
Abstract
Introduction
T
NRF2 is controlled by a complex array of transcriptional regulators and post-translational modifications that ensure proper transcriptional activity, under basal conditions and under adaptation to environmental changes (15). Most studies have focused on the role of the eletrophile and redox sensor Kelch-like ECH-associated protein 1 (KEAP1) to adjust NRF2 protein levels to metabolic demands. KEAP1 interacts with two regions of NRF2 (amino-acid sequences DLG and ETGE) located at the Neh2 N-terminal domain to direct ubiquitination by the Cullin-3/Rbx1 complex and proteasome degradation of NRF2 (8, 26, 51, 56). On eletrophile modification or oxidation of KEAP1, the interaction with NRF2 is disrupted. Then, NRF2 escapes degradation, enters the nucleus, and targets ARE genes to increase the capacity of antioxidant and biotransformation reactions (31, 48). While the KEAP1 regulation elegantly explains how NRF2 levels adjust to metabolic demands, it does not clarify the role of NRF2 under basal homeostatic conditions or when KEAP1 activity is limiting (see Discussion). Moreover, heterozygous deletion in the Keap1 gene has different effects on the NRF2 transcriptional signatures of the forestomach, intestine, and liver (32), further supporting the existence of other layers of tissue-specific NRF2 regulation.
We identify a completely novel regulation of nuclear factor (erythroid-derived 2)-like 2 (NRF2) by the canonical WNT signaling pathway that is redox independent and not connected with β-Catenin. At the mechanistic level, we show for the first time that NRF2 associates with Axin1 in a WNT-3A-regulated signalosome. Here, NRF2 is phosphorylated by GSK-3 and targeted for ubiquitin/proteasome degradation by β-TrCP. At the functional level, the relevance of this new pathway is demonstrated in mice with a conditional deletion of Axin1 in the liver. Depletion of Axin1 in these mice results in upregulation of the NRF2-dependent antioxidant metabolism most prominently in hepatocytes of the perivenous zone.
We have previously reported that glycogen synthase kinase-3 (GSK-3) phosphorylates NRF2 in the Neh6 domain (amino-acid sequence DSGISL) to create a recognition site for the E3 ligase adapter SCF/β-TrCP (38, 39). This, in turn, leads to ubiquitin-proteasome degradation of NRF2 through a Cullin-1/Rbx1 complex. The two prototypic mechanisms of regulation of GSK-3/β-TrCP comprise the insulin and WNT pathways, where GSK-3 is regulated via phosphorylation or protein complex formation, respectively, but the participation of these pathways in NRF2 regulation has been hardly addressed.
Regarding insulin, it has been suggested that the protein tyrosine phosphatase 1B modulates GSK3β/NRF2 and Insulin-like Growth Factor-1 Receptor signaling pathways in a model of acetaminophen-induced hepatotoxicity (33). Two recent studies showed that loss of a downstream regulator of insulin signaling, the tumor suppressor phosphatase and tensin homolog (PTEN), results in inhibition of GSK-3 and subsequent induction of NRF2, leading to enhanced cholangiocyte expansion in mice (46) and endometrial carcinogenesis in humans (41).
The experimental evidence connecting NRF2 and WNT signaling is even scarcer despite the fact that WNT is instrumental in acquisition and maintenance of the metabolic signatures of hepatocytes (2, 21, 34, 49). In this study, we have analyzed this connection and its relevance in antioxidant metabolism of hepatocytes taking β-Catenin as a qualified control. In the absence of canonical WNT signaling, β-Catenin is recruited to a multi-protein complex composed by the scaffold proteins Axin1 and adenomatous polyposis coli and the kinases casein kinase 1 and GSK-3β (45). This protein complex phosphorylates β-Catenin on conserved N-terminal Ser and Thr residues, thereby marking it for recognition by SCF/β-TrCP and Cullin-1/Rbx1-mediated ubiquitination and proteasomal degradation. WNT binding to its receptors Frizzled and LRP5/6 at the cell surface results in the disassembly of β-Catenin from the Axin1-GSK-3β complex, leading to accumulation of dephosphorylated β-Catenin, nuclear import, binding to T-cell factor/lymphoid enhancer factor (TCF/LEF), and upregulation of downstream genes (11).
Here, we report that NRF2 participates in the formation of a protein complex with Axin1/GSK-3/β-TrCP that is disrupted by WNT-3A, a prototypic canonical WNT ligand, and leads to upregulation of the NRF2 transcriptional signature. This new signaling axis is KEAP1 independent and, therefore, controls antioxidant metabolism of hepatocytes under basal eletrophile pressure.
Results
The canonical WNT pathway regulates the NRF2/ARE axis
We first analyzed the regulation of NRF2 by WNT-3A. In isolated mouse hepatocytes (Fig. 1A–E), WNT-3A-conditioned medium (CM) (see Materials and Methods) induced a time-dependent increase in the messenger RNA levels of two well-established WNT/β-Catenin targets, Axin2 and Gs (coding glutamine synthetase [GS]), as expected, but, very interestingly, there was a parallel upregulation of genes traditionally considered targets of NRF2, such as Hmox1 (coding heme oxygenase-1 [HO-1]), Nqo1 (NAD(P)H:quinone oxidoreductase 1), and Gpx1 (glutathione peroxidase 1). At the protein level, WNT-3A not only increased β-Catenin and GS, but it also led to a comparable increase in NRF2 and HO-1 (Fig. 1F, G). Recombinant WNT-3A also induced a dose-dependent and time-dependent increase in β-Catenin, NRF2, and HO-1 protein levels (Supplementary Fig. S1; Supplementary Data are available online at
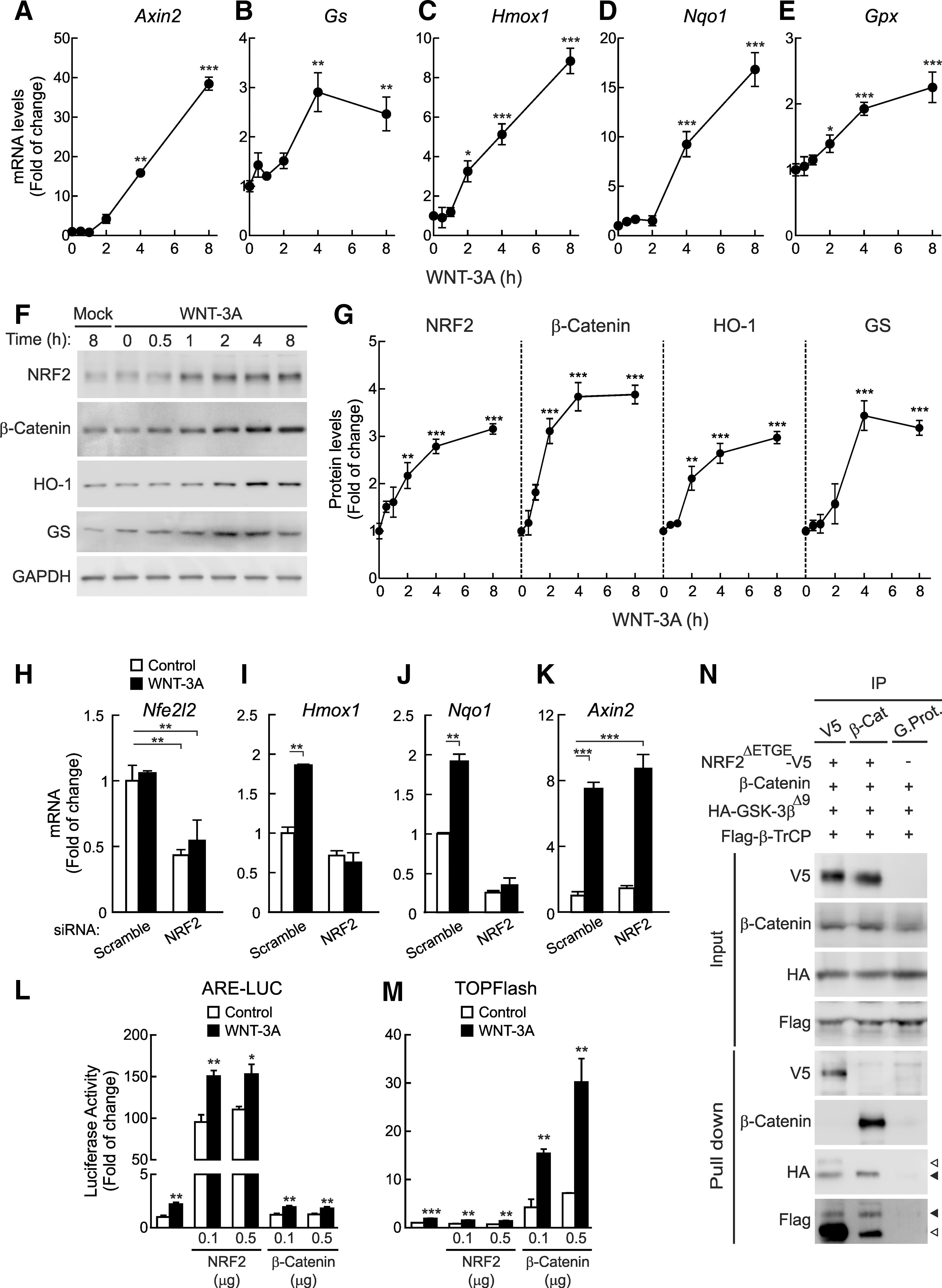
To formally demonstrate a role of NRF2 in induction of antioxidant enzymes by WNT-3A, we knocked down Nrf2 (Nfe2l2) expression in mouse hepatocytes with interfering RNA (Fig. 1H–K). Knockdown of Nrf2 rendered hepatocytes completely unresponsive to WNT-3A-dependent induction of Hmox1 and Nqo1, while Axin2 expression was unaffected. Altogether, these results demonstrate that the WNT canonical pathway regulates the NRF2/ARE axis.
To further determine whether there might be a functional interference between NRF2 and β-Catenin, we used their respective luciferase reporters ARE-LUC and TOPFlash. HEK293T cells were co-transfected with either of these reporters plus pTK-Renilla as control and different amounts of expression vectors for NRF2ΔETGE-V5 or β-Catenin. NRF2ΔETGE-V5 is a stable mutant version of NRF2 with an extended stability, because it lacks the high-affinity binding site for KEAP1 (30, 38, 39). Then, cells were treated with WNT-3A for 16 h. As shown in Figure 1L and M, over-expression of NRF2 but not β-Catenin induced the ARE reporter and WNT-3A led to a similar increase of ARE activity. On the other hand, over-expression of β-Catenin but not NRF2 induced the TOPFlash reporter and WNT-3A led to a similar induction. We also analyzed whether NRF2 and β-Catenin might be a part of a common protein complex. As shown in Figure 1N, immunoprecipitation of either NRF2 or β-Catenin did not pull down the other. As a positive control, we observed that two well-known NRF2 and β-Catenin interacting proteins, GSK-3β and β-TrCP, co-immunoprecipitated with NRF2 or with β-Catenin. Therefore, our results suggest that these two transcription factors are not associated in a common protein complex and that WNT-3A targets them independently.
WNT regulates NRF2 through a KEAP1-independent but GSK-3/β-TrCP-dependent mechanism
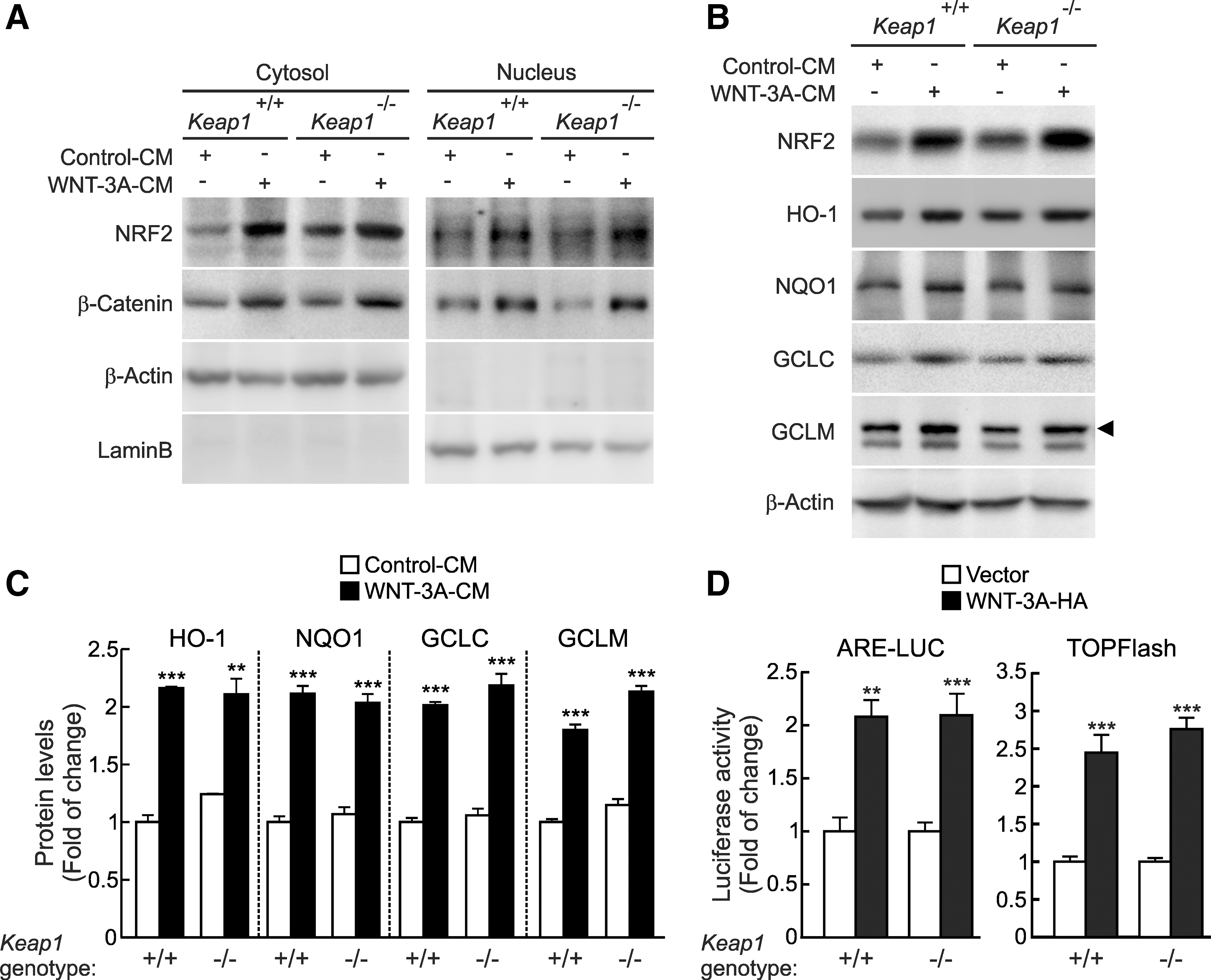
Considering that KEAP1 is the redox-dependent controller of NRF2 stability, we studied whether this E3 ligase adapter might participate in WNT signaling to NRF2 by using mouse embryonic fibroblasts (MEFs) derived from KEAP1-deficient (Keap1 −/−) and wild-type (Keap1 +/+) mice as a control. WNT-3A (4 h) increased the protein levels of NRF2 in wild-type MEFs in both the cytosolic and nuclear fractions in parallel to the increase in β-Catenin used as a control (Fig. 2A). As previously reported, the baseline NRF2 protein levels in Keap1 −/− MEFs were modestly higher than in Keap1 +/+ MEFs due to the disruption of KEAP1-mediated degradation of NRF2 (38). However, more relevant, WNT-3A also increased NRF2 levels in Keap1 −/− MEFs. Moreover, on WNT-3A stimulation, we found an induction of approximately two-fold in the NRF2-regulated enzymes HO-1, NQO1, GCLC, and GCLM in both cell lines (Fig. 2B, C). As an alternative approach, we further explored the effect of WNT-3A on the NRF2 transcriptional activity in Keap1 +/+ and Keap1 −/− MEFs transfected with the NRF2 luciferase reporter ARE-LUC/pTK-Renilla and the β-Catenin reporter TOP/FOPFlash as control (Fig. 2D). Regardless of the genotype, WNT-3A led to a similar increase in ARE-LUC activity. These results further confirm that WNT-3A stabilizes NRF2 protein levels by a KEAP1-independent mechanism.
Since GSK-3 plays a key role in the WNT signaling pathway, we studied whether GSK-3α or GSK-3β might be involved in the upregulation of NRF2 mediated by WNT-3A. For this purpose, we knocked down each GSK-3 isoform with short interfering RNAs (siRNAs). HEK293T cells were transfected with an expression vector for NRF2-V5 and then with siRNA against GSK-3α, GSK-3β, or both isoforms (Fig. 3A). Partial knockdown of either protein isoform (about 50%–60% for GSK-3α, and 60%–70% for GSK-3β) was enough to increase NRF2 protein levels, and this effect was maximal when both isoforms were knocked down together. WNT-3A also increased NRF2 protein levels and, in this case, knockdown of GSK-3α, β, or both did not lead to a further effect. A similar effect was observed for β-Catenin, used as a control.
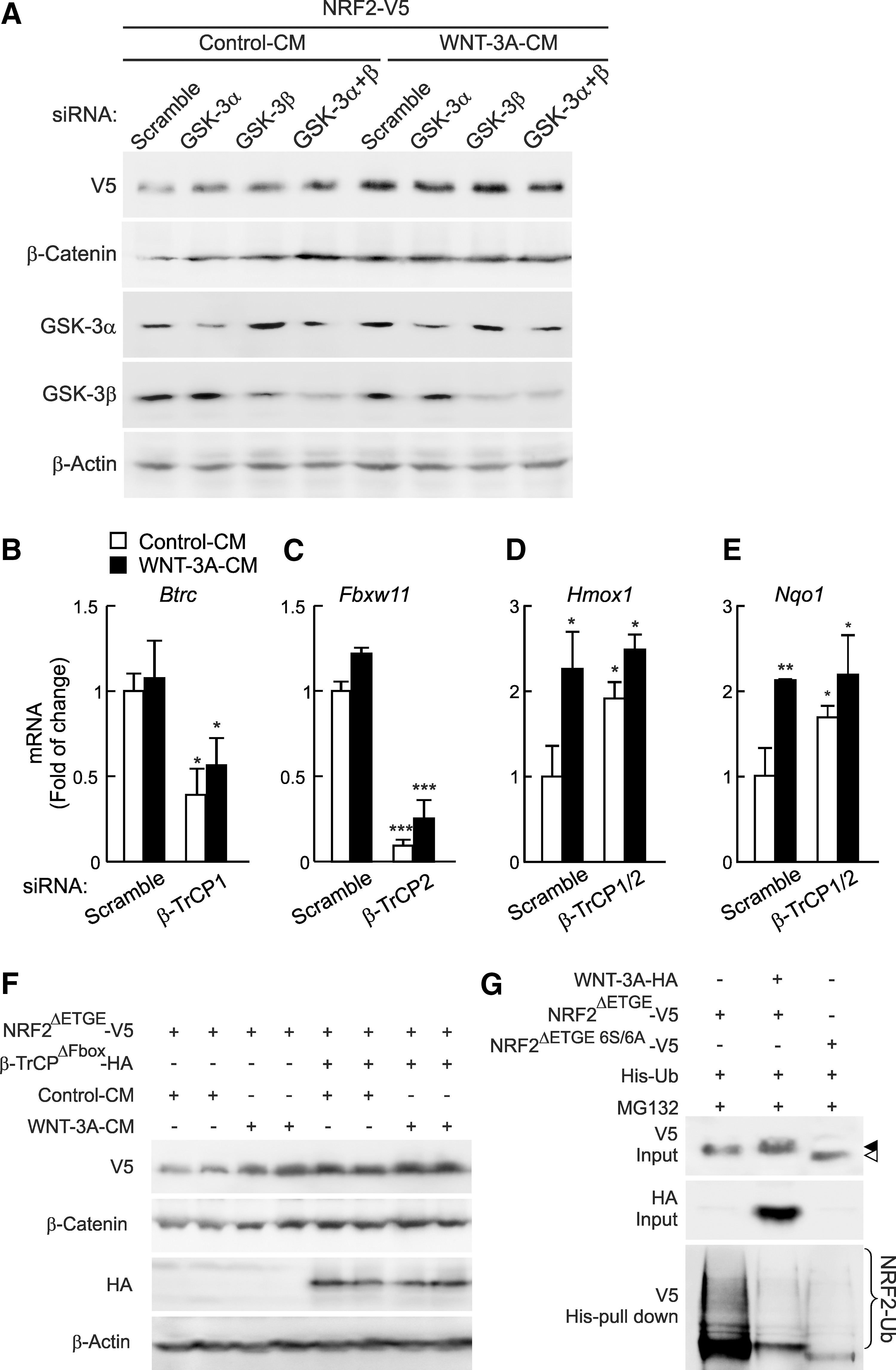
We have previously reported that the E3 ligase adapter SCF/β-TrCP targets NRF2 for proteasome degradation when it had been previously phosphorylated by GSK-3. Therefore, to test the involvement of β-TrCP in the context of WNT signaling to NRF2, we knocked down β-TrCP1 and β-TrCP2 with siRNA in mouse hepatocytes (Fig. 3B–E). Reduction of β-TrCP1/2 levels led to a statistically significant increase in the basal mRNA levels of HO-1 and NQO1, which were close to saturation under WNT-3A induction. This outcome is consistent with a role of this E3 ligase as a negative regulator of NRF2 (7, 38, 39). As an alternative approach, we expressed a dominant-negative mutant (β-TrCPΔFbox), which lacks the Fbox motif and cannot form functional complexes with Cullin-1/Rbx1 but competes with endogenous β-TrCP for binding to its substrates (54). To avoid a potential interference with KEAP1, we used the KEAP1-insensitive NRF2 mutant (NRF2ΔETGE-V5). HEK293T cells were co-transfected with NRF2ΔETGE-V5 and β-TrCPΔFbox-HA, and after 24 h were treated with WNT-3A- or control-CM for 3 h (Fig. 3F). When NRF2 stabilization was achieved by the presence of β-TrCPΔFbox, WNT-3A did not promote an additional increase in NRF2, indicating that the inactivation of β-TrCP is involved in the stabilization of NRF2 mediated by WNT-3A. Similar results were found with stabilization of β-Catenin, carried as a control.
Further evidence for the role of WNT-3A in the GSK-3/β-TrCP-mediated regulation of NRF2 was obtained by analyzing the pattern of NRF2 ubiquitination (Fig. 3G). HEK293T cells were co-transfected with plasmids expressing NRF2ΔETGE-V5, His-Ubiquitin, and either the control vector or an expression plasmid for HA-tagged WNT-3A. Since NRF2ΔETGE-V5 is not targeted by KEAP1, ubiquitination of this protein rests on other E3 ligase adaptors such as β-TrCP (7, 38, 39). As a control for the GSK-3-independent NRF2-ubiquitination, we also examined the levels of ubiquitinated NRF2ΔETGE 6S/6A-V5, a mutant that is insensitive to both KEAP1 and GSK-3/β-TrCP-mediated degradation pathways (38, 39). WNT-3A led to a significant reduction in the ubiquitination of NRF2ΔETGE to levels comparable to those of NRF2ΔETGE 6S/6A, indicating that WNT-3A is blocking NRF2 degradation through the inhibition of GSK-3/β-TrCP pathway. We could still detect residual polyubiquitinated NRF2 in the presence of WNT-3A and in NRF2ΔETGE 6S/6A, indicating the existence of other possible proteasome degradation motifs apart from those involving GSK-3 pathways (7). Taken together, these findings demonstrate that the WNT canonical pathway regulates NRF2 through a mechanism which implies the inhibition of GSK-3/β-TrCP axis.
Axin1 is involved in the regulation of NRF2
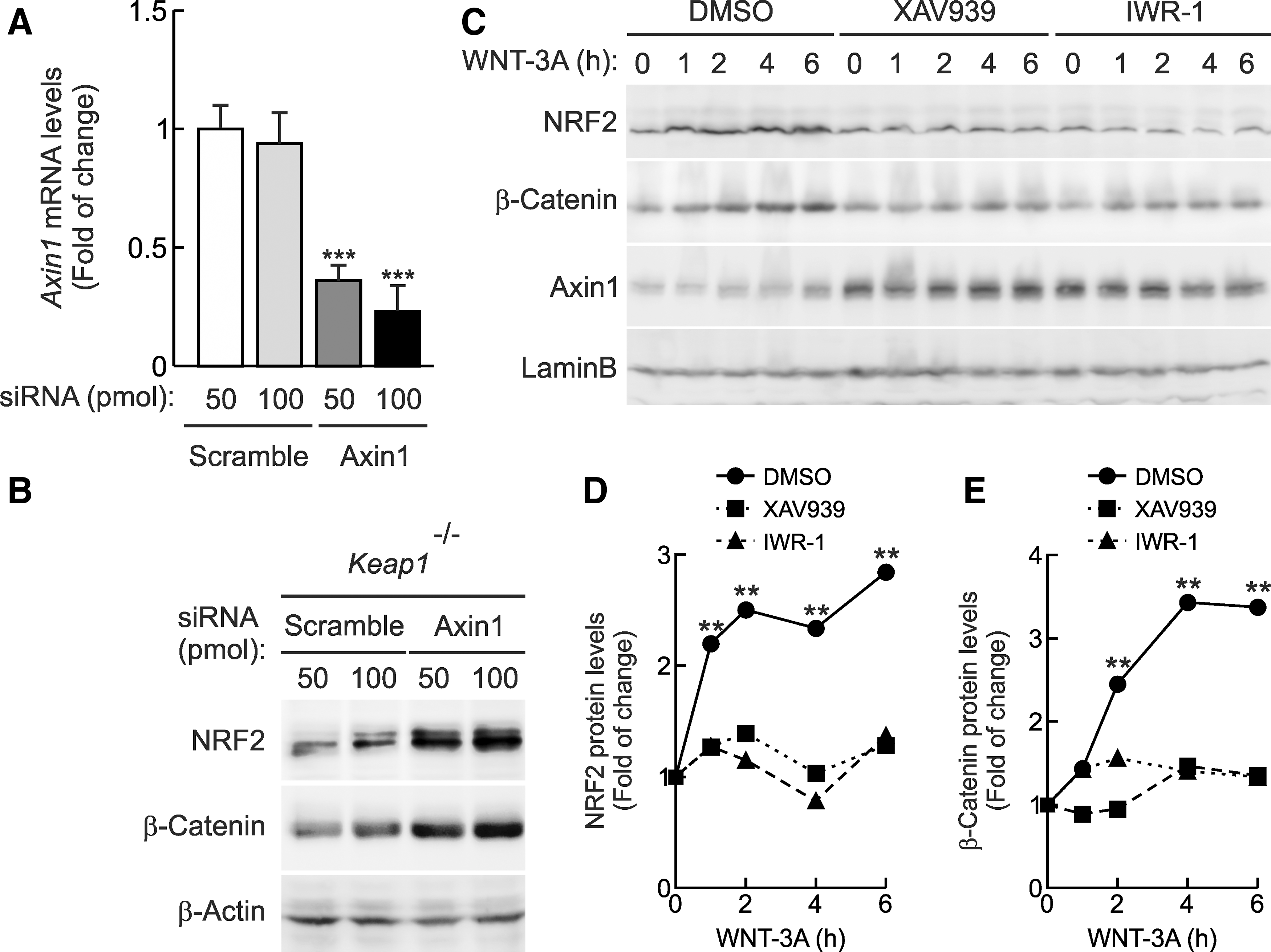
Axin1 is an essential element of the WNT signalosome that scaffolds specific kinases and substrates together (36). To study the possible connection between NRF2 and Axin1, first we knocked down the expression of Axin1 in Keap1 −/− MEFs. In line with the previous data, siRNA-mediated depletion of Axin1 resulted in high NRF2 and β-Catenin protein levels (Fig. 4A, B). Then, we used two recently characterized small-molecule WNT inhibitors named XAV939 and IWR-1, which inhibit Tankyrase-dependent degradation of Axin1 (6, 19). HEK293T cells were treated for 16 h with vehicle (DMSO), XAV939 (10 μM), or IWR-1 (20 μM) and then with WNT-3A-CM for the time points indicated in Figure 4C–E. As expected, Axin1 protein levels were stabilized in HEK293T cells treated with XAV939 or IWR-1. Interestingly, both WNT inhibitors blocked NRF2- as well as β-Catenin-induction, in response to WNT-3A. Similar results were obtained in mouse hepatocytes (Supplementary Fig. S4). Overall, these data show that genetic inactivation or pharmacologic stabilization of Axin1 inversely modulates NRF2 protein levels.
These results suggested that NRF2 might be a part of a protein complex with Axin1 as is the case for β-Catenin. To determine whether endogenous NRF2 and Axin1 proteins could be physically associated, we used a co-immunoprecipitation assay in mouse hepatocytes. Cells were treated with MG132 (40 μM) for 3 h to prevent NRF2 and β-Catenin degradation and maintain the protein complexes. Both NRF2 and β-Catenin associated with Axin1 in the same samples (data not shown) and, very importantly, WNT-3A disrupted the Axin1/NRF2 association (Fig. 5A).
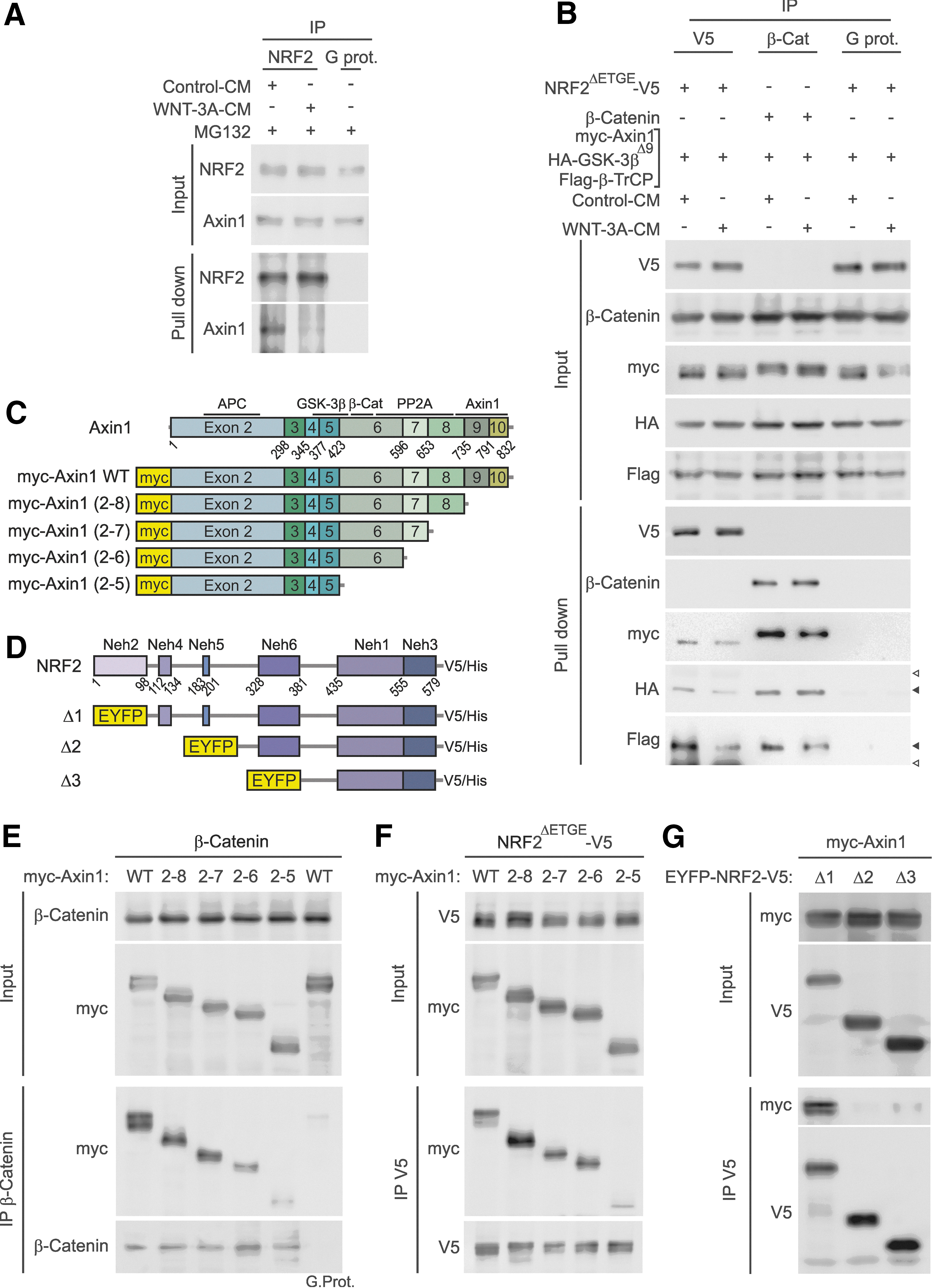
Molecular details of Axin1 and NRF2 association were further explored in HEK293T cells. In Figure 5B, KEAP1-insensitive NRF2ΔETGE-V5 or β-Catenin, as control, were co-transfected with myc-Axin1, HA-GSK-3βΔ9, and β-TrCP. Then, cells were maintained in low-serum medium for 16 h and finally treated with WNT-3A. Cellular lysates were immunoprecipitated with anti-V5 for NRF2 or anti-β-Catenin. In line with the results obtained in hepatocytes, we found that Axin1 interacts not only with NRF2 and β-Catenin but also with GSK-3β and β-TrCP. Moreover, these interactions were reduced by WNT-3A, suggesting that this factor disassembles the protein complex. Taken together, these data indicate that NRF2 is bound to Axin1 and released from it on WNT-3A stimulation.
Characterization of the regions of interaction between NRF2 and Axin1
We generated four myc-tagged amino-terminal deletion mutants of mouse Axin1 by consecutive exon deletion from the 3′-end (Fig. 5C). Then, HEK293T cells were co-transfected with these Axin1 deletion mutants and either β-Catenin, as control (Fig. 5E), or NRF2ΔETGE-V5 (Fig. 5F). Co-immunoprecipitation experiments revealed that the Axin1/β-Catenin complex is formed only when exon 6 of Axin1 is present. Regarding NRF2ΔETGE-V5, when cellular lysates were pulled down with anti-V5 antibody, we detected that, similar to β-Catenin, loss of exon 6 resulted in a dramatic reduction of the amount of Axin1 bound to NRF2.
In additional experiments, we determined the region of NRF2 required for Axin1 binding by the use of fusion proteins carrying enhanced yellow fluorescent protein (EYFP) and progressive N-terminal truncations of NRF2, as schematically indicated in Figure 5D. The EYFP-NRF2 chimera Δ1 lacks the Neh2 domain required for binding to KEAP1; it retains the ability to bind Axin1, and this interaction is lost in the presence of WNT-3A (Supplementary Fig. S5). As shown in Figure 5G, immunoprecipitation of NRF2 chimeras revealed that the region comprising the Neh4 and Neh5 domains (i.e., EYFP-NRF2Δ2-V5) is required for binding to Axin1.
Conditional disruption of Axin1 in the liver results in increased NRF2 transcriptional activity
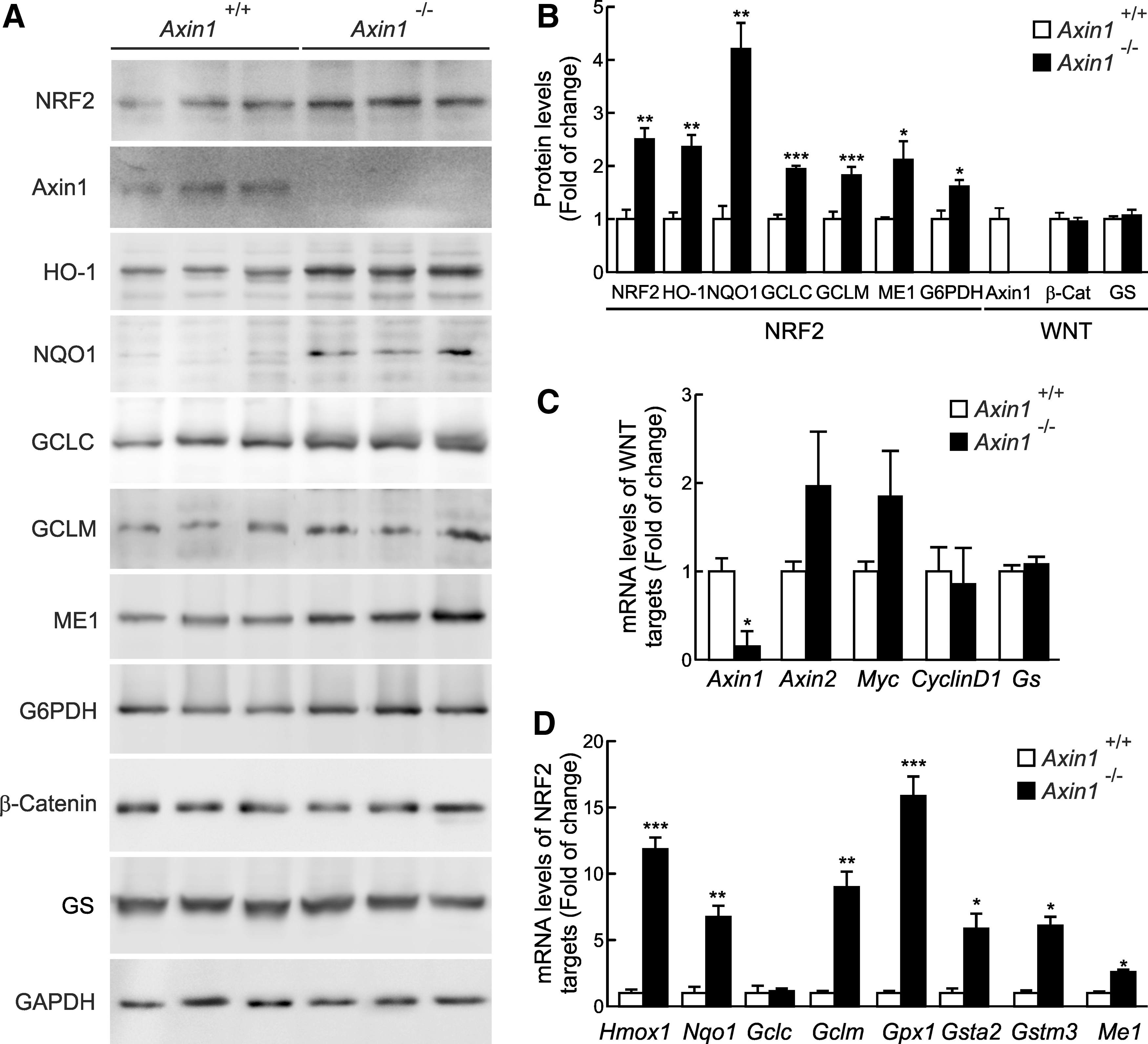
We analyzed NRF2 function in the liver of transgenic mice with a conditional liver-specific depletion of Axin1 (Axin1 −/−) (see Materials and Methods). As shown in Figure 6A and B, these mice showed a dramatic depletion of Axin1. Furthermore, in agreement with (10), total β-Catenin and GS protein levels were not substantially altered. Regarding NRF2, the depletion of Axin1 resulted in upregulation of the NRF2 signature as determined by increased NRF2 protein levels and a two- to four-fold induction of six enzymes regulated by this transcription factor: HO-1, NQO1, GCLC, GCLM, ME1 (malic enzyme 1), and G6PDH (glucose 6 phosphate dehydrogenase). In addition, quantitative real time PCR (qRT-PCR) was used to analyze messenger RNA levels of well-known NRF2 and WNT-specific targets. Levels of Axin1 mRNA were strongly reduced, while there was a modest increase in Axin2 and Myc gene expression and no change in CyclinD1 and Gs (Fig. 6C). This modest increase in β-Catenin-regulated genes is comparable to that reported in the initial characterization of these mice (10) and suggests a compensatory mechanism, maybe mediated in part through Axin2 upregulation. However, more importantly for our study, with the exception of Gclc, mRNA levels of the NRF2-targeted enzymes Hmox1, Nqo1, Gclm, Gpx, Gsta2, Gstm3, and Me1 were increased in Axin1 −/− livers (Fig. 6D). These results demonstrate that the basal NRF2 transcriptional signature is inversely dependent on the availability of Axin1 in the liver.
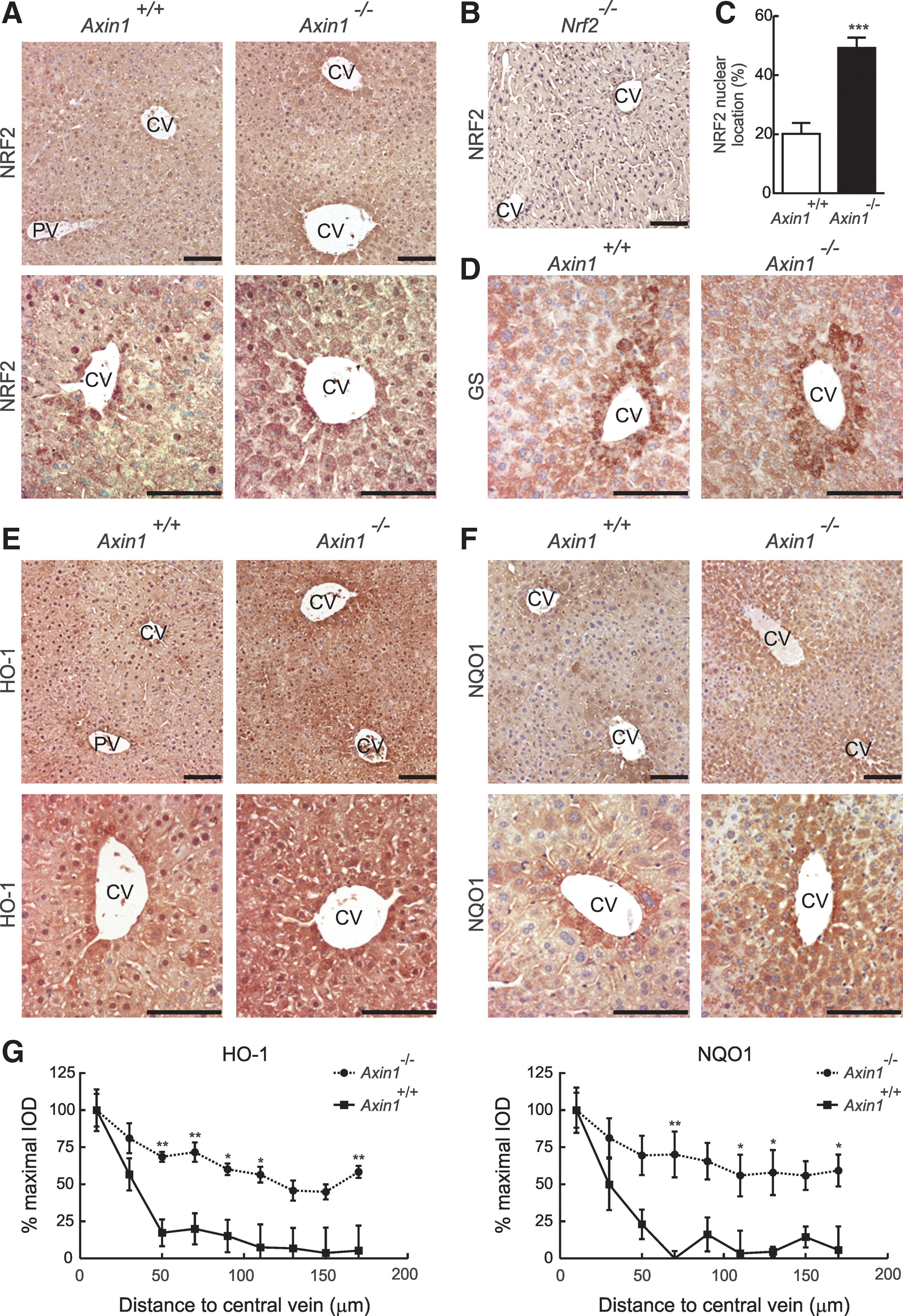
We sought to determine whether Axin1 −/− mice show alterations in the phenotypic distribution of enzymes regulated transcriptionally by NRF2. Five-micrometers-thick sections of paraffin-embedded livers were first stained with an anti-NRF2 antibody. Figure 7A shows low and high magnification of NRF2 staining in representative perivenous zones, and Figure 7B shows a negative control in the liver of Nrf2 −/− mice. We found that NRF2 was cytosolic and nuclear in Axin1 +/+ mice. In Axin1 −/− mice, NRF2 staining was increased, suggesting not only in agreement with Figure 6 that it was upregulated in the absence of Axin1, but also we counted more hepatocytes with a nuclear accumulation of NRF2 (Fig. 7C). GS distribution was not altered in Axin1 −/− livers, as expected (10), indicating that other elements of the WNT pathway may be responsible for regulation of ammonia detoxification (Fig. 7D). However, two enzymes transcriptionally regulated by NRF2, HO-1 (Fig. 7E, G, left panel), and NQO1 (Fig. 7F, G, right panel), which in Axin1 +/+ livers were expressed around the central vein, exhibited exacerbated expression in the Axin1 −/− livers, covering the entire hepatic lobule or expanding toward the periportal zone. Overall, these results demonstrate that the disruption of Axin1 results in upregulation of NRF2 and its target genes in vivo and that NRF2 participates in liver zonation of antioxidant metabolism.
Discussion
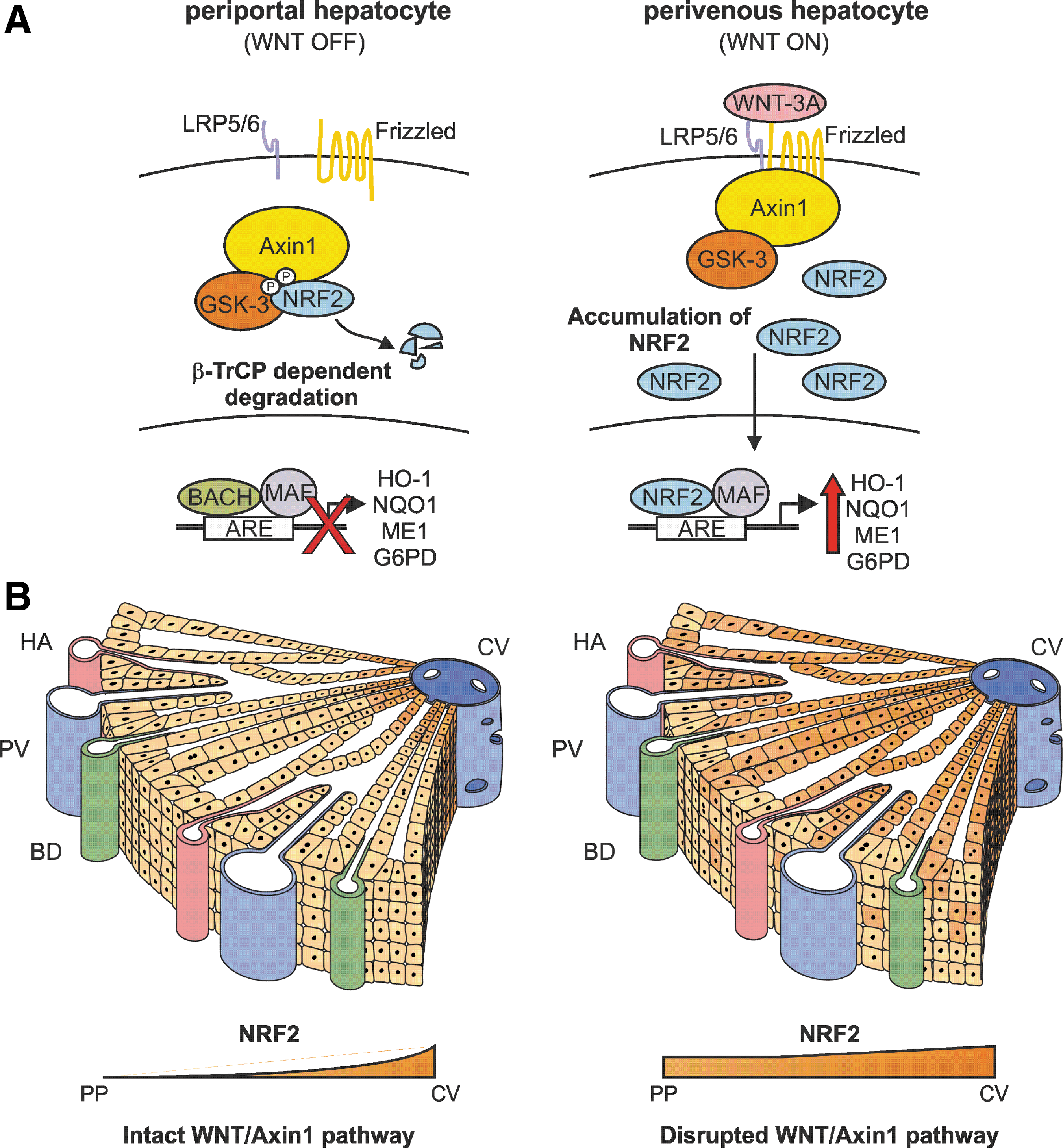
In this study, we report a completely unexplored link between the canonical WNT-3A signaling pathway and the regulation of antioxidant metabolism by transcription factor NRF2. Figure 8A describes the elements identified in this new signaling pathway. The canonical WNT pathway has been traditionally connected with development and maintenance of differentiated cell phenotypes, including those of hepatocytes. It is, therefore, very interesting that at least in the liver it exerts a part of this function through the regulation of NRF2. Indeed, while WNT signaling has been demonstrated to regulate antioxidant metabolism through the β-Catenin-dependent upregulation of some antioxidant genes (13, 35), it is now clear that it also targets the NRF2 signature. This is done in a β-Catenin-independent manner (Fig. 1), further suggesting that these transcription factors, although regulated by WNT, operate independently.
Most signaling pathways lead to production of reactive oxygen species that might contribute to regulation of NRF2 by interfering with the redox sensor KEAP1. In the case of WNT, a role for this redox modulation was reported through the RAC1 GTPase-dependent activation of NADPH oxidase 1 (NOX1) (22). In addition, the Wilms tumor gene on X chromosome (WTX) inhibits degradation of NRF2 through competitive binding to KEAP1 (5). These observations indicated the importance of determining whether WNT was somehow modulating the KEAP1/NRF2 response. We found that the WNT regulation of NRF2 was independent of KEAP1, because (i) WNT-3A stabilized the KEAP1-insensitive mutant NRF2ΔETGE-V5 and (ii) WNT-3A stabilized endogenous NRF2 in MEFs from Keap1 −/− mice.
On the other hand, we found that Axin1 binding, GSK-3β phosphorylation, and β-TrCP ubiquitination of NRF2 were disrupted by WNT-3A and, therefore, established the basis to study this novel mechanism of NRF2 regulation. We mapped the region of interaction between NRF2 and Axin1 to the exon 6 of Axin1. This is a critical scaffolding segment of Axin1, termed the central region, that enables Axin1 to interact with several proteins, including transcription factors β-Catenin, MYC, Smad3, and p53 as well as protein kinases GSK-3β, CK1, MKK1, and MKK4, among others (36). This region is natively unfolded, and it has been suggested to undergo a sort of “induced fit” to accommodate specific groups of proteins. Thus, our study suggests that Axin1 scaffolds a complex with NRF2, GSK-3β, and β-TrCP to downregulate NRF2. The existence of several proteins which compete for binding to Axin1 in the same region suggests that Axin1 levels may be limiting to control specific WNT pathways.
In addition to the well-characterized KEAP1/NRF2 system, it is becoming clear that other regulatory mechanisms also operate on NRF2. In fact, graded reduction in expression of Keap1, achieved in mouse liver by several genetic techniques, leads to graded increase NRF2 levels (47, 55), suggesting that KEAP1 operates close to saturation and that an increase in NRF2 overloads this mechanism. Formal proof of this hypothesis is lacking, as, to our knowledge, a comparison of turnovers of KEAP1 and NRF2 under different experimental conditions has not yet been reported. In any case, the modest induction of NRF2 that escapes KEAP1 regulation has a physiological meaning. As an example, in pancreatic cancer, a modest K-Ras-dependent upregulation of less than two-fold in NRF2 transcription causes an increase in the NRF2 signature (9). Here, we extend the importance of modest increases in NRF2 to the modulation of antioxidant metabolism in the liver, and we propose that NRF2 levels appear to be fine-tuned not only by the relative levels of KEAP1 but also by other NRF2 interacting proteins which now include Axin1.
The molecular mechanisms responsible for maintenance of metabolic zonation in the liver are poorly understood. Oxygen gradients and hormones that run across the liver lobule participate in acquisition of periportal versus perivenous phenotypes of hepatocytes, and at least β-Catenin has been also involved in the upregulation of some antioxidant enzymes (2). The implication of NRF2 has not yet been studied, but very recently, it has been reported that Nrf2 −/− mice develop a congenital intrahepatic shunt which alters hepatic oxygen and protein expression gradients (44). Here, by making use of liver-specific floxed Axin1 −/− mice, we found a substantial increase in the transcriptional activity of NRF2 as determined by increased protein and mRNA levels of HO-1, NQO1, GCLM, ME1, and G6PDH. Moreover, compared with control livers, NRF2, HO-1, and NQO1 expression expanded to hepatocytes located beyond the initial rows of the hepatic central vein. In summary, these results indicate that WNT signaling to NRF2 participates in maintenance of the perivenous hepatocyte phenotype. Figure 8B proposes a mechanism used by WNT/Axin1 to control zonation of NRF2 signature in the liver. It is interesting that other WNT-regulated transcription factors do not have this effect. Thus, c-MYC, a crucial element in APC-related tumorigenesis, does not seem to participate in liver zonation of ammonia detoxification (4). These facts suggest that Axin1 scaffolds several protein complexes with different functions.
It may seem somewhat intriguing that NRF2-mediated antioxidant metabolism is more noticeable in the perivenous zone, rather than in the periportal zone, which is more exposed to delivery of oxygen from the hepatic artery and xenobiotics from the portal vein. A possible explanation to this paradox is again the distinction between KEAP1-dependent regulation of NRF2, adapting NRF2 levels to transient demands, which should impact the periportal region, and the effect of WNT signaling that may be more related to the basal physiology of topologically different hepatocytes. There is evidence of basal low oxygen tension in the perivenous zone, with increased expression of hypoxia-inducible factor-1α, 2α, and 3α (23). Considering that hypoxia may lead to release of mitochondria-generated reactive oxygen species, a WNT-regulated developmental control of NRF2 might provide a KEAP1-independent layer of regulation to this zone.
The finding of an Axin1/NRF2 complex and its relevance in liver metabolism has important consequences for therapy of liver diseases and probably others. Several pharmacological pipelines are being developed to inhibit WNT, particularly in cancer therapy (37). These drugs should be now tested for add-on effect on NRF2. On the other hand, WNT activators are also being considered and these should also provide a KEAP1-independent or complementary mechanism of NRF2 activation Here, we have used Axin1 stabilizers, acting as Tankyrase inhibitors (28), to inhibit NRF2 induction by WNT. Together, these studies open new therapeutic possibilities by targeting WNT/β-Catenin and, in our case, WNT/NRF2.
Materials and Methods
Cell culture and reagents
Hepatocytes from neonatal mice were obtained as previously reported (14) and grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 20 mM HEPES, and 80 μg/ml gentamycin. HEK293T cells were grown in DMEM supplemented with 10% fetal bovine serum and 80 μg/ml gentamycin. MEFs from Keap1 −/− mice and Keap1 +/+ littermates were kindly provided by Dr. Ken Itoh (Center for Advanced Medical Research, Hirosaki University Graduate School of Medicine, Japan). Conditioned media from L-Control (Control-CM) and L-WNT-3A (WNT-3A-CM) confluent cells (grown in DMEM supplemented with 10% fetal bovine serum, 0.5 U/ml penicillin, and 0.5 μg/ml streptomycin) was collected and passed through a 0.2 μm filter. Transient transfections were performed with calcium phosphate, using reagents from Sigma-Aldrich, or with TransFectin™ lipid reagent from Bio-Rad. MG132, XAV939, and IWR-1 were from Sigma-Aldrich. Recombinant WNT-3A was from Cell Guidance Systems.
Conditional Axin1 mutant mice
All animal experiments were performed according to UK Home Office regulations. Details of the generation of Axin1 −/− mice are described in (10). Briefly, Ah-Cre mice were intercrossed with mice carrying LoxP-flanked Axin1 (Axin1fl /fl) and Rosa 26 LacZ reporter alleles (LacZ) to generate Axin1 −/− mice. Cre activity was induced by four intraperitoneal injections of β-naphthoflavone (BNF; 80 mg/kg body wt) over 4 days. Littermates were used as controls (Axin1 +/+). Three months after injections of BNF, the animals were sacrificed and liver samples were snap frozen in liquid nitrogen and stored at −80°C. For immunoblots, livers were homogenized in lysis buffer (50 mM HEPES, 1% Triton X-100, 10 mM EDTA, 50 mM sodium pyrophosphate, 1 mM phenylmethylsulfonyl fluoride, 0.1 M sodium fluoride, 10 mM sodium orthovanadate, and 1 μg/ml leupeptin) using the Brinkman PT 10/35 Polytron (American Laboratory Trading, Inc.). Extracts were kept ice cold at all times. Liver extracts were cleared by microcentrifugation at 40,000 g (40 min, 4°C). The supernatants were aliquoted and stored at −80°C. For mRNA, livers were homogenized in TRIzol reagent using the Brinkman PT 10/35 Polytron.
Plasmids
Expression vectors pcDNA3.1-mNRF2-V5/HisB and pcDNA3.1-mNRF2ΔETGE-V5/HisB were provided by Dr. John D. Hayes (Biomedical Research Institute, Ninewells Hospital and Medical School, University of Dundee). pCGN-HA-GSK-3βΔ9 was provided by Dr. Akira Kikuchi (Department of Biochemistry, Faculty of Medicine, Hiroshima University). pcDNA3-Flag-β-TrCP2 was provided by Dr. Tomoki Chiba (Department of Molecular Biology, University of Tsukuba). Plasmid encoding β-TrCPΔFbox (pcDNA3-β-TrCPΔFbox-HA) was provided by Dr. Serge Y. Fuchs (Department of Animal Biology, University of Pennsylvania, Philadelphia, PA). Chimeric deletion constructs of the pEYFP-mNRF2-V5 were generated as described in (39, 40). pcDNA3.1-mNRF2ΔETGE 6S/6A-V5/HisB was previously described in (38). Expression vector for WNT-3A (pCCBS-Hygro-WNT-3A-HA) was provided by Dr. Stuart A. Aaronson (Department of Oncological Sciences, Mount Sinai School of Medicine, New York, NY). Plasmid encoding mouse myc-Axin1 (pCMV5-myc-Axin1) was kindly provided by Dr. Sheng-Cai Lin (Department of Biochemistry, Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China). Expression vectors for the myc-Axin1 deletion mutants were generated by polymerase chain reaction (PCR) using pCMV5-myc-Axin1 as a template with the following primers: Common forward, 5′ TAAC
Analysis of mRNA levels
Total RNA extraction, reverse transcription, and quantitative PCR were done as detailed in (39). See Supplementary Table S1 for primer sequences. Data analysis is based on the ΔΔCt method with normalization of the raw data to housekeeping genes as described in the manufacturer's manual (Applied Biosystems). All PCRs were performed at least in triplicate.
Immunoblotting
Immunoblots were performed as in (39). The primary antibodies are described in Supplementary Table S2. Proteins were detected by enhanced chemiluminescence (GE Healthcare).
siRNA assays
siRNA used to knock down mouse NRF2, mouse β-TrCP1, mouse β-TrCP2, human GSK-3α, human GSK3β and mouse Axin1 expression, and control scrambled siRNA sequence are described in Supplementary Table S3. Immortalized hepatocytes were seeded in six-well plates (50,000 cells/well in 2 ml medium). When 40%–50% confluence was reached, cells were transfected with 25 nM siRNAs following DharmaFECT General Transfection Protocol (Dharmacon). After 48 h, cells were used for experiments. HEK293T cells were seeded in six-well plates (200,000 cells/well in 2 ml medium) before being transfected using calcium phosphate and the appropriate NRF2 expression plasmids. To knock down GSK-3 isoforms, we performed siRNA transfection during two consecutive days. On the first day, we knocked down GSK-3 using 80 ng of Silencer Select validated siRNA with 30 μl of siPORT reagent, and on the second day we used 40 ng of Silencer Select validated siRNA with 15 μl of siPORT reagent. Twenty four hours later, the cells were collected and NRF2 and GSK-3 levels were analyzed. Keap1 −/− MEFs were seeded in six-well plates (200,000 cells/well in 2 ml medium) at 16 h before transfection. To knock down Axin1, we performed siRNA transfection during two consecutive days. On the first day, we knocked down Axin1 using 50 or 100 pmol of Silencer Select validated siRNA with 20 μl of RNAimax Lipofectamine (Life Technologies), and on the second day we used 50 or 100 pmol of Silencer Select validated siRNA with 20 μl of RNAimax Lipofectamine. Twenty four hours later, the cells were harvested for analysis.
Luciferase assays
Transient transfections of HEK293T or Keap1 −/− cells were performed with the expression vectors ARE-LUC (a gift of Dr J. Alam, Department of Molecular Genetics, Ochsner Clinic Foundation, USA), or TOPFlash and FOPFlash, as indicated. pTK-Renilla were used as an internal control vector (Promega). Luciferase assays were performed as described in (39).
In vivo ubiquitination assay
This was carried out using the method of Treier et al. (50). HEK293T cells were transfected with His-tagged Ubiquitin along with the indicated plasmids. Sixteen hours later, the transfected cells were washed with prewarmed phosphate-buffered saline and scraped into 0.4 ml of phosphate-buffered saline. Whole-cell lysates were prepared from 80 μl of the cell suspension and are referred to as the “input” fraction. His-tagged protein was purified from the remainder of the cell suspension as follows: The cell suspension was lysed by the addition to 1 ml of buffer A (6 M guanidine:HCl, 10 mM Tris in 0.1 M phosphate buffer, pH 8.0) supplemented with 5 mM imidazole. The resulting lysate was sonicated, to reduce viscosity. Then, 60 μl of Probond™ resin (Invitrogen) was added, and the mixture was rotated for 4 h at 25°C. Thereafter, the beads were washed sequentially with buffer A supplemented with 0.1% (v/v) Triton X-100, buffer B (8 M urea, 10 mM Tris in 0.1 M phosphate buffer, pH 8.0) supplemented with 0.1% Triton X-100, buffer C (8 M urea, 10 mM Tris in 0.1 M phosphate buffer, pH 6.5) supplemented with 0.2% Triton X-100, and finally, buffer C supplemented with 0.1% Triton X-100. Bound material was eluted from the beads by suspension in 50 μl of modified Laemmli sample buffer (20 mM Tris-Cl, pH 6.8, 10% [v/v] glycerol, 0.8% [w/v] SDS, 0.1% [w/v] bromophenol blue, 0.72 M 2-mercaptoethanol and 300 mM imidazole) followed by boiling for 4 min. The suspension was centrifuged (16,000 g, 1 min, 20°C), and the resulting supernatant was collected and is referred to as the “pull down” fraction.
Immunohistochemistry
Liver tissues from Axin1+/+ and Axin1 −/− mice were always dissected from the same part of the liver and fixed in ice-cold 10% neutral buffered formalin for no longer than 24 h before being processed into paraffin wax according to standard procedures. Tissue sections (5 μm thick) were used. The Streptavidin Biotin Peroxidase method was performed as follows: Sections were mounted on slides coated with 3-aminopropyltriethoxy-silane (Sigma Chemical Co.), hydrated after deparaffinization, and incubated for 10 min in 0.3% H2O2 in phosphate-buffered saline (PBS) to reduce endogenous peroxidase activity. For antigen retrieval, sections were submitted twice to microwave pretreatment for 2.5 min at 800 W in sodium citrate buffer 0.01 M (pH 6) and subsequently blocked with Tris-buffered saline containing 1% bovine serum albumin. All sections were incubated for 16–20 h at 4°C with the primary antibody (diluted in PBS containing 1% serum albumin). Sections were washed in PBS to remove unbound primary antibodies and then incubated with a broad spectrum second antibody solution at room temperature. After incubations, the sections were washed and submitted to the supersensitive Streptavidin Biotin Peroxidase (Histostain-SP IHC kit, DAB sprectum, 95-9643; Life Technologies). The immunoreaction was developed by incubation with 3, 3-diaminobenzidine (DAB; Sigma-Aldrich) with H2O2 and diluted in phosphate buffer for peroxidase kits. The sections were counterstained with Harris' hematoxylin, dehydrated in ethanol, and mounted in DePex (Thermo Fisher Scientific). The primary antibodies and optimal dilutions used for these studies were as follows: anti-mouse NRF2 (1:50; generated in Cuadrado's laboratory), anti-GS (1:1000; Sigma-Aldrich), anti-HO-1 (1:50; Millipore), and anti-NQO1 (1:50; Abcam).
Subcellular fractionation
Keap1+/+ and Keap1 −/− MEFs were seeded in p100 plates (2×106 cells per plate). Cells were treated with Control or WNT-3A-CM for 4 h. Cytosolic and nuclear fractions were prepared as previously described (12). The supernatants from cytosolic and nuclear fractions were resolved in SDS-PAGE and immunoblotted with the indicated antibodies.
Co-immunoprecipitation
Assays were performed as indicated in (39). The samples were resolved by SDS-PAGE and immunoblotted. Mouse IgG TrueBlot (eBiosciences) was used as peroxidase-conjugated secondary antibody (1:10,000 dilution) to avoid interference with the 55 kDa heavy and 23 kDa light chains of the immunoprecipitating antibody.
Image analyses and statistics
Different band intensities (arbitrary units) corresponding to immunoblot detection of protein samples were quantified using the MCID software (MCID). Student's t-test, one-way, and two-way analysis of variance were used to assess differences between groups. Unless indicated, all experiments were performed at least thrice with similar results. The values in the graphs correspond to the mean of at least three samples. Results are expressed as mean±SEM.
Footnotes
Acknowledgments
This work was funded by SAF2013-43271-R of the Spanish Ministry of Economy and Competitiveness. The authors thank Carmen Sánchez-Palomo (Departamento de Anatomía, Histología y Neurociencia Facultad Medicina, UAM, Madrid) for technical assistance.
Author Disclosure Statement
The authors declare that there are no conflicts of interest.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.