
Abstract
Introduction
Angiogenesis
A
Under physiological conditions, angiogenesis is finely regulated. Typically, normal angiogenesis occurs in the embryo, where it establishes the primary vasculature for growing and developing organs (38). Angiogenesis occurs in the adult during the ovarian cycle and in physiological repair processes such as wound healing (70). Lack of regulation of angiogenesis leads to pathological conditions. Healing of larger wounds depends on wound angiogenesis (108). Cutaneous wound healing is a multi-stage process that orchestrates the reconstitution of the defective dermal and epidermal layers of the skin. During the proliferative phase, the wound microvasculature is reconstructed to re-establish the nutrient supply to regenerating tissue, promote fibroplasia, and prevent sustained tissue hypoxia. Failure to do so will disallow tissue growth (44). Induction of angiogenesis enhances healing rate, particularly under conditions such as diabetes, where peripheral blood supply is limited. Seminal works by Dr. Judah Folkman have helped us to understand that tumor formation is dependent on angiogenesis (37). Angiogenesis plays a pivotal role in tumor growth, progression, invasiveness, and metastasis (31). For tumors to develop in size and metastatic potential, they must make an “angiogenic switch.” Angiogenesis is thought to be driven largely by hypoxia in tumors and is particularly relevant to metastatic spread, as primary tumor cells enter the circulation via these new blood vessels (40). Further, the endothelial angiogenic cells can enhance the invasive and metastatic potential of cancer cells (59, 137). Studies showed that in vitro co culture of endothelial cells (ECs) with prostate cancer cells resulted in TGF-β/MMP-9 signaling, leading to increased invasion of prostate cancer cells (137). Moderate hypoxia serves as a productive micro-environmental cue, causing the induction of key angiogenic cytokines such as the vascular endothelial growth factor (VEGF-A) (26). Thus, moderate hypoxic conditions are present in tumors, developing embryo and during the initial stages of wound healing driving angiogenesis (131).
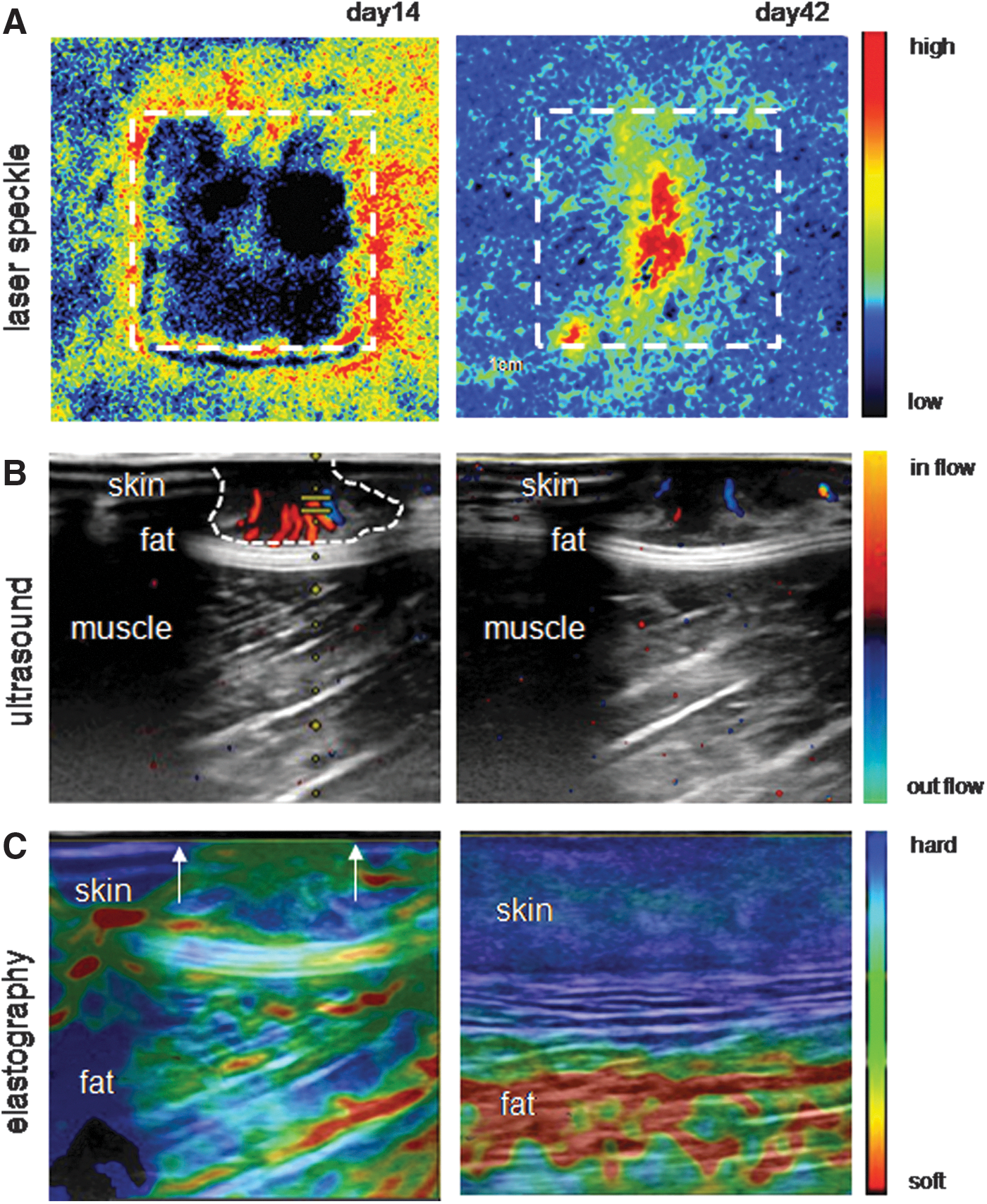
Wound angiogenesis can be studied by emerging noninvasive imaging technologies. Laser speckle provides information regarding blood perfusion in a 2D area; ultrasound provides information regarding the wound depth and angiogenesis; whereas the healing can be monitored by tissue elastography (43) (Fig. 1). These tools have enabled the repeated study of the healing tissue without the need for biopsies, providing unprecedented insights into the dynamics of cutaneous wound angiogenesis.
Angiogenesis, vasculogenesis, and vascular mimicry refer to development of new blood vessels; each process is physiologically distinct from the other. Angiogenesis comprises of two mechanisms: endothelial sprouting and intussusceptive microvascular growth. In the initial step of angiogenesis, ECs start migrating and proliferating (105). Therefore, at the onset of angiogenesis, regulators associated with blood vessel homeostasis need to be transiently suppressed. Indeed, transient disruption of the integrity of cell-to-cell binding is required to induce angiogenesis (133). Vasculogenesis refers to in situ differentiation and growth of blood vessels from mesodermal-derived hemangioblasts. Vasculogenic mimicry is a process in which highly aggressive and metastatic cancerous cells are able to form highly patterned vascular channels that are lined externally by tumor cells, without the requirement of ECs (89). Blood vessel development through any of these processes requires the process of epithelial–mesenchymal transition (EMT).
EMT is a reversible dedifferentiation process that leads epithelial cells to dedifferentiate, generating cells with mesenchymal features. Epithelial cells lose cell polarity, cell-to-cell contact and gain migratory and invasive characteristics. This process is characterized by the loss of epithelial traits and the acquisition of mesenchymal phenotypes (62, 130). Maintenance of healthy tissue is dependent on a fine balance between pro-growth and growth arrest factors. In case of tissue insult or during a pathogenic condition, these breaking mechanisms transiently disengage themselves. One such breaking mechanism is afforded by microRNAs or miRs, which confer post-transcriptional silencing of gene expression.
microRNAs
MicroRNAs (miRNAs/miRs) are short noncoding RNAs of ∼21–23 nucleotides in length. The database of miRNAs, miRBase, enlists 35,828 mature miRNA sequences across 223 species with 2588 miRNAs in human [miRBase (46); Version 21]. miRs are primarily involved in post-transcriptional gene silencing. They bind to target mRNA transcripts, leading to translational repression or degradation of mRNA transcripts. This pairing between miR and mRNA is usually of partial complementarity, resulting in a single miR targeting numerous mRNA transcripts. A single miR, on an average, is predicted to target around 200 mRNA transcripts (73). What makes this regulatory network even more interesting is the observation that a single mRNA is targeted by more than one miR depending on the length of the 3′-UTR of the mRNA.
miR genes are transcribed by RNA polymerase II or RNA polymerase III into primary miRNA transcripts (pri-miRNA) (10, 78). On transcription, primary miRNAs fold into hairpins. miRNA hairpins are reported to be different from the hairpins found in mRNAs and other types of noncoding RNAs (95). Based on their genomic position, they are either located in the form of independent genes, referred to as intergenic miRs, or situated in introns of protein coding genes, referred to as intronic miRs. It is often assumed that intronic miRs are processed from spliced introns. However, in certain cases, the miR hairpin are actually cleaved first, followed by splicing of the severed mRNA (68). The intronic miRs are reported to be co-transcribed with their host protein coding genes. Thus, intronic miRs are co-regulated with the coding genes. Intergenic miRs are regulated independently of other genes. Unlike protein coding genes and similar to some tRNA genes, miRs might be transcribed together; these miRs are termed clustered miRs. Clustered miRs can be intronic or intergenic. After transcription, these miRs are then individually processed.
miRs transcribed by RNA POL II are 5′ capped and polyadenylated. The RNase III enzyme Drosha and the double-stranded RNA-binding domain (dsRBD) protein DiGeorge syndrome critical region gene 8 (DGCR8, Pasha in Drosophila melanogaster) form the nuclear microprocessor complex that recognizes the stem loop structure of the pri-miRNA and cleaves at the stem of the hairpin, thus producing an approximately 70 nt stem loop miR precursor (pre-miRNA). Exportin-5 in complex with Ran-GTP then transports the pre-miRNA to the cytoplasm (150). Exportin-5 recognizes the pre-miRNA independent of its sequence or the loop structure. A defined length of the double-stranded stem and the 3′ overhangs are important for successful binding to Exportin-5, ensuring the export of only correctly processed pre-miRNAs (86). A schematic representation of the miR biogenesis has been shown in (Fig. 2).
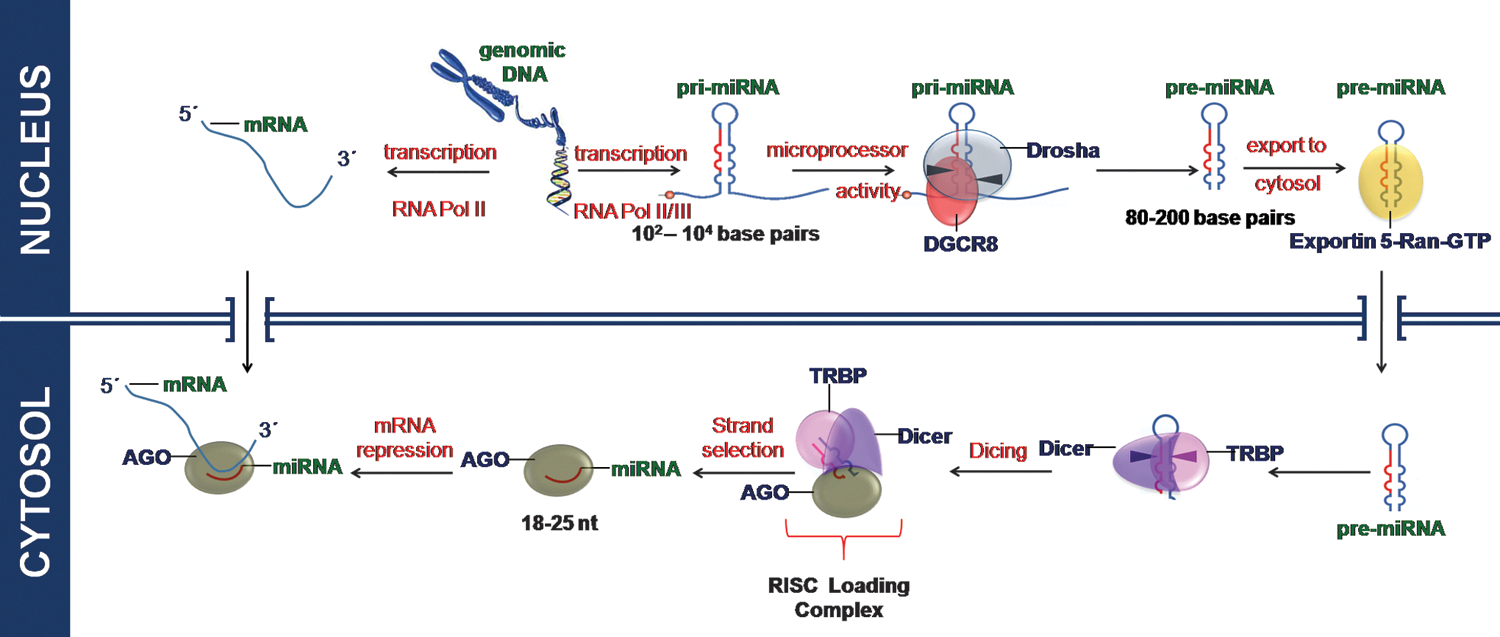
Some of the intronic miRs are believed to bypass the pri-miRNA stage and emerge straight as pre-miRNAs not requiring the Drosha processing step. Such intronic miRs are termed mirtrons. In a conventional mirtron locus, the resultant small RNAs begin and end precisely with splice donor and splice acceptor sites. In other words, both ends of the pre-miRNA are generated by the splicing reaction. Mirtrons were originally recognized in flies and worms, but similar loci (i.e., short hairpin introns associated with small RNA arising from introns) were later found in rodents, primates (3, 8), chicken (42), and rice (153).
On reaching the cytoplasm, another RNase III enzyme termed Dicer generates a 21–25 nt long miR duplex intermediate with 5′ phosphates and 2 nt 3′ overhangs, referred to as miRNA/miRNA* (miRNA star). For functional activity, the double-stranded duplex is separated using helicase associated with the RISC complex into the functional guide strand. This strand is complementary to the target mRNA transcript. The other strand called the passenger strand is subsequently degraded. Following the thermodynamic asymmetry rule (64, 113), one strand of the intermediate double-stranded RNA, typically with a relatively lower stability of base pairing at the 5′ end, is incorporated into RISC complex by binding of the miRNA to a member of the Argonaute protein family. The opposite strand, the miRNA*, is degraded; however, there are also reports of functional miRNA* sequences, especially under distinct cellular conditions and in different tissues (54, 114). In some cases, both the strands are retained and are incorporated into the RISC complex; in such cases, miRNA arising from 5′-end of the precursor miRNA (pre-miRNA) is termed miRNA-5p and that arising from 3′-end of the pre-miRNA is termed miRNA-3p.
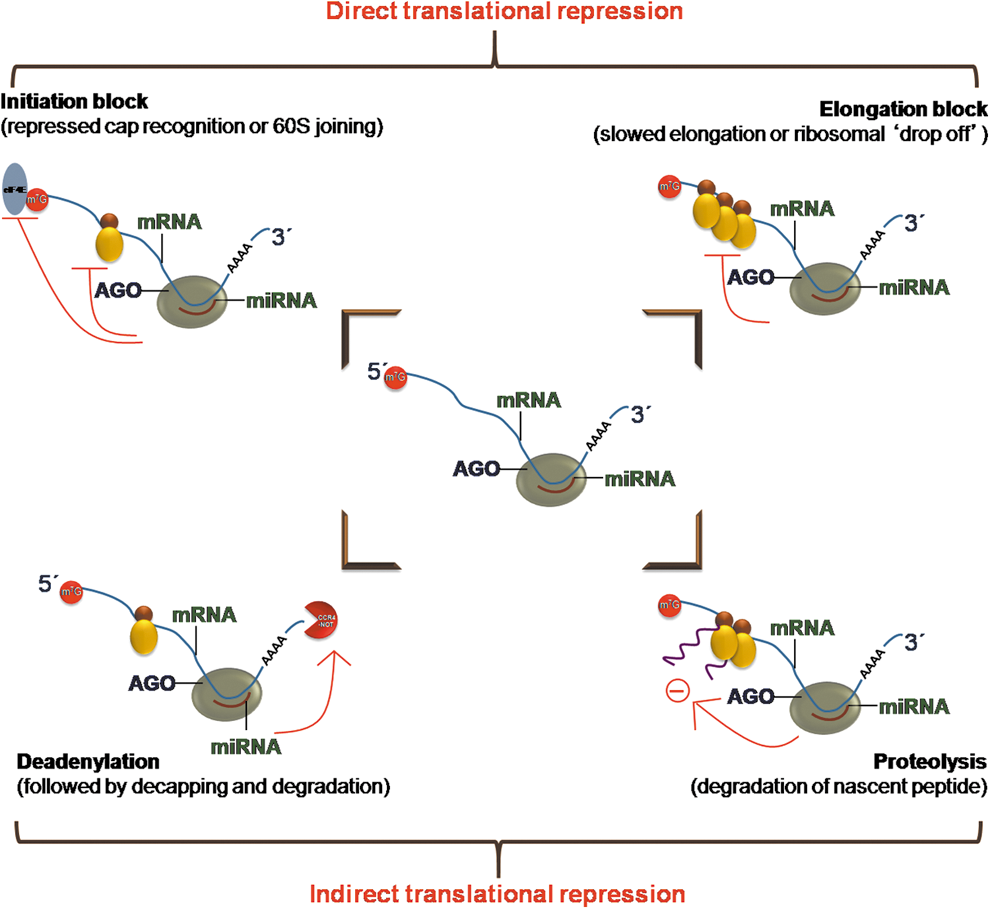
Within the RISC complexes, miRs guide Argonaute proteins to fully or partially complementary miR targets, which are then silenced post-transcriptionally. The mechanism of translational repression by miRs can be mediated through ribosomal dropoff during translation, translational initiation blockage, decapping of mRNA, or degradradation of mRNA transcript (32) (Fig. 3).
microRNAs in angiogenesis
Significance of miR in the regulation of mammalian vascular biology was established from studies involved in blocking miR biogenesis to deplete the miR pools of vascular tissues and cells (74, 127). In 2008, our group first reported on the regulation of angiogenesis by miRs (123) and since then, we have studied how miRs regulate wound healing (6, 9, 16, 27, 106, 107, 117, 122) and angiogenesis (17 –19, 108, 115, 116, 123). The class of miR is often designated by prefixing its function to miR. For example, oxymiRs are miRs dysregulated in response to the state of tissue oxygenation (118). Similarly, miRs that collectively act on pathways modulating angiogenesis are collectively referred to as angiomiRs.
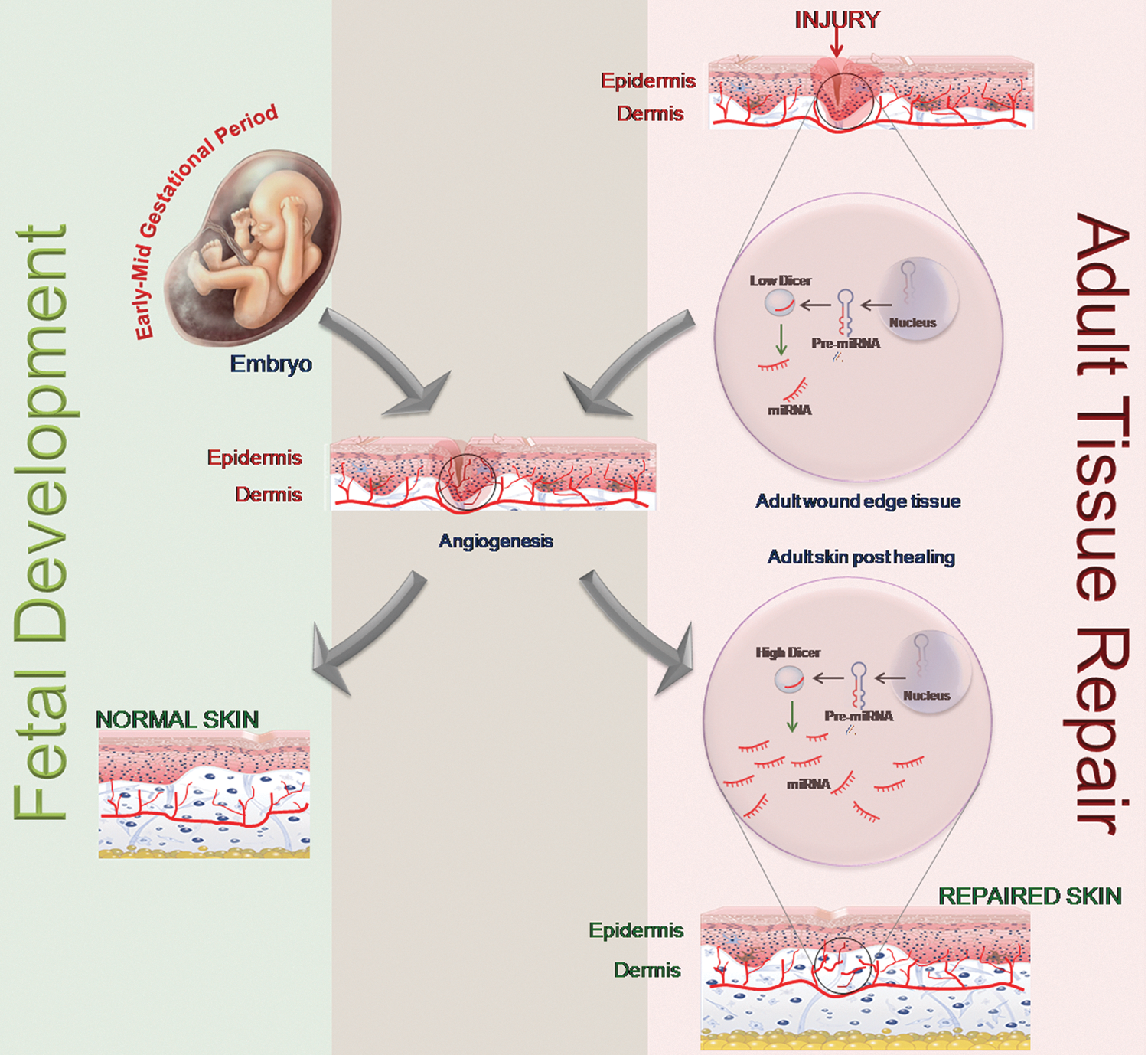
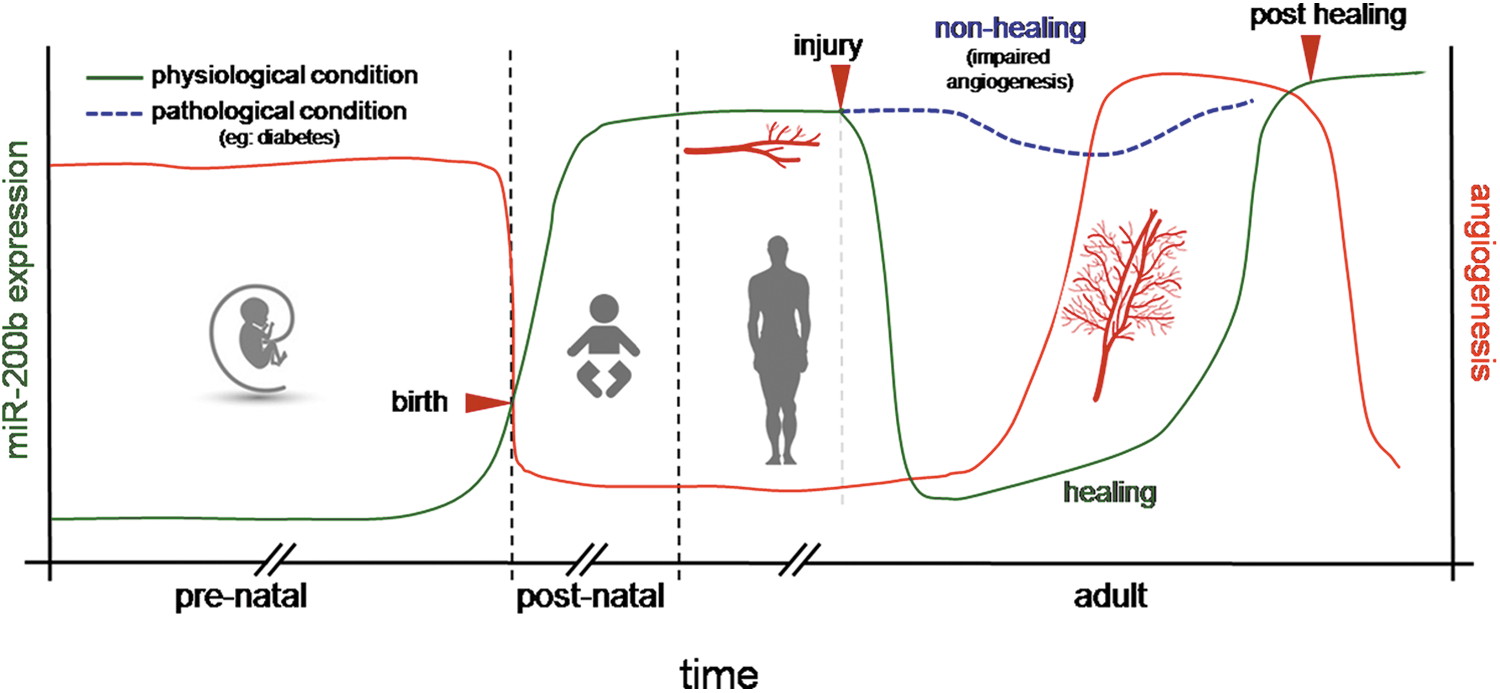
Dicer represents a key enzyme involved in miR biogenesis (60). Early embryonic lethality, observed in dicer knockout mice, has been suggested to be a consequence of defective blood vessel formation and maintenance. This observation underscores the fact that post-transcriptional gene silencing using miRs is required for angiogenesis. miRs can influence angiogenesis by regulating angiogenic growth factors, promoting cell migration. As explained in the previous sections, the regulation by miRs takes place at the translational level by regulating the translation of mRNAs of angiogenic factors. In the initial stages of embryonic development, the expression of dicer remains low. As the embryo grows, regulation and synchronization of various genes becomes important, and, hence, the endogenous braking mechanism of miRs sets in with dicer expression. The dicer gene is significantly expressed throughout the mouse embryonic tissues as early as day 11 and remains constant through day 17 (146). Starting from embryonic day 11.5, virtually all homozygous dicer knockout embryos were found to be growth retarded and underdeveloped as compared with their wild-type or heterozygous litter mates. The embryos that were still viable at this stage, however, had thin and sub-optimally developed blood vessels. This observation supports the fact that miRs are required for blood vessel development during embryogenesis (146). Dicer-null zebrafish embryos exhibit severe defects most prominently in gastrulation, brain morphogenesis, and cardiac development associated with a disrupted blood circulation (41, 141). Angiogenesis during fetal development and in adult dermal wounds shares a common platform based on their miR expression pool. The expression level of miRs in the wound edge tissue during the initial stage of healing is lower compared with that in the post-healing phase. Low miR abundance facilitates angiogenesis during the development as well as repair phase (Fig. 4).
miRs involved in angiogenic pathways can be broadly classified into two types, that is, pro-angiogenic miRs and anti-angiogenic miRs (Table 1). The pro-angiogenic miRs assist in the process of angiogenesis by desilencing angiogenic growth factors, promoting cell migration and EMT. However, anti-angiomiRs antagonize these events. It has to be cautiously noted that both these classes are equally important in ensuring healthy angiogenesis. During initial stages, the levels of anti-angiogenic miRs are repressed and levels of pro-angiogenic miRs are elevated so that the process of angiogenesis can resume. In the later phase, once blood vessels are formed, the pro angiogenic miRs go down and anti-angiogenic miRs revert to prevent uncontrolled angiogenesis and establish homeostasis.
miRs belonging to miR-17∼92 cluster and miR-126 are known to promote angiogenesis. miR-126 is a key positive regulator of angiogenic signaling in ECs and of vascular integrity in vivo (36, 75, 136). Knockdown of miR-126 during zebrafish embryogenesis or deletion of miR-126 in mice resulted in defects in vascular development. miR-126 mutant mice displayed diminished angiogenesis and increased mortality after coronary ligation, a model for myocardial infarction (136). ECs deficient in miR-126 failed to respond to angiogenic factors, including VEGF-A, epidermal growth factor (EGF), and basic fibroblast growth factor (bFGF) (36, 75, 136). miR-126 functions by directly repressing negative regulators of the VEGF pathway, including the Sprouty-related protein SPRED1 and phosphoinositol-3 kinase regulatory subunit 2 (PIK3R2/p85-beta) (75). The clusters of miRNAs miR-17 through miR-92 (miR-17–92), which are transcribed as a polycistron that is stimulated by Myc, exert their proangiogenic effect by targeting the secreted factors, namely thrombospondin 1 (TSP1) and connective tissue growth factor, both of which inhibit angiogenesis.
On the other hand, examples of anti-angiogenic miRs include miR-24. miR-24 inhibit angiogenesis by targeting of the endothelium-enriched transcription factor GATA-2 and the p21-activated kinase PAK4 (35). AntagomiR are synthetic RNA molecules complementary to an miRNA of interest. AntagomiR binds to miRNA and prevents it from binding to its target mRNA. Systemic use of antagomiR against miR-24 in the mouse ischemic myocardium resulted in improved myocardial angiogenesis and cardiac function. The anti-angiogenic miR-100 also regulates the vascular response to myocardial infarction. miR-100 expression is reduced in mouse ischemic muscles, further antagomiR-mediated miR-100 inhibition showed a therapeutic potential. miR-100 reduced mTOR expression and, consequently, attenuated cellular proliferation (47). More pro- and anti-angiomiRs have been detailed in Table 1. In this work, we review the role of anti angiogenic miR-200b and its importance as a regulator of inducible adult angiogenesis. One of the areas in angiogenesis that has seen significant development in recent years has been the study of post-transcriptional gene regulation of miRNAs, and temporal regulation of miR to fine tune angiogenic pathway has drawn surging attention. Recents reports on the role of miR-200b in tumor angiogenesis and wound angiogenesis have greatly expanded our view on the overall regulation of angiogenic pathways.
miR-200b Is Transiently Turned Down to Switch on Inducible Wound Angiogenesis
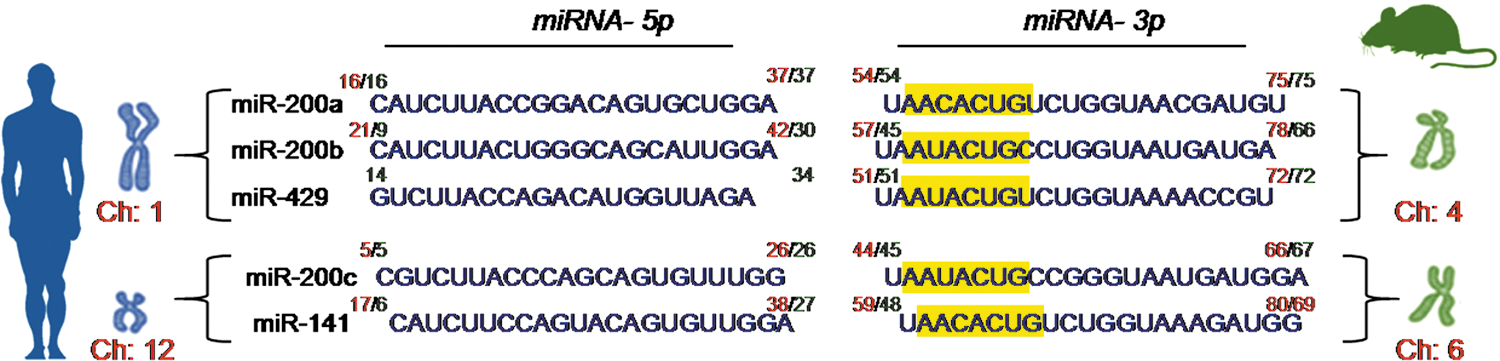
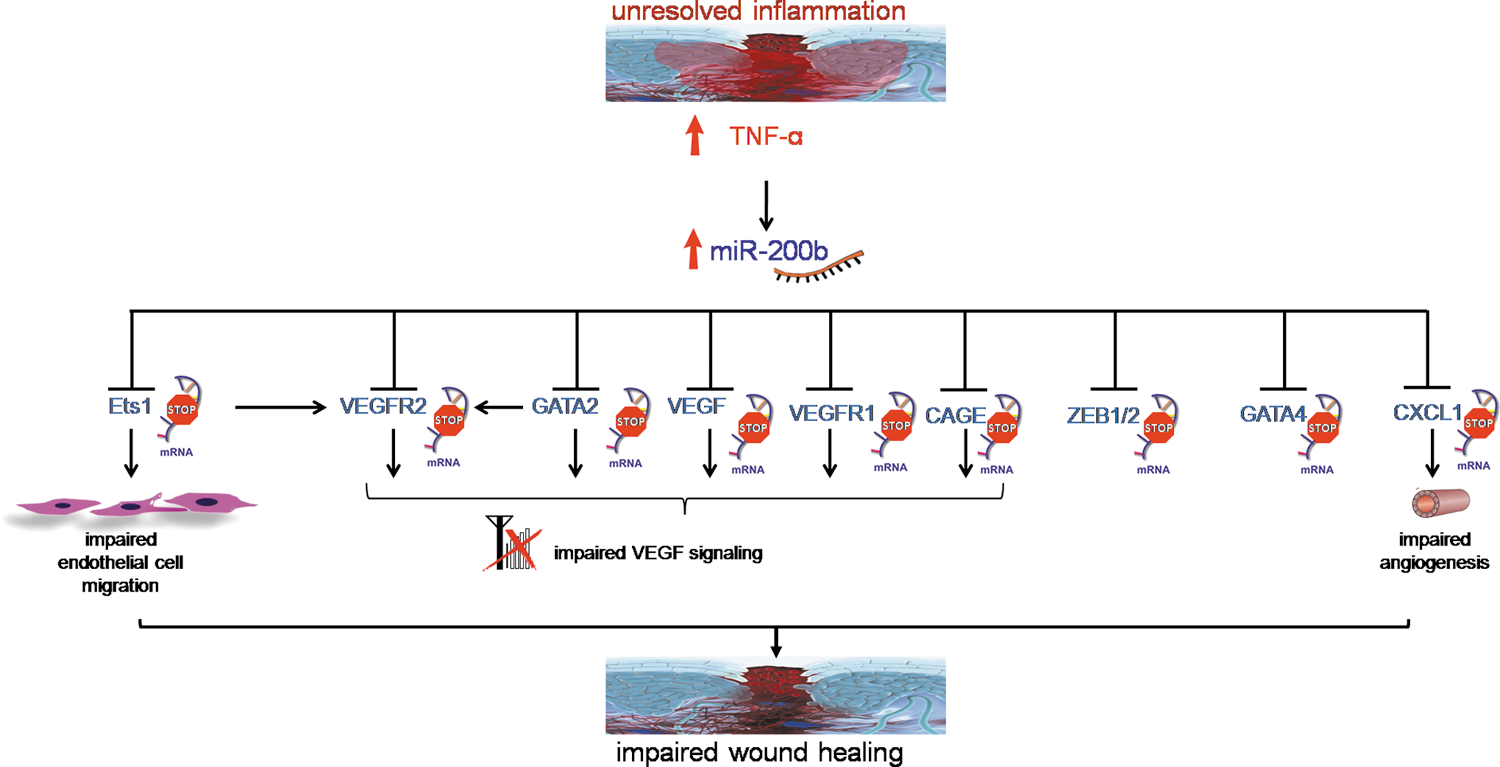
miR-200b belongs to the miR-200 family that is organized into two groups based on a single nucleotide difference in their seed sequence (group A: miR-141 and -200a; group B: miR-200b, -200c, and -429). In human, the miR-200 family is transcribed from two chromosomal clusters: miR-200b/a/429 from chromosome 1, and miR-200c/141 from chromosome 12. The mature miR-200b arises from the 3′- arm of pre-miR-200b. The miR is conserved across many species, including human and mouse (Fig. 5). It is anti-angiogenic and, thus, its expression must be silenced to initiate inducible angiogenesis (17). Downregulation of endothelial miR-200b helps in cutaneous wound angiogenesis (19). Injury transiently suppresses miR-200b expression for angiogenesis to occur postwounding. Inability to suppress miR-200b expression leads to impaired angiogenesis (Fig. 6). EMT has been reported to contribute to the formation of angiogenic blood vessels. miR-200b draws extraordinary significance, as it can target the transcription factors involved in EMT. Dysregulation of miR-200b has been described as playing a critical role in the EMT and metastasis in cancers such as breast, gastric, and pancreatic carcinomas. Moreover, miR-200b can silence a number of angiogenic growth factors and its receptors by directly targeting their mRNA transcripts (Fig. 7). Thus, miR-200b represents a critical hub in the regulation of inducible adult angiogenesis.
Angiogenic Transcription Factors and miR-200b
miR-200b and GATA
GATA factors are zinc finger DNA-binding proteins that control the development of diverse tissues by activating or repressing transcription. GATA-1, GATA-2, and GATA-3 are reported as hematopoietic GATA factors (98). GATA-1 functions to promote erythrocyte, megakaryocyte, mast cell, and eosinophil development and GATA-3 functions to promote specific aspects of T-cell lymphopoiesis (12). GATA-2 is uniquely essential for the genesis and/or function of hematopoietic stem/progenitor cells (12). GATA-2 is the most abundantly expressed GATA factor in microvascular ECs (125). GATA-2 is important in development of Flk-1+/Tal1+hemangioblast-like cells and in the induction of endothelial-specific genes (85). GATA-2 serves as a transcriptional activator (29). Transcription factor GATA-2 binds to the promoter of genes comprising GATA-2 binding elements, resulting in the transcriptional activation of the gene.
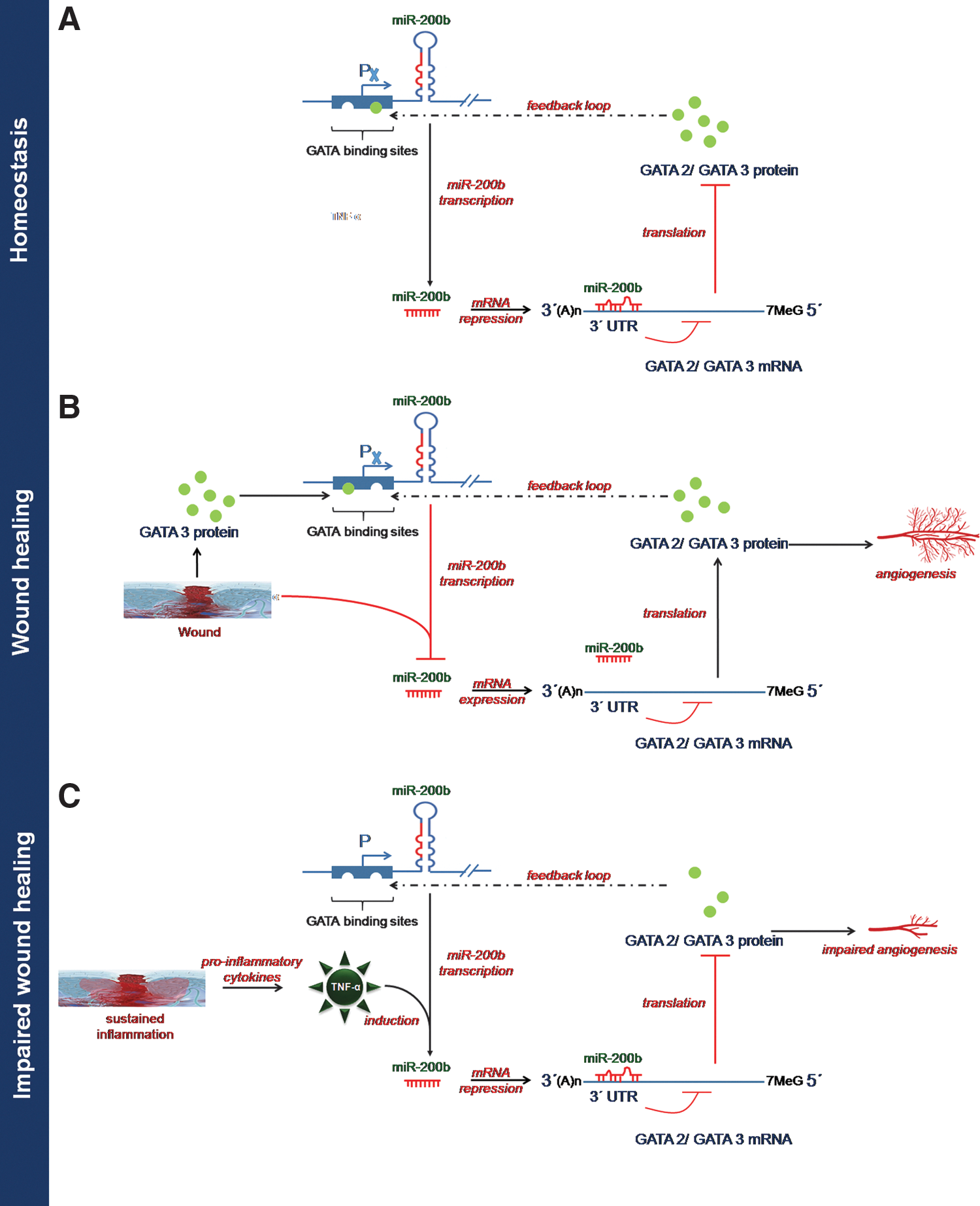
The miR-200b promoter contains multiple conserved GATA-binding sites (148). Occupancy of such sites by GATA-3 suppresses expression of miR-200b (148). GATA-3 binds directly to the promoter region of the miR-200b-200a-429 cluster in human H322 lung cancer cells. It is to be noted that miR-200b directly targets GATA-2, GATA-3, and GATA-6, thus forming a feedback loop (19, 147, 148). Overexpression of GATA-2 in ECs rescued the angiostatic effect of miR-200b in vitro. Downregulation of miR-200b derepressed GATA-2 expression to switch on wound angiogenesis, which was disrupted in diabetic wounds. We have reported that sustained presence of tumor necrosis factor-α (TNF-α), a pro-inflammatory cytokine, caused loss of endothelial GATA-2 (19). Treatment of ECs with TNF-α, abundant in diabetic wounds, induced miR-200b expression, silenced GATA-2, and suppressed angiogenesis (19). The diabetic tissue, thus, seems to be resistant to inducible adult angiogenesis. The interaction of miR-200b with GATA has been shown in Figure 8.
GATA-4 is an important transcription factor involved in several developmental processes of the heart, such as cardiac myocyte proliferation, differentiation, and survival. Normal and infarcted hearts treated with GATA-4 showed a significant increase in myocardial capillary density, indicating that GATA-4 regulates angiogenesis in the adult heart and, when upregulated, is sufficient to induce formation of new capillaries (111). Overexpression of miR-200b leads to the downregulation of GATA-4 mRNA and a decrease in GATA-4 protein levels (149).
miR-200b and ZEB
The ZEB family comprises two proteins, ZEB1 and ZEB2. ZEB1 is a trans-repressor supporting anti-apoptotic response (83, 88, 129). ZEB1 knockdown induced apoptosis in human lung cancer cell line, H460 cells. Further, ZEB1 knockdown inhibited lung cancer cells (NSCLC) growth in soft agar colony formation assay. However, the ability of transformed cells to grow under anchorage-independent condition is the most reliable predictor for tumorigenicity and metastatic potential. Strong growth inhibitory effect of ZEB1 knockdown in anchorage-independent condition suggests that ZEB1 expression contributes to maintaining aggressive phenotype of lung cancer cells (129). ZEB1 negatively regulates pathological angiogenesis by regulating endothelial invasiveness by acting as a transcriptional attenuator of matrix metalloproteinase 1 (MMP-1). ZEB1 also activates R-Ras, another class of angiogenic regulator, to suppress angiogenesis (57). Vascular expression of ZEB1 is compromised in diabetics (104). More interestingly, miR-200b downregulates the expression of ZEB1 and ZEB2 by interacting with 3'-UTR of ZEB1 and ZEB2 mRNA (11, 45). The miR-200 family is known to induce apoptosis (88, 112), and, thus, this is mediated via ZEB1 silencing. The promoter region of miR-200b has two E-box motifs to which ZEB1 and ZEB2 can bind. Binding of ZEB1/2 to the site suppresses the expression of miR-200b, establishing a negative feedback loop (11). These results also signify a double-negative feedback loop between miR-200b and ZEB1/ZEB2, and they allow the maintenance of the EMT phenotype.
miR-200b and p53
The tumor suppressor protein p53 has been shown to negatively regulate angiogenesis. Wild-type p53 can repress VEGF-A transcription in vitro (94). p53 repress VEGF-A transcription by direct binding and sequestration of transcriptional regulator SP1 and/or binding to the E2F transcription factor, forming a transcriptional repressor complex for VEGF-A expression (100, 103, 151). The other two p53 family members, p63 and p73, have a similar structure and mechanism of regulation and are relevant regarding their effect on angiogenesis. In cell lines that contain wild-type p53, p73 seems to increase VEGF-A expression, exerting a proangiogenic phenotype. The two major isoforms of p63, TAp63γ and dNp63α, have been shown to have opposite effects on the VEGF promoter. Similar to p53, TAp63γ represses VEGF-A expression by interacting with HIF-1α and mediating its proteasomal degradation. Conversely, dNp63α seems to have a dominant-negative effect on p53 and TAp63γ and induces VEGF-A expression by stabilizing HIF-1α and possibly contributing to the recruitment of p300/CBP to promote target gene expression (119). The promoters of both miR-200 chromosomal loci contain p53 family binding sites. Chromatin immunoprecipitation with various members of the p53 family show strong enrichment of p73 and p63 with the miR-200b/a/429 p53 family binding site (71). Silencing p53 in diabetic wounds is known to improve wound closure and angiogenesis (63, 96). p53 activation results in miR-200b transactivation and expression (66). Thus, p53 can suppress angiogenesis via a two pronged mechanism, including direct repression of VEGF-A and also by increasing miR-200b levels, which can, in turn, target VEGF-A.
miR-200b and NF-κB
NF-κB has been shown to respond to a variety of metabolic stress signals, including hypoxia (110), which is necessary for the process of angiogenesis to begin. Inhibition of NF-κB activity decreased VEGF expression (121). Overexpression of NF-κB contributes to VEGF-induced angiogenesis through upregulation of VEGF-A mRNA expression in many tumors. In vitro studies showed that overexpression of miR-200b and miR-200c dampens NF-κB activity through the TLR4-mediated pathway (139). Using luciferase reporter assay of p65 subunit of NF-κB, the authors found miR-200b and miR-200c reduced NF-κB activity. MyD88, which is involved in the TLR4 pathway, was found to be the target of miR-200b (139). The NF-κB pathway plays a critical role in the induction of EMT induced by the overexpression of platelet-derived growth factor (PDGF)-D in PC3 cells. The loss of miR-200, especially miR-200b, is, in part, responsible for the induction of EMT. It is well known that NF-κB plays an important role in mediating the processes of EMT induced by different factors through the upregulation of transcription repressor function of ZEB1 and ZEB2, which, in turn, repress the expression of miR-200 family by the binding to the E-box sequence of the miR-200b promoter.
miR-200b and ETS
The Ets factor Ets-1 is enriched in the developing blood vessels of the chicken, and antisense oligonucleotides have been shown to inhibit angiogenesis when delivered to the chicken chorioallantoic membrane (140). Ets-1 in tissues is associated with increased expression of VEGF-A. Ets-1 regulates the expression of several downstream targets in ECs that promote an angiogenic phenotype, including the VEGF receptors (VEGF-R1 and VEGF-R2), urokinase, and several MMPs. Studies support a role for Ets-1 expression in the development of tumor angiogenesis. In ovarian cancer, for example, Ets-1 expression strongly correlates with the degree of angiogenesis in the primary tumor and the development of metastatic lesions [24]. Ets-1 is a key transcription factor that is known to support angiogenesis. Pro-angiogenic stimuli such as VEGF-A, angiotensin II, and FGF induce Ets-1 expression. miR-200b targets Ets-1(17). Overexpression of Ets-1 reverses the phenotypic changes caused by miR-200b mimics, further supporting the notion that miR-200b inhibited the angiogenic response via silencing of Ets-1. Cancer/testis antigen cancer-associated gene (CAGE) is a member of Ets-family transcription factors (24). CAGE is expressed in a variety of cancers but not in normal tissues except for the testis (23). Downregulation of CAGE leads to the decreased expression of PAI-1, which is a TGF-β-responsive protein and is involved in angiogenesis (67). PAI-1 is involved in retinal angiogenesis and forms a member of VEGF-mediated angiogenesis as VEGF-A induces PAI-1 expression. Furthermore, miR-200b silences CAGE by binding to the 3′-UTR of CAGE (67).
miR-200b and p300
p300 is a transcriptional co-activator that possesses intrinsic histone acetyltransferase activity (97). p300 also drives a broad angiogenic transcription program that results in the development of abundant blood vessels. HIF-1 is activated by p300 and is an important transcriptional regulators of VEGF-A (2). p300 levels also lead to the upregulation of miRs that repress angiogenic transcription (120). miR-200 may regulate p300, a histone aceylator and transcription coactivator in malignancies (55). In pancreatic ductal adenocarcinoma, six p300 targeting miRNAs, including miR-200b, were found to be downregulated in the highly metastatic group (91). However, the molecular mechanism of miR-200b-mediated p300 regulation is not known. Effects of p300 are mediated by its capacity to control the expression of a number of transcription factors (21). Such p300-mediated action of miR-200b may potentially affect gene expression of multiple vasoactive factors.
miR-200b Silences Angiogenic Growth Factors
miR-200b silences VEGF
VEGF plays an important role in the regulation of blood flow and vascular permeability in angiogenesis. VEGF-A is an important member of the VEGF family. The reduction of VEGF-A diminishes vascularity and decreases scar formation in adult wounds (143). Although the underlying mechanisms remain obsure, VEGF-A may directly stimulate both ECs and fibroblasts. VEGF-A receptors on fibroblasts are responsible for induced proliferation of keloid fibroblasts (144). VEGF-A exerts its biological activity predominantly through transmembrane receptors linked to tyrosine kinase domains. Many different cell types, fibroblasts, ECs, macrophages, and keratinocytes are able to produce VEGF-A, and mainly the latter two are responsible for the VEGF-A production during wound healing (7). Anti-VEGF strategies inhibit the formation of granulation tissue in the wound (53), indicating an important function of VEGF-A in angiogenesis that occurs during the proliferative phase. Low oxygen tension (hypoxia), as occurs during tissue injury, constitutes a significant inducer of the production of this growth factor (1).
In clear cell renal cell carcinoma, a strong negative correlation is noted between expression of VEGF-A and the miR-200 family (82). Employing ELISA and luciferase reporter assays, VEGF-A was validated to be a direct target of miR-200b (25). Downregulation of miR-200b caused aberrant expression of VEGF in diabetic ECs. Delivery of miR-200b mimic prevented diabetes-induced, VEGF-mediated functional changes in the endothelial and retinal cells (90). In addition, miR-200b mimic delivery downregulated endothelial VEGF-A expression. Interestingly, blocking miR-200b expression in diabetic wounds restored VEGF-A levels. Finally, elevated miR-200b was associated with loss of endothelial VEGF-A in human diabetic wound-edge tissue.
Although the VEGF-A gene is strongly induced by hypoxia and cytokines at the level of transcription, it is also regulated at the post-transcriptional level by RNA-binding proteins and miRNAs such as AUF1, HuR, miR-15b, miR-16, miR-20a/b, and miR-200b (20, 34, 56, 58). AUF1 is an RNA-binding protein. AUF1 represses expression of VEGF-A in macrophages by binding to its 3′-UTR (34). Infiltration of macrophages is essential for neovessel formation (51). Studies in mice have shown that macrophages infiltrate and drill tunnels using their proteolytic activity, which can serve as a scaffold during neovascularization (93). The human (Hu) antigen R (HuR) is a ubiquitously expressed member of the Hu family of RNA-binding proteins (87). ARE elements on mRNA primarily serve as the binding site for HuR. Binding of HuR to mRNA leads to stabilization of mRNA (84). HuR directly binds to VEGF-A mRNA (20, 69). In the absence of HuR, the basal expression of VEGF-A mRNA and polypeptide was significantly suppressed due to lack of mRNA stabilization by HuR. It was observed that the Hur binding site and miR-200b binding site on VEGF-A 3-UTR overlap, thus HuR and the miR-200b-RISC complex competitively regulate VEGF-A. The ability of miR-200b to suppress VEGF-A expression was antagonized by HuR. Importantly, more Ago-2-containing RISC complex was associated with VEGF-A mRNA in the absence of HuR (20). Post-transcriptional gene regulation by myeloid HuR is critical in tumor angiogenesis. Subcutaneous implants in myeloid HuR KO mice show compromised vascular density, vessel branching, vascular leakage, and attenuated tumor growth, suggesting that HuR promotes the pro-angiogenic phenotype in macrophages that enhance tumor growth. HuR enhances angiogenesis by antagonizing the anti-angiogenic effect of miR-200b in zebrafish embryos (20). Thus, a complex interplay between RBPs and miRNAs allow specificity, precision, and robustness of post-transcriptional gene regulation.
Besides VEGF-A, its receptors VEGF-R1 and VEGF-R2 also play a vital role in angiogenesis. The VEGF-R1 receptor is required for normal blood vessel development during embryogenesis, since homozygous deletion of VEGFR-1 is lethal in mice at embryonic day E8.5 due to severe malformation of the vasculature (39). VEGFR-2 regulates EC migration, proliferation, differentiation and survival, as well as vessel permeability and dilation (13).VEGF-R2 is found in stromal cells and plays a role in wound repair (152). miR-200b targets VEGF receptors VEGF-R1 (109) and VEGF-R2 (19). Thus, miR-200b can regulate angiogenesis by targeting both VEGF-A and its receptors.
Mir-200b silences PDGF
The PDGF family of ligands are closely related to VEGF-A and may have evolved from a common gene (52). The homodimer of PDGF-B recruits perivascular cells during vasculogenesis, possibly through the generation of reactive oxygen species and subsequent activation of extracellular-regulated kinase 1, 2 (ERK 1, 2) (76). Endothelial-derived PDGF-B dimer also induces progenitor cell migration and expansion during vascular development and is critical during vascular bed formation by mesangial progenitor cells (50, 81). VEGF ligands can also bind and activate PDGF pathways, a process important during mesenchymal stem cell-associated vasculogenesis (5). PDGF pathways have been used to control neovascularization in various animal models. For example, nanofibrous scaffolds incorporated with PDGF activate cytokine signaling and improve angiogenesis during wound repair in rats (61).
As discussed in an earlier section, the processes of EMT have been linked with cell migration and invasion during angiogenesis. PDGF-D, which is a member of the PDGF family, is a potent angiogenic growth factor. PDGF-D induces cellular transformation and promotes tumor growth by accelerating the proliferation rate of the tumour cells, and by stimulation of tumour neovascularization (79). Expression of miR-200b in PC3 PDGF-D cells (PC3 cells overexpressing PDGF-D) led to the reversal of the EMT phenotype. In this study, it was noted that the transfection of PC3 PDGF-D cells with miR-200b inhibited cell migration and invasion (72). It is widely recognized that the metastatic process of cancer cells requires cell detachment from the site of origin, intravasation, translocation through blood and lymphatic vessels, extravasation, attachment to the secondary site, and colonization. Moreover, cell detachment from basement membrane and re-attachment play critical roles during cell migration and invasion as well as in tumor cell metastasis. PC3 PDGF-D cells exhibit significant enhancement in cell detachment from culture surface and attachment to the culture surface. More importantly, transfection with miR-200b remarkably attenuated the ability of PC3 PDGF-D cells to attach and detach from the culture surface. LNCaP cells (prostate adenocarcinoma cells) with a lower invasive capacity show reduced expression of endogenous PDGF-D and an increased level of miR-200b; whereas increased expression of PDGF-D in LNCaP cells resulted in lower miR-200b levels (72).
miR-200b silences IL-8 and CXCL1
Interleukin-8 (IL-8) and CXCL1 are potent proangiogenic cytokines that are overexpressed in several types of tumors. IL-8 and CXCL1 mediate their pro-angiogenic effect in an autocrine fashion through endothelial CXCR2 receptors (92). Beyond being a potent proangiogenic cytokine, IL-8 also has known roles in promoting tumor self-seeding, chemoresistance, and metastasis in various models (65). miR-200b can directly affect angiogenesis by targeting EC production of IL-8 and CXCL1. There were significant reductions in the expression of these pro-angiogenic factor after transfection with either miR-200a or -200b in ECs (101).
Epigenetic Regulation of miR-200b
Current developments recognize that epigenetic mechanisms are involved in the regulation of miR-200 expression (134, 142). Both loci of miR-200 family have CpG islands, 1–200 bp downstream of miR-200b and 234–371 bp upstream of miR-200c. During early tumor development, CpG islands of miR-200b are unmethylated, leading to expression of miR-200b. However, as the tumors progress, there is gain of CpG promoter methylation, associated with lowered expression of miR-200b and increased metastasis. Lower levels of miR-200c have been associated with a higher frequency of lymph node metastases in nonsmall cell lung cancer patients (15), and miR-200 downregulation is observed in metastatic lymph nodes (4). TGF-β induction of EMT demonstrated similar results. Unmethylated CpG islands are associated with strong expression of the miR-200. However, on induction of EMT by TGF-β treatment, there is a progressive gain of CpG methylation in both the miR-200 loci, associated with a reduced expression of the miR-200b. Most notably, when these cells underwent mesenchymal-to-epithelial transition (MET) phenotype after TGF-β withdrawal from the medium, the previously methylated CpG islands recovered the original unmethylated CpG status, and the expression of the miR-200 was restored. miR-200 hypermethylation-associated inactivation in the TGF-β-induced EMT was accompanied by increased ZEB1 expression, loss of E-cadherin, and the acquisition of a spindle-like shape, a mesenchymal phenotype marked by vimentin expressing. Subsequent acquisition of an MET phenotype on TGF-β lowering led to reduced ZEB1 and vimentin expression (28).
Conclusion
Angiogenesis in adults is a highly regulated phenomenon. The process needs to be initiated when required and silenced once the formation of blood vessels is completed. Failure in either of the two steps mentioned earlier results in faulty angiogenesis. For example, in diabetic retinopathy, failure to stop angiogenesis results in bleeding, cloudy vision, and destruction of retina. Pharmaceutical angiogenic inhibitors such as semoxind (SU5416), orantinib (SU6668), and others are under active clinical trials. These compounds target single gene products or metabolites. miRs, on the other hand, can target a number of genes. To establish a compound for traditional drugs, many molecules are required to be synthesized and screened to generate a single drug candidate. However, screening approaches for miRs/antagomiRs to be used as a drug can have faster translational value for the following reasons: (i) the short sequence of the miRNA and (ii) specificity to its target. It has been highlighted, in this review, that miR-200b can simultaneously regulate various factors involved with angiogenesis. However, it is to be noted that the promiscuity of miR that makes it target many genes in a patho-physiological pathway can also result in more side effects. This can, however, be tackled by base modification of miR sequence to render them more specific to target mRNA. The short sequence of miRs provide relative easy access for their synthetic modification and delivery. Being endogenous molecules, miRs have reduced side effects associated with them, which might not be the case with pharmaceutical inhibitors.
miR-200b is a potent angiomiR. It is anti-angiogenic. As discussed here, miR-200b can regulate expression at the post-transcriptional level for various angiogenic growth factors and transcription factors. Further, many of these factors also regulate miR-200b expression at the transcriptional level. Such interactions of miR-200b with various angiogenic factors justify its role as a hub miR modulating angiogenic outcomes. The role of miR-200b in regulating epithelial-to-mesenchymal transformation through ZEB family of transcription factors further strengthens its significance in wound healing and tumor biology, as in both cases EMT plays a critical role. Multiple regulatory features of miR-200b mediated by regulating the expression of angiogenic factors can thus be utilized toward therapeutic intervention. Delivery of miR-200b mimic may be valuable under conditions where regression of angiogenesis is necessary. Similarly, silencing the level of miR-200b through antagomiR delivery can help in conditions where angiogenesis is required, as in the case of wound healing.
Footnotes
Acknowledgments
This work was supported by National Institutes of Health awards DK076566 to SR and R01GM077185, R01GM069589, R01NR013898, and R01GM108014 to CKS.