
Abstract
Introduction
A
Fatty acid (FA)–stimulated and redox-stimulated insulin releases have not been fully understood as well as acute lipotoxicity, instantly decreasing insulin secretion in pancreatic β-cells. We describe a feedback antioxidant mechanism based on redox signaling initiated by FA β-oxidation, and promoted plus amplified by mitochondrial phospholipase iPLA2γ. Not only the antioxidant synergy of iPLA2γ with mitochondrial UCP2 is demonstrated, but also the iPLA2γ role in the amplifying mechanism, since further free FAs cleaved by iPLA2γ serve as “messengers” for G-protein–coupled receptor 40 (GPR40). Consequently, the iPLA2γ/UCP2 synergy regulates glucose-stimulated, redox-, and FA-stimulated insulin release in pancreatic β-cells.
Superoxide formation is an inevitable side reaction at Complex I and III of mitochondrial respiratory chain (24) and in 2-oxoacid dehydrogenases (41, 46). Mitochondrial superoxide formation increases with an increasing substrate (NADH) load, represented by increasing glucose in pancreatic β-cells (10). Similarly, in numerous situations of local or global electron transfer retardation within the respiratory chain, superoxide production is specifically elevated. This serves for redox signaling, for example, during initiation of hypoxic gene expression remodeling (27).
Mitochondrial H+ pumping is usually tightly coupled to the H+ backflow via the ATP synthase. Since any uncoupling of this accelerates electron transfer within the respiratory chain (and hence respiration), the superoxide formation is attenuated by mitochondrial uncoupling. This represents the key mechanism exerted by UCP2, although it slightly attenuates ATP synthesis. In pancreatic β-cells, the increase in oxidative phosphorylation (OXPHOS) substantiates the canonical mechanism of glucose sensing. The increasing ATP/ADP ratio at higher glucose initiates the glucose-stimulated insulin secretion (GSIS) (5, 26, 47).
By shifting ROS homeostasis, UCP2 may participate in redox signaling in β-cells (31, 48), which may be easily transmitted due to the low capacity of redox buffers (23). H2O2-responsive gene expression is manifested for both major differentiation factors of β-cells, PDX-1 and MafA (47). Impaired antioxidant defense leading to chronic oxidative stress may affect insulin secretion machinery that is finely tuned for optimum GSIS in β-cells, as recognized in type 2 diabetes patients (16, 39, 40) and rodent diabetic models (30, 33). ROS may further accelerate diabetic development by promoting apoptosis, thus decreasing β-cell mass (51). Consequently, oxidative stress serves as a mediator of β-cell remission.
The function of UCP1 (12) and recombinant UCP2 (6, 7, 20, 53) is essentially dependent on its anionic transport substrates, nonesterified fatty acids (FAs) (6, 7, 18, 20, 53). However, FAs augment GSIS in β-cells, when exposed for hours (8, 15, 19), but chronically excessive saturated FAs suppress insulin secretion (32, 43, 52), the phenomenon termed lipotoxicity (15, 19). As a simplifying scheme, UCP2 might counteract acute lipotoxicity arising from oxidative stress due to the incoming FAs. Nevertheless, its role should be further clarified.
The role of phospholipases A2 (PLA2) (21, 22, 25, 34, 35, 38) residing in (such as iPLA2β) (34) or recruited to mitochondria of pancreatic β-cells should also be explained in relation to their activation. PLA2 may amplify lipotoxicity, but in concert with UCP2 a hypothetical synergic antioxidant activity may prevail, such as with mitochondrial iPLA2γ in heart (25) and lung tissues (21). Both iPLA2γ and iPLA2β belong to the group VI of PLA2s (38) ascribed to the cytosolic Ca2+-
The FAs were also implicated in the insulin release in pancreatic β-cells. Although the mechanism is not decisively elucidated (15), the G-protein–coupled receptor 40 (GPR40) has been recently implicated in insulin-stimulating responses (11). In addition, the threshold FA concentration, above which acute lipotoxicity is exerted and below which insulin release is stimulated, is not known.
In this work, we aimed at studying how UCP2-mediated antioxidant action may be initiated and regulated and at elucidating its functional relationship with PLA2s. We revealed the antioxidant synergy of UCP and iPLA2γ and describe in detail its impact on acute lipotoxicity and insulin secretion in model pancreatic β-cells, insulinoma INS-1E cells.
Results
Uncoupling manifested by respiration increase after tert-butylhydroperoxide addition to INS-1E cells is absent in cells silenced for UCP2 or iPLA2γ
Nontransfected INS-1E cells with glucose lowered to 3 mM still exhibited a substantial respiration of 23.6±6.3 pmol O2·s−1×10−6 cells (n=49; number of preparations N=10). The corresponding state may be regarded as the phosphorylating state-3, although with low substrate levels available for OXPHOS. Tert-butylhydroperoxide (TBHP) addition resulted in the acceleration of respiration, so that the differences (ΔTBHPJO2) between respiration rates before (J0 O2) and after TBHP exhibited a saturating dependence on glucose.
INS-1E cells transfected with nontargeting microRNA (miRNA) (ntgINS-1E cells) and with glucose lowered to 3 mM exhibited virtually unchanged respiration of J0
O2=25.3±6.3 pmol O2·s−1×10−6cells (n=356; N=51) and responded to TBHP in the same way (Fig. 1A). UCP2-silenced cells (Supplementary Fig. S1A, C; Supplementary Data are available online at
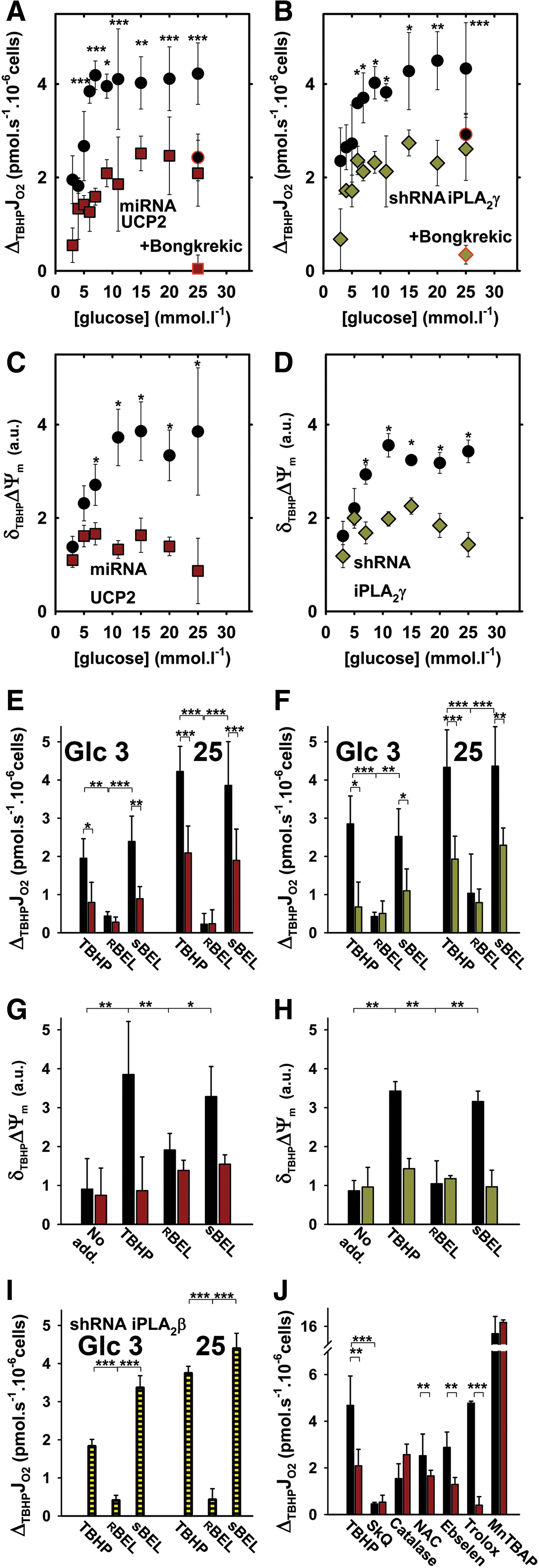
In addition, iPLA2γ-silenced cells (Supplementary Fig. S1B, C) with glucose lowered to 3 mM did not significantly change respiration (J0 O2=19.4±3.9 pmol O2·s−1×10−6 cells; n=116; N=23; compared with own ntg shRNA controls having J0 O2=22.4±3.3 pmol O2·s−1×10−6 cells; n=117; N=24). They also exhibited suppressed respiratory responses to TBHP addition with increasing glucose and complete inhibition by bongkrekic acid (Fig. 1B).
To verify that the observed respiratory elevations represent a mild mitochondrial uncoupling, we monitored the IMM potential decrease (δTBHPΔΨm) at various glucose concentrations, after TBHP addition to UCP2- (Fig. 1C) and iPLA2γ-silenced INS-1E cells (Fig. 1D). We found the same pattern as for respiration increases. Since the accelerated respiration, together with simultaneous ΔΨm decline, strictly defines mitochondrial uncoupling, we conclude that TBHP induces uncoupling in INS-1E cells in a UCP2- and iPLA2γ-dependent manner.
The iPLA2γ contribution also stems from the sensitivity to its stereo-selective inhibitor
TBHP-induced uncoupling is prevented by antioxidants
As expected, UCP2-dependent TBHP-induced mitochondrial uncoupling in ntgINS-1E cells at 25 mM glucose was completely prevented by matrix-targeted antioxidant SkQ1 and substantially blocked by catalase overexpression (Fig. 1J). Among nonmitochondrial antioxidants, only N-acetyl-
iPLA2γ is directly activated by TBHP or H2O2
Reconstituted recombinant iPLA2γ was reversibly activated by H2O2 (Fig. 2A–E) with a half-maximum activation constant AC50 corresponding to ∼1.5 nmol H2O2 per nmol of enzyme (Fig. 2E). The H2O2-induced FA release in proteoliposomes containing iPLA2γ was fully reversed by membrane-permeable dithiothreitol (DTT) and partially by membrane-impermeable glutathione (Fig. 2D, E). These results indicate that iPLA2γ activation by H2O2 is dependent on reversible oxidation of accessible cysteine residues. In cells, AC50 was 200 μM for TBHP (Supplementary Fig. S2A).
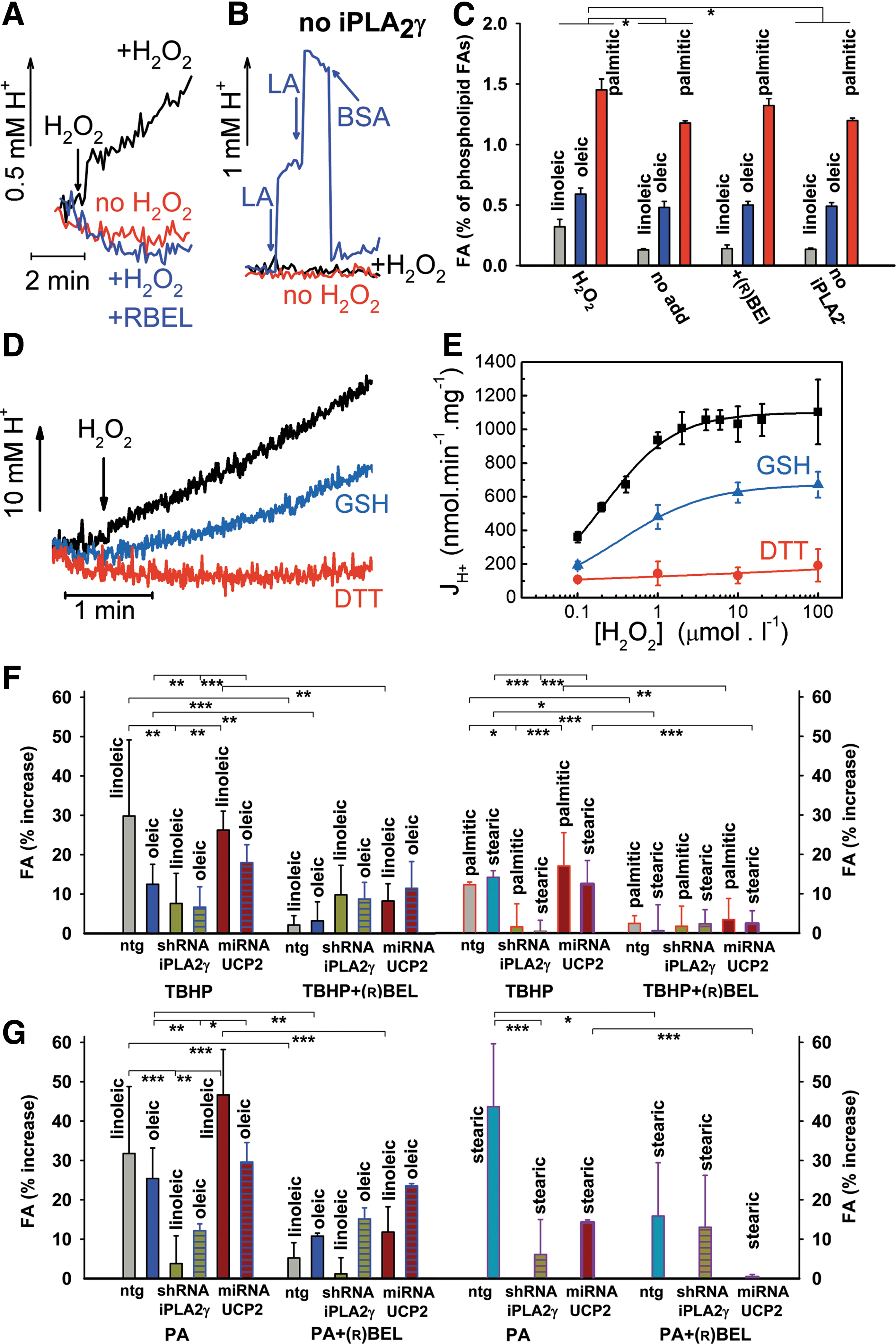
Gas chromatography/mass spectrometry quantification of released FAs from iPLA2γ-containing proteoliposomes after 60 min at 30°C had indicated a release of approximately 1 nmol−1·min·(mg protein)−1 (1.5% of phospholipid FAs) of free palmitic acid (PA), oleic, or linoleic acid, which was enhanced by H2O2, but not with
Decrease of superoxide release into mitochondrial matrix on TBHP addition is absent in INS-1E cells silenced for UCP2 or iPLA2γ
We further investigated whether the observed TBHP-induced uncoupling attenuates mitochondrial superoxide production. We employed MitoSOX Red fluorescence (10), measuring the in situ surplus superoxide release into the mitochondrial matrix as the rate Jm of fluorescence changes in regions of interests identical to the matrix (Supplementary Movie S1). We have evaluated (Fig. 3A, D) how Jm is altered after the immediate TBHP addition to ntgINS-1E cells pre-equilibrated with 25 mM glucose for 10 min (after 16-h preincubation with 3 mM glucose). In spite of the pro-oxidant nature of TBHP, Jm rates decreased to a minimum on TBHP addition (Fig. 3A, D). Such attenuation of mitochondrial superoxide production ceased in the presence of
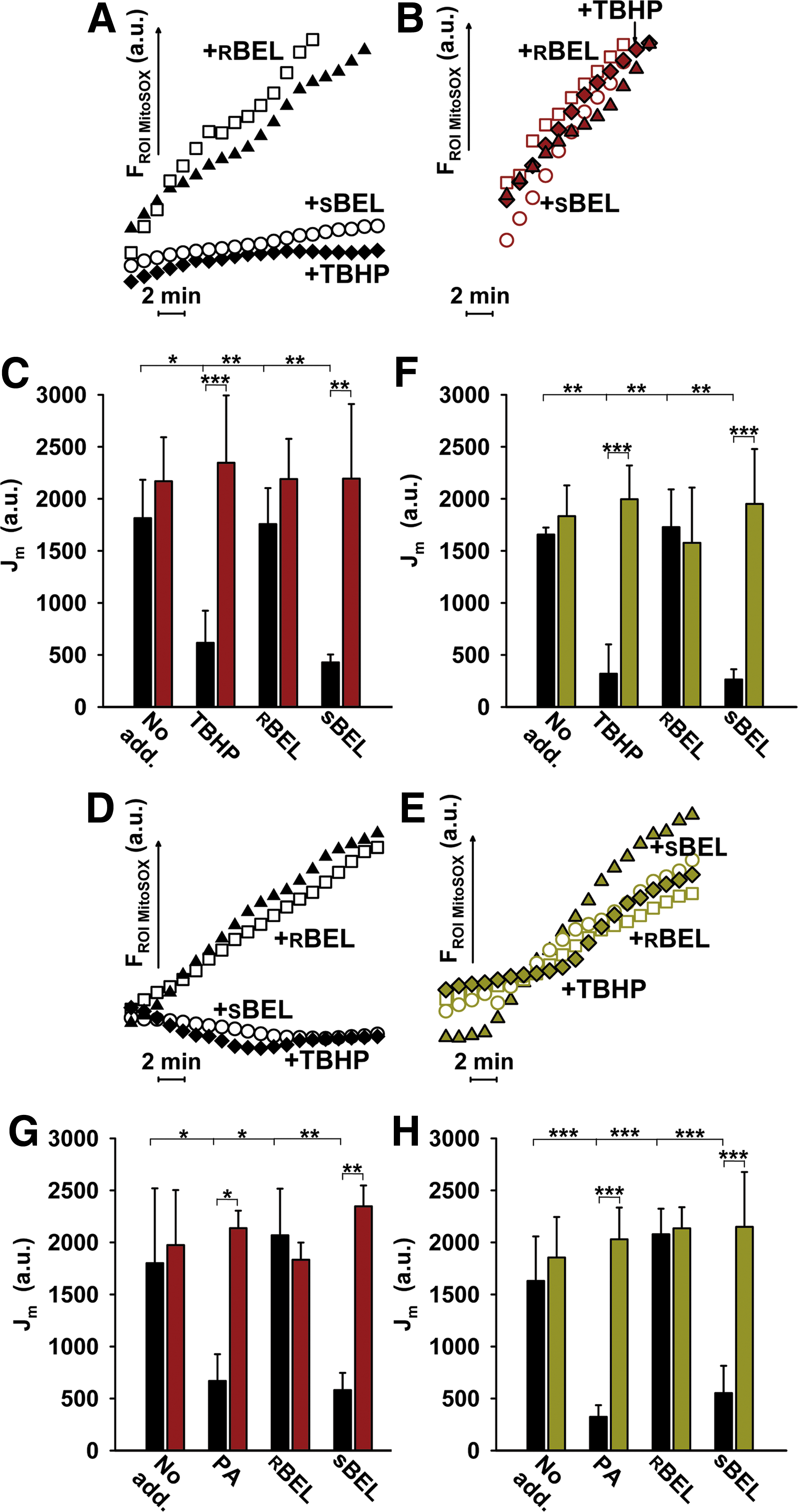
We conclude that iPLA2γ and UCP2 synergy provides a suppression of mitochondrial superoxide production by mild uncoupling initiated by the TBHP-mediated activation of iPLA2γ. Consequently, we predict that in pancreatic β-cells the H2O2-activated iPLA2γ/UCP2 synergy provides an antioxidant mechanism, enabling a feedback downregulation of oxidative stress.
iPLA2γ activation during β-oxidation of palmitate in INS-1E cells
Mitochondrial iPLA2γ was also activated in ntgINS-1E cells by a matrix ROS/H2O2 burst, resulting from the ongoing β
Altogether, these data suggest that FAs instantly cleaved off phospholipids by iPLA2γ to determine the UCP2-mediated uncoupling, and that UCP2 requires such a specific nascent FA delivery (for UCP1 see Ref. 11). This is because no response to PA addition took place in the presence of
A similar antioxidant effect in the mitochondrial matrix (Fig. 3G, H) as seen with TBHP activation of iPLA2γ was found for iPLA2γ activation by H2O2, resulting from the pro-oxidant PA β-oxidation (Fig. 5C). The superoxide release (Jm) rates declined to a minimum on the PA addition (Fig. 3G, H). Such attenuation of mitochondrial superoxide production ceased in the presence of
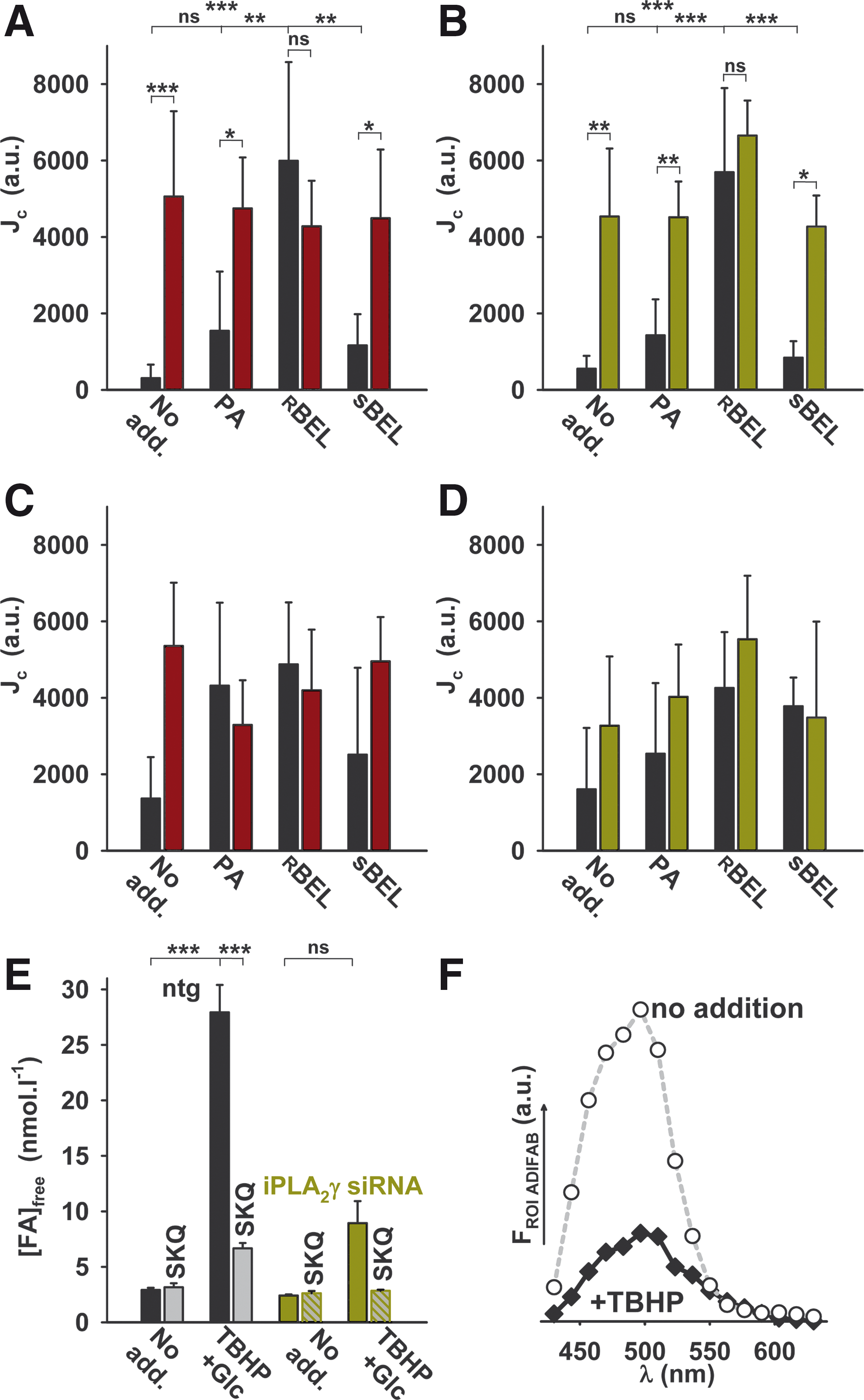
FA-induced uncoupling is prevented by antioxidants
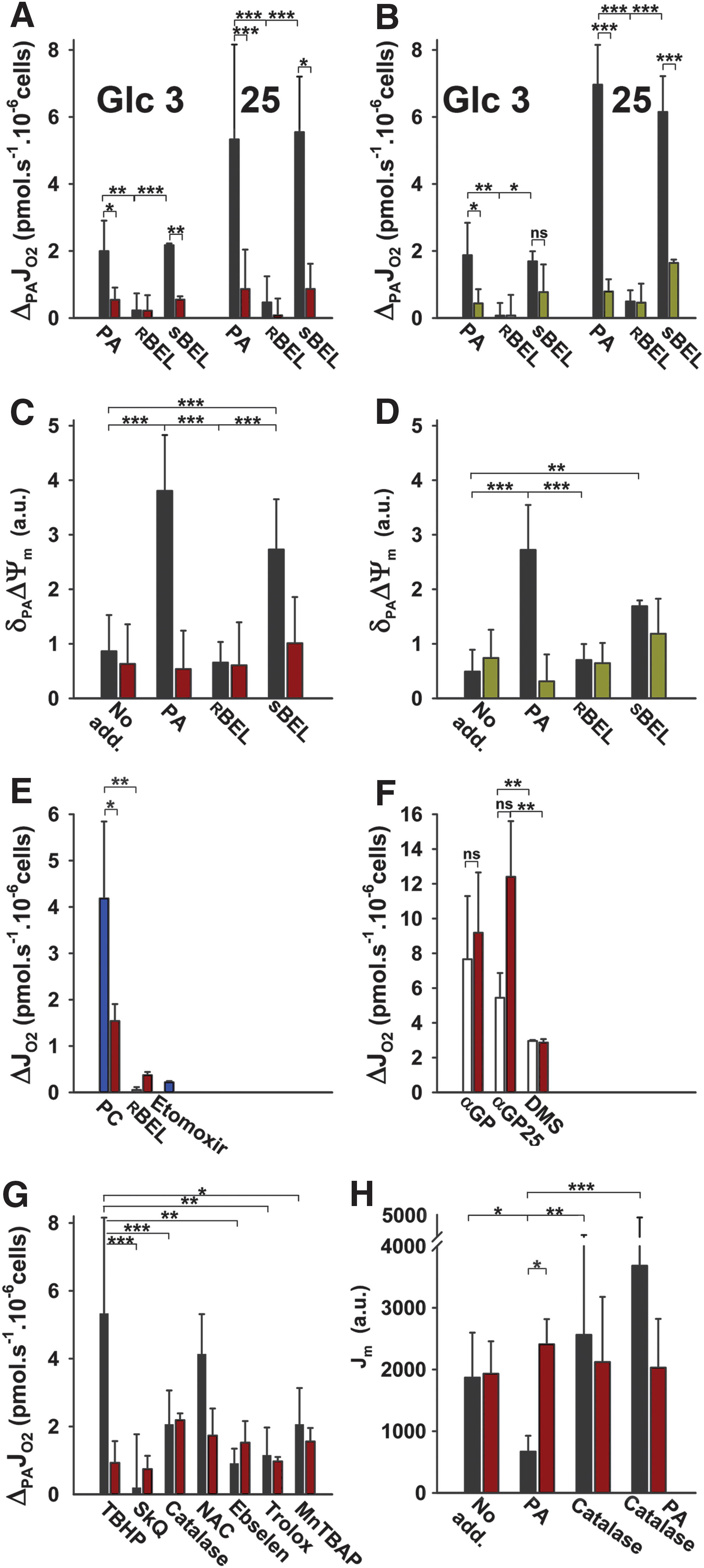
The PA-induced iPLA2γ/UCP2-dependent mitochondrial uncoupling in INS-1E cells at 25 mM glucose was prevented by the matrix-targeted antioxidant SkQ1, by ebselen but not by N-acetyl-
Concomitant redox changes in the cytosol of INS-1E cells
We have also studied redox changes in the cytosol of ntgINS-1E cells and confirmed that both PA β-oxidation (Fig. 5A–D) and, as expected, TBHP additions (not shown) oxidize cytosol, when rates of oxidation (Jc) were indicated by the cytosolic ROS fluorescent probe CellROX immediately after insults (Fig. 5C, D). However, when detected ∼15 min after PA addition, the cytosolic oxidation rates Jc were faster in UCP2- (Fig. 5A) and iPLA2γ-silenced cells (Fig. 5B) when compared with ntgINS-1E cells. This difference ceased in the presence of
Mitochondrial iPLA2γ-cleaved free FAs are accessible for plasma membrane
A confocal microscopy assay with the extracellular fluorescent free FA indicator ADIFAB2 demonstrated FA binding to the extracellular ADIFAB2, which ceased in iPLA2γ-silenced cells (Fig. 5E, F). This demonstrates that FAs cleaved by iPLA2γ in mitochondria can be exported to bind to the extracellular ADIFAB2 and therefore are also accessible to the entire cell cytosol and plasma membrane.
Role of UCP2/iPLA2γ synergy in insulin release
Next, we set to analyze in detail the behavior of the glucose sensor. INS-1E cells with glucose lowered to 3 mM responded to the increasing glucose by elevating the respiration in a saturated manner till the final 25 mM glucose (49). ntgINS-1E cells responded in the same way (Fig. 6A). With the UCP2 silencing, beginning at 9 mM glucose, this relationship was shifted to higher respiration increases (Fig. 6A), indicating that mitochondria lacking UCP2 respond to a higher extent on GSIS. It is known that not only respiration but also ΔΨm increases after glucose addition (49), and this was observed in ntgINS-1E cells (Fig. 6C). Again, these ΔΨm elevations at GSIS were higher in UCP2-silenced cells (Fig. 6C). In contrast, both respiratory (Fig. 6B) and ΔΨm responses (Fig. 6D) to glucose in iPLA2γ-silenced cells were identical to their corresponding ntg controls. We conclude that UCP2 presence in ntgINS-1E cells diminishes respiratory and ΔΨm responses to the added glucose. These responses are intimately inherent to the glucose-sensing function of pancreatic β-cells. Hence, the UCP2 presence sacrifices a small portion of the glucose-sensing capacity (function range) for benefits of antioxidant protection. The simplest reasoning predicts that UCP2 does it via a mild uncoupling of mitochondria at state-3 (i.e., at phosphorylating state). This is supported by the observed, slightly increased respiration on UCP2 silencing.
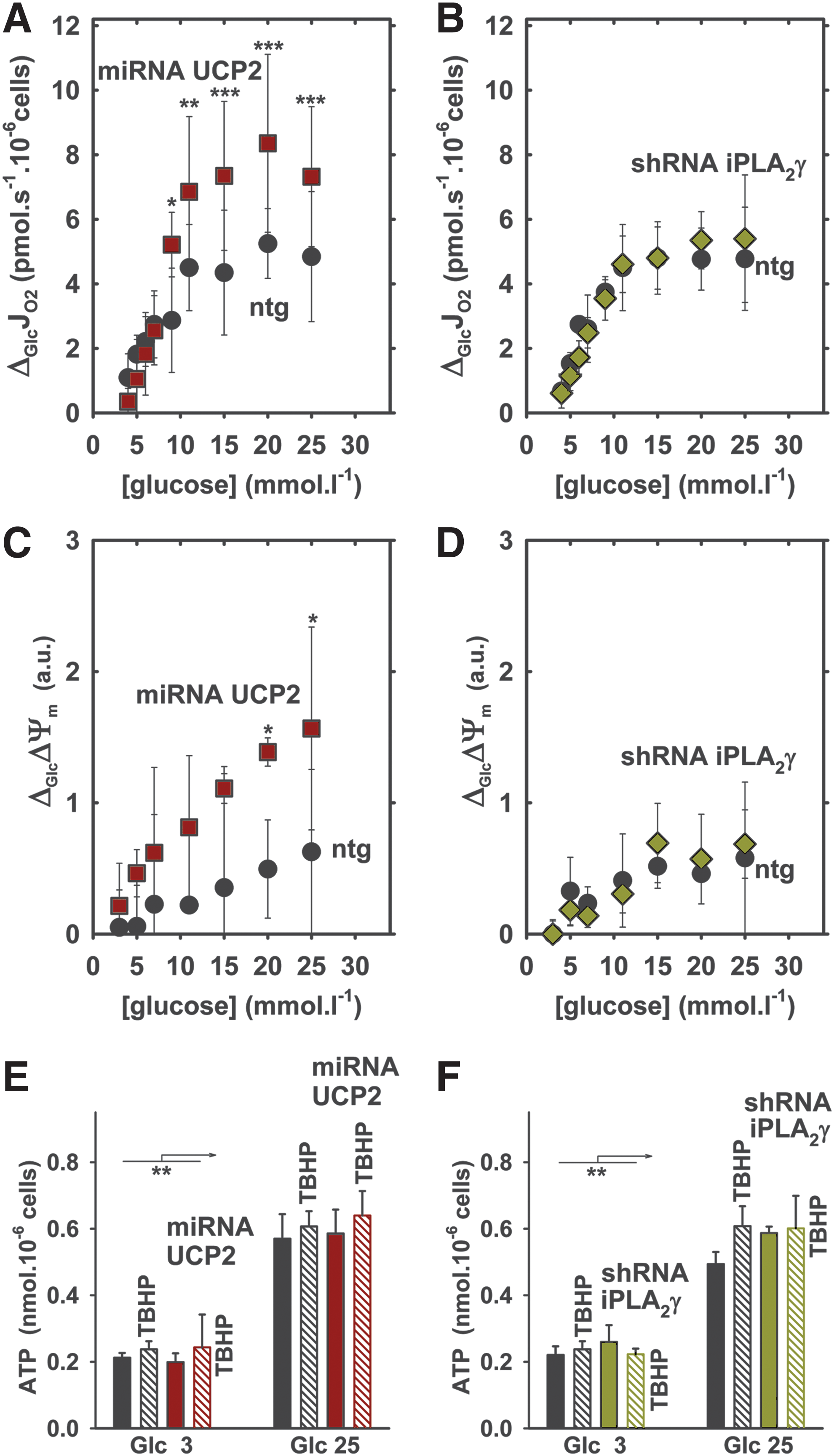
However, the rates of glucose-stimulated insulin release in UCP2- or iPLA2γ-silenced INS-1E cells were equal to ntgINS-1E controls (Fig. 7A). In the presence of TBHP addition, nonlinear (biphasic) insulin release was initially faster and attained higher magnitudes 10–60 min after glucose addition to UCP2- and iPLA2γ-silenced INS-1E cells (Fig. 7B), demonstrating that unchanged and lower-magnitude insulin release rates in ntgINS-1E controls are depressed (45) in the presence of a pro-oxidant due to the iPLA2γ/UCP2-initiated uncoupling, similarly as previously observed (48). Hence, a portion of maximum possible insulin-secreting capacity is sacrificed for benefits of antioxidant protection. But independently of TBHP presence, ATP cell levels were equal 60 min after glucose addition, to all studied cells, to ntgINS-1E, UCP2- (Fig. 6E) and iPLA2γ-silenced cells (Fig. 6F). This implies that mild uncoupling is not able to substantially block OXPHOS, hence ATP synthesis in the long term. It should be noted, however, that TBHP alone accelerated insulin release in the absence of glucose, causing the redox-stimulated insulin secretion (RSIS) (cf. dashed fits in Fig. 7B vs. 7A).
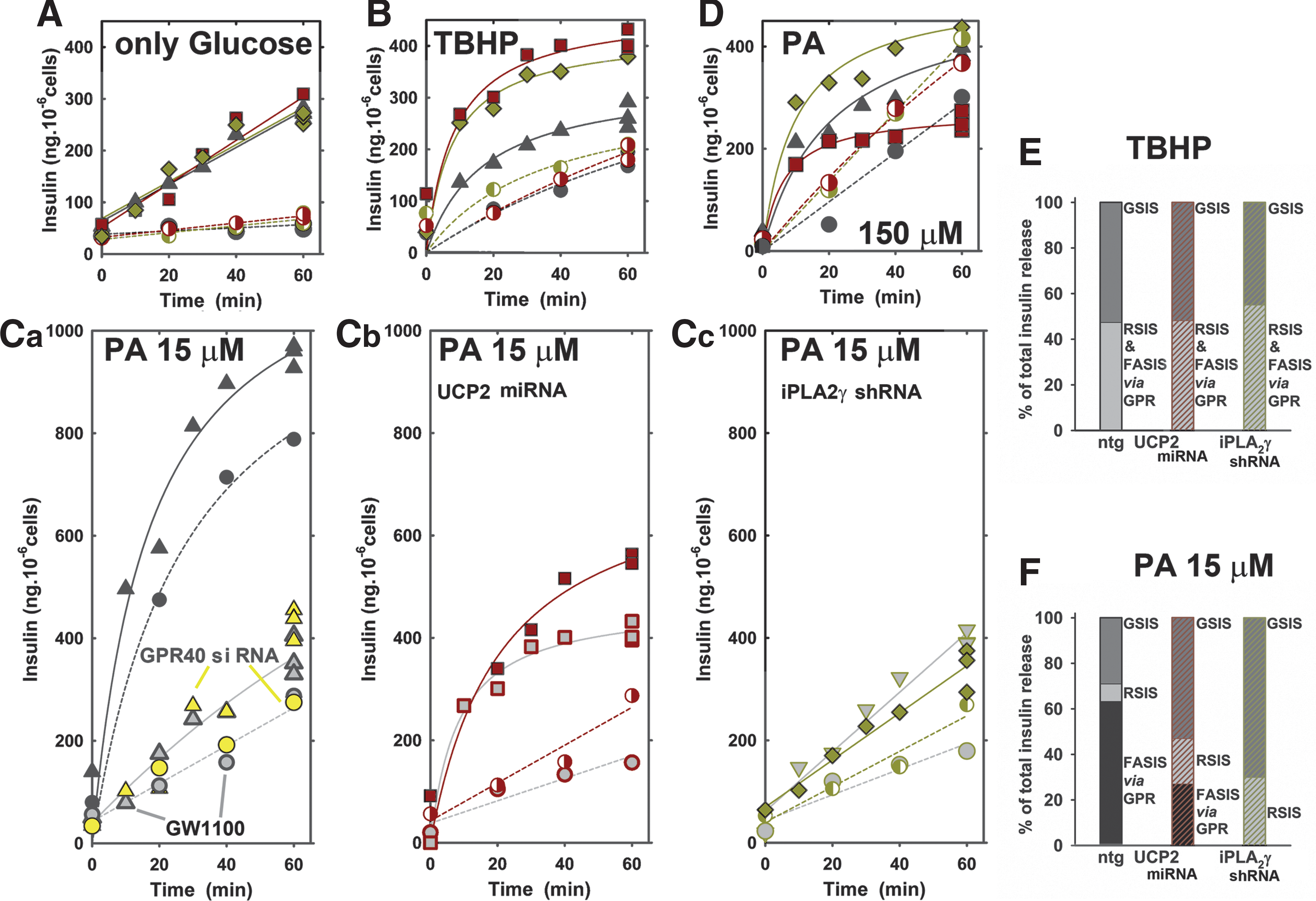
Similar to TBHP, but to a much higher extent, externally added PA extremely elevated insulin release (also together with H2O2/ROS generated during its pro-oxidant β-oxidation). PA induced higher rates of insulin release and its nonlinear time course even in the absence of glucose in ntgINS-1E cells (Fig. 7Ca, black dashed fit). This was not altered in cells with silenced phospholipase D (Supplementary Fig. S4). Surprisingly, in UCP2- and iPLA2γ-silenced INS-1E cells, PA promoted much less of the glucose-insensitive insulin release, regarding to an extent and also rates, for iPLA2γ-silenced cells (Fig. 7Cb, Cc, dark red dashed or green dashed fit). At 15 μM PA (∼4 pmol×10−6 cells), glucose only slightly further increased the already fast insulin release in ntgINS-1E and also the relatively slower insulin release in iPLA2γ-silenced cells (Fig. 7Cb, Cc dark red or green fit). In UCP2-silenced cells, 15 μM PA with glucose promoted an insulin release extent, attaining ∼50% of levels in ntgINS-1E cells after 60 min. It should be noted that at 15 μM PA, the majority of PA is bound to the albumin present and partitioned into membranes, whereas only ∼1.3 nM PA was free in our experiment, representing a concentration comparable to normal plasma levels. The time course was similar to that with the TBPH pro-oxidant insult (Fig. 7Cb vs. 7B). Similar results were obtained with 30 μM PA (not shown). To simulate acute lipotoxicity, we used a much higher PA (150 μM; exceeding 200 nM free PA). Here, the net GSIS was reduced down to 25% (Fig. 7D), indicating such an acute lipotoxicity that overcomes the stimulating effect of PA. As a result, the insulin release in the absence of PA was equal to the one with PA in UCP2- and iPLA2γ-silenced cells.
Redox-activated iPLA2γ supplies FAs for GPR40-stimulated insulin release
Surprisingly, the GPR40, previously indicated to act in insulin-stimulating responses (11), turned out to be a major player in excessive rates of PA–insulin release in ntgINS1-E cells (Fig. 7Ca). This conclusion stems from the finding that in GPR40-silenced cells (Fig. 7Ca, yellow symbols) and in ntgINS1-E cells subjected to the GPR40 antagonist GW1100 (Fig. 7Ca, gray symbols) the PA-induced insulin release was profoundly inhibited to the levels induced by TBHP (Fig. 7B) in both the absence and presence of glucose. In UCP2-silenced cells, where intermediate PA-induced insulin release rates versus ntg controls were assessed, GW1100 also slightly inhibited the PA-induced insulin release (Fig. 7Cb). In contrast, in iPLA2γ-silenced INS-1E cells, GW1100 had no effect in the presence of glucose and its effect was very small in the absence of glucose (Fig. 7Cc).
Taken together, we interpret these results as follows: In the situation where, promoted by sole FA β-oxidation, ROS activate insulin release in the absence (the net RSIS; Fig. 7E, F) or presence of glucose (RSIS plus GSIS, Fig. 7E, F), the lack of iPLA2γ evidently causes that no further FAs are cleaved, in contrast to the ongoing cleavage in ntg or UCP2-silenced cells. In ntg cells, it is not the added PA that stimulates GPR40, but FAs cleaved off phospholipids by iPLA2γ. Lysophosphatidic acid is excluded from GPR40 stimulation and subsequent insulin secretion, since silencing of phospholipase D did not change the observed fatty acid–stimulated insulin secretion (FASIS; Supplementary Fig. S4).
Discussion
Chronic lipotoxicity is a situation of persistent oxidative stress in pancreatic β-cells originating from the long-term intake of excessive FAs and lipid peroxidation products, which leads to GSIS impairment (15, 19). At low doses, FAs, however, stimulate insulin release (Fig. 7C) (15). Studying immortalized but noncancer pancreatic β-cells, INS-1E cells (37), we demonstrated that a mild acute oxidative stress originates from the β-oxidation of FAs. This elevates superoxide in the mitochondrial matrix as well as H2O2 in the cytosol and simultaneously initiates a feedback downregulating mechanism that is capable of counteracting such a repeatable ROS burst given by postprandial physiological acute FA intake. Moreover, a different spectrum of FAs cleaved off mitochondrial phospholipids by iPLA2γ can spread within the cell serving as another messenger (as revealed by the ADIFAB2 assay, Fig. 5E, F).
The antioxidant mechanism is provided by a synergy between H2O2-activated mitochondrial iPLA2γ and UCP2. The latter provides mild uncoupling, which attenuates the otherwise excessive superoxide production, the one resulting from β
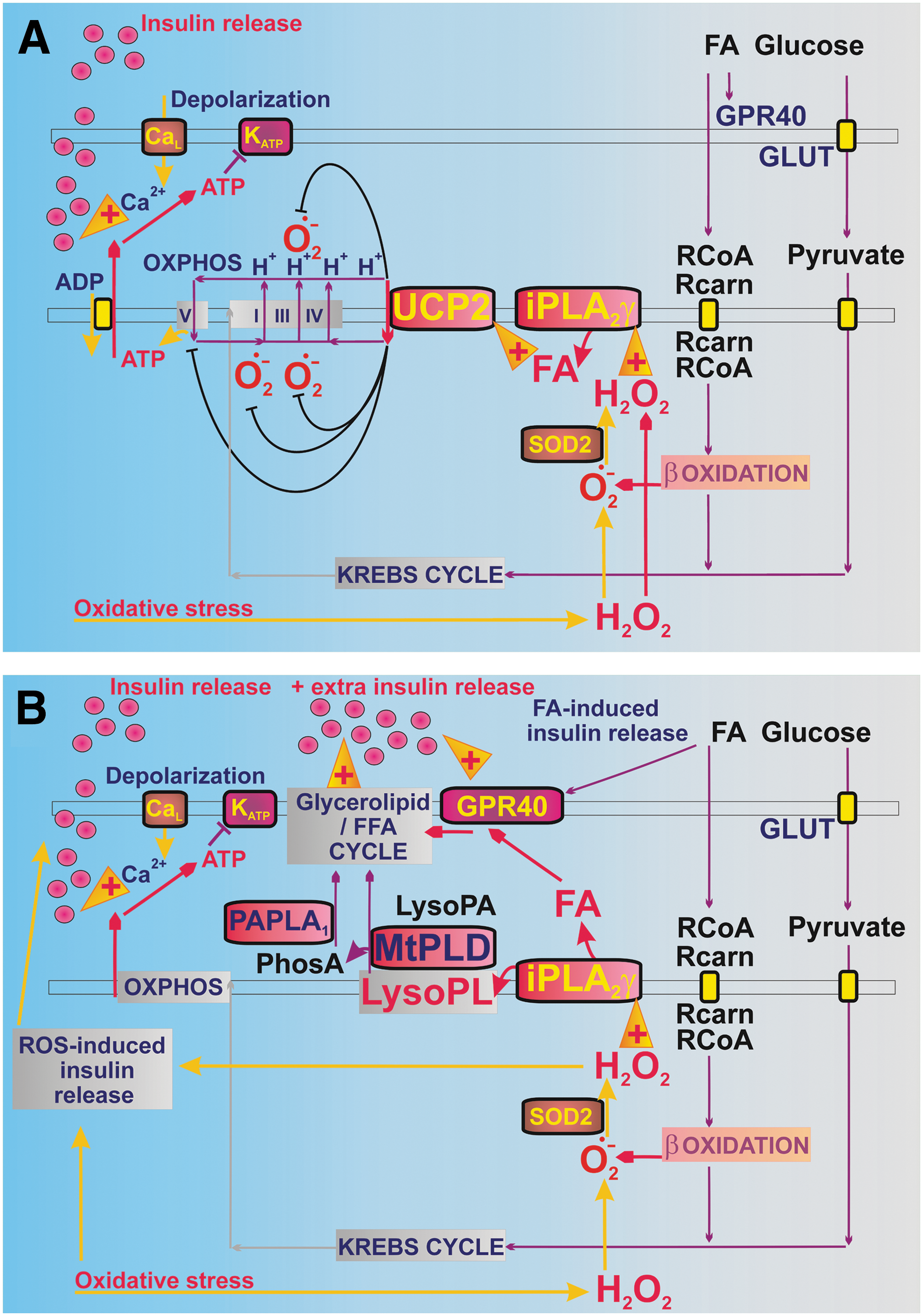
The substantial extent of respiration and insulin release (48, 54) are sacrificed and utilized for such antioxidant function. The respiration extent of the β-cell glucose sensor corresponds to ∼30% of state-3 respiration (Fig. 6A), whereas ∼15% is sacrificed for antioxidant mild uncoupling (Fig. 6A, differences between ntg and UCP2–silenced cells). Hence, till 50% of maximum sensor capacity is utilized for the antioxidant role. Deriving from elevations of insulin secreted after 40–60 min with TBHP in UCP2-silenced versus ntg cells (Fig. 7A, B), till 50% insulin surplus released in the absence of the antioxidant mechanism is also sacrificed for antioxidant protection. Nevertheless, the pro-oxidant impulse enhances insulin release rates in the absence of glucose (Fig. 7B, C) (31, 42). Details of insulin release at the glucose or FA intake should be further studied within the more complex system of the Langerhans islets possessing numerous autocrine and paracrine interrelationships. They evoke certain limitations of our in vitro system used.
Rather complex results were obtained for FASIS or FASIS combined with GSIS (Fig. 7Ca, E, F). At low (15 μM, corresponding to 1.3 nM free) PA, the overall FASIS+GSIS is lower in UCP2- and iPLA2γ-silenced cells versus ntg cells, in spite of the tighter mitochondrial coupling versus ntg cells. The two components are involved in FASIS. The first one stems from the insulin release activated via the GPR40 pathway (11) (Fig. 7C). The second one represents the net RSIS and is solely manifested in iPLA2γ-silenced cells without glucose. The GPR40 pathway of insulin secretion can be activated and is strongly amplified (Fig. 8B) by FAs cleaved off phospholipids by iPLA2γ. Lysophospholipids remaining after the iPLA2γ reaction could be hypothetically cleaved by specific mitochondrial phospholipase D (mtPLD) (14), but as demonstrated this does not affect FASIS (Supplementary Fig. S4). FAs are involved in the glycerol lipid/FA cycle signaling stimulating insulin secretion (44).
The iPLA2γ is most likely initially activated in vivo by the pro-oxidant β-oxidation of FAs incoming from the blood. During FA β-oxidation, activation of iPLA2γ by H2O2 burst arises from the electron-transfer flavoprotein:ubiquinone oxidoreductase (41, 46, 50) and SODs activities. The situation resembles a self-accelerating cycle, since more active iPLA2γ provides more FAs for further β-oxidation, which further elevates ROS/H2O2. Besides iPLA2γ, these surplus ROS/H2O2 themselves trigger insulin secretion and, in parallel, persistently cleaved FAs promote GPR40 activation. All these contributors add on the resulting overstimulation of insulin secretion (Fig. 7Ca), which is permanent (unlike with a single bolus of TBHP) until the added PA is metabolized. In its intermediate state, certain overstimulation is also possible with ablated UCP2 but logically not with ablated iPLA2γ. We estimated that at 15 μM PA plus glucose, the insulin release induced during 60 min consists of ∼30% net GSIS, <10% net RSIS, and 60% GPR40 pathway (Fig. 7F). The activation of the GPR40 pathway vanished with ablated iPLA2γ. Without UCP2, hence at a more coupled state without antioxidant protection, the net GSIS increases to >50% and RSIS is doubled (Fig. 7F). Similar 50% net GSIS existed with TBHP (Fig. 7E), inducing the net RSIS in the absence of glucose.
We also conclude that UCP2-mediated mild uncoupling is initiated only when free FAs are directly cleaved off the phospholipids at the UCP2 vicinity (7) by iPLA2γ (for UCP1 see Ref. 12). In other words, only the nascent FAs are capable of initiating UCP2-mediated uncoupling and a simple PA addition to cells is insufficient (Fig. 4B). FAs are considered the UCP2 cycling/wobbling substrates/cofactors, respectively, essential for their uncoupling function (6, 7, 12, 18, 20, 53).
Demonstrating UCP2/iPLA2γ antioxidant synergy and initiation of UCP2-mediated mild uncoupling by a nascent FA, we have resolved some contradictions that have been reported during the past decade for UCP2 role in pancreatic β-cells. Various reports of ROS-activated UCP2 function (1, 28) can be alternatively explained by the ROS-initiated iPLA2γ activation providing FAs for UCP2. Previously, a mild uncoupling in mitochondria isolated from INS-1E cells was also linked to UCP2, while accounting for approximately 30% of H+ leak (1, 2). Unlike negative demonstrations (13), we now show that UCP2 is activated exclusively by nascent FAs. This ensures that the net glucose sensing in pancreatic β-cells is not substantially affected by mild uncoupling, unless acute lipotoxicity and/or excessive oxidative stress activate the nascent FA delivery toward UCP2 by iPLA2γ. One would predict that the lack of either UCP2 or iPLA2γ must result in an easier reach of the oxidative stress threshold from which a pathological outcome is initiated. These characteristics were reported for UCP2 ablation (29, 42, 48).
The unique feature of the iPLA2γ protective role is represented by the fact that iPLA2γ is not constitutively active. Hence, without redox signaling or excessive oxidative stress (such as resulting in acute lipotoxicity), iPLA2γ does not contribute to mitochondrial uncoupling and to the diminished insulin release (Fig. 7A, B). In turn, when activated, its synergy with UCP2 leads to the antioxidant protection at the expense of insulin release. Unless redox activated, no other mitochondria-localized PLA2 isoform can substitute iPLA2γ in these roles (21, 25). We excluded redox activation of iPLA2β, previously found in β-cell mitochondria (34).
In conclusion, our results elucidated iPLA2γ- plus UCP2-dependent regulation of mitochondrial superoxide production and insulin release in pancreatic β-cells. We describe a mechanism of free nascent FA-mediated antioxidant function, due to the synergic action of iPLA2γ and UCP2, counteracting mild sub-threshold lipotoxicity in pancreatic β-cells. The iPLA2γ-cleaved FAs may also participate in the lipid-mediated signaling such as in amplification of FA-induced insulin release via the GPR40 pathway.
The revealed redox activation of iPLA2γ and the requirement of nascent FAs for UCP2-mediated uncoupling ensure that the glucose sensing in pancreatic β-cells is not substantially affected by mild uncoupling, unless acute lipotoxicity and/or excessive oxidative stress activate the iPLA2γ/UCP2 antioxidant synergy. This synergy developed as an inherent antioxidant, cytoprotective, and specifically anti-lipotoxic mechanism that is protective before reaching a critical threshold and provides defense against long-term dysbalanced conditions, such as lipotoxicity. When these pro-oxidant impulses (stimulating otherwise antioxidant protection) are compromised, the inevitable self-accelerating vicious cycle leads to permanent oxidative stress, cell dysbalance, and the progression of type-2 diabetes pathology (15, 23). This progression has to be further elucidated and extended to future translational research.
Materials and Methods
Chemicals
Cell cultivation
Rat insulinoma INS-1E cells were a kind gift from Prof. Maechler, University of Geneva (37). Cells were cultivated with 11 mM glucose in RPMI 1640 medium with 2 mM
Cell transfections and silencing
A BLOCK-iT Pol II miR RNAi system (Life Technologies, Carlsbad, CA) has been employed to express miRNAs against UCP2. The two miRNA sequences, 5′-TACAGA GTCGTAGAGGCCAATGTTTTGGCCACTGACTGACAT TGGCCTACGACTCTGTA-3′ and 5′-ATTTCGGGCAAC ATTGGGAGAGTTTTGGCCACTGACTGACTCTCCCAA TTGCCCGAAAT-3′, were designed using the BLOCK-iT RNAi Designer and annealed into double-strand oligonucleotides, which were inserted into the linearized miRNA expression vector pcDNA6.2-GW/EmGFP-miR. Individual miRNAs were chained up into tandem constructs. The vector with UCP2-miRNA or control miRNA plasmids was cloned by Gateway BP/LR reaction into pLenti6.2/V5-DEST expression vector, and final constructs were validated by DNA sequence analysis. Lentiviral expression plasmids were co-transfected with the ViraPower™ Packaging Mix (Life Technologies) into the 293LTV cells using Lipofectamine 2000 (Life Technologies). The lentiviral stock was used to transduce INS-1E cells, followed by selection of a stably transduced cell line by blasticidin and verification by a green fluorescent protein (GFP) inherent cytosolic reporter.
For catalase overexpression (17), pZeoSV2(+) vector bearing human catalase sequence (gift from C. Glorieux and Prof. J. Verrax, Université Catholique de Louvain, Belgium) was transiently transfected into INS-1E cells using Lipofectamine 2000 (2.5 ml·mg−1 DNA).
To silence iPLA2γ or iPLA2β, pGFP-V-RS vectors (Origene, Rockville, MD) as specific expression cassettes were used with inserted 29-mer shRNAs, 5′-CAAACACTGGC ACTCTTCAAGCAACCATGTCAAGAGCATGGTTGCT TGAAGAGTGCCAGTGTTTG-3′ or 5′-CAGTAGTGTCA CCAACTTGTTCTCAAACCTCAAGAGGGTTTGAGAAC AAGTTGGTGACACTACTG-3′, respectively. The same vectors with inserted scrambled shRNA expression cassettes served as negative controls. INS-1E cells were transiently transfected using Lipofectamine 2000 (2.5 ml·mg−1 DNA) and verified by the GFP cytosolic reporter. For GPR40 (FFAR1) or phospholipase PLD6 silencing, we used Silencer Select siRNAs (Life Technologies), cat. No. 4390771, IDs s141419 and s141420; or IDs s142381 and s142382. They were used in comparison versus negative control siRNA (cat. number 4390843). RNA oligonucleotides (20+20 pmol of siGPR40 vs. 40 pmol of negative control) were used.
High-resolution respirometry and fluorescent assays for ΔΨm and ATP monitoring
Cells were trypsinized, counted by a Countess Automated Cell Counter (Life Technologies), and adjusted to 2×106 cells/ml. Respiration was detected by an Oxygraph 2k (cells at 37°C, isolated mitochondria at 30°C; Oroboros, Innsbruck, Austria). Rates taken 3 min after sequential additions of glucose, TBHP, or PA (when applied) were corrected for KCN-insensitive respiration. Five micromolars of tetramethylrhodamine ethyl ester (TMRE; Life Technologies) added immediately before the assay were used for ΔΨm monitoring on an RF 5301 PC spectrofluorometer (Shimadzu, Tokyo, Japan) in a stirred 2 ml cuvette with excitation at 561 nm (3 nm slit) and emission at 579 nm (5 nm slit). Trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP) was titrated to a maximum (respiration) or added twice (ΔΨm). ATP quantification was performed using ATP Assay bioluminescence kit HSII (Roche, Basel, Switzerland) and the spectrofluorometer at 557 nm (5 nm slit).
Confocal microscopy
A confocal inverted fluorescent microscope Leica TCS SP2 AOBS was employed, with a PL APO 100×/1.40–0.70 oil immersion objective (pinhole 1 Airy unit) and a sample chamber set to 37°C supplied with 5% CO2. Cells were seeded on poly-
Confocal microscopy monitoring of cytosolic ROS and extracellular FAs
Cytosolic ROS monitoring was ensured by 10 μM CellROX (Life Technologies) after 30 min loading, while excited at 633 nm with a 1.2 mW HeNe laser and collecting emission between 650 and 700 nm. An ADIFAB2 FA indicator (FFA Sciences, San Diego, CA) was used to monitor extracellular free FAs, while excited with a 405 nm diode laser and collecting emission between 430 and 645 nm.
Confocal microscopy monitoring of superoxide released into mitochondrial matrix
A triphenylphosphonium-conjugated dihydroethidine, MitoSOX Red (Life Technologies), was used to monitor rates (Jm) of in situ surplus superoxide release into the mitochondrial matrix (10). The surplus represents the portion of superoxide not neutralized by the matrix Mn-SOD. Using rates, any variations in initial background fluorescence are eliminated and the method is feasible for semiquantification of matrix-released superoxide even at low or collapsed ΔΨm (10). Excitation was at 488 nm by a 20 mW Argon laser, with emission collected between 550 and 650 nm, while the GFP reporter was checked between 508 and 516 nm. Cells were loaded with 4 μM MitoSOX for 15 min. A series of confocal images were taken every 30 s for 20 min (Supplementary Movie S1). Regions of interest corresponding to mitochondria were selected using the Ellipse software (ViDiTo, Košice, Slovakia). Changes of integrated fluorescence intensity were quantified from plots of fluorescence in the selected areas versus time.
Assay for insulin release
Cells were seeded at 0.4×106 cells/well in poly-
SDS-PAGE and Western blotting
Silencing was verified by SDS-PAGE followed by Western blotting on mitochondria isolated from cells and using primary antibodies against UCP2 (No. 662047; Merck Millipore) or in cell lysates for iPLA2γ (PNPLA8 N-terminal., AP4706a; Abgent, San Diego, CA); otherwise, ATP-synthase β-subunit (ATPB, ab14730; Abcam, Cambridge, MA), TIM23 (611222; BD Biosciences, Franklin Lakes, NJ), β-actin (ab3280; Abcam), and horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit secondary antibody, respectively (Sigma) were used. Blots were visualized using an LAS-1000 Imager, and quantitative image analysis was performed with ImageJ software.
iPLA2γ reconstitution
The recombinant human iPLA2γ was reconstituted into liposomes by adopting published protocols (20). The enzyme was purchased (PNPLA8; Novus Biologicals, Littleton, CO) or was cell expressed from a vector encoding His-tagged enzyme (GeneCopoeia, Rockville, MD) and affinity purified using a nickel resin column (Qiagen, Germantown, MD). Ten microgram per milliliter of Novus protein was dissolved in 1% n-lauroylsarcosine, passed through a Sephadex G-25-300 column, and equilibrated with an assay medium plus 0.1% n-octylpentaoxyethyelene (Bachem, Bubendorf, Switzerland). One hundred microgram per milliliter of the affinity-purified protein had to be treated with 10 mM DTT and dialyzed against the assay medium, in order to reversibly modify the protein to its reduced form. A Sephadex G-25-300 column (0.5% n-octylpentaoxyethyelene) was used to remove DTT. Aliquots in μg·(mg lipid)−1 (0.04 Novus protein; 0.4–0.8 affinity-purified protein) were mixed with phospholipids (Escherichia coli total lipid extract supplemented with 20% bovine heart cardiolipin; Avanti Polar Lipids, Alabaster, AL), to yield 28% cardiolipin, 46% phosphatidylethanolamine, 12% phosphatidylglycerol plus 14% other lipid, and 1 mg·(mg lipid)−1 n-octylpentaoxyethyelene. Proteoliposomes were formed by detergent removal using Bio-Beads SM-2 (Bio-Rad, Hercules, CA). Both the intraliposomal and assay medium contained Na+ salts of TES (50 mM), SO4 2− (80 mM), and EDTA (2 mM), pH 7.5. Sulfopropylquinolinium-based monitoring of FA release into the liposome interior or GC/MS analyses of cleaved FAs was performed.
Quantification of free FAs
Free FAs released in liposomes or cells were quantified using GC/MS (6890 gas chromatograph, 5973 mass spectrometer; Agilent Technologies, Palo Alto, CA). Reaction mixtures were extracted with acidic 2-propanol/n-heptane, treated with ether/diazomethane at room temperature for 20 min, and evaporated under argon. The methylated FAs were reconstituted in 100 μl of n-heptane and analyzed by GC/MS. FAs were identified/quantified in comparison with spectra of purified standards. FA total content in phospholipids was evaluated after hydrolysis by 0.5 M NaOH at 60°C for 30 min, and after acidification and extraction.
Footnotes
Acknowledgments
This project was supported by grants of the Grant Agency of the Czech Republic (P303/11/P320 to J.J.; 15-15-02051S to M.J.; 13-02033 and 13-06666 to P.J.) and by the research project RVO67985823. The authors gratefully acknowledge the excellent technical assistance of Lenka Josková with cell cultivation, Jana Vaicová with immunoblotting and ELISA, and Jitka Smiková with plasmid isolations. The authors express their gratitude to Prof. Libor Vítek, M.D., Ph.D., from the First Faculty of Medicine, Charles University, Prague, for kindly providing them with GC/MS and to Prof. Vladimir Skulachev, from Moscow University, Russia, for providing them with SkQ1.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.