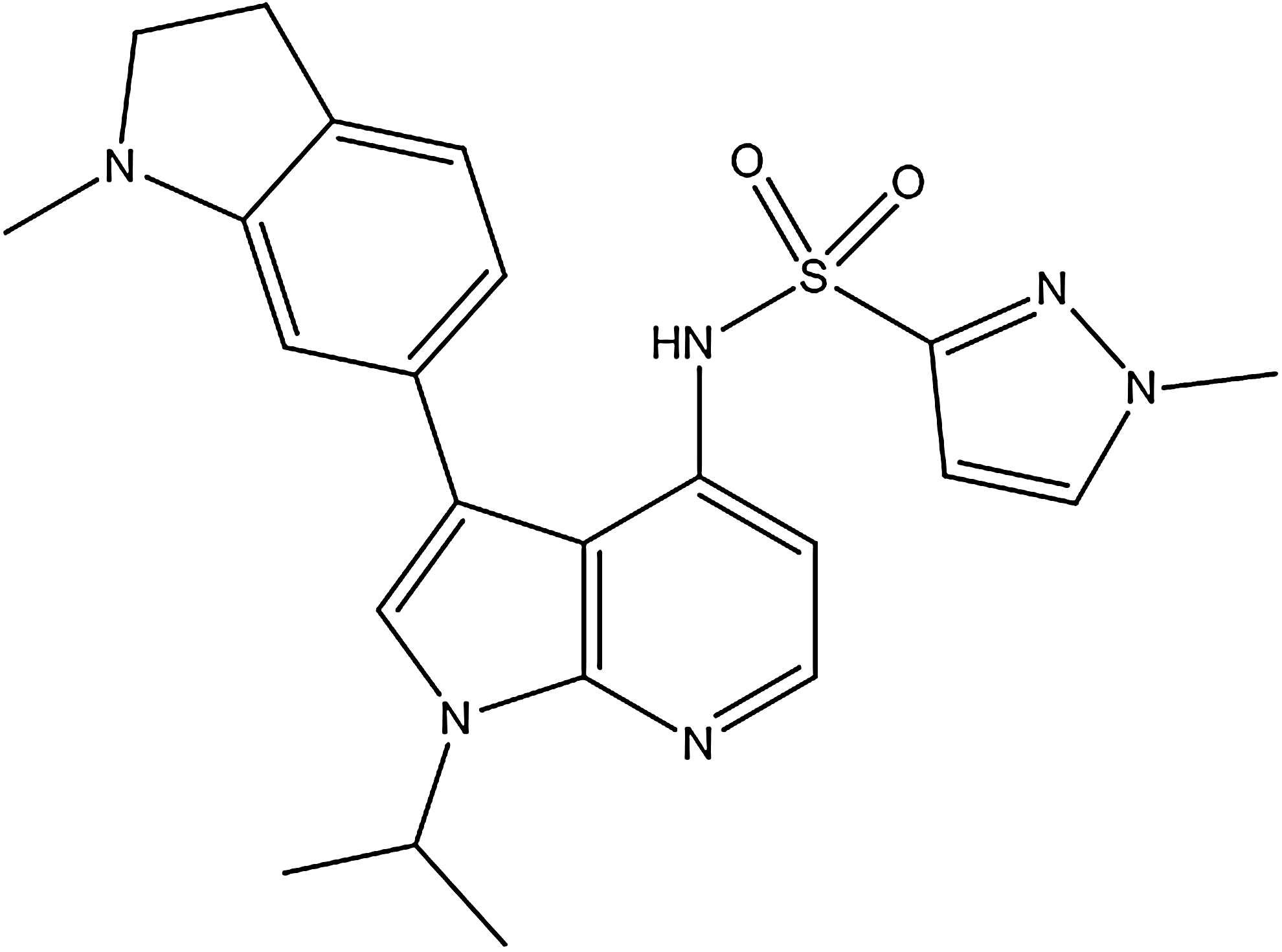
A number of assays were developed to assess the pharmacological activity of test compounds on the NOX2 enzyme and to counter screen for nonspecific and off-target effects that may manifest as apparent inhibitory activity in the enzyme assays. The primary assay utilized a semirecombinant NOX2 enzyme system using baby hamster kidney (BHK) cell membranes expressing gp91phox and purified recombinant cytosolic proteins. The enzyme was activated with arachidonic acid in combination with the horseradish peroxidase (HRP)/Amplex Red method of ROS detection. This assay format was chosen for a number of reasons: a high signal:noise ratio, the possibility to use a recombinant cell-free assay system rather than a whole-cell-based assay to increase the chances of identifying direct inhibitors of the NOX2 enzyme, and the possibility of miniaturization of the assay to a 1536-well plate format.
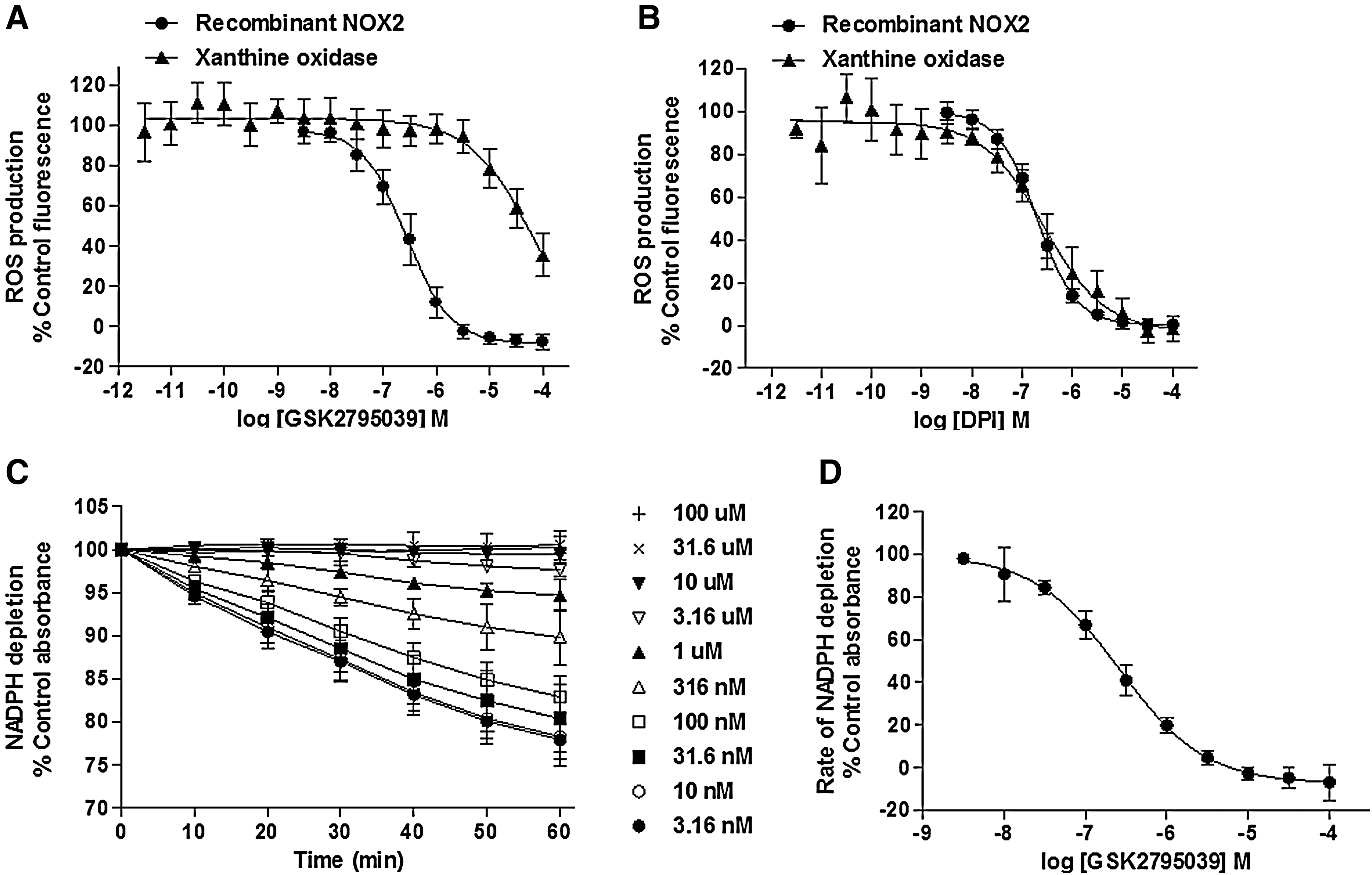
As shown in Figure 2A and B and summarized in Table 1, GSK2795039 and the positive control DPI fully inhibited NOX2-mediated activation of HRP/Amplex Red with pIC50 values of 6.57±0.17 and 7.07±0.25, respectively. To confirm that this activity was not due to inhibition of HRP activity or direct scavenging of ROS, we used additional semirecombinant cell-free NOX2 enzyme assays using the ROS detection reagents, Oxyburst Green and water-soluble tetrazolium salt1 (WST-1), whereas a complementary assay monitored the time-dependent depletion of the enzyme substrate, NADPH. WST-1 is a formazan salt specifically reduced by the superoxide anion. Its use for detection of NOX activity was elegantly validated by Tan and Berridge (61) who highlighted its improved sensitivity over cytochrome C for measuring NOX activity. Specificity for NOX2-dependent superoxide detection by Oxyburst Green and WST-1 assays is documented in Supplementary Figure S1 (Supplementary Data are available online at
Data are expressed as mean±standard deviation (n). Symbol (<) indicates that compound was inactive and/or showed less than 50% inhibition at top concentrations.
Indicates that compound showed less than 50% inhibition at top concentrations in some replicate experiments, and a value of 4 was used to calculate the population mean.
Indicates that interference of the test compound with the reagent was observed potentially obscuring a real pharmacological effect.
DPI, diphenyleneiodonium; HRP, horseradish peroxidase; ND, not done; NOX2, NADPH oxidase 2; PKCB II, protein kinase C beta2; ROS, reactive oxygen species; WST-1, water-soluble tetrazolium salt1.
The measurement of substrate depletion in the NADPH absorbance assay is a key assay technique to characterize NOX inhibitors because it is not prone to the potential artifacts commonly associated with ROS detection methodologies. As shown in Figure 2C, the concentration of NADPH measured via absorbance at 340 nm declined in a linear manner over 60 min in the presence of activated NOX2 enzyme. Both GSK2795039 and DPI concentration dependently decreased the rate of NADPH utilization with pIC50 values of 6.60±0.13 and 7.55±0.24, respectively (Fig. 2C, D and Table 1).
GSK2795039 and DPI were also tested for inhibitory activity against another oxidase enzyme, purified xanthine oxidase, using the HRP/Amplex Red method of ROS detection. In contrast to DPI, which inhibited xanthine oxidase with a pIC50 of 6.57±0.21, GSK2795039 was only a weak inhibitor with a pIC50 of 4.54±0.16 exhibiting >100-fold selectivity for NOX2 over xanthine oxidase (Fig. 2A, B, and Table 1). In addition, GSK2795039 only weakly inhibited endothelial nitric oxide synthase (eNOS) (<50% when tested at 100 μM, Table 2).
pIC50 values for GSK2795039 were generated in the DPPH assay to determine electron accepting properties in comparison with N-acetylcysteine and DPI. Inhibitory activity against recombinant eNOS was determined in comparison with the positive control L-NMMA. Cytotoxic effects of GSK2795039 were determined via measurement of ATP levels in HEK 293 cells following 24-h compound treatment. Where less than 50% effect was observed (i.e., pIC50<4) at the highest concentration of compound, the % inhibition values are shown. Data are expressed as mean values or mean±standard deviation (n).
DPPH, 2,2-diphenyl-1-picrylhydrazyl; eNOS, endothelial nitric oxide synthase; HEK, human embryonic kidney; L-NMMA, NG-Monomethyl-
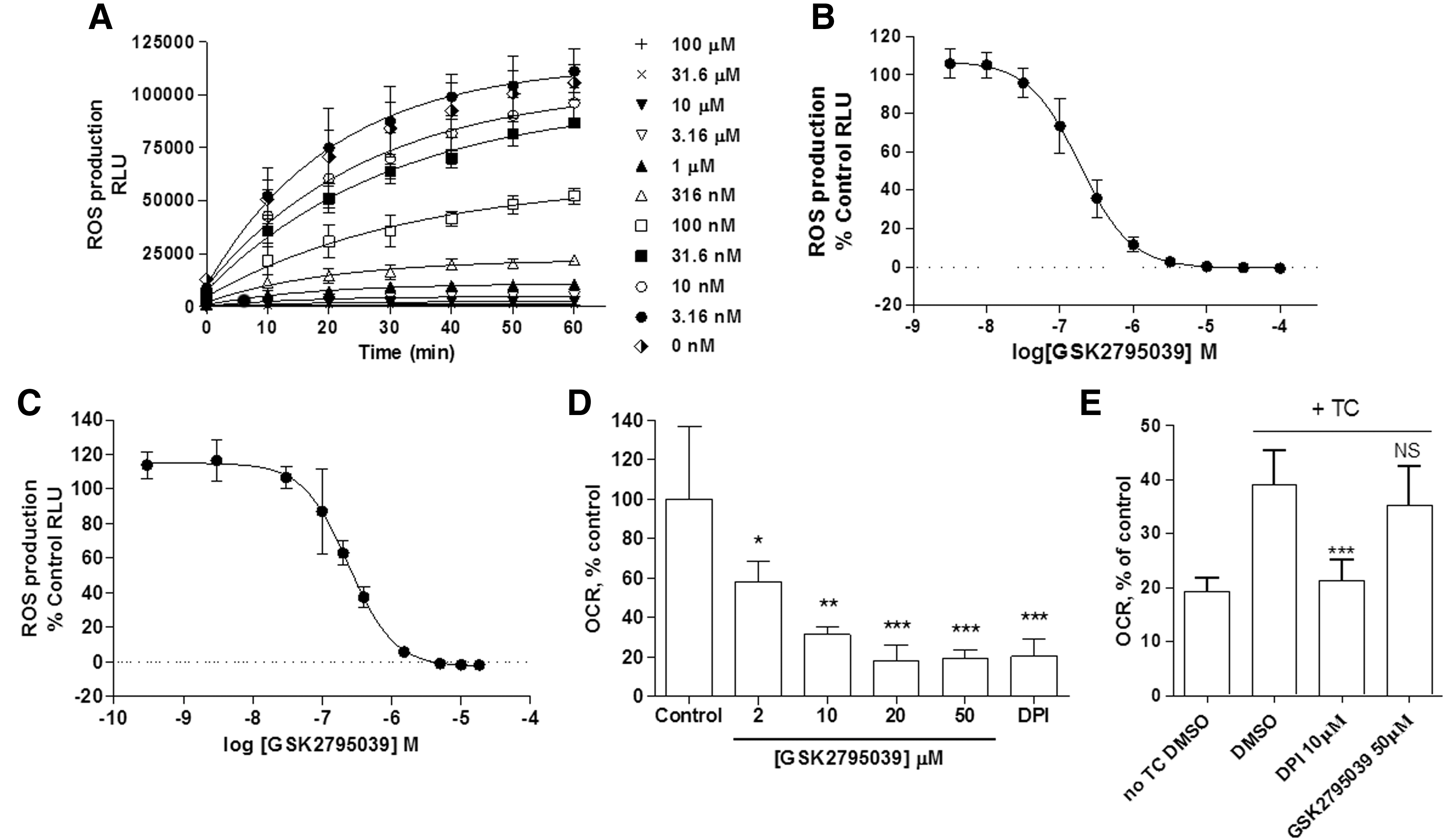
To evaluate the cellular activity of GSK2795039, differentiated HL60 cells and human peripheral blood mononucleated cells (PBMCs) were stimulated with phorbol 12-myristate 13-acetate (PMA) in the presence and absence of compound, using L-012 or Oxyburst Green to detect ROS production. PMA (12 nM) stimulation of HL60 cells caused a time-dependent increase in L-012 luminescence over 60 min that was fully inhibited by GSK2795039 and DPI with pIC50 values of 6.74±0.17 and 6.84±0.22, respectively (Fig. 3A, B, and Table 3). Similar pIC50 values (6.73±0.16 and 6.49±0.18, respectively) were obtained when Oxyburst Green was used as the ROS detection reagent. In PBMCs, GSK2795039 inhibited ROS production as measured with L-012 (Fig. 3C) with a pIC50 value of 6.60±0.08 (n=4); an effect consistent with potent inhibition of the NOX2 enzyme. We also evaluated potential cytotoxic effects of GSK2795039 in cultured cells (human embryonic kidney [HEK] 293 cells), but did not observe any significant toxicity as measured by cellular ATP levels, up to 100 μM following a 24-h incubation (Table 3 and Supplementary Fig. S2).
Data are expressed as mean±standard deviation (n). Symbol (<) indicates that compound was inactive and/or showed less than 50% inhibition at top concentrations.
Indicates that compound showed less than 50% inhibition at top concentrations in some replicate experiments, and a value of 4 was used to calculate the population mean.
Indicates that interference of the test compound with the reagent was observed potentially obscuring a real pharmacological effect.
PMA, phorbol 12-myristate 13-acetate.
Given the potential for pharmacological agents to block either the production or the detection of ROS in whole cells through nonspecific pathways, we also assessed the activity of GSK2795039 in a number of additional assays. First, since the primary pathway for NOX2 stimulation following addition of PMA is through direct activation of protein kinase C (PKC), it was important to demonstrate lack of pharmacological activity on the activity of the PKC enzyme. As shown in Table 1, GSK2795039 did not show any inhibitory activity at PKC beta 2 (PKCB II) up to 25 μM. Second, to confirm that the apparent inhibitory activity in whole cells was not due to either antioxidant activity or a nonspecific direct effect on the L-012 or Oxyburst reagents, we assessed the effect of the compound on the rate of NOX2 substrate utilization by performing oxygen consumption experiments. In neutrophils, NOX assembly and activation trigger the respiratory burst, during which the rate of oxygen consumption is directly related to NOX activity and can be measured using an oximeter. As shown in Figure 3D, GSK2795039 demonstrated a concentration-dependent suppression of oxygen consumption with a maximum effect observed at 20 μM, equivalent to the level of inhibition exhibited by the positive control, 10 μM DPI.
To characterize the activity of GSK2795039 against other NOX isoforms (Table 4), cell-based assays for NOX1, NOX3, NOX4, and NOX5 were used as in Seredenina et al. (53). For NOX2, we measured ROS production by differentiated PLB-985 cells (63). All NOX isoforms generated ROS upon activation or stimulation that could be detected using either HRP/Amplex Red or the SOD-inhibitable WST-1 assay. Intriguingly, the two probes generated different profiles of NOX inhibition. In the WST-1 assay, GSK2795039 produced complete inhibition of NOX2 with a pIC50 of 5.54±0.25, but was inactive on the other NOX isoforms (up to 100 μM, Table 4). In contrast, inhibitory activity was observed across all isoforms in the HRP/Amplex Red assay. Interestingly, maximal inhibition was ∼70% for NOX1, NOX3, NOX4, and NOX5 and H2O2 alone, while complete maximal inhibition (>95%) was observed for NOX2 (Table 4). This submaximal activity against some NOX isoforms as well as H2O2 alone is most likely due to an off-target effect of GSK2795039 on the HRP/Amplex Red. It is known that reducing agents such as ascorbate interfere with HRP-dependent assays (16). To address the possibility that GSK2795039 has electron donor properties, we performed the colorimetric 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay (34). In the presence of a substance that can donate a hydrogen atom, the violet radical gives rise to its yellow-colored reduced form. Electron donor properties were confirmed for GSK2795039 and N-acetylcysteine, but not DPI, with pIC50 values of 4.32±0.03 and 4.75±0.36, respectively (Table 3 and Supplementary Fig. S2). Thus, HRP/Amplex Red assay should not be recommended when testing small molecules even with weak reducing activities such as GSK2795039. The selectivity of GSK2795039 for NOX2 over other isoforms was confirmed by measuring oxygen consumption in NOX4-expressing cells. As shown in Figure 3E, DPI fully inhibited NOX4-mediated oxygen consumption, whereas GSK2795039 did not have any effect up to 50 μM.
The values determined for a control assay using H2O2 instead of cells are also shown. Data are expressed as mean±standard deviation (n).
NA, not applicable.
Enzyme mode of action studies
To understand both the mode of action of GSK2795039 at the enzyme and the apparent selectivity for NOX2 over other NOX isoforms, we explored two potential modes of action: (i) oxidation of cysteine residues in gp91phox. Compounds able to oxidize dithiols to form a disulfide complex such as phenylarsine oxide (PAO) (39) and gliotoxin (67) are specific NOX2 inhibitors. They act through a 369CysGlyCys371 region in the NOX2 sequence, which is absent in NOX1, NOX3, NOX4, and NOX5 (33) and is necessary for NOX2 function, probably by mediating the binding of the p67phox subunit (6). When the NOX2 complex is not assembled, the cysteine thiol groups are exposed and gp91phox is more sensitive to thiol-oxidizing compounds. We therefore incubated different concentrations of GSK2795039 and PAO before and after addition of recombinant cytosolic subunits in a semirecombinant NOX2 assay using PLB-985 cell membranes and WST-1 as the detection reagent.
PAO was less active when added afterward, that is, when cysteines were protected (Fig. 4B), while GSK2795039 was similarly active in both conditions (Fig. 4A), suggesting that GSK2795039 does not act through oxidation of thiols. (ii) Competition for the NADPH binding site of NOX2. We considered this possibility based on the fact that the sequences of NADPH binding sites differ significantly among NOX isoforms (Supplementary Fig. S3). Addition of increasing concentrations of NADPH decreased the NOX2 inhibitory potency of GSK2795039. This dependency on the substrate concentration is indicative of a competitive mode of action. The Michaelis–Menten and Lineweaver–Burk plots shown in Figure 4D and E graphically display that similar maximum enzyme velocity (Vmax) can be reached while the apparent Michaelis constant (Km) decreases (Supplementary Fig. S4). Increasing concentrations of NADPH did not affect DPI inhibition (Fig. 4C) and both Vmax and Km simultaneously decreased with increased concentration of DPI, consistent with an uncompetitive mode of action for NADPH (48) (Supplementary Fig. S4).
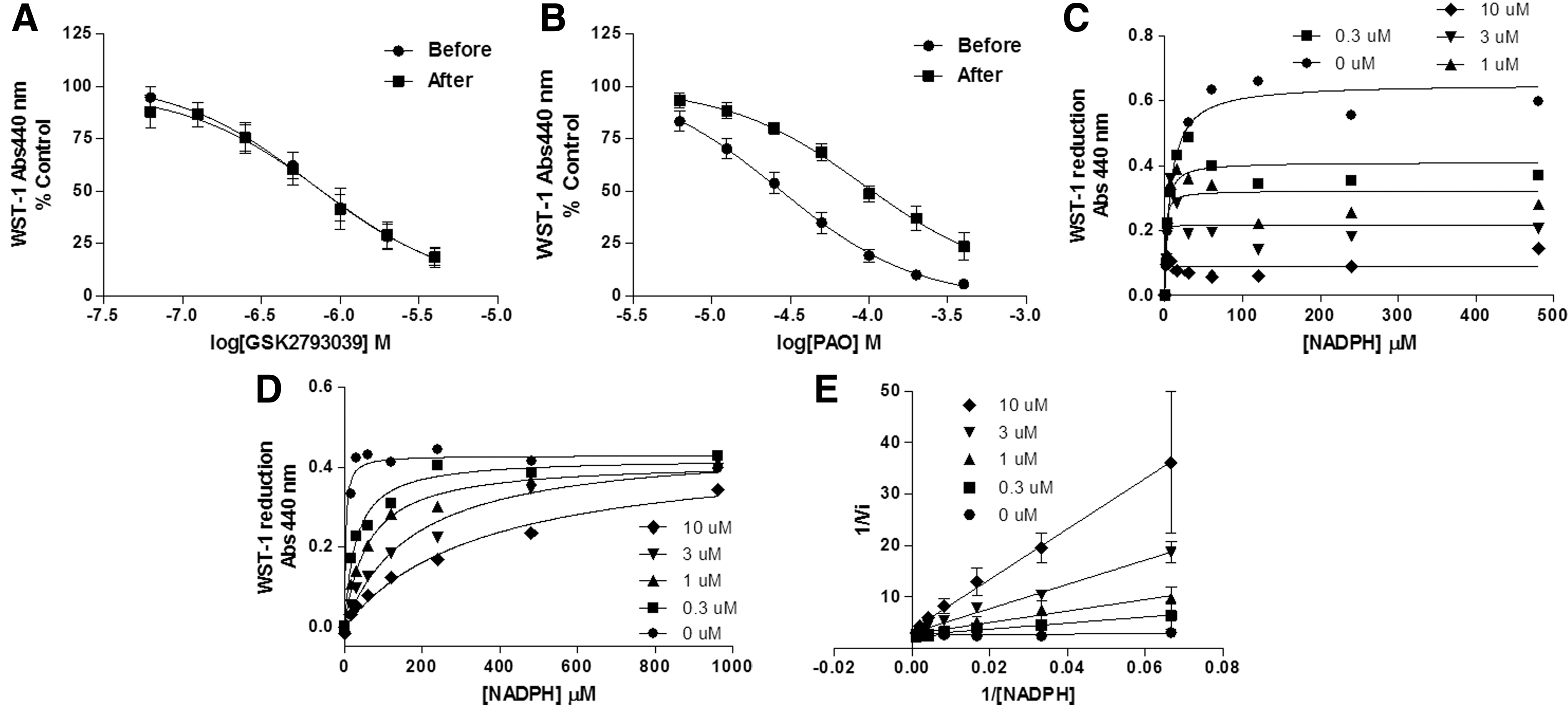
Further evidence in support of a competitive mode of action of GSK2795039 was gained by conducting studies in the NADPH depletion assay at a range of NADPH concentrations (25 μM–1 mM) (Supplementary Fig. S5). For DPI, its inhibitory potency was independent of the NADPH concentration used in the assay, while the measured pIC50 values of both suramin and GSK2795039 varied with NADPH concentration. For GSK2795039, the pIC50 values were 6.35±0.12 at 25 μM NADPH and 5.64±0.05 at 1 mM NADPH, consistent with an NADPH competitive mode of action.
Comparison of GSK2795039 with other reported NOX2 inhibitors
The pharmacological profile of GSK2795039 was compared against a number of other reported NOX inhibitors in the various assays described (summarized in Tables 1 and 2). Apocynin, naloxone, Shionogi 2 (21), and VAS2870, all showed some inhibitory effects in whole-cell assays (Table 2). However, they showed little or no inhibitory activity in the semirecombinant cell-free NOX2 assays regardless of whether enzyme activity was measured via HRP/Amplex Red, Oxyburst ROS detection, or via NADPH utilization (Table 1). Therefore, the apparent activity in HL60 cells is unlikely to be due to a direct effect on the NOX2 complex. Indeed, for Shionogi 2, it is likely that inhibition of PKCB II (pIC50 7.6±0.27) is responsible for the apparent activity in HL60 cells. However, this mechanism of action cannot account for the inhibitory activity of naloxone, VAS2870, and apocynin since they were not found to inhibit PKCB II. Suramin, DPI, and celastrol inhibited ROS production and NADPH utilization in the recombinant NOX2 enzyme system in Amplex Red/HRP, Oxyburst, and NADPH assays (Table 1), but suramin was inactive in whole cells (Table 2), perhaps due to lack of cell membrane penetration (60). Celastrol showed inhibitory activity in cell-free NOX2 assays as well as in HL60 cells. DPI, consistent with its known mode of action, that is, inhibition of most flavoproteins, also showed consistent inhibitory activity across all five NOX2 assays and the xanthine oxidase assay. ML171 showed apparent inhibition in both HL60 cell-based assay readouts (Table 2) and in the semirecombinant cell-free system using HRP/Amplex Red and Oxyburst as the detection methods (Table 1), but did not show any effect in the NADPH depletion assay. This is likely due to assay interference as previously reported for this compound (53).
Systemic administration of GSK2795039 in mice inhibits the NOX2 enzyme in vivo
The pharmacokinetic properties (summarized in Table 5) of GSK2795039 were determined in rat and mouse. Following intravenous (i.v.) infusion, GSK2795039 had a moderate-to-high clearance rate in rat and mouse of 54 and 95 ml/min/kg with half lives of 2 and 0.2 h, respectively. GSK2795039 was moderately brain penetrant with brain:blood ratios of 0.83 and 0.49 in rat and mouse, respectively, after intraperitoneal (i.p.) administration. In rat, following oral administration (p.o.) at 3 mg/kg, the oral bioavailability was poor (3%) with a peak blood concentration (Cmax) of only 46 nM in blood. In mouse, oral bioavailability was higher and varied between 10% and 54% when dosed at 5 and 100 mg/kg, respectively, with associated blood Cmax values of 0.2 and 11 μM.
Blood clearance, half-life, volume of distribution, oral Cmax and Tmax were estimated using a noncompartmental model. The oral bioavailability was estimated using results obtained from intravenous (1 mg/kg) and oral profile studies (3, 5 or 100 mg/kg as shown in parenthesis). Cmax and Tmax values were determined following oral dosing at the doses (mg/kg) indicated in parenthesis. The brain:blood ratios (median value) were determined at 1 hour following intraperitoneal injection at 100 mg/kg.
Cmax, peak blood concentration; Tmax, time to reach Cmax.
GSK2795039 was further evaluated in mice to determine whether systemic administration inhibited the NOX2 enzyme in different tissues in vivo. NOX2 activity was assessed following the induction of inflammation in the hind paw of mice via injection of complete Freund's adjuvant (CFA), which is known to trigger neutrophil migration to the paw with subsequent localized NOX2-dependent respiratory burst (43, 50, 63). Enzyme activity was monitored 24 h after CFA injection into the plantar surface via systemic administration of L-012 (25 mg/kg). Activated L-012 reagent was detected by whole animal luminescence imaging. As shown in Figure 5A, the L-012-derived chemiluminescence in the inflamed paw reached its maximum at ∼10 min after L-012 injection and decreased gradually over the next 15 min, presumably due to clearance of L-012 from the circulation. In gp91phox-deficient mice, the L-012 signal in the inflamed paw was nearly abolished compared with wild-type mice (Fig. 5A). This suggests that NOX2 is the main enzyme responsible for the production of superoxide in the inflamed paw.
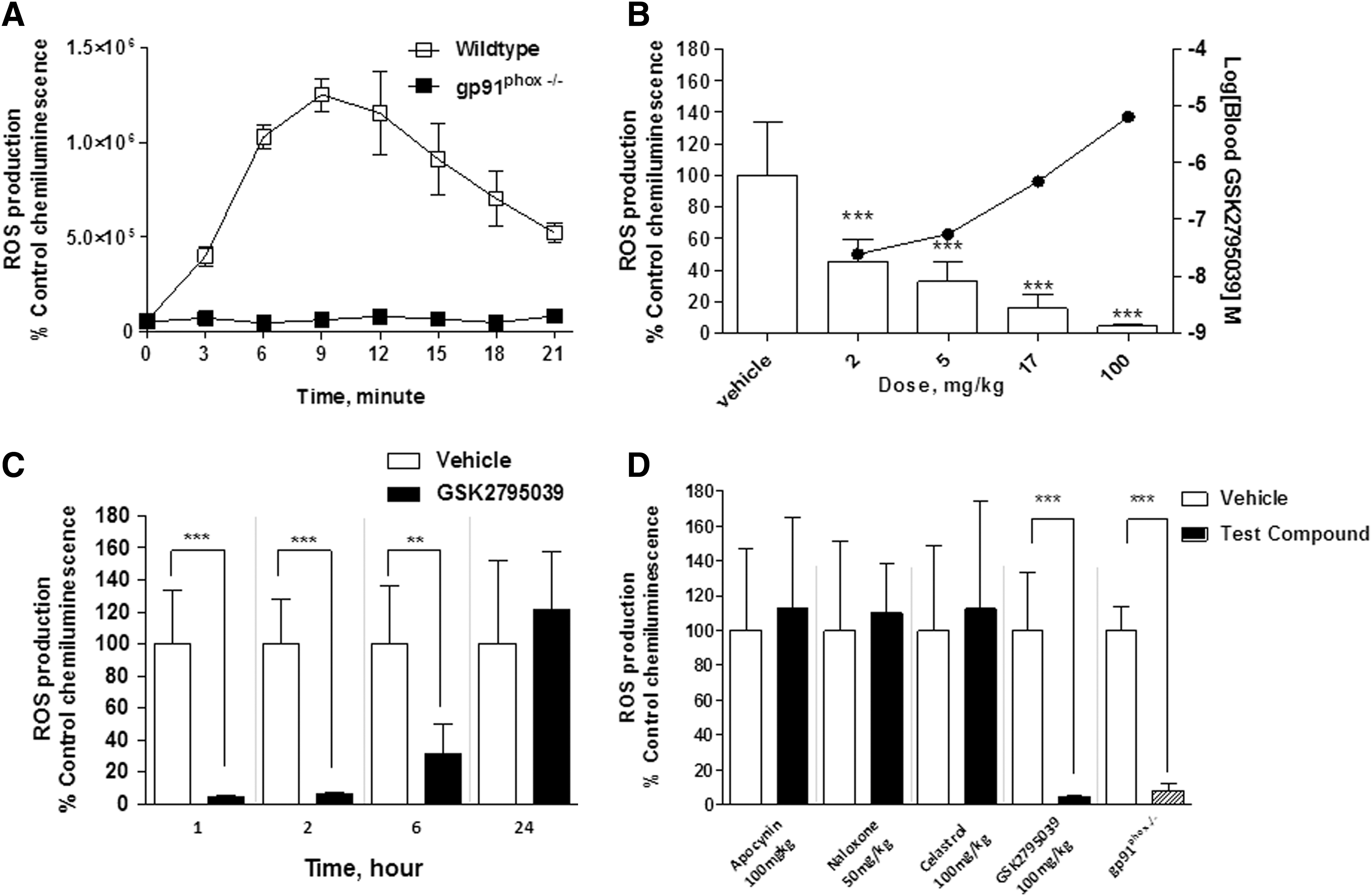
Following i.p. administration, GSK2795039 dose-dependently inhibited the L-012 signal in the inflamed paw with ∼50% inhibition observed at 2 mg/kg and almost complete suppression of enzyme activity at 100 mg/kg compared with vehicle-treated animals (Fig. 5B). Following a single 100 mg/kg i.p. dose, complete inhibition of enzyme activity in the paw was observed at 1 and 2 h postdose (Fig. 5C). Significant inhibition of enzyme activity was still observed up to 6 h postdose (Fig. 5C), while 24 h postdose enzyme activity was similar to vehicle-treated animals. The concentration of GSK2795039 in blood measured at 1 h postdose was 7.9 μM and was below the limit of quantification at 24 h postdose. To compare other NOX2 inhibitors with GSK2795039, we also evaluated apocynin (100 mg/kg, i.p.), celastrol (100 mg/kg, p.o.), and naloxone (50 mg/kg, subcutaneous [s.c.]) in the paw inflammation assay. As shown in Figure 5D, no reduction in NOX2 enzyme activity was observed following treatment with any of these compounds.
GSK2795039 is active in a mouse model of acute pancreatitis
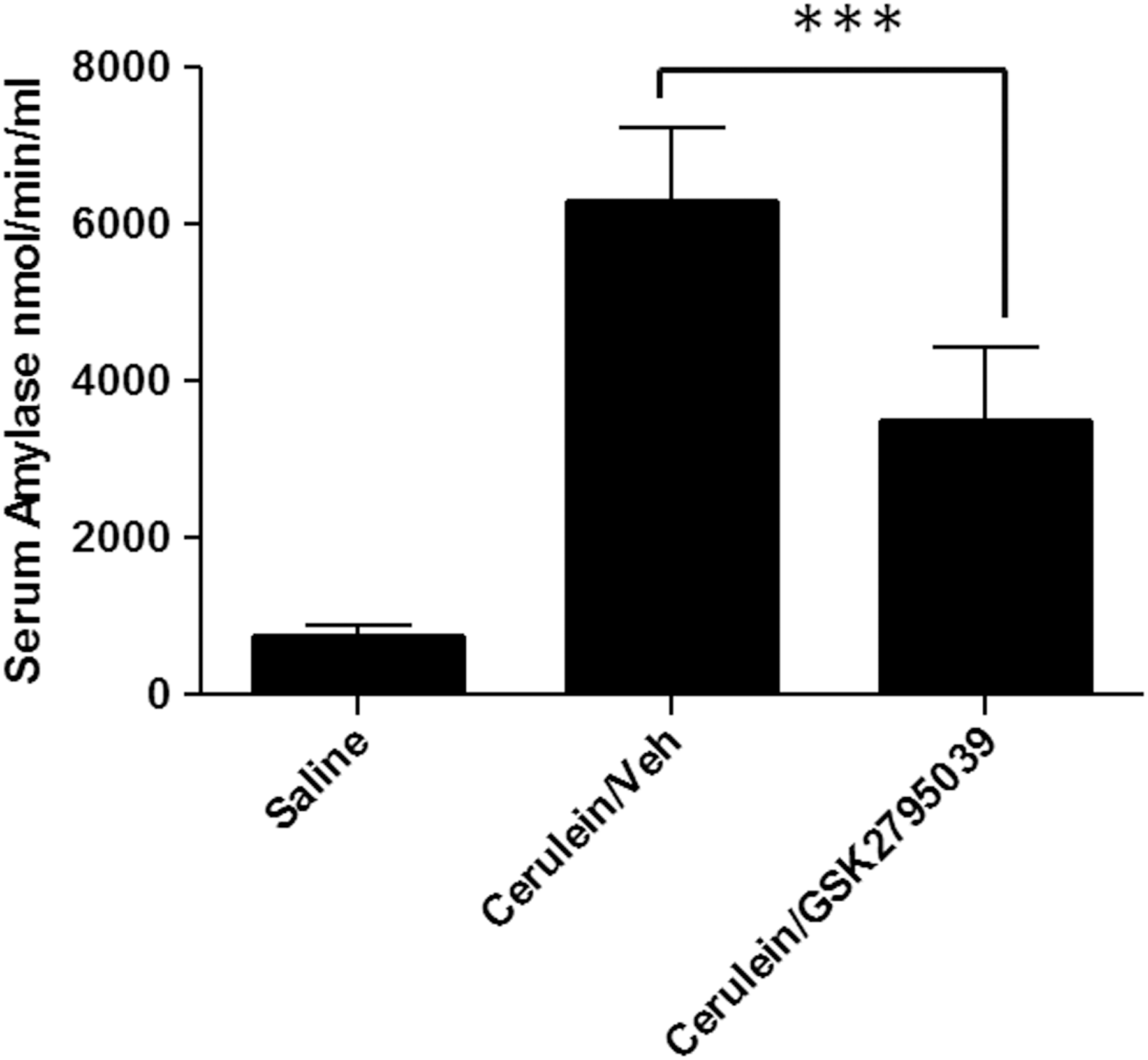
To evaluate whether the in vivo inhibition of NOX2 by GSK2795039 also resulted in biological effects consistent with the known in vivo function of the NOX2 enzyme, we assessed the effect of GSK2795039 in the cerulein model of acute pancreatitis in mice. We chose this model because gp91phox knockout mice, but not myeloperoxidase-deficient mice, were shown to be protected (26). GSK2795039 (100 mg/kg) was administered twice via i.p. injection; the initial dose given 1 h before the first of 6 hourly injections of cerulein (50 μg/kg), the second dose given 4 h later (i.e., at the time of the fourth cerulein injection). The mice were sacrificed 1 h after the last injection of cerulein and levels of serum amylase, the marker for pancreatic cell necrosis, were quantified. As shown in Figure 6, GSK2795039 caused ∼50% reduction in the level of serum amylase activity similar to what was previously reported for gp91phox knockout mice (26).
Discussion
In this study, we describe the pharmacological profile of GSK2795039, the first small molecule to demonstrate inhibition of the NOX2 enzyme following systemic administration in vivo. To enable the discovery of GSK2795039, several assays were developed to measure NOX2 activity in different biological systems using mechanistically diverse detection methodologies. To unequivocally confirm blockade of NOX2 enzyme activity in a biological system, it is critical to demonstrate that a compound inhibits not only the catalytic formation of ROS, as measured using multiple ROS detection reagents, but that it also blocks the utilization of substrate in both semirecombinant cell-free enzyme and whole-cell-based systems. As such, we have compiled a pharmacology data set sufficient to claim that GSK2795039 is a true NOX2 inhibitor.
In the semirecombinant system, GSK2795039 is a potent inhibitor of NOX2-dependent ROS production (as measured using WST-1, HRP/Amplex Red, and Oxyburst detection methods) and NADPH utilization. In HL60 cells, human PBMCs, and polymorphonuclear leukocytes (PMNs), it is a potent inhibitor of NOX2-dependent ROS production as measured by L-012 chemiluminescence, HRP/Amplex Red fluorescence, Oxyburst fluorescence, and WST-1 absorbance. In PMNs following PMA stimulation, it blocks utilization of oxygen, the substrate of the reaction catalyzed by NOX2. In neutrophils, activated NOX2 contributes >95% of the total oxygen consumption (11) as evidenced by oxygen consumption measurements of neutrophils from chronic granulomatous patients who have genetic loss of function of NOX2 (9, 51).
The NOX activity assays measuring substrate utilization, that is, oxygen consumption in whole cells and NADPH depletion in cell-free assays, are of utmost importance in defining GSK2795039 as a direct inhibitor of NOX2. In terms of orthogonal assays, GSK2795039 is not cytotoxic and does not inhibit PKC nor xanthine oxidase at concentrations efficacious for NOX2 inhibition.
The rationale for using such a range of assays was to address the difficulties associated with the use of any one assay in isolation, in particular with regard to the potential nonspecific effects of test compounds. This is illustrated simply by the profile of the Shionogi 2 compound, which initially appeared to be a potent inhibitor of ROS production in HL60 cells, but was inactive in the cell-free semirecombinant NOX2 enzyme assay. It acts in fact as a potent PKC inhibitor, thereby blocking the PKC-dependent translocation of NOX2 subunits upon PMA stimulation.
Our study is also representative of the fact that ROS detection assays using fluorescent or luminescent ROS sensitive reagents are susceptible to interference and prone to detecting nonspecific effects of compounds [reviewed in Maghzal et al. (42)]. It is only by using a variety of assays that we demonstrated the selectivity of GSK2795039 for NOX2. A superficial interpretation of the results of the HRP/Amplex Red detection, for example, would suggest apparent nonselective pharmacological activity across all NOX isoforms. As the compound showed inhibition of HRP/Amplex Red when activated by H2O2 alone (Table 4), a nonspecific effect of GSK2795039 on the HRP/Amplex Red was most likely. Although we did not address the chemistry of the interaction between GSK2795039, Amplex Red, and HRP, it is possible that adding a small molecule with one-electron reducing capacity diverts compound I of HRP (or other heme-containing peroxidases) from its normal cycle (16, 19). The fact that GSK2795039 is inactive in NOX4-dependent oxygen consumption as well as in the WST-1 assay for NOX1, NOX3, NOX4, and NOX5 confirms GSK2795039 selectivity for NOX2. However, this stresses the fact that future studies using GSK2795039 should avoid the use of HRP/Amplex Red assay and that extreme care should be taken in the interpretation of results when small molecules are used in peroxidase-dependent ROS detection methods (69).
In addition to the in vitro characterization presented here, we have shown that GSK2795039 is orally available and can be measured in the blood and central nervous system, suggesting that it can cross the blood–brain barrier. It fully inhibits NOX2 enzyme activity in vivo following systemic dosing in mice. Importantly, the level of inhibition achieved at the highest dose was complete and equivalent to that observed in gp91phox-deficient mice as measured by in vivo L-012 chemiluminescence. The potent activity in vivo confirms the bioavailability of the compound and the validity of the cell-free and cell-based assays used in defining NOX2 activity, despite the potential flaws inherent in these assays. For example, in the cell-free semirecombinant assay, the use of cell membranes from BHK cells overexpressing gp91phox/p22phox or the use of arachidonic acid as the mode of enzyme activation may have generated an enzyme system that was not representative of the enzyme composition or activation mechanism, respectively, in vivo. Second, in the cell-based assay, we only evaluated compounds for activity against the enzyme when activated by PMA, which may not accurately recapitulate the dynamics of enzyme activation in vivo. In the paw inflammation paradigm, we can assume that the NOX2 enzyme is assembled and activated in a more physiologically relevant way, and as such, it is an important confirmation that GSK2795039 is active at the endogenous enzyme in this in vivo biological context. Importantly, the paw inflammation studies also demonstrate that inhibition of NOX2 by GSK2795039 is reversible since 24 h after administration, when the levels of compound in blood were undetectable, ROS production induced by CFA was similar to that in vehicle-treated animals.
The paw inflammation model enabled us to fully characterize the relationship between dose, blood exposure, and NOX2 enzyme inhibition in vivo over time, a critical data set required to enable the appropriate design and interpretation of studies aimed at evaluating the effect of GSK2795039 in more complex in vivo systems. The other reported NOX2 inhibitors, celastrol, naloxone, and apocynin, did not demonstrate significant inhibition of ROS production in the paw inflammation model. As such, this is the first direct demonstration of pharmacological inhibition of NOX2 in vivo with a small molecule inhibitor.
We have also used an acute pancreatitis model to show that GSK2795039 has in vivo physiological effects consistent with NOX2 inhibition since GSK2795039 recapitulates the effect of genetic ablation of gp91phox in the same mouse model. We also observed that GSK2795039 (100 mg/kg) was well tolerated in rodents, with no obvious adverse effects following 5 days of twice daily dosing via the p.o. or s.c. dose routes (data not shown), and is therefore a suitable tool molecule to further characterize NOX2 biology.
Mechanistic evaluation in the cell-free NOX2 semirecombinant assay using WST-1 revealed a competitive inhibition of GSK2795039 toward NADPH and excluded a potential action on intracellular cysteines, a known inhibitory mode of action specific for NOX2 (6). This finding was confirmed when NADPH depletion was used as readout. This is an important finding because it indicates that NADPH binding sites are sufficiently divergent among NOX isoforms to allow isoform-specific inhibition. Future structural analysis of the gp91phox NADPH binding site by cocrystallization with GSK2795039 may reveal a specific pocket.
Among other NOX inhibitors tested in this study, only DPI, celastrol, and suramin showed inhibition across the cell-free assays independently of assay format, in agreement with previous reports suggesting that DPI is an irreversible inhibitor of NOX2 (14), that celastrol inhibits NOX2 directly and via binding to the p47phox subunit (30), and that suramin also acts via binding to the NADPH binding site (21). While DPI and celastrol also demonstrated inhibitory activity in the cell-based assays, suramin was not active, perhaps due to lack of cell penetrance as has previously been suggested (60). The other reported NOX inhibitors, apocynin, naloxone, Shionogi 2, VAS2870, and ML171, showed similar activity in the cell-based assays used here to the previously described whole-cell systems (3, 12, 23, 31), although the potency of apocynin was rather weak. However, they were mostly inactive in the cell-free assays with the exception of ML171, which showed activity in the HRP/Amplex Red and Oxyburst assays, but not the NADPH depletion assay, and Shionogi 2, which showed weak activity only in the Oxyburst assay. Notwithstanding their potential therapeutic or pharmacological value, this highlights the fact that none of these compounds act directly on the NOX2 complex, but rather have indirect modes of action on NOX2-derived ROS. For example, the reported pharmacology of VAS2870 is complex with activity in cell-free assays only observed if added before addition of sodium dodecyl sulfate (SDS) to induce enzyme assembly (2). In the cell-free assay used here, the enzyme components were preassembled and activated with arachidonic acid before addition of compound, followed by NADPH. Therefore, the assay may not have been optimal to detect its activity if the mode of action is via inhibition of subunit assembly. The reported pharmacology of naloxone at NOX2 is also complex as it is proposed to bind to the gp91phox subunit of NOX2 and inhibit enzyme activity by modulation of p47phox subunit translocation (65). The method for the cell-free assay used here differs from what was previously used to demonstrate naloxone activity because we used a fully recombinant enzyme activated with arachidonic acid, whereas the assay reported by Wang et al. (65) used PMA-activated neutrophil membranes in a semirecombinant system. In the case of Shionogi 2, however, we found that it is a potent PKC inhibitor, consistent with previous reports (21), and this likely explains its apparent inhibitory activity in HL60 cells. For ML171, no mode of action at the NOX2 enzyme has been described, but it has recently been shown that it is most likely not an NOX inhibitor (53).
While variations in the configuration of the in vitro assays may explain apparent pharmacological differences between GSK2795039 and other reported NOX inhibitors, the paw inflammation assay clearly highlights that GSK2795039 is distinct in terms of its in vivo pharmacological activity. The doses of apocynin (100 mg/kg), naloxone (50 mg/kg), and celastrol (100 mg/kg) used here were selected on the basis of doses reported to be efficacious in various disease models (13, 17, 35, 40, 55). None of these three compounds showed NOX2 inhibition in the paw inflammation model. There are a number of potential explanations for the observed lack of activity. First, it is possible that the mechanism of NOX2 enzyme activation differs between different in vivo biological systems, which may therefore have different sensitivities to inhibitors with alternative modes of action at the enzyme. It is also possible that the levels of tissue exposure, specifically in the inflamed paw, were not sufficient to elicit a significant effect at the enzyme. Alternatively, it is possible that the pharmacology shown at least in the cell-based HL60 assay does not translate to the in vivo system. Either way, our results raise the possibility that the reported biological effects of these molecules in disease models may be driven by additional pharmacology beyond NOX2. We did not evaluate VAS2870 in the paw inflammation model because we found that the compound was rapidly degraded in blood and hence unsuitable for in vivo experiments, nor did we test suramin due to its lack of cell penetration. While we have not profiled all the reported NOX inhibitors, for example, fulvene-5 (8) and imipramine (44) have also been reported to inhibit NOX enzymes, GSK2795039 is the only NOX inhibitor tested here that demonstrates a convincing in vitro pharmacological profile combined with in vivo pharmacological activity at the NOX2 enzyme.
Only celastrol and DPI were active in all assays and may be bona fide NOX inhibitors in vitro; however, they may not be suitable tools to test NOX enzyme function in vivo. As we have shown, celastrol does not appear to inhibit NOX2 in vivo at the doses tested here. DPI, as well as being a nonspecific irreversible inhibitor of flavin-dependent enzymes, is limited in terms of its in vivo utility by its high toxicity in rodents (LD50<10 mg/kg) (20, 49). Consistent with the observed NOX2 enzyme inhibition following systemic dosing, we have also demonstrated that GSK2795039 has the same biological effect as gp91phox gene ablation in a rodent model of acute pancreatitis (26).
In summary, we report here the discovery of a novel small molecule NOX2 inhibitor, GSK2795039, which inhibits enzyme activity in both cell-free and whole-cell systems. Importantly, GSK2795039 was bioavailable after systemic administration and showed in vivo target engagement in peripheral tissues, thus supporting the utility of GSK2795039 as a tool NOX2 inhibitor to further evaluate the biology of the NOX2 enzyme and the therapeutic potential of NOX2 inhibitors in acute and chronic inflammatory diseases, such as pulmonary inflammation (29) and neurodegenerative diseases (46), where genetic deletion of NOX2 has been shown to be protective.
Materials and Methods
Reagents
GSK2795039, N-(1-Isopropyl-3-(1-methylindolin-6-yl)-1H-pyrrolo[2,3-b]pyridin-4-yl)-1-methyl-1H-pyrazole-3-sulfonamide, was synthesized by the Neural Pathways Discovery Performance Unit, GlaxoSmithKline (Biopolis, Singapore). All purified recombinant cell-free NOX subunits (p47phox, p67phox, and Rac2), BacMam viruses (gp91phox and p22phox), and DNA plasmids harboring NOX cDNAs (Rac1 and p22phox) were generated by GlaxoSmithKline Platform Technology & Sciences (Stevenage, United Kingdom). NOX isoform-specific cells were generated by the University of Geneva (Geneva, Switzerland); for detailed description, see Seredenina et al. (53) and references therein. The human biological samples, PBMCs, were used in accordance with the terms of approval by the Human Biological Sample Users committee, GlaxoSmithKline. PMNs were used in accordance with the terms of the Ethics Commission of the Research on Human Beings of the Geneva University Hospitals and informed consents. Sources of other reagents are indicated where appropriate.
Animals
Male Sprague-Dawley (SD) rats (Charles River Laboratories, Inc., and Animal Resources Centre), C57BL6/J mice (Biological Resource Centre), and gp91phox knockout mice (Jackson Laboratories) were used in this study. Animals were housed in groups of five at controlled temperature (20°C±1°C) and humidity (40%±2%) with a 12-h light/12-h dark cycle and allowed ad libitum access to water and food. All studies were conducted in accordance with the GlaxoSmithKline Policy on the Care, Welfare, and Treatment of Laboratory Animals. Animal experiment protocols were reviewed and approved by the Singapore National Advisory Committee at the Biological Resource Centre, Biopolis.
Cell-free recombinant NOX2 enzyme assays (BHK cell membranes)
Cell-free NOX2 enzyme preparations were prepared using membranes from either transduced BHK/AC9 cells (European Collection of Cell Cultures) (18) or from differentiated PLB-985 myeloid cells. To generate the BHK cell membranes coexpressing human gp91phox and p22phox, full-length cDNAs were cloned into BacMam vectors. BacMam viruses were prepared from Sf9 cells as previously described and used to transduce BHK cells (18). Homogenized cells expressing gp91phox/p22phox were collected by high-speed centrifugation, resuspended in 10% sucrose buffer, then layered on top of a 40% sucrose buffer solution. Following centrifugation at 28,500 rpm for 60 min, the interface layer was collected and further centrifuged to isolate cell membranes. Cytosolic proteins used to reconstitute NOX2 enzyme in BHK cells were full-length N-terminal His-tagged human p47phox and human p67phox, and human Rac2 proteins were expressed individually in Sf9 cells via baculovirus transduction and purified using Ni-NTA affinity resin (Qiagen). Fractions were further purified by size exclusion chromatography on Superdex 200 (GE Healthcare Life Sciences).
The potency of test compounds to inhibit ROS production by the reconstituted recombinant NOX2 enzyme was measured using either HRP/Amplex Red or Oxyburst Green (Life Technologies) detection reagents. Enzyme activity was also measured by quantification of NADPH utilization. For HRP/Amplex Red-mediated ROS detection, the activated NOX2 enzyme mixture (20 nM Rac2, 100 nM p47phox, 150 nM p67phox, 0.0046 μg/μl gp91phox-p22phox-BHK membranes, and 20 μM arachidonic acid) was prepared in assay buffer (10 μM FAD, 1 mM EGTA, 4 mM MgCl2, 1 μM GTPγS in 50 mM KPO4 at pH 7) supplemented with 2 U/ml HRP. Test compounds diluted in assay buffer were incubated with the enzyme mixture for 15 min at room temperature before addition of the enzyme substrate and detection mix (50 μM NADPH, 25 U/ml catalase beads, and 20 μM Amplex Red final concentrations in assay buffer) to yield a final assay volume of 25 μl and dimethyl sulfoxide (DMSO) concentration of 1%. At 5 min after addition of substrate, fluorescence was detected at λEx/λEm=560 nm/590 nm using a BioTek spectrophotometer. For Oxyburst Green detection of ROS, the assay was run using the same conditions as for the HRP/Amplex Red assay, except that 250 μg/ml Oxyburst Green was used in place of Amplex Red in the enzyme substrate mix, and HRP was omitted. The enzyme reaction was incubated for 90 min. Fluorescence was detected at λEx/λEm=485 nM/530 nM at t=0 and t=90 min using a BioTek spectrophotometer to calculate the enzyme rate over 90 min. Test compound pIC50 values for both assays were determined from concentration response curves using a four-parameter logistic curve fitting model.
To measure NADPH utilization (i.e., substrate depletion), test compounds were incubated with the same NOX2 enzyme mixture for 25 min at room temperature. NADPH (50 μM) was added and absorbance measured at 340 nm using a microplate spectrophotometer (BioTek) at 10-min intervals to determine the rate of NADPH utilization [(Abs t=60−Abs t=0)/60 min]. The pIC50 values were calculated from concentration response curves using a four-parameter logistic curve fitting model. To conduct enzyme mode of action studies in the NADPH depletion format, assay conditions were exactly as above, except that NADPH concentrations were varied between 25 and 1000 μM.
Cell-free recombinant NOX2 enzyme assays (PLB-985 cell membranes)
PLB-985 myeloid cells were cultured in RPMI medium supplemented with fetal bovine serum (FBS; 10%), penicillin (100 U/ml), and streptomycin (100 μg/ml) and grown at 37°C in air with 5% CO2. The cells were differentiated into granulocyte-like cells by the addition of DMSO (1.25%) to the medium for 3–5 days. To prepare NOX2-containing membranes, differentiated PLB-985 cells from wild-type (64) and NOX2 knockout (68) were used. Approximately 107 cells were homogenized in 1.5 ml of a sonication buffer containing phosphate-buffered saline (PBS) (0.1×), sucrose (11%), NaCl (120 mM), and EGTA (1 mM) supplemented with protease inhibitors (Complete Mini; Roche) and sonicated twice for 45 s (level 4, Branson Sonifier 250). Cell debris were removed by 10-min centrifugation at 800 g. The supernatant was laid on a sucrose gradient consisting of a bottom layer of 1.5 ml sucrose (40%) and an upper layer of 1.5 ml sucrose (17%). Ultracentrifugation was performed at 150,000 g in an SW60 rotor for 30 min at 4°C. Following separation, the upper cytosolic fraction was discarded and the cloudy membrane fraction was kept, protein content was measured using the standard Bradford procedure, and was stored in small aliquots at −80°C.
Measurement of ROS generation by NOX2-containing membranes was performed as previously described (58, 53) with slight modification such as the use of WST-1 absorbance or Oxyburst Green fluorescence as readouts. Briefly, 1 μl of various concentrations of compounds dissolved in DMSO was incubated in 80 μl of a mix containing NOX2 membranes (1 μg of protein), 0.5 μg p67Np47N chimeric protein (a fusion of the N-termini of human p67phox [residues 1–210] and p47phox [residues 1–286]), 0.4 μg Rac1Q61L, 0.025 μM FAD, 90 μM SDS, and 1 mM WST-1 or 250 μg/ml Oxyburst Green assay in PBS. After 5 min at room temperature, the reaction was initiated by addition of 20 μl NADPH (final 60 μM). For WST-1, absorbance was immediately measured at 440 nm in a FlexStation® 3 Multimode Microplate Reader (Molecular Devices) at 37°C for 30–60 min. Oxyburst Green fluorescence was measured in a BMG Fluostar Optima Microplate Reader with excitation 485 nm and emission 530 nm after 90 min.
For enzyme mode of action studies, the enzyme assay was initiated by the addition of different concentrations of NADPH (0–960 μM). ROS generation was measured using WST-1 over the first 30 min to calculate an enzyme rate. Michaelis–Menten and Lineweaver–Burk analyses were performed in GraphPad Prism 6.
For the evaluation of potential thiol-oxidizing activity of test compounds, cell membranes and recombinant proteins were mixed with compounds immediately or after 5 min of preincubation. After another 5 min, 60 μM NADPH was added. WST-1 absorbance was measured after 1 h. Test compound pIC50 values for both assays were determined from concentration response curves using a four-parameter logistic curve fitting model.
Xanthine oxidase, eNOS, and PKCB II assay
Test compounds were incubated with 5 mU/ml bovine-derived xanthine oxidase (Life Technologies) for 15 min at room temperature, followed by the addition of substrate and detection mix (final concentrations of 0.2 U/ml HRP, 5 μM hypoxanthine, and 50 μM Amplex Red) in 18 μl assay volume. Fluorescence (λEx/λEm=535 nm/590 nm) was detected every minute using an Envision spectrophotometer (PerkinElmer, Inc.) for 10 min to determine the enzyme rate. The pIC50 values for test compounds were calculated from concentration response curves using a four-parameter logistic curve fitting model.
The activity of test compounds to inhibit recombinant human PKCB II (ProQinase) was determined using the IMAP Screening Express and Progressive Binding Bead System (Molecular Devices) with a fluorescein-labeled 5-FAM-KRREILSRRPSYR-NH2 peptide (FAM; Molecular Devices). Fluorescence polarization measurements were taken at λEx/λEm=485 nm/535 nm with a 505 nm dichroic mirror using an Acquest plate reader (Molecular Devices). The pIC50 values for test compounds were calculated from concentration response curves using a four-parameter logistic curve fitting model.
The eNOS assay was run at CEREP.
DPPH assay
For the DPPH radical scavenging assay, 2 μl of serial dilutions of compounds in DMSO was mixed with 180 μl of DPPH in methanol (40 μg/ml) in each well of a 96-well plate. The plate was kept in the dark for 15 min, after which the absorbance of the solution was measured at 540 nm in a FlexStation 3 microplate reader. N-acetylcysteine and DPI were used as positive and negative controls, respectively.
Cellular ATP assay
Potential cytotoxic effects of test compounds were evaluated in HEK 293 cells. Cells (20,000 cells/well) were cultured for 24 h in the presence or absence of test compound. Cells were then washed and ATP levels quantified using a Cell Titre-Glo assay kit (Promega).
NOX2 HL60 and PBMC-based assays
The activity of test compounds to inhibit endogenous NOX2 in whole cells was performed using differentiated HL60 cells (American Type Culture Collection) and human PBMCs. HL60 cells were differentiated into neutrophil-like cells by culturing in medium containing Iscove's modified Dulbecco's media (Life Technologies), 20% heat-inactivated FBS (Life Technologies), and 1.3% DMSO for 6 days. Compounds were preincubated with 10,000 cells in Hank's balanced salt solution (HBSS) containing 0.1% glucose and 0.01% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), then stimulated with 12 nM PMA. Production of ROS was monitored with 125 μM L-012 (Wako Pure Chemical Industries Ltd.) in a 50 μl final assay volume every 10 min up to 60 min using a Microbeta Trilux luminescence counter (PerkinElmer, Inc.). For the calculation of pIC50 values, the luminescence signal at 60 min was used and concentration response curves were analyzed using a four-parameter logistic curve fitting model. An alternative ROS detection assay was established in HL60 cells using Oxyburst Green. This assay used the same conditions as for the L-012 assay, except for the following changes: 1 μg/ml Oxyburst was used in place of L-012; 50,000 cells per well; 40 nM PMA; and 120-min incubation time. Fluorescence was detected at the end of the incubation at λEx/λEm=485 nm/530 nm using a BioTek spectrophotometer. For the calculation of pIC50 values, concentration response curves were analyzed using a four-parameter logistic curve fitting model.
Human PBMCs were isolated from the blood of healthy donors (StemEasy) using Lymphoprep density gradient medium (Axis-Shield Plc.) and resuspended at 5×105 cells/ml in HBSS buffer. Cells (25,000) were incubated with test compound, 400 μM L-012, and 0.01% CHAPS for 15 min at room temperature. PMA (127 nM) was added to produce an assay volume of 200 μl, and chemiluminescence was detected 30 min later using an Infinite 1000 spectrophotometer (Tecan). The pIC50 values for test compounds were calculated from concentration response curves using a four-parameter logistic curve fitting model.
NOX isoform cell-based assays
To compare the pharmacology of test compounds across the NOX isoforms, NOX1–5-expressing cells were generated as previously described. Briefly, NOX1-expressing cells were generated by transduction of Chinese hamster ovary cells with human NOX1, NOXO1, NOXA1, and p22phox (30); the NOX3-expressing tetracycline-inducible HEK T-Rex cell line was generated using NOX3, NOXO1, NOXA1, and p22phox (63); and NOX4 and NOX5-expressing cell lines were generated as previously described (54). PLB-985 cells, differentiated into granulocyte-like cells by the addition of DMSO (1.25%) to the medium for 72 h, were used for the NOX2 isoform (64).
ROS were measured using HRP-dependent Amplex Red (Life Technologies) or a colorimetric probe, WST-1 (Dojindo Molecular Technologies, Inc.). At least three independent experiments were performed for all measurements. For Amplex Red/HRP assay, cells were preincubated with the test compound for 10 min at room temperature in PerkinElmer black 96-well microplates at 50,000 cells per well, after which the reaction mixture was added to a final concentration of 25 μM of Amplex Red and 0.05 U/ml of HRP in HBSS. NOX enzymes were activated via 100 nM PMA (NOX1, NOX2), 1 μg/ml tetracycline for 24-h activation of inducible genes (NOX3, NOX4), and 1 μM Ca2+ ionophore ionomycin (NOX5). Fluorescence measurements were then performed using a FluoSTAR OPTIMA plate reader (BMG Labtech) every minute for 60 min at 37°C with λEx/λEm=570 nm/580 nm.
For WST-1 assay, the test compound was preincubated with cells for 10 min at room temperature in a 96-well plate using 100,000 cells per well, as described above, after which the reaction mixture containing 2 mM WST-1 plus stimuli was added. Absorbance was measured at 440 nm at a single point 45–60 min after incubation of the plate at 37°C. The pIC50 values for test compounds were calculated from concentration response curves using a four-parameter logistic curve fitting model.
Oxygen consumption in human PMNs and NOX4 HEK cells
Human PMNs were isolated from fresh whole blood collected from healthy volunteers as described previously (45) using Ficoll Plaque™ Plus (GE Healthcare Life Sciences) and resuspended in HBSS buffer. A Clarke electrode oximeter was prewarmed to 37°C in saturated KCl, then stabilized in HBSS for 15 min. PMNs (5×106 cells) were prewarmed for 5 min at 37°C with and without test compounds. Cells were then placed into the oximeter and oxygen consumption recorded for 5 min, followed by the addition of 2 μM PMA and a further 20-min recording. The oxygen content of air-saturated buffer was 240 nmol/ml. The oxygen consumption rate (OCR) was calculated from the slope of the oxygen consumption curve between 5 and 10 min.
Oxygen consumption in NOX4 T-Rex HEK cells was evaluated using the Seahorse XF24 Extracellular Flux Analyzer. NOX4 T-Rex HEK cells (50,000 cells/well) were plated onto poly-
CFA-induced paw inflammation
Paw inflammation was induced in male C57BL6/J and gp91phox knockout mice by a single injection of 30 μl CFA (Sigma-Aldrich) by s.c. injection into the plantar surface of the left hind paw. Animals were anesthetized with isoflurane at 24 h after CFA injection. Each animal was then administered 25 mg/kg L-012 in 0.9% NaCl at a dose volume of 5 ml/kg via i.p. injection and immediately placed in a Xenogen IVIS imaging system (PerkinElmer, Inc.). Chemiluminescence in the inflamed paw was quantified with a 7-s exposure time at 0, 3, 6, 9, 12, and 15 min postinjection. The sum of chemiluminescence measured at all time points was calculated as the total chemiluminescent signal.
Test compounds were administered before L-012 injection. Apocynin (100 mg/kg, 30-min pretreatment, suspended in 2% DMSO in corn oil, dose volume of 6.6 ml/kg) and GSK2795039 (2–100 mg/kg, 60-min pretreatment, dissolved in 20% DMSO, 20% Tween 80, 60% polyethylene glycol 200, dose volume of 2 ml/kg) were administered by i.p. injection, naloxone (50 mg/kg, 15-min pretreatment, dissolved in saline, dose volume of 5 ml/kg) by s.c. injection, and celastrol (10 mg/kg, 60-min pretreatment, suspended in 10% DMSO in 1% methylcellulose, dose volume of 5 ml/kg) by oral gavage. Chemiluminescence values for treated animals were normalized to values measured in vehicle control animals. Tail vein blood samples were collected after the IVIS imaging session for the determination of the test compound blood concentration.
Pharmacokinetic study
The pharmacokinetic parameters of GSK2795039 were determined in male SD rats (350–450 g; Animal Resources Centre) and male C57BL6/J mice (10- to 20-week-old; Biological Resource Centre, Biopolis) via p.o. and i.v. dosing. SD rats were prepared with a cannula inserted via the femoral vein into the vena cava (for compound administration) and via the jugular vein (for blood sampling). To determine the pharmacokinetics following i.v. administration, cannulated rats were administered an i.v. infusion of GSK2795039 in vehicle (2% DMSO, 10% hydroxy-β-cyclodextrin, 0.9% NaCl) at a dose of 1 mg/kg and at a rate of 10 ml/kg/h through the femoral cannula for 1 h. In mice, a single bolus injection of 1 mg/kg GSK2795039 was administered in the same vehicle via the tail vein.
To determine the pharmacokinetics following oral administration in rat, 3 mg/kg GSK2795039 was administered in 1% methylcellulose via oral gavage at a dose volume of 10 ml/kg. In mice, 100 mg/kg GSK2795039 was administered in 1% methylcellulose either via oral gavage or i.p. injection at a dose volume of 10 ml/kg. Blood samples were collected from the tail vein at various time points from 0 to 360 min. Brain tissues were also collected to determine the brain penetration of GSK2795039. The concentration of GSK2795039 in blood and tissue samples was determined by quantitative liquid chromatography, followed by mass spectrometry.
Pharmacokinetic modeling was performed using WinNonlin (Pharsight Corporation) using a noncompartmental model. Parameters obtained included Cmax, blood clearance (Clb), volume of distribution (Vss), and half-life (t1/2). The oral bioavailability (%Fpo) was estimated by (AUCpo/dosepo)/(AUCiv/doseiv)×100%.
Mouse acute pancreatitis model
Acute pancreatitis was induced in male C57BL6/J mice using the pancreatic secretagogue, cerulein. Mice were administered 6 hourly s.c. injections of cerulein (50 μg/kg) or saline control, animals were sacrificed 1 h after the last cerulein injection, and blood samples collected. Serum was prepared via centrifugation of blood samples at 10,000 rpm for 5 min. GSK2795039 (100 mg/kg) or vehicle (20% DMSO, 20% Tween 80, 60% polyethylene glycol 200 vehicle) was administered twice during the protocol via i.p. dosing at a dose volume of 2 ml/kg. The first dose was 1 h before the first cerulein injection, and the second dose was 4 h after the first dose of compound (i.e., dosing concomitantly with the fourth cerulein injection). Amylase enzyme levels were quantified in serum samples using an amylase activity assay kit (Sigma-Aldrich).
Statistics
All graphs were prepared using GraphPad Prism™ 5 or 6 and all data are expressed as mean±standard deviation. pIC50 values for test compounds, except PKCB II, were calculated from concentration response curves using the XLfit (IDBS) add-in to Excel or GraphPad Prism 5. pIC50 values for test compounds in PKCB II were analyzed using ActivityBase XE (IDBS). Statistical analysis was performed using Statistica™ 8 (StatSoft). Data obtained from the PMN oxygen consumption assay were analyzed using a one-way ANOVA, followed by the post hoc Dunnett test to compare each group with control. Data obtained from CFA-induced paw inflammation assay were analyzed using a repeated measure ANOVA, followed by the post hoc Fisher least significant difference test. Data obtained from the acute pancreatitis assay were analyzed using a two-tailed t-test.
Footnotes
Acknowledgments
The authors would like to thank Gregory Gatto and Krista Goodman from the Metabolic Pathways and Cardiovascular Therapeutic Area (GlaxoSmithKline, USA) for their contributions to the NOX2 enzymology and lead identification, respectively; Charlotte Ashby, Philip Hardwicke, Kelvin Nurse, Rob Tanner, Oxana Polyakova, Ben Schwarz, and Monique Murray-Thompson in the Platform Technology & Sciences Department (GlaxoSmithKline, United Kingdom and USA) for their support to prepare recombinant proteins; and Andy Lockhart in the Neurodegeneration Discovery Performance Unit (GlaxoSmithKline, United Kingdom) for his support on human PBMC work. Tamara Seredenina and Vincent Jaquet are funded by the European Community Framework Programme FP7/2007-2013 under grant 278611 (Neurinox).
Author Disclosure Statement
Kazufumi Hirano, Woei Shin Chen, Angela A. Dunne, Adeline Chueng L.W., Sumitra Ramachandran, Angela Bridges, Laiq Chaudry, Gary Pettman, Craig Allan, Sarah Duncan, Kiew Ching Lee, Jean Lim, May Thu Ma, Agnes B. Ong, Nicole Y. Ye, Shabina Nasir, Sri Mulyanidewi, Chiu Cheong Aw, Pamela P. Oon, Shihua Liao, Dizheng Li, Douglas G. Johns, Neil D. Miller, Ceri H. Davies, Edward R. Browne, Yasuji Matsuoka, Deborah W. Chen, and Anthony Richard Rutter are employees of GlaxoSmithKline.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.