
Abstract
Introduction and Brief Overview of Mitochondrial Biogenesis
M
As the replacement of any cellular component is metabolically expensive, mitochondrial biogenesis is particularly costly, because it may involve the synthesis of hundreds or, in some cases, thousands of new proteins. As a result, mitochondrial biogenesis is strictly controlled by intra- or extracellular signals communicating energy imbalance from increased energy demand, decreased energy production, or both. Mitochondrial biogenesis can be induced by exercise, fasting, cold exposure (thermogenesis), oxidative stress, and inflammatory cell stress. Depending on the stimulus, the program is executed through a variety of signaling pathways that converge on a handful of coactivators and nuclear transcription factors (including the peroxisome proliferator-activated receptor gamma-1 coactivator family [PGC-1α, PGC-1β, and PRC] and nuclear respiratory factors [NRF-1 and NRF-2]) (94). PGC-1α, in particular, has been identified as an important coordinator of the biogenesis response and has been found to orchestrate a wide variety of anti-inflammatory and metabolic nuclear genes; those most directly related to mitochondrial biogenesis in inflammation are covered here, including the NRF-1, NRF-2 (also called GA-binding protein A [GABPA]), and nuclear factor erythroid 2-related factor 2 (Nfe2l2 or Nrf2) transcription factors (16, 120, 129).
These transcription factors and coactivators coordinate the complex bigenomic programs of biogenesis by participating in feedback loops for the precise regulation of mitochondrial quality control (such as reactive oxygen species [ROS], Ca2+, and anti-inflammatory signaling [reviewed in Dominy and Puigserver (24)]) and by modulating specific gene expressions at various regulatory levels within the process. For instance, NRF-1 and NRF-2 upregulate the transcription of many nuclear-encoded mitochondrial proteins, which are transported across the mitochondrial membranes by processes that are themselves modulated by external stress stimuli and some of the same central transcription factors (45). These imported proteins serve as the building blocks for mitochondrial proliferation, while those same central coactivators and transcription factors upregulate expression of mitochondrial DNA (mtDNA)-binding proteins (mitochondrial transcription factor A [mtTFA], B1 [mtTFB1], B2 [mtTFB2], and DNA polymerase [Pol] γ). The mtDNA-binding proteins also undergo mitochondrial importation, where they directly activate mitochondrial transcription and replication (93, 94).
The induction of host inflammatory processes has a direct impact on mitochondrial function and quality control; mitochondrial biogenesis, in particular, is upregulated in response to both mitochondrial damage and the concomitant increases in energetic demand associated with severe inflammation. However, inflammatory processes have a multitude of different effects on various immune and somatic cell types that depend on the nature, severity, and timing of the inflammatory stimulus. The host responses to these stimuli (i.e., the production of pro-inflammatory cytokines, chemokines, and/or anti-inflammatory factors) also impact mitochondrial function and energy balance and so, in turn, have been found to participate both directly and indirectly in the regulation of mitochondrial biogenesis programming over the inflammatory cycle. Finally, the consequences of inflammation on mitochondria themselves signal for adaptive modulation of the inflammatory response. In this study, we briefly review the intersection of innate inflammation and mitochondrial biogenesis by examining proinflammatory effects on mitochondrial function and biogenesis and, conversely, mitochondrial feedback on counter-inflammation, with an emphasis on redox signals that influence cellular and tissue homeostasis during the course of the inflammatory cycle.
Impacts of Inflammation on Mitochondrial Function
Oxidative phosphorylation and impairment of mitochondrial respiration
The impact of inflammation on mitochondrial function has often been modeled by exposure to bacterial endotoxin; as early as the late 1960s, it was known that gram-negative lipopolysaccharide (LPS) exposure decreased respiration in both the isolated mitochondria (46, 97) and in various tissues in vivo (68, 69, 115). Animal models of bacterial sepsis have confirmed that mitochondrial function and/or metabolic profiles are also significantly impacted by septic inflammation, with the most profound changes typically seen early in the host response (4, 9, 52, 62, 63). Finally, in the most relevant model of all, in human patients with sepsis from various pathogens and sources, there is also clear evidence of proinflammatory mitochondrial dysfunction (Fig. 1). Importantly, the degree of mitochondrial dysfunction appears to be roughly associated with clinical outcomes, with nonsurvivors of severe sepsis demonstrating an early decrease in the ATP/ADP ratio in skeletal muscle (8), impaired fatty acid utilization (58), and later a failed induction of upregulation of mitochondrial biogenesis markers when compared with survivors (13).
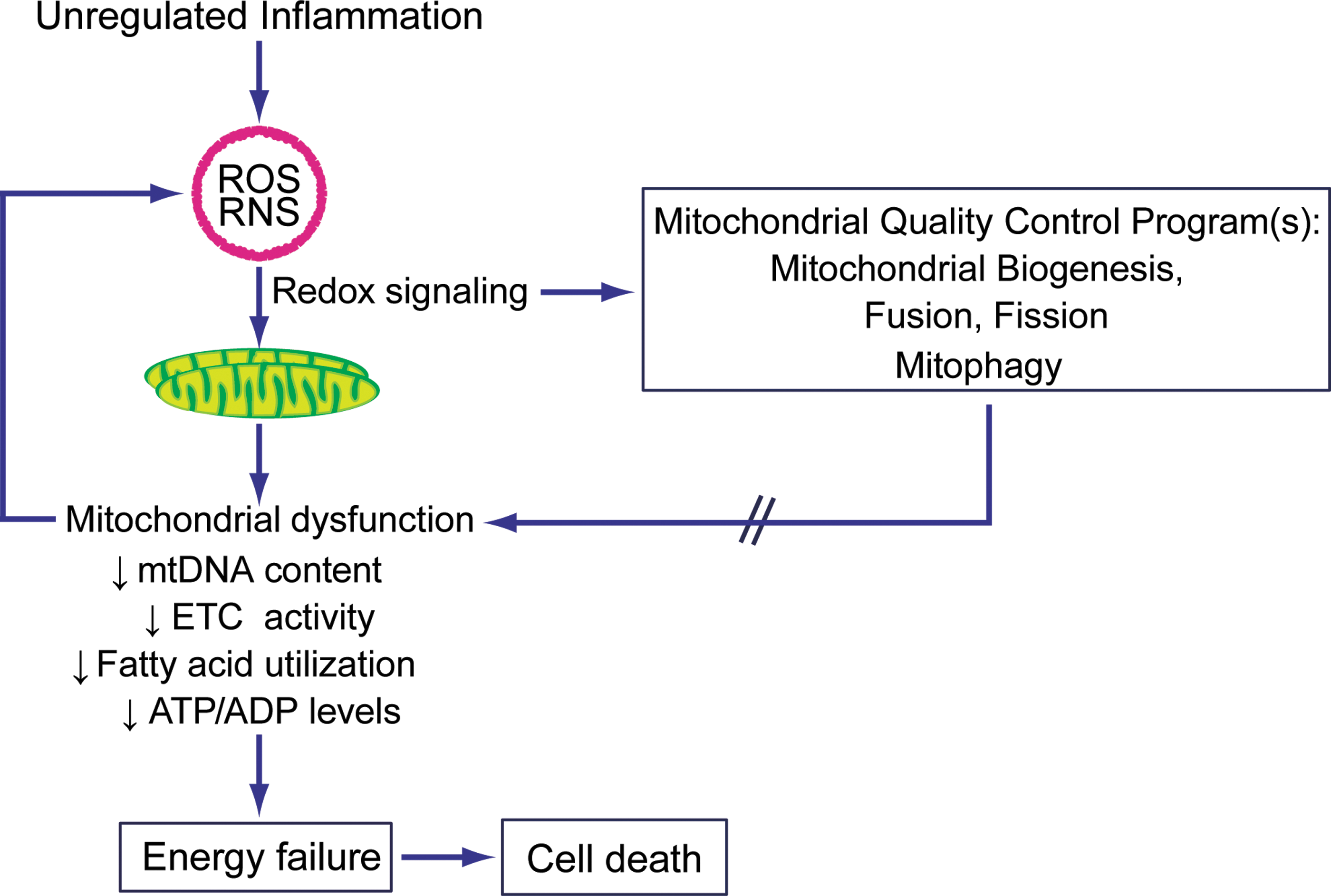
There are clear advantages and limitations associated with various experimental models of acute inflammation (22); actual pathogenic inflammation within an intact organism involves far more complex stimuli and responses than a time-limited exposure to one or a mixture of inflammatory mediators. Although LPS administration models are simple and reliably produce profound biological effects that lend themselves to mechanistic investigations of the nature of proinflammatory mitochondrial dysfunction and recovery during the resolution phase, the doses of LPS required for mitochondrial damage are supraphysiological and confounded by serious hypotension and tissue ischemia. Animal and human models of bacterial infection have fewer controlled variables, but allow examination of the effects of systemic-level inflammation on mitochondrial function, as well as on survival and other outcomes. Nonetheless, these various models show that the reduction of mitochondrial oxygen consumption in early inflammation is accompanied by decreased ATP or ATP/ADP levels (4, 11, 27) corresponding to nitric oxide synthase 2 (NOS2) induction (6, 115), which may result in, for example, complex I inhibition (9). There is also evidence of mitochondrial dysfunction through mtDNA depletion (109) due to cytochrome c oxidase (COX) inhibition (6, 31, 62, 63) and resulting in decreased State 3 respiration in several tissues (11, 111). Also, while decreased electron transport chain (ETC) protein activities certainly contribute to impaired respiration, it has also been suggested that ETC protein levels are also decreased early after endotoxin/tumor necrosis factor-α (TNF-α) exposure (11, 111). These relatively rapid changes in the mitochondrial protein content may reflect protein disposal mechanisms and/or the activation of selective mitochondrial autophagy (mitophagy) along with perhaps altered antibody peptide recognition due to ROS- or NO-induced post-translational protein modifications (62).
In any event, inflammation-induced mitochondrial dysfunction is a common, but sometimes subtle feature of inflammatory tissue damage in a variety of cell types and inflammatory models. Although profound and permanent respiratory impairment is incompatible with cellular survival, conservation of this response across models and organisms suggests that some type of temporary energetic conservation mechanism in early inflammation may be activated for its adaptive advantages (103). On the tissue level, decreased ATP availability in areas of localized inflammation may prevent pathogenic hijacking of host cell energetic infrastructure and help contain infection, preventing systemic spread (62). More globally, the example of a hibernating myocardium in response to ischemic threat may be analogous to the temporary downregulation of mitochondrial respiration in the systemic inflammation of sepsis, but just as the hibernating myocardium is not sustainable indefinitely, decreased mitochondrial respiration in sepsis eventually results in irreversible energetic deficits that lead to cell death (104).
Reactive oxygen and nitrogen species
In the setting of such metabolic stasis, the temporary decrease in oxidative phosphorylation not only potentially conserves limited mitochondrial substrate for respiration but it may also mitigate pathologic mitochondrial ROS production. This aspect may be of particular importance in the setting of inflammation, where systemic oxygen (or carbon substrate) supply is not thought to be a limiting factor (32) (notwithstanding limitations in local areas of hypoperfusion [reviewed in Trzeciak and Rivers (119)]). Small amounts of ROS are generated in the course of normal oxidative phosphorylation (primarily by complexes I and III of the ETC) (82); however, the production of both ROS and reactive nitrogen species (RNS) is increased in the setting of mitochondrial damage, such as that encountered in severe inflammation. Inflammatory ROS/RNS production may eventually overwhelm the cellular antioxidant capacity and lead to damage to proteins, lipids, and DNA by oxidation or nitration. Oxidative damage to cellular and mitochondrial components, particularly to the ETC complexes (10, 18, 71), leads to further dysfunction and ROS/RNS production.
Inflammation-induced changes in mitochondrial function and ROS/RNS production vary depending on the intensity and duration of the inflammatory stimulus. Low-level ROS production is important in redox signaling and in prosurvival cellular adaptations (95), particularly evident is H2O2 leakage from mitochondria as a retrograde signal to nuclear-targeted cytosolic pathways that warn the cell of internal oxidative stress. On the other hand, more profound energetic deficits and ROS production not only lead to damage of cellular components but also to loss of ROS signal localization (1). If repair mechanisms become overwhelmed, the intrinsic apoptotic cascade may be triggered in the setting of only moderate inflammation (132). In severe inflammation, mitochondrial dysfunction and ROS/RNS-induced damage may lead to extensive cellular necrosis and release of cellular contents, including mitochondria and mitochondrial fragments, which act as further inflammatory stimuli (91, 122).
Inflammatory Regulation of Mitochondrial Biogenesis
Since the level of mitochondrial dysfunction, ROS/RNS production, and mitochondrial damage induced by the host response depends on the magnitude and timing of that stimulus, it follows that successful restoration of a fully functional mitochondrial population is dependent on the timely upregulation of mitochondrial quality control programs (Fig. 2). Following acute inflammation and an initial decrease of mitochondrial function, there may be a restoration of respiratory capacity (42, 111). Data from animal studies and septic patients show that evidence of mitochondrial recovery is predictive of (9, 33) and/or significantly associated with clinical recovery (8, 13, 55). In this study, we have focused on the induction of mitochondrial biogenesis pathways in inflammation, but all of the quality control elements are important for sepsis resolution (104).
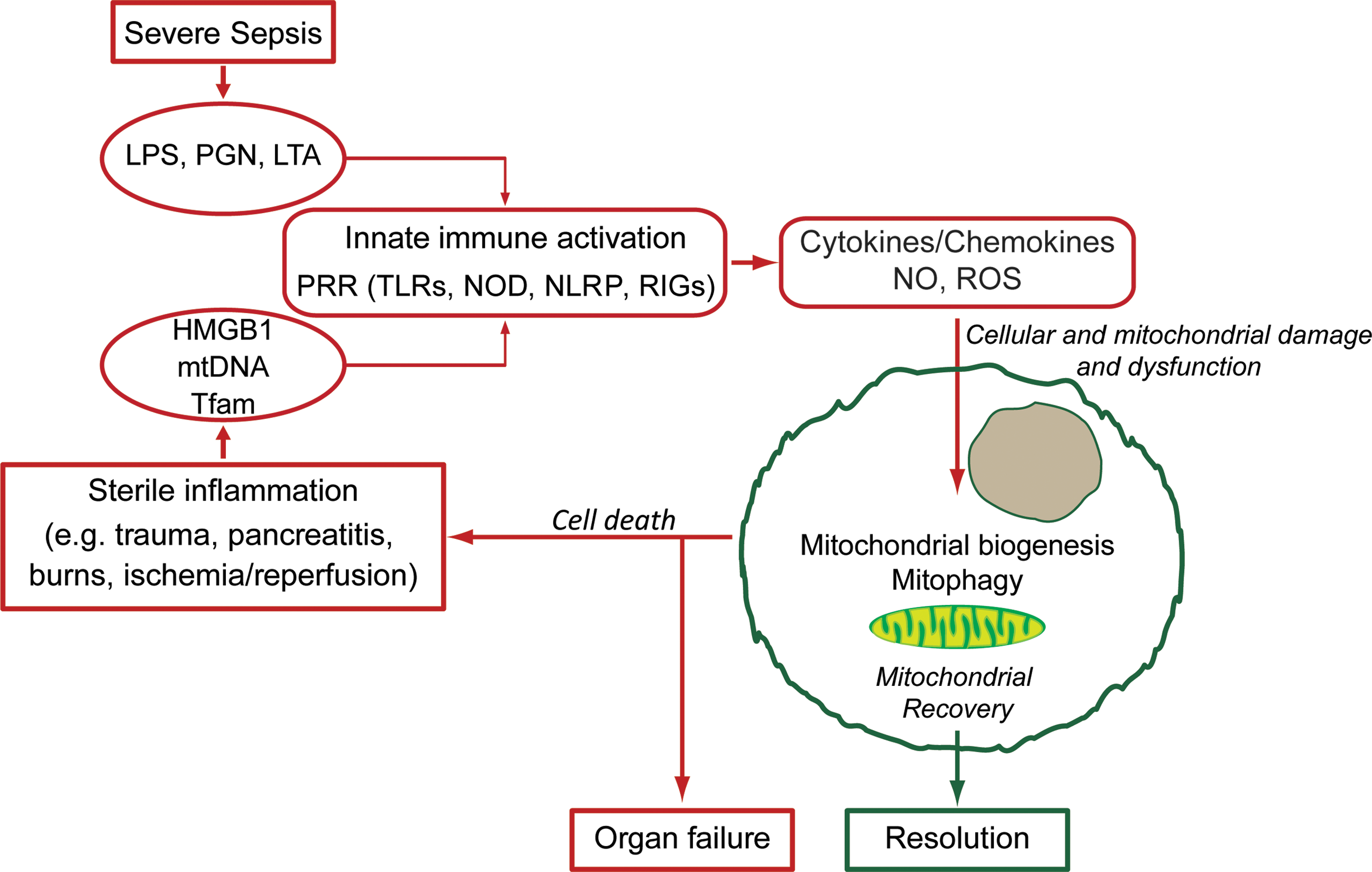
For nonimmune cells (immune cells are discussed later), mitochondrial dysfunction in the early phase of septic inflammation is stimulated, in part, by circulating proinflammatory cytokines like TNF-α (96) and a subset of the interleukins. A plethora of pathways participate in regulating the mitochondrial response (Fig. 3), and many of the intermediate cellular transcription factors and coactivators can also participate in regulating adaptive responses and/or amplifying the inflammatory signal. The inhibition of respiration and initiation of mitochondrial damage also initiate retrograde signaling to the nucleus for the genetic activation of mitochondrial quality control programs, pro- and anti-inflammatory elements, and antioxidant enzymes. Each is discussed briefly below.
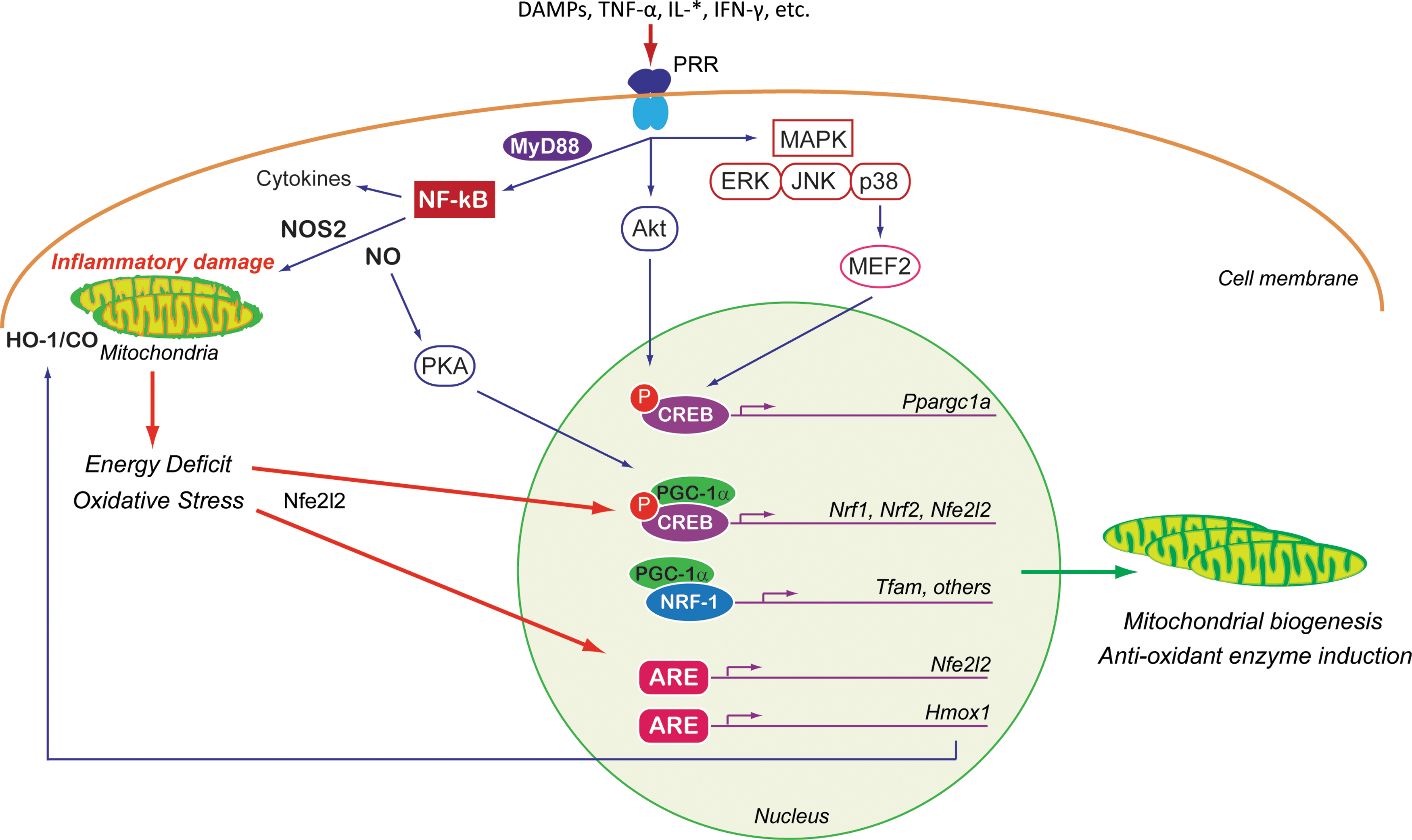
Pathways and mediators of inflammatory mitochondrial biogenesis
Toll-like receptors, inflammatory mediators, and biogenesis gene regulation
The innate immune response is activated through the recognition of microbial antigens (pathogen-associated molecular patterns [PAMPs]) or intrinsic factors released into the circulation (alarmins) (74). Together these factors, called danger-associated molecular patterns (DAMPs), are sensed by cellular pattern recognition receptors (PRRs) such as toll-like receptors (TLRs). In general, PRR activation upregulates the release of early-phase inflammatory protein mediators (TNF-α, interleukins [IL-*], interferon gamma [IFN-γ], etc.), growth factors, hormones (cortisol and adrenaline), and ROS/RNS. These are far from being the only mediators of immune signaling [reviewed in Castellheim et al. (14)], but the pathways by which these modulate inflammatory responses in nonimmune cells are relatively well studied; these are encapsulated here. The adrenergic and hormonal responses to inflammation certainly also lead to adaptations in mitochondrial function and quality control (37, 117, 125, 127), but are not covered here in detail.
The circulating inflammatory proteins released by cellular components of the innate immune system provide important information to nonimmune cells through cell-type-specific arrays of inflammatory protein receptors and PRRs (including TLRs) for the recognition of relevant DAMPs (17). Many of these receptors (TLRs as well as PRRs for TNF-α, IFN-γ, and interleukins [specifically IL-6]) activate one or several well-defined inflammatory signaling pathways. The first of these is via the NF-κB family of transcription factors, which activates pathways that contribute to inflammatory, apoptotic, proliferative, and tumorigenic processes. NF-κB is normally present in the cell in an inhibited state until myeloid differentiation primary response protein (MyD88)-dependent or -independent phosphorylation of the NF-κB inhibitor (IκB); NF-κB subunits are then free to translocate to the nucleus to regulate transcription in combination with other transcription factors (39). Many targets of TLR signaling through NF-κB are proinflammatory and induce further oxidative stress (114). The p65 subunit may even suppress the PGC-1α activity and related metabolic activity in some tissues such as the heart (3). On the other hand, LPS-stimulated TLR4 signaling, operating through NF-κB, can promote (cAMP response element-binding protein [CREB]) upregulation of PGC-1α, NRF-1, NRF-2, and other components of the mitochondrial biogenesis framework in both CREB-dependent (110, 112) and CREB-independent pathways (113). Evidence also suggests that parkin, best known for its role in mitophagy regulation in the setting of Parkinson's disease and other chronic inflammatory states, may also regulate mitochondrial biogenesis (35, 92). There is recent evidence that parkin binds to mtDNA via mtTFA, enhancing transcription (56, 88). Parkin has also been found to regulate degradation of PARkin-interacting substrate (PARIS, ZNF746) by ubiquitination, which then leads to decreased repression of Ppargcla transcription (102). Intriguingly, in an LPS model, parkin was increased by a MyD88/NF-κB-dependent pathway, suggesting a potential for parkin-modulated mitochondrial biogenesis in acute inflammation (118).
Another group of pathways commonly activated by TLRs or other inflammatory receptors are those involving the mitogen-activated protein kinases (MAPKs) and protein kinase B (PKB/Akt). Core MAPK signaling pathways include extracellular signal-related kinases (ERK), p38, and c-Jun NH2-terminal kinases (JNK). The ERKs, p38s, and JNKs phosphorylate a vast array of other kinases and transcription factors (57); activities specifically relevant to the mitochondrial function include activation of activating protein-1 (AP-1) by all three core pathways (100) and p38 activation of myocyte enhancer factor-2 (MEF2) (43). AP-1 participates in apoptosis regulation, cell differentiation, and proliferation, and MEF2 participates in a positive feedback loop (which is enhanced by Ca2+ signaling), increasing PGC-1α expression (44). Akt/PKB is a prosurvival kinase that also has a role in controlling many inflammatory response processes, including protein synthesis, chemotaxis regulation, apoptosis, and cell metabolism (14). In the context of mitochondrial biogenesis, Akt phosphorylates and activates the NRF-1 transcription factor, resulting in induction of mtTFA (80), and it stimulates the expression of several biogenesis genes via the CREB/CBP nuclear transduction pathway (26). However, Akt can also be proinflammatory through the regulation of NF-κB expression (21) and, when chronically overexpressed, has been shown to lead to decreased PGC-1α levels in the heart (19).
Nitric oxide
One of the most important NF-κB-dependent responses in sepsis is the upregulation of NOS2 (iNOS) in both immune and nonimmune cells. TNF-α, IL-1β, IFN-γ, and platelet-activating factor (PAF) can also activate NOS2 expression individually or in concert in many cell types (53, 116), acting through a variety of other transcription factors, including interferon regulatory factor-1 (IRF-1), signal transducer and activator of transcription-1a (STAT-1a), CREB, and AP-1 (77). Because NOS2 is regulated mainly at the transcriptional level and not by calcium, it can in some cells produce very high levels of NO, which is important to antimicrobial defense, but also cytotoxic to the host cell (34). In the setting of oxidative stress, NO will react with superoxide to produce peroxynitrite (ONOO−), a highly RNS that can lead to damage of mitochondrial ETC components by chemical nitration and hydroxylation (10). At high levels, NO also inhibits complex I, cytochrome c, and COX (6, 10, 59).
Apart from those relatively direct effects on mitochondrial function, NO also functions as an intracellular signal. The effect most relevant here is that NO stimulates mitochondrial biogenesis via the induction of the PGC-1α coactivator (41, 73, 84). There is also evidence that S-nitros(yl)ation of certain heat shock chaperone proteins by endogenous NO production is associated with induction of mitochondrial biogenesis in murine sepsis (108). In addition, Nfe2l2-mediated antioxidant gene expression is dependent on NO production in cardiomyoblasts (54), neuroblastoma cells (23), endothelial cells (7), and others. NO also modulates inflammatory signals—NO leads to suppression of NF-κB [via the inhibitory protein IκB (78)] and upregulates endothelin-1 (ET-1), a proinflammatory mediator, for instance, in the liver in sepsis (30).
Heme oxygenase-1 and carbon monoxide
The induction of heme catabolism during a variety of cell stresses ranging from hypoxia to inflammation has been recognized for many years. This is the function of the two isoforms of the heme oxygenases (HO) (90). HO-1 upregulation is seen consistently with LPS exposure in most tissues (12, 27, 36, 85). In addition, NO plays a role in HO-1 expression in the liver during sepsis (30). Heme metabolism by HO-1 produces carbon monoxide (CO) as a product, which through its interactions with reduced transition metals, is itself a source of oxidative stress and ROS signaling. Otherwise, HO-1 and CO exert anti-inflammatory effects (decreased TNF-α, IL-1β, and macrophage inflammatory protein-1 β; and increased IL-10) (75, 81, 126), thus providing negative feedback control on the inflammatory response and facilitating the proresolution state. Of mention, in the setting of chronic metabolic inflammation, the loss of HO-1 in hepatic tissue has been reported to reduce steatosis and toxicity—the mechanism underlying these findings is unexplained as yet (51). In addition to these adaptive processes, HO-1/CO increases mitochondrial biogenesis and antioxidant gene expression via upregulated Nfe2l2 activation (65, 80, 124). Nfe2l2, itself a redox-sensitive transcription factor, is capable of promoting further expression of HO-1 (2).
Energy deficit and oxidative stress
Apart from the mechanisms of induction of mitochondrial biogenesis that are relatively specific to inflammatory states, the energetic deficits and oxidative stress induced by inflammation activate more general pathways for mitochondrial quality control recruited by the cell stress response. These will be mentioned only briefly as more complete detailed reviews of signaling and mitochondrial biogenesis in response to cellular energetic sensing can be found elsewhere (94, 131).
Cellular energy deficits and mitochondrial dysfunction may become manifest by an increase in the AMP/ATP or the NAD+/NADH ratios. Increased AMP/ATP induces PGC-1α phosphorylation via AMPK signaling (50); a rise in NAD+increases deacetylation of PGC-1α via sirtuin 1 (SIRT1) (86). As mentioned, PGC-1α is a key regulator of mitochondrial biogenesis, and it also modulates anti-inflammatory (28) and antioxidant pathways (16, 64, 106). Phosphorylation of PGC-1α increases its translocation into the nucleus and coactivation of the mitochondrial biogenesis transcriptional process (5); SIRT1 deacetylation increases the activity of the PGC-1α protein and its regulation of nuclear and mitochondrial gene transcription (38). Finally, CREB1, an intermediate transcription factor that also has a number of roles in regulating gene transcription (often in partnership with PGC-1α) in response to metabolic needs (128), also helps mediate ROS signaling (106).
This overview leads us to a brief discussion of mitochondrial biogenesis in response to oxidative stress. Mitochondrial mass has been shown to increase in cells under external sublethal oxidative stress (61). The generation of ROS, primarily in the form of relatively diffusible H2O2 and through oxidation of various proteins, can provide signal transduction communicating the mitochondrial oxidative state, as feedback for control of nuclear programs for antioxidant defense or mitochondrial quality control. Even in the absence of a clear inflammatory stimulus, oxidative stress can upregulate many of the transcription factors already discussed and induce some of the mitochondrial biogenesis signaling pathways mentioned. These include increased CREB promoter binding (106), AMPK activation of PGC-1α and-1β (49), and expression of NRF-1 (80) and NRF-2. Additionally, oxidative stress induces the HO-1/CO system via Akt/PKB, activating Nfe2l2 and ultimately enhancing NRF-1 expression resulting in mitochondrial biogenesis and an increase in mitochondrial mass in the mouse heart (79).
Inflammation and mitochondrial biogenesis in immune cells
A perhaps more neglected component of the mitochondrial biogenesis response to inflammatory stimuli is the role of mitochondrial biogenesis in immune effector cells for the support of increased cellular energy requirements, as well as for cell proliferation and differentiation. This is addressed briefly below and leads naturally to novel concepts of how inflammatory damage to mitochondria modulates subsequent pro- or anti-inflammatory processes. Sepsis induces both proliferation and apoptosis in immune cells (87). Increased respiration and mitochondrial proliferation have been reported in circulating peripheral immune cells of septic patients, although different results have been reported (105). In vitro and animal models support this finding: AMPK stimulation in macrophages induces autophagy and improved clearance of mycobacterium, and also increased PGC-1α and mtDNA as markers for accompanying mitochondrial biogenesis (130). During activation of murine T cells, T-cell receptor activation increases mitochondrial mass and mtDNA content also via AMPK (20). Moreover, IL-15 influences mitochondrial biogenesis to provide for spare respiratory capacity for maintenance of the memory phase of immunity (CD8+ T memory cells) following clonal expansion in response to infection (121).
Mitochondrial dysfunction and damage as modulators of inflammation
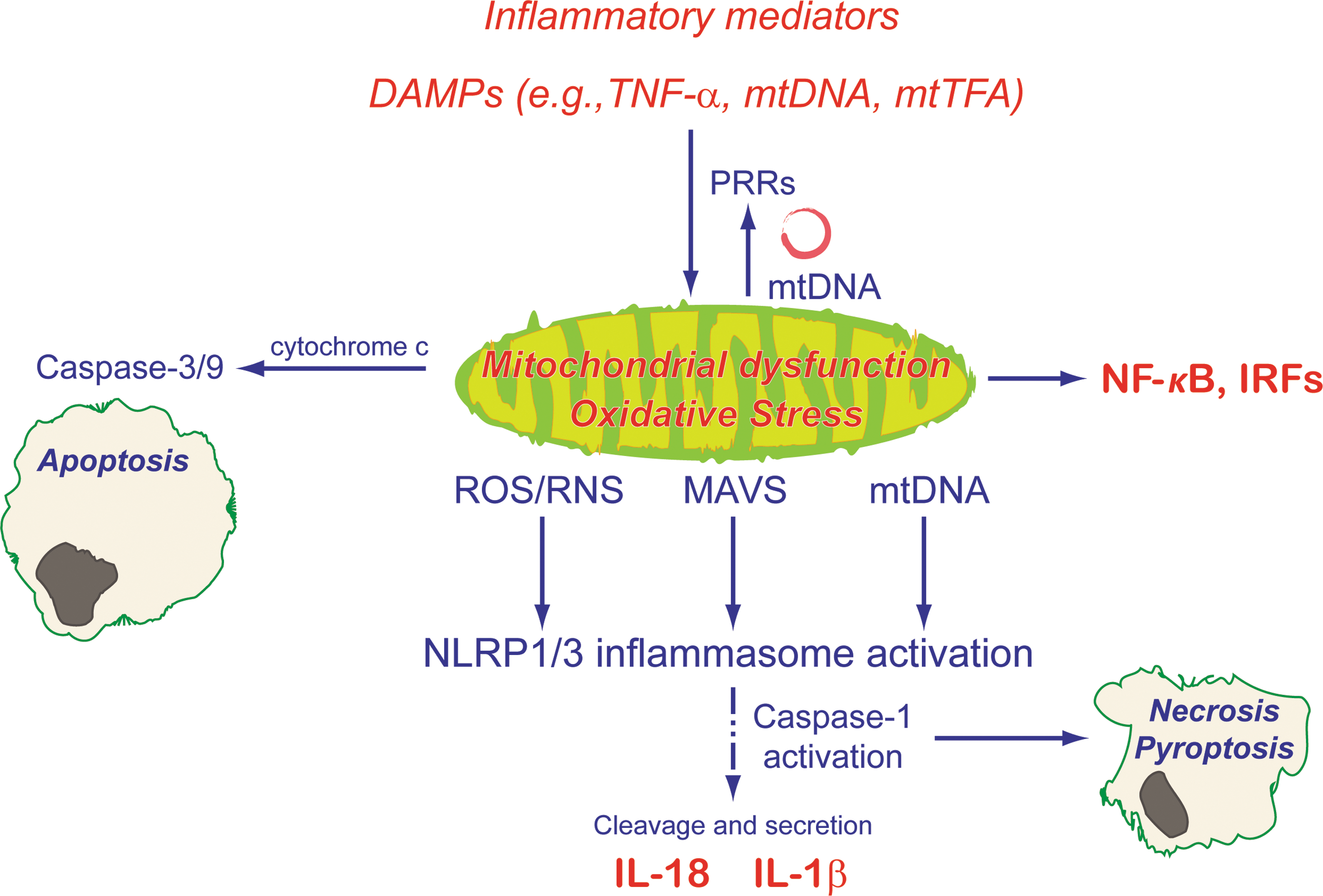
The mitochondrial dysfunction induced by inflammatory stress is also known to have a signaling role in modulating further inflammation. Severe stress induces cell death and the extracellular release of varying amounts and types of intracellular products depending on the specific cell death pathway (91). These extracellular DAMPs, including mitochondrial DAMPs [e.g., mtDNA (134), cytochrome c (29), mtTFA (15), high-mobility group box 1 (HMGB1) (83, 123)], ATP (40), and other mitochondria-derived peptides [e.g., humanin, which has a cDNA sequence virtually identical to mitochondrial 16S rRNA (47)], interact with PRRs to trigger pro- or anti-inflammatory responses as reviewed above (67, 98). Additionally, the signaling activities of many cytokines and cell death-associated proteins are redox regulated through cysteine modifications and are therefore sensitive to inflammatory oxidative stress (89).
Inflammatory mitochondrial dysfunction also induces intracellular inflammatory signaling (Fig. 4); mitochondrial dysfunction increases oxidative stress and also leads to membrane permeability transition (MPT) and mtDNA translocation to the cytoplasm (72). The presence of cytoplasmic mtDNA upregulates NF-κB through TLR9 signaling (133). Cytosolic mtDNA and increased ROS—via thioredoxin-interacting protein (135)—initiate NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome assembly (25, 101); this allows caspase-1 activation with subsequent proinflammatory IL-1β and IL-18 release (66). Macroautophagy, specifically mitophagy, is a negative regulator of inflammasome activation (72, 136). Impaired autophagy reduces clearance of damaged mitochondria, resulting in increased ROS production and mtDNA accumulation in the cytoplasm, leading to inflammasome activation and inflammatory mediator release. The translocation of mtDNA into the cytoplasm itself seems to be partially dependent on inflammasome activation (72). More recent evidence also supports roles for both mitochondrial antiviral signaling protein [MAVS; previously known to induce NF-κB (99) and IRFs (60)] (107) and mitochondrial Ca2+ signaling in NLRP3 inflammasome activation (48, 70).
Summary and Conclusions
In summary, mitochondria not only play a vital role in cellular homeostasis but also in the response to environmental stimuli. Initially, inflammation-induced mitochondrial dysfunction results in decreased oxidative phosphorylation and increased oxidative stress. Host recovery is dependent, in part, on the upregulation of mitochondrial quality control programs. Mitochondrial biogenesis, one component of these programs, is tightly controlled by specific coactivators and transcription factors that regulate the expression of components of the nuclear and mitochondrial genome. These are found to be stimulated by various elements of the inflammatory response; in nonimmune cells, early-phase inflammatory protein mediators interact with PRRs to activate the NF-κB, MAPK, or PKB/Akt pathways. A shared component of these pathways is increased expression or activity of the core coactivators and/or transcription factors controlling mitochondrial biogenesis (PGC-1α, NRF-1, NRF-2, and Nfe2l2). Inflammatory upregulation of NOS2-induced NO, although directly impacting negatively on mitochondrial function as a reactive species, also functions as a signaling molecule; NO upregulates mitochondrial biogenesis via PGC-1α, participates in Nfe2l2-mediated antioxidant gene expression, and itself has a role in inflammatory mediation. Similarly, NO and other components of the host inflammatory response induce the HO-1/CO system, which stimulates both mitochondrial biogenesis and antioxidant genes. More generic energy-sensing and redox-sensitive pathways (such as AMPK, SIRT1, CREB, and HO-1/CO) also modulate mitochondrial biogenesis in the setting of inflammatory stress. Finally, inflammatory mitochondrial dysfunction and oxidative stress can initiate further inflammatory responses through DAMP interaction with PRRs and by inflammasome activation. In short, the host response depends profoundly on a complex network of pro- and anti-inflammatory pathways that impact, and are impacted by, mitochondrial dysfunction. Recovery of mitochondrial function and cell redox status is vital to cellular survival and organ function in unregulated acute inflammatory states such as severe sepsis.
Footnotes
Acknowledgment
This work was supported, in part, by the NIH P01 HL108801-04, Project 2 (CAP).