
Abstract
Introduction
T
RR, relative risk; CV, cruciferous vegetables; OR, odds ratio; HR, hazard ratio.
Phylogeny, Molecular Genetics, and Biochemistry of Glucosinolate-Containing Plant Species
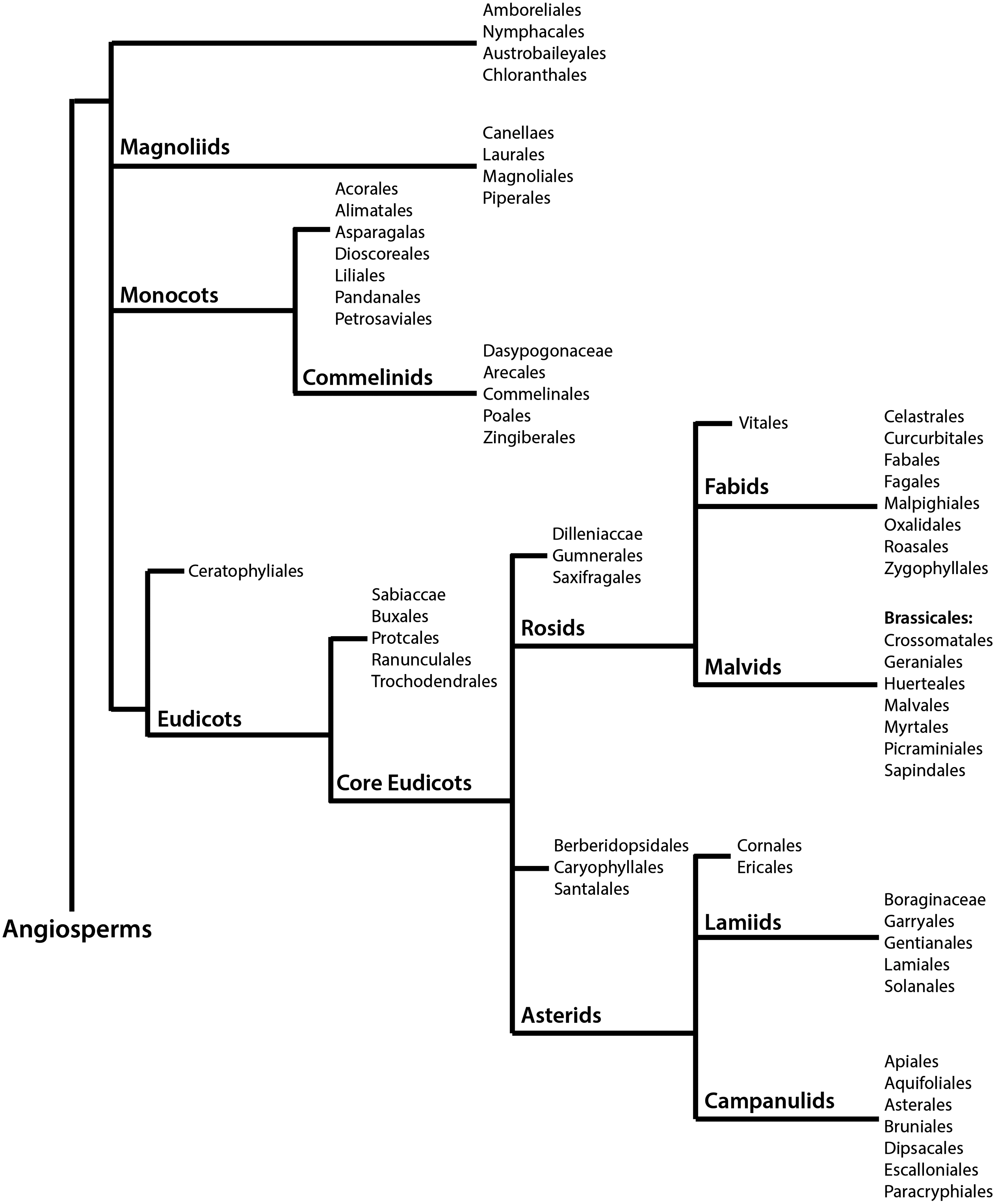
Glucosinolates are a class of secondary compounds present in angiosperms of the order Brassicales, which comprises 18 families, 398 genera, and ∼4450 species (100). Figure 1 summarizes the phylogeny of angiosperms, with the Brassicales belonging to the malvids group of the monophyletic clade rosids, in accordance to the Angiosperm Phylogeny Group (APG) III system. The APG III system of flowering plant classification is the third version of a molecular-based plant taxonomy system published in 2009 by the APG (278). Overall, Brassicales contain ∼2.2% of the eudicot diversity, with their earliest fossil known from the Turonian (89.5 millions years ago) (180). The most important, and perhaps most extensively studied glucosinolate-producing family is the Brassicaceae, which comprises 49 tribes, including the tribes Brassiceae and Arabideae (190). Plants belonging to the Brassicaceae family exhibit species-specific profiles of glucosinolates, a class of compounds thought to function as a part of a defence mechanism against pathogens and insects (9). Difficulty in establishing evolutionary trends and utilizing morphology in phylogenetic studies has been a challenge due to homoplasy in the family (6, 281). The first comprehensive phylogenetic study by Beilstein et al. used a sample of 101 genera within the family to assess the chloroplast gene ndhF for an intergeneric relationship (19). This study placed the genera into clades (recognized as tribes) grouped into three main lineages (I-III). Currently, lineage I include 15 tribes, lineage II has been expanded to include 25 tribes, and lineage III now comprises seven tribes (82, 88, 295). Substantial support for this system has been provided using an internal transcribed spacer of nuclear ribosomal DNA (ITS)-based phylogeny, nuclear alcohol dehydrogenase, chalcone synthase, and plastidic maturase sequence data (14, 150).The tribes Brassiceae and Arabideae are a part of lineage II, with floral monosymmetry evolved independently several times within this lineage (27).
Advances in the understanding of glucosinolate biosynthesis through studies in Arabidopsis thaliana and Brassica sp. have suggested that aliphatic glucosinolates (including glucoraphanin) are derived from methionine, tryptophan, and phenylalanine (191, 192). A comparison between Brassica oleracea L. var. italic, possessing a Brassica C.genome, and Arabidopsis genomes showed sequence similarity and gene order and content colinearity in specific chromosomal segments (222). The synthesis of glucosinolates is determined by a simple genetic system containing two distinct sets of genes, one determining sidechain elongation and the second involved in the chemical modification of the sidechains (Fig. 2A).
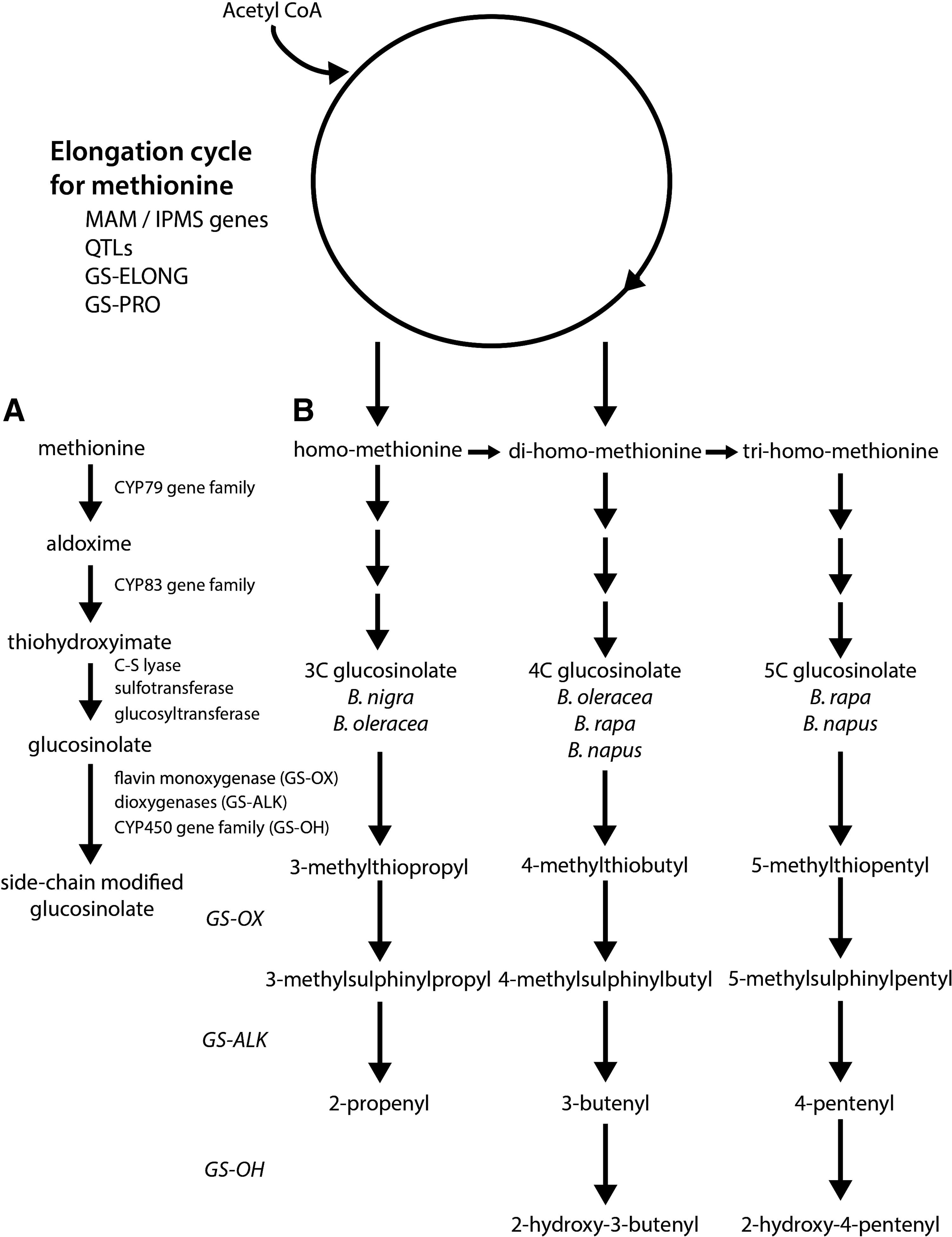
Many of the glucosinolates found in Brassica vegetables are derived from chain elongated forms of methionine or phenylalanine (286). Figure 2B provides a genetic model of methionine-derived glucosinolate biosynthesis, with the total level and nature of the glucosinolate determined early in the process, and the initial entry of methionine into the pathway catalyzed by methylthioalkylmalate (MAM) synthase genes at the GS-ELONG loci (58, 76). MAM synthases catalyze the condensation of acetyl CoA as the methyl donor, with an α-keto acid derived by amino-acid transanimation to result in elongated methionine. Different members of this family may catalyze different number of rounds of elongation, with genetic variation at the GS-ELONG and GS-PRO loci enabling selection for different glucosinolate profiles, while allelic variation at the quantitative trait loci determines overall amount (286). These initiating factors have been observed in both Arabidopsis and Brassica (58, 242). In B. oleracea, these genes determine the length of the side chain with either one or two rounds of methionine elongation to produce 3C or 4C glucosinolates, respectively (242). Li and Quiros demonstrated that glucosinolate synthesis was independently regulated by genes GSL-PRO which determines the synthesis of 3C glucosinolates, and GSL-ELONG which determines synthesis of 4C glucosinolates (164). Furthermore, MAM and a homologue protein of Arabidposis AOP2 were confirmed to be useful markers for breeding of high glucoraphanin varieties of B. oleracea. Importantly, it was shown that the glucosinolate biosynthesis pathway may be manipulated toward the synthesis of glucoraphanin (precursor to sulforaphane) by increasing the expression level of GSL-ELONG and reducing expression of GSL-PRO. Studies in A. thaliana showed that the overexpression of R2R3-MYB transcription factors (known regulators of glucosinolate biosynthesis), including AtMYB28, AtMYB29, and/or AtMYB76, resulted in the upregulation of the glucosinolate biosynthetic genes and an increase in selected classes of glucosinolates (90, 91, 109, 264). Although genes involved in the regulation of glucosinolate biosynthesis have not yet been functionally identified in Brassica species, Araki et al. suggest that gene homologues of AtMYB28 and AtMYB29 are important in B. oleracea (9).
After biosynthesis, methionine-derived glucosinolates may undergo a number of side chain modifications, including hydroxylation, methoxylation, oxidation, desaturation, conjugation with benzoic acid, and glucosylation (286). For example, methylthioalkyl glucosinolates may be converted to methylsulphinylalkyl glucosinolates by flavin monoxygenases at the GLS-OX loci (99). In turn, these glucosinolates may be modified by 2-oxogluturate-dependent dioxygenases at the GLS-ALK loci and by an unknown enzyme at the GLS-OH loci to form alkenyl and hydroxyl-alkenylglucosinolates, respectively (98). Due to the considerable variation at these loci in B. oleracea, selection for specific glucosinolate profiles may be possible with the genetic background being a major factor in determining glucoraphanin concentration and composition (Table 2). Brown et al. concluded that the percentage of glucosinolate variability in broccoli was attributable to genotype for individual compounds, including 54.2% for glucoraphanin (25). High glucosinolate varieties of broccoli have been specifically bred to accumulate levels that are significantly higher than regular broccoli (242). The best example of deliberate breeding of such broccoli was achieved by crossing a standard cultivar with B.villosa, a wild form of B. oleracea from Sicily, which accumulates high levels of 3-methylthiopropyl glucosinolate (85). This high glucosinolate variety of broccoli was shown to deliver about four times the amount of sulforaphane to the systemic circulation than standard cultivars, with sulforaphane metabolites consumed by GSTM1-positive subjects measured to be 107.5 μM after consumption of regular broccoli compared with 345.8 μM in subjects within the group who consumed the high-glucosinolate variety.
fw, fresh weight; dw, dry weight.
Pharmacokinetics and Bioavailability of Dietary Sulforaphane
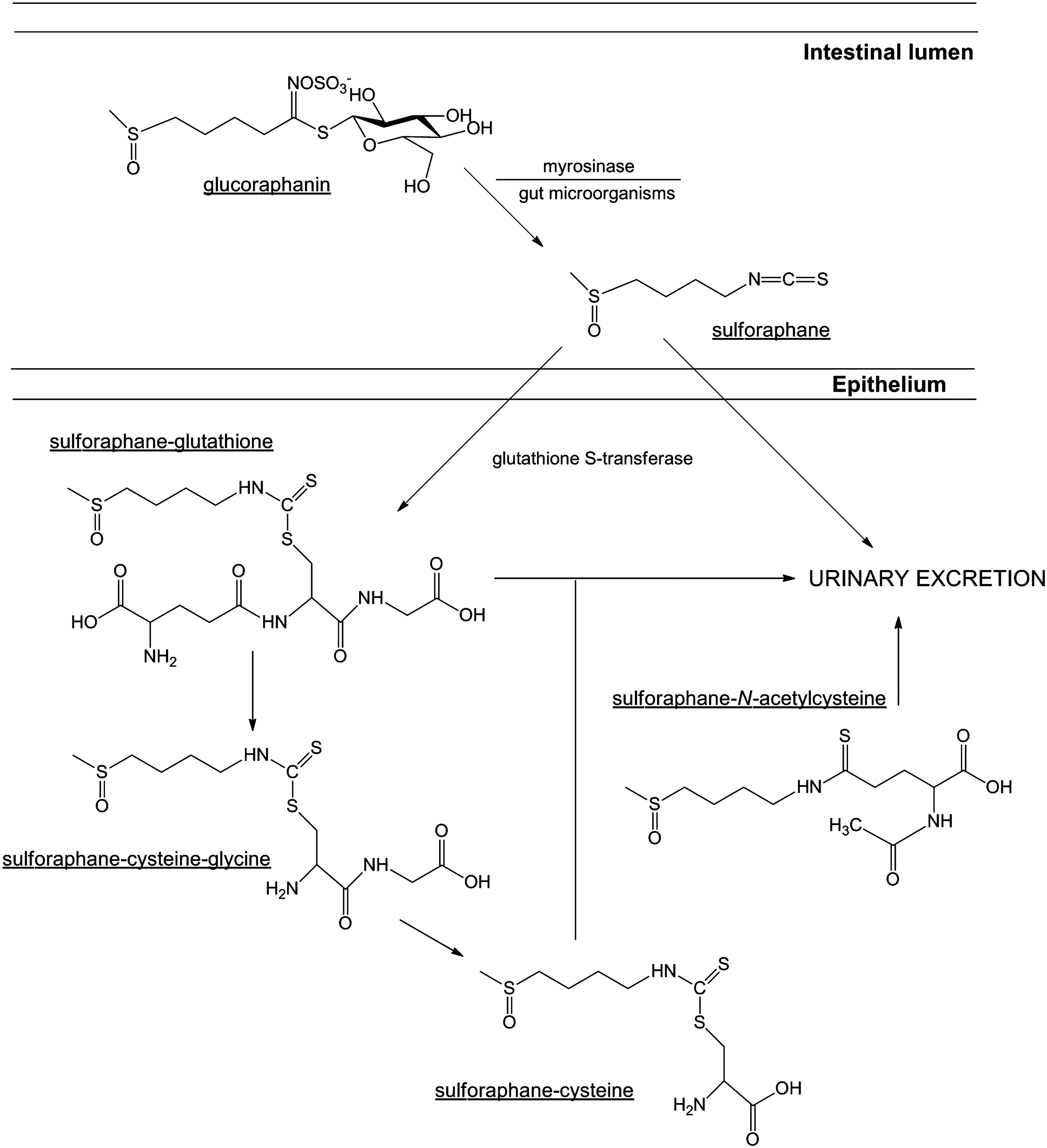
The significant accumulation of the glucosinolate, glucoraphanin [4-methylsulfinylbutyl glucosinolate] in broccoli has been shown to be important as the precursor of the bioactive isothiocyanate, sulforaphane (Fig. 3). It is rapidly absorbed, metabolized, and excreted, with ∼80% appearing in the urine within 12–24 h after consumption and/or administration (54). There are many factors that may affect the bioavailability, and therefore overall therapeutic benefit, of dietary sulforaphane, including pharmacokinetic properties, genetic variation, and food preparation (42). Hydrolytic conversion of glucoraphanin to sulforaphane through the action of physical damage to the plant occurs either by the action of plant-derived myrosinase (intracellular broccoli thioglucosidase) (70) or by the microbiota of the human colon (48, 251, 252). Approximately, 60%–80% of glucoraphanin is converted to sulforaphane (131), with most broccoli varieties possessing between 0.1 and 30 μmol/g of glucoraphanin (Table 2). After rapid diffusion into the cells of the intestinal epithelium due to its lipophilic nature and low molecular weight, sulforaphane undergoes metabolism via the mercapturic acid pathway (218). This process involves its initial conjugation with glutathione, rapidly catalyzed by important glutathione S-transferase (GST) enzymes (272).
As previously described, GST enzymes are important in sulforaphane metabolism by their ability to conjugate the isothiocyanate to glutathione, leading to its excretion (272). The GST isoforms, GST-M1 and GST-T1 have been reported to have the greatest activity on sulforaphane (151, 315). Genetic polymorphisms, as well as geographical and ethnic variations in genotype frequencies have been described for both GST-M1 and GST-T1, including null mutations, which result in the absence of a functional gene product (101). Interestingly, the frequency of a homozygous null genotype for both isoforms is relatively high, with the frequency of the GST-M1 null genotype varying between 39% and 63%, and the GST-T1 null genotype between 10% and 21% for Caucasian populations, but it may be as high as 64% in subjects of Asian descent (53). The implication of a homozygous null genotype has been shown to be an important variable that determines the biodistribution, and therefore the protective effect of broccoli consumption in human populations. Epidemiological studies, however, have reported conflicting results with regard to the association between genetic polymorphisms in GST isoforms and cancer risk. For example, some studies have concluded that GST-M1-positive subjects gain greater cancer protection from either broccoli consumption or total cruciferous vegetable consumption than those who possess the GST-M1 null genotype (129, 266, 292). In contrast, others report that consumption of cruciferous vegetables leads to a greater protective effect in GST-M1 and GST-T1 null subjects than those with functional GST enzymes (173, 245). This variation, however, may be explained through the innate biological function of GST enzymes and their role in the metabolism and subsequent elimination of various compounds after exposure. Specifically, molecular epidemiological studies involved in elucidating the association between GST-M1 and GST-T1 genotypes with cancer susceptibility have shown that those who carry the homozygous deletion appear to have a small increase in the magnitude of risk (odds ratio of <2); however, other factors (i.e., cigarette smoking) further enhance the risk (odds ratio of 3–5) (228). This is accounted to their ability to regulate the conjugation of carcinogenic compounds, including benzo(a)pyrene and styrene-7,8-oxide by GST-M1, and ethylene oxide and methyl bromide by GST-T1, to excretable hydrophilic metabolites (10, 59, 97, 316). Through the inability to express functional GST enzymes, individuals possessing the homozygous null genotype have a limited capacity to efficiently detoxify potentially carcinogenic exposures. Thus, the consumption of cruciferous vegetables may have limited protection in such subjects, as chronic exposure to carcinogens causes a reduction in their preventative activity. Conversely, studies that report an enhanced protective effect of sulforaphane in GST-M1 and GST-T1 null genotypes hypothesize that due to the inability to efficiently metabolize sulforaphane for urinary excretion, exposure of target tissues to the protective compound and/or metabolites is higher and prolonged (170).
The sulforaphane metabolites produced in such a pathway are distributed throughout the body and accumulate in different tissues, with unpublished data from Franklin and co-workers after a whole body autoradiographic study in rats suggesting that high concentrations of isothiocyanate metabolites are present in the gastrointestinal tract, liver, kidneys, and blood (26). The basis for the distribution of sulforaphane is the high degree of binding to glutathione, and its capacity to drive passive diffusion (47, 108). Due to analytical limitations, a few studies have successfully measured the distribution of sulforaphane and its metabolites in humans. Results from human studies demonstrated that 74% (±29%) of sulforaphane from broccoli extracts may be absorbed in the jejunum, with a portion returning to the intestinal lumen of jejunum as sulforaphane-glutathione (218).The amount of sulforaphane metabolites in plasma, however, may reflect the amount of sulforaphane exposed to tissues, and are, therefore, considered important biomarkers in the determination of distribution (55). In plasma, more than 50% of total sulforaphane metabolites were sulforaphane-glutathione with free sulforaphane, although other metabolites, including sulforaphane-N-acetylcysteine, were present in quantifiable amounts (121). For example, in rats, after a single dose of (50 μmol) sulforaphane, detectable levels of the compound were evident after 1 h, peaking at ∼20 μM at 4 h with a half life of ∼2.2 h (114). Ye et al. have shown a rapid absorption and appearance of isothiocyanates and their metabolites in the blood of human subjects, with this level observed to decline after first-order kinetics (indicating rapid distribution and/or metabolism (305). These subjects were given a single dose of 200 μmol broccoli sprouts isothiocyanate preparation, with isothiocyanate plasma concentrations peaking between 0.943 and 2.27 μM 1 h post exposure, with a half life calculated at 1.77 h (±0.13 h). Subsequent accumulation after distribution within target tissue is also an important aspect in the context of sulforaphane and its ability to elicit chemopreventive and anticancer effects. In an in vivo study in mice given 300 or 600 ppm sulforaphane, accumulated sulforaphane and sulforaphane-glutathione plasma concentrations were recorded at 124–254 nM and 579–770 nM, respectively (115). Concentrations within the small intestine were also measured, and they were between 3–13 nmol/g of tissue and 14–32 nmol/g of tissue, respectively (equivalent to ∼3–30 μM of total sulforaphane). Interestingly, the accumulation of sulforaphane within the colonic tissue of treated mice corresponded with decreased adenoma formation. Detoxification genes NQO1 and heme oxygenase-1 (HO-1) were also detected in the removed tissue. In addition, the rapid passage of a bolus of preformed sulforaphane in rats did not achieve equal distribution across all tissues, with the highest levels reached in the stomach wall (284). Sulforaphane distribution was decreased in more distal parts of the digestive tract, with the rectum accumulating ∼1% of that found in the stomach. At 24 h post administration, however, levels were low and approximately equal in all evaluated tissue, indicating the rapidity of sulforaphane metabolism and excretion. A study of human breast tissue after consumption of broccoli sprouts, however, showed that the tissue concentration of isothiocyanates failed to be different from plasma levels (51). The kidney is the major organ involved in the conversion of glutathione conjugates into the corresponding N-acetyl-S-cysteine conjugates (176). The process of N-acetylation is important for the subsequent excretion of isothiocyanates from the body. Excretion of sulforaphane and its metabolites have been shown to follow first-order kinetics, with most data indicating clearance from the body within 72 h of administration (251, 305).
Glucoraphanin is relatively stable under chemical and thermal conditions, and, therefore, hydrolysis is mainly enzymatic (myrosinase mediated) (286). Cooking and/or blanching (during freezing process) of cruciferous vegetables inactivates myrosinase, and has been shown to decrease the bioavailability of sulforaphane (47, 239, 287). In general, results suggest that only about 30%–50% of the initial administered dose is excreted after these preparation processes (127, 240). Boiling for more than 1 min, or steaming for more than 4–5 min has been shown to lead to the loss of myrosinase activity (291). Conaway et al. performed a study to assess the metabolic fate of glucosinolates after ingestion of steamed and fresh broccoli in 12 male subjects in a crossover design (47). Results of this study indicate that the bioavailability of sulforaphane from fresh broccoli is approximately thrice higher than that from cooked broccoli. Interestingly, coadministration of semi-purified glucoraphanin with a myrosinase source was reported to increase the bioavailability of the isothiocyanate (54). In addition, the concentration of glucoraphanin varies widely among development stages of the plant, and between different parts of the plant, which may also influence sulforaphane bioavailability (45, 210, 226). For example, two varieties of broccoli, a purple-sprouting broccoli and a green-sprouting broccoli, were cultivated to compare the amount of sulforaphane and its metabolites within commercial samples (234). In the initial samples, it was shown that there were quantitative differences between organs, with the seeds, edible sprouts, and florets determined to possess the most intact sulforaphane (in descending order). Furthermore, these two varieties produced higher sulforaphane levels compared with those within the commercial samples. Higher sulforaphane concentrations were also found in the initial steps of the digestion process, with the concentration of sulforaphane and its subsequent metabolites decreased in accordance with the amount of precursor glucoraphanin.
The concentration of sulforaphane required to observe therapeutic activity has not yet been determined in human clinical trials, with rough estimates based on the active dose in animal models (108). The amount of dietary glucoraphanin that is converted to bioavailable sulforaphane is typically calculated as 20% of the overall consumed amount in humans (250 –252). For example, the typical sulforaphane concentration that has been shown to inhibit the growth of human cancer xenografts in mice is ∼4.4 mg/kg per day (133). This corresponds to 308 mg of sulforaphane daily administered to a 70 kg person (108).
Chemopreventive Activity Against the Initiation of Carcinogenesis
Chemoprevention, which refers to the use of a nontoxic natural or synthetic chemical that possesses the capacity to intervene in multistage carcinogenesis, has emerged as a promising approach to reduce the risk of the development and progression of malignancy (163). Microarray analyses in cell lines, animal tissue, and human biopsy samples have shown the capacity of sulforaphane to modulate global gene expression, especially resulting in the differential expression of genes that are important in chemoprevention (Table 3). In general, results indicate that sulforaphane affects the expression of genes involved in xenobiotic metabolism, antioxidation, cell cycle regulation, apoptotic pathways, and stress response (115, 179, 306).
LSF, sulforaphane; qRT-PCR, quantitative real-time reverse-transcription polymerase chain reaction; ppm, parts per million; sqRT-PCR, semi-quantitative real-time reverse-transcription polymerase chain reaction; AKR, aldo-keto reductase; SILAC, stable isotope labeling by amino acids in cell culture.
Evidence suggests that sulforaphane, its metabolites, and synthetic analogues possess the capacity to inhibit the malignant transformation of various cell types, and limit cancer progression after carcinogen exposure (71, 92). For example, sulforaphane and its conjugate metabolite N-acetylcysteine was shown to limit the malignant progression of lung adenomas in A/J mice exposed to tobacco carcinogens (49). Prevention of mutagenesis by sulforaphane was also demonstrated in mice given a single application of the sulfur mustard analogue, 2-(chloroethyl) ethyl sulphide (CEES) (1). Abel et al. reported that a single topical treatment with sulforaphane induced the production of phase II detoxification enzymes, with an increase in the epidermal levels of the regulatory subunit of glutamate-cysteine ligase, and reduced glutathione. Furthermore, sulforaphane treatment limited the CEES-induced increase in mutation frequency in the skin, which was measured at 4 days post exposure. A reduction in the number, size, and development of mammary tumors in rats after exposure to the carcinogen dimethylbenz[a]anthracene has also been observed with sulforaphane treatment (73, 312). Furthermore, sulforaphane possessed an ability to decrease the amount of DNA-adduct formation in normal mammary cells exposed to polycyclic aromatic hydrocarbons (259). Inhibition of DNA-adduct formation has also been displayed in human bladder cells in vitro and in mouse bladder tissue in vivo, after treatment with sulforaphane and exposure to the bladder carcinogen, 4-aminobiphenyl (60). This inhibition was dependent on the activation of an important chemoprotective signaling pathway, nuclear factor erythroid 2-related factor 2 (Nrf2), within the epithelium, which is the main site for bladder cancer development. Exposure to benzo[a]pyrene results in the induction of pulmonary carcinogenesis in mice via oxidative damage, with sulforaphane treatment found to decrease the production of H2O2 (134). Results suggest that sulforaphane leads to the activation of apoptotic pathways in this experimental model of chemical lung carcinogenesis, included the increased release of cytochrome c from mitochondria, enhanced caspase-3 activity leading to DNA fragmentation, and a decrease in Bcl-2 expression coupled with an increase in the expression of Bax. Interestingly, sulforaphane was shown to protect primary dermal fibroblasts and keratinocytes against oxidative stress caused by UVA radiation under basal conditions and in the presence of the photosensitising drug, 6-thioguanine (20). In this study, sulforaphane treatment resulted in an ∼50% reduction in the formation of reactive oxygen species (ROS). Furthermore, the protective properties of sulforaphane were shown to be caused by an increase in Nrf2-depednent cytoprotective responses, including those involved in the induction of gene transcription and expression of important metabolic and detoxification enzymes.
The chemopreventive activity of sulforaphane in carcinogenesis is related to the inhibition of phase I enzymes, which are responsible for the activation of pro-carcinogens (15, 313) and the induction of phase II enzymes that are responsible for mutagen elimination (314). Phase I enzymes such as cytochrome p450 (CYP) are important in the transformation and bioactivation of pro-carcinogens to carcinogens (297). Through common enzymatic processes, electrophilic intermediates covalently bind to nucleophilic sites in important macromolecules (77). Adduct formation, coupled with inadequate reparation, may, in turn, cause miscoding and mutations in critical genes to initiate carcinogenesis. Multiple studies have reported the capacity of sulforaphane to inhibit the catalytic activity of phase I enzymes (15, 181). Specifically, Barcelo et al. reported the dose-dependent inhibition of both cytochrome p450-1A1 and -2B1/2 enzymatic activity in rat hepatocytes after sulforaphane treatment (15). Similar results were also found in human hepatocytes, with sulforaphane significantly decreasing the expression of CYP1A1, 1A2, and CYP3A4 genes (96). Evidence also suggests that sulforaphane is an effective antagonist of the human steroid and xenobiotic receptor (SXR), which is an important transcriptional factor that regulates the expression of CYP3A genes (318). In primary rat hepatocytes, sulforaphane caused a significant inhibition of the microsomal ethoxyresorufin-O-deethylase (EROD) activity (a selective marker for CYP1A1 and 1A2) and P-benzoquinone dioxime (BQD) activity (a marker of CYP3As) (158).
Cells possess innate protective mechanisms in order to minimize damage caused by highly reactive metabolites, with the induction of phase II enzymes that are of major importance in detoxification. These enzymes include GST, NAD(P)H:quinine oxidoreductase (NQO-1), and UDP-glucuronosyltransferase (UGT) (138). Phase II enzymes are potent antioxidants that have a relatively long half life, and have the capacity to conjugate endogenous substrates such as glutathione to phase I metabolites in order to limit further biotransformation and result in enhanced elimination and excretion (35). Evidence suggests that sulforaphane is a potent inducer of phase II antioxidant enzymes (61). For example, sulforaphane significantly reduced the number and level of DNA adducts after exposure to methyl-6-phenylimidazo[4,5-b]pyridine in a dose-dependent manner in hepatocytes due to the induction of GST-A1 and UGT-1A1 mRNA expression (11). NQO-1 and GST activity was also shown to be enhanced in the forestomach, duodenum, and bladder of rats treated with 40 μmol of sulforaphane per kg (197), while an increased dose of 200–1000 μmol of sulforaphane per kg increased activity in the liver, colon, and pancreas (183). Jones and Brooks demonstrated an increase in enzymatic activity in prostate, liver, kidney, and bladder tissues after administration of sulforaphane (128). Human clinical studies also provide evidence for the induction of phase II enzymes by sulforaphane after broccoli consumption (Table 4).
CV, cruciferous vegetables; mRNA, messenger RNA.
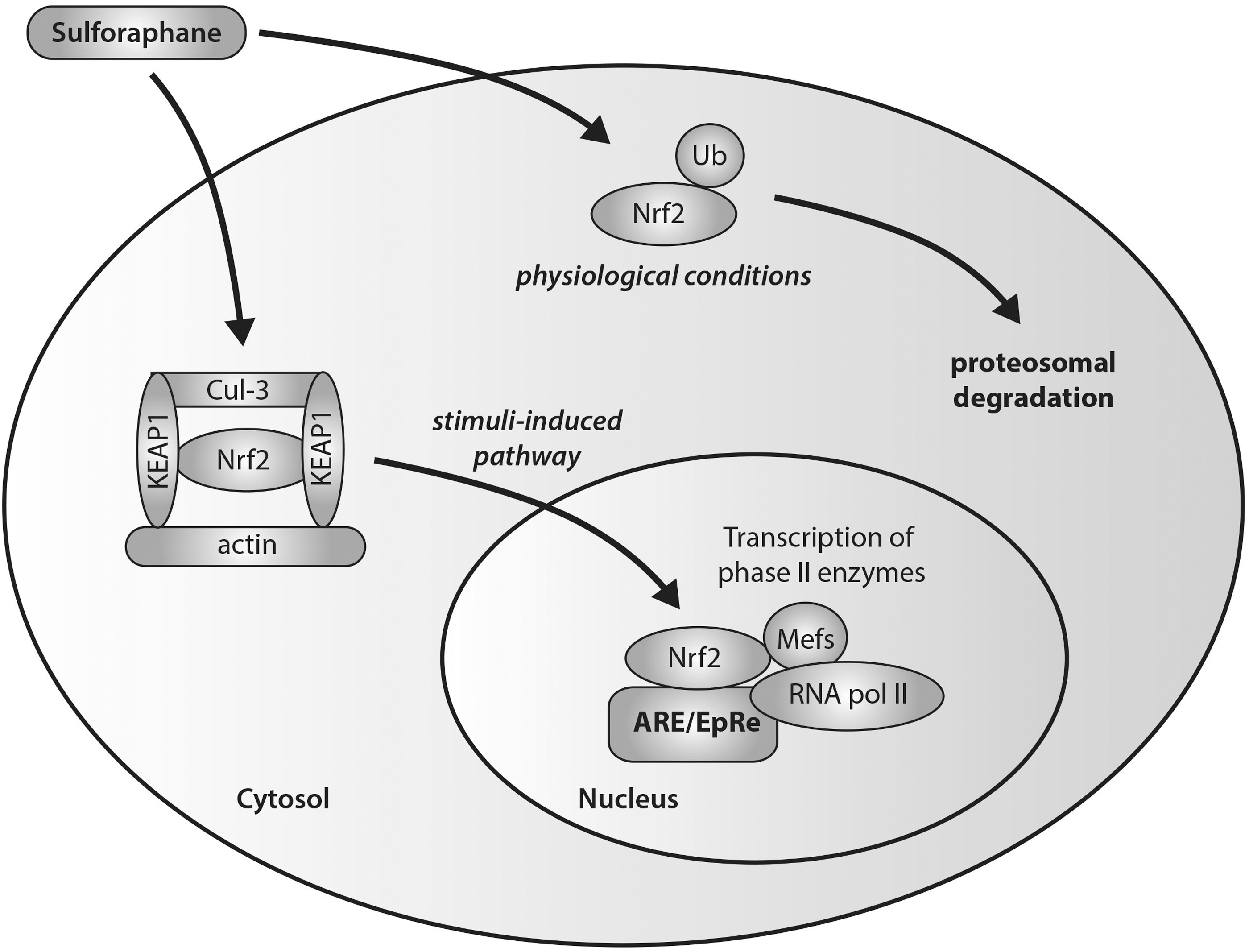
The induction of phase II enzymes by sulforaphane has been shown to be associated with a disruption of Nrf2-Keap1 interactions and increased translocation of Nrf2 (Fig. 4) (117, 148, 206). In unstimulated cells, Kelch-like ECH-associated protein 1 (Keap1) mediates the function of Nrf2 through sequestration mechanisms in order to bind it to Cul-3-dependent ubiquitinase for subsequent ubiquitination and targeted proteosomal degradation. After stimulation due to environmental insult, the Nrf2-Keap1 complex is disrupted with conformational changes, leading to a switch in ubiquitination from Nrf2 to Keap1, and the nuclear translocation of Nrf2. Activation of Nrf2 in the nucleus occurs through its heterodimerization with small Maf transcription factors to form a complex that binds to the antioxidant/electrophile response element (ARE/EpRE) that is found in the promoters of many phase II enzymes. Sulforaphane has the ability to induce nuclear translocation of Nrf2 through the disruption of the Nrf2-Keap1 complex via the degradative loss of Keap1 via conformational changes (62). Specifically, sulforaphane has been demonstrated, with the use of spectroscopic evidence, to react with the thiol groups of Keap1 to form thionoacyl adducts (62, 110). This specific modification of Keap1 released Nrf2 from sequestration, promoting the subsequent activation of ARE-driven gene expression. Conceptual proof of this action by sulforaphane has been shown through experiments in Nrf2 knockout mice (279). Thimmulappa et al. generated a transcriptional profile of the small intestine of wild-type (Nrf2+/+) and knockout (Nrf2−/−) mice treated with sulforaphane. Numerous genes were found to be regulated by Nrf2, including the previously reported phase II xenobiotic metabolizing enzymes, as well as antioxidative and cytoprotective proteins that are important in limiting cancer progression.
Anticancer Properties of Sulforaphane Limiting Tumor Progression
In addition, evidence suggests that sulforaphane possesses the capacity to limit the progression of tumor development through a number of mechanisms, including activation of apoptosis, NFκB pathway inhibition, and cell cycle arrest induction (Fig. 5).
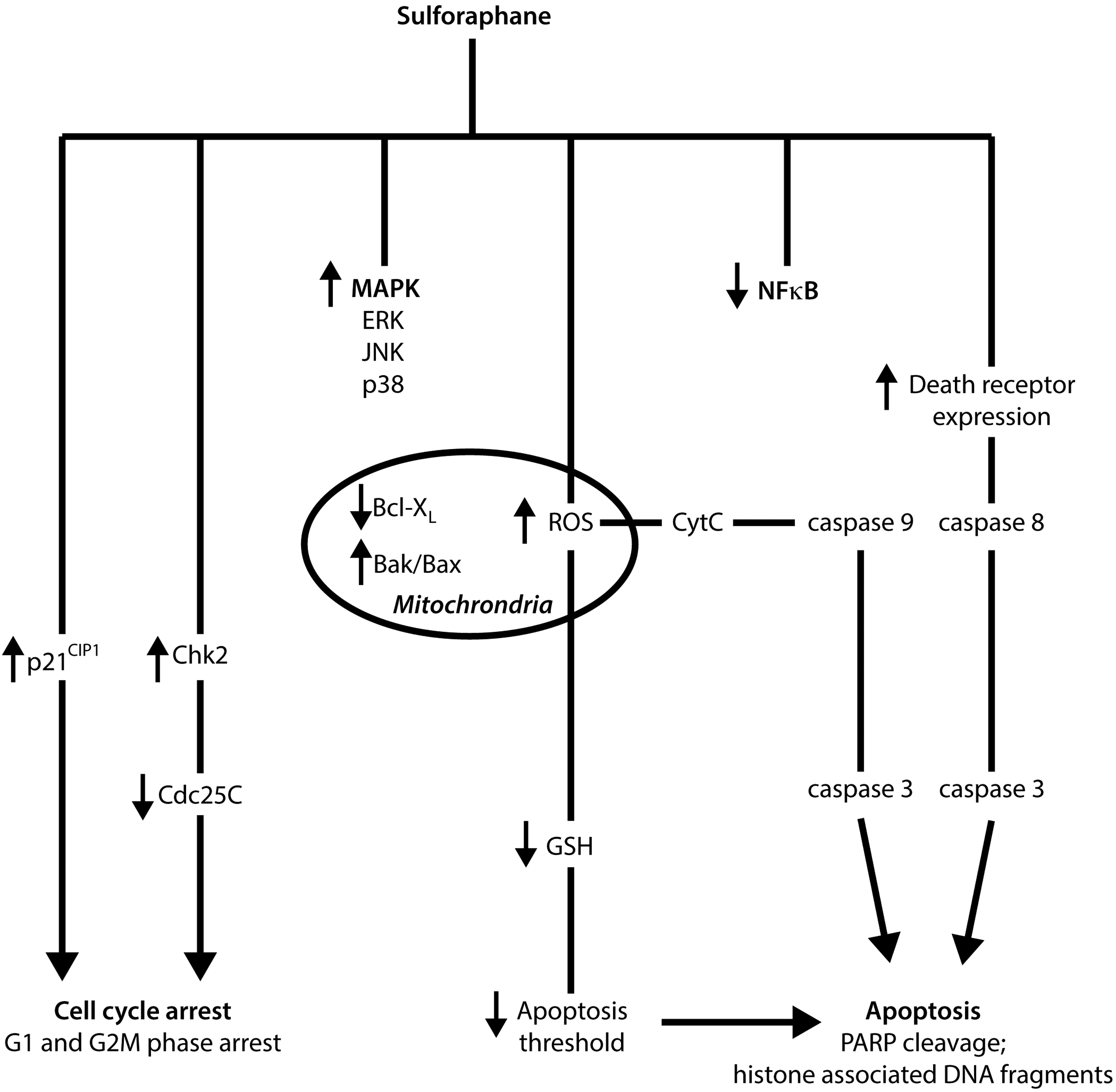
Apoptosis or programmed cell death is an important and selective mechanism in the regulation of cell proliferation in both physiological and pathological conditions (229). The activation of apoptotic pathways in malignant cells is a major focus for current cancer therapeutic research, with evidence suggesting that sulforaphane possesses the capacity to induce apoptosis through the activation of several proapoptotic pathways (238). For example, sulforaphane induced apoptotsis in both human cervical HeLa cancer and hepatocellular HepG2 carcinoma cell lines in vitro, demonstrated by the formation of apoptotic bodies and the accumulation of cells in the sub-G1 phase (215). Administration of 10 μM sulforaphane reduced cell viability and induced apoptosis in prostate DU145 cancer cells as indicated by the cleavage of poly(ADP-ribose) polymerase (PARP) and increased release of histone-associated DNA fragments (38). In HCT-116 colon cancer cells, 15 μM sulforaphane induced activation of pro-apoptotic caspase-7 and caspase-9, independent of p53 expression (212). Recently, sulforaphane-induced apoptotis was also associated with the activation of caspase-8 and caspase-9, the initiating caspases that are important in both extrinsic and intrinsic apoptotic pathways in human bladder 5637 cancer cells (214). Park et al. also reported the ability of sulforaphane to affect important molecular targets that are intimately involved in apoptotic pathways, including the downregulation of anti-apoptotic Bcl-2 and Bcl-XL gene expression, the upregulated expression of pro-apoptotic Bax, proteolytic activation of caspase-3, and the degradation/cleavage of PARP (215). The activation of Bax, the downregulation of the inhibitors of apoptosis (IAP) protein family, and the induction of apoptotic protease activating factor-1 have been shown to be involved in the regulation of sulforaphane-induced cell death (39). A study using Jurkat T-leukemia cells suggests that vulnerability to sulforaphane-mediated apoptosis was dependent on cell-cycle mechanisms, with cells most sensitive to sulforaphane-induced apoptosis in the G1 phase, less sensitive in the G2/M phase, and least sensitive during the S phase (78).
The proposed initiating signal of sulforaphane-mediated apoptosis is the formation of ROS and the disruption of mitochondrial membrane potential, leading to cytosolic release of cytochrome c via both death-receptor and mitochondrial caspase cascade pathways, as observed in human prostate cancer PC3 cells (258). More specifically, sulforaphane was capable of inducing apoptosis through the activation of the ROS-dependent, caspase-3 in multiple tumor necrosis factor-α-resistant leukemia cell lines (194). Interestingly, administration of the antioxidant N-acetylcysteine and the overexpression of catalase resulted in the reversal of sulforaphane-induced ROS formation in the same study. Singh and co-workers thus indicated a necessity for the conjugation of sulforaphane with glutathione during metabolism, in order to deplete intracellular concentrations of glutathione and potentially lower the oxidative stress threshold of cells. In general, high concentrations of sulforaphane are required in order to induce ROS formation. For example, mitochondrial ROS generation and disruption of mitochondrial membrane potential leading to the formation of acidic vesicular organelles and autophagy in PC3 and LNCap cells may only be observed after administration of 40 μM sulforaphane (105). This response has unique morphological effects and was shown to possess the ability to inhibit the release of mitochondrial cytochrome c and apoptosis. Therefore, it has been suggested that ROS production after sulforaphane treatment has the capacity to influence cell death in an alternative pathway to apoptosis.
While activation of apoptosis by sulforaphane in the human breast cancer MDA-MB-231 cells was reported to be initiated through the induction of Fas ligand, which resulted in the activation of caspase-8, caspase-3, and PARP, sulforaphane induced apoptosis in the human breast cancer cell lines, MDA-MB-468, MCF-7, and T47 via mechanisms involved with decreased Bcl-2 expression, release of cytochrome c into the cytosol, activation of caspase-9 and caspase-3 (but not caspase-8), and poly(ADP-ribose) polyermase cleavage (219). Taken together, these findings indicate that sulforaphane has an innate ability to modulate both extrinsic and intrinsic apoptotic pathways, via the production of ROS and regulation of gene expression.
In contrast to the well-documented induction of apoptotic pathways by sulforaphane, recent studies have indicated that sulforaphane may also possess the capacity to cause autophagy in cancer cells. The significance of these findings is displayed in the publication by Herman-Antosiewicz et al., which associated the induction of autophagy with the inhibition of cytochrome c release and, ultimately, apoptosis (106). In this study, treatment of prostate cancer cell lines, PC-3 and LNCap with sulforaphane resulted in the upregulation, processing, and recruitment to autophagosomes of microtubule-associated protein light chain 3 (LC3). In addition, inhibition of autophagy (incubation of cells with 3-methyladenine) potentiated the proapoptotic effects of sulforaphane in human colon cancer cells (207). Similar results were also demonstrated in human breast cancer cell lines, MCF-7 and MDA-MB-231 (136). Investigation providing mechanistic insights indicates that autophagy and cell death signaling after sulforaphane treatment of pancreatic cells are independent pathways which depend on ROS production (204). Pharmacologic inhibition of autophagy in vivo validated cell culture studies, with most suggesting that autophagy is a cytoprotective mechanism against sulforaphane-induced apoptosis (290).
Inhibition of cellular growth may also be caused by an irreversible arrest during the cell cycle, with sulforaphane shown to possess such inhibitory effects on cells (84, 257). A significant increase in G2/M cell cycle arrest was observed in LNCap prostate cancer cells after sulforaphane incubation in a concentration- and time-dependent manner (107). A reduction in cell viability after the induction of G2/M cell cycle arrest after treatment of DU145 prostate cancer cells with 10 μM sulforaphane has also been demonstrated (38). In human ovarian PA-1 cancer cells treated with sulforaphane, an accumulation in the G2/M (metaphase) phase was reported through the downregulation of CDC2, as well as through the downregulation and dissociation of the cyclinB1/CDC2 complex (31). A decrease in protein levels of cyclin B1, cell division cycle Cdc25B, Cdc25C, and an accumulation of Tyr-15-phosphorylated (inactive) Cdk1 has also shown to be important in sulforaphane-mediated cell cycle arrest in PC3 cells after incubation with 20 μM sulforaphane (257). In addition, sulforaphane treatment resulted in a rapid and sustained phosphorylation of Cdc25C at Ser-216, which was a result of Chk2 activation, leading to translocation of Cdc25C from the nucleus to the cytoplasm, and increased binding to 14–3-3beta. It is thus proposed that Chk2-mediated phosphorylation of Cdc25C is an important regulation mechanism in irreversible sulforaphane-induced G2/M arrest.
The tumor suppressor and cell cycle inhibitor protein p21 also appears to play an important role in sulforaphane-induced cell cycle arrest with its induction observed in a number of studies irrespective of cell type and expression of p53. Sulforaphane treatment of p53 negative colon cancer cell lines (HT-29 and Caco-2) was shown to induce p21 expression (216). In LNCap prostate cancer cells, induction of p53 and p21 was observed after 20 μM sulforaphane treatment (199). Interestingly, cell cycle arrest in LNCap cells occurred after the p21 expression induction but not p53. In this same study, induction of p21 expression by sulforaphane was shown to occur in p53-null PC3 prostate cancer cells, suggesting a possible p53-independent regulatory pathway. Treatment of acute lymphoblastic leukemia cells from human patients with sulforaphane resulted in dose-dependent apoptosis and G2/M cell cycle arrest (274). These anticancer mechanisms were associated with the activation of caspases (3, 8, and 9), inactivation of PARP, p53-independent upregulation of p21, and inhibition of the CDC2/cyclin B1 complex. Colon tissue surgically removed from three human subjects treated with sulforaphane for 2 h exhibited a strong induction of p21 in cancer tissue, with expression failing to be significant in the normal tissue of two patients (282).
Although G2/M arrest is the predominant stage of cell cycle arrest induced by sulforaphane (84, 216), arrest at other phases of the cell cycle has been observed. For example, a G1 cell cycle arrest was shown to occur in HT-29 cells concomitant with an increase in p21CIP1, and a decrease in cyclin D1, cyclin A, and c-myc (254). G1/S cell cycle arrest has also been reported in LNCap and DU145 cells (37, 293). Evidence suggests that the phase in which cell cycle arrest occurs after sulforaphane treatment is dependent on administered concentration and incubation time. In human colon adenocarcinoma Caco-2 cells, G2/M cell cycle arrest was observed at a dose of 20 μM sulforaphane (120). Conversely, administration of sulforaphane at concentrations >20 μM induced an accumulation of sub-G1 cells and the loss of mitochondrial membrane potential. Pappa et al. demonstrated reversible G2/M cycle arrest and cytostatic growth of p53 wild-type 40–16 colon cancer cells after transient exposure till 6 h (211). Treatment with sulforaphane for longer than 12 h, however, resulted in irreversible G2/M arrest and subsequent apoptosis. Interestingly, cytostatic growth effects observed with 12 h of exposure was sustained till 72 h post sulforaphane removal and the IC50 calculated was comparable to cells transiently exposed to sulforaphane for 72 h.
Mitogen-activated protein kinases (MAPKs), including the extracellular signal-regulated kinases (ERKs), c-Jun NH2-terminal kinases (JNK), and p38, are believed to be involved in carcinogenesis and tumor progression (41). The activation of the MAPK/ERK pathway has been reported after sulforaphane treatment in a number of cell lines, including PC3 cells, through the activation of the activator protein-1 (AP-1) transcription factor that is involved in the regulation of cell death (303). The modulatory effect on AP-1 transcription is dependent on the concentration of sulforaphane administered. In HT-29 colon cancer cells, activation of AP-1 luciferase activity occurred at low concentrations of sulforaphane treatment (≤35 μM), while activity was inhibited at high concentrations (≥50 μM) (124). In addition, cyclin D1 levels increased at lower concentrations, and decreased at high concentrations. Interestingly, however, is that despite the difference in modulation after varying concentrations of sulforaphane, cell viability decreased in a dose-dependent manner. Administration of 50 μM sulforaphane in cells corresponded to activation of the 46-kDa isoform of phospho-JNK (p46-JNK) but not the p54-JNK isoform, indicating the importance of the 54-kDa isoform for persistent activation of AP-1. Consistent with this report, Shen et al. also observed a decrease in cell viability and the activation of MAPK pathways (including, ERK, JNK, and p38) (254). Furthermore, activated JNK was shown to decrease cyclin D1 expression at high concentrations of administered sulforaphane. The treatment of Caco-2 human colon adenocarcinoma cells with sulforaphane was shown to induce ERK activation, but failed to significantly increase the activation of JNK and p38 (120). In another study, DU-145 prostate cancer cells were exposed to sulforaphane with the activation of the JNK pathway shown to be important in cell death induction (38). Activation of p38 was also shown to be important in the upregulation of Nrf2-ARE-driven enzymes, and the downregulation of pro-inflammatory COX-2 in human bladder T24 cancer cells (247). The MAPK pathway is, therefore, an important mechanism activated by sulforaphane, and indirectly contributes to cell death and cell viability regulation, as well as to Nrf2-Keap1 interactions and the transcription of phase II antioxidant enzymes.
Nuclear factor B (NFκB) and its active role in inflammation, cancer cell survival, and progression has been widely reported, with the ability to bind to the promoter of many pro-inflammatory genes, including inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), and tumor necrosis factor (TNF) (137). After incubation of PC3 cells with 20 μM sulforaphane for 1 h, a reduction in nuclear localization of p65-NFκB was observed (39). In addition, inhibition of NFκB transcriptional activity in PC3 cells resulted in the suppression of the nuclear translocation of p65 and the decreased gene expression of NFκB-regulated VEGF, cyclin D1, and Bcl-xL (302). The decreased nuclear translocation and activation of p65-NFκB was attributed to the inhibition of IκB kinase (IKK) phosphorylation, which is necessary for IκB degradation, and subsequent release of inactive NFκB to become active. Similar effects are observed in HT-29 cells in a dose-dependent manner (125). Interestingly, an initial increase in NFκB activity at 6–12 h post sulforaphane treatment in LNCap cells has also been observed, followed by inhibition at the 24 h time point (39). In this same study, the modulation of the IAP family, downstream factors that are shown to be upregulated by NFκB activation, was directly proportional to the level of NFκB activity. Studies also suggest that sulforaphane has the capacity to reduce the DNA binding ability of NFκB directly, with two mechanisms proposed (36). The first involves the modification of NFκB subunits via thiol-dependent interactions to cause dithiocarbamate formation and the direct binding to essential cysteine (Cys) residues of NFκB, thereby decreasing the capacity to bind to nuclear DNA (104). The other suggests that sulforaphane may interact with glutathione and other redox regulators such as thioredoxin or redox factor-1 (Ref-1), which, in turn, indirectly interferes with NFκB-DNA binding. Sulforaphane has been identified as an inhibitor of the thioredoxin/thioredoxin reductase (TrxR) enzymatic activity in vitro (103). Findings also suggest that the inhibition of 12-O-Tetradecanoylphorbol-13-acetate (TPA)-induced matrix metalloproteinase-9 (MMP-9) expression and cell invasion by sulforaphane is mediated by the suppression of the NFκB pathway in human breast MCF-7 cancer cells (162). Furthermore, the selective inhibition of NFκB by sulforaphane has been demonstrated in receptor activator of nuclear factor kappa-B ligand (RANKL)-induced osteoclastogenesis through the interaction with the thiol groups of NFκB (145). Coactivators of NFκB may also be affected by sulforaphane, including CCAAT-enhancer-binding proteins, cAMP response element-binding protein, and AP-1 (299). Woo and Kwon demonstrated the capacity of sulforaphane to inhibit such coactivators, and, ultimately, its indirect ability to downregulate the expression of pro-inflammatory COX-2. Despite the evidence, the inactivation of the NFκB pathway by sulforaphane requires further elucidation to determine its impact as an important molecular chemopreventative mechanism.
The regulation of endogenous receptor expression in selected cells may also be an important chemopreventative mechanism of sulforaphane. For example, sulforaphane has been shown to inhibit the expression of estrogen receptor alpha (ERα) in the human MCF-7 breast cancer cell line due to an inhibition of ERα mRNA transcription as well as due to increased proteosome-mediated degradation (225). These data suggest that sulforaphane has the potential to inhibit cancer cell proliferation caused by aberrant hormone ER receptor expression in MCF-7 cells. In addition, sulforaphane significantly increased the reactivation of ERα expression in ER-negative breast cancer cells, and was consistently correlated with ERα promoter hypomethylation and hyperacetylation (185). Treatment of Caco-2 cells with sulforaphane for 48 h also led to the downregulation of serotonin receptor 5-HT to undetectable levels as compared with the control (182).
The ability of sulforaphane to sensitize drug-resistant cancer cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis has been demonstrated in multiple studies, with reports demonstrating sulforaphane-enhanced TRAIL-induced apoptosis in human osteosarcoma cells (Saos2 and MG63) and hepatoma cells (143, 182). The induction of cell death in various tumor cell lines by sulforaphane was coupled with increased p53, activated caspase-3 proteins, and decreased hypoxia-inducible factor-1α activation under hypoxic conditions (which mediates resistance to TRAIL) (123). It was also shown that sulforphane may mediate sensitization to TRAIL-induced apoptosis through the upregulation of death receptor 5 (DR5) mRNA and protein expression (182).
Recently, the ability of sulforaphane to modulate toll-like receptor (TLR) activation and signaling has been implicated as an additional chemopreventive property. TLRs recognize specific patterns derived from invading microorganisms and pathogens, or damaged cells and tissues to elicit the innate and/or adaptive immune response (153). Zhu et al. observed that sulforaphane caused inhibition of TLR3, with the ability to modulate NFκB signaling and downstream gene expression, including the downregulation of IL-8 and TNF-α (320). Sulforaphane has also been reported to form adducts with cysteine residues in the extracellular domain of TLR4, which results in the inhibition of TLR4 dimerization in a thiol-dependent manner (307). In addition, sulforaphane interfered with the binding of lipopolysaccharide to myeloid differentiation 2 (MD2) by its ability to preferentially bind to the cysteine 133 residue in the hydrophobic pocket of MD2 (154). Although this inhibition is important for the attenuation of the inflammatory response (an important factor in the development of cancer), modulation of downstream mechanisms to TLR signaling may also limit the progression of cancer. Specifically, sulforaphane inhibited the expression of intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 through TLR4-dependent pathway in cultured endothelial cells (246). Blocking the expression of adhesion molecules is important in limiting cancer cell invasion and metastasis (147, 178).
Targeting Cancer Stem Cells with Sulforaphane
Most recently, tumor heterogeneity has been considered to arise through the aberrant differentiation of cancer cells, as well as a result of continuing mutagenesis (230). The existence of cancer cells with stem cell-like properties in a number of cancer types has recently been identified (23, 34, 63, 177, 209, 256). It is proposed that the growth of a tumor is driven by a small population of cancer cells, which have the ability to undergo self-renewal, and may be responsible for tumor relapse, metastasis, and resistance (155, 230, 317).
A number of studies have suggested that sulforaphane possesses the capacity to target cancer stem cells through direct and indirect mechanisms, alone or in combination with other anticancer compounds (168). As previously described, sulforaphane is able to attenuate NFκB activity and nuclear translocation of the NFκB subunit, resulting in decreased expression of NFκB-regulated genes (125, 302). Kallifatidis et al. demonstrated the ability of sulforaphane to abrogate the resistance of pancreatic tumor-initiating cells to TRAIL by interfering with TRAIL-activated NFκB signaling (133). Specifically, sulforaphane reduced the DNA binding capacity of transactivation-competent NFκB dimers that were found in a tumor initiating cell-enriched cell population. Thus, sulforaphane was shown to impair the expression of NFκB target genes with antiapoptotic effects.
In addition, the sonic hedgehog (SHH) signaling pathway is linked to NFκB signaling, with reports of an overexpression of SHH activated by NFκB in pancreatic cancer (203). After sulforaphane treatment, inhibition of SHH signaling has been demonstrated, and may contribute to the regulation of the self-renewal capacity of human pancreatic cancer stem cells (167, 233). In an in vitro model, sulforaphane was shown to inhibit the SHH signaling pathway, and reduced the expression of Smo, Gli1, and Gli2 (233). Sulforaphane was also shown to inhibit the nuclear translocation and transcriptional activity of Gli1 and Gli2 in a dose-dependent manner. After treatment for 1 week, the formation of human pancreatic cancer stem cell-derived spheres was significantly inhibited, suggesting the clonogenic depletion of the cancer stem cells. Similarly, sulforaphane treatment resulted in a significant reduction in the tumor growth of orthotopically impanted primary pancreatic cancer stem cells isolated from human pancreatic tumors into the pancreas of mice in vivo (167). Through the modulation of SHH signaling, decreased expression of downstream target genes (i.e., Nanog, Oct-4, VEGF, and ZEB-1) was also demonstrated.
The epithelial-mesenchymal transition (EMT), which is of critical importance in tumorgenesis and metastasis (118), has also been shown to be modulated after sulforaphane treatment with the downregulation of EMT markers, including ZEB-1, Twist-1, and vimentin (267). Consistent with this finding, sulforaphane was shown to inhibit the EMT process via COX2/MMP2,9/ZEB1, Snail, and miR-200c/ZEB1 pathways, and possessed the ability to suppress metastasis in human bladder cancer cells (249). Several studies have also shown that sulforaphane may inhibit the pro-survival PI3K/Akt pathway, which has been implicated in cellular survival and growth, and resistance (33, 83, 253). Sulforaphane alone and in combination with quercetin suppressed the growth of pancreatic cancer stem cells derived from pancreatic cancer cell lines in vitro, through the inhibition of the PI3K/Akt and MAPK/ERK pathways (237).
β-catenin, an important protein subunit of the cadherin protein complex and that functions as an intracellular signal transducer in the Wnt signaling pathway, has been shown to be important in the self-renewal of cancer stem cells and in the EMT process. In the human cervical carcinoma (HeLa) and hepatocarcinoma (HepG2) cell lines, sulforaphane has displayed a capacity to induce the downregulation of β-catenin (215). Li et al. evaluated the effect of sulforaphane on breast cancer stem cells with profound implications (169). Sulforaphane (1–5 μM) decreased aldehyde dehydrogenase (ADH)-positive cell population by 65%–80%, and reduced the size and number of primary mammospheres by 8- to 125-fold and 45%–75%, respectively, in vitro. A daily injection with 50 mg/kg sulforaphane for 2 weeks in nonobese diabetic/severe combined immunodeficient xenograft mice reduced the ADH-positive cell population by >50%. Sulforphane successfully eliminated breast cancer stem cells in vivo, thereby limiting tumor growth after implantation of primary tumor cells into secondary mice. Through Western blotting analysis and β-catenin reporter assay results, sulforaphane was shown to promote β-catenin phosphorylation and its subsequent degradation. It was thus proposed that the inhibition of breast cancer stem cells was caused, at least in part, by the downregulation of the Wnt/β-catenin self-renewal pathway.
Due to the finding that sulforaphane may possess therapeutic potential against cancer stem cells, its use in combination with clinically relevant drugs that limit tumor growth through its toxicity to differentiated stem cells may overcome limitations observed in current cancer management strategies, including resistance and reoccurrence (168). Kallifatidis et al. also demonstrated that sulforaphane enhanced the toxicity of various cytotoxic compounds, including cisplatin, gemcitabine, doxorubicin, and 5-flurouracil, toward pancreatic and prostate cancer stem cells (132). Sulforaphane increased the chemotherapeutic effects on self-renewal and ALDH activity, while limiting toxicity in normal cells. Significantly, the combination therapy abrogated tumor-initiating potential in vivo, with no adverse effects reported. Another study suggested that sulforaphane had the ability to potentiate the drug efficacy of imatinib by limiting leukemia cancer stem cells through the downregulation of β-catenin (171).
Epigenetic Modulation by Sulforaphane to Exert Anticancer Effects
The modulation of the epigenome is critical in the development and progression of cancer, contributing to the malignant transformation of cells through the ability to regulate gene expression without modifying the underlying DNA sequence (196). Dysregulated epigenetic processes, including aberrant DNA methylation, histone modifications, and post-transcriptional regulation of gene expression by noncoding microRNA, are implicated in multistage carcinogenesis, and may be regarded as promising targets for cancer prevention and management strategies (18). Dietary isothiocyanates, including sulforaphane and their subsequent metabolites, have displayed a capacity to regulate gene expression through epigenetic mechanisms, with further studies required in order to completely elucidate their impact in cancer chemoprevention (Fig. 6).
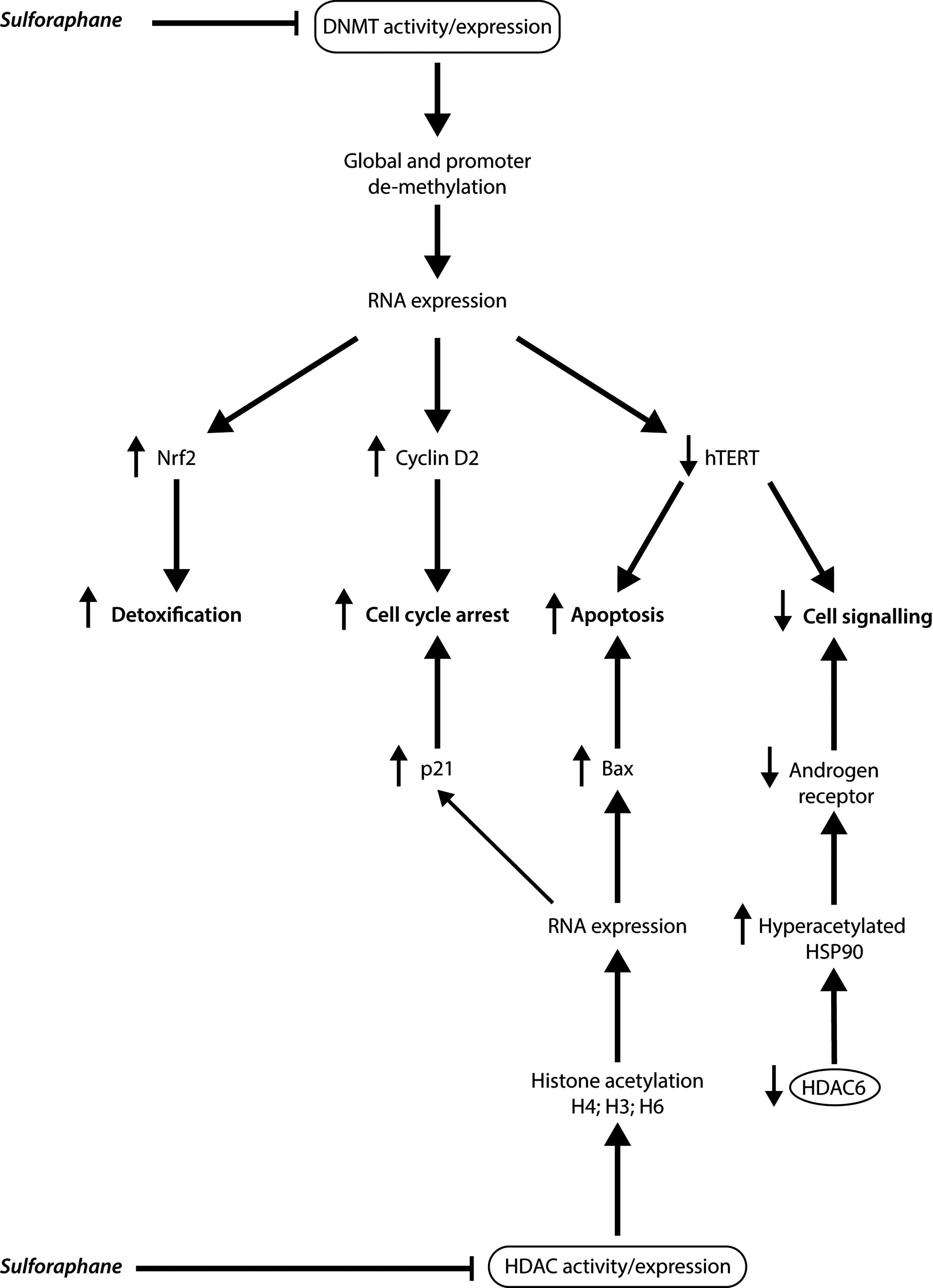
In general, the addition of acetyl groups to histones by histone acetyltransferase (HAT) promote gene expression through the ability to create an “open” chromatin conformation, allowing the transcription machinery to access DNA (304). Conversely, removal of acetyl groups by (histone deacetylase enzymes) HDACs results in a “closed” conformation, repressing transcription and inhibiting gene expression. An increase in HDAC activity and expression has been demonstrated in a number of cancer types, and may result in a repression of gene transcription that results in the deregulation of differentiation, cell cycle, and apoptotic mechanisms (68). In addition, important tumor suppressor genes, including p21, are observed to be a target for HDAC-mediated transcriptional silencing. HDAC inhibition has also been reported to disrupt the cell cycle in the G2 phase, allowing cells to prematurely enter the M phase, as well as to directly interfere with the mitotic spindle checkpoint (235).
HDAC inhibition was first reported by Myzak et al., who observed that sulforaphane increased TOPflash reporter activity without altering protein levels of β-catenin or HDAC1, indicating that the activity of HDAC itself was altered (200). Previous results had shown that TOPflash reporter activity may be used as an indirect measure of HDAC activity, with an increase in reporter activity corresponding to a decrease in HDAC activity in cells (22). In cytoplasmic and nuclear extracts from human embryonic kidney 293 (HEK293) cells treated with sulforaphane, HDAC activity was attenuated compared with untreated cells, global histone acetylation was increased, and activation of TOPflash was observed after its association with acetylated histones H3 and H4 (200). In addition, results from this study displayed the importance of sulforaphane metabolites (sulforaphane-N-acetylcysteine, SFN-NAC, and sulforaphane-cysteine, SFN-Cys) to produce a concentration-dependent inhibition of HDAC activity. It is proposed that SFN-Cys is the ideal dietary HDAC inhibitor, due to its formation during sulforaphane metabolism, and its regeneration from SFN-NAC (56, 57). Sulforaphane treatment also markedly increased p21Cip1/Waf1 protein expression due to an increase in acetylated histone H4 bound to the promoter region of p21. Similar results were observed in three prostate epithelial cell lines (BPH-1, LNCap, and PC3) with HDAC inhibition significant at a concentration of 15 μM sulforaphane (199). Interestingly, 15 μM sulforaphane also increased acetylated histone H4 association with the pro-apoptotic bax promoter and regulated bax mRNA expression, indicating that sulforaphane may directly mediate bax induction through chromatin remodeling. After a single oral gavage dose of sulforaphane in Apcmin mice, significant inhibition of HDAC activity in the mouse colonic mucosa was observed in vivo (198). HDAC activity was decreased by 50% with SFN-NAC and by ∼65% with sulforaphane in comparison to untreated mice. Both sulforaphane and its metabolite produced an increase in acetylated histone H3 and histone H4 in the colon, with a ∼two-fold increase at both the 6 and 24 h time points before returning to baseline levels. In Apcmin mice, sulforaphane suppressed tumor multiplicity, with the average tumor yield lowered by ∼50% in all regions of the intestine. Similar to the results from the in vitro studies, sulforaphane induced global histone acetylation, with a marked increase in acetylated histone H2 associated with the promoter regions of both the p21 and bax genes. Moreover, in human subjects, HDAC acitivity was significantly inhibited in the peripheral blood mononuclear cells of all three subjects, with detection at 3 h post consumption of 68g broccoli sprouts (201). In two of the subjects, HDAC inhibition was observed at the 6 h time point, with levels returning to baseline by 24–48 h post consumption. In the third subject, HDAC activity at baseline was lower than expected, and, therefore, the highest HDAC activity was observed at the 48 h time point. In continuation, healthy subjects consumed either the 68 g of broccoli sprouts or six supplements (∼3 g of freeze-dried broccoli sprouts) to assess isothiocyanate metabolite levels and HDAC activity (44). In addition to confirming that active myrosine is required for sulforaphane metabolism, inhibition of HDAC activity was measured and statistically significant at 12 and 48 h post consumption of either the sprouts or the supplementation, when compared with baseline HDAC activity. Further human studies are required in order to determine the significance of HDAC inhibition in disease, and the ability to provide therapeutic benefit via decreased HDAC activity.
Studies involved in the evaluation of sulforaphane as an HDAC inhibitor, and those aimed at providing insights into the mechanisms associated with sulforaphane-regulated inhibition of HDAC activity, have focused on the roles of HDAC3 and HDAC6 as important targets (43, 89, 223, 224). After the incubation of HCT116 human colon cancer cells with 15 μM sulforaphane, a significant reduction in HDAC1, HDAC2, HDAC3, and HDAC8 was observed compared with vehicle-treated cells at 36 h post administration (223). Among the class I HDACs, HDAC3 protein expression was the most susceptible to sulforaphane-induced loss, with cells treated with 35 μM reporting a reduction of HDAC3 expression by more than 95%. HDAC4 and HDAC6 were also shown to be reduced after 24 h. Overexpression of either HDAC3 or HDAC6 was shown to inhibit sulforaphane-induced histone H4-K12 acetylation (important in the regulation of gene expression). The silencing mediator for retinoid and thyroid hormone receptors (SMRT) was phosphorylated in the nucleus within 6 h of sulforaphane treatment, and was, subsequently, implicated in HDAC3 turnover mechanisms. Sulforaphane-induced association of HDAC3/SMRT complexes coincided with increased binding of HDAC3 to 14–3-3 and peptidyl-prolyl cis/trans isomerise 1 (PIN1), ultimately directing HDAC3 for degradation. Further investigations led to the finding that sulforaphane metabolites may interact with the allosteric site between HDAC3 and SMRT, providing new insights into the dissociation of HDAC3/SMRT complex in colon cancer cells (224). Sulforaphane and its metabolites were also shown to alter the acetylation status of key DNA repair protein CtIP, with a significant increase at 6 h post treatment. The induction of double-strand breaks (measured by γH2AX immunofluorescence) increased after sulforaphane treatment of cancer cells, but failed to effect noncancer controls. Collectively, this evidence suggests that sulforaphane may also compromise DNA repair mechanisms in cancer cells with selectivity.
In prostate cancer cells, sulforaphane treatment increased acetylation of two HDAC6 target proteins: heat shock protein 90 (HSP90) and α-tubulin (89). Despite unchanged HDAC6 protein levels at early time points, HSP90 became hyperacetylated and dissociation of the androgen receptor (AR) from HSP90 was observed. At later time points, AR protein levels declined after sulforaphane treatment, indicating the ability of sulforaphane to destabilize AR. Importantly, incubation of recombinant HDAC6 with sulforaphane inhibited HDAC6 deacetylase activity, with sulforaphane leading to reduced HDAC6 protein levels at the 16 h time point in prostate cancer cells. Clarke et al. demonstrated the ability of sulforaphane to preferentially induce apoptosis and cell cycle arrest in the benign prostatic hyperplasia (BPH-1) cell line and the LNCap and PC3 prostate cancer cell lines, as well as the induction of phase II enzymes (43). At 24 h post incubation, HDAC inhibition was observed in all cells lines, with LNCap cells with a significant inhibition compared with healthy controls. By the 48 h time point, sulforaphane induced sustained HDAC inhibition in all cancer cell lines. Specifically, HDAC2 and HDAC3 protein levels were decreased in BPH-1 cells and HDAC3 in both LNCap and PC3 cell lines at 48 h post treatment. HDAC4 protein expression was decreased in all cell lines at one or both time points. HDAC6 showed the most significant and consistent decrease in protein levels in all prostate cancer cell lines at both 24 and 48 h time points, with this decrease not observed in healthy prostate cells (PrEC). Interestingly, overexpression of HDAC6 protected PC3 cells from the sulforaphane-induced decrease in cell viability, indicating the likely importance of HDAC6 in sulforaphane-mediated chemoprevention in prostate cancer.
The modulation of patterns in DNA methylation are observed during cancer development and progression, and is characterized by global- and site-specific DNA hypomethylation as well as gene-specific promoter hypermethylation (17, 220). While DNA hypomethylation may contribute to genome instability and increased expression of oncogenes, DNA hypermethylation may lead to the inhibition of tumor suppressor genes, transcription factors, and genes involved in cell cycle regulation and apoptosis. DNA methylation patterns are mediated by DNA methyltransferases (DNMTs), and an overexpression of DNMTs is observed in a number of cancers, including leukemic, gastric, lung, and prostate camcer (69, 172, 193, 195).
Meeran et al. first observed the significant inhibition of DNMT1 and DNMT3a expression by sulforaphane in a dose-dependent manner in human breast cancer cells (MCF-7 and MDA-MB-231 cell lines), and to a lesser extent in normal MCF10A cells (186). Specifically, 10 μM sulforaphane in 6 days inhibited DNMT1 and DNMT3a expression by 48% and 78%, respectively. Ultimately, this study aimed at assessing the telomerase activity and human telomerase reverse transcriptase (hTERT; important catalytic component of telomerase) in human breast cancer, and the effect of sulforaphane on hTERT expression. Sulforaphane treatment led to the significant downregulation of hTERT in breast cancer cells, with a negligible inhibitory activity in the normal control, indicating specificity. Investigation into the molecular mechanisms of sulforaphane-induced inhibition of hTERT expression found significant demethylation of CpGs in the CTCF binding region on the hTERT regulatory region in both breast cancer cell lines after treatment with sulforaphane. It is known that the transcription repressor CTCF binds to exon 1 of the hTERT gene and, in turn, reduces the expression of hTERT. Treatment with sulforaphane was shown to increase CTCF binding to the hTERT exon 1 binding site to subsequently inhibit hTERT expression. It is suggested that the downregulation of hTERT expression facilitated the induction of cellular apoptosis in the cancer cell lines.
Similar results in prostate cancer cell lines established the ability of sulforaphane to significantly decrease DNMT1 and DNMT3a mRNA expression (113). Decreased global methylation in LNCap cells after treatment with sulforaphane for 24 h was also observed. In prostate cancer, cyclin D2 silencing has been observed to be associated with cancer progression, with restoration of cyclin D2 inducing cell death in prostate LNCap cancer cells (149). Investigation of the promoter methylation status of cyclin D2 in prostate cancer cells reported a significant decrease in methylated CpG sites in the region after sulforaphane incubation. In particular, sulforaphane significantly decreased methylation at the binding site of the c-Myc transcription factor (observed to be hypermethylated in untreated tumor cells). Due to the demethylation of the cyclin D2 promoter, sulforaphane increased cyclin D2 mRNA expression in a dose-dependent manner. Conversely, treatment of colon cancer cells with sulforaphane in varying concentrations failed to impact abnormal methylation patterns in critical genes involved in colon carcinogenesis in vitro (16). However, Barrera et al. suggest that their finding may be due to heterogeneity in cancer, with some cell lines and/or tissue affected by sulforaphane-induced gene-specific methylation, while others are more resistant.
Recently, assessment on the DNA methylation profile in normal prostate epithelial cells (PrEC), androgen-dependent (LNCap) and androgen-independent (PC3) prostate cancer cells was shown to be altered depending on the cell line (298). Untreated LNCap and PC3 cells had significantly higher baseline expression of DNMT1, DNMT3a, and DNMT3b compared with untreated PrEC cells. While sulforaphane treatment of PrEC and LNCap cells decreased DNMT1 and DNMT3b gene expression, the expression of all three DNMTs was downregulated in the PC3 cell line. Hierarchical clustering analysis showed that 64% of the 54,876 probes and 49% of the 78,272 probes in LNCap cells displayed hypermethylation relative to PrEC cells (298). This represented 10,315 and 8013 genes differentially methylated in LNCap and PC3 genes, respectively, compared with the normal control. Functional annotation analyses indicated that genes with altered methylation profiles in the prostate cancer cell lines were genes involved in cancer progression, and associated with cell migration, cell adhesion, cell–cell signalling, as well as transcription regulation (298). After sulforaphane treatment, 2472 (in PrEC cells), 3508 (in LNCap cells) and 6778 (in PC3 cells) differentially methylated genes were detected. Interestingly, sulforaphane altered methylation in distinct sets of genes in each of the cell lines (298). In PrEC cells, genes involved in transcription, apoptosis, and chromatin organization/modification were altered with sulforaphane treatment. Functional annotation analysis in LNCap cells found that two general categories of genes were affected: (1) genes associated with cell movement, including cell migration, adhesion, and localization, and (2) genes associated with the immune response, including inflammation, leukocyte activation, and immune regulation. Sulforaphane treatment in PC3 cells altered genes shared both PrEC and LNCap cell lines, including those involved in transcription, apoptosis, cell migration, and immune response (298). Perhaps the most interesting finding was the reversal of the methylation profiles of 1509 (14.6%) genes (out of 10,315 genes) after sulforaphane treatment in LNCap cells. Many of these genes are known to be dysregulated or are highly involved in cancer progression, including transforming growth factor-β1 receptor type I (TGFBR1), C-C chemokine receptor type 4 (CCR4), C-X-C chemokine receptor receptor type 4 (CXCR4), and cysteine-rich angiogenic inducer 61 (CYR61). Overall, this study demonstrated the capacity of sulforaphane to modulate DNA methylation and regulate gene expression in order to act as a chemopreventive compound in prostate cancer in vitro (298). Further studies are required in order to enhance understanding of the epigenetic modulatory mechanisms of sulforaphane.
As previously described, sulforaphane was first identified as a potent inducer of phase II detoxifying enzymes via the disruption of Nrf2-Keap1 interactions, and an increased translocation and therefore activation of Nrf2 (117, 148, 206). Zhang et al. aimed at investigating the potential of sulforaphane to reactive the expression of Nrf2 through epigenetic regulation (310). In prostate cancer TRAMP C1cells, Nrf2 transcription was significantly inhibited when the first five CpG regions of the Nrf2 gene promoter were hypermethylated (309). Treatment with sulforaphane (1.0 and 2.5 μM) reduced the level of methylation to 56.0% and 38.7%, respectively. Demethylation of the promoter region was shown to result in the transcription activation of Nrf2, with increased mRNA and protein expression measured. This study also demonstrated decreased protein levels of DNMT1 and DNMT3a after sulforaphane treatment in a dose-dependent manner in TRAMP C1 cells. In addition, sulforaphane decreased protein levels of HDAC1, HDAC4, HDAC5, and HDAC7, indicating HDAC inhibitory activity. The global level of acetylated histone 3 was also highly induced by sulforaphane treatment. Comparable to this study, sulforaphane significantly inhibited 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced mouse skin epidermal (JB6) transformation via its ability to enhance nuclear translocation of Nrf2 and increase expression of Nrf2-target antioxidant genes, including HO-1, NQO1, and UGT1A1 (273). Bisulfite sequencing determined that 15 CpG regions in the Nrf2 gene promoter were hypermethylated in the untreated cells. The ratio of methylated CpG regions was decreased to 68.7% by 2.5 μM sulforaphane. Furthermore, sulforaphane decreased the protein expression of DNMT1, DNMT3a, and DNMT3b in a concentration-dependent manner in TPA-induced JB6 cells post treatment (5 day period). In addition, treatment with either 2.5 or 5.0 μM sulforaphane significantly inhibited relative HDAC activity by 50%, with reduced protein expression levels of HDAC1, HDAC2, HDAC3, and HDAC4 observed. These findings suggest that sulforaphane has the ability to restore expression and activation of Nrf2 via a number of epigenetic regulatory mechanisms.
MicroRNAs (miRNAs) are short noncoding RNAs, 18 to 25 nucleotides in length, that regulate gene expression post-translation through binding to 3′ untranslated regions of target mRNA (261). Bioinformatic studies predict that miRNAs regulate almost 30% of all mammalian genes, and are dysregulated in the majority of human cancers (29). The assessment of sulforaphane in the regulation of miRNA expression is limited in the literature, and further investigation is required in order to complete understanding. The treatment of nontransformed human colonic epithelial cells with sulforaphane resulted in significant changes in miRNA expression profiles (15 upregulated and three downregulated) as analyzed by quantitative real-time reverse-transcription polymerase chain reaction (260). Global downregulation of miRNA expression was demonstrated from colorectal tumor tissue. Focusing on miRNA expression changes in both cell lines, the upregulation of miR-23b and miR-27b was significant due to a previous observation of decreased levels in clinical tumor tissue (241). MiR-23b has been shown to be critical in tumor suppression and regulation of EMT (30, 311). Conversely, downregulation of miR-155 (a well-known oncogenic miRNA) by sulforaphane was also identified as important (280). In addition, the expression of miR-200c, which has been identified in the regulation of EMT in human bladder cancer (3), was significantly induced by sulforaphane in the human bladder T24 cancer cell line (248).
In basal-like ductal carcinoma in situ (DCIS) lesions, 68 miRNAs were significantly dysregulated in tissue compared with controls via microarray analysis (166). The downregulation of miRNA-140 expression was indicated to be the most reproducible miRNA signature in the lesions that was implicated in tumor suppression. In a subsequent analysis of 22 DCIS samples, Li et al. observed the miR-140 loss in all tumors irrespective of histological grade and breast malignancy type. Also demonstrated was the finding that the 72 miRNAs were found to be differentially expressed between normal mammary epithelial stem cells (isolated from MCF-10A cells) and stem-like cells isolated from the DCIS lesions (denoted as MCF10DCIS cells). Interestingly, miR-140 was one of the most significantly downregulated in cancer stem-like cells, with a 13.55-fold difference compared with normal stem cells. Treatment of DCIS tissue with sulforaphane was shown to activate miR-140 expression and downregulation of SOX9 and ALDH1 (important in stem cell regulation, and targets of miR-140 in DCIS stem-like cells), resulting in a decrease in stem-like cell frequency in vitro and a significant inhibition of tumor growth in vivo. Further characterization of exosomal trafficking of a stem-like population in basal-like DCIS lesions resulted in the finding that the exosomal miRNA expression pattern in DCIS stem-like cells possessed an inverse trend in comparison to nontumorigenic MCF10A stem cells (165). For example, miR-21 and miR-29 were secreted in high levels in the DCIS stem-like cells, with normal stem cells secreting much lower levels. In the context of sulforaphane, treatment resulted in increased exosomal miR-140 (with the capacity to transfer into breast cancer cells) and decreased miR-21 and miR-29. Collectively, this evidence suggests that sulforaphane may have the capacity to inhibit DCIS stem cell signaling in neighboring cells through epigenetic mechanisms by increasing exosomal miR-140 secretion in the tumor microenvironment.
Sulforaphane in Human Clinical Trials
Multiple commercially developed sulforaphane supplements are currently available; however, the difficulty in manufacturing a potent and bioavailable formula has proved to be difficult, with an intrinsic instability of the sulforaphane molecule preventing this method of delivery (64). Manufacturing a sulforaphane-yielding supplement requires the ability to retain both the glucoraphanin precursor and the myrosinase enzyme for subsequent metabolism and transformation to the bioactive isothiocyanate (111). A number of phase I and II clinical trials on sulforaphane, however, are in progress or have been completed to assess its safety, tolerance, pharmacokinetics, and therapeutic benefit in healthy human subjects and in the context of cancer (Table 5).
BSE, broccoli sprout extract.
Residents of Qidong are at high risk for the development of hepatocellular carcinoma, which was in part due to long-term exposure to aflatoxin-contaminated food, and the airborne carcinogen, phenanthrene (67). An inverse association between the level of sulforaphane metabolites and carcinogen-related markers, including aflatoxin-DNA adducts, was demonstrated (139). It was also reported that sulforaphane increased the excretion of airborne pollutants in individuals consuming the broccoli extract beverage, with the administration of a broccoli sprout-infused beverage containing 400 μM glucoraphanin nightly for 2 weeks causing no adverse effects and being well tolerated in 200 subjects (140). Although promising, results displayed significant variability in the bioavailability of the active compound. Consistent with this finding, Fahey et al. found that administration of a sulforaphane-rich broccoli sprout extract to two distinct populations (Chinese and Baltimoreans) resulted in varied bioavailability between individuals in both populations, ranging from 1% to 40% (72). In a recent intervention study, the total sulforaphane metabolite concentration in plasma was the highest (>2 μM) at 3 h in human subjects who consumed fresh broccoli sprouts (40g) in the first phase of study, compared with that measured after administration of a commercially available broccoli supplementation in the second phase (42).
A randomized, placebo-controlled double-blind Phase I clinical trial involving healthy volunteers and the administration of glucoraphanin or isothiocyanate as the sulforaphane source examined parameters of safety, tolerance, and pharmacokinetics (250). No significant toxicity was observed with the dose concentrations administered in this study. Although the plasma concentration of total bioactive sulforaphane may be measured in order to provide a crude estimate of bioavailability and therapeutically significant dosage, accumulation within the tumor site itself may not be significant. For example, a pilot study detected an accumulation of sulforaphane in human breast tissue after consumption of broccoli extract containing 200 μM sulforaphane ∼1 h before elective reduction mammoplasty (51). Mean epithelial-/stromal-enriched breast tissue dithiocarbamate concentration was 1.45±1.12 pmol/mg in the right breast, and 2.00±1.95 pmol/mg in the left breast. In comparison, the plasma concentration post sulforaphane administration was measured at 0.92±0.72 μM.
Future Directions and Combinatorial Cancer Management Strategies, Including Sulforaphane
Although the chemopreventive properties of sulforaphane have been widely investigated and extensively reported, the difficulty in the administration of a dose that may confer therapeutic significance limits its potential for clinical use. In addition, the estimated consumption required to observe benefits in limiting cancer development and progression has been shown to be unrealistic in the average diet. Limitations in the bioavailability of the compound after administration and/or consumption may also become problematic in human trials as genetic variance and patient compliance become factors. The use of a combination of anticancer compounds that affect different functional pathways may possess the capacity to generate additive or synergistic activity. In theory, this is an attractive approach for the prevention and/or treatment of complex disease, including cancer.
Current chemotherapeutic agents fail to adequately treat malignancy with multivariable dose-restricting factors, including systemic toxicity and multi-drug resistance limiting therapeutic benefits and long-term remission rates. Interestingly, a number of studies have aimed at using sulforaphane in combination with a range of cytotoxic compounds in order to enhance drug efficacy, and, therefore, limit adverse effects observed in chronic administration and high concentration regimens. To demonstrate, sulforaphane was shown to enhance the antitumor activity of oxaliplatin and synergistically activate apoptotic pathways in the colorectal cancer cell line, Caco-2 (135). Fimognari et al. investigated the effects of sulforaphane in combination with doxorubicin on cell viability and apoptosis in fibroblasts characterized by a different p53 status (wild-type, knockout, and with a mutation at codon 220) (79, 80). This mutation has been implicated in reduced efficacy and drug resistance in osteosarcomas and breast cancer treated with doxorubicin (28, 87). Sulforaphane was shown to restore chemosensitivity and to induce apoptosis in doxorubicin-resistant p53 mutated and p53 knockout cells, irrespective of p53 status. The induction of apoptosis was caspase-3 dependent and caspase-8 independent. Interestingly, proliferation of salivary gland adenoid cystic carcinoma high metastatic cell line (ACC-M) and low metastasis cell line (ACC-2) observed to be relatively resistant to a classic chemotherapeutic agent, 5-fluroracil, was inhibited with a synergistic combination using sulforaphane (294). A decreased expression of NFκB p65 protein was also demonstrated, with results more significant in ACC-M cells. Although effective in acute promyelocyctic leukemia (APL), arsenic trioxide fails to adequately treat non-APL blood cancers (65). Significantly, sulforaphane enhanced arsenic trioxide-mediated cytotoxicity and apoptosis in a panel of leukemic cell lines, with a dramatic increase in the number of ROS compared with treatment with either agent alone. Continuing with this combination, Doudican et al. co-treatment of multiple myeloma cells resulted in elevated expression of the molecular chaperone HSP90, along with increased protein kinase RNA-like endoplasmic reticulum kinase (PERK), and important makers of unfolded protein response (UPR) activation (66). Sulforaphane in combination with arsenic trioxide effectively disrupted protein homeostasis through the production of ROS and induction of apoptotic processes. Synergistic effects in pancreatic cancer stem cells were demonstrated when sulforaphane was administered in combination with sorafenib (multikinase inhibitor in clinical trials) (227). It was proposed that these effects were mediated by the reduction of sorafenib-induced NFκB binding by sulforaphane. Although treatment with sulforaphane at varying concentrations resulted in a dose- and time-dependent decrease in cell viability of MCF-7 cells, a combination of sulforaphane and gemcitabine demonstrated the capacity of sulforaphane to enhance the growth inhibitory effects of gemcitabine at sub-lethal doses (116). Administration of sub-lethal doses of sulforaphane (5–10 μM) when used in various combinations with lower doses of gemcitabine (5–10 mM) resulted in a decrease in cell viability that was more pronounced than either of the compounds alone and the combination index (CI) was found to be <1, indicting synergistic effects.
In addition, interactions with other known chemopreventive and antioxidant dietary components have also observed synergistic activity after administration in a number of models and systems. For example, the administration of sulforaphane and resveratrol (polyphenolic compound) inhibited cell proliferation and migration, reduced cell viability, induced lactate dehydrogenase release, decreased pro-survival Akt phosphorylation, and increased caspase-3 activation in human U251 glioma cells (126). A combination of sulforaphane and the flavonoid apigenin was shown to modulate gene expression of phase II detoxifying enzymes (including GST and UGT) in the human epithelial colorectal adenocarcinoma cell line, Caco-2 (275). This combination resulted in a synergistic induction of UGT1A1 mRNA by approximately 12-fold. The indole, 3,3′-diindolylmethane (DIM) also derived from cruciferous vegetables has been shown to have cytostatic mechanisms in human colon cancer cell lines (213). In this same study, sulforaphane (10 μM) in combination with DIM (10 μM) resulted in strong G2/M cell cycle arrest, which was not observed with either compound alone. Nair et al. also demonstrated a synergistic effect after administration of sulforaphane and the catechin epigallocatechin-3-gallate in HT-29 AP-1 human colon carcinoma cells (202). This combination dramatically enhanced transcriptional activation of the Ap-1 reporter, with analysis finding that the CI was <1. Cell viability assays showed that low-dose combinations decreased cell viability to 70%, and to 40% with high-dose combinations at 48 h post treatment. Pretreatment with 100 ng/ml of TSA (a potent HDAC inhibitor) potentiated (by 88-fold) the synergism observed with the low-dose combination on the AP-1 reporter transcriptional activation. Pancreatic stem cells and their self-renewal capacity may also be modulated with a combination of sulforaphane and the dietary polyphenol, quercetin (267, 319). The synergistic effect of this combination has also been observed in melanoma B16F10 cells, with the suppression of proliferation and migration due to an implicated decrease in MMP-9 expression (221).
Recent data published by Grandhi et al. reported a novel combinatorial nano-based delivery of chemopreventative agents in the suppression of pancreatic carcinogenesis induced by N-nitroso-bis(2-oxopropyl)amine in hamsters (95). A combination of aspirin and curcumin in a solid lipid nanoparticle formulation, and sulforaphane in solution was administered to hamsters by daily oral gavage, which resulted in a decrease in the effective inhibitory dosages by a factor of 10 as compared with these compounds in free forms. This combination of chemopreventive compounds reduced tumor incidence, tumor multiplicity, and severity of histologic lesions.
Conclusion
The importance of a diet that is rich in cruciferous vegetable and the role of isothiocyanates, including sulforaphane, in promoting good health has been extensively studied and consistently demonstrated in a wide variety of models and systems. Understanding the phylogeny of glucosinolate-producing plants, in conjunction with the molecular genetics of the Brassicaceae family may enable us to further enhance bioavailability of the bioactive compounds through the development of cultivars with significantly higher amounts of glucoraphanin. Bioavailability and therefore pharmacokinetics of sulforaphane are also dependent on a range of other factors, including active myrosinase content, preparation, and human genetic patterns. Although commercially available as a supplement, sulforaphane has yet to be FDA approved for the treatment of human disease, including cancer. However, the chemopreventive properties of sulforaphane, and its capacity to be selectively toxic to malignant cells and impart these effects through a number of mechanisms, provide rationale to completely elucidate and evaluate its potential as an anti-cancer compound alone, and in combination with clinically relevant therapeutic and management strategies.
Footnotes
Acknowledgments
The support of the Australian Institute of Nuclear Science and Engineering (AINSE) is acknowledged. TCK was the recipient of AINSE awards. TCK was supported by an Australian Research Council Future Fellowship, and the Epigenomic Medicine Laboratory was supported by McCord Research. This study was supported in part by the Victorian Government's Operational Infrastructure Support Program.
Author Disclosure Statement
Epigenomic Medicine Laboratory (TCK) receives funding from McCord Research, which has a commercial interest in sulforaphane. Stephanie Tortorella, Simon G Royce, and Paul V Licciardi have no conflict of interest.