
Abstract
Introduction
C
The multisystemic character of CF has led to a number of investigations aimed at identifying CFTR functions. This transmembrane protein is a dependent and multifunctional cyclic adenosine monophosphate (cAMP) low chloride conductance channel (94). CFTR belongs to the adenosine triphosphate (ATP)-binding cassette family and as such intervenes in the transport of a great variety of substrates, such as sugars, proteins, metals, and medications (153). CFTR interactions with other transmembrane conductance chloride and sodium channels underscore its major role in intra- and extracellular hydroelectrolyte balance (153). CFTR is also implicated in the regulation of the transport of bicarbonate ion, nitrate, formate, acetate, and glutathione (GSH) (52, 114, 155). Noteworthy, the glutathione/glutathione disulfide (GSH/GSSG) couple constitutes the most important pool of cellular redox systems (31, 47). The implication of oxidative stress (OxS) in the complex pathophysiology of CF is widely accepted (159). This involvement has been described predominantly in respiratory pathways in relation to chronic pulmonary infection, inflammation, lipid peroxidation, and the intensification of the metabolism (52, 62, 161). However, little is known about the role of OxS in the intestinal metabolism of CF. In addition, it is important to stress that various antioxidant supplementation studies on CF patients have for the most part led to mixed results that have not allowed any conclusions to be drawn as to whether their efficacy is significant with regard to pulmonary disease and patient's quality of life (52).
Essential fatty acid (EFA) imbalance in CF patients has been known for many decades, long before the discovery of the gene involved. It is characterized by a decrease in linoleic acid and docosahexaenoic acid (DHA) and an increase in arachidonic acid (AA) (23, 116, 143). These changes were for a long time mainly attributed to intestinal malabsorption secondary to exocrine pancreatic insufficiency (143). Over the last few years, other mechanisms have been proposed such as inadequate dietary EFA consumption, an intensification of the β-oxidation of polyunsaturated fatty acids (PUFAs), an increase in the production of proinflammatory eicosanoids, a rise in the peroxidation of PUFAs, an impairment of desaturases or hepatic lipase activity, and finally, the possibility of an intrinsically defective EFA metabolism in CF epithelial cells (107, 116, 158). The severity of the CF genotype has been correlated with the importance of these lipid disorders that could contribute to the inflammatory status observed in this pathology (151). A link between OxS and fatty acids (FAs) is known as well. The latter have been described as modulators of reactive oxygen species (ROS) generation at the level of the mitochondria, phagocytic neutrophils, and endoplasmic reticulum (ER) (137). The capacity to increase OxS by reducing antioxidant enzymatic activities has also been reported (137). Some studies on DHA and/or eicosapentaenoic acid supplementation associated with antioxidant vitamin supplementation have succeeded in re-establishing EFA imbalance in in vitro (116) and in vivo (107) models and humans (161). However, other studies have proven to be unsuccessful (23). To this day, lipid disorders observed in CF patients remain a concern and still raise a host of questions. In fact, various studies show that despite the intake of gastroresistant pancreatic extracts associated with adequate dietary intake, OxS and FA abnormalities persist (108, 159). A parallel plasma FA and ROS concentrations increase (161), given a rise to speculation about tolerance of the high-fat diet recommended for the nutritional care of patients. These findings reaffirm the need to understand CF OxS mechanism and lipid disorders particularly in the intestine that constitutes the first interface with alimentation.
OxS and Inflammation in CF
The redox homeostasis results from the balance between the production of pro-oxidants and their effective neutralization by antioxidant defense systems (150). OxS has thus been defined as a disequilibrium of pro- and antioxidative balance in favor of the former (63). The pro-oxidants consist of the ROS, reactive nitrogen species, and reactive chlorine species. ROS fill a prominent place in this group (150) and are the intermediates generated by oxygen during oxidation reactions (117). They consist of free radicals, such as superoxide anions (O2 −), hydroxyl radicals (OH•), and nonradical species, such as singlet oxygen (1O2) and hydrogen peroxide (H2O2), which react as precursors of free radicals with a wide range of cellular macromolecules and reactive substances to form strong free radicals (54). Notably, O2 − is formed directly from oxygen molecules (O2) by the gain of electron and is therefore considered “primary radical.” Its source is essentially mitochondrial, but during enzymatic process or catalysis by various metals, it generates the so-called “secondary” radicals (117).
The production of ROS, at moderate rates, is a physiological phenomenon for cell survival, given its potential in essential protection of hosts from pathogen invasion, apoptosis regulation, and transcription factor activation. In contrast, their overproduction leads to pathological effects in view of deleterious oxidative changes in cellular lipids, proteins, and DNA, as well as the formation of inflammatory proteins (109).
Overproduction of ROS and especially O2 − most often results from the excessive stimulation of NADPH oxidases (NOX) through inflammation mediators (e.g., tumor necrosis factor α and interleukin [IL]-1β), mitochondrial respiratory chain, and xanthine oxidase (92).
In an infectious context, ROS may be generated in cell membranes of macrophages and neutrophils following the excessive activation of NOX (92). This pathway is also strongly implicated in CF where infection and chronic neutrophilia are major features giving rise to ROS, thereby resulting in cell signaling (29). Xanthine oxidase is also capable of reducing O2 − from electrons derived from the oxidation of nicotinamide adenine dinucleotide (NADH) to NAD+ using flavine adenine dinucleotide (FAD) (68). Various enzymes (e.g., dehydrogenases, cyclooxygenase-2, lipoxygenases, peroxidases) are implicated in the production of ROS, and xenobiotics play a decisive role in the magnitude of this production (83). Thus, the long-term antibiotic treatment proposed in CF patients could be indexed in this context.
The dismutation of O2 − by mitochondrial Mn-SOD produces H2O2, which in the absence of degradation generates OH• via Fenton reactions and/or Haber–Weiss (83). The latter play a major role in the occurrence of OxS-related deleterious changes and in activation of the kinase cascade (45). The integrity of the antioxidant defense system of mitochondria appears as one of the “hubs” of the redox homeostasis. Its defect reported in CF therefore increases the susceptibility of patients to stress and could be an advantageous therapeutic target. H2O2 and O2 − can be generated in the cytosol not only from peroxisomes but also from the ER via the cytochrome P450 (29). Importantly, ROS production can come from external stimuli, such as ionizing radiation, UV radiation, smoking, and air pollution (83).
On the other hand, antioxidant defense systems bring together all the molecules capable of inhibiting the production of ROS, limiting their propagation or provoking their destruction (117). The antioxidant defense systems are divided into three groups: enzymes (superoxide dismutase [SOD], catalase [CAT], glutathione peroxidase [GPx], and glutathione reductase [GRx]), redox systems (glutaredoxin and thioredoxin, represented by the two redox couples GSH/GSSG and thioredoxin/thioredoxin oxidized [Trx(SH)2/Trx-SS], respectively, as well as various proteins such as heme oxygenase, quinone reductase, and proteins that chelate iron [transferrin and hemosiderin] and copper [ceruloplasmin and albumin]), and antioxidants or scavengers of free radicals (vitamin E, vitamin C, carotenoids, uric acid) (59, 79, 109).
In CF, malabsorption of fat-soluble vitamins exposes patients to vitamin E deficiency (α- and γ-tocopherol), which plays an essential role in protecting PUFA membrane against lipid peroxidation. It interacts with the peroxyl radicals to form tocopheryl radical (63). These are taken into account by the vitamin C and ascorbyl generating radicals, which will ultimately be neutralized by GSH (123).
The ubiquinone or coenzyme Q10 (CoQ10) is a lipid-soluble antioxidant that could be affected by fat malabsorption. It is a potent inhibitor of lipid peroxidation, which is found in inner mitochondrial membrane and it acts synergistically with vitamin E (63). The deficit of these two major antioxidants increases the vulnerability of lipid membranes, including mitochondrial and lipoproteins of CF in response to ROS adverse actions.
GSH deficiency is widely reported in CF because of the involvement of CFTR in its transmembrane transport. GSH is the first line of defense against free radicals, and its reduced form (GSH or γ-glutamyl-cysteinyl-glycine) is the most important in the control of thiol redox status (15, 104). Various GSH properties are attributed to the prevention of oxidation of the thiol groups (120), the free radical scavenging, metal chelation, and the DNA protection (117). GSH serves as a substrate for GPx while CAT converts H2O2 into H2O and GSSG. GSH also prevents oxidation and alkylation of Keap1 residues. In this context, it is worth recalling that if under nonoxidative states, nuclear factor (erythroid-derived 2)-like 2 (Nrf2) is normally located in the cytosol through its association with Keap1; during OxS, cysteine residues within Keap1 are oxidized, dissociating Nrf2 from Keap1, which allows nuclear translocation of Nrf2 where it binds to genes containing the antioxidant response element (15). The mitochondrial GSH preserves the integrity of mitochondrial proteins and lipids while controlling ROS production (103). The couple GSH/GSSG is recognized as the major redox couple acting as redox buffer (109) and its homeostasis disruption raises oxidative susceptibility of CF patients.
Cysteine (Cys) is the precursor of GSH and plays a critical role in the synthesis, structure, and protein functions, such as the formation of selenoproteins and GPx (19, 78). An elevated plasma level of its oxidized form (cystine or CySS) has been associated with the induction of the markers for chronic fibrotic lung diseases including CF (56, 126).
Trx(SH)2/Trx-SS is the main partner of GSH/GSSG in redox regulation. It is highly responsible for the ubiquitous reduction and repair of oxidative damage of protein thiol groups (5). This couple participates in lipid peroxides degradation, reduces peroxiredoxins involved in the reduction of H2O2, regenerates the ascorbyl radical, and is involved in redox regulation of transcription factors nuclear factor-κB (NF-κB) and activator protein-1 (AP-1) (5). Its action, with respect to AP-1, involves the reduction of oxidized forms of redox effector factor 1, which is responsible for the redox regulation of AP-1 and NF-κB, through the regulation of activation and dissociation of I-κB. Trx(SH)2 is regenerated by NADPH under the action of thioredoxin reductase (109).
Pyridine coenzymes (NADH/NAD+, NADPH/NADP+, FADH2/FAD) are also involved in cellular redox processes. NADH and FADH2 deliver electrons to the mitochondrial respiratory chain, while NADPH is involved in antioxidant defense by playing a central role in the reduction of GSSG and Trx-SS. The conversion of NADH to NADPH via nicotinamide nucleotide transhydrogenase of the inner mitochondrial membrane is a critical step in the antioxidant role of coenzymes. Under physiological conditions, it maintains mitochondrial NADPH redox homeostasis (29).
Recently, recognition of ROS as secondary messengers in signaling translation could redefine OxS instead as a disturbance of redox control state and redox signaling (31, 57, 81). This disturbance may be caused by metallic ions. Alternatively, redox-cycling agents may be accompanied with a modification in redox equilibrium. The protective capacity of antioxidants with regard to this disturbance could not be demonstrated at the end of various large-scale human intervention studies (67). GSH/GSSG, Trx(SH)2/Trx-SS, and Cys/CySS couples, as previously described, constitute the three major redox couples (32). There is no redox balance in these couples that could represent control hubs in various redox processes (31, 82).
These redox couples are found at variable redox potentials in different cell compartments. The mitochondria possess the lowest redox status as a result of its alkaline pH, followed by the nucleus, cytoplasm, ER, and finally extracellular space (39). The presence of the GSH/GSSG couple has been reported in the mitochondria, nucleus, ER, and extracellular space. The Trx(SH)2/Trx-SS couple is present in the nucleus; Trx1 has been located in the cytoplasm and Trx2 in the mitochondria. As for the Cys/CySS couple, it is found in the cytoplasm and extracellular space. Physiological oxidative stimulation can therefore affect a specific couple in a well-determined compartment (67).
The presence of OxS markers is regularly associated with CFTR defects. Various causes have been reported and include the stress of the ER, innate defect in the metabolism of GSH, intestinal fat malabsorption, abnormalities in antioxidant vitamin transport, and inflammation (8, 52, 119, 147, 153).
In severe homozygous CF subjects, misfolded proteins are sequestered in the ER and accumulate between the ER and the Golgi apparatus (102). They interact with the calcium-dependent chaperones, thereby causing calcium homeostasis alterations (3). These abnormalities contribute to ER stress installation resulting in the activation of specific pathways, including the unfolded protein response path that reduces protein synthesis and the transcription of chaperone gene (89). Consequently, the ER-associated degradation becomes dysfunctional and ineffective in eliminating defective proteins by the proteasome system, leading to an overproduction of ROS and apoptosis (89). Additionally, the accumulation of defective CFTR proteins in the ER contributes to endogenous activation of NF-κB with direct implications on calcium leak and ROS generation (153). The elevation of intracellular calcium is closely associated with phospholipase A2 (PLA2) activation that increases the release of AA from cell membranes, which contributes to the rise in EFA imbalance reported in CF. Indeed, AA serves as a substrate for the lipoxygenase system, conducting to raised ROS generation, lipid peroxidation, and eicosanoid synthesis, along with a fall in mitochondrial membrane potential, ATP production, and other oxidative damage to mitochondria (21, 131, 146). In the CF context, mitochondrial ROS overproduction is associated with marked depletion of GSH and redox imbalance (137). The innate defect in GSH metabolism favors constant antioxidant deficit while promoting PUFA oxidative damage in lipid membranes and circulating lipoproteins, which could explain mitochondrial membrane fluidity changes in CF (137).
Evidently, the exaggerated ROS availability via these various mechanisms will activate various signaling pathways involving growth factors, protein kinases, and transcription factors (NF-κB and AP-1) of inflammation, culminating in the induction of target gene of inflammation (92), which is consistent with the excessive production of proinflammatory cytokines and endogenous NF-κB activation in the absence of bacterial infection in CF. This inflammatory phenomenon was also observed in heterozygous patients with a single allelic CFTR mutation (131).
Whether the activation of transcription factors and their translocation to the nucleus require oxidative signal in the cytoplasm, their DNA link to the gene regulation of oxidative and inflammatory components requires on the contrary, reduction status inside the nucleus ensured by Trx (SH)2 and the redox effector factor 1 (1, 16).
Pathophysiology of respiratory dysfunctions
The presence of OxS and inflammation markers in the respiratory pathways of CF patients has been widely reported in the literature. Chronic infections of the respiratory pathways, by various pathogens, such as Staphylococcus aureus, Hemophilus influenzae, and Pseudomonas aeruginosa, have for a long time been considered the starting point first for inflammation and OxS in the respiratory pathways (20). Recent studies have, however, highlighted an increase in the level of expression of proinflammatory molecules outside any infection, as well as pulmonary neutrophil infiltration in 24-week-old fetuses (153). This finding suggests instead the existence of an intrinsic inflammatory process linked to CFTR mutation and resulting from a constitutive activation of NF-κB in CF epithelial cells (153). This inflammation of slight intensity is thought to be increased by chronic infections. The increase in the concentration of ceramides in the lungs and trachea of CFTR-deficient mice and CF patients has also been reported, even though the exact mechanism by which they trigger inflammation still remains unknown (12). CF inflammation is characterized by neutrophilia, a significant source of IL-8 production (159) and very probably IL-17, for which a link to inflammatory neutrophilia and mucus excess is increasingly suggested (18). Neutrophil migration in the CF bronchial lumen is accompanied by the release of massive quantities of ROS, including O2 −, H2O2, and OH•, mainly by activating the NOX system (37). Type II alveolar epithelial cells and bronchial ciliated cells are also capable of producing significant quantities of ROS via two NOX isoforms (DUOX1 and DUOX2) expressed at the apical membrane of these cells (48). In addition, disequilibrium of the redox balance in bronchial epithelial cells is suggested and three potential mechanisms are proposed: a defect in GSH homeostasis (74, 132), an impairment of nitrogen monoxide metabolism (60, 61), and disequilibrium of the intracellular production of H2O2 because of the Nrf2 dysfunction (24, 128). The respiratory manifestations of CF have been associated with low GSH levels in bronchial epithelial liquid compared with healthy subjects. In the latter, the increase in GSH concentration is thought to be a defense mechanism against oxidative damage induced by inflammatory stimuli (37). However, the origin of this GSH deficit remains to be elucidated, as it could result from iterative bronchial infections or simple CFTR dysfunction (37). In any event, the absence or malfunction of CFTR, therefore, seems to lead to redox disequilibrium in epithelial cells and extracellular liquids as well as abnormal ROS production (25). On the other hand, mitochondria, excellent ROS producers and targets via the mitochondrial chain, could participate in this state of stress (45). Lipid peroxidation and protein modifications are biological markers of oxidative damage sustained by epithelial cells and extracellular fluids from the respiratory pathways (86, 140). In fact, high plasma levels of 8-isoPGF2α (produced by the nonenzymatic oxidation of AA) have been reported over the course of respiratory exacerbation periods in CF (160). Markers of protein damage evolve along with the importance of neutrophilia and respiratory dysfunction (77).
Hepatic and pancreatic pathophysiology
In CF, the role of OxS and inflammation has been described more in respiratory pathways than in hepatic and pancreatic dysfunctions. However, we know that focal biliary cirrhosis, a characteristic lesion found in CF patients, associates with inflammation, biliary proliferation, and fibrosis (106). As for pancreatic inflammation, it has been described as mild with a small number of neutrophils and macrophages in acinous tissues (106). The role of OxS in the pathophysiology of pancreatitis has been mentioned by certain authors outside the specific context of CF. They suggest an important role for ROS in the development of pancreatic damage as observed in acute or chronic pancreatitis. The accumulation of ROS in the pancreas is believed to result not only from their production over the course of acute pancreatitis but also from a reduction in endogenous antioxidant capacities (51). ROS could also play the role of a second messenger or chemoattractant for inflammatory cells that would contribute to the production of aggravating inflammatory cytokines as well as pancreatitis (124). The severity of pancreatic involvement correlated with oxidative damage is thought to be alleviated by the use of antioxidant (99).
OxS and intestinal metabolism in CF
Potential origins of OxS in the intestinal CF
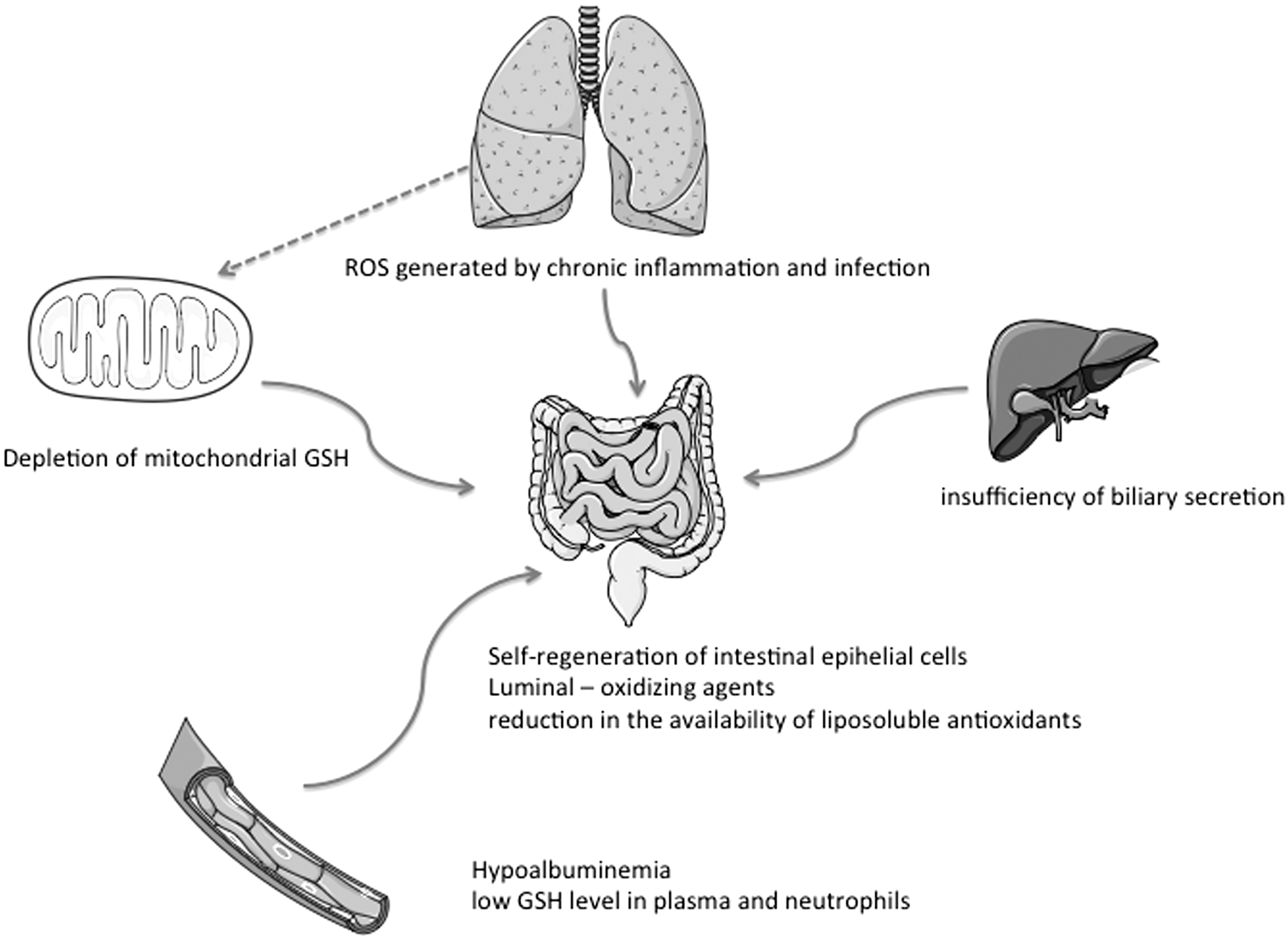
Gastrointestinal epithelial tissue is very proliferative and self-regenerates every 4–5 days (30). The passage of intestinal cells from proliferation to differentiation or apoptosis is accompanied by an increase in the oxidation potential of intracellular GSH/GSSG redox and extracellular Cys/CySS couples (81, 135). In vitro studies report that the Cys/CySS ratio modulates the proliferative potential of Caco-2/15 cells (6) and therefore probably influences their redox status. Located at the interface between the digestive lumen and the body, the intestinal epithelium is subject to oxidative damage induced by luminal-oxidizing agents (7, 31). The intestine and pancreas are the two organs with the greatest number of CFTR transcripts after birth (142). The known role of CFTR in GSH transport could therefore suggest this antioxidant's homeostatic abnormalities in CF intestinal epithelial cells (114). Studies report low levels of GSH in the plasma and neutrophils of CF patients, which suggest GSH homeostasis dysregulation (52, 147). A number of studies reveal a depletion of mitochondrial GSH caused by a CFTR mutation in the lungs. This depletion can reach 43% in human pulmonary epithelial cells and 85% in CFTR knockout mice (152). Maldigestion and intestinal malabsorption, characteristic of CF, also lead to a considerable reduction in the availability of liposoluble antioxidants, such as vitamin E, carotenoids, CoQ10, and FAs, in favor of redox disequilibrium (8, 73, 119). Thus, an innate defect in GSH metabolism coupled with insufficient absorption of liposoluble antioxidants would contribute to a failure in antioxidant defense and to an exacerbation of OxS (52). This malabsorption state is in part related to an insufficiency of biliary secretion that also participates in intestinal luminal redox balance (10). Resulting malnutrition could in itself be the source of stress because of the hypoalbuminemia with which it is associated. In fact, certain authors report that albumin is thought to be the plasma protein that is the most susceptible to damage from OxS, which is believed to be the potential link among hypoalbuminemia, inflammation, and cardiovascular risks (70). ROS from respiratory pathways submitted to chronic inflammation and infection could also infiltrate the intestine of CF patients via the systemic pathway. It has been demonstrated that mitochondrial ROS production is dependent on its internal redox equilibrium (137), and it increases considerably when there is a depletion of GSH in the mitochondrial matrix. The attendant redox disequilibrium means a reduction in the capacity of Gpx to reduce intramitochondrial H2O2. A relationship has been established between 50% depletion in GSH in the matrix and an increase in H2O2 production (64). Other studies have demonstrated that this depletion of mitochondrial GSH increased the sensitivity of cells to oxidative damage and apoptosis (28, 53). Defective CFTR could therefore contribute to redox disequilibrium of the mitochondria in CF exocrine epithelial cells, including intestinal cells. Little is known to date about the redox profile of the mitochondria in the intestine of CF patients. Its characterization and knowledge about its impact on intestinal metabolism could improve the understanding of the intestinal disorders observed in these patients. The degree to which OxS is implicated could be elucidated and therefore lead to an optimization of therapeutic protocols. Recurrent digestive disorders in CF patients could impair intestinal microbiota implicated in intraluminal antioxidant defense (31). Recent studies on microbiota in CF infants have revealed a concordance of microbial genera found in the intestinal and respiratory microbiota of these patients as well as growth in diversity over time (101). For some bacterial genera, intestinal colonization preceded appearance in the respiratory tract (101). This demonstrates that the digestive pathway should be given more consideration as a potential infectious gateway in these patients, given the repercussions on pro-oxidant/antioxidant balance (Fig. 1).
Intestinal antioxidant defense system
Intestinal antioxidant defense is as well organized in the intestinal lumen as in the epithelium and the plasma environment. This defense involves various factors whose intricate actions ensure intestinal redox equilibrium, namely diet, biliary secretion, intestinal microbiota, redox couples, and their related enzymes.
Intestinal lumen
A deficiency in the secretion of biliary and pancreatic lipase is well known in CF and constitutes the principal etiology of intestinal malabsorption (52). GSH from the intestinal lumen comes from the diet, de novo synthesis, and biliary secretion, which corresponds to 50% of hepatic GSH (10). Luminal GSH is involved in trapping divalent metals, maintaining mucus fluidity, capturing peroxidized lipids, and reducing their lymphatic transport (9, 36, 75). Any abnormality in biliary secretion could therefore influence intestinal luminal redox equilibrium by disturbing GSH metabolism. Gpx3 participates in the cytoprotection of intestinal mucus against luminal oxidizing agents (7, 31). In addition, the GSH/GSSG closely participates in the predominant role of Cys/CySS couple in luminal redox equilibrium. This Cys comes from the diet, thiol–disulfide exchange reactions, a reduction of CySS and GSH hydrolysis, which provide about 40%. Cys is considered to be the determinant of luminal redox potential in the gut (35).
Intestinal microbiota consisting of 500–1000 bacterial species prevents colonization by pathogens, supports intestinal nutrition, and regulates the mucosal immune system. In addition, this microbiota produces hydrogen sulfide in millimolar concentrations that is capable of preventing the inhibition of mitochondrial cytochrome oxidase, ROS production, GSH redox disequilibrium, and OxS (95). These various functions underscore the participation of intestinal microbiota in intestinal antioxidant defense.
Intestinal epithelium
Antioxidant defense of the intestinal epithelium is for the most part ensured by GSH via GSH-dependent enzymes. GSH is the predominant thiol at the intracellular level, of which it is the determinant of redox potential (31) where it is present in an essentially reduced form (63). Cellular GSH consists of various localized pools in the cytosol, mitochondria, ER, and nucleus (11, 39, 85). There is a difference in redox potential between these compartments based on their metabolic and biological specificities (11). GSH-derived enzymes such as Grx and Gpx are involved in intestinal antioxidant defense at various levels. Isoenzymes from Grx1 and Grx2, found in the cytosol and mitochondria, participate in GSSG reduction (46). Grx3 is implicated in various regulatory processes, such as embryo development, immune response, and cardiac physiology. Its implication is believed to occur through direct and specific interaction with other proteins in response to ROS-induced signaling (69). The four isoenzymes of Gpx, including Gpx2 (specific to the gut), reduce various forms of H2O2. Gpx1 and Gpx2 are two related cytosolic enzymes in terms of their structure and specificity for H2O2 and FA hydroperoxides. Gpx1 is also located in the mitochondria (26). Gpx3, in addition to the two preceding types of peroxides, reduces phospholipid (PL) but not cholesterol hydroperoxides. Gpx4 is present in cytosol, mitochondria, and the nucleus and reduces PL hydroperoxides, cholesterol, and thymine. Gpx4 is involved in the prevention of lipid peroxidation in synergy with vitamin E (26). Intestinal Trx(SH)2 is well known for its role in intestinal immune response and innate immunity. Strongly expressed in intestinal mucus, it is involved in the antimicrobial defense of human β-defensin 1 at the epithelial level in humans and is in fact recognized as being a physiological mediator that catalyzes the reduction of this peptide (138). Intestinal Trx(SH)2 also participates in antioxidant defense and the regulation of redox equilibrium by CySS reduction (58, 67). CoQ10, as previously described, is a powerful inhibitor of lipid peroxidation (63), which exerts its antioxidant action in the mitochondria and also in lipid membranes by trapping oxygen radicals. The mitochondria, membrane PL, serum PUFAs, and lipoproteins such as low-density lipoprotein are then protected from OxS (119). The combination of these different antioxidant actions could contribute toward preserving the redox equilibrium of the cell itself.
Extracellular compartment (plasma and interstitial environment)
The two redox couples found in this compartment are GSH/GSSG and Cys/CySS (31, 67). Their redox status in the compartment is highly more oxidized than in cellular cytoplasm. It is so well oxidized that CySS is considered the major determinant of redox potential in this compartment (36). The redox status of extracellular GSH/GSSG is considered to be the best reflection of tissular antioxidant defense, whereas that of extracellular Cys/CySS appears to regulate cell functions (80, 110). In fact, associations have been established between the oxidized status of Cys/CySS and the presence of various cardiometabolic pathologies (56). The implication of GSH in the reduction of CySS in this compartment therefore appears essential for the prevention of these chronic pathologies. The predominant role of GSH in the organization of antioxidant defense in general and of the intestine in particular highlights the importance of a functional CFTR protein in maintaining the homeostasis of exocrine epithelial cells.
Mitochondrial Functions and CF
β-Oxidation FAs and oxidative phosphorylation
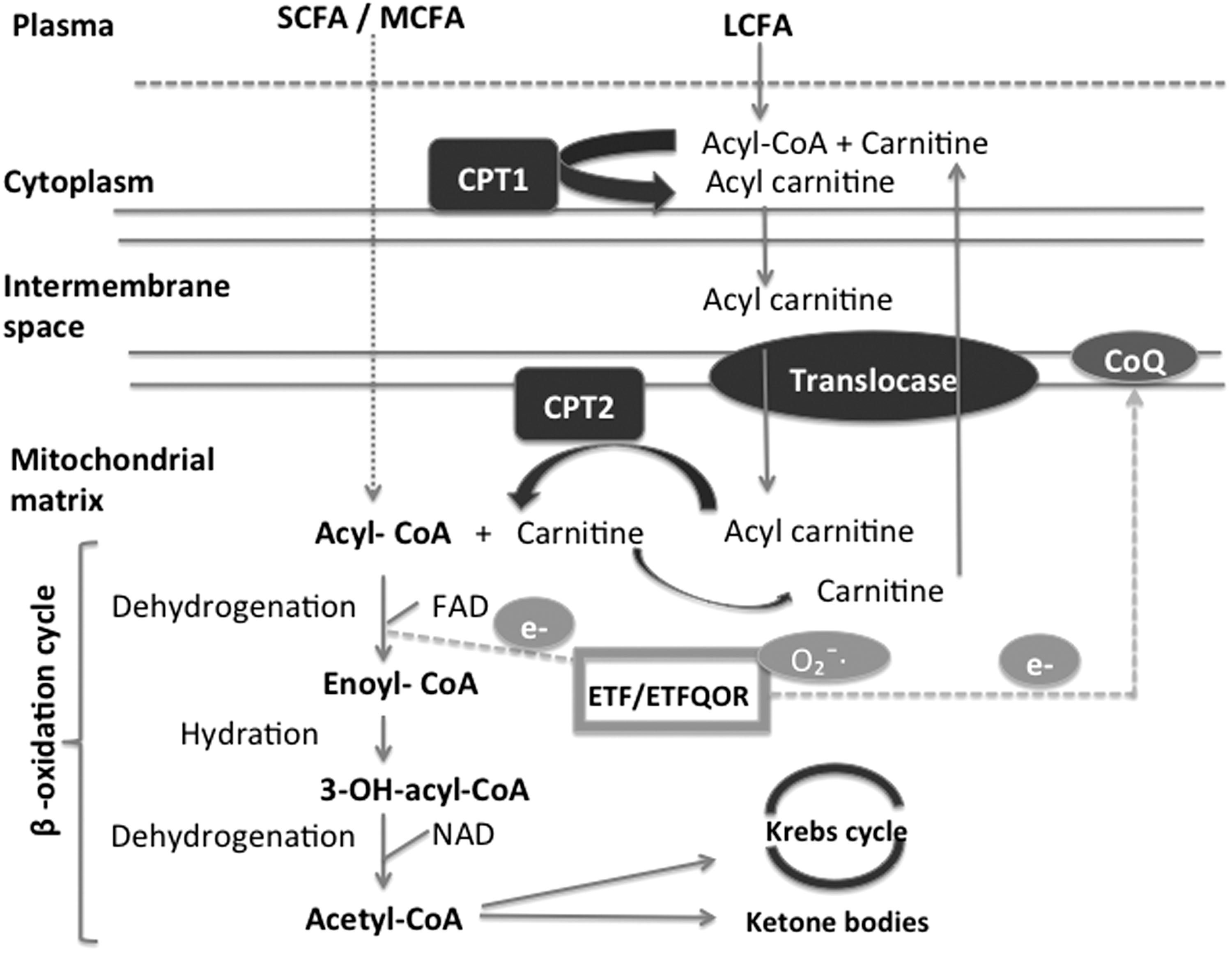
Mitochondria are oval-shaped organelles, which constitute a dynamic network in the cytoplasm of all cells. They are surrounded by two membranes consisting of PLs and separated by an intermembrane space. They possess their own genetic material and numerous functionalities, the most important of which is the production of energy by FA β-oxidation and oxidative phosphorylation (91). Short- and medium-chain FAs penetrate directly into the mitochondria, whereas long-chain FAs are first activated in cytosolic acyl-CoA under the action of acyl-CoA synthetase. This enzyme belongs to the acyl thiokinase family of cytoplasmic enzymes that bind FAs to coenzyme A by using energy from the hydrolysis of ATP in adenosine monophosphate and pyrophosphate. Acyl-CoA then enters the carnitine cycle and is transformed into acyl-carnitine while passing through mitochondrial membranes before being retransformed into acyl-CoA in the mitochondrial matrix. Acyl CoA is oxidized into acetyl-CoA via the β-oxidation cycle. This cycle, still called Linen's spiral, consists of a succession of four enzymatic reactions. The first is dehydrogenation by acyl-CoA dehydrogenase, which has FAD as a cofactor. This reaction results in trans-Δ-enoyl-CoA and FADH2 and involves electron-transferring flavoprotein (ETF). The identification of the mitochondrial acyl-CoA dehydrogenases was followed by the identification of the ETF as their common electron acceptor (100). It is now recognized that electron transfer flavoprotein:ubiquinone oxidoreductase (ETF-QOR), a component of the mitochondrial respiratory chain, together with ETF forms a short pathway that transfers electrons from 11 different mitochondrial flavoprotein dehydrogenases to the ubiquinone pool (CoQ10) in the mitochondrial inner membrane (127, 133, 141). The electrons can then passed to complex III (CIII) of the respiratory chain, cytochrome c, complex IV (CIV), and finally molecular oxygen (154). CoQ10 serves as a link between the β-oxidation of FAs and the mitochondrial respiratory chain. The ETF/ETF-QOR system has also been described as a likely source of O2 −/H2O2 production-like sites of the respiratory chain. Some studies have also reported that Multiple Acyl-CoA-dehydrogenase deficiency is associated with changes in protein structure of ETF-QOR, and the patients present with a secondary CoQ10 deficiency and increased rates of mitochondrial ROS. The production of these ROS would be linked to the defective protein that promotes ETF-QOR electron leakage reacting with molecular oxygen (33). In the context of CF, ETF-QOR characteristics are not well known and are worth exploring because of the known CI inhibition and potential deficiency of CoQ10. Indeed, changes in the gene ETF dehydrogenase that encodes the protein ETF-QOR have been associated with CoQ10 deficiency and also with a deficiency in the respiratory chain (33).
The second reaction consists of enoyl-CoA hydration by enoyl-CoA hydratase and results in 3-OH-acyl-CoA. It is followed by a second reaction of dehydrogenation by 3-OH-acyl-CoA dehydrogenase with NAD as a cofactor. The final reaction involves splitting by 3-ketothiolase into acetyl-CoA, the final product of the β-oxidation cycle, and also NADH and FADH2 (91, 98).
This acetyl-CoA can be completely degraded by the Krebs cycle (with CO2, NADH, and FADH2 production) or results in the formation of ketone bodies in the liver (91). NADH and FADH2 derived from β-oxidation and the Krebs cycle provide various complexes of the oxidative phosphorylation system with electrons (45) (Fig. 2).
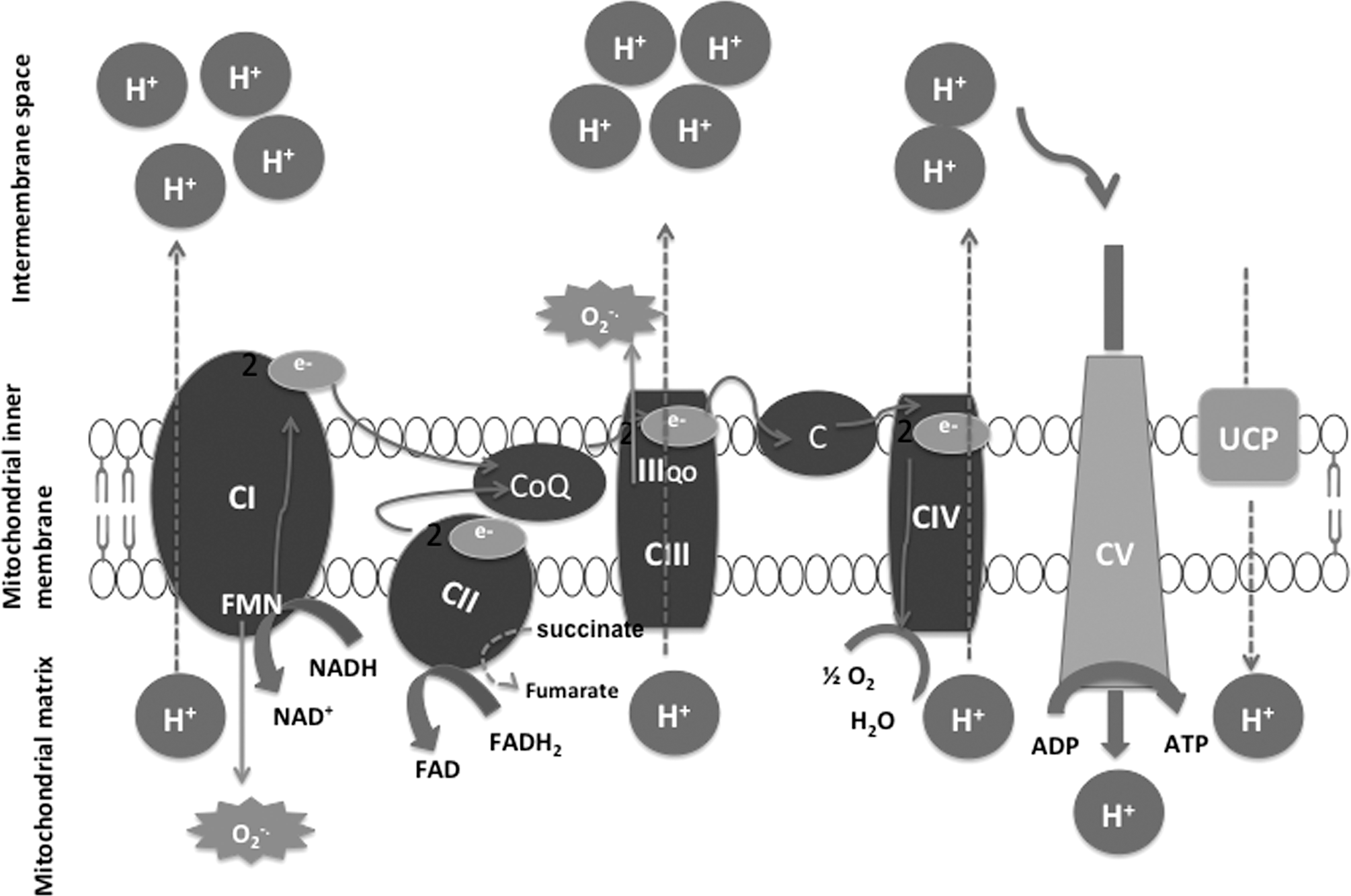
The mitochondrial respiratory chain is a metabolic pathway that generates energy for adenosine diphosphate (ADP) phosphorylation into ATP. This energy comes from the oxidation of hydrogen transport enzymes (FAD, NAD, and CoQ10) and electrons (cytochrome c). The mitochondrial respiratory chain functions at the level of the internal mitochondrial membrane and consists of five enzyme complexes (136): (i) CI or NADH ubiquinone oxidoreductase has flavin mononucleotide and iron–sulfur centers as cofactors. Its substrate is hydrogen provided by NADH from which it catalyzes oxidation. This hydrogen is then transported toward CoQ10 located in lipid membranes to ensure its reduction. (ii) CII or succinate ubiquinone oxidoreductase has the particularity of also belonging to both the mitochondrial respiratory chain and the Krebs cycle. It has FAD and iron–sulfur centers as cofactors. Its substrate, succinate, is oxidized into fumarate and hydrogen transported toward CoQ10. (iii) CIII or ubiquinone-cytochrome c oxidoreductase has cytochrome b and iron–sulfur centers as cofactors. Its substrate is composed of hydrogen atoms brought by reduced CoQ10 from which it catalyzes oxidation. Their electrons are then transported toward cytochrome c. (iv) CIV or cytochrome c oxidase has cytochrome with the nuclei of two heme iron moieties (a and a3) as a cofactor. Each nucleus is associated with a copper atom. Its substrate is made of electrons brought by cytochrome c from the intermembrane space from which it catalyzes oxidization. These electrons are transported by the enzyme toward the oxygen of the respiration that diffuses vessels toward the mitochondrial matrix to form water. (v) CV or mitochondrial ATPase pumps protons toward the matrix space from the intermembrane space into which they have been displaced by the various preceding oxido-reduction reactions from this space. It uses the energy to this effect generated by the other enzyme complexes for the phosphorylation of ADP into ATP. The return of protons toward the mitochondrial matrix can also be done outside ATPase via decoupling proteins (UCP1, UCP2, and UCP3), thereby resulting in the dissipation of the energy in the form of heat (129) (Fig. 3).
The ATP generated by the mitochondria is used in the functioning of various organs, such as the heart, liver and muscles (98); and certain structures like CFTR protein. In fact, the opening and closing mechanism of the CFTR channel involves ATP and cAMP (76, 153). This underlines the need for a stable mitochondrial metabolism to allow the optimal functioning of the CFTR protein.
Abnormalities in the mitochondrial respiratory chain, particularly the selective loss of the functionality of CI, have been reported in the context of CF. This inhibition of CI coupled with the overoxidation of NADH is presented by some authors as a consequence of intramitochondrial redox disequilibrium resulting from ROS overproduction and mitochondrial GSH depletion (84). Other studies report that this functional loss of CI in CF cells is believed to stem from the weak expression of the mitochondrial gene coding for the MTND4 subunit of CI, as the subunit is responsible for the assembly and activity of the complex (149). They underline in this regard the association that exists between defective CFTR and the functional inhibition of CI. Variations in the kinetics of CI among CF patients, heterozygous subjects, and controlled subjects had been suggested after in vitro study findings before the discover of the CFTR gene (139). In fact, CI kinetics is reduced in CF patients. The authors suggest the existence of an association between the CF gene and mitochondrial CI after having eliminated a post-translational glycosylation difference in the enzyme, a difference in the interaction of PLs from the internal mitochondrial membrane on the binding site of the enzyme, and finally a difference in the effect of mitochondrial calcium on the Michaelis constant (139). The probable association of CF with intracellular membrane abnormalities in the same context is supported by certain authors (38). There is scant mention of the dysfunction of other mitochondrial respiratory chain enzymes in the context of CF in the literature (4). It should be noted that these studies have been carried out for the most part on cutaneous fibroblasts and white blood cells. The characteristics of the mitochondrial respiratory chain in the intestine of CF patients remain to be discovered.
The mitochondrial respiratory chain generates ROS in weak or moderate proportions over the course of its physiological development (97). The production of O2 − has thus been reported at the level of CI, CII, and CIII.
As mentioned before, CI constitutes a major entry point for electrons to the respiratory chain in mitochondria (71). This complex oxidizes NADH to NAD+, by the reduced flavin mononucleotide (88), reduces ubiquinone to ubiquinol, and uses the redox potential difference to translocate protons across the mitochondrial inner membrane. It also represents a substantial source of cellular ROS (72, 113). In the presence of NADH, mitochondria reduce O2 to predominantly O2 − that dismutates rapidly to produce H2O2 (88). Noteworthy, the NAD+/NADH ratio determines the number of CI molecules (that have a fully reduced flavin) and the binding site occupancy, thereby explaining how the composition of the mitochondrial NAD+ pool may affect ROS production (14). Overall, these observations establish the direct links between NAD(P)H oxidation state and ROS production in mitochondria (87) and they especially specify that the CI flavin-site mechanism is behind many of the functional characteristics of O2 − and H2O2 production by mitochondria. The binding site of ubiquinone was also identified as the source of production of O2 − in the presence of a proton motive force and a significant pH gradient during the reverse flow of electrons from the pool of reduced ubiquinone, which supports ATP synthesis and many transport processes (148). Some studies have shown that the flavin site of CI is well capable of producing O2 −/H2O2 at high levels in the β-oxidation of FAs and in the reverse electron flow from the pool of reduced CoQ10 (122). Indeed, inhibition of CII leads to diminished O2 − production during the reverse flow of electrons (42). The central CIII of the respiratory chain also seems to contribute to the production of O2 − through its catalytic active site and during the oxidation of ubiquinone passing through the auto-oxidation stage of the semiubiquinone controlled by a delicate balance among membrane potential, reduced availability, and oxidized forms of ubiquinone and the redox status of CIII (55, 65). However, in CF, a more complete investigation is required to obtain a better understanding of mitochondrion contribution, ROS production, and the etiopathology.
O2 − from CI is released in the mitochondrial matrix where it is converted by Mn-SOD into H2O2 and O2 accounted for by GSH, CAT, and Trx(SH)2. That of CIII is released into the matrix and intermembrane space (64).
Non-mitochondrial ROS are thought to originate from the degradation of purine, the degradation of very-long-chain and branched-chain FAs in the peroxisome, the activity of plasma membrane NOX, and finally hydroxylation reactions catalyzed by microsomal cytochrome P450 (137). The production of ROS and nitrogen species is a physiological phenomenon strictly regulated by enzymes such as nitric oxide synthase and the isoforms of NOX (150). Mitochondria equipped with a high-level antioxidant defense system inhibit the harmful action of ROS through Mn-SOD, matrix GSH, Gpx, Trx(SH)2, and CAT (63, 97, 137).
Within a context of OxS with a disequilibrium of the redox balance, pro-oxidant agents increase, whereas antioxidant defense decrease (63, 159). This oxidative environment leads to apoptosis, an increase in synthesis and secretion, impairment of ionic transport systems, including chlorine secretion and dysregulation of cytosolic calcium (20, 115). Some authors report the existence of an association of ROS production, defective mitochondria, and mitochondrial GSH loss (64, 137). The exact chronology of these different events, however, remains to be established. By considering the known role of CFTR in GSH transport and the low GSH content in bronchial epithelial fluid even in the absence of infection, it is conceivable that, within the context of CF, low mitochondrial GSH content would be earlier than the other two events. Mitochondrial GSH cannot be synthesized de novo. It is pumped by the mitochondria from GSH contained in the cytosol, which explains the similarity of GSH concentrations in these two compartments (67). The absence of CFTR protein responsible for GSH transport from the intestinal lumen to the cytosol could lead to low cytosolic and therefore low mitochondrial GSH content. This would reduce the antioxidant capacities of the mitochondria in the face of a physiological production of ROS and expose it to the occurrence of various types of damages.
The impact of OxS on mitochondrial metabolism is well known and has been described by various authors outside the CF context. Lipid peroxidation induced in vitro by the iron/ascorbate couple leads to a disruption of mitochondrial function in Caco2/15 intestinal epithelial cell line (145). The dysregulation results in an increase in 8-OH deoxyguanosine (evidence of DNA) lesions and a decrease in its repairing enzyme (146). Studies report that intracellular OxS induces mitochondrial DNA (mtDNA) lesions (66) that would seem to precede cell nuclear damage (115). As a result of this high sensitivity to OxS, quantification of mtDNA damage in the blood has been proposed as a more sensitive marker of systemic OxS (22). A decrease in ATP production, a dysregulation of calcium homeostasis, and an increase in cytochrome c, apoptosis-inducing factor, and mitochondrial transcription factors have also been mentioned (146). Other in vivo studies performed on rat hepatocytes were submitted to OxS report; in addition to the aforementioned disorders, a significant decrease in mitochondrial antioxidative enzymes (Mn-SOD, Gpx, GSH), dehydrogenase activities, and respiratory chain complexes. Similarly, a decrease in membrane potential, the transitory permeability of the mitochondrial membrane with pore opening, and triggering of inflammatory responses have also been reported (145).
Role of FAs in the modulation of ROS production
Dietary FAs following digestion and intestinal absorption are transported in the blood in the form of chylomicrons (17). In plasma, the action of lipoprotein lipase gives rise to nonesterified FAs, a fraction of which binds to albumin. The unbound fraction, free FAs, is captured by adipose tissue, muscles, and peripheral tissue. Chylomicron residues go to the liver (17).
The intracellular mechanisms of FAs are variable and mainly occur in cell membranes, mitochondria (β-oxidation), and the nucleus. FAs enter the composition of the PLs of the membranes of all the body's cells and play a structural role through their capacity to modulate membrane permeability. They also influence ratios between PLs and membrane proteins (17, 143). Membrane PLs can be synthesized de novo or undergo a modification of their FA composition via a remodeling mechanism. This remodeling consists of the hydrolysis of a PL FA under the action of PLA1 and PLA2. It results in the release of signaling molecules, such as ceramides, diglycerides, and lysoderivatives as well as the synthesis of eicosanoids (inflammation mediators) when the FA that is released is AA (17). These membrane FAs also regulate protein channels and functions. They are for this reason considered to be important signaling regulators (143). Their nuclear actions result in the modulation of gene expression (17, 143).
The role of FAs in the genesis of inflammation is well known. CFTR dysfunction has been associated with an increase in the expression of PLA2, an increase in the expression and activity of cyclooxegnase-2, resulting in an increase in prostaglandin E2 production (13, 105). This PLA2 increase would seem to justify the increase in the release of AA from membrane cells and therefore the constant inflammation found in CF. Overall, these abnormalities reflect the structural membrane disruption as described in oxidative denaturation of membrane lipids.
Lipid peroxidation is a chain reaction that starts when OH• or O2 − tear a hydrogen atom from unsaturated FA esters of lipid membranes (117), thereby altering membrane potential and permeability, and functionality of membrane enzymes and receptors (27). Lipid peroxidation generates hydroperoxides from unstable lipids, a process catalyzed by various compounds, such as transition metals. The products of this degradation include malondialdehyde that alters membrane fluidity and function, and 4-hydroxy-2-nonenal (HNE) and 4-oxo-nonenal (NEB) that react with DNA to form adducts responsible for mutagenic chemical modifications of DNA bases (41, 93). In the absence of inhibitor of lipid peroxidation (e.g., CoQ10 and vitamin E) as would likely be the case in CF, lipid peroxidation products form protein adducts and activates various signaling pathways with a cascade of kinases. They can also form a cytosolic glutathionyl-4-hydroxy-2-nonenal (GS-HNE) complex whose persistence in the cytoplasm (due to antibody binding protein1 Ral or RLIP76) contributes to OxS and thus to inflammation. The RLIP76 is a protein that catalyzes the ATP-dependent transport of GSH and GS-HNE and allows their excretion into the extracellular medium (118). Nevertheless, fat-soluble antioxidants and GSH are key players in the prevention of lipid peroxidation and adverse effects of its metabolites.
The implication of FAs in the modulation of ROS production was suggested following various reports of association that exists between an increase in free FAs, an increase in ROS levels, and a decrease in antioxidant enzymatic activities. ROS cell production induced by free FAs is believed to take place in the mitochondria, ER, and phagocytic neutrophils via the NOX system for which the arachidonoyl-sn-glycero-3-phosphoinositol are particularly known as stimulators of O2 − generation (137, 159).
FAs have numerous interactions with the mitochondria and can modulate mitochondrial ROS production directly by β-oxidation through a decoupling effect (115). The literature reports that the implication of FAs at the level of the mitochondrial respiratory chain has a dual effect (115, 137). In fact, they can block intracellular OxS or, on the other hand, inhibit the respiratory chain and favor an increase in the generation of H2O2 when their doses are elevated (115). The mechanisms by which FAs interfere with the respiratory chain vary and have been described outside the CF context (137): (i) Inhibition/reinforcement of reverse electron transport from the CoQ10 pool to CI: This transport is associated with O2
− production and requires a high transmembrane (Δψ) potential. Free FAs through their protonophoric effect reduce O2
− production by CI via Δψ reduction. On the other hand, β-oxidation of acyl CoA could reinforce reverse electron transport and thereby increase O2
− production by CI. An in vitro study conducted on vascular cells that used oleic acid due to its reduced sensitivity to oxidation revealed that this FA inhibits ROS production without, however, inducing Δψ modifications. The authors reject the inhibition of ROS production via an FA decoupling effect (115). (ii) Inhibition of electron transport through CI and CIII: Free FAs exert a partial inhibition of electron transport through CI and CIII, thereby favoring their loss and allowing the reduction of O2 into O2
−. (iii) Possible interference with the GSH system: Free FAs could inhibit GSH regeneration from GSSG and thereby prevent the neutralization of H2O2 by Gpx. However, some authors report the capacity of oleic acid to stimulate Gpx activation via the activation of the epidermal growth factor receptor (115). (iv) Modulation of internal mitochondrial membrane fluidity: Free FAs are able to modify the fluidity of the internal mitochondrial membrane and thereby facilitate O2 access to electron donor sites, thereby increasing O2
− production. (v) Detachment of cardiolipin bound to cytochrome c: Free FAs are able to detach cardiolipin bound to cytochrome c on the external aspect of the internal mitochondrial membrane, which results in a reinforcement of the reduction status of electron transporters located upstream.
These different mechanisms, while being described outside the CF context, highlight the capacity of FAs to modulate ROS production in vitro by acting at different steps of the mitochondrial respiratory chain. A much more recent in vivo study conducted on renal tubules of recently evolved type 1 diabetic rats shows FA oxidation and not the respiratory chain as the source of ROS hyperproduction by the mitochondria in this context (130). The authors identify ETF as the main site of electron leakage. These variable data justify an exploration of mitochondrial metabolism in the specific context of CF where the exact origin of OxS and the link to FA disequilibrium have yet to be elucidated.
Probable existence of a deterioration in mitochondrial functions in CF
Some authors have reported specific characteristics of the mitochondria in CF. In vitro studies on CF respiratory epithelial cells (ΔF508 mutation) have highlighted a fragmented and dispersed mitochondrial network, a depolarized and therefore reduced transmembrane potential, and a reduction in calcium sequestration capacity by the mitochondria (2). This mitochondrial capture is mediated by the mitochondrial calcium uniporter whose functioning depends on the intensity of the Δψ membrane potential (2).
This Δψ depolarization could also be incriminated in low mitochondrial GSH content since it pumps cytosolic GSH (67). Another study, however, shows that mitochondrial GSH depletion is not due to defective transport, although a reduction in mitochondrial GSH transporters was observed (84). The authors of this study report GSH depletion associated with ROS overproduction and the oxidative environment that reigns within the mitochondria, the consequence being Cl inhibition with overoxidation of NADH, Δψ depolarization, and IL-8 secretion anomaly by CF cells. This Δψ depolarization has also been described as an initial step in the apoptosis process (137). Mitochondrial ROS overproduction has also been associated with modifications to the fluidity of the internal mitochondrial membrane leading to an increase in O2 capture (137).
The incorporation EFAs in membrane PLs seems to be influenced by chlorine channels, including CFTR. Studies report a deficit in incorporating linoleic acid in the PLs of CF epithelial membranes (13).
These various reported associations, the known effects of OxS on the mitochondria and their similarity to the described characteristics as well as the impact of CFTR deficit on the structure and functioning of cell membranes, suggest a probable oxidative origin for mitochondrial metabolism disorders in CF. The lipid abnormalities observed in CF could also be related to this oxidative context given the interconnectedness of both metabolisms (mitochondria and FAs) (Table 1).
AIF, apoptosis inducing factor; ATP, adenosine triphosphate; CI, complex I; Gpx, glutathione peroxidase; GSH, glutathione; MtDNA, mitochondrial DNA; ROS, reactive oxygen species; SOD, superoxide dismutase.
Antioxidant and/or FA Intervention Studies in CF
Antioxidants
There has been renewed interest in antioxidants in CF following various observational studies that reported an association of low levels of antioxidants, increased OxS markers, and impaired clinical condition in patients (8, 62, 90, 157, 159). Their incorporation into patient management protocols in the form of routine supplementation has therefore been widespread. However, certain studies aimed at evaluating the effects of this type of supplementation in CF patients have revealed generally low levels of antioxidants (CoQ 10, α-tocopherol, vitamin A, GSH) (8, 62, 77, 90, 119, 159), more or less casting doubt on their efficacy. This was the motivation for various intervention studies aimed at supplementing patients over a determined period and evaluating the impact on their antioxidant profile. These ad hoc supplementations consisted for the most part of vitamins (E or A). In certain cases, they were vitamin complexes with minerals, such as selenium. The findings of these studies were mixed. In fact, antioxidant serum levels could be normal, increased, or low in supplemented patients within the same study (73). The increase in antioxidant vitamin serum levels occurred with respect to the other liposoluble vitamins (D and K). Respiratory function barely improved, and the negligible reduction in urinary 8-isoprostanes meant any impact of this supplementation on OxS was unlikely (134, 161). Some authors also reported that a resistance to oxidation correlated with α-tocopherol levels in low-density lipoprotein and a reduction in malondialdehyde (156) (Table 2).
CoQ, coenzyme Q.
Considering the lack of efficacy of antioxidants, one might assume that, in CF, OxS goes far beyond intestinal fat and fat-soluble vitamin malabsorptions. It seems to result from more complex mechanisms other than dietary intake deficit since increased plasma antioxidants coexist with the presence of OxS markers (43, 161). The failure to fully eliminate ROS production by adequate supplementation could partly be explained by the persistence of OxS (159). In particular, homeostasis dysregulation of the major redox couples appears as a very likely explanation especially as CFTR−/−-induced GSH deficit may profoundly disrupt signaling and redox control concomitantly with or without changes in pro-oxidant/antioxidant balance (67). Both the involvement of these redox couples and the alterations in the regulation of inflammation by various transcription factors justify further mechanism exploration in the context of CF.
Fatty acids
The aim of routine administration of gastro-resistant pancreatic extracts combined with a high-fat high-calorie diet is to improve FA imbalance in CF patients. However, various studies show the limited impact of this routine therapy on patients' clinical conditions. In fact, a negligible improvement in anthropometric measures, an increase in saturated FAs and monounsaturated FAs, an increase in DHA/AA ratio, and onset of diabetes-type hepatic and metabolic complications have been reported (34, 49, 77, 108). Some authors also mention significantly low levels of vitamin E associated with significantly high levels of oxysterols in patients with pancreatic insufficiency or diabetes. These two groups of patients presented more markers of impaired FA profiles (77).
FA supplementation studies have been carried out in both animals and humans. In in vivo studies, supplementation most often consisted of DHA. This intake corrected DHA levels with a significant DHA/AA ratio increase in most tissues and membrane PLs. In some cases, this was associated with n-6 PUFA pathway impairment in CF target organs and the changes observed in long-chain PUFA levels varied according to tissue and mouse type (107). Other studies mention that DHA administration led to a reduction in eicosanoid levels in cftr−/− mice previously exposed to P. aeruginosa lipopolysaccharides as well as a negligible decrease in tumor necrosis factor α (50).
Clinical studies on supplementation have revealed certain interactions between FAs and other biological constants. They have reported that the FA profile of CF patients depends on their clinical, nutritional, respiratory, and oxidative conditions (43). In fact, FA plasma concentrations significantly influence the increase of 8-iso-Prostaglandin F2 α concentrations and the improvement of respiratory functions (161). An adequate nutritional intake in CF patients aimed at enhancing FA plasma concentrations is believed to lead to similar responses from an oxidative and respiratory aspect (161). Similarly, antibiotherapy administered over the course of pulmonary exacerbations leads to an increase in FAs, vitamins A and E, and lipid peroxidation markers (43, 161). The adverse effects of OxS on respiratory function could therefore be inhibited by effective antioxidant supplementation (161). These findings are, however, contradicted by other authors who demonstrated the absence of a correlation in EFAD, nutritional and clinical conditions, and vitamin E and zinc serum levels. They state that this deficiency is thought to be linked more to the genotype than to lipid peroxidation (144).
These studies highlight the association of FA imbalance with an increase in OxS markers and an impairment of antioxidant defense mechanisms in CF patients. Routine supplementation appears to be of little benefit in the long term in view of persistent underlying abnormalities and the onset of metabolic complications. As the results of ad hoc supplementations vary, objective conclusions cannot be made. It is, however, admitted that the administration of FAs enhances their plasma content while increasing OxS markers. Nevertheless, there is little consensus on the benefit of this correction to the clinical profile of CF patients (Table 3).
In vivo studies.
AA, arachidonic acid; DGLA, dihomo-γ-linolenic acid; DHA, docosahexaenoic acid; FSV, fat-soluble vitamins; GLP, glycerophospholipids; HFHCD, high-fat high-calorie diet; LA, linoleic acid; LPS, lipopolysaccharide; MUFA, monounsatured fatty acid; PERT, pancreatic enzyme replacement therapy; PUFAs, polyunsatured fatty acids; SFA, satured fatty acid.
Conclusion
The existence of FA imbalance in CF patients as well as the implication of OxS in the physiopathology of certain disorders encountered in this disease has been demonstrated. The close link that exists between FA metabolism and the endogenous genesis of ROS, particularly at the mitochondrial level, has been widely reported in the literature. The similarity that exists between the characteristics of the mitochondria in CF and the context of OxS could rightfully suggest a probable oxidative origin for mitochondrial metabolism disorders in CF. The innate GSH transport defect in CF patients could contribute to generating an overall oxidative environment at the mitochondrial level. On the other hand, the disruption of mitochondrial redox balance, by having an impact on mitochondrial functioning, could influence FA metabolism. The low impact of a high-fat high-calorie diet on correcting lipid profile, improving patient clinical condition, and preventing metabolic decompensations are a concern. In addition, an increase in OxS markers with this type of diet without correction by antioxidant vitamin supplementation raises concerns. Considering re-establishing mitochondrial redox balance and judging its impact on lipid profile would be a promising option. Investigating the role of OxS in the modulation of lipid intestinal metabolism in CF patients could help elucidate persistent FA imbalance despite the intake of gastro-resistant pancreatic extracts and a high-fat diet.
Footnotes
Acknowledgments
The authors thank Mrs. Schohraya Spahis for her technical assistance. The current work was supported by research grants from the Canadian Cystic Fibrosis Foundation and the JA deSève Research Chair in Nutrition (E.L.).