
Abstract
Introduction: Oxidative Stress and Aging
A
Formation and Fate of Oxidized Proteins
Short overview on chemistry of protein oxidation
The reactions of ROS with proteins lead to oxidative damage. The targets for oxidant reactions are either the protein backbones, resulting in fragmentation reactions, or the amino-acid side chains, leading to the formation of a variety of different oxidation products.
Oxidative protein modifications cause changes in the protein structure and a partial unfolding of the protein. Both the unfolding and the direct oxidation of functional amino-acid side chains may lead to an impaired protein function.
Damage of the protein backbone
Radical-induced reactions often rapidly lead to backbone damage. In general, an oxidative attack on the backbone first results in the abstraction of the hydrogen atom from the α-carbon, leading to formation of a stabilized carbon-centered radical. This radical may react with other carbon-centered radicals or with O2. The latter results in the formation of a peroxyl radical (121). Peroxyl radicals may either undergo an elimination reaction, by which HO2 • is released, or generate hydroperoxides through hydrogen abstraction from another molecule. Both paths finally lead to protein backbone fragmentation (35).
Damage to amino-acid side chains
Since there are 20 different amino acids, the oxidation of amino-acid side chains is a more complex mechanism than the protein backbone oxidation and leads to the formation of several oxidation products. Therefore, only a brief overview on possible reactions will be given [for an extensive review, see Refs. (36, 147)]. Similar to backbone damage, oxidation of aliphatic amino-acid side chains often leads to the formation of carbon-centered radicals. There are various fates for these radicals. For instance, they may undergo dimerization due to the reaction with other carbon-centered radicals or they may be repaired by thiols, generating thiyl radicals. However, the predominantly occurring reaction under aerobic conditions is the formation of peroxyl radicals (121). As a result, alkoxyl radicals, hydroperoxides, alcohols, and carbonyls may be generated (36).
A good example for oxidation of aromatic amino-acid residues is tyrosine. Oxidative damage of tyrosine often leads to generation of tyrosyl radicals. These radicals may dimerize, generating dityrosine, a process involved in crosslinkage and protein aggregation (2). Further important products of tyrosine oxidation are 3-chlorotyrosine, 3,4-dihydroxyphenylalanine, and 3-nitrotyrosine (3-NT) (36). The latter are generated due to the reaction with ONOO−.
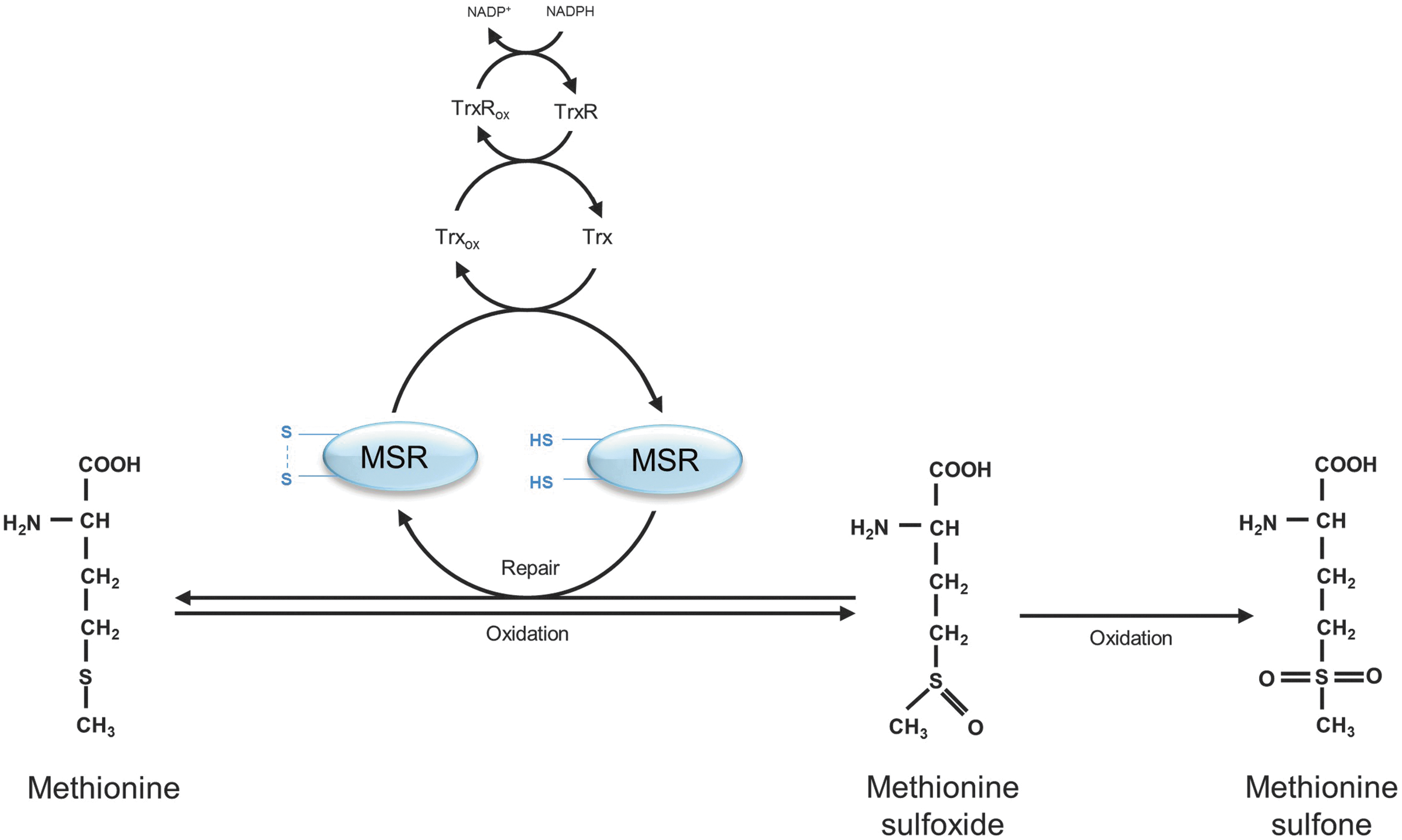
Sulfur-containing amino-acid side chains are the most sensitive targets for oxidation. The oxidation of cysteine leads to the formation of thiyl radicals. There are several fates for these thiyl radicals. Dimerization reactions give rise to disulfides, generating cystine. In addition, mixed dimers may be formed due to the reaction of thiyl radicals with other species. The reaction of thiyl radicals with O2 leads to the formation of peroxyl radicals and gives rise to oxyacids (sulfenic acid, sulfinic acid, and sulfonic acid) (11, 36). Methionine sulfoxides (MeSOs) are one of the main products, resulting from methionine oxidation (Fig. 1). They are generated in S- or R-stereoisomers. MeSOs may form sulfones after further oxidation (36) or may be reduced by methionine sulfoxide reductases (MSRs) (Fig. 1) (see Repair and Degradation of Oxidized Proteins section).
Methods to measure protein oxidation products
Two of the most commonly measured oxidative protein modifications are protein carbonyls and 3-NT, which are used to assess the cellular status of protein oxidation.
Protein carbonyls are generated due to the oxidation of proline, arginine, lysine, threonine, and other amino-acid residues and due to the oxidation of the protein backbone. Moreover, they might be the result of secondary reactions of amino acids (cysteine, histidine, and lysine) with reactive carbonyl compounds (ketones, aldehydes), emerged in lipid peroxidation or glycation/glycoxidation reactions (33). These secondary reactions may lead to the formation of advanced glycation and lipoxidation end products (AGEs/ALEs). Protein carbonyls are chemically stable and easy to detect after modification with dinitrophenylhydrazine by various methods of immunostaining; for example, immunoblot, ELISA, and fluorescence microscopy [for a review, see Ref. (154)].
The determination of 3-NT levels is widely used to investigate the role of oxidative protein modification. There are several methods available for measurement of 3-NT, including methods of immunostaining (e.g., ELISA), HPLC, and mass spectroscopy (62, 158).
Physiological formation of oxidized proteins
ROS are generated in a highly controlled way under physiological conditions. They are produced in the mitochondrial electron transport chain (mETC), which is responsible for cellular respiration, as well as in reactions of the cytochrome P450 system, involved in drug metabolism. In addition, many other enzymes are known to produce ROS, such as lipoxygenases, prostaglandin sythases, ribonucleotide reductases, xanthine oxidases, and NADPH oxidases. Besides the damaging effects of ROS, in low concentrations they may also be useful metabolites and carry out regulatory functions. Some ROS, such as H2O2, have the ability to act as second messengers due to their specificity to interact with effectors in signaling pathways. Other ROS, such as HO•, are highly reactive and interact rapidly with any target near their generation site. Thus, they do not possess the specificity to act as second messengers [for a detailed review, see Refs. (43, 101)].
Since oxidative protein modifications result in protein unfolding and the impairment of protein function, this might negatively affect cellular metabolism. However, inactivation of proteins due to protein oxidation may also have regulatory functions (100). Similar to other post-translational protein modifications, such as phosphorylation and acetylation, oxidation also seems to be involved in the regulating of a large number of metabolic processes. Especially oxidation of sulfur-containing amino acids methionine and cysteine seems to be important, as these are the only oxidative protein modifications that are reversible (see Repair and Degradation of Oxidized Proteins section). In many cases, regulatory protein oxidation is performed by enzymes, such as peroxidases, which use H2O2 as a substrate [for a review, see Refs. (43, 101)].
The function of transcription factors, protein kinases and protein phosphatases is regulated by oxidation of cysteine. Protein tyrosine phosphatases (PTPs) constitute one group of proteins that is very sensitive for oxidation. They contain catalytic cysteine residues in their active site. Oxidation of these cysteine residues leads to disulfide bond formation. Due to oxidation, the PTP is inactivated and, thus, not able to dephosphorylate the target protein kinases (127). Consequently, reversible cysteine oxidation of PTPs is implicated in tyrosine phosphorylation-dependent pathways.
Similar to cysteine oxidation, the oxidation of methionine and formation of MeSOs is often a protein inactivating post-translational protein modification. For example, the oxidation of a methionine residue of calmodulin, a protein involved in immune response, metabolism, and inflammation, leads to loss of the calcium-binding ability, responsible for activation of target enzymes. However, methionine residue oxidation may also trigger the protein activity, while reduction via MSRs (see Repair and Degradation of Oxidized Proteins section) leads to inactivation (38). To give an example, calmodulin kinase II, important for calcium homeostasis, is usually activated due to binding of calcium and calmodulin, but, in addition, shows a calcium and calmodulin-independent activation after methionine oxidation (39, 73).
Pathophysiological formation of oxidized proteins
Endogenous changes, such as inflammation, or exogenous influences, for instance UV radiation, gamma radiation, and xenobiotics, may increase ROS formation, either directly or by promoting endogenous ROS generators. As a consequence, antioxidant defenses may be overwhelmed, resulting in a cellular state referred to as oxidative stress and in enhanced macromolecule damage, including oxidative damage to proteins. One initial oxidation event may lead not only to multiple oxidative protein modifications within a protein, but also to oxidation of further proteins and other macromolecules. Thus, protein oxidation may give rise to uncontrollable chain reactions (106, 122). To protect the cells from the overall consequences of protein oxidation, there exists an effective and fast degradation and repair system to remove oxidized proteins. However, this system may be overloaded during increased protein oxidation. Therefore, oxidized proteins accumulate in the cell.
The accumulation of oxidized proteins results in crosslinking reactions and the increased formation of nondegradable protein aggregates. Protein aggregates are initially the result of hydrophobic interactions due to the increased surface hydrophobicity of oxidized proteins, resulting from their partial unfolding (52, 128). Furthermore, the oxidized proteins in these aggregates become chemically crosslinked by covalent reactions; for example, dityrosine formation, interaction of two carbon-centered radicals, disulfide formation, and the reaction of protein carbonyls with lysine and arginine residues (see Short Overview on Chemistry of Protein Oxidation section).
Protein aggregates are insoluble and nondegradable (79). Therefore, protein aggregates accumulate within the cell, which seems to have various consequences for the cellular metabolism and viability. It was shown that the accumulation of protein aggregates leads to inhibition of the proteasomal system and, thus, to further aggregation (66, 141) (see Repair and Degradation of Oxidized Proteins section).
Protein aggregates may accumulate in various structures, such as inclusion bodies, lysosomes, aggresomes, or plaques. “Protein aggregate” is, therefore, a very simple term that only describes the existence of crosslinked proteins. Plaques are extracellular occurring protein aggregates. Inclusion bodies describe intracellular accumulations of protein aggregates. The term aggresome was described in 1998 by the group of Kopito as a “pericentriolar, membrane-free, cytoplasmic inclusion containing misfolded, ubiquitinated proteins ensheeted in a cage of intermediate filaments, formed specifically at the microtubule organization center (MTOC)” (79). Thus, the term describes the cellular location and the composition of the aggregates, as well as the involvement of filaments. In general, aggresome formation seems to be a protective mechanism, because the smaller intermediate aggregates have a larger surface and, therefore, a higher potential for interaction with cellular macromolecules and membranes compared with mature aggregates caged in aggresomes (125). Aggresomes might contain several forms of oxidized proteins and otherwise misfolded proteins, which might be ubiquitinated but for some reason are not degraded by the 26S proteasome.
Existing protein aggregates may undergo further direct oxidation reactions or secondary reactions with products of lipid peroxidation, for example, 4-hydroxynonenal (HNE) and malondialdehyde (45), resulting in the formation of highly crosslinked fluorescent material referred to as lipofuscin (see Pathophysiological Roles of Accumulated and Aggregated Proteins section).
Repair and degradation of oxidized proteins
Repair of oxidized proteins
The majority of oxidative protein modifications are nonrepairable, making a fast and effective degradation system essential to protect the cells from accumulation of oxidized proteins and related consequences. However, a few enzymes exist, which are able to repair modifications of sulfur containing amino-acid residues (disulfides and MeSOs). These enzymes are of biological importance due to their involvement in regulatory functions of disulfide and MeSO formation (see Physiological Formation of Oxidized Proteins section).
Disulfides may be repaired either by thioredoxin (Trx) or by the protein disulfide isomerase (PDI) (24, 160). Both enzymes contain cysteine residues in their active sites, which are able to reduce the disulfides being oxidized themselves, resulting in the formation of intramolecular disulfides. Afterward, Trxs may be recycled by thioredoxin reductase (TrxR) using NADPH. PDIs may be reduced by glutathione.
A group of enzymes, called MSRs, is able to reduce the two stereoisomers of MeSO. MSRAs, which are able to reduce S-stereoisomers, reduce free MeSO and MeSO that are embedded in proteins. In contrast, MSRBs are able to reduce R-stereoisomers and almost exclusively in proteins (38). Similar to Trxs and PDIs, reduction of MeSO via MSRs is catalyzed by cysteine residues, which may form intramolecular disulfides (Fig. 1). Next to these catalytic cysteines, there are other cysteine residues in the active site of MSR, referred to as recycling cysteines, which are besides the Trx/TrxR system involved in the reduction of the catalytic cysteines (Fig. 1) (105).
Degradation by the proteasomal system
As mentioned earlier, degradation systems have a large share in the removal of protein oxidation products. The most important system responsible for degradation of oxidized proteins is the proteasomal system and, in particular, the 20S proteasome (Fig. 2) [for a review, see Refs. (34, 82)]. The 20S proteasome is a barrel-shaped multi-enzyme complex (Fig. 2), with a molecular weight of 700 kDa. It is composed of four homologous rings. Each ring is divided into seven subunits. The inner rings, which form the catalytic center of the 20S proteasome, consist of seven different β-subunits (β1–β7). Three of them possess a proteolytic activity. β1 has a peptidyl-glutamyl-peptide-hydrolizing activity, β2 has a trypsin-like activity, and β5 possesses a chymotrypsin-like activity. The outer rings consist of seven different α-subunits (α1–α7) that have a function in recognition and access of the substrates to the catalytic center of the 20S proteasome (83).
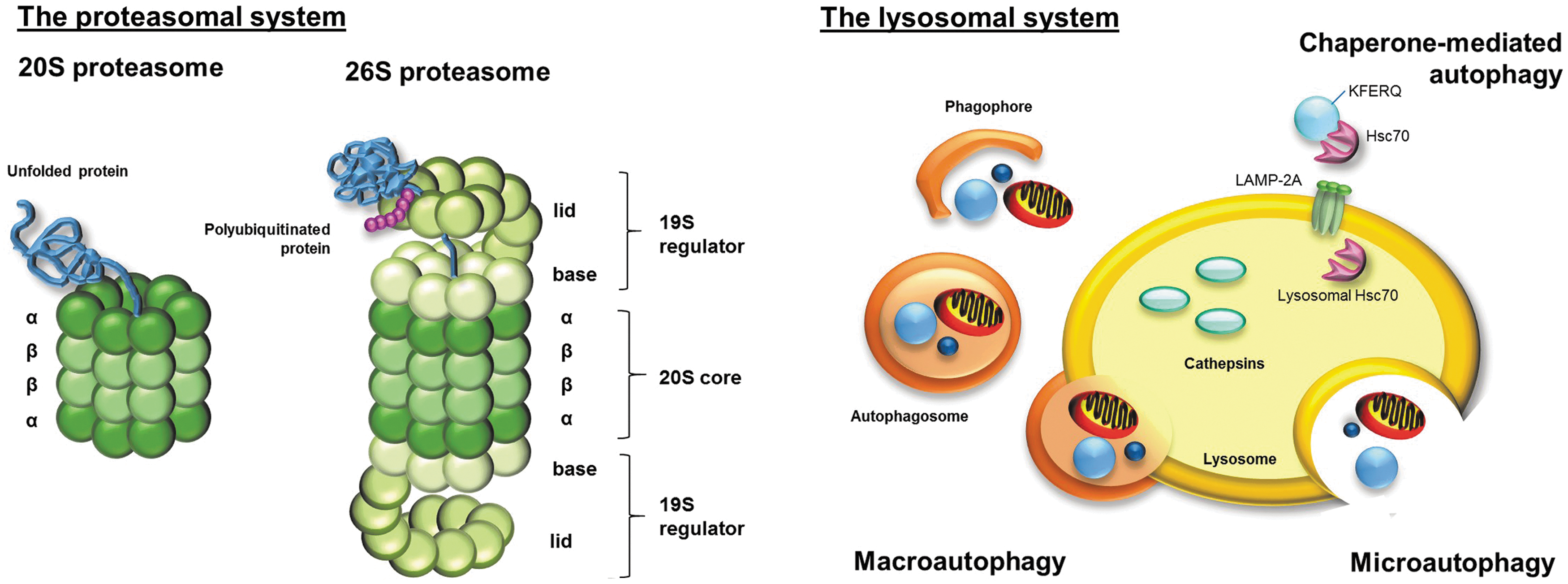
Another proteasomal form is the 2 MDa large 26S proteasome, which consists of the 20S core and two bound 19S regulators (Fig. 2). In contrast to the 20S proteasome, the 26S proteasome degrades substrate proteins in an ATP-dependent way and it requires a polyubiquitin sequence to recognize the substrate proteins. Due to the involvement of the ubiquitin-targeting machinery in degradation of 26S substrate proteins, this system is also referred to as the ubiquitin-proteasome system (UPS) (81, 96). The 19S regulator (also known as PA700) consists of some 19 monomers (Rpt- and Rpn-subunits). The Rpn-subunits functions in recognition of the polyubiquitinated substrate proteins. The Rpt-subunits have an ATPase activity. ATP hydrolysis causes the substrate intake through the resulting conformational change of the 26S proteasome and, in addition, the unfolding of the substrate proteins (83). The major function of 26S proteasome is the degradation of functional proteins, for example, responsible for cell cycle regulation, apoptosis, and signal transduction, which are no longer needed in the cellular metabolism and, therefore, marked by the ubiquitination machinery for degradation [for a review, see Refs. (96, 119)]. Moreover, it is postulated that newly synthesized, but misfolded proteins are degraded by the 26S proteasome (93).
For a long time, it was a controversial issue whether oxidized proteins are degraded by 20S or 26S proteasomes. However, the predominant role of the 20S proteasome was confirmed by many research groups. On the one hand, oxidized proteins seems to be degraded independently from ATP and without the necessity of a polyubiquitin-tag (34, 56, 57, 87, 140). In contrast to the 26S proteasome, the 20S proteasome is able to recognize unfolded proteins and, therefore, oxidized proteins, due to their increased surface hydrophobicity (Fig. 2) (52, 95, 128). On the other hand, the 20S proteasome seems to be more resistant to oxidative stress than the 26S proteasome (137, 138). It is very likely that during oxidative stress, the 26S proteasome dissociates into the 20S core and the 19S regulators. The increased availability of the 20S proteasome results in an elevated degradation of oxidized proteins. Hsp70 seems to be involved in this process by keeping the 19S regulator in its active form until re-association to the 26S proteasome (55) (Fig. 2).
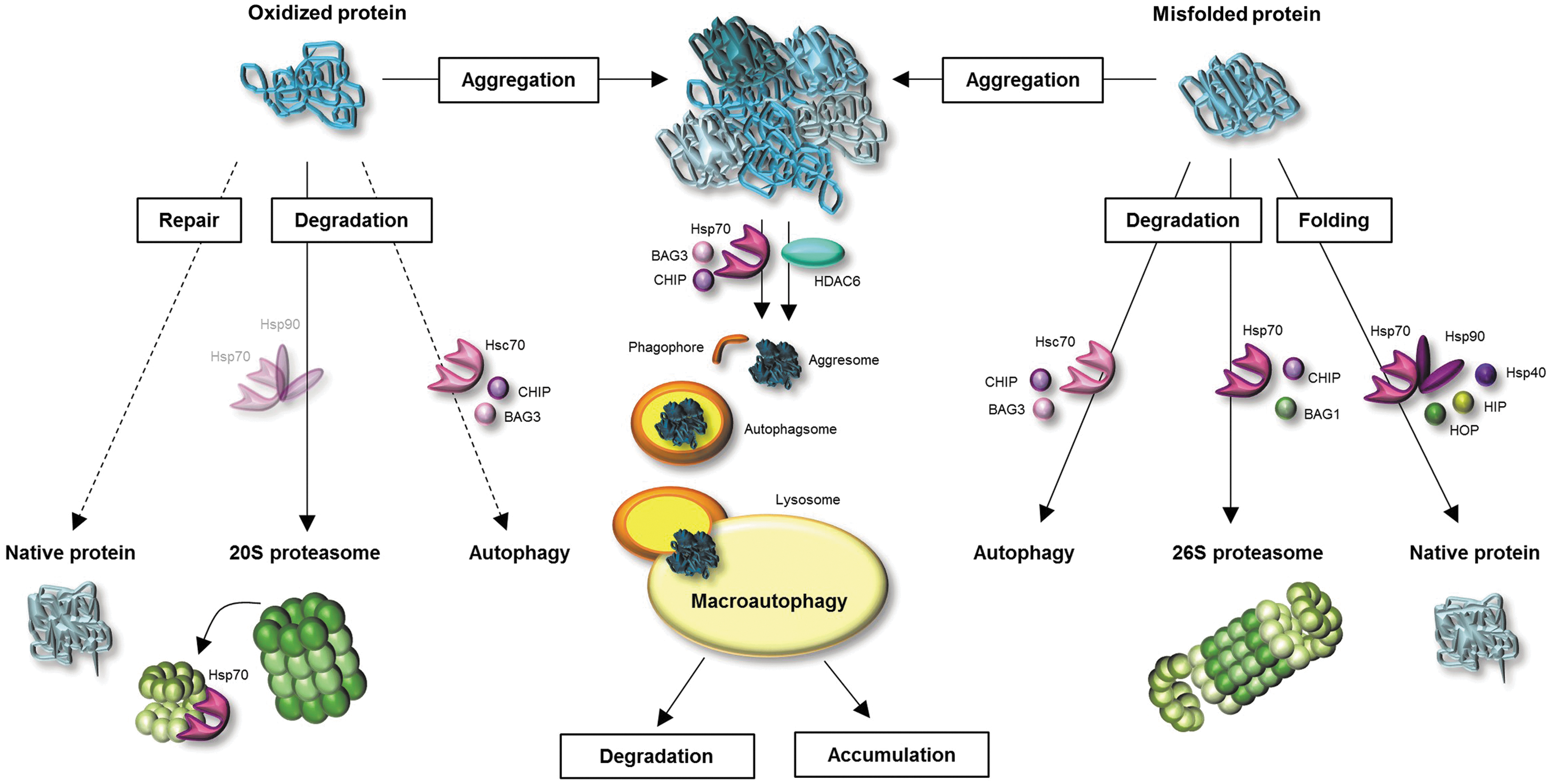
Heat shock proteins (HSPs) are of importance for protein pool maintenance. Due to their function as molecular chaperones, they are involved in refolding of misfolded and damaged proteins (Fig. 3). Although oxidized proteins cannot be repaired by HSPs, due to covalent modifications of oxidized proteins, chaperones have other important functions during oxidative stress. Thus, they are possibly involved in the functionality of the proteasomal degradation system. For example, Conconi et al. showed that Hsp90 leads to an enhanced activity of the 20 proteasome after FeCl3-induced ROS formation in vitro (25). Whittier et al. showed that Hsp90 is selectively involved in 20S proteasome-related degradation of oxidized calmodulin, but not in degradation of native calmodulin (159). In addition, a direct participation of HSPs (Hsp70 and Hsp90) in the degradation by the 26S proteasome is postulated (Fig. 3). HSP-bound substrate proteins are polyubiquitinated by the ubiquitin E3 ligase CHIP, which is an important co-chaperone of Hsp70 and Hsp90 (117). A further co-chaperone BAG1 mediates binding of HSP-substrate complex to the 26S proteasome (4). However, the degree to which HSPs are involved in degradation of oxidized proteins by the 20S proteasome and exact mechanisms of the involvement have not yet been clarified.
Involvement of the lysosomal system in the degradation of oxidized proteins
Besides the proteasomal degradation system, there exists a further degradation machinery that might be involved in proteolysis of oxidized proteins: the lysosomal system (Fig. 2). Lysosomes contain a wide range of degrading enzymes that are responsible not only for degradation of proteins (cathepsins) but also for carbohydrates and lipids. Due to the high activity of hydrolyzing enzymes in the lysosomes, the uptake of material into the lysosomes is strongly regulated. However, the lysosomal degradation of proteins is a less selective process than the proteasomal degradation.
Lysosomal uptake is accomplished via three different processes: phagocytosis, endocytosis, and autophagy. In particular, autophagy is important for degradation of endogenous proteins. There are three types of autophagy: microautophagy, macroautophagy, and chaperon-mediated autophagy (CMA) (Fig. 2) (152). Direct intake of degradable material into lysosomes is called microautophagy (115). In contrast, macroautophagy describes the inclusion of organelles and soluble proteins, first into double-membrane structures (phagophores), generating double-membrane vesicles, called autophagosomes, which afterward merge with the lysosomes (65, 76). Compared with microautophagy and macroautophagy, substrate uptake via CMA is a more substrate-specific process. Proteins, containing a KFERQ-pentapeptide motive, may be selectively recognized by the molecular chaperone Hsc70. Afterward, the Hsc70 co-chaperones CHIP and BAG3 mediate ubiquitination and transport of Hsc70-bound substrate protein to the lysosomal membrane. The substrate protein is translocated into the lysosomal lumen via LAMP-2A receptor and lysosomal Hsc70 (Fig. 2) (29, 32, 150). Even if there is evidence for an upregulation of LAMP-2A during oxidative stress, indicating a role of CMA in degradation of oxidized proteins (89), the selective degradation of oxidized proteins is not confirmed. Nevertheless, it is possible that oxidation-related unfolding might expose the KFERQ motive for selective recognition.
Macroautophagy does not seem to have a large share in degradation of oxidized proteins, due to the fact that the selectivity of this process for oxidized proteins is still under discussion. However, the role of macroautophagy in the lysosomal uptake of protein aggregates, also called aggrephagy (94), seems to be proved (Fig. 3). According to this idea, the accumulation of oxidized proteins in pericentriolar aggresomes seems to be an intermediate step in the removal of soluble oxidized proteins, followed by aggresome degradation (44, 79, 162). In 2000, Kopito describes that aggresome formation may facilitate the lysosomal degradation of aggregates (92). Thus, protein aggregates, transported to the MTOC, might be included into phagophores, generating autophagosomes, which fuse with lysosomes. The aggresome-autophagy pathway is considered a protective “back up” mechanism when the molecular chaperones and the proteasomal system are overwhelmed. The transport of protein aggregates to the aggresomes is realized either by histone deacetylase 6 (HDAC6) or by Hsp70 and co-chaperone BAG3 (94) (Fig. 3). Protein aggregates are coupled to dynein motor complex and along microtubules, they are transported to the MTOC (125).
Protein Oxidation in Human Aging
Model systems demonstrating protein oxidation in cellular senescence and aging
The investigation of the role of oxidized proteins in human aging is very difficult, on the one hand, due to the low availability of human material, on the other hand due to the difficulty to determine whether changes are a result of aging or of other processes, such as diseases, environmental exposures, or lifestyle. Since it is assumed that aging first takes place at a cellular level, human cells are often used to study processes which contribute to aging and the role of protein oxidation in aging. Differentiation is conducted between three cellular model systems: (i) in vitro aged primary cells, also referred to as postmitotic senescent cells; (ii) cells aged in vitro due to chronic oxidant treatment; and (iii) primary cells from old donors (in vivo aged) (Fig. 4).
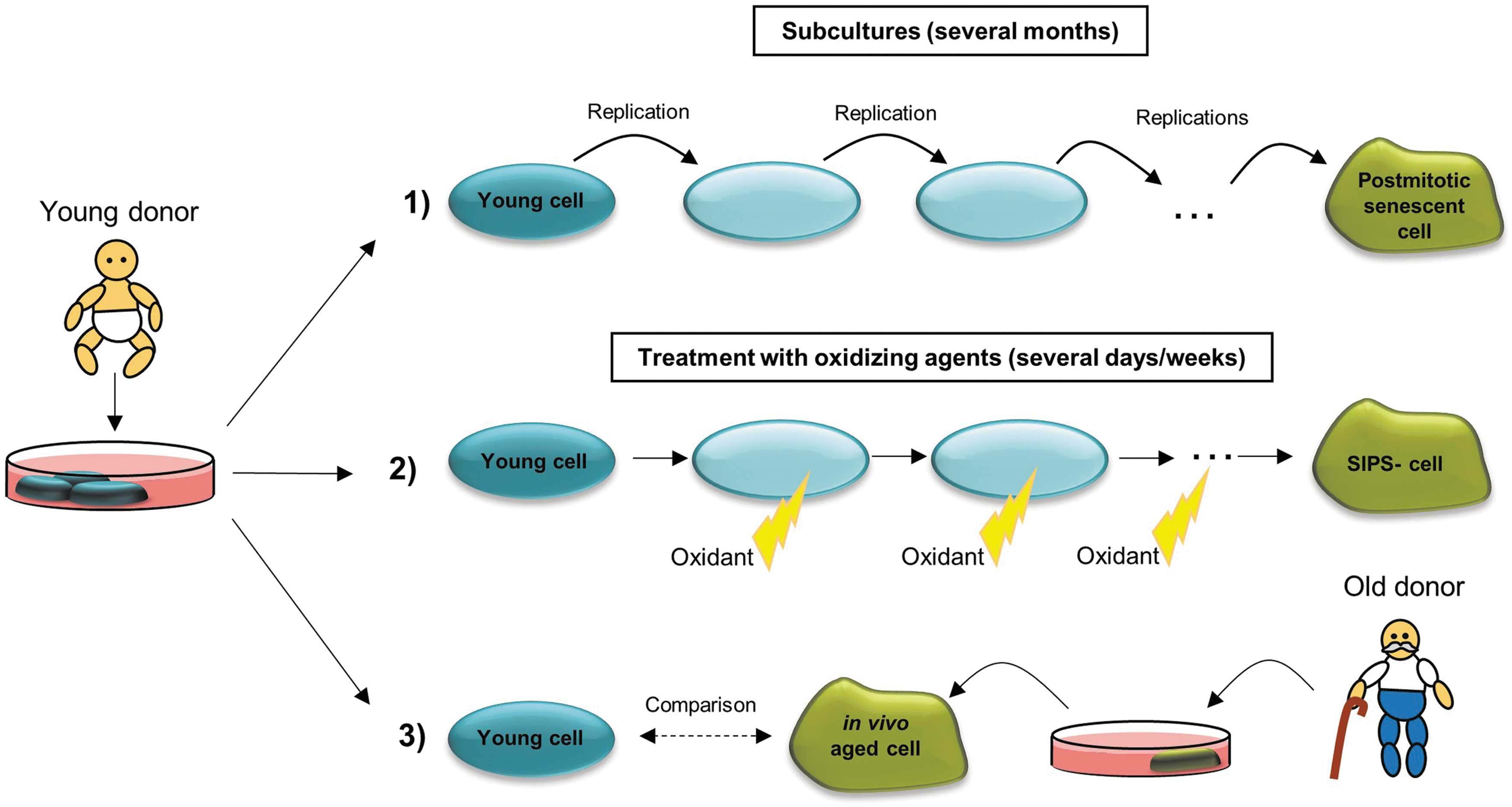
Cellular senescence defines the state when primary mitotic cells reach their replicative limit. To differentiate, organismal senescence describes the biological aging of the whole organism. The cellular replicative limit, also called “Hayflick limit,” was first postulated by Hayflick and Moorhead (59). They described the characteristic of primary cells, grown in culture, only to experience a finite number of cell divisions. Cells that achieve the replicative limit undergo no further division cycles and are referred to as senescent cells. These cells do not die immediately and can, therefore, be used as a model for aging of nondividing cells. Senescent cells are characterized by changes in morphology (increased cell surface and volume), short telomeres, growth arrest, apoptosis resistance, and altered gene expression (20). The occurrence of senescent cells was also observed in vivo in mitotic tissues of humans and primates, and it was shown that the number of senescent cells increases with age (20, 70, 77). Baker et al. have shown that the removal of senescent cells may delay the formation of age-related diseases (6). Thus, the usage of in vitro aged senescent cells in aging research may reflect organismal aging.
To study cellular aging without waiting for the replicative limit, which lasts several months, there is the possibility of modeling the characteristic phenotype of senescent cells due to stress-induced premature senescence (SIPS) [for a review, see Ref. (153)]. This premature aging may be induced via repeated treatments of young primary cells with nonlethal concentrations of H2O2, paraquat, or other stressors (23, 66, 67, 69, 84). The oxidant treatment is repeated every day or every 2 days, leading to chronic oxidative stress, which seems to be the main reason for premature aging (153).
One further cellular model system in aging research represents the isolation and cultivation of primary cells from old donors, although several difficulties have to be considered. The properties of these in vivo aged primary cells may be compared with those of young primary cells.
Although the usage of different cellular model systems seems to be a good tool for investigating aging at the cellular level, it is a matter of debate whether in vitro results are representative for what happens in vivo. Therefore, short living non-mammalians, such as Caenorhabditis elegans, are used as model organisms for aging studies.
In Saccharomyces cerevisiae, two types of aging can be studied: the replicative and the chronological aging. Yeast cells divide by asymmetric budding, building a daughter cell that is smaller than the mother cell. To analyze replicative aging, the daughter cells should be manually separated from the mother cells. The mother cells go only through a limited number of cell divisions (around 20–25) before they enter a short post-replicative phase and die. It is hypothesized that the replicative aging may reflect aging of proliferating cells in complex organisms. In contrast, the chronological aging may be used as a model for aging of nondividing cells. In chronological aging, also referred to as conditional senescence, the cells are limited in one nutrient and, therefore, leave the cell cycle and stop dividing. The chronological lifespan describes the time of survival of nondividing yeast cells [for a review, see Ref. (104)].
The nematode C. elegans is being used since the 1960s as a model organism in research. Since the genome is completely sequenced (27) and there is the possibility to work with techniques, such as RNAi [86% of the 20,000 genes can be knocked down (86)] and cloning, it is an extensively used model for studying molecular and developmental biology. C. elegans has a maximum lifespan of 20–25 days, which is a great advantage in aging research. Next to the short lifespan, there is a further advantage for studying aging in C. elegans: Since they are self-fertilizing hermaphrodites, there are no inbreeding effects on lifespan, which would lead to potential experimental complications (49). An adult C. elegans consists of exactly 959 somatic cells, all of which are postmitotic. Therefore, C. elegans is frequently used to study age-related cellular alterations (72), such as the accumulation of oxidized proteins. It was shown that with age there is an accumulation of fluorescent material (47, 50), comparable to lipofuscin, which is a marker for accumulation of oxidized protein during human aging (see Pathophysiological Roles of Accumulated and Aggregated Proteins section). For studying oxidation-related aging processes, oxidation-sensitive mutants, for example, mev-1, are often used. mev-1 encodes for a subunit of complex II of mETC; mutation of mev-1 leads to increased mitochondrial ROS generation and to decreased lifespan (49).
To achieve a better comparability to human aging, there are also studies in which mammals, such as rodents (102, 142, 144) and monkeys (118, 144, 163), were used to study the aging process and protein oxidation in aging.
Accumulation of oxidized proteins during aging
In humans, ROS are generated during the whole life, either endogenously or through exposure to exogenous ROS generators. As a consequence, oxidized proteins are also generated constantly during lifetime. Aging is accompanied by accumulation of damaged DNA, misfolded proteins, and oxidized proteins (124, 143, 148). The accumulation of oxidized proteins is limited in proliferating cells due to the fact that permanent cell divisions lead to a dilution of damaged molecules. In contrast, postmitotic cells have the potential to accumulate oxidatively damaged proteins. It is still unclear whether the age-related accumulation of oxidized proteins is a product of increased protein oxidation, decreased degradation of oxidized proteins, or a combination of both mechanisms.
An age-related impaired functionality of the proteasomal system has already been described. Friguet and colleagues showed an age-related increase of post-translational oxidative modifications (HNE and glycoxidaton adducts) of proteasome subunits and decreased peptidase activities in peripheral blood lymphocytes and in human lens cells (18, 21, 155). In addition, some authors described an age-related decline of the expression of proteasomal subunits (19, 88, 97, 130), which finally leads to a decreased proteasome formation. A reduced content of 19S regulators was observed in aged rat muscle cells (41). Thus, the ability of the 19S regulators to bind the 20S core and, therefore, the 26S proteasome formation is decreased. This might result in a decreased protein turnover. Reduced 20S activity, in turn, gives an explanation for increased accumulation of oxidized proteins. In addition, 20S subunits may be oxidized and activity of the 20S proteasome seems to be reduced during aging (26, 88, 114, 130).
The age-related impairment of the lysosomal system, described in “The Mitochondrial-Lysosomal Axis Theory of Aging,” defined by Brunk and Terman (16), seems to be a further explanation for age-related accumulation of oxidized proteins. With aging, there is a dysfunctional regulation of lysosomal pH, decreased lysosomal stability, as a consequence of the high sensitivity of lysosomal membrane toward oxidative damage, and an impaired protein targeting to the lysosomes (31). This may lead to a decreased lysosomal degradation of oxidized proteins. In addition, accompanied with aging, a reduced CMA activity was reported as the result of decreased levels of lysosomal membrane receptors (30, 89). However, probably only a small part of oxidized proteins is degraded by lysosomal proteases. A greater contribution to increased accumulation of oxidized proteins seems to come from the elevated ROS formation due to lysosomal impairment, which is, in particular, a result of age-related lipofuscin accumulation in lysosomes (see Pathophysiological Roles of Accumulated and Aggregated Proteins section).
Pathophysiological roles of accumulated and aggregated proteins
Due to the accumulation of oxidized proteins during aging, there is an extensive formation of protein aggregates over the whole lifespan. As mentioned earlier, these aggregates undergo secondary modifications, for example, due to lipid peroxidation products, resulting in the formation of highly crosslinked oxidized material. One important example is lipofuscin, also called “age-related fluorophore,” “age pigment,” or “ceroid.” It consists of approximately 30%–70% crosslinked proteins and 20%–50% lipids, but carbohydrates were also identified to be a component of lipofuscin (9, 37).
Lipofuscin accumulation is thought to be one of the most important factors limiting the lifespan of a cell (67). Lipofuscin is not degradable by the proteasomal or the lysosomal system. Therefore, with age, lipofuscin accumulates in postmitotic and slowly dividing cells. However, not only the amount of lipofuscin increases during aging, but also the rate of lipofuscin accumulation is positively correlated with the rate of aging (72, 118). Thus, liposfuscin is widely used as a biomarker of aging. Although no specific antibodies against lipofuscin are available, the content of lipofuscin is easy to detect due to its autofluorescence (85).
Liposfuscin accumulates, especially in the lysosomal lumen (84). One possible mechanism of lipofuscin formation in lysosomes was proposed by Brunk and Terman (15). ROS, generated as a consequence of age-related oxidative stress, diffuse into the lysosomes, filled with autophagocytosed material, including protein aggregates (92). Since there is a high amount of iron in lysosomes, released during intralysosomal degradation of metalloproteins, highly reactive HO• are generated via Fenton reaction. These radicals may react with protein aggregates and other macromolecules, which are engulfed in the lysosomes, resulting in crosslinkage and lipofuscin formation. However, low amounts of lipofuscin are also found in the cytosol. Cytosolic lipofuscin may be, on the one hand, a result of lysosomal membrane permeabilization (LMP), which is induced due to a variety of chemicals; for example, ROS, generated in an intralysosomal iron-catalyzed Fenton reaction (5, 129, 151). As a consequence of LMP, lysosomal content is released into the cytosol; this may finally lead to cell death [for a review, see Ref. (12)]. It is hypothesized that the accumulation of lipofuscin may be a possible reason for LMP (15); in fact, accumulation of lipofuscin leads to increased ROS formation. However, currently there is no evidence for this theory.
Although the bulk of lipofuscin is located in the lysosomes, it is not clarified whether it is exclusively generated in the lysosomes or additionally in the cytosol. Höhn et al. have shown that neither macroautophagy nor lysosomal activity is necessary for lipofuscin formation (69). Thus, lipofuscin accumulates in the cytosol after inhibition of the macroautophagy pathway.
Lipofuscin is able to inhibit the proteasome. Therefore, in the presence of high amounts of lipofuscin, degradation of oxidized proteins is decreased, resulting in further intracellular accumulation of oxidized proteins and their aggregation (Fig. 5). The mechanism of proteasome inhibition was proposed by Höhn et al. (66). The 20S proteasome binds to hydrophobic patches of unfolded oxidized proteins, which are located on the lipofuscin surface. Although degradation is impossible, because of crosslinks in the lipofuscin structure, 20S proteasome remains bound to the surface, repeatedly trying to degrade the structure, and is, therefore, not available for degradation of other substrates. Moreover, impairment of autophagy and lysosomal degradation due to the lysosomal accumulation of lipofuscin is proposed by Brunk and Terman (15) (Fig. 5). As a consequence, this might cause decreased lysosomal degradation of protein aggregates and organelles, such as impaired mitochondria, finally resulting in increased ROS generation, protein oxidation, further aggregation, and more lipofuscin formation (Fig. 5).
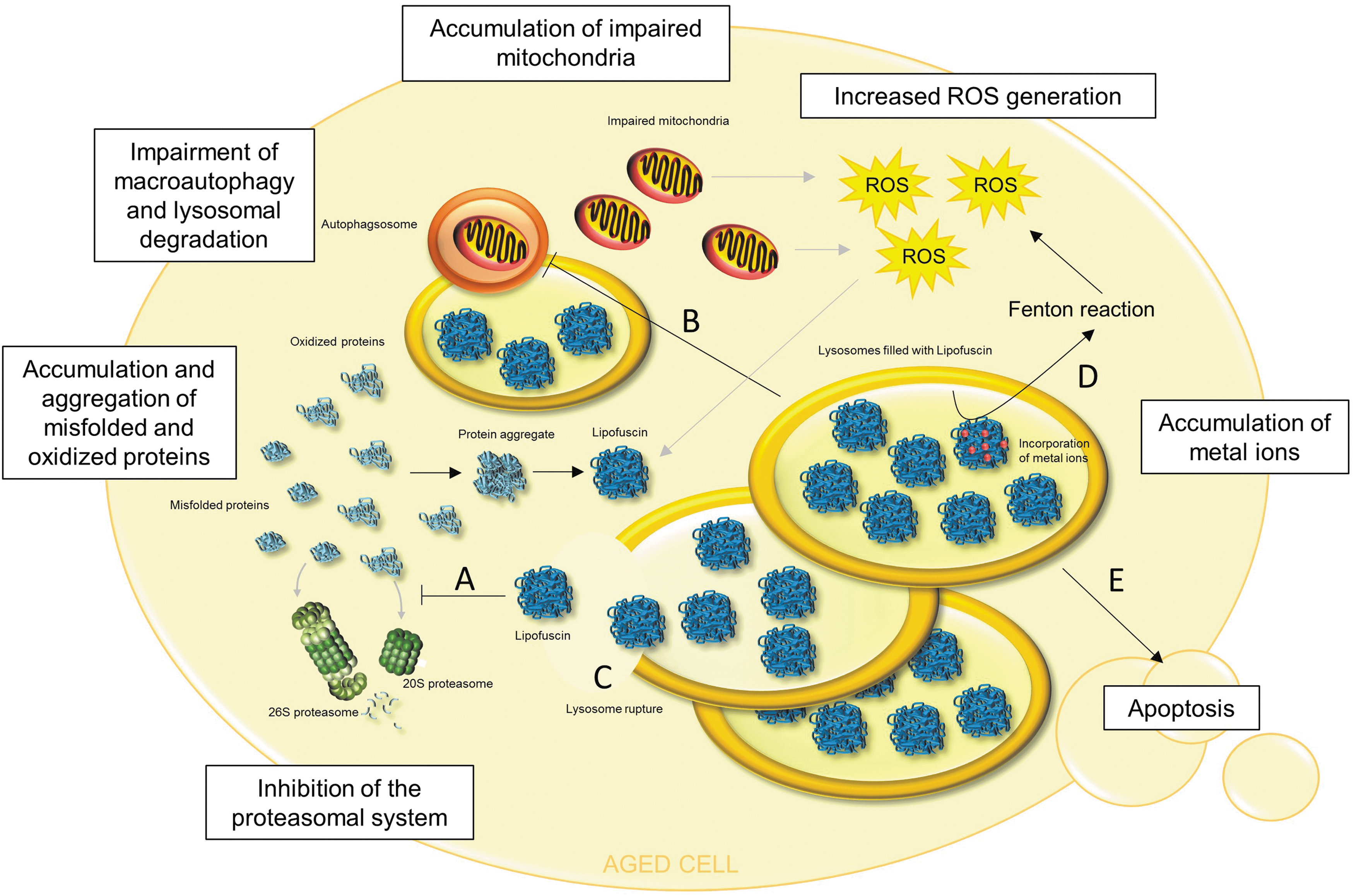
In addition to proteins, lipids, and carbohydrates, lipofuscin contains approximately 2% of metals, including Fe, Cu, Zn, Al, Mn, and Ca (80). Especially the ability of lipofuscin to incorporate iron was shown to contribute to intracellular ROS generation via Fenton reaction and, therefore, to elevated protein oxidation and lipofuscin formation (67) (Fig. 5).
Cells that are overloaded with cytosolic and lysosomal lipofuscin undergo apoptotic cell death. One possible mechanism by which protein aggregates and, in particular, lipofuscin lead to apoptosis is proposed by Powell et al. (132). Lipofuscin-related inhibition of the proteasomal system inhibits the degradation of proapoptotic proteins, including c-jun, Bax, and p27, leading to the initiation of the apoptotic cascade. In addition, proteasome inhibition blocks activation of NFκB signal transduction. NFκB, usually located in the cytosol, is inhibited by binding of IκB. In cellular stress situations, IκB is phosphorylated, ubiquitinated, and degraded by the 26S proteasome. Afterward, NFκB is translocated into the nucleus where it activates genes, in particular responsible for expression of anti-apoptotic proteins. Therefore, proteasomal inhibition leads to apoptosis (65). Castro et al. have shown that the presence of protein aggregates is sufficient to stop cell division (22), and Höhn et al. have demonstrated the induction of apoptosis due to enhanced cytosolic protein aggregate accumulation (67).
Finally, it is important to note that accumulation of protein aggregates with age is not only caused by oxidative stress. Many other processes lead to the aggregation of proteins, such as an impairment of 26S proteasome and lysosomal system. Another important cause, often involved in age-related neurodegenerative diseases (NDs), could be attributed to mutations, which may lead to abnormal fording of proteins and, therefore, to aggregation.
Protein Oxidation in Age-Related Disease
Protein oxidation in NDs
The nervous system seems to be very susceptible to oxidative damage due to the high oxygen consumption (20%–25% of the oxygen consumption of the whole body). Furthermore, the brain has a high content of polyunsaturated fatty acids, which are very susceptible to oxidation, and of iron, which is responsible for the generation of highly reactive ROS via the Fenton reaction [for a review, see Refs. (42, 48, 109)]. In addition, neurons are postmitotic cells and are, therefore, prone to accumulation of damaged molecules. Thus, various markers of oxidative stress, including the accumulation of oxidatively damaged macromolecules, are often elevated in NDs. These facts suggest a possible involvement of oxidative stress and oxidative damage, including protein oxidation, in the pathogenesis of NDs.
To investigate the role of protein oxidation in NDs, in most cases, post-mortem tissues of patients with NDs were analyzed, showing in general an increased content of protein carbonyls and 3-NT as markers for protein oxidation. Moreover, some studies have demonstrated in detail which proteins are oxidatively damaged (110). Oxidized proteins detected in NDs can be divided into different functional groups: (i) glycolysis and energy metabolism, (ii) mitochondrial proteins, (iii) cytoskeleton, (iv) chaperones, and (v) members of the UPS. The oxidative modification of proteins involved in proteostasis (group iv and v) and their impairment may result in accumulation of nonfunctional proteins. Since NDs are associated with the accumulation of aggregated proteins and protein aggregation is often a result of an imbalance between protein unfolding and the removal of these proteins, the impairment of the proteasomal system seems to play a major role in development of NDs.
Alzheimer's disease
Alzheimer's disease (AD) is an irreversible progressive brain disease that is characterized by progressive loss of memory and other cognitive functions (74). AD is the main reason for dementia in older people, and it mostly occurs after an age of 65. The formation of amyloid plaques (APs) and neurofibrillary tangles (NFTs) is characteristic for the disease. APs and NFTs are products of the accumulations of abnormal folded and aggregated beta-amyloid (Aβ) and hyperphosphorylated tau proteins (75).
Tau, the term derived from “tubulin associated unit,” is a protein that is highly expressed in the human brain and which functions in binding and stabilization of microtubules (91, 107). Phosphorylation ensures the biological activity of tau. Tau turnover is catalyzed by the 20S proteasome (54). However, in AD, tau undergoes hyperphosphorylation, which reduces the proteasomal susceptibility of the protein (131). Hyperphosphorylation results in conformational changes of tau. It twists into pairs of helical filaments, which again twist into tangles. Thus, NFTs consist of aggregated and hyperphosphorylated tau proteins, displaying one of the most important markers for AD. Since microtubules are not able to function correctly, aggregation of tau finally leads to an impaired neuronal transport (17, 91).
APs, also called senile plaques, display a further important histological marker of AD. APs consist mainly of aggregated Aβ, and emerge due to an alternative enzymatic cleavage of the amyloid precursor protein (APP). Usually, α- and γ-secretase cleave the APP into the short fragments sAPPα and P3. In AD, the cleavage is carried out by the β- and γ-secretase, leading to the generation of the sAPPβ and Aβ. The latter accumulates in the extracellular space between the neurons. It is known that the formation of APs may be promoted by mutations of three different genes: APP, PS1, and PS2. The transmembrane proteins presenilin-1 (PS1) and presenilin-2 (PS2) are a part of the γ-secretase complex and are involved in the activity of the secretase.
There is evidence supporting the role of oxidative stress in AD, including increased levels of markers for DNA oxidation, lipid peroxidation, and protein oxidation as well as altered activities and expression of antioxidant enzymes (164). Especially the fact that oxidative stress is increased already in the stage of mild cognitive impairment, an early stage of dementia, may indicate a crucial role of oxidative stress during initiation and development of AD (133). It was shown that APs contribute to the formation of oxidative stress in AD brains. For example, Aβ is able to interact with Cu2+ and Fe3+ via its metal-binding domain and it catalyzes the reduction of Cu2+ and Fe3+ and the generation of H2O2 (78, 126), which may be converted to HO• in a Fenton reaction. As mentioned earlier, in AD brains, there are increased levels of protein carbonyls and 3-NT (3, 61, 143), which serve as reliable markers for protein oxidation. Especially in AD hippocampus and inferior parietal lobule, regions that are rich in APs, protein carbonyl levels were significantly higher than in the same regions of control brains (61).
Parkinson's disease
Parkinson's disease (PD) is a movement disorder, associated with a progressive loss of dopaminergic neurons in the substantia nigra pars compacta and with a decrease of neurons in the locus coeruleus. It is the second most prevalent ND after AD. PD is associated with the occurrence of intraneuronal cytoplasmic inclusion bodies, referred to as Lewy bodies. Components of these Lewy bodies are synphilin-1, α-synuclein, parkin, members of the UPS, and others. Although α-synuclein is the major constituent of Lewy bodies, its function is not known. It is highly expressed in the brain and is associated with membrane and vesicular structures of presynaptic terminals (157). Thus, it may be involved in regulation of dopamine release (1). α-synuclein may form oligomeric and amyloid fibrils, which represents the major component of the Lewy bodies. Synphilin-1 is an α-synuclein binding protein. Parkin is an ubiquitin E3 ligase that seems to be involved in turnover of α-synuclein. In most cases, PD occurs sporadic; however, there are some cases of PD that are familial linked. It has been shown that missense mutations in α-synuclein genes, as well as gene duplications and triplications increase the aggregation of α-synuclein and may cause familial PD. PD is characterized by a decreased activity of mitochondrial complex I, which leads to an increased mitochondrial ROS generation and elevated levels of oxidatively damaged lipids, DNA, and proteins. Thus, there is evidence for the role of oxidative stress in pathogenesis of PD.
It is believed that decreased activity of mitochondrial complex I may contribute to aggregation of α-synuclein (10, 108). Furthermore, nitration of tyrosine residues of α-synuclein may play a role in α-synuclein aggregation and its deposition in Lewy bodies (51, 64). Other oxidative modifications are also known to contribute to α-synuclein aggregation and pathogenesis of PD. For example, oxidation of dopamine leads to the generation of oxidized species, such as dopaminochrome, which may interact with α-synuclein. These dopamine adducts on α-synuclein are able to stabilize the toxic α-synuclein protofibrils and inhibit the further aggregation into less toxic mature fibrils (112, 123).
Huntington's disease
Huntington's disease (HD) is an autosomal dominant inherited ND, characterized by cognitive impairment, neuropsychiatric symptoms, and premature death. Further symptoms, which are often associated, are motor skill impairments, such as uncontrollable twitching and spasms. HD is associated with aggregation of the protein Huntingtin. This aggregation is a result of a mutation-driven prolongation of a CAG-motive in the Huntington gene. These abnormal Huntingtin proteins tend to aggregate, leading to the formation of inclusion bodies. Similar to other NDs, markers of oxidative stress are also increased in HD (13). For example, elevated levels of 3-NT in the cortex and striatum have been shown (14).
Protein oxidation in other age-related diseases
Atherosclerosis
Atherosclerosis is a chronic inflammatory disease that is characterized by the accumulation of modified low-density lipoproteins (LDL) in the arterial wall, which leads to proliferation of different cell types. It is one of the major causes for mortality in the Western world (149). The risk to become diseased increases with age. Other risk factors are, for example, increased plasma cholesterol levels, smoking, hypertension, and diabetes. The pathogenesis of atherosclerosis is initiated by an endothelial dysfunction of arteries, resulting in an extravasation of (modified) LDLs, responsible for cholesterol and triglycerides transport, into the subendothelial space (Fig. 6). These LDLs become further modified, for example, by oxidation. Expression of adhesion molecule VCAM-1 is increased, and monocytes and lymphocytes are recruited. Monocytes differentiate into macrophages. Phagocytosis of modified LDLs by macrophages results in intracellular lipid accumulation and foam cell formation, what appears as a “fatty streak.” Furthermore, smooth muscle cells, which are usually localized in other layers of the artery wall (media and adventitia), proliferate and migrate into the intima, forming a fibrous capsule over the “fatty streak.” This process can progress unnoticed for years until the atherosclerotic plaque becomes ruptured, exposing the core of the plaque in the arterial lumen, leading to fatal or nonfatal thrombosis (40, 149).
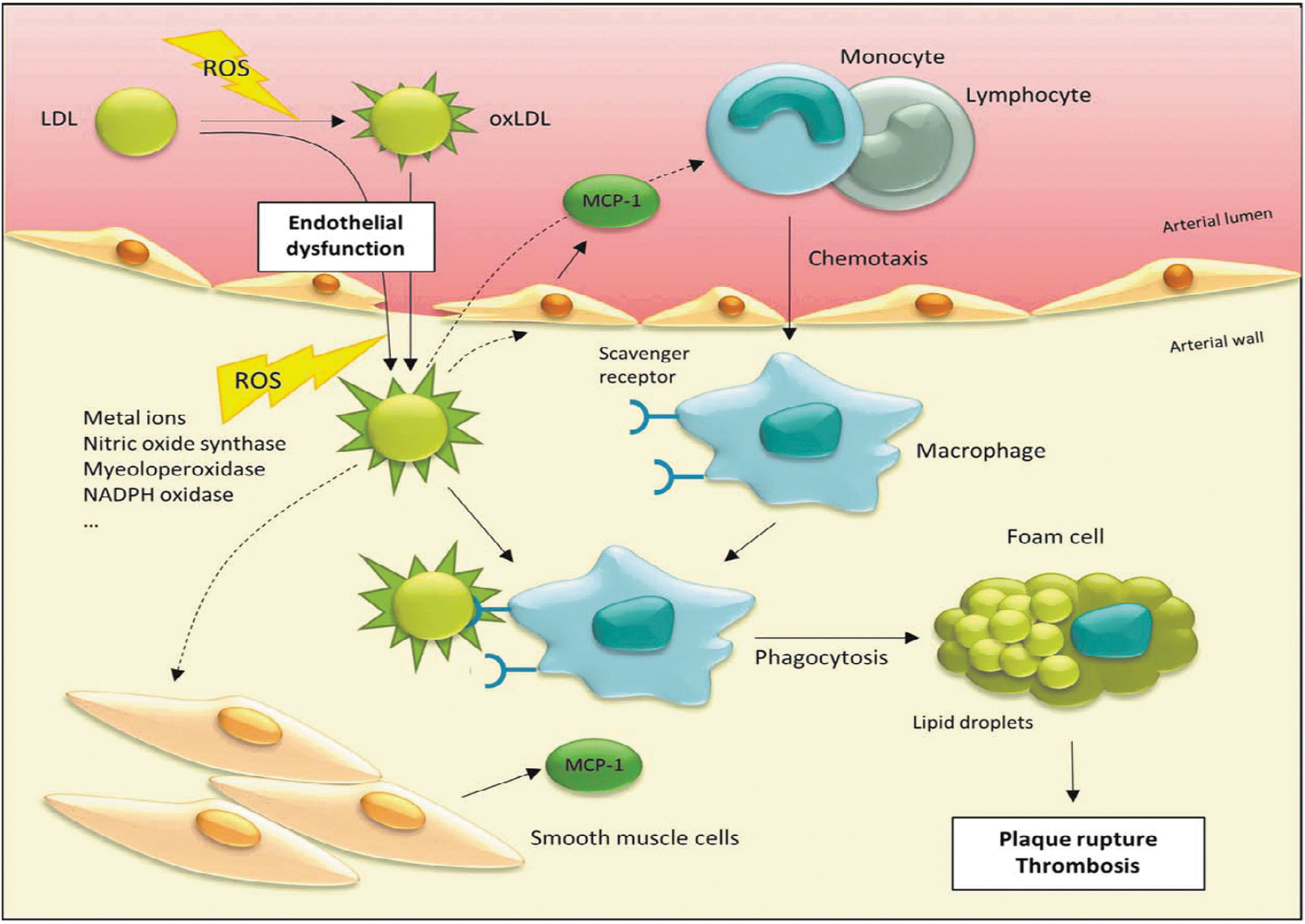
The oxidation of LDL is thought to play a central role in development of artherosclerosis (Fig. 6), although it is widely unknown where, how, and to what extent oxidized LDL (oxLDL) is generated during atherogenesis. It was shown that oxLDL is involved in recruitment of monocytes and lymphocytes (113, 134); for example, via stimulating endothelial cells and smooth muscle cells to secrete monocyte chemotactic protein-1 (MCP-1) (Fig. 6) (120, 136). Furthermore, oxLDL is involved in foam cell formation (103, 116). One major source for ROS that is responsible for subendothelial oxidation processes is the NADPH oxidase complex of leukocytes, which converts O2 into O2 •−. Studies with mice deficient in NADPH oxidase complex activity show either no changes or a decrease in lesion development (7, 90). Other ROS generators in the arterial wall are enzymes, such as xanthine oxidase, nitric oxide synthase, myeoloperoxidase and liopxygenase, and redox-active metal ions (156) (Fig. 6). LDL, oxidized by HO•, exhibits increased levels of ortho-tyrosine and meta-tyrosine, products of phenylalanine residue oxidation. Increased levels of these oxidative modifications of LDL were not found in early atherosclerosis, but in advanced atherosclerosis (99). NO is generated by endothelial cells and nitric oxide synthase, and it mediates a variety of potentially anti-atherogenic effects. However, the highly reactive ONOO−, a product of a reaction of NO and O2 •−, generates 3-NT modifications on LDL (98). Myeloperoxidase generates HOCl, which also may react with tyrosine residues, generating 3-chlorotyrosine. LDLs of atherosclerotic plaques contain elevated levels of 3-NT and 3-chorotyrosine (46). Furthermore, these oxidative protein modifications seem to contribute to foam cell formation (53, 60).
These results show a probable participation of protein oxidation in the conversion of native LDL into oxLDL and thus in foam cell formation and development of atherosclerosis. However, it is important to mention that oxLDL is not only a result of protein oxidation; there are also ongoing lipid peroxidation processes, leading to modification of LDL apoproteins (149).
Age-related macular degeneration
Age-related macular degeneration (AMD) is characterized by the progessive loss of vision, which results from the degeneration of the macula. The macula, consisting of rods and cones, is a part of the retina, that functions in high resolution and color vision (161). Similar to other age-related diseases, markers of oxidative stress, including oxidative damage to DNA, lipids, and proteins, are also increased in AMD. It is postulated that the cumulative oxidative damage may contribute to pathogenesis of AMD (8). The retina seems to be very susceptible to oxidative stress. Besides the exposure to visible light, there is a high consumption of oxygen and a high content of polyunsaturated fatty acids, comparable to neuronal tissues. Oxidative protein modifications were found in retina (139) and drusen (28). Drusen are accumulations of extracellular material in the macula. The increased levels of drusen in the macula are correlated with AMD. Analyses of drusen from AMD patients show, in particular, an increased occurrence of protein crosslinks, which are a result of reactions with products of lipid peroxidation and glycation/glycoxidation (28). Furthermore, there is evidence indicating the connection between the development of AMD and the occurrence of lipofuscin. Lipofuscin accumulates in the lysosomes of the retinal pigment epithelium (RPE). RPE cells are responsible for phagocytosis and lysosomal degradation of the discs. In this degradation process, the fluorophore A2E is generated, which accumulates in the lysosomes and represents a preliminary stage of lipofuscin formation and a component of lipofuscin in the macula (161). It was demonstrated that humans with Stargardt disease, which accumulate A2E in the RPE, show degeneration of the macula and loss of vision, similar to AMD (111, 135).
Conclusion
The investigation of the role of potentially contributing factors to the aging process is a very difficult task, as aging is a complex process involving a whole array of contributing factors. However, a multitude of data indicate that protein oxidation, accumulation of oxidized proteins, and protein aggregation as well as the impairment of the proteasomal system play a major role in the aging process and in the development of some, if not all, age-related diseases. In particular, the accumulation of lipofuscin during aging is an indication of the involvement of protein oxidation in the aging process. More importantly, the accumulation of lipofuscin has serious consequences for cell functionality and metabolism and is an important factor determining the lifespan of a cell. Accumulation and aggregation of oxidized proteins and impairment of the proteasomal system are two mechanisms that are interdependent. It remains to be clarified whether an early intervention on the process of oxidation-related protein aggregation would potentially slow down the aging process or the progression of diseases related to aging.