
Abstract
Introduction
H
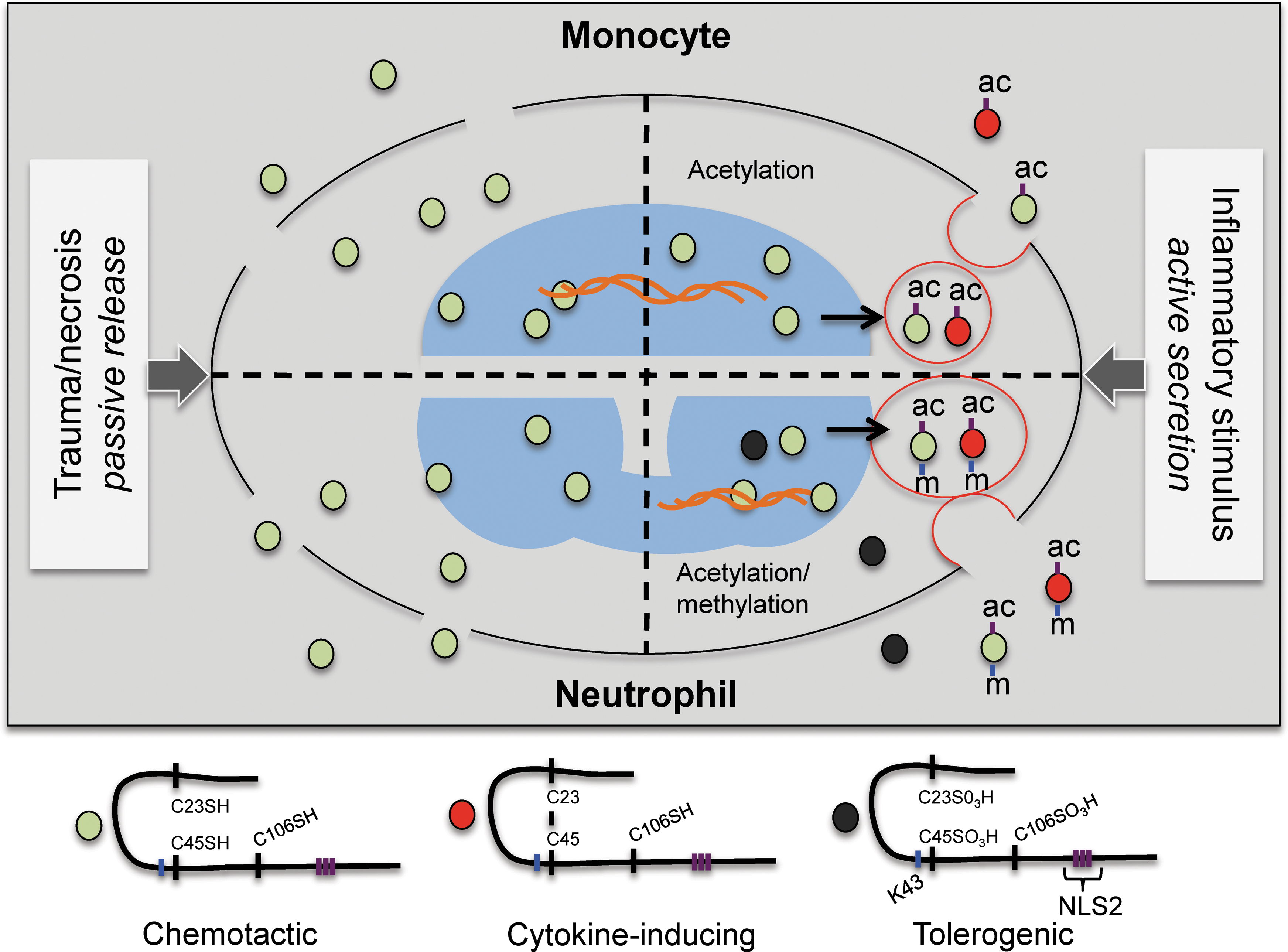
Structurally, HMGB1 consists of two DNA binding domains termed box A and box B, and an acidic C-terminal tail consisting of a continuous stretch of glutamic and aspartic residues. The active release and the extracellular functions of HMGB1 are currently believed to be regulated by specific post-translational modification (PTM) events. Studies of HMGB1 release have mainly been performed in cells of myeloid origin and have demonstrated that stimulatory molecules such as lipopolysaccharide (LPS) or pro-inflammatory cytokines induce cytoplasmic HMGB1 translocation followed by active release through nonclassical secretory vesicles (13, 43).
Post-translational modifications (PTM) of high mobility group box protein 1 (HMGB1) regulate both its release and function. This work provides the first characterization of HMGB1 PTMs in a cohort of patients with chronic inflammation. Our study indicates that HMGB1 is actively released into the inflammatory joints of juvenile idiopathic arthritis patients and that neutrophils may contribute to the high levels of HMGB1 recorded in the synovial fluid. We show evidence that HMGB1 has multiple inflammatory functions in the chronically inflamed joint, including chemotaxis and cytokine induction. Finally, our data highlight the complexity of ongoing chronic inflammatory processes in arthritic joints.
Hyperacetylation of lysine residues within the two nonclassical nuclear location sequences (NLS) is the key event regulating the nuclear and cytoplasmic shuttling and precedes active release from macrophages (6). Lysine mutagenesis leads to nuclear retention, and pan-inhibition of histone deacetylases (HDACs) induces spontaneous translocation of HMGB1 to the cytoplasm, as demonstrated in in vitro systems (6, 11, 49). Inflammasome activation and JAK/STAT signal transduction have also been reported to induce hyperacetylation of HMGB1 NLS regions, resulting in cytoplasmic translocation and subsequent release (22, 24, 25, 27). Further supporting that active HMGB1 release is acetylation dependent is the fact that HMGB1 acetylation patterns change during an inflammatory process, shown in vivo in a model of acetaminophen (APAP) liver toxicity (3). In addition, a monomethylation of the lysine in position 43 (K43) has been associated with cytoplasmic translocation and release of HMGB1 in neutrophils, suggesting the existence of cell-specific PTMs of HMGB1 (18).
The pathogenic and extracellular inflammatory effects of HMGB1 have been assigned to activation through Toll-like receptors (TLRs), the receptor for advanced glycated end-products (RAGE), and CXCR4 (20, 31, 36, 39, 48). Early studies reported that the cytokine activity is localized to the first 20 amino acids (89–108) of box B and that the cysteine in position 106 (C106) is critical in activation of TLR4 (23, 45). A cysteine-to-alanine mutation of C106 eliminates the ability of HMGB1 to interact with and activate TLR4 (45). We and other groups have further demonstrated that cysteine redox is fundamental for determination of the extracellular functions of HMGB1 (40, 46).
The cytokine-inducing function of HMGB1 requires the formation of an intramolecular disulfide bond between the cysteines in box A (C23 and C45), whereas C106 must be in its reduced thiol state (40, 46). Reduction of the intramolecular disulfide bridge to thiols or oxidation of all cysteines to sulfonic acids completely abolishes the cytokine-inducing effect of HMGB1 (46). HMGB1 cysteine redox status also defines the chemotactic effect of HMGB1. Induction of migration, as demonstrated using 3T3 fibroblasts, requires that all three cysteines are in the reduced, thiol form. Oxidation of the cysteines dose dependently abolishes the migration effects induced by HMGB1 (40). A recent study reported, both in vitro and in vivo, that the chemotactic effect of HMGB1 is also dependent on a heterodimer formation with CXCL12 that synergistically activates CXCR4 (36). The heterodimer formation is cysteine redox dependent and only occurs when all three HMGB1 cysteines have reduced thiol side-chains (36, 40).
To date, questions regarding the structure-to-function relationship of HMGB1 have mainly been investigated in vitro. HMGB1-specific PTMs have only been investigated in vivo in APAP overdose patients and in pre-experimental models thereof (1, 3 –5). As a consequence of the initial hepatocellular injury and cell death caused by APAP toxic metabolites, a hepatic inflammatory response is triggered. Measurement of acetylated HMGB1 in sera has been reported to out-perform the standard clinically liver injury marker alanine aminotransferase as both a diagnostic and prognostic marker. Interestingly, acetylated HMGB1 levels may be used to successfully predict disease severity and thus in determination of treatment strategies. Patients requiring liver transplantation and patients with a fatal outcome have significantly elevated levels of acetylated HMGB1, both compared with healthy controls and with patients with a more benign outcome (1, 3). APAP overdose reflects an acute inflammatory process, whereas HMGB1 has been implicated in many autoimmune diseases and thus in chronic inflammatory processes.
To date, there are no reports regarding patterns of HMGB1 PTMs during chronic inflammatory conditions. High HMGB1 levels in synovial fluid (SF) and synovial tissue of patients with rheumatoid arthritis (RA) are well known (16, 21, 38). In addition, intra-articular HMGB1 injections induce destructive arthritis in mice and antibodies targeting HMGB1 ameliorate disease activity in several models of arthritis (16, 30, 34). We have recently reported increased levels of HMGB1 in SF of juvenile idiopathic arthritis (JIA) patients (35). However, measurement of total HMGB1 levels without distinguishing between the different redox isoforms does not adequately reveal the functional contribution of HMGB1 to the studied inflammatory condition, nor whether the increased HMGB1 levels reflect increased cell death or activation of inflammatory cells.
The aim of our study was to characterize HMGB1-specific PTMs, thereby defining the inflammatory properties of HMGB1 in SF from JIA patients as well as the different associated release mechanisms. This will further increase our understanding of the pathogenic contribution of HMGB1 in chronic inflammatory conditions in general and in arthritis in particular. We determined that HMGB1 is actively secreted in the arthritic joint and exists in both the migration-inducing, fully reduced form and the cytokine-inducing, disulfide form. Furthermore, our results indicate that neutrophils may be an important source of secreted HMGB1.
Results
HMGB1 levels do not correlate with conventional cell death
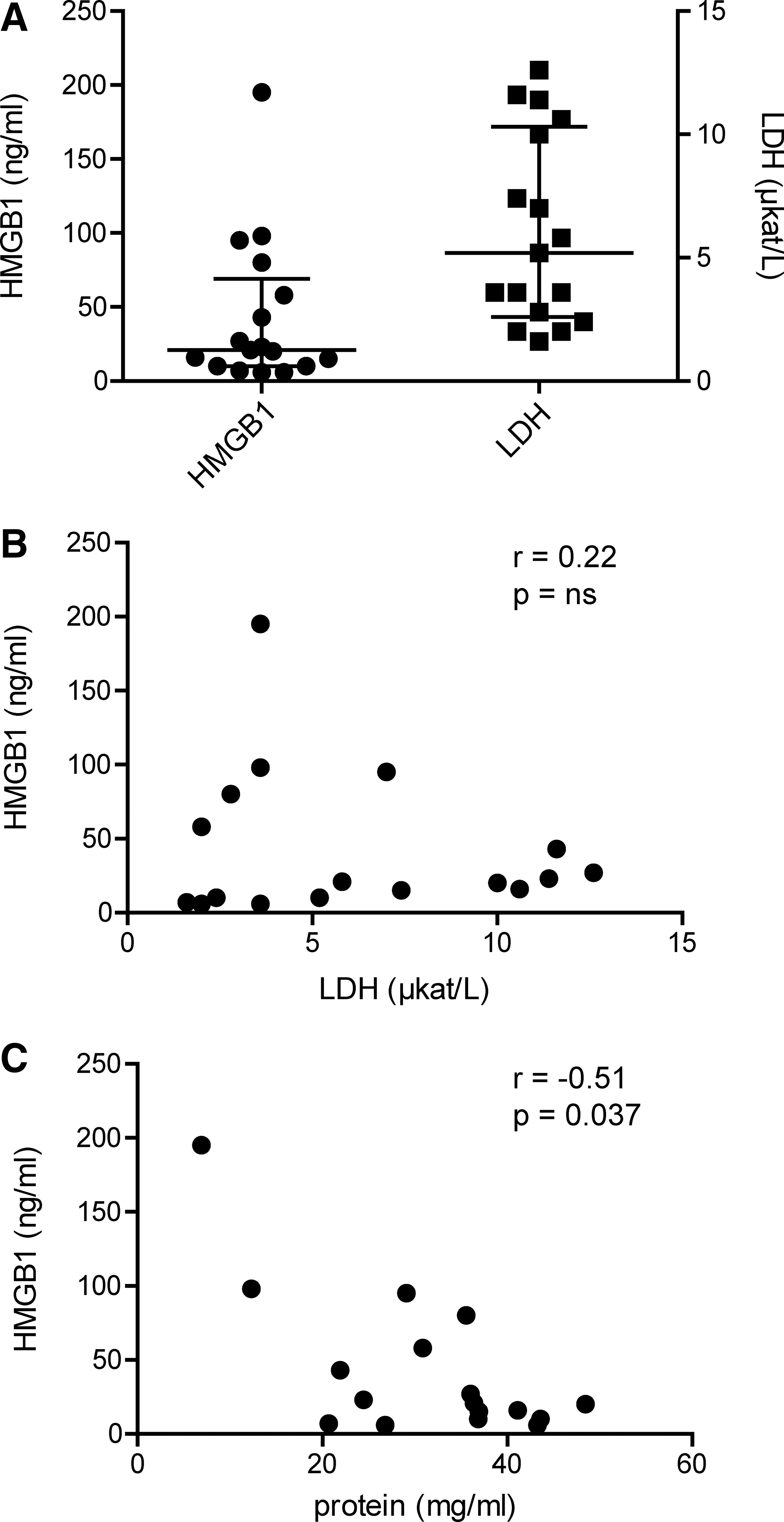
We first investigated whether HMGB1 release was caused by increased cell death as a result of the ongoing joint inflammation. Levels of lactate dehydrogenase (LDH) and HMGB1 in the SF of 17 JIA patients were measured, and the median levels for LDH and HMGB1 were 5.2 (IQR 2.6–10.3) μkat/L and 21 (IQR 10–69) ng/ml, respectively (Fig. 1A). We could not detect any significant correlation (r=0.22; p-value=0.39) between levels of HMGB1 and LDH in SF (Fig. 1B). Furthermore, a rather weakly negative but still statistically significant correlation (r = −0.51; p-value=0.037) was observed between total protein and HMGB1 levels in SF (Fig. 1C). Collectively, these data suggest that HMGB1 is actively released into the SF of JIA patients and is not merely a product of increased cell death.
HMGB1 release is associated with NLS2 hyperacetylation in JIA
To unambiguously define PTMs known to affect HMGB1 release and function, we isolated HMGB1 from SF and performed immunoprecipitation followed by de novo sequencing using liquid chromatography tandem mass-spectrometry (LC-MS/MS) (Figs. 2 and 3). Area under the curve (AUC) for peptide fragments (precursor ions) containing the known regulatory PTMs was extracted from the first round of MS. A second round of MS (MS/MS) validated both peptide sequences and modifications. A correlation matrix analysis for the total HMGB1 levels and AUCs for PTMs was performed. p-values were corrected for multiple testing with a high stringency (Holm's method) to avoid false-positive data (Fig. 4B).
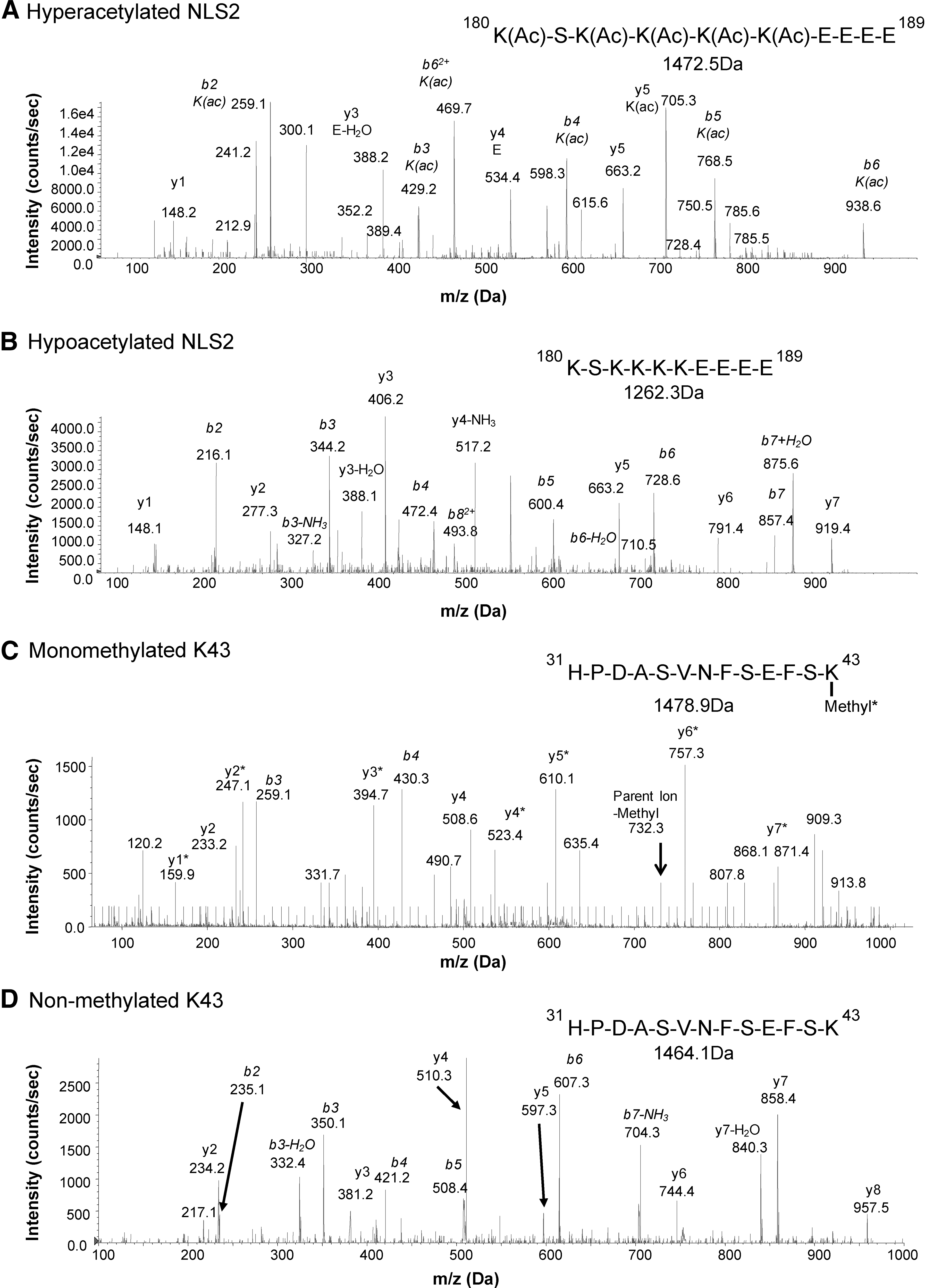
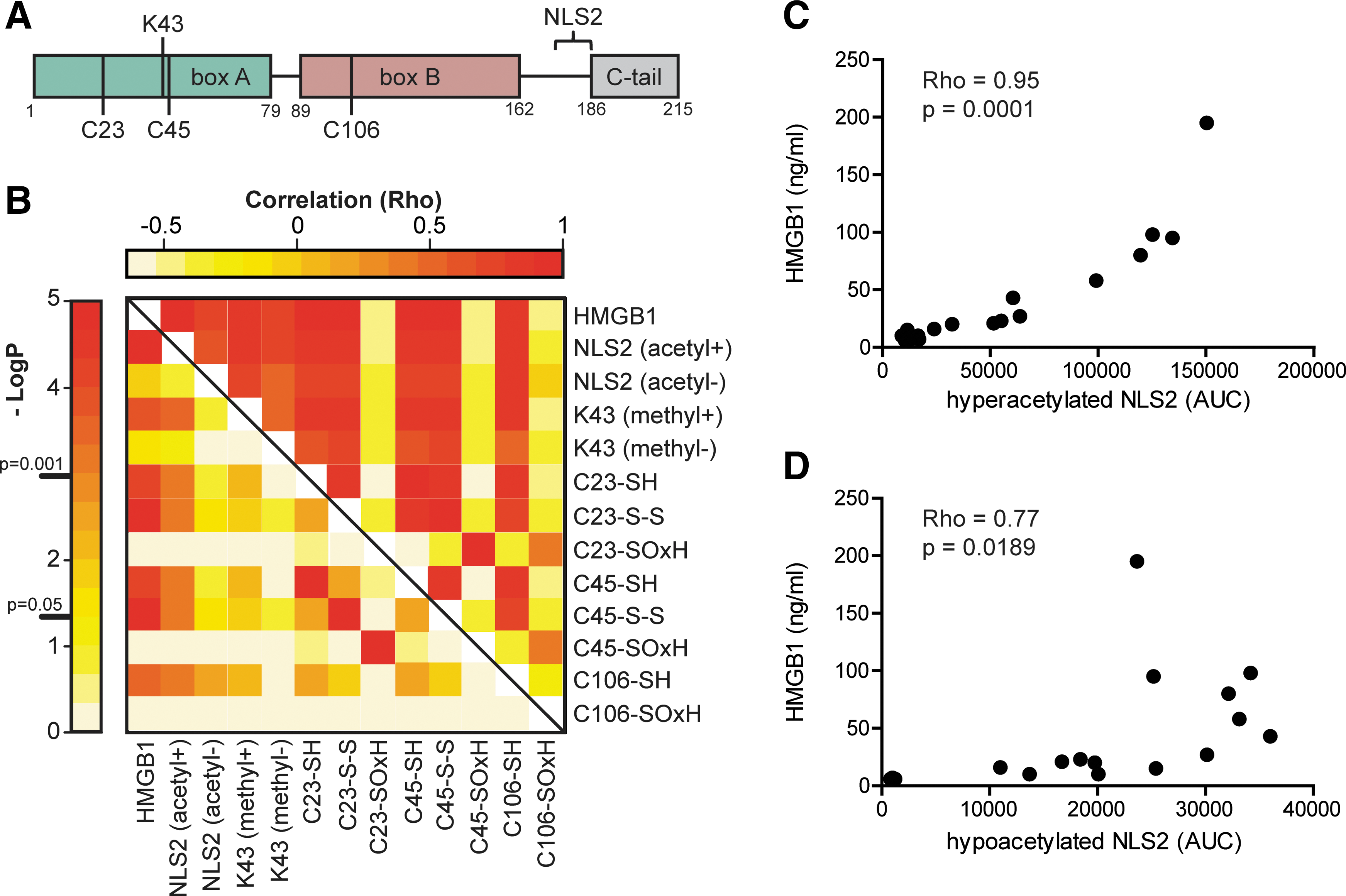
Active release of HMGB1 is considered a result of acetylation of the two nonclassical NLS regions followed by a nuclear-to-cytoplasmic translocation and subsequent release. Mass-spectrometric analysis revealed that hyperacetylated lysines in the NLS2 region (K180, K182, K183, K184, and K185) were present in all patients (Figs. 2A and 4C). As expected, a strong positive correlation between total HMGB1 levels and AUCs for the hyperacetylated NLS2 (Rho=0.95; p-value=0.00001) peptides was detected (Fig. 4C), confirming that HMGB1 was actively released in SF of the studied JIA patients. However, a weak but still significant correlation between AUCs for hypoacetylated NLS2 traces and total HMGB1 levels (Rho=0.77; p-value=0.019) was also detected (Figs. 2B and 4D). In macrophages, acetylation of NLS2 and the active release of HMGB1 have also been described as a result of inflammasome activation followed by caspase-1 activation, in a manner resembling IL-1β secretion (22, 25, 27). However, IL-1β was not detected by enzyme-linked immunosorbent assay (ELISA) in any of the samples (data not included).
Neutrophils as a source of extracellular HMGB1 in the inflammatory joint
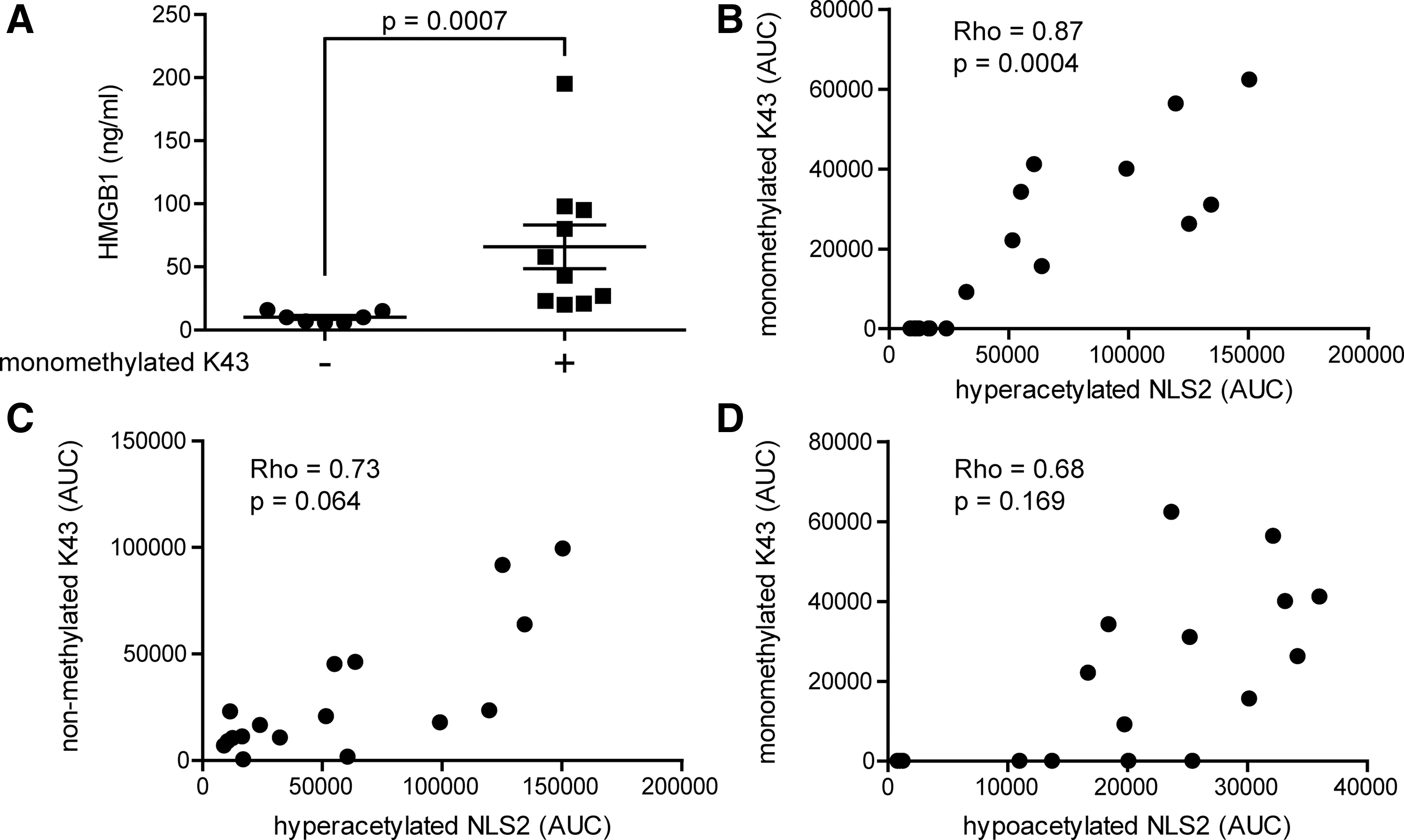
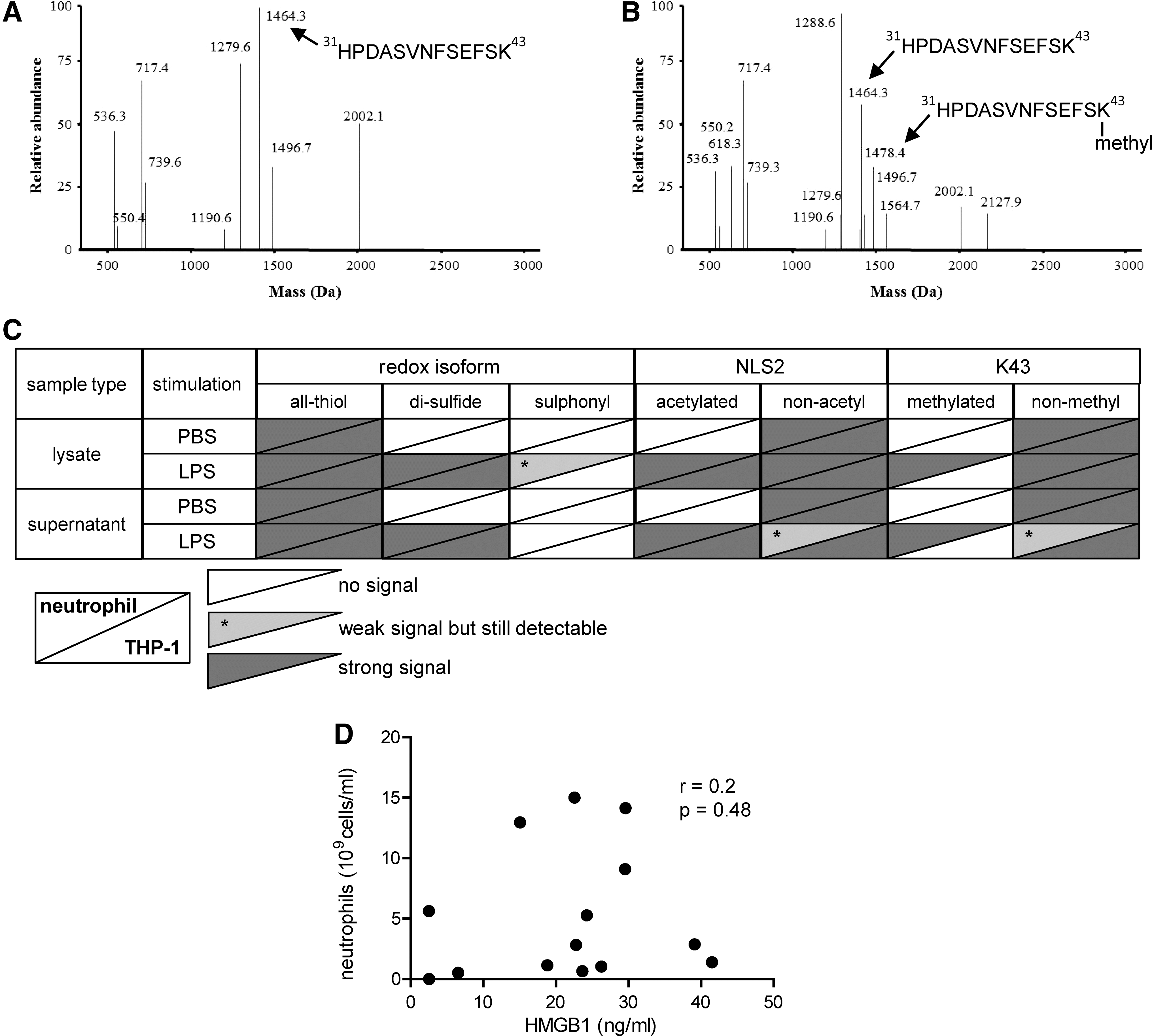
Neutrophils are highly abundant in the joints of patients with inflammatory joint diseases, comprising as much as 90% of immune cells present in the inflamed joint. Activated neutrophils undergoing NETosis are known to release HMGB1 in vitro, and a suggested monomethylation of K43 is associated with this process (18, 26). Mass-spectrometric traces of K43 monomethylation were detected in 10 out of 17 patients, and the total HMGB1 levels recorded in these individuals were significantly higher (p-value=0.0007) as compared with those with no traces of monomethylation (Figs. 2C and 5A). The abundance of the monomethylated K43 peptide correlated strongly with hyperacetylated HMGB1 (Rho=0.89; p-value=0.0004); whereas the abundance of nonmethylated K43 peptide did not (Rho=0.73; p-value=0.0637), indicating that neutrophils may concurrently monomethylate K43 and hyperacetylate NLS2 during active release of HMGB1 (Figs. 2D and 5B, C). Moreover, increased levels of monomethylated K43 did not correlate with hypoacetylated NLS2 (Rho=0.68; p-value=0.169), indicating that cells passively releasing HMGB1 do not monomethylate K43 (Fig. 5D). To further investigate whether only neutrophils are capable of monomethylating of the K43 residue, we stimulated isolated neutrophils or THP-1 cells with LPS and investigated HMGB1 PTMs in both cell lysates and supernatants. Mass-spectrometric analysis indicates that neutrophils but not THP-1 cells monomethylate K43 when stimulated with LPS (Fig. 6A, C) but not when stimulated with PBS (Fig. 6B). A second round of MS validated the peptide sequences, and the monomethylation on the K43 residue in LPS stimulated neutrophils (data not shown). LPS stimulation induced acetylation of the NLS2 lysines in both THP-1 and neutrophils (Fig. 6C). Unexpectedly, neutrophil concentrations did not correlate with levels of total HMGB1 (r=0.2; p=0.48) in the SF of JIA patients (Fig. 6D).
Diverse redox isoforms are present in the inflammatory joint of JIA patients
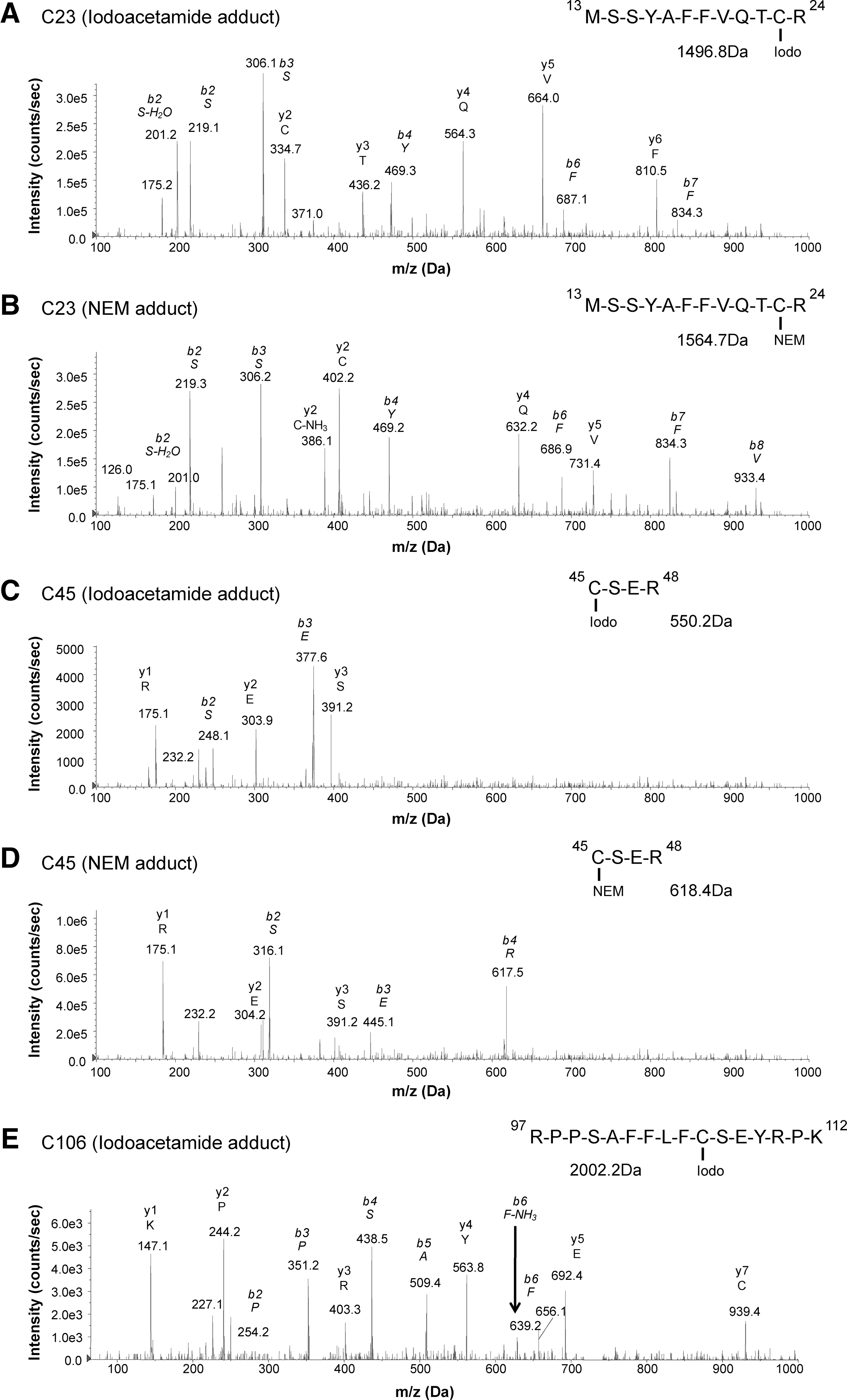
HMGB1 is a redox-sensitive protein, and in vitro redox manipulation of the three intra-molecular cysteine residues has a great impact on its inflammatory function. Fully reduced HMGB1 is known to have chemotactic properties, whereas the cytokine-inducing (via TLR4 activation) isoform contains an intra-molecular disulfide bond between the box A cysteines (HMGB1C23-C45) and a reduced C106 (C106h) (2, 40, 46). To define different redox states of the cysteine residues, we utilized a two-step alkylation processing (iodoacetamide-DTT-NEM) of isolated HMGB1 before tryptic digestion (25, 27). This allowed us to define whether cysteines were present as reduced thiols (iodoacetamide adduct) or as a part of disulfide bonds (NEM adduct). A fraction of the isolated HMGB1 was not alkylated, and the presence of sulfenic (SOH), sulfinic (SO2H), or sulfonic (SO3H) acid cysteine modifications (denoted as SOxH) could be defined.
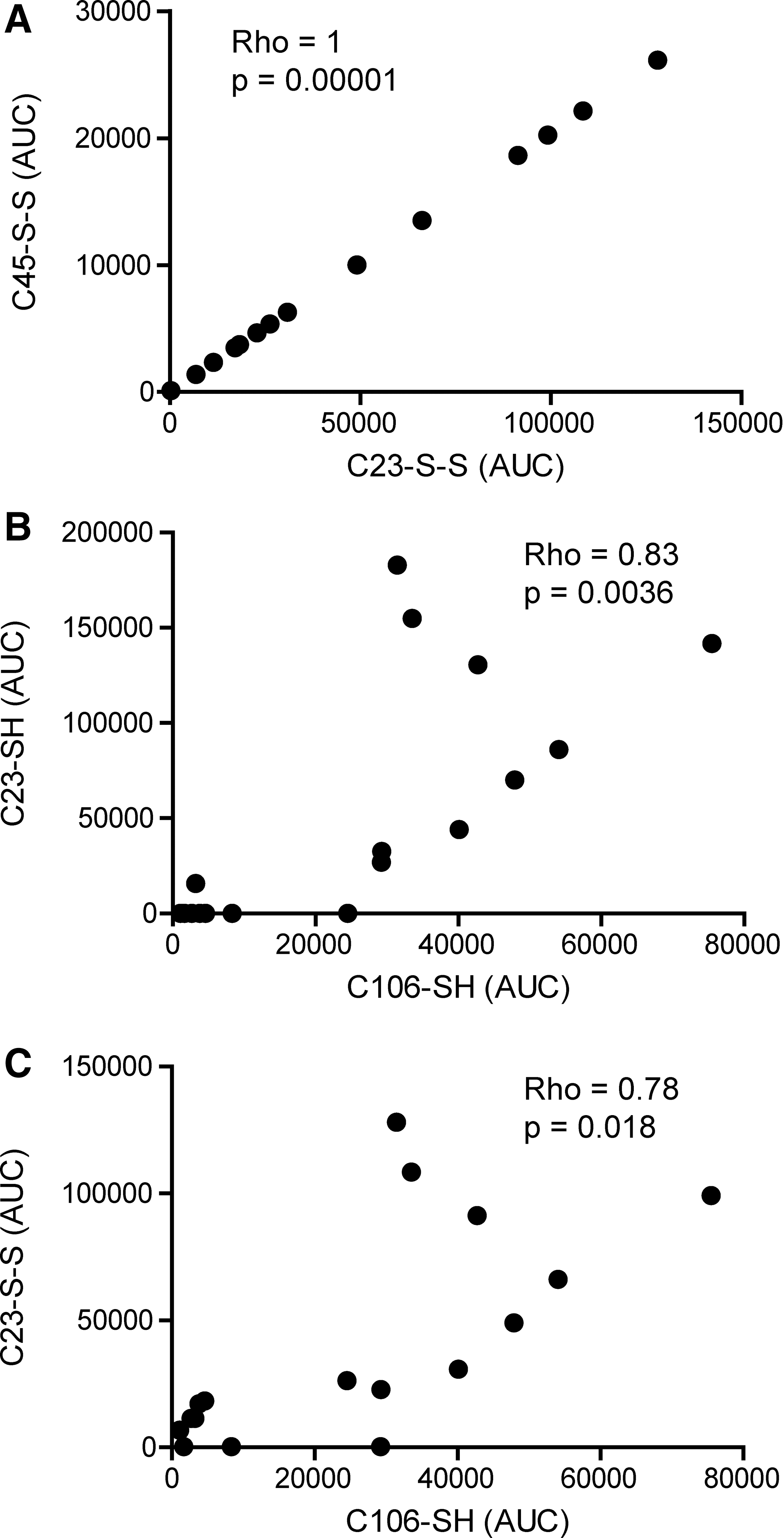
Reduced cysteine traces were detectable in 10 out of 17 (C23iodo), 10 out of 17 (C45iodo), and 17 out of 17 (C106iodo) patients (Fig. 3A, C, E and Table 1). Traces characteristic of disulfide bridge formation were present in 14 out of 17 (C23nem), 14 out of 17 (C45nem), and 0 out of 17 (C106nem) patients, indicating that C106 does not form disulfide bridges (Fig. 3B, D and Table 1). A higher degree of cysteine oxidation was detectable in 9 out of 17 (C23-SOxH), 9 out of 17 (C45-SOxH), and 17 out of 17 (C106-SOxH) patients (Table 1). Only 2 out of 17 patients had detectable traces of all three cysteine modifications investigated (Table 1). Empirically, the box A cysteines (C23 and C45) are expected to follow the same redox pattern whereas C106 has not been described to form either intra- or inter-molecular disulfide bridges. As expected, the abundance of C23nem and C45nem adducts correlated positively (Rho=1; p-value=0.00001), implying that C23 and C45 form disulfide bridges to the same extent (Fig. 7A). The same pattern was evident for iodoacetamide and SOxH cysteine modifications with regard to C23 and C45 (Fig. 4B).
Patients are arranged after increasing oxidative cysteine status. Mass-spectrometric traces of peptides with indicated cysteine redox status are denoted by (+). The absence of mass-spectrometric traces of an indicated cysteine redox status is represented by (−). HMGB1, high mobility group box protein 1; JIA, juvenile idiopathic arthritis; undiff., undifferentiated.
C106iodo adducts correlated positively with both C23/C45iodo (Rho=0.83; p-value=0.0036) and C23/C45nem peptide modifications (Rho=0.78; p-value 0.018), indicating the presence of both the chemoattractant and cytokine-inducing (HMGB1C23-C45) isoforms in the SF (Fig. 7B, C). In contrast, C106-SOxH traces did not correlate with any of the other redox isoforms, suggesting that C106 may be further oxidized irrespective of the box A cysteines redox status (Fig. 4B).
Chemoattractant and cytokine-inducing redox isoforms of HMGB1 are actively released in SF
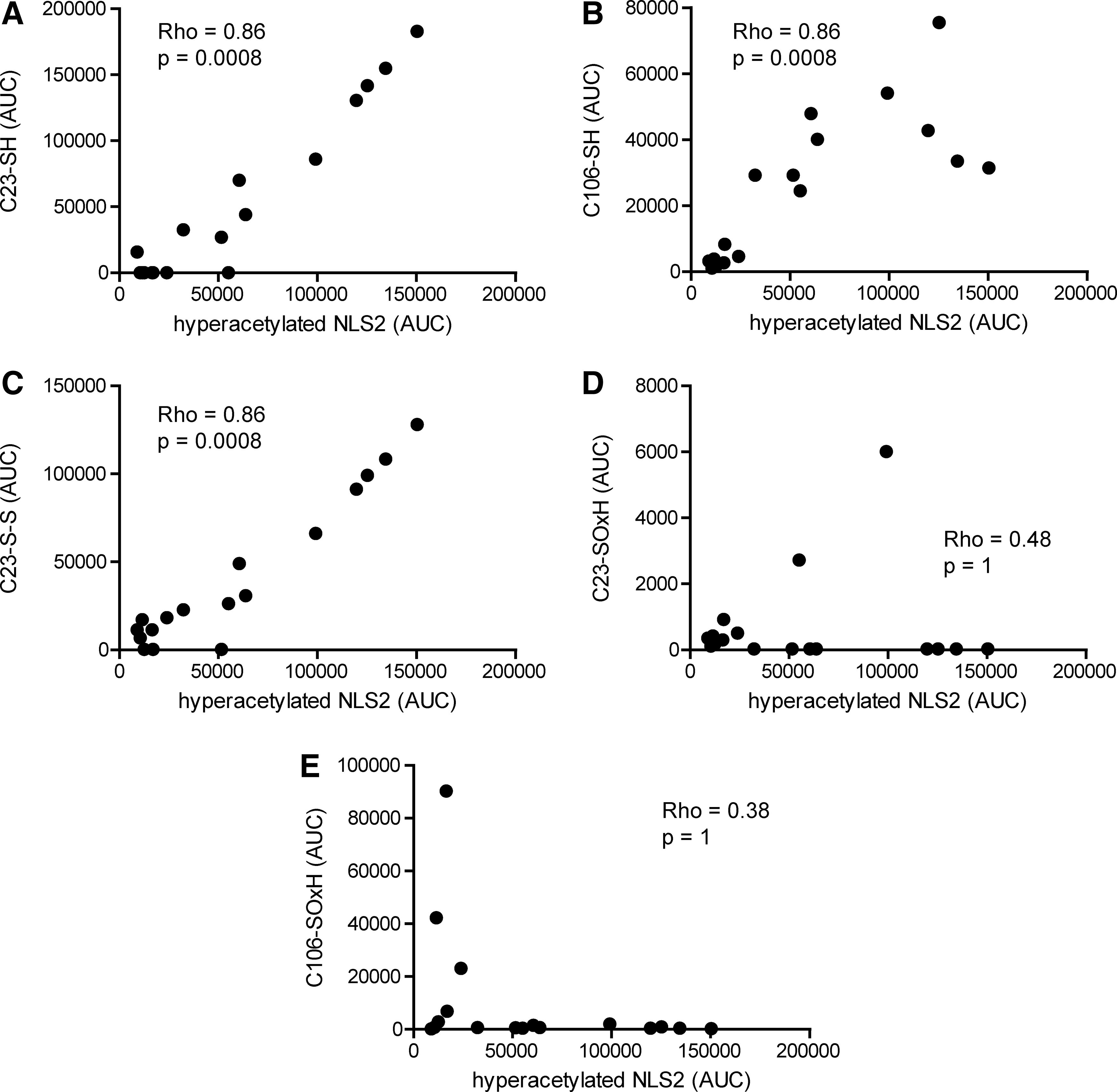
We next studied the inflammatory features of the actively released HMGB1. NLS2 hyperacetylation significantly correlated with the LC-MS traces for C23iodo, C45iodo, and C106iodo peptide AUCs (Rho=0.86; p-value=0.0008), indicating that actively released HMGB1 was present in a chemoattractant isoform (Figs. 4B and 8A, B). Furthermore, hyperacetylated NLS2 traces also significantly correlated to C23nem and C45nem peptides (Rho=0.86; p-value=0.0008), implying that the cysteines within box A were also present as a disulfide bond and thus likely to form the cytokine-inducing redox isoform of HMGB1 (Figs. 4B and 8C). More advanced oxidization, that is, C23SOxH (Rho=0.48; p-value 1), C45SOxH (Rho=0.48; p-value=1), and C106SOxH (Rho=0.38; p-value=1), did not correlate with hyperacetylated NLS2 (Figs. 4B and 8D, E).
Besides its intrinsic inflammatory functions, HMGB1 also has the ability to synergistically potentiate the function of other endogenous inflammatory mediators, including CXCL-12 and IL-1β (12, 36, 42). The levels of CXCL-12 in the SF of this patient cohort were in similarity to IL-1β undetectable by ELISA. We also analyzed levels of additional inflammatory mediators, including MCP-1, IP-10, RANTES, IL-6, and IL-8. Confirming our previously reported results, no correlation was recorded with total HMGB1 levels (data not included) (35). In addition, no correlation was noted with any traces of HMGB1 PTMs (data not included).
Discussion
During inflammation, changes in redox status of the tissue microenvironment are evident and cysteine redox modifications are known to impact protein structure and function by the formation or breaking of disulfide bridges. Recent studies regarding the extracellular functions of HMGB1 have revealed that redox modifications of its cysteine residues are key regulatory PTMs affecting the inflammatory properties of HMGB1 (40, 44, 46). The majority of the studies have been performed in in vitro cell systems, and no previous report has investigated the presence of HMGB1 PTMs known to regulate HMGB1 release and function in clinical samples from patients with a chronic inflammatory disease. In this study, we reveal strong indications that the main HMGB1 release occurring in SF from chronic inflammatory JIA patients is regulated by NLS2 hyperacetylation and that neutrophils might be strong contributors to the high levels of HMGB1 detected. Furthermore, our results also indicate that such actively released HMGB1 has both chemotactic and cytokine-inducing properties.
Hyperacetylation of lysines within HMGB1 NLS regions has been demonstrated to regulate intracellular shuttling of HMGB1 and has been used as a marker for active HMGB1 secretion (6, 13, 24, 25, 27). The fact that HMGB1 contains 43 acetylation-susceptible lysines poses technical constrains on investigation of HMGB1 release mechanisms. Even in unstimulated cells, a number of lysines outside of the NLS regions are acetylated, making anti-acetyl lysine immunoblot detection-based methods unsuitable for defining acetylation patterns and regulation of HMGB1 release (24). By utilizing LC-MS/MS, we could detect an NLS2-specific hyperacetylation and define that this acetylation pattern positively correlated with synovial HMGB1 levels. This suggests that HMGB1 in SF from JIA patients is mainly actively released through an acetylation-dependent mechanism.
Increased extracellular HMGB1 in JIA SF could also be a consequence of increased cell death within the inflamed joint. However, our data indicate that such passive release is not the major cause of extracellular HMGB1, as the recorded levels of the commonly used cell death marker, LDH, did not correlate with the recorded HMGB1 levels. Neither did total HMGB1 levels correlate with total protein levels.
Intracellular protein acetylation is mainly regulated by histone acetyltransferases and HDACs (15). Pharmacological HDAC inhibition has been demonstrated to induce HMGB1 release both in vitro and in vivo in monocytes, hepatocytes, and, more recently, in hippocampal-entorhinal slice cultures (6, 11, 24, 49). With regard to HMGB1 as a pathogenic mediator in chronic arthritis, HDAC inhibitors are paradoxically under investigation in RA and systemic onset JIA (soJIA) as an alternative treatment strategy for conventional therapy nonresponders (8, 17, 41). However, the broad systemic effects on transcriptional control of HDAC inhibition may outweigh the negative effect of an expected local increase in HMGB1 levels. Additional studies are warranted to define the effects of systemic HDAC inhibition and its influence on HMGB1 release both locally and systemically.
Most of the previously published studies investigating active HMGB1 release mechanisms suggest that it is mainly cells of the myeloid lineage that actively secrete significant levels of HMGB1. Other studies suggest that nonimmune cells can also actively release HMGB1, but most likely at lower levels. Synovial fibroblasts isolated from RA patients have been reported to secrete HMGB1 in response to hypoxia, and extranuclear HMGB1 expression co-localized with hypoxic areas in immunostainings of RA synovial tissues (16). Whether the hypoxia-induced HMGB1 release is an apoptosis-related, passive event or an activation consequence of synovial fibroblasts is still unclear. Interestingly, hypoxia-induced release of HMGB1 is reported as both an acetylation-dependent (hepatocytes) and a nonacetylation-dependent (U937 cells) event (11, 16). This may imply that, first, different cell types may have different release mechanisms and, second, it is possible that different environmental conditions may induce diverse active release mechanisms. Nevertheless, lysine hyperacetylation of the NLS regions is associated with active HMGB1 release and when interpreting our results we have regarded acetylation of the NLS2 sequence as evidence of active HMGB1 secretion.
We and others have previously demonstrated that myeloid cells release significant amounts of NLS hyperacetylated HMGB1 as a result of inflammasome activation (25, 27). The inflammasome is a large multiprotein caspase-1-activating complex that regulates maturation and release of IL-1β and IL-18. Increased levels of IL-1β are implicated in the pathogenesis of arthritis and IL-1 receptor blockade is used as a treatment regimen for certain types of arthritis, including JIA. We thus analyzed the IL-1β content in our patient samples but could not detect measurable levels of IL-1β. This might have several explanations: Detectable IL-1β levels have mainly been described in the systemic onset form of JIA (SoJIA) patients, and our small cohort is mainly composed of oligoarthritic JIA patients; IL-1β is a potent cytokine and may have been released in amounts not detectable by our ELISA. Indeed, very few JIA patients have detectable levels of IL-1β but may still benefit from IL1 receptor blockade, emphasizing the potency of IL-1β in the pathogenesis of diseases (29). Furthermore, it is also likely that inflammasome activation is not the only mechanism through which HMGB1 release occurs in an acetylation-dependent fashion during chronic inflammatory diseases.
We also demonstrated a positive correlation between hypoacetylated NLS2, although much weaker than for hyperacetylated NLS2, and total HMGB1 levels. This might be explained in two ways: that passive release of HMGB1 due to cell death is contributing to, although not being the dominant source of, increased total HMGB1 levels. Despite the lack of a direct positive correlation between high total HMGB1 levels and high LDH activity, cell death is a feature of inflammatory activity and will result in passively released, hypoacetylated HMGB1. Another possibility is that other mechanisms leading to active release, independent of acetylation, occur in the inflamed joint, as previously discussed for hypoxia-induced release of HMGB1.
Post-translational methylation of the lysine in position 43 is suggested to regulate the affinity of HMGB1 to DNA, thereby also impacting its release from neutrophils. In this study by Ito et al., the cytoplasmic translocation of HMGB1 in neutrophils was only compared with lymphocytes and monomethylated K43 was suggested to be a neutrophil-specific feature (18). Neutrophils are highly abundant in inflamed joints and taken together with our finding of significantly elevated levels of K43 monomethylated HMGB1, we believe that neutrophil activation strongly contributes to the increased SF levels of HMGB1. The fact that LPS challenged neutrophils monomethylate K43 (Fig. 6A) further supports this conclusion. In contrast, neutrophil counts did not correlate with HMGB1 levels in the SF (Fig. 6D). This unexpected finding might be explained by differences in activation and the short lifespan of neutrophils, in addition to the complexity of SF. Thus, our data taken together with the report by Ito et al. demonstrate that neither monocytes nor lymphocytes monomethylate K43. Other cell types may, however, still be capable of methylating the K43 residue.
With the emerging knowledge regarding regulation of HMGB1 extracellular functions, defining HMGB1 cysteine redox properties is fundamental in the investigation of HMGB1 and its role in inflammatory conditions. The redox state of HMGB1 can change during different phases of ongoing inflammation and cysteine redox changes hence act as a molecular switch that defines the extracellular functions of HMGB1. The amount of reactive oxygen species produced from HMGB1 secreting cells can regulate and define the HMGB1 cysteine redox properties, but it is also likely that the redox state of the extracellular microenvironment is capable of regulating HMGB1 functions (10, 27).
Manipulation of redox signaling affects inflammasome activation and hence provides a link between redox signaling and the potential hyperacetylation and active release of HMGB1 (33, 37). However, a direct connection between HMGB1 hyperacetylation and redox modifications remains to be elucidated. Similarly, the mode of cell death also influences the redox status of the passively released HMGB1. Apoptotic cells have been reported to have a sulphonyl C106 side-chain and have been proposed to induce immunological tolerance (19). HMGB1 from pyroptotic cells has both chemotactic and cytokine-inducing properties, whereas HMGB1 from necrotic cells only has chemotactic functions (25, 27, 40). Our in vivo data clearly demonstrate that multiple redox isoforms, and therefore HMGB1 with different functional features, are present in SF from chronic inflammatory JIA patients.
Detection of total HMGB1 and of HMGB1-specific PTMs has attracted attention as potential biomarkers of inflammatory conditions. An increase in both total HMGB1 and especially hyperacetylated serum HMGB1 is associated with a worse prognosis in APAP-intoxicated patients. In addition, hyperacetylated HMGB1 may be used as a prognostic biomarker and may even define whether APAP-intoxicated patients will die or require liver transplant (1, 3). High levels of serum HMGB1 and in particular the cytokine-inducing redox isoform have recently been shown in patients with early macrophage activation syndrome. When treated with the apoptotic inducer Etoposide, HMGB1 levels drastically decreased and the redox isoform mainly shifted toward the “inactive” sulfonyl isoform (28). The complex mixture of hyperacetylated and hypoacetylated HMGB1, K43 monomethylated HMGB1 and of different HMGB1 cysteine redox isoforms detected in each investigated patient in our studied cohort indicates that the HMGB1 patterns may be different during chronic inflammatory conditions as compared with acute inflammatory conditions. Our analysed JIA cohort is, however, too small to allow any conclusions regarding total HMGB1, acetylated HMGB1, and redox isoform HMGB1 measurements and their usefulness as prognostic or diagnostic biomarkers. Our study would need to be repeated with much larger patient cohorts of clinically well-defined patients. Such studies will be greatly facilitated when other, less time-consuming, methods than LC-MS/MS are available for large-scale analysis of HMGB1 isoforms.
In summary, our findings suggest that HMGB1 is actively released in the chronically inflamed joints of JIA patients and that the active release is mainly acetylation dependent, although as yet unidentified alternative active release pathways may be present. We have also demonstrated that high levels of HMGB1 are associated with a monomethylated K43, which emphasizes a role of neutrophils as a significant contributor to the recorded HMGB1 levels. In addition, HMGB1 in the SF had multiple inflammatory properties concomitantly in each patient, as defined by analysis of the HMGB1 redox isoforms (Fig. 9). Our study further supports the fact that measuring total HMGB1 levels is not sufficient to determine its functions during disease pathogenesis. Detailed analysis of HMGB1 isoforms will convey additional knowledge of the biological importance and will be of relevance for the development of HMGB1-targeted therapies and biomarkers for potential treatment stratification.
Materials and Methods
Juvenile arthritis patients
In this study, SF from 17 JIA patients with active disease was collected at Astrid Lindgren's Children Hospital. Median patient age was 11 years (range: 3–18). Mean disease duration was 30.11 months (range: 0–180). Patients were enrolled according to ILAR criteria, and JIA subtype distributions were as follows: oligoarthritis (70%), polyarthritis (18%), and undifferentiated (12%) (Table 1). The SF was collected in citrate tubes, 40 μM-filtered, centrifuged to obtain cell-free SF, and stored at −80°C until use. Informed consent was given by both parents and patients. The study was approved by the North Ethical Committee in Stockholm, Sweden.
Isolation and preparation of SF HMGB1
SF (150 μl) was diluted to 1 ml with 0.9% saline and precleared with 50 μl protein G-sepharose beads (GE Healthcare). The precleared SF was incubated with 5 μg anti-HMGB1 antibody (Abcam; ab18256), and HMGB1 was immunoprecipitated for 16 h at 4°C. Antibody and HMGB1 complexes were isolated with 50 μl protein-G beads for an additional hour. To cap cysteine redox isoforms, isolated HMGB1 was subjected to 10 mM iodoacetamide (Sigma) for 1 h at 4°C, followed by 30 mM DTT (Sigma) for 30 min and 90 mM of NEM (Sigma) for 15 min at room temperature. Alkylated HMGB1 was then subjected to either GluC (New England Biolabs) or trypsin (Promega) proteolytic digestion according to the manufacturer's instructions. Generated peptides were desalted by C18 ziptips (Millipore) before being subjected to LC-MS/MS (25, 27).
Liquid chromatography and tandem mass-spectrometry
All chemicals and solvents used were of the highest available grade (Sigma). HMGB1-specific PTM characterization was determined as previously described using an AB Sciex QTRAP 5500 (Sciex, Inc.) equipped with a NanoSpray II source by in-line liquid chromatography using a U3000 HPLC System (Dionex), connected to a 180 μm×20 mm nanoAcquity UPLC C18 trap column and a 75 μm×15 cm nanoAcquity UPLC BEH130 C18 column (Waters) via reducing unions. A gradient from 0.05% TFA (v/v) to 50% ACN/0.08% TFA (v/v) over 40 min was applied at a flow rate of 200 nl/min (3, 14). The ion spray potential was set to 2200–3500 V, the nebulizer gas to 19, and the interface heater to 150°C. The limit of detection for each peptide of interest was designated to be the amount of peptide detectable on the column after serial dilution with a peak height greater than thrice the background signal. AUC for each peptide of interest was determined as a function of peak height over the time-dependent elution. The extracted ion counts for the full-length and truncated peptide were normalized for total ion count. The lower limit of quantification (LLOQ) and upper limit of quantification (ULOQ) were, respectively, the lowest and highest concentrations of extracted and digested HMGB1 with acceptable accuracy and precision. The acceptance criterion of precision was 15% coefficient of variation, or 20% for the low QC and LLOQ, while the acceptance criterion of accuracy was 85%–115% or 80%–120% for the low QC and LLOQ, which was determined by extraction of each of the nine calibration peptide standards (three replicates) or three QC (six replicates) in three independent batches. A batch was deemed acceptable if ≥4 of the 6 QC replicates were within 85%–115% of the set value, in accordance with biomarker assay validation guidelines and our previously published LC-MS/MS assay validation protocols (32).
LDH and protein levels
LDH levels were measured by LD reagent according to the manufacturer's instructions (Beckman Coulter). Briefly, the reaction of L-Lactate and NAD to pyruvate and NADH is catalyzed by LDH. The production of NADH is directly proportional to LDH activity and was followed spectophotometrically at A340nm. Protein levels were determined using Bradford protein assay (Bio-Rad).
Isolation of neutrophils and white blood cell count
Neutrophils were isolated from a healthy donor by 3% dextran sedimentation. Neutrophil purity was above 90% as determined by flow cytometry. Isolated neutrophils were seeded in Opti-MEM (Life-technologies) supplemented with penicillin-streptamycin (Sigma) and stimulated with 100 ng/ml ultrapure LPS (Invivogen) or PBS for 2 h (Fig. 6A–C). THP-1 cells were stimulated with 1 μg/ml LPS or PBS for 20 h. Supernatant-derived and intracellular HMGB1 were isolated and analyzed by LC-MS/MS as previously described for SF. For correlation with total HMGB1 levels, SF was obtained from 14 separate JIA patients; white blood cell count was analyzed; and neutrophil count was assumed to be equal to the granulocyte count.
Detection of HMGB1 and other inflammatory mediators
All analyses were performed according to the manufacturer's instructions. SF HMGB1 levels were quantified by HMGB1 ELISA kit II (IBL-international) with a detection limit of 0.3 ng/ml. IL-1β and CXCL-12 was measured by ELISA (R&D Systems). SF levels of IL-6, IL-8, monocyte chemotactic protein-1 (MCP-1), and interferon gamma-induced protein (IP-10), regulated on activation normal T cell expressed and secreted (RANTES), were measured using Human Soluble Protein Flex Sets and Human Soluble Protein Master Buffer Kits (BD Biosciences). The lowest level of detection for the selected cytokines was 20 pg/ml.
Statistical analysis
Modifications below the detectable levels were substituted with the lowest detectable quantity to preserve the power to detect correlations between factors. This is a conservative estimate of the actual level of modification, as the actual quantity is lower than the entered quantity, and will therefore not inflate relationships between factors. Correlation between total HMGB1 levels and AUCs from specific modifications was calculated using Spearman rank implemented in R (version 2.9.2) and Rcmdr (version 1.5–4). The p-values were stringently corrected for multiple testing using Holm's method. Correlation matrix heat map was generated in the gplots package implemented in R (version 2.9.2). For heatmap, the correlation coefficient was represented by Spearman's Rho-value and the significance level is represented by the negative Log10 of the p-value (logarithmic scale where 2=0.01, 3=0.001, etc.). Other statistical analysis was performed using GraphPad Prism version 5.0 (GraphPad Software v5.0, Inc.). Correlations between LDH, protein, and HMGB1 levels were analyzed by Spearman's correlation test. A comparison of HMGB1 levels in samples with or without the presence of monomethylated K43 was analyzed by Mann–Whitney U-test. A p-value of<0.05 was considered significant.
Footnotes
Acknowledgments
The authors would like to thank Hanna Schierbeck for technical assistance and RA Harris for linguistic advice. This work was supported in part by grants from the Swedish Association against Rheumatism, the Swedish Science Council, Berth von Kantzows foundation, Erik and Edith Fernström foundation, King Gustaf V's Foundation, and the Stockholm County Council.
Author Disclosure Statement
No competing financial interests exist.