
Abstract
Neurotransmitter Glutamate
I
Under physiological conditions, release and uptake of neurotransmitter glutamate occurs in a tightly controlled manner within the boundaries of a tripartite glutamate synapse that consists of pre- and post-synaptic neuronal terminals as well as synapse-associated astrocytes (4) (Fig. 1). Glutamate release from pre-synaptic vesicles is primarily triggered by the depolarization of synaptosomes via N-type voltage-gated Ca2+ channels. After acting upon post-synaptic receptors that include N-methyl-
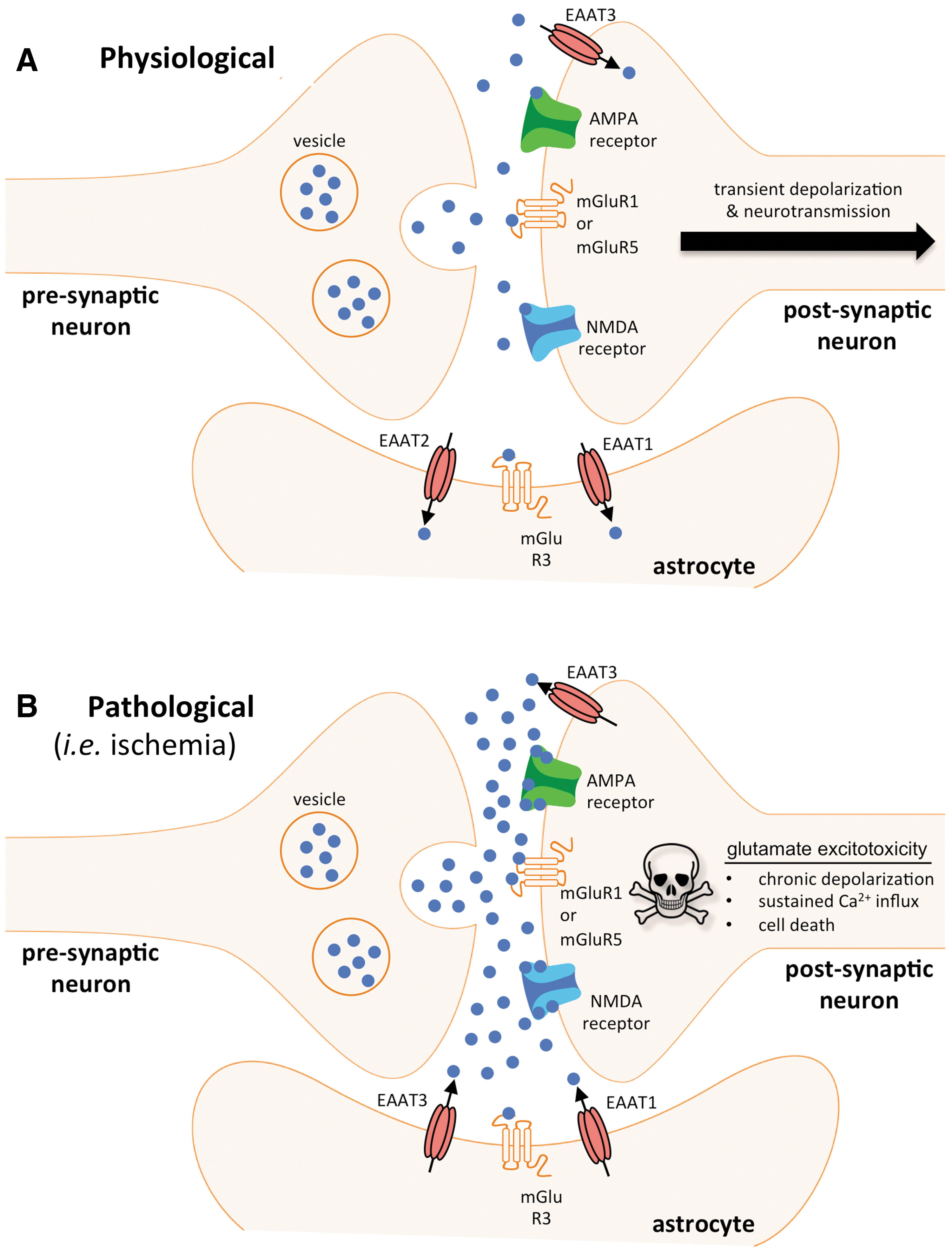
Hallmarks of Ischemic Stroke Pathophysiology and Glutamate Release
In spite of only accounting for 2% of total body weight and not performing the metabolically demanding mechanical work of skeletal muscle, the energy demands of the brain are among the highest of all organs (73). Under normal physiological conditions the metabolic activity of the brain requires 15% of cardiac output (800 ml/min) and 20–25% of total oxygen (49 ml/min) intake (73, 81). The majority (∼60%) of aerobic oxidation of glucose in brain tissue is requisite for the maintenance and restoration of ionic gradients used for neuronal membrane depolarization and repolarization (81). To that end, neurons preferentially derive their energy for functional activity from the oxidation of glucose. Unlike astrocytes, neurons lack glycogen stores. Glycogen synthase in neurons is highly phosphorylated, rendering it inactivated and targeted for proteaosome-dependent degradation. Furthermore, neurons do not express detectable levels of glycogen phosphorylase, the key enzyme for glycogen degradation (10, 95, 98). Without glycogen stores, adenosine triphosphate (ATP) levels in neurons are rapidly depleted during ischemic stroke when blood supply carrying oxygen and glucose to brain tissue is arrested. Within 10 min of cerebral ischemia, intrasynaptosomal ATP levels acutely drop by >95%, with the ATP to adenosine diphosphate ratio declining from 6.06±0.20 under physiological conditions to 0.11±0.02 during ischemia (77). During this time, ATP-dependent ion pumps (i.e., Na+/K+ adenosine triphosphatase) fail to maintain transmembrane ion gradients resulting in Ca2+ influx through voltage-sensitive Ca2+ channels. Elevated intracellular Ca2+ is known to trigger a rapid pre-synaptic glutamate efflux that results in excessive activation of NMDA receptors, further perturbation of ion homeostasis, cell swelling and necrotic death (46). The resulting over-activation of NMDA receptor mediates the formation of neurotoxic receptor/protein complexes (90). Activated NMDA receptor is linked to nitric oxide synthase via postsynaptic density-95 and causes neurotoxic nitric oxide production (78). Excitotoxic activation of calpains (30) only serves to exacerbate the problem by cleaving the Na+/Ca2+ exchanger, thereby preventing Ca2+ extrusion and contributing to lethal Ca2+ overload (8, 9). Stroke-associated anoxia further potentiates ionic imbalance via transient receptor potential melastatin (TRPM) channels. Calcium permeable TRPM7 is activated during cerebral ischemia and has been proposed to mediate neural cell death by oxidative stress-dependent mechanisms (1, 2).
In addition to Ca2+-dependent mechanisms, there are also Ca2+-independent mechanisms that exacerbate the neurotoxic accumulation of glutamate at the synapse. The depolarizing loss of Na+ and K+ gradients across neuronal and glial membranes due to ATP depletion leads to reverse transport by EAATs. EAAT uptake of extracellular glutamate occurs against a large concentration gradient that is coupled to the co-transport of three Na+, one H+, and anti-transport of one K+ ion. Under conditions of acute ischemic stroke when ionic gradients are disrupted, EAAT transporter function is reversed such that glutamate moves down its concentration gradient, from intracellular to extracellular space. Beyond the acute response for EAATs to reverse glutamate transport during stroke, cerebral ischemia produces long-term changes in glutamate transporter expression as well. Twenty-four hours after transient middle cerebral artery occlusion (tMCAO), messenger ribonucleic acid (mRNA) and protein expression of astrocyte glutamate transporter EAAT2 is reduced >50% in the stroke-affected cortex as compared to control (no stroke) cortex (42).
The major source of non-synaptic extracellular glutamate in the brain is the cystine/glutamate antiporter (system xc−). System xc− is predominantly expressed in microglia, where the extracellular export of glutamate is coupled to the import of cystine (28). Once imported, cystine is converted into cysteine as the rate-limiting substrate required for synthesis of the endogenous antioxidant glutathione. However, under conditions of oxygen glucose deprivation (OGD), system xc− can contribute to the pathological release of extracellular glutamate as well (7). Indeed, recent evidence supports that the pharmacological inhibition of system xc− attenuates OGD-induced neural cell death in vitro, and ischemic cell death in cerebral cortex organotypic slices (86).
Taken together, the aforementioned Ca2+-dependent and independent mechanisms contribute to a rapid and sustained rise in extracellular glutamate concentration after the onset of cerebral ischemia. Dialysis studies in rats subjected to tMCAO report a 227% increase in extracellular glutamate after only 2-min of cerebral ischemia (96). After 1 h of MCAO, extracellular glutamate levels increase more than 31.5-fold over baseline where they are sustained for up to 3 h (96).
Pre-clinical in vivo testing has identified a threshold-type relationship between extracellular glutamate release and cerebral blood flow (CBF). In cats subjected to global ischemia, excessive glutamate release was reported to occur in cerebral cortex when relative CBF decreased to 36% of baseline (82). Importantly, this value is within the range of clinical CBF thresholds for patients with large-vessel stroke that developed cerebral infarctions (27–44% of baseline CBF) (53). These outcomes highlight the sensitivity of brain tissue to ischemic injury and glutamate-mediated excitotoxicity. In the context of acute ischemic stroke, glutamate release and neuronal cell death occur not only at the site of CBF occlusion, but also in adjacent tissues (i.e., penumbra) where low-flow is encountered. In totality, the pathological consequence of brain injury caused by acute ischemic stroke is particularly devastating to neurons that are highly sensitive to metabolic disturbance. Supratentorial cerebral ischemia, for example, is estimated to result in the loss of 1.9 million neurons, and 14 billion synapses per minute (80).
Therapeutic Targeting of Glutamate-Mediated Neuronal Cell Death
As scientific research to elucidate glutamate's contribution to stroke pathophysiology has grown considerably over the past 30 years, so have research efforts on therapeutic strategies that target glutamate-mediated neuronal cell death. While many of theses interventions have shown promise in cell culture and pre-clinical animal studies, none to date have translated into clinical success. Indeed, neuroprotective agents as a whole have largely failed in clinical stroke trials (19, 31, 67) (summarized in Table 1).
AMPA,
Glutamate receptors have been one of the most intensely investigated targets for neuroprotection against ischemic stroke (12). Literature is replete with examples of both competitive and non-competitive ionotropic antagonists showing promise in pre-clinical models of ischemic stroke, but failing in clinical trials (39). Selfotel is a competitive NMDA antagonist that showed great potential in attenuating neural cell death in animal stroke models (79, 84, 92). However, in clinical trials (Acute Stroke Strials Involving Selfotel Treatment [ASSIST]) there were no significant differences in post-stroke neurological outcomes between patients treated with Selfotel and placebo controls. Furthermore, in instances of severe stroke, mortality was higher in patients administered Slefotel as compared to placebo (26). The ASSIST trials were discontinued early (25). Non-competitive NMDA antagonists such as dizocilipine (MK-801) and dextromethorphan showed early promise in attenuating stroke-induced lesion volume in rats (13, 88). However MK-801 failed to protect against focal ischemic stroke in a primate pre-clinical trial (5), and clinical trials to test both drugs were terminated early because of inefficacy and phencyclidine-like side effects such as hallucinations and agitation (24). Similar fates in clinical trials have been met by other non-competitive NMDA antagonists. Eliprodil, a non-competitive NMDA antagonist that blocks the NMDA receptor by interaction with a modulatory polyamine site (21), reduced stroke lesion size by >50% in rat model of embolic stroke (50). However, phase III clinical testing of elipdrodil was stopped early when no efficacy was found in stroke patients treated with eliprodil or a placebo (27).
In spite of early promise for AMPA receptor antagonists to succeed where NMDA antagonists had failed (15, 100), they have yet to succeed in clinical trials. YM-872, a water-soluble AMPA receptor antagonist improved neurological outcomes and reduced stroke-induced lesion in multiple rat models of ischemic stroke (41, 83, 91). Clinical trials to demonstrate efficacy, however, have stalled. Multicenter, placebo-controlled, Phase II clinical trials to test YM-872 within 6 h of acute ischemic stroke symptoms were started in December of 2000. The YM-872 trials were reportedly completed in January of 2003 (clinicaltrials.gov identifiers NCT00044070 and NCT00044057), but to date no results have been published.
Therapeutic targeting of metabotropic glutamate receptors for neuroprotection against ischemic stroke is also being investigated (14). Unlike the direct activating mechanisms of their ionotropic counterparts, metabotropic glutamate receptors indirectly modulate excitatory neurotransmission by G-protein-coupled proteins that in turn affect enzyme activity and ion channel conductance. Metabotropic glutamate receptors are divided into three groups, with Group 1 increasing NMDA receptor activity, and Groups 2 and 3 decreasing NMDA receptor activity. In a rat model of transient acute ischemic stroke, intravenous delivery of a selective Group 1 metabotropic glutamate receptor antagonist (YM-202074) following ischemic stroke reduced lesion volume by 37% in the stroke-affected hemisphere (44). As of the time of this writing, a review of clinicaltrials.gov website reveals that no clinical trials to date have tested metabotropic glutamate receptor antagonists against stroke.
Efforts to intercede against glutamate receptor-mediated ionic imbalance and neurotoxicity have also focused on testing specific blockers of ion channels. A systematic review of 29 clinical trials to test a range of calcium antagonists that included isradipine, nimodipine, and flunarizine in patients with acute ischemic stroke failed to demonstrate reduced mortality or improvement in functional outcomes following calcium antagonist treatment (38). In some cases, calcium antagonist use (i.e., flunarizine) resulted in statistically significant worsening of outcomes. Furthermore, adverse events were reported more often in patients (N=7522) treated with calcium antagonists than in the control groups (38). Clinical outcomes with other sodium and potassium channel blockers have not fared better. Sipatrigine is a sodium channel blocker that reduced stroke lesion volume by >60% when given intravenously at doses of 30–50 mg/kg in rat models of ischemic stroke (18, 49, 89). In clinical testing, however, no benefit was observed in stroke patients administered sipatrigine within 12 h of symptom onset and trials were discontinued due to deleterious neuropsychiatric effects including reduced consciousness, agitation, confusion, and hallucinations (54). MaxiPost (BMS-204352), a fluoro-oxindole developed by Bristol-Myers Squibb, is a positive modulator of Maxi-K potassium channels (33). Intravenous delivery of MaxiPost in rats at a concentration of 0.03 mg/kg reduced infarct volume by >20%. While safely tolerated by patients in Phase II trials, MaxiPost showed no efficacy compared to placebo controls in protecting against stroke in Phase III trials that included 1978 patients receiving treatment within 6 h of stroke symptom onset (Potassium channel Opening Stroke Trial) (40).
Another mechanism actively investigated for neuroprotection against glutamate-induced excitotoxicity is focused on improved glutamate clearance in the synaptic cleft by EAATs. In 2001 it was reported that antisense knockdown of glial glutamate transporter EAAT2 in brain exacerbates neuronal damage and cell loss in a mouse model of traumatic brain injury (68), suggesting a critical role for glutamate clearance following brain injury. Since then, research efforts have focused on identifying small molecule inducers of EAAT2 expression. In 2007, Chu et al. reported that intraperitoneal injection of the beta-lactam antibiotic Ceftriaxone (200 mg/kg daily for 1 week) increased EAAT2 gene and protein expression in brain (20). Furthermore, prophylactic delivery of Ceftriaxone attenuated ischemic stroke-induced lesion volume and down-regulated pro-inflammatory cytokine expression following transient cerebral ischemia (20). As EAATs are also implicated in glutamate-mediated excitotoxicity by reverse transport during ischemic stroke, there has been noted concern by authors that overexpression of EAAT2 may worsen excitotoxic glutamate release and injury in ATP depleted brain (20). In line with this theory, Weller et al. reported in 2008 that adenoviral overexpression of EAAT2 in astrocytes increased expression and activity of EAAT2 and protected neurons from cell death under conditions of moderate, but not severe OGD (97). More recently, another small molecule inducer of EAAT2, pyridazine derivative LDN/OSU-0212320, has been shown to protect neurons from glutamate-induced excitotoxicity in mixed neuron and astrocyte primary culture conditions (45). Notably, culture conditions in this study did not model OGD pathology as in acute ischemic stroke. Small molecule inducers of EAATs have yet to be tested in clinical trials.
Taken together, neuroprotective strategies that include those that target glutamate-mediated excitotoxicity are among hundreds of others (64, 70) that target different aspects of the ischemic injury cascade (Fig. 2) (48, 66, 99). To date, over 1000 experimental treatments that showed promise in pre-clinical testing to protect against acute ischemic stroke injury have failed to translate into clinical success (60). This translational failure has led some to question whether neuroprotection for ischemic stroke is an attainable goal at all (51), or if pre-clinical models are simply unable to bridge the translational gap between bench research and clinical care (69). Despite these setbacks, research of neuroprotective strategies and agents continues as knowledge gleaned from past failure is leveraged to test new hypotheses and refine our understanding of ischemic stroke pathophysiology. Most apparent from our historical failure in clinical trials is that despite our best efforts we have been unable to intervene against ischemic stroke at any single downstream target in the ischemic injury cascade (Fig. 2, i.e., ionic imbalance, glutamate release, or receptor-mediated excitotoxicity). Indeed, the only Food and Drug Administration-approved therapeutic intervention for stroke patients remains the clot busting tissue plasminogen activator (tPA). tPA acts on the very upstream problem of energetic failure by restoration of blood flow. In that light, it appears neuroprotective agents that act against downstream targets without addressing the underlying problem of energetic failure may be wholly ineffective. On the other hand, tPA is not without its own limitations. With a limited window of therapeutic opportunity due to risk of hemorrhagic transformation, <5% of ischemic stroke patients are eligible for tPA use.
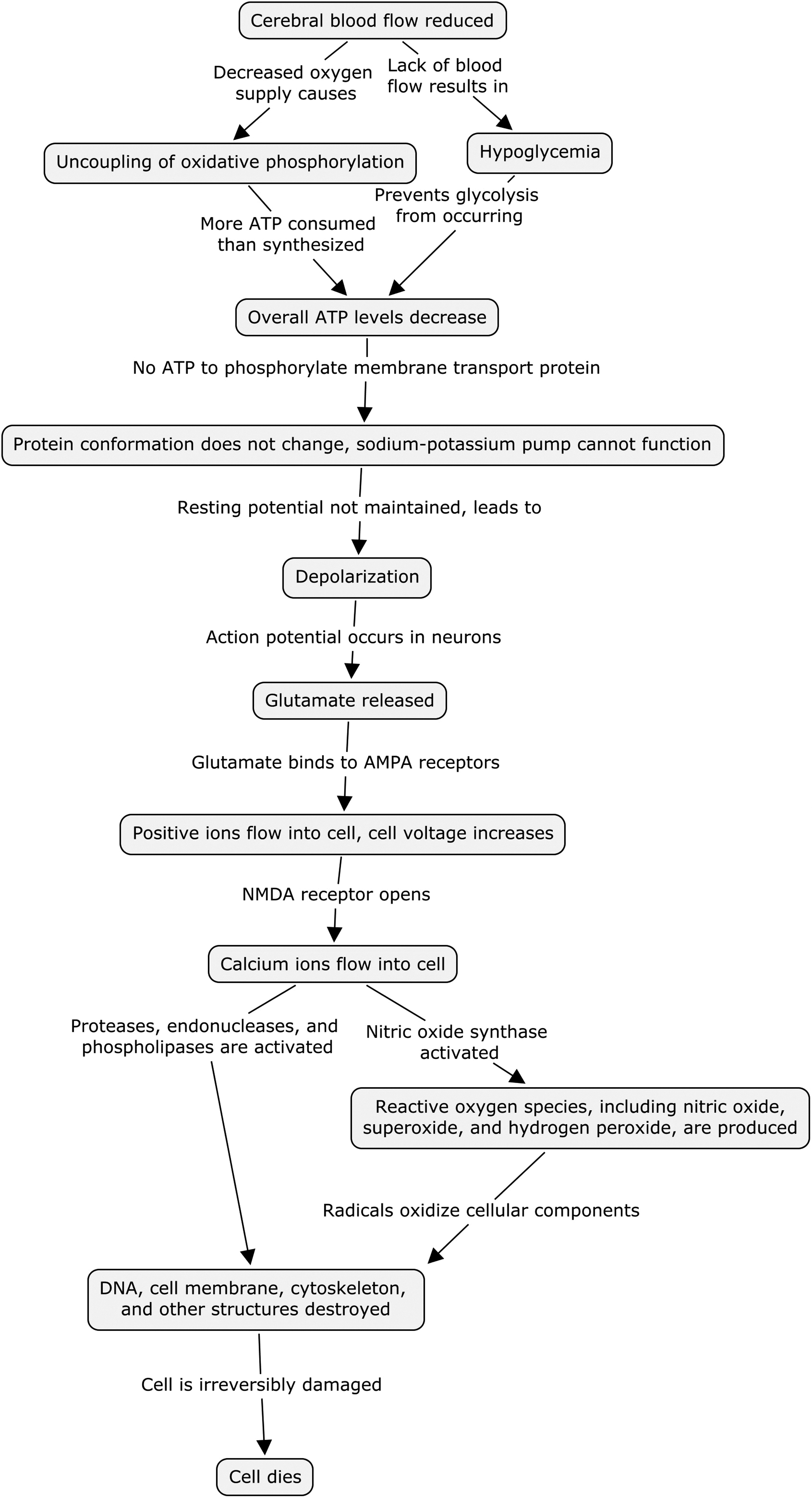
Turning a Neurotoxin into a Cell Survival Factor
Our laboratory recently identified a novel therapeutic opportunity that addresses both downstream glutamate-mediated excitotoxicity and upstream energetic failure. In 2010 we reported on the specific contribution of stroke-induced focal brain hypoxia in brain injury (74) where normobaric oxygen (NBO) and hyperbaric oxygen (HBO) therapy were used to correct pO2 in the ischemic stroke-affected brain. During cerebral ischemia (90 min MCAO), correction of ischemic stroke-induced hypoxia by NBO and HBO protected brain tissue from injury. In rats subjected to 90 min of MCAO, NBO (100% O2 at 1ATA) and HBO (100% O2 at 2ATA) reduced the stroke lesion volume by >50% after 48h post-reperfusion. The opposite effect was observed, however, when NBO and HBO were applied after 90 min of cerebral ischemia. Upon reperfusion that restored CBF and pO2, NBO and HBO increased the size of stroke lesions with evidence of exacerbated oxidative stress. This outcome was consistent with clinical trials that failed to demonstrate protection for HBO when applied in the days and weeks after resolution of cerebral ischemia (3, 59, 76).
But how did correction of stroke-induced hypoxia with NBO and HBO protect the brain during ischemia? After all, we did not correct for stroke-induced hypoglycemia which in the hypermetabolic brain would still contribute to energetic failure and cell death. To answer this question, we queried the entire mRNA transcriptome and uncovered the oxygen-dependent up-regulation of a novel neuroprotective target—glutamate oxaloacetate transaminase (GOT). GOT catalyzes the transfer of the amino group from glutamate to the four-carbon tri-carboxylic acid (TCA) cycle intermediate oxaloacetate to generate aspartate and the five-carbon TCA cycle intermediate alpha-ketoglutarate (Fig. 3) (34, 87). That GOT was up-regulated when stroke-induced hypoxia was corrected led to the exciting hypothesis that GOT transamination could (1) enable extracellular glutamate clearance from the ischemic site, as well as (2) feed cellular respiration to generate energy in otherwise glucose-starved, energy-deprived neurons.
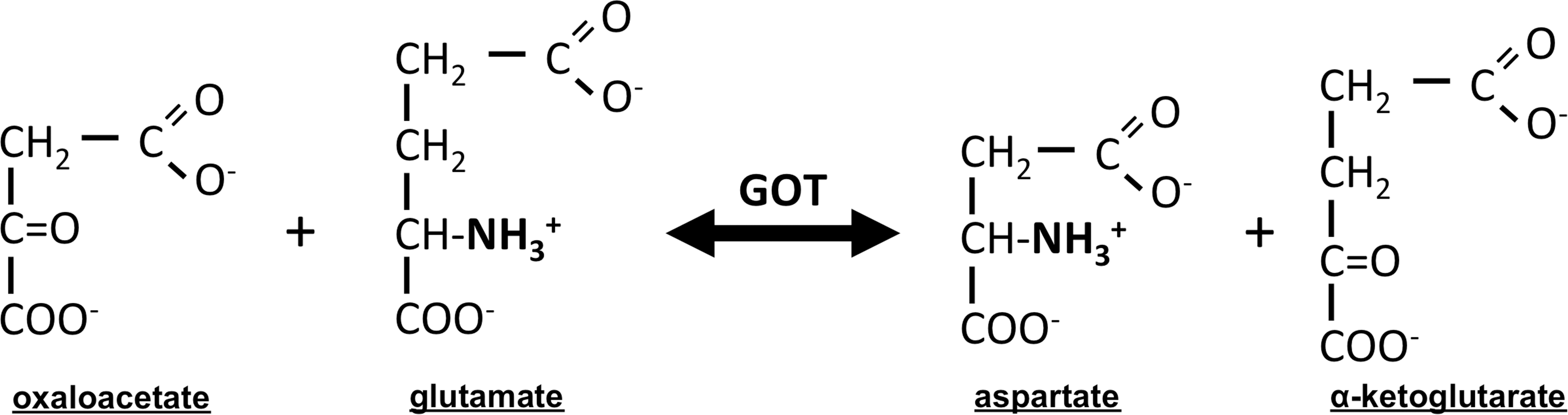
Importantly, a formative figure in cell metabolism had already made some striking observations as they relate to glutamate metabolism in the brain. In 1935, 2 years prior to publishing the discovery of the TCA cycle, Krebs reported that glutamate was the only amino acid to increase respiration in rabbit brain cortex in the absence of glucose (47). He noted that respiration in brain slices “falls off quickly if no suitable substrate is added,” and that “glutamic acid is the only amino-acid which I have found capable of maintaining the respiration of brain” (47). Before glutamate was known as a neurotransmitter, Krebs' work led many to believe that the role of glutamate in brain tissue was related to cell metabolism (75). In 1963, Krebs extrapolated upon his early experiments and reported that in the absence of glucose extracellular glutamate was readily metabolized, with 80–90% converted into aspartate by transamination in rat brain homogenate (34). Interestingly, the addition of glucose and pyruvate inhibited the transamination reaction and subsequent formation of aspartate.
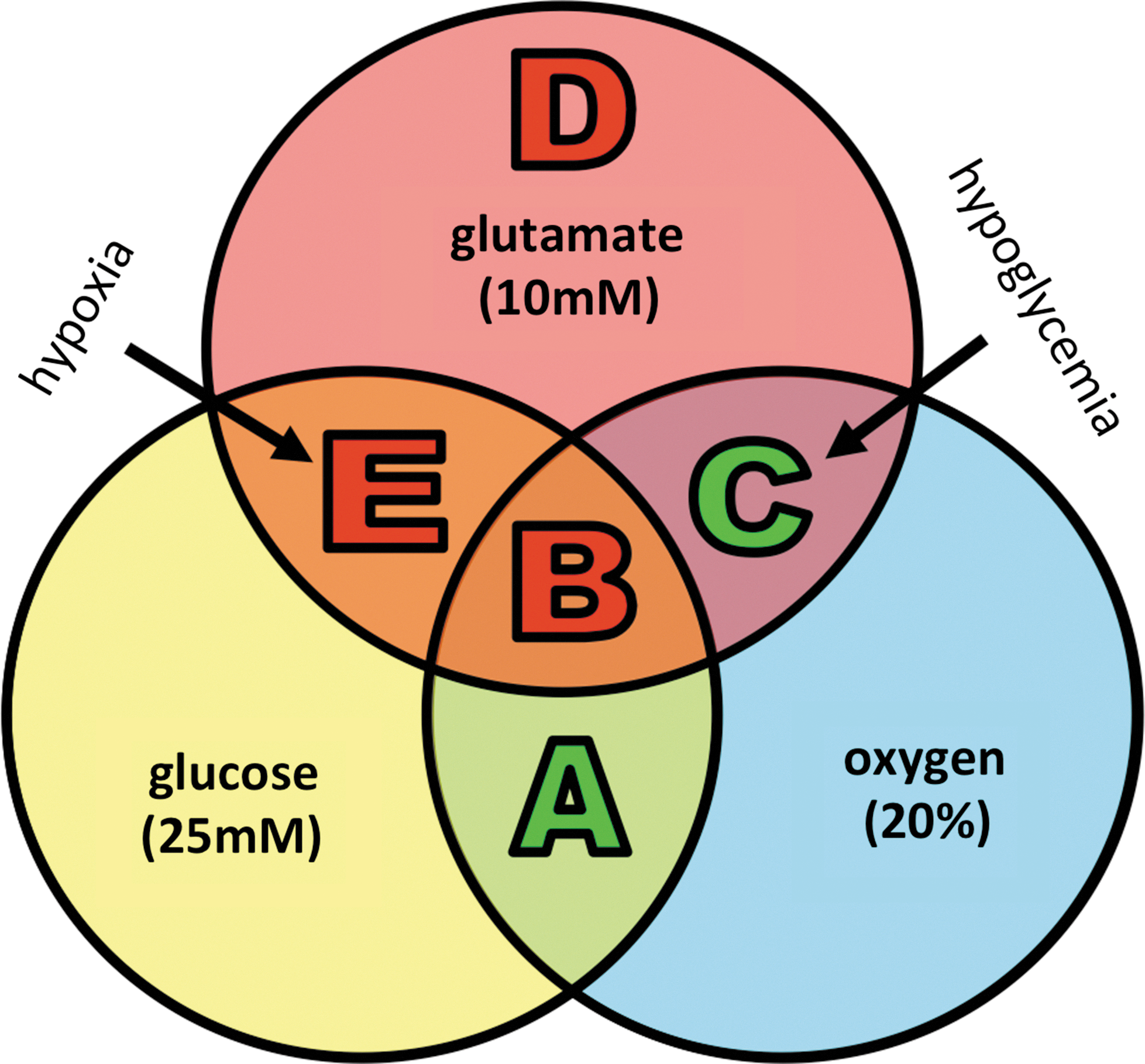
Retrospective review of Krebs' work supported our hypothesis that extracellular glutamate could be used by neurons as metabolic fuel in the absence of glucose. To test this hypothesis directly, we devised a simple in vitro experiment in primary cultured neurons (Fig. 4) (72). Under normoxic (21% O2) and normoglycemic (25 mM glucose in culture media) conditions, primary cultured neurons thrive. In order to model oxidative toxicity in vitro, extracellular glutamate is commonly added to culture media (73). As we and others have previously reported (43, 55), extracellular glutamate (10 mM) caused profound neural cell death under normoglycemic (25 mM) and normoxic (20% O2) conditions with more than 75% of cells dead by 24 h (72). However, when glucose was removed from media but other culture conditions remained the same, extracellular glutamate (10 mM) no longer induced neural cell death as it had under normoglycemic conditions. In fact, neural cell viability in the presence of extracellular glutamate but absence of glucose was sustained at the same level as observed under normoglycemic and normoxic conditions in the absence of glutamate. Under hypoxic (0.5% O2) and hypoglycemic (no glucose in culture media) conditions as in acute ischemic stroke, neural cells died in the presence of extracellular glutamate (10 mM). Importantly, this experiment uncovered a glucose and oxygen dependent role reversal for otherwise neurotoxic extracellular glutamate to become a cell survival factor (Fig. 4). Next, we sought to test whether this role reversal was dependent on GOT as suggested by the work of Krebs and our in vivo gene expression data (74).
To do this, we returned first to our in vitro culture model where transfection of HT4 neural cells with GOT small interfering ribonucleic acid (siRNA) was employed to knockdown GOT expression and activity (72). With our siRNA approach we achieved 75% knockdown of GOT mRNA expression and more than 50% lower enzymatic activity in neural cells. When these cells were incubated under culture conditions where extracellular glutamate had prevented neural cell death previously (normoxia and hypoglycemia), knockdown of GOT expression and activity resulted in neural cell death. This outcome suggests that GOT is required for the therapeutic switch of glutamate from neurotoxin to survival factor (72). To test the significance of GOT protection against ischemic stroke, we returned to our murine stroke model. When stroke-induced hypoxia was corrected with HBO, we documented increased GOT activity, and lower glutamate levels in stroke-affected cortex as compared to room air controls. Furthermore, ATP levels in stroke-affected cortex were significantly depleted under room air conditions, but preserved when hypoxia was corrected by HBO. To determine the effect of GOT on stroke outcome, we over-expressed and knocked down GOT expression prior to experimental ischemic stroke. GOT over-expression significantly attenuated stroke-induced lesion volume compared to scramble siRNA controls as measured by magnetic resonance imaging (MRI) at 48 h post-stroke. In mice subjected to GOT knockdown, stroke severity was exacerbated. Five of six mice died prior to 48 h MRI and the stroke-induced lesion volume of the sole survivor was dramatically larger than scramble controls and mice with over-expression of GOT in stroke-affected cortex (72).
GOT as a Therapeutic Target
Under conditions of ischemic stroke, the inability to envision that glutamate could assume the role of a survival factor represents a critical barrier to develop the greater significance of glutamate in stroke biology. The fact that correction of hypoxia in an ischemic setting can promote glutamate clearance and sustain cellular energetics (72) suggests that GOT may enable anaplerosis (refilling) of TCA cycle intermediates to sustain neural cell energetics in the absence of glucose during stroke (Fig. 5). Experiments are currently in progress to directly test whether GOT enables a truncated TCA cycle during ischemic stroke. While the new paradigm of GOT orchestrating a functional switch of extracellular glutamate from an inducer of cell death to energy substrate is an exciting, testable hypothesis; the looming question of translational relevance remains if therapeutic efficacy is dependent on correction of stroke-induced hypoxia. Like many predecessors, HBO therapy has thus far failed in clinical testing despite early promise in pre-clinical trials (3, 59, 76). In that light, we specifically characterized a therapeutic window of opportunity for which oxygen therapy could benefit ischemic stroke patients (73, 74). However, as with tPA, this window of opportunity appears to be critically short. Furthermore, efficacious use of supplemental oxygen therapy may require real-time monitoring of CBF as oxygen therapy during spontaneous reperfusion worsens stroke outcome (74).
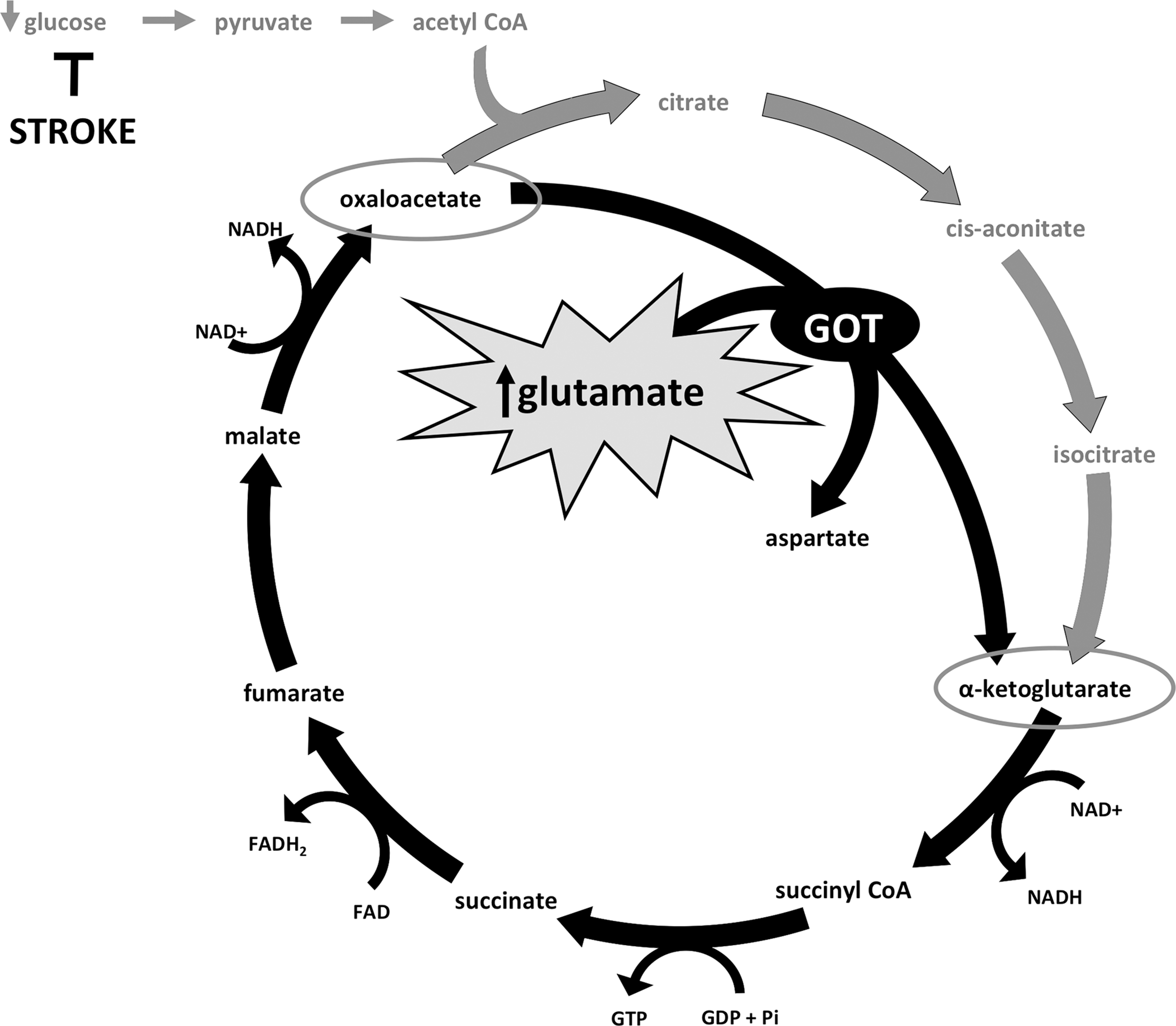
Fortunately, oxygen therapy may not be requisite for GOT to enable neuroprotection against glutamate-mediated excitotoxicity during ischemic stroke. In 2011, Campos et al. reported on the glutamate scavenging properties of GOT in blood (16). In rats subjected to transient focal ischemia of the middle cerebral artery, an intravenous bolus of oxaloacetate (3.5 mg/100 g) was effective in scavenging glutamate in blood and reducing stroke-induced infarct volume in brain. Prior to this, intravenous oxaloacetate had also been shown to lower glutamate levels in cerebrospinal fluid (32) and improve somatosensory evoked potentials in stroke-affected brain (56). While these studies did not measure GOT activity in brain directly, the basis of protection was suggested to be oxaloacetate-dependent activation of GOT in blood and subsequent scavenging of glutamate (16, 32). Even though scavenging addresses downstream clearance of extracellular glutamate to prevent excitotoxicity, it fails to address the upstream problem of energy depletion in the brain. Our concept moves beyond GOT minimizing the levels of glutamate as a neurotoxin at the ischemic site to actually metabolizing the scavenged glutamate to sustain cellular energetics in the brain. Notably, without blood to deliver oxygen to sustain cellular respiration via the electron transport chain, GOT-mediated anaplerosis of TCA cycle intermediates would not be effective. However, neither would GOT-mediated scavenging without blood flow to enable glutamate diffusion from brain to blood (17). In that light, blood-born GOT that enables scavenging or brain-born GOT that enables anaplerosis is likely to occur in the ischemic penumbra, where low blood flow enables some metabolic activity. Indeed, it was in the ischemic penumbra of the primary somatosensory cortex that we observed GOT over-expression to protect against focal cerebral ischemia and GOT knockdown to exacerbate injury (72).
Conclusions and Future Directions
Like the thousands of neuroprotective targets that came before it, GOT holds exciting promise for therapeutic efficacy on the basis of pre-clinical studies alone. While it has yet to be tested in clinical trials, GOT holds unique therapeutic potential to protect against ischemic stroke for its ability to (1) clear otherwise excitotoxic extracellular glutamate and (2) metabolize it into TCA cycle intermediates in the absence of glucose. Beyond oxygen therapy with aforementioned logistic challenges that preclude clinical translation, research efforts are seeking to identify therapeutic strategies that increase GOT expression and activity at the time of stroke. Recently, intravenous human recombinant GOT delivered in a rodent model of ischemic stroke was shown to effectively scavenge and lower brain glutamate levels similarly to oxaloacetate (65). While too bulky to cross the blood–brain barrier alone, that recombinant human GOT in blood was effective in lowering brain glutamate levels holds promise for next generation targeted delivery systems currently under development (63). There may also exist opportunity for small molecules to target GOT with drug-like effects. We recently reported that a natural phytoestrogen isoflavone from red clover, biochanin A, induces GOT expression, increases activity, and protects against stroke injury (71). Importantly, phytoestrogen isoflavones from red clover have been shown to protect human cortical neurons from glutamate-induced toxicity (61). As a safe, naturally occurring compound, Biochanin A is a small molecule that could be quickly translated to clinical study.
Footnotes
Acknowledgment
Supported in part by NIH grant NS085272 to S.K. and C.R.