
Abstract
Introduction
T
In neural cells, the sigma 1 receptor (σ1R) binds to NMDAR NR1 subunits and it co-operates with the redox-regulated histidine triad nucleotide-binding protein 1 (HINT1) protein to bring mu-opioid receptor (MOR) signaling under the regulation of the NMDAR, thereby contributing to tolerance. In this protein assembly, opioids promote the binding of the redox sensor PKCγ to the HINT1 histidine residues, via nitric oxide (NO) and zinc metabolism, whereby the kinase recruits NMDAR activity proportional to MOR signaling. In naïve mice, the σ1R antagonists disrupt σ1R-NR1 interaction and uncouple the NMDAR from MOR activity, enhancing morphine analgesia and reducing the development of acute opioid tolerance. In mice rendered tolerant to morphine, σ1R antagonists promote the inhibition of NMDARs via Ca2+-CaM and they then increase the strength of the MOR signaling, rescuing morphine analgesia from tolerance. Thus, selective σ1R antagonists could be therapeutically exploited as adjuvants of opioid analgesia, reducing the risk of adverse effects.
The sigma 1 receptor (σ1R) has been proposed as a tonic anti-opioid system (39) that modulates the activity-induced sensitization in nociceptive pathways (8). The σ1Rs are widely expressed in nervous tissue, presenting high levels in areas that are associated with pain control (28). Whereas σ1R agonists facilitate nociception (27, 69), σ1R antagonists reduce the allodynia and hyperalgesia that accompany neuropathy in different animal models, improving the activity of opioids against nociceptive stimuli (8, 52, 53, 70). The σ1R was initially considered a type of opioid receptor (35); however, the σ1R lacks glycosylation, and its molecular structure suggests a different class of regulatory function, most likely that of chaperones (21). The σ1R constitutes a unique class of linear proteins that only has two transmembrane (TM) domains (3), with both N and C terminal sequences projecting to the same side, cytosol (59), or extracellular space (4), similar to the hairpin-like structure of caveolins, which are non-neural scaffold proteins (42).
The σ1R activity is modulated through a series of endogenous and exogenous substances. The pharmacology of the σ1R is complex, with exogenous ligands showing different profiles depending on the system under study (38). Notwithstanding this drawback, σ1R ligands are of therapeutic interest for the treatment of neurological diseases (31), substance abuse syndromes (46), and NMDAR-related neuropsychiatric disorders (22) or as adjuvants of opioid analgesia (25, 39, 64). According to the anti-opioid function of the σ1R (39), σ1R antagonists enhance the analgesic effect of systemic morphine, which is prevented by σ1R agonists, and also restore morphine analgesia in tolerant mice (64). As expected, σ1R−/− mice exhibit an increased response to morphine antinociception that cannot be regulated by σ1R ligands (57). Importantly, the opioid effects that are enhanced by σ1R antagonists are those regulated by the NMDAR/NOS/CaMKII pathway (70); thus, σ1R ligands do not modify morphine-induced hyperlocomotion or gastrointestinal transit inhibition. The positive features of the highly selective σ1R antagonist S1RA make this drug a good candidate for the treatment of neuropathic pain (53), and this treatment has satisfactorily completed phase I safety and pharmacokinetic evaluation in humans (1).
The σ1R ligands modulate NMDAR functions both in vivo and in vitro (36, 41, 55). Indeed, in cellular expression systems and in vitro assays, the σ1R displays calcium-dependent binding with NMDAR NR1 subunits (55). Because σ1Rs also associate with MORs (25), it is possible that these proteins regulate opioid function within the protein assembly that, via the histidine triad nucleotide-binding protein 1 (HINT1), redox signaling and zinc metabolism, support the MOR-NMDAR physical association and functional cross-regulation (48 –50).
Within this background, this study analyzed the potential role of σ1Rs in the cross-regulation between MORs and NMDARs in the mesencephalic periaqueductal grey (PAG) matter, a supraspinal region that regulates spinal nociceptive signals. The σ1Rs associate with NMDAR NR1 subunits, and σ1R antagonists promote the binding of negative regulators of NMDAR activity. The negative feedback that NMDARs display on MOR signaling requires the nitric oxide (NO)- and zinc-dependent recruitment of PKCγ to the HINT1 proteins followed by σ1R-NR1 binding to the MOR-HINT1 complex. Consequently, σ1R antagonists uncouple NMDAR effects from MOR signaling, thereby enhancing morphine analgesia and reducing the development of opioid tolerance.
Results
Organization of the σ1R receptor in the cell membrane
In the nervous tissue, the σ1R exists as long and short isoforms (21, 58). The long form of σ1R comprises 223 amino-acid residues, while the short form contains 106 amino acids. The σ1R has a hairpin structure with a short N terminal sequence before the first hydrophobic TM domain that leads to the loop region, followed by the second TM domain and the long C-terminal domain (cd). The short form of σ1R contains a clipped C-terminal sequence that differs from that of the long form in the four last residues. There is some controversy regarding the potential arrangement of σ1R in the plasma membrane, specifically with respect to whether both the N- and C-terminal sequences are directed to the cytosolic side leaving the loop in the extracellular milieu, or in the opposite orientation with the loop facing the cytosolic side (4, 44) (Fig. 1A).
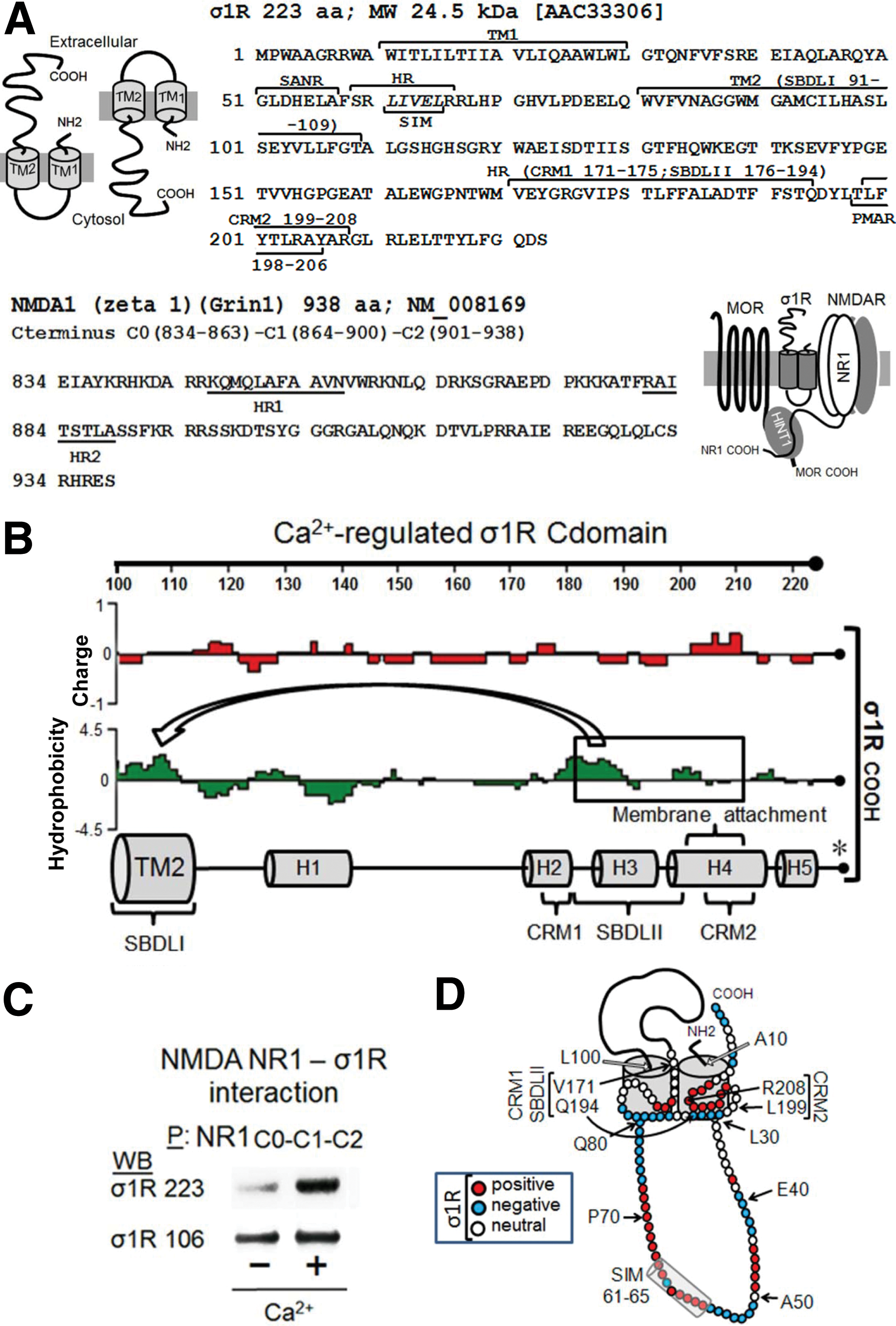
The σ1R in the endoplasmic reticulum membrane binds Bip in a calcium-dependent manner, and this binding is retained through the cd region (residues 112–223; σ1R) (43). In cell expression systems, σ1R shows a calcium-dependent interaction with the NR1 subunit of NMDAR, and both proteins can be co-immunoprecipitated from brain synaptosomes (4, 55). In in vitro assays, σ1R binds to the NR1 cytosolic C-terminal region C0-C1-C2 (55). The σ1R loop has a SUMO-interacting motif (SIM) domain (LIVEL: 61–65) that is typical of intracellular interactions (Fig. 1A and Supplementary Fig. S1; Supplementary Data are available online at
Regarding the arrangement of σ1R in the cell membrane, we examined the calcium-dependent binding of σ1Rs to NR1 subunits through peptide interference (Fig. 2 and Supplementary Fig. S2). This approach suggested that NR1 C0 (839–853) and NR1 C1 (864–878) bind to the σ1R. Peptide mapping of the hydrophobic regions in the NR1 C0 and C1 segments greatly enhanced the interaction between the σ1Rs and NR1 subunits, suggesting that both of the NR1 hydrophobic domains interact and that σ1R disrupts this interaction to achieve NR1 binding. It is therefore possible that peptide 9 interferes with σ1R binding in the NR1 region of the 883 residue; however, this interference could be masked by peptide 9 better exposing the NR1 C0-C1 domain for interaction with third-part proteins. Peptide interference indicated that two regions of the σ1R loop could be implicated in its interaction with NR1 subunits; the first region lies between residues 40 and 60, and the second lies between residues 71 and 80. Mapping the σ1Rcd (174–223) indicated that the HR1 region of NR1 C0 does not interact with σ1R 184–203 sequence, and then the NR1 C0 region that is most likely involved in σ1R binding is 839–848. Notably, peptides mapping the σ1Rcd hydrophobic helical regions H3 and H4 impaired the binding of this receptor to the NR1 subunit. The interaction of these hydrophobic regions with the TM1 and TM2 domains is essential to maintain the conformation of the σ1R as being able to bind to the NR1 C0-C1 domains (43, 44). Thus, an excess of these peptides would remove the σ1R H2 and H3 regions from the TM domains, disorganizing the structure of the σ1R and consequently reducing its interaction with the NR1 subunit (Supplementary Fig. S2).
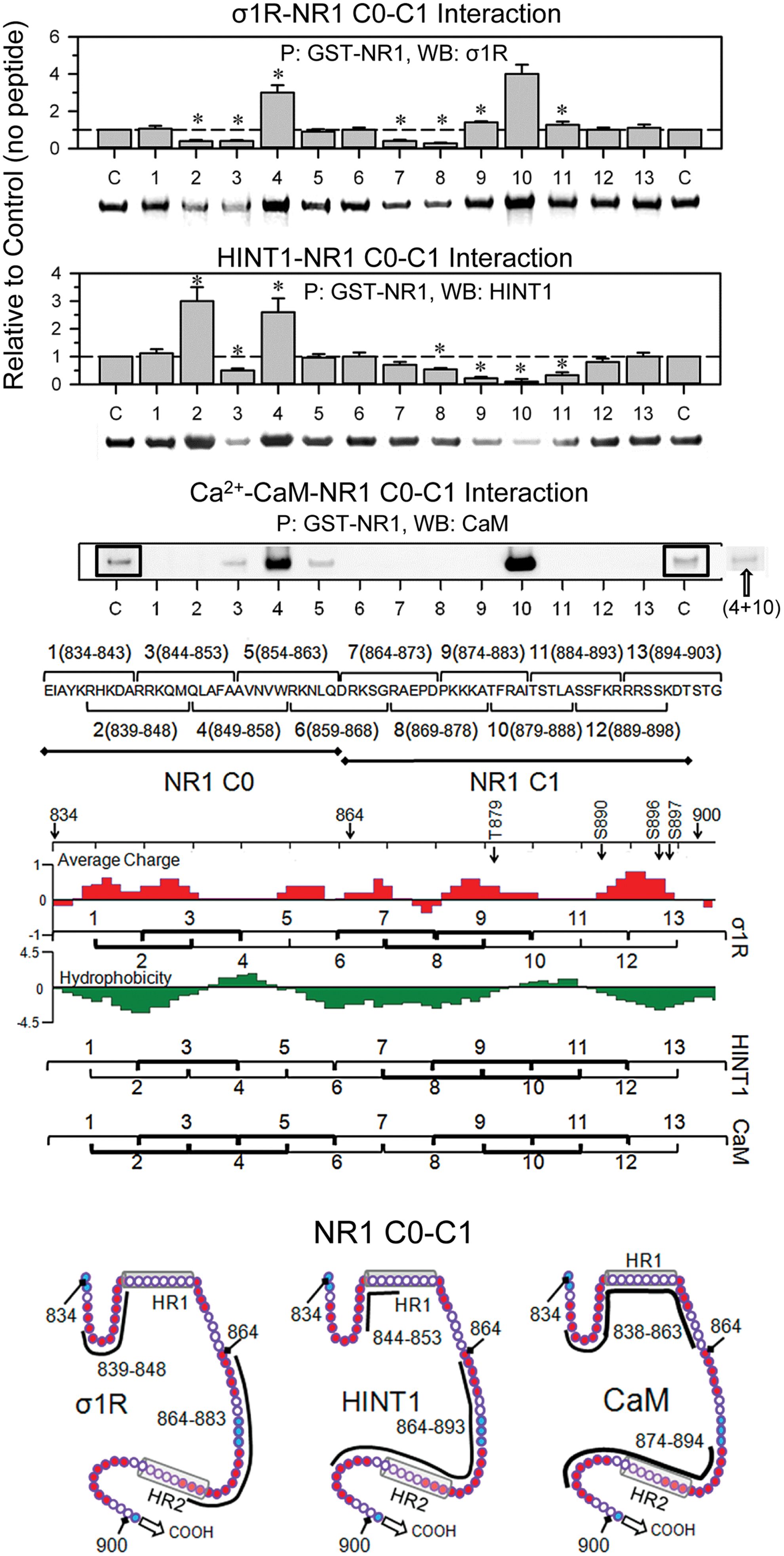
A similar approach revealed that the HINT1-binding region on NR1 C0-C1 overlaps with the σ1R-binding region. Thus, peptides mapping to the NR1 C0 hydrophobic region enhanced the HINT1-NR1 interaction. However, peptides mapping to the corresponding region in the NR1 C1 segment diminished this interaction. These observations indicate that HINT1 unfolds the NR1 but primarily binds to the NR1 C1 hydrophobic region, ignoring the hydrophobic region on the NR1 C0 segment. In the absence of peptides 4 and 10, the σ1R and HINT1 proteins exhibited noticeable binding to NR1 C0-C1, whereas the binding of Ca2+-CaM was certainly weak. It is possible that the disruption of the NR1 intrahydrophobic interaction through Ca2+-CaM is weaker than that achieved by σ1R or HINT1; thus, the Ca2+-CaM binding to NR1 subunits likely requires the assistance of third-party proteins. This possibility has been previously suggested through the binding of the cytoskeletal protein α-actinin2 with the C0 region of the NR1 subunits (40). The NR1 subunit contains two putative regulatory sites for Ca2+-CaM, with one site in the C0 segment and the other in the C1 region (11), and the disruption of the NR1 C0-C1 hydrophobic interaction through either peptide 4 or 10 exposed one of the NR1 sites for Ca2+-CaM binding. Accordingly, the presence of both peptides eliminated Ca2+-CaM binding to the NR1 subunits (Fig. 2).
The juxtaposition of the σ1R loop and NR1 C0-C1 regions revealed complementary charges between the loop emerging from TM1 and the upper half of the NR1 C1 segment, followed by putative hydrophobic interactions between the SIM-containing sequence 58–66 and the NR1 hydrophobic region S890 (Fig. 3A). In addition, the preincubation of small ubiquitin-related modifier 1 (SUMO1) with σ1R impaired subsequent binding to the NR1 subunits, and SUMO1 weakly disrupted the existing σ1R-NR1 association (Fig. 3B). Thus, the SIM-containing loop is involved in σ1R binding to NR1, and this region is hardly available during their association, reflecting the interaction of the SIM-associated negative region with complementary charges in the NR1 C1 segment, subsequently weakening the possible SUMO-SIM interaction (24).
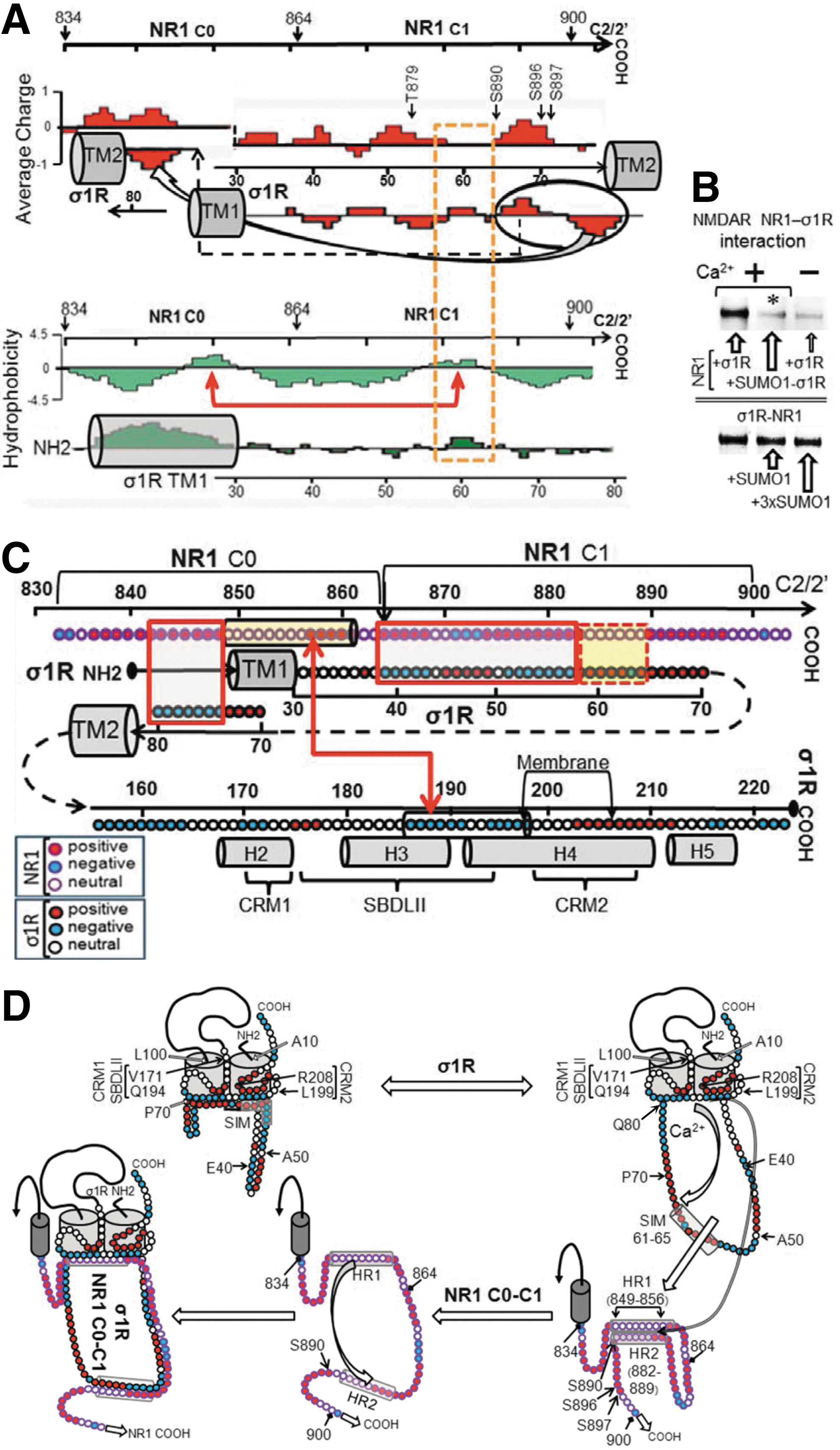
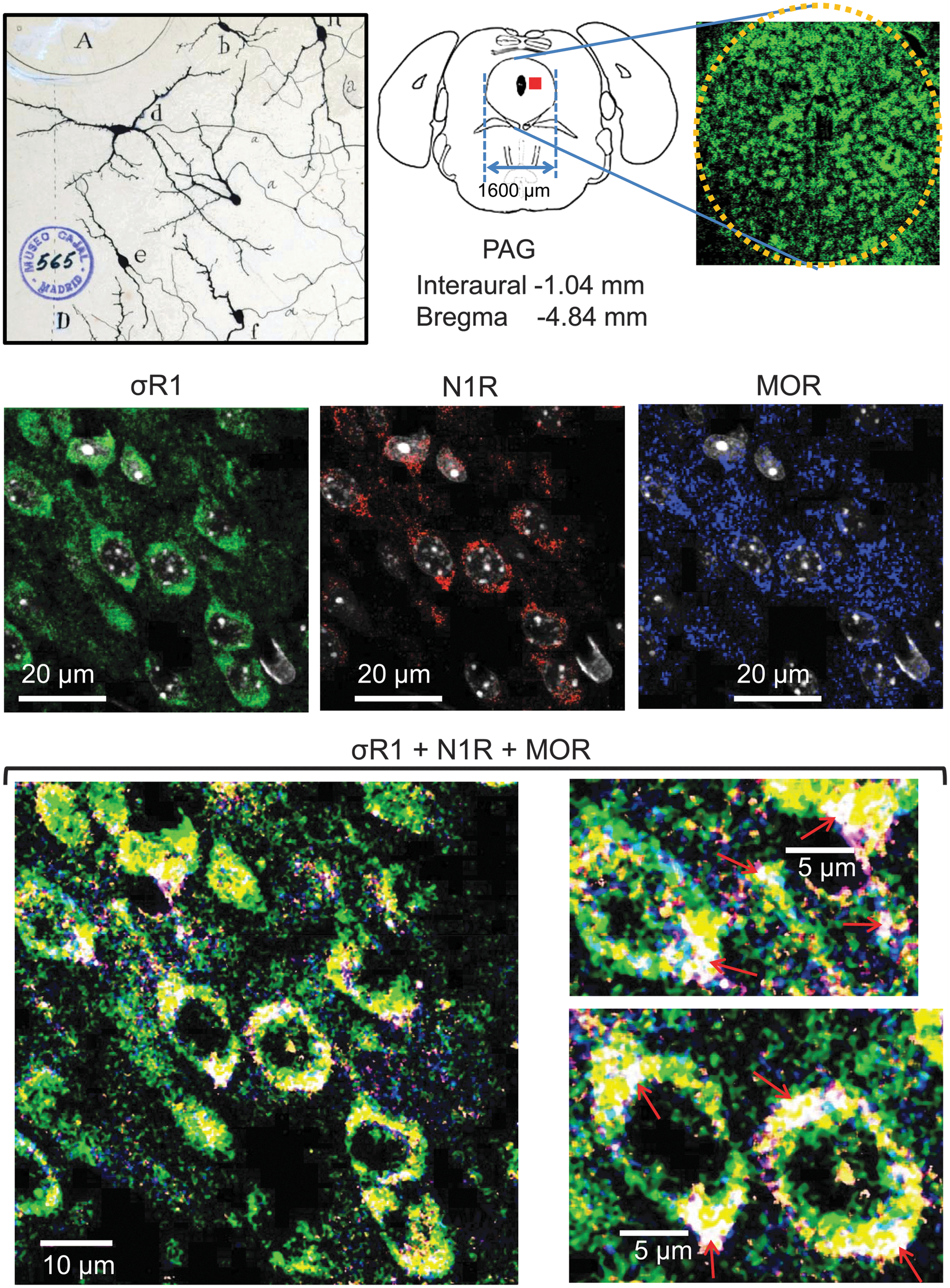
Considering our (Fig. 3C) and other (4, 55) data, we constructed the model of the σ1R-NR1 interaction that is shown in Figure 3D, in which σ1R is arranged in the plasma membrane with the SIM-containing loop facing the cytosol and the N-terminal sequence facing the extracellular space. The orientation of the short C-terminal sequence that is outside of the TM2 is tentatively orientated to the extracellular space; however, this sequence could also be facing the cytosolic side. The region of the σ1Rcd that is potentially regulated by calcium (SBDLII) is located on the cytosolic surface of the cell membrane. Thus, our arrangement of the σ1R in its interaction with the NMDAR NR1 subunit maintains the essential described features for σ1Rs in the cell membrane (4, 44) but with the cd and loop hydrophobic regions, which show complementary charges, situated on the cytosolic side of the membrane. In the absence of calcium, the interaction between the σ1R and the NR1 subunit is occluded. The positive calcium ions bind to the hydrophobic region of the σ1Rcd and neutralize the negative charge that is required to bind the SIM-containing positive region of the loop. Thus, the calcium-bound σ1Rcd region can now disrupt the internal hydrophobic interaction NR1 C0 (HR1)-NR1 C1 (HR2), exposing the required binding surface to the σ1R loop. We could detect these signaling proteins in mouse PAG neural cells using antibodies against the MOR, σ1R, and NMDAR NR1 subunits. Immunohistochemical analysis revealed a high degree of co-localization in the lateral PAG between the NR1 subunit and its regulator, the σ1R. In this region, the MOR showed a discrete co-localization with these proteins. The triple co-localization of the MOR, σ1R, and NR1 subunits could be observed as a series of white spots located at the cell periphery and also in fibers. This distribution/co-localization is compatible with σ1Rs regulating MOR and NMDAR function (Fig. 4 and Supplementary Fig. S3).
These observations situate σ1R in the protein assembly that controls MOR activity via NMDARs. Thus, we addressed whether the antinociception and molecular changes that are produced by morphine in the presence of σ1R antagonists fit this model.
Antagonists of σ1Rs enhance morphine-evoked supraspinal antinociception
The MOR in the mesencephalic PAG plays the most relevant role in the antinociception produced by opioids when injected by the intracerebroventricular (icv) route. The icv administration of all the substances studied circumvents the possibility that the drugs reach receptors beyond the brain. Subsequently, the capacity of morphine to produce supraspinal antinociception, and that of the studied drugs to modulate this effect, was assessed through the warm water tail flick test (see Materials and Methods section). The distribution of the thermal stimulus over two-thirds of the tail and the cut-off of 10 s protect the mice from tissue damage, as well as prevent the particular thermal sensitivity of a discrete point of the tail from compromising the data. Moreover, the mice practically do not anticipate their responses in using this analgesic test when they are studied several times during a time-course study.
The icv administration of the highly selective σ1R antagonist S1RA [E-52862; (53)] at 10 or 30 min before morphine treatment (3 nmol, icv) increased the analgesic activity when evaluated at 30 min postopioid injection. S1RA given 1 h or longer before morphine produced no such effect. Therefore, 30 min was selected to study the effect of σ1R ligands on the capacity of morphine to promote supraspinal analgesia in the thermal tail-flick test. We have reported a model for the detection of σ1R antagonist activity, and S1RA is likely the most potent and pure σ1R antagonist available (55). S1RA at 3 nmol increased the analgesic activity of morphine; other antagonists, such as BD1047 and NE100, also performed well in this paradigm, whereas BD1063 weakly increased morphine analgesia. The agonist PRE084 did not affect morphine analgesia but prevented S1RA from enhancing opioid antinociception (Fig. 5A).
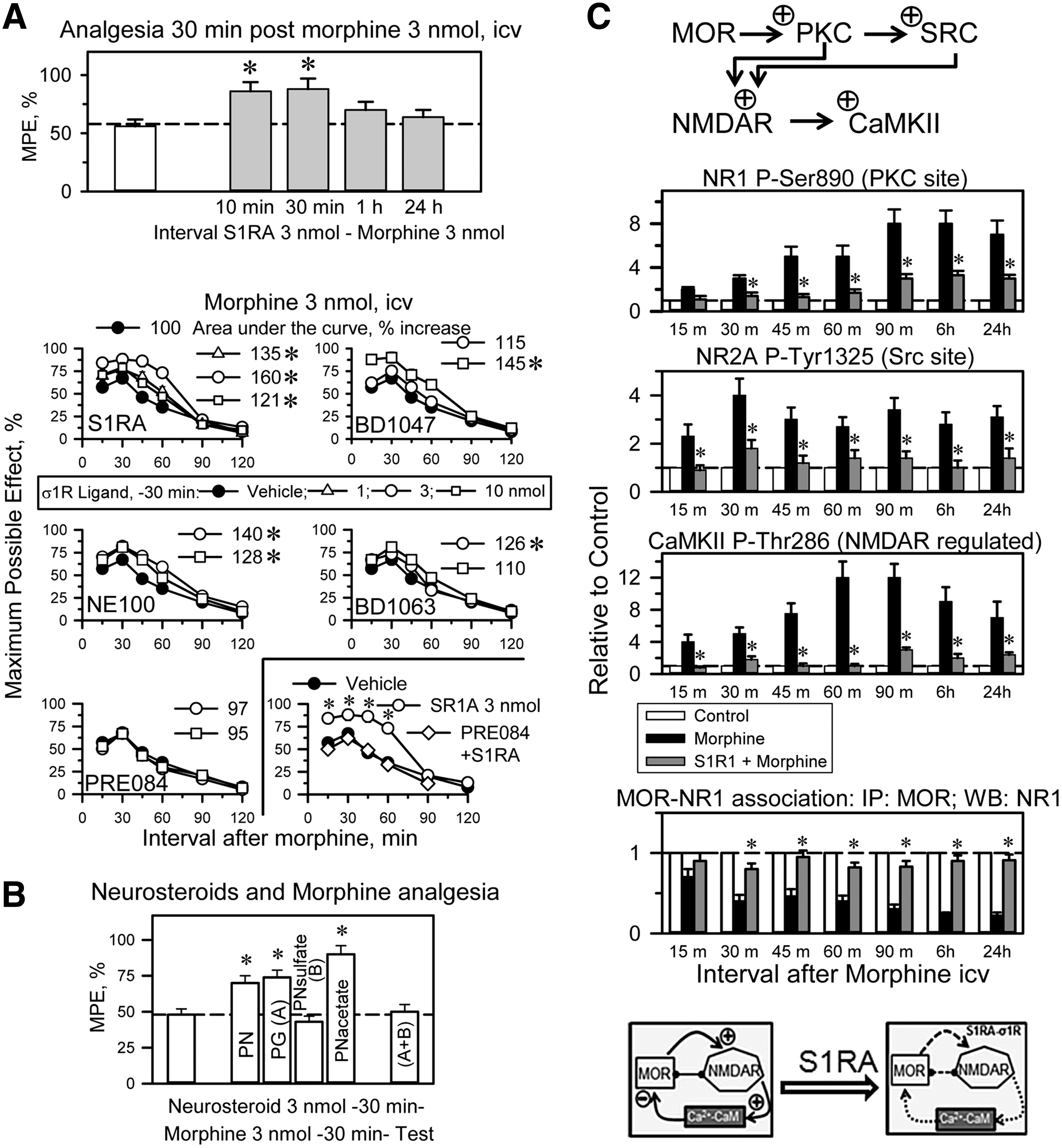
Neurosteroids, putative endogenous regulators of the σ1R, were also studied. Pregnenolone is a σ1R agonist, and its metabolite progesterone is a σ1R antagonist. Pregnenolone and its acetate derivative enhance morphine antinociception; however, the sulfate form of pregnenolone does not show this effect. Progesterone enhances morphine antinociception, an effect that is reduced by pregnenolone sulfate (Fig. 5B), suggesting that pregnenolone, but not its sulfate form, is rapidly converted into progesterone.
Morphine increases NMDAR activity: effect of σ1R antagonism
At the molecular level, the icv administration of 10 nmol morphine increased the function of the NMDAR-CaMKII pathway. This opioid increased the phosphorylation of NR1 subunits (PKC on S890) and NR2 subunits (Src on Y1325). Moreover, morphine increased the T286 autophosphorylation of the Ca2+- and CaM-dependent serine and threonine kinase CaMKII, an NMDAR effector. As a result of the MOR-induced activation of NMDARs, the MOR-NR1 association was weakened, and these changes persisted beyond the morphine analgesic time course (49). The presence of 3 nmol S1RA, administered 30 min before morphine treatment, nearly abolished CaMKII activation, reduced NR1/NR2 phosphorylation, and preserved the MOR-NR1 inhibitory association during the intervals that corresponded to the morphine analgesic time-course and beyond (Fig. 5C and Supplementary Fig. S4).
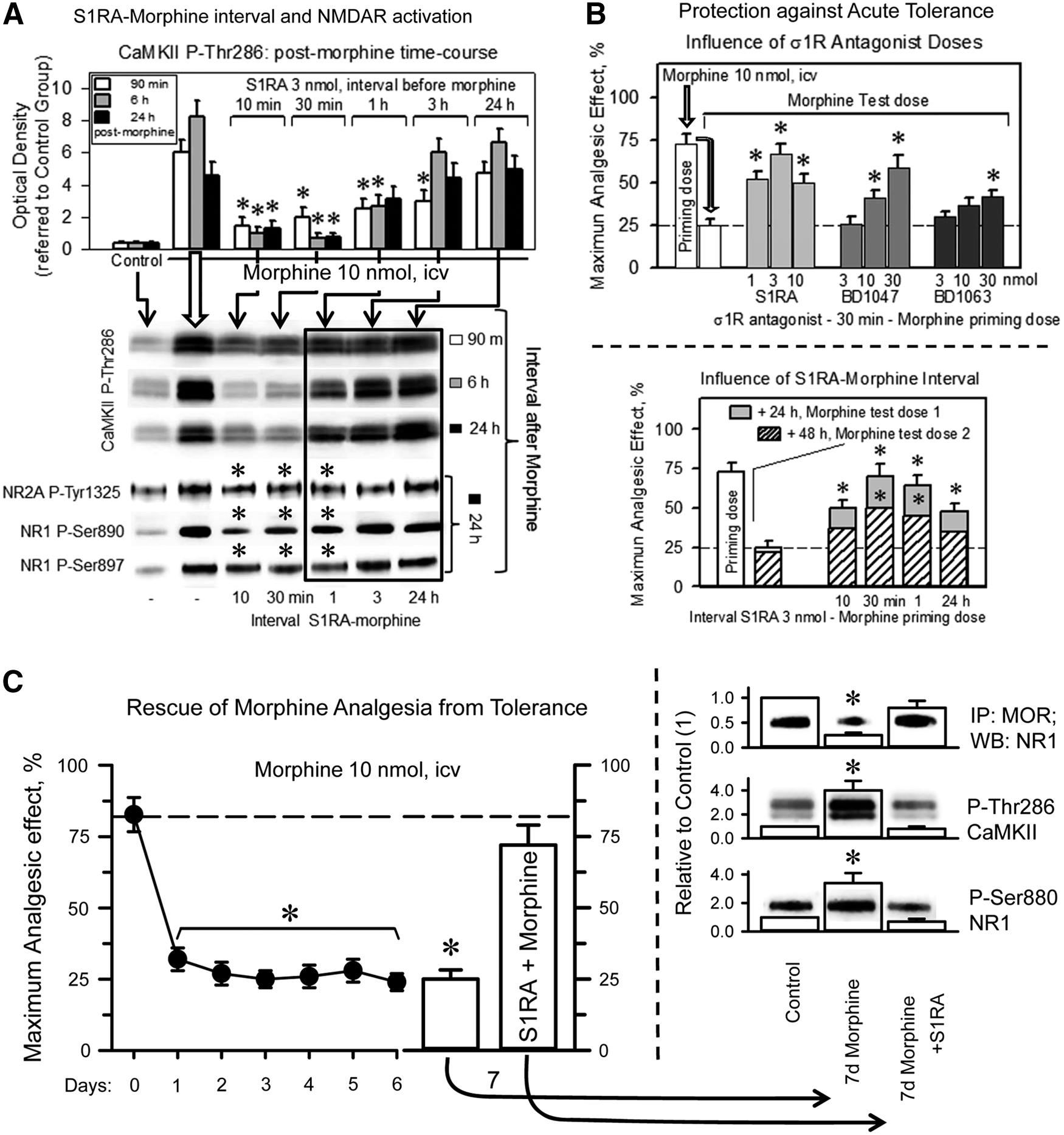
The capacity of S1RA to enhance morphine antinociception or alter MOR-induced NMDAR changes decreased as the S1RA-morphine interval increased (Fig. 6A). The σ1R antagonists S1RA, BD1047, and, to a minor extent, BD1063, when given before the first dose of morphine (priming dose), protected the analgesic effects of a subsequent dose of morphine (test dose) against acute tolerance (Fig. 6B). In this paradigm, the S1RA-morphine intervals of 30 min and 1 h were more successful than that of 10 min. S1RA icv-injected between 1 and 24 h before morphine neither enhanced morphine analgesia nor prevented the activation of MOR-coupled NMDARs (Figs. 5A and 6A); however, the σ1R antagonist still moderately decreased the antinociceptive acute tolerance (Fig. 6B). In a previous study, systemic S1RA was found to restore morphine analgesia in mice rendered tolerant by repeated subcutaneous administration of this opioid (64). Thus, we addressed whether S1RA was capable of such positive effects on mice that had received a daily icv dose of 10 nmol morphine for 7 days (Fig. 6C). Antinociception was evaluated at 30 min after the delivery of each morphine dose. The analgesic response diminished during the chronic morphine protocol; however, on day 7, the icv administration of 3 nmol S1RA 20 min before morphine enabled the opioid to produce an analgesic effect comparable to that of the first 10 nmol dose delivered on day 0 of the assay. At the molecular level, chronic morphine weakened the association of MORs with NMDAR NR1 subunits, whereas it enhanced the PKC-mediated phosphorylation of NR1 serine 890 and the activating autophosphorylation of CaMKII. These changes revealed an increase in NMDAR activity that returned to control levels when the chronic morphine-tolerant mice received a single icv dose of S1RA (Fig. 6C). Thus, the rescue of morphine analgesia from tolerance correlated with the deactivation of NMDARs.
The σ1Rs controls the interaction of NR1 subunits with NMDAR inhibitors and the redox-regulated HINT1 protein
The MOR C terminus interacts with HINT1, which carries the regulator of the G-protein signaling RGSZ2-nNOS complex (2, 15, 20). The RGSZ2 binds to the HINT1 protein in a zinc-independent manner and prevents the entrance of NMDAR NR1 subunits. Thus, activated MORs release the RGSZ2-negative control on nNOS and stimulate the production of NO to release zinc ions from RGSZ2 zinc domain (54) (Fig. 7A). This activity promotes the zinc-dependent binding of the redox sensor PKCγ to HINT1 proteins through zinc ions bridging HINT1 histidines with the cysteines of the PKCγ regulatory domain (15, 51), and both RGSZ2 and PKCγ bind simultaneously to the redox-regulated scaffold HINT1 protein (Fig. 7A). The MORs provide activated GαGTP subunits that bind to the RGS domain of RGSZ2, exposing the HINT1 protein to the effect of PKCγ (48) (Fig. 7A). The phosphorylation of HINT1 is the switch that releases RGSZ2, bringing NMDARs under MOR control. Within the MOR-HINT1-σ1R-NR1 protein assembly, the NMDAR displays low activity (63). PKCγ increases the σ1R-NR1 interaction and weakens that of HINT1 with NR1 subunits (Fig. 7A). This concatenated process situates NMDARs close to MORs to build together the negative feedback on opioid signaling.
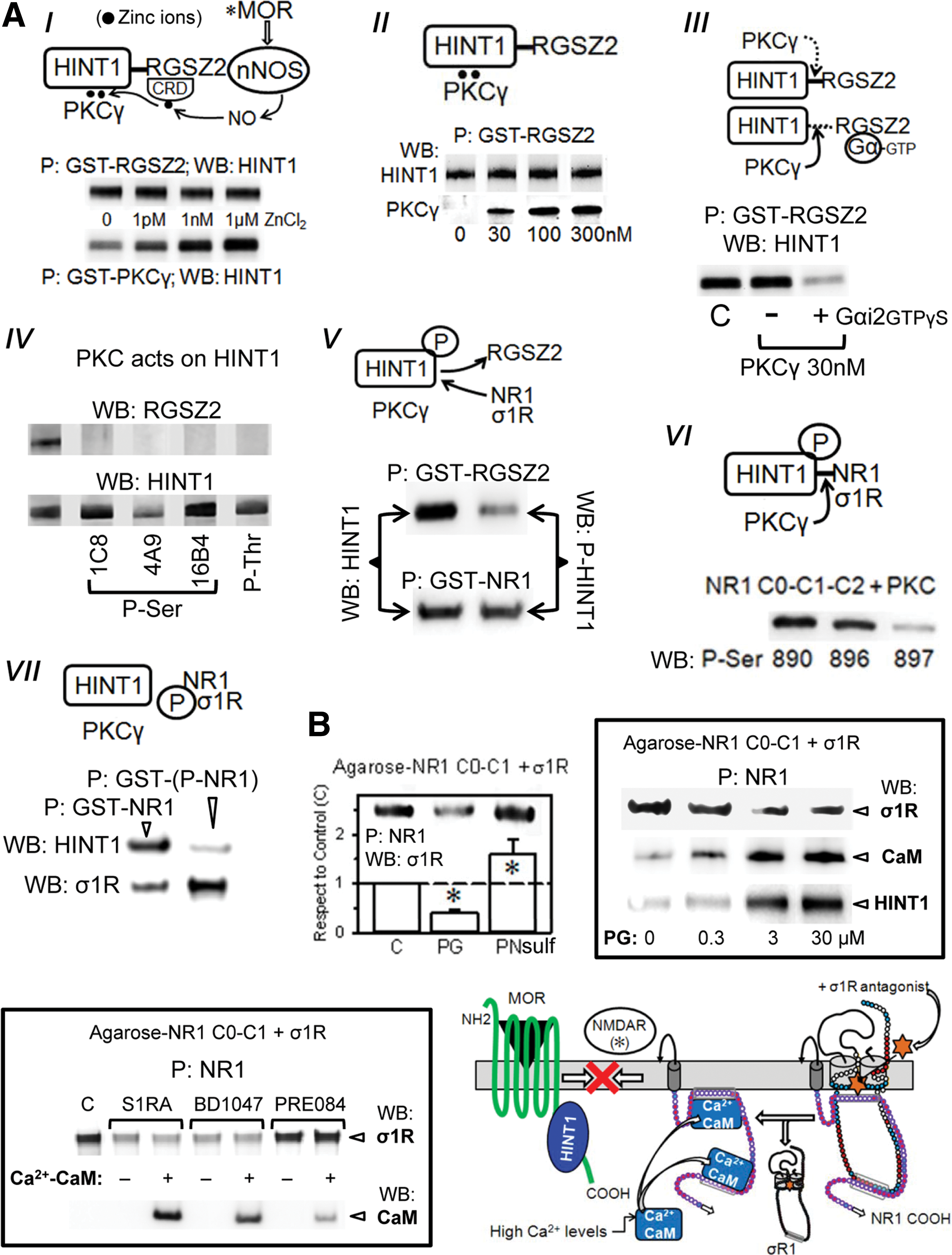
The binding of the σ1R to the NR1 subunit covers a region that is critical for the high-affinity binding of negative regulators of NMDAR function, such as HINT1 and Ca2+-CaM (11, 63). The in vitro assays indicated that pregnenolone sulfate enhanced and progesterone diminished the association of NR1 with the long isoform of the σ1R. On the reduction of σ1Rs binding to NR1 subunits, the NMDAR subunit was made available for CaM (in the presence of peptide 4 and 2.5 mM CaCl2) or HINT1 binding. This pattern was also observed for the exogenous ligands of the σ1R receptor, and while the antagonists S1RA and BD1047 stimulated CaM binding, the agonist PRE084 did not (Fig. 7B). In addition, the peptide interference assay showed that σ1Rcd peptides 7 and 10 could couple to the steroid-binding domain, reducing the σ1R-NR1 interaction. It is possible that these peptides and σ1R antagonists share a common mechanism that alters the structure of the σ1R and then diminishes its affinity for NR1 binding (Supplementary Fig. S2). Thus, by affecting the interaction of σ1Rs with NR1 subunits, σ1R antagonists regulate the activity of NMDARs.
The σ1R is essential for MOR-NMDAR cross-regulation
In σ1R−/− mice, the opioids showed an enhanced capacity to produce antinociception, and in these mice, the antinociceptive peak effect of an icv dose of 3 nmol morphine was comparable to that produced by 10 nmol morphine in wild-type mice (Fig. 8A). The administration of an agonist of NMDARs, NMDA, to wild-type mice significantly reduced the capacity of morphine to produce antinociception; however, NMDA failed to do so in σ1R−/− mice. Because MOR-NMDAR cross-regulation is a redox-regulated process that depends on NO and zinc metabolism (51, 54), in wild-type mice, the inhibition of NOS enhanced morphine antinociception and prevented NMDA from reducing the antinociceptive capacity of morphine; however, this approach was not effective in the σ1R−/− mice (Fig. 8A). At the molecular level, the inhibition of NOS prevented morphine from recruiting PKCγ in the MOR environment, and then opioid- and PKCγ-triggered activation of the NMDAR-CaMKII pathway, as well as the weakening of MOR-NR1 interaction were not observed (Supplementary Fig. S5). Synaptosomal membranes from mice that were treated in vivo with NOS inhibitors responded to SNAP, NO donor, or ZnCl2, recruiting PKCγ to the MOR environment. This effect was prevented by the co-incubation of the PAG membranes with TPEN, a zinc chelator. These observations indicate that the in vivo administration of NOS inhibitors specifically prevented morphine from stimulating NO production but left operative the NO-mediated removal of zinc ions from zinc fingers. The MOR-coupled HINT1 protein binds PKCγ through zinc ions (54). Accordingly, in PAG synaptosomes from HINT1−/− mice, SNAP or zinc ions did not stimulate PKCγ arrival at the MOR. The absence of σ1Rs greatly impaired the arrival of PKCγ to MORs, suggesting an altered MOR-HINT1 relationship, but some response to SNAP and zinc ions could still be observed (Fig. 8B).
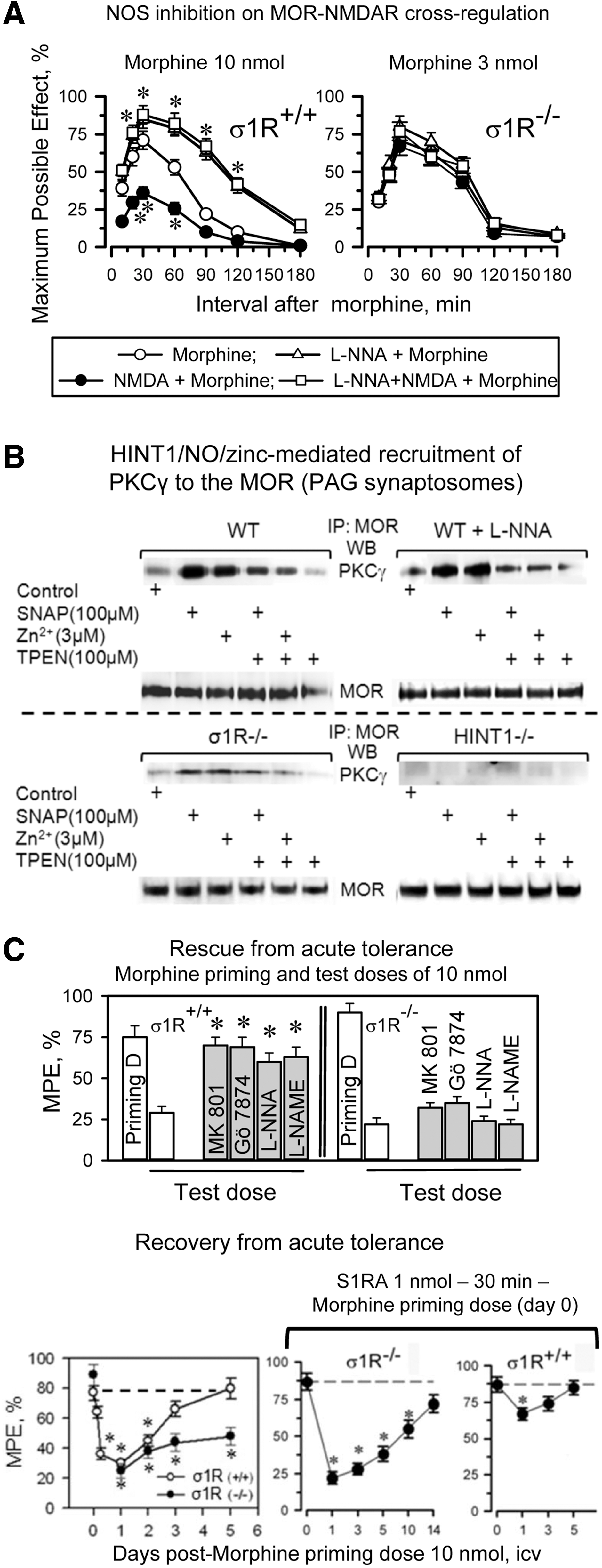
The analgesic acute tolerance that an icv-dose of morphine produces in wild-type mice can be reverted by NMDAR antagonists, PKC inhibitors, and NOS inhibitors. However, this effect could not be produced in σ1R−/− mice (Fig. 8C). In σ1R−/− mice, the NMDAR-mediated regulation of MOR signaling is impaired, and this control is apparently taken beyond the receptor level, most likely at the effectors. Thus, in σ1R−/− mice, this protective desensitization affects not only MORs but also other GPCRs, such as α2-adrenoceptors and cannabinoids (Supplementary Fig. S6A). Recovery from this state of heterologous tolerance requires longer periods than those that are produced by MOR-coupled NMDAR activity, and in σ1R−/− mice, the restoration of analgesic effects could not be accelerated σ1R antagonists (Fig. 8C).
The earlier observations indicate that the σ1R plays an essential role in bringing MOR signaling under the negative control of NMDAR activity. Thus, we analyzed whether the deletion of σ1R influences the MOR-HINT1-NR1 association. Indeed, in σ1R−/− mice, the MOR-NR1 association was greatly diminished, most likely because HINT1 primarily associates with NR1 subunits (Fig. 9A). Thus, in the absence of σ1Rs, the MOR-NMDAR cross-regulation is impaired, and morphine produced almost no recruitment of NMDAR activity, that is, CaMKII autophosphorylation or weakening of the residual MOR-NR1 association, molecular events that were not altered by the administration of S1RA (Fig. 9B and Supplementary Fig. S6B). Interestingly, the in vivo icv-injection of the recombinant σ1R restored the negative influence of NMDAR activation and diminished MOR-mediated morphine analgesia in σ1R−/− mice, and this effect was associated with a decrease in the binding of HINT1 proteins to NR1 subunits (Fig. 9C).
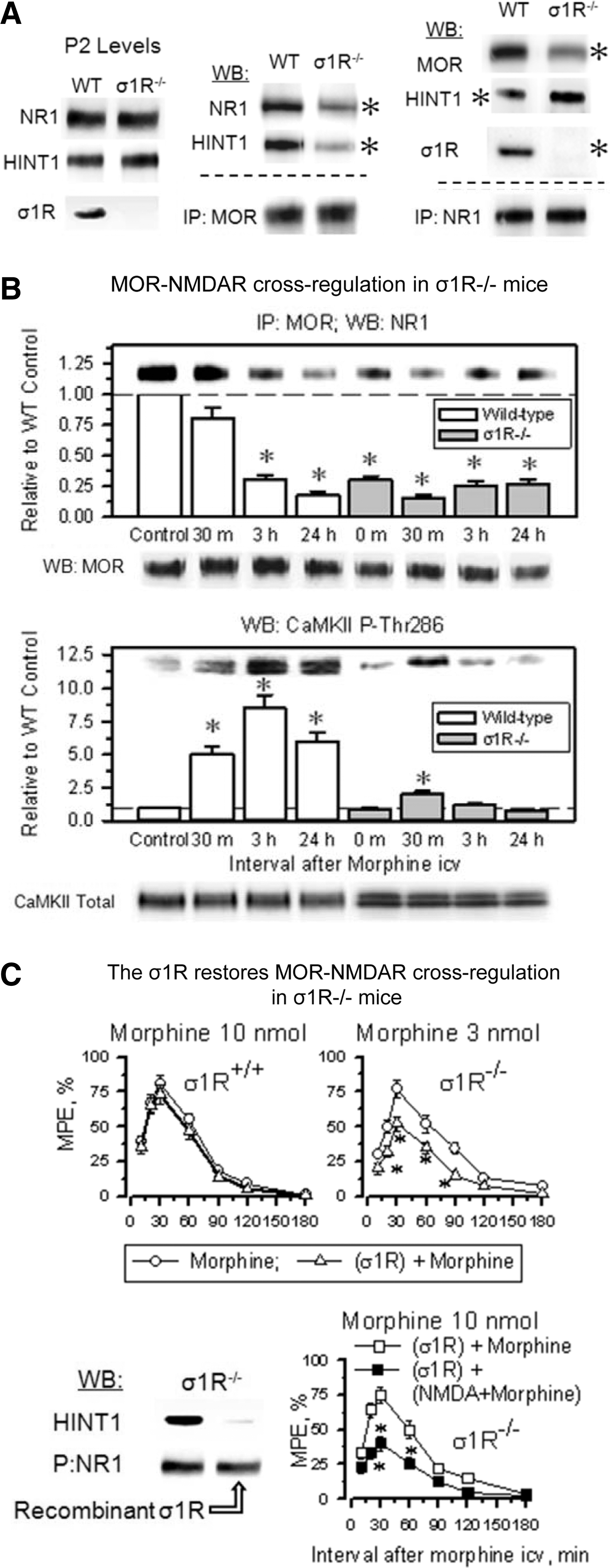
The σ1R prevents the swapping of HINT1 proteins between MOR and NR1 subunits
The administration of S1RA at 10 min or 30 min before morphine administration reduced the opioid-induced phosphorylation of the NR1/NR2 subunits and promoted an early re-association of NR1 subunits under MOR-HINT1-negative control, thereby limiting the activity of the NMDAR-CaMKII pathway (Figs. 5C, 6A and 10A). However, longer S1RA-morphine intervals, that is, 1, 3, and 24 h, brought about a delayed re-association between the morphine-activated MORs and NR1 subunits (Fig. 10A). Interestingly, in wild-type mice, morphine alone promoted certain transfer of HINT1 proteins from MORs to NR1 subunits (Fig. 10A and Supplementary Fig. S7), and S1RA given before morphine greatly increased this translocation. Thus, when S1RA was given from 1 to 24 h before morphine, the transfer of HINT1 proteins increased. Under these circumstances, PKC still acts on NR1 C1 S890/896/897 (Fig. 6A); however, the kinase is now with HINT1 at the NMDAR side and hardly reaches the MOR to promote those changes that are responsible for acute analgesic tolerance. Thus, the MOR-NMDAR physical and functional uncoupling was observed for HINT1−/− mice (48) and for σ1R−/− mice in this study.
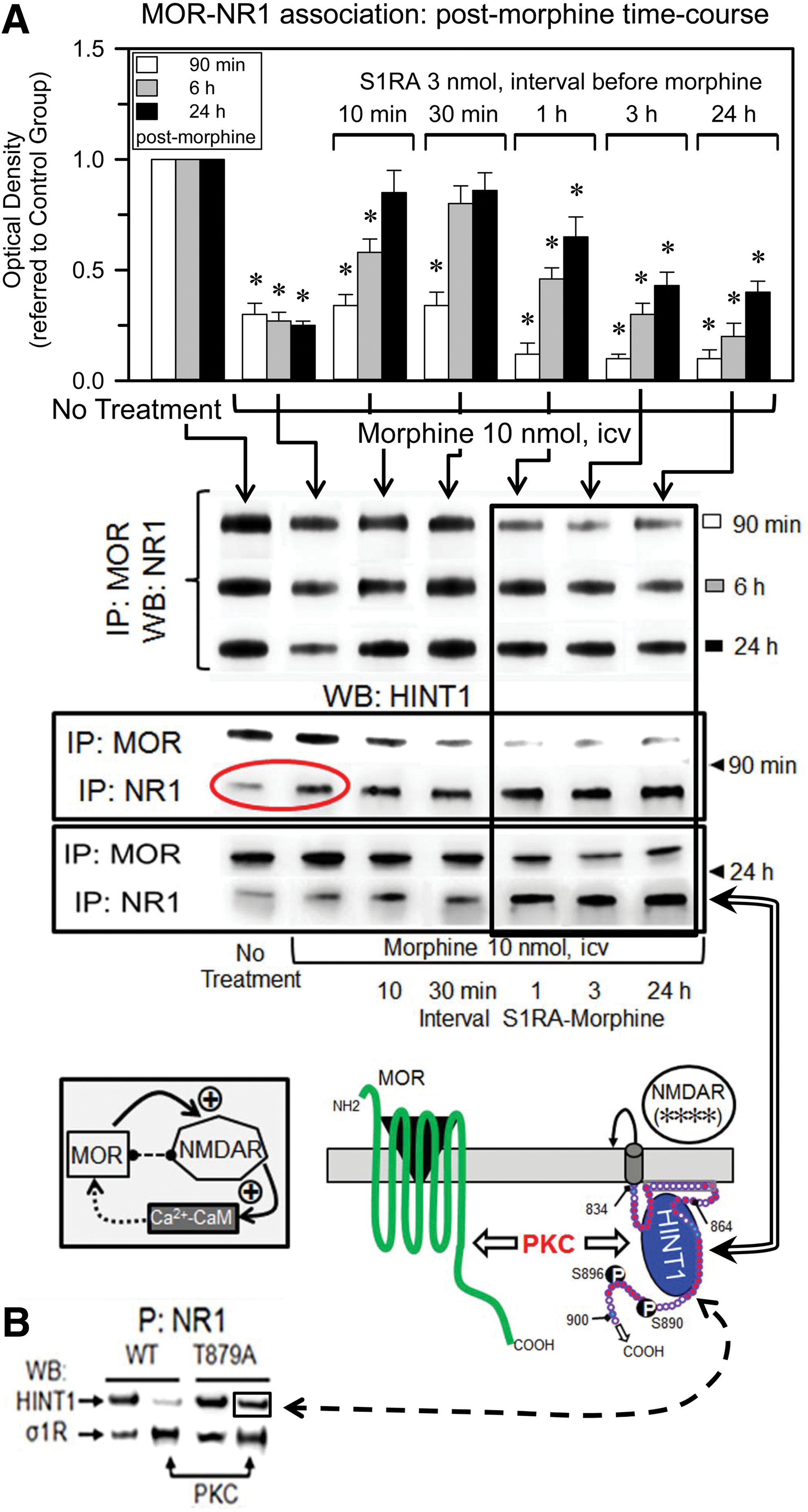
In the absence of morphine, σ1R antagonists disrupt σ1R-NR1 binding, which facilitates the association of NR1 subunits with HINT1 (Fig. 7A). Notably, several hours after S1RA administration and in the absence of morphine challenge, the basal association between MORs and NR1s increased (Supplementary Fig. S8). It is under these circumstances that morphine promotes the long-term transfer of HINT1 proteins to NMDARs (Fig. 10A). Regarding the NR1-HINT1 interaction, in vitro PKC acts on NR1 C1 T879 and Ser 890, 896 (60), thereby abolishing HINT1 binding (Fig. 7A). However, ex vivo data suggest that HINT1 binds to NR1 subunits in the presence of the serine phosphorylation of C1 segment, such as S890 (Figs. 5C and 10A). Within the S1RA-induced MOR-NMDAR complex and before morphine activates PKCγ, HINT1 could bind to the NR1 region of T879. Thus, we evaluated whether HINT1 binds to PKCγ-phosphorylated T879A NR1 subunits, and the results indicate that this binding is indeed possible (Fig. 10B). Interestingly, PKCγ increased the binding of NR1 subunits to σ1Rs, indicating that in the absence of σ1R antagonists, the phosphorylation of C1 segment could increase the ability of σ1Rs to remove MOR-HINT1 binding to NR1 subunits.
Discussion
The antinociceptive effects of opioids are related to their capacity to diminish calcium-dependent neurotransmitter release. Thus, to prevent opioids from producing an excessive reduction of neuronal excitability, NMDARs are recruited to the MOR environment, where they become activated to restrain opioid signaling (45, 61). For this control to be effective, the negative feedback must be proportional to the power of MOR signaling. The disruption of this balance could provoke a disproportionate NMDAR function, leading to cell damage or to an excessive MOR activation, which negatively affects cell homeostasis. The results of this study indicate that σ1Rs are necessary to establish the NMDAR control on MOR signaling by facilitating the MOR-HINT1-σ1R-NMDAR protein assembly. In this context, the σ1R cooperates with the HINT1 protein to equilibrate the negative influence of NMDARs to the strength of the MOR signals. This process is redox regulated, and NOS inhibition or the targeted deletion of either HINT1 or σ1R increases the MOR-mediated analgesic effects that elude regulation through NMDAR activity (20, 48, 57; and the present study). Interestingly, σ1R antagonists affect MOR function comparable to the effect of the removal of σ1R/HINT1 proteins, that is, the impairment of the capacity of MORs to trigger NMDAR-mediated restrain on opioid signaling. Notwithstanding, the experimental use of σ1R antagonists in wild-type mice was successful in increasing MOR-mediated analgesic effects without promoting tolerance. Therefore, this cellular mechanism can be exploited without triggering an excessive MOR function, which could recruit alternative systems to ultimately induce MOR hypofunction.
The σ1R as a ligand-regulated chaperone in its interaction with different proteins could adopt different conformations. Thus, in its particular binding to NMDAR NR1 subunits at the cell membrane, the cellular and in vitro assays suggest that the σ1R loop faces the cytosolic side (4, 44, 55). The NR1 C terminal sequence is a target of the calcium-binding protein CaM, which applies negative feedback to down-regulate the gating of the NMDAR calcium channel in response to high levels of cytosolic Ca2+ (11). The NR1 C1 amino-acid sequence is essentially a positively charged domain that includes the binding site for the σ1R loop that overlaps with that of the negatively charged HINT1 protein (34) and with the Ca2+-CaM binding domain. The σ1R loop binds specific regions on NR1 C0-C1 segments, while the σ1Rcd interacts with the NR1 C0 region. The σ1R has an SIM that could potentially bind a series of sumoylated proteins in the MOR environment, such as the RGSZ2 (16). The relevance of these SUMO-SIM regulatory interactions warrants further study.
In response to opioids, such as morphine, the σ1R is essential to bring the NMDARs under the control of MOR-HINT1 complexes. This process is exquisitely regulated, begins with the activation of MORs, and requires the redox-regulated HINT1 protein, the redox zinc switch RGSZ2 protein, and redox sensor proteins, such as PKC and Raf-1 (15, 47, 50, 51, 54). In the absence of MOR activation, the HINT1 protein carries the RGSZ2-nNOS assemblage that blocks MOR interaction with NMDARs. Opioids release this barrier and enable the arrival of the NMDAR to the MOR-HINT1 complex through NO and the zinc-dependent binding of PKCγ to the HINT1 protein (47, 50, 54). Within the MOR-NMDAR interaction, HINT1 binds the Ca2+-CaM site in the NR1 C1 segment and inhibits NMDAR activity, whereas the σ1R covers the C0 and the upper region of the C1 segment and reduces the overall affinity of NR1 subunits toward HINT1 proteins (60). To build up the negative feedback on opioid signaling, PKCγ phosphorylates the NR1 C1 Ca2+-CaM site to release the NMDAR from the HINT1 inhibitory influence. The NMDAR is now ready to collaborate with the MOR to increase PKCγ and CaMKII activities and make operative the control on opioid signaling (14). Whether the activity of the MORs reaches a certain threshold, PKCγ increases the formation of reactive oxygen species (ROS) by acting on NOX/NADPH, consolidating the long-term PKCγ activation that is required to regulate the Raf-1/MAPK cascade and enhancing NMDAR function. Thus, NADPH/ROS production and the sustained activation of PKCγ triggered by opioids are essential to develop and maintain NMDAR-mediated tolerance to the analgesic effects of morphine (10, 23).
In the absence of HINT1, the MOR-NMDAR cross-regulation is disrupted, and PKCγ cannot reach specific residues in the MOR C-terminal or the third internal loop, as required to desensitize responses to opioids and, consequently, the capacity of morphine to produce antinociception increases (6, 9, 48). In σ1R−/− mice, MORs and NMDARs are also molecularly and functionally disconnected because the HINT1 protein swaps MORs with NMDAR NR1 subunits. The cytosolic calcium levels regulate the permeation of Ca2+ ions through the NMDAR pore; thus, σ1R binds to the NR1 subunit in a calcium-dependent manner, blocking the entrance of Ca2+-CaM and also weakening that of HINT1 proteins (55). In the presence of Ca2+-CaM, the removal of σ1Rs facilitates its inhibitory binding to the NR1 subunit to reduce NMDAR calcium fluxes. However, reductions in the cytosolic calcium levels diminish the interaction of NR1 subunits with σ1Rs and the presence of Ca2+-CaM, thereby favoring NR1 binding to HINT1 proteins. Under these circumstances, MOR-coupled HINT1 binds tightly to Ca2+-CaM domains on NR1 subunits, producing some inhibition of NMDAR function (20, 63). Then, opioid-activated PKCγ binds to HINT1 and, acting on NR1 S890/896, separates NR1-HINT1-PKCγ from the MOR. The transfer of the PKCγ scaffold, the HINT1 protein, to NR1 subunits situates this kinase activity apart from MORs. Accordingly, the in vivo administration of recombinant σ1Rs to σ1R−/− mice removed the HINT1 binding from NR1 subunits and restored the negative influence of NMDARs on MOR-mediated antinociception.
The role of σ1Rs in the function of at NMDAR suggests that σ1R ligands could regulate the strength of MOR signaling. Indeed, it has been consistently reported that σ1R agonists reduce and σ1R antagonists enhance morphine analgesia (25, 39). In in vitro assays, agonists enhanced and antagonists reduced the association of σ1Rs with NR1 subunits; however, the reducing effects of σ1R agonists on opioid analgesia are not so evident. The release of endogenous σ1R agonists, most likely neurosteroids, could mask the effect of the exogenous. The selective σ1R antagonist S1RA induces a two-fold reduction in the analgesic ED50s of different opioids against thermal stimuli (64, 70). The effect of icv S1RA on morphine supraspinal analgesia depended on the interval between the administration of these drugs, showing an optimal effect at ∼10–30 min and nearly no effect after 1 h. When S1RA was administered shortly before morphine, the σ1R antagonist reduced the capacity of the opioid to activate the NMDAR/CaMKII pathway in the MOR environment. Consequently, the negative feedback of NMDARs on MOR signaling was impaired, and morphine analgesia increased from the initial intervals postopioid with almost no development of acute tolerance.
Because the NMDAR contributes to the recruitment and activation of PKCγ in the postsynapse (5), when used at intervals that reduced the morphine-induced activation of NMDARs, antagonists of σ1Rs also weakened that of PKCγ. The extension of the S1RA-morphine interval for 3 or 24 h did not potentiate morphine analgesia, but some protection against acute tolerance was still observed. The molecular data suggest that the administration of S1RA long before morphine treatment increases the capacity of the opioid to swap the HINT1 proteins from MORs to NR1 subunits. In σ1R−/− mice, the HINT1 protein, to the detriment of its association with MORs, was mostly found at NR1 subunits. This observation suggests that S1RA alters the conformation of the σ1R (7, 33) and then removes its regulatory binding to the NR1 subunit. At this point, two scenarios could account for the influence of the S1RA-morphine interval on morphine analgesia and NMDAR function: In the first scenario, Ca2+ levels are provided by morphine-activated PLCβ, and S1RA, by disrupting σ1R-NR1 binding, facilitates the access of Ca2+-CaM to reduce the activation that MOR-PKCγ promotes on NMDARs. The reduction of NMDAR function diminishes the cellular influx of Ca2+ and the local formation of Ca2+-CaM, and on clearance of the σ1R antagonist the σ1Rs bind again to NR1 subunits, enabling in the absence of HINT1-bound RGSZ2 the inhibitory re-association of NMDARs with MOR-HINT1 complexes. Thus, S1RA removes the negative influence of NMDARs on MOR function; enhances morphine analgesia in naïve mice; as well as recovers the effects of morphine in mice chronically treated with the opioid. The other scenario considers the absence of MOR activation, low Ca2+, and, consequently, low Ca2+-CaM levels; in these circumstances S1RA, by removing the σ1R, promotes the tight binding of RGSZ2-free MOR-HINT1 complexes to NR1 subunits. When morphine recruits PKCγ to the HINT1 protein, the absence of σ1Rs facilitates the swap of HINT1 from MORs to NR1 subunits. The activation of MORs increases, via PLCβ, calcium levels and that of Ca2+-CaM; however, PKCγ acting on NR1 S890 prevents Ca2+-CaM and HINT1 inhibitory binding to CaM binding site on the NR1 subunit while not abrogating that of HINT1 to the upstream region of the NR1 C1 segment. In these circumstances, when bound to HINT1-NR1, PKCγ cannot transmit its negative influence on the MOR cytosolic residues, and the morphine analgesic tolerance develops at a slower rate. The clearance of the σ1R antagonist S1RA, together with the increases in local calcium, allows σ1R to disrupt NR1-HINT1 interaction and re-establish σ1R-NR1 and MOR-NR1 complexes. Thus, the S1RA- and calcium-dependent status of the σ1R-NR1 association determines whether HINT1 swaps partners between MOR and NR1 subunits.
Morphine alone also promoted the transfer of HINT1 proteins, but this effect was limited in extent and reversible in the short term. In the absence of the σ1R antagonist, the presence of calcium rapidly restored σ1R-NR1 binding and the regulation of HINT1 through MORs. Thus, HINT1 swapping is under physiological regulation by endogenous agonists of the σ1R (19). Notably, the neuroactive steroid pregnenolone is released in response to drugs that act at GPCR-NMDAR complexes, such as cannabinoids and opioids (62). Pregnenolone displays agonist activity at σ1Rs, whereas its metabolite progesterone acts as an antagonist (37). However, pregnenolone is rapidly converted into progesterone, and its effects are mostly associated with the antagonism of σ1Rs (65). To delay this metabolism, pregnenolone is sulfated by pregnenolone sulfotransferase (SULT2B1a), and this enzyme is induced in response to events that are associated with NMDAR activity, such as AMPA receptors and NO (30).
Thus, σ1R agonists promote and antagonists reduce the activity of NMDARs, and these effects have been documented in electrophysiological studies as changes in NMDAR currents (36, 68) and variations in the PKC-mediated phosphorylation of NR1 subunits (26, 27). These profiles of neurosteroids on NMDAR activity when coupled with the opioid-mediated activation of MOR most likely contribute to the regulation of their analgesic effects. Thus, pregnenolone sulfate would promote NMDAR control of MOR activity, and if the strength of MOR signaling produces an NMDAR activity that compromises cell homeostasis, then pregnenolone could be converted into the σ1R antagonist progesterone to uncouple NMDARs from the activating influence of MORs.
In addition, in neuropathic pain where NMDARs become over activated, the antagonists of σ1Rs could disconnect the origin of such activation and/or promote the binding of negative regulators of NMDAR function, thereby demonstrating a therapeutic potential (8, 52, 53, 70).
Materials and Methods
In vitro interactions between recombinant proteins: pull-down of recombinant proteins and phosphorylation assays
A series of recombinant proteins were obtained (see Supplementary Materials and Methods section), and having demonstrated that the σ1R, NR1 C0-C1-C2, or HINT1 proteins did not bind to GST (100 nM, Z02039; GenScript Co., Piscataway, NJ) (55, 56), we determined the association of σ1R with NMDAR NR1 subunits. The NR1 C-terminal sequence C0-C1-C2 or mutated NR1 T879A was immobilized through covalent attachment to NHS-activated sepharose 4 fast flow (#17-0906-01; GE Healthcare, Barcelona, Spain) according to the manufacturer's instructions. The HINT1 protein (200 nM) and σ1R variants (100 nM) were incubated either alone (negative control) or together with the immobilized proteins in 400 μl of a buffer containing 50 mM Tris-HCl, pH 7.4 and 0.2% CHAPS and mixed by rotation for 30 min at RT. The influence of Ca2+ on this association was evaluated after the addition of 2.5 mM CaCl2 to the media. After incubation, the pellets were obtained by centrifugation, washed thrice, solubilized in 2×Laemmli buffer, and analyzed by Western blotting.
The regions involved in the interaction of NR1 C0-C1 with σ1R, HINT1, or Calmodulin were investigated through peptide interference of binding using 13 peptides (overlapping five residues) covering C0-C1 region of NR1. The interaction σ1R-NR1 was also analyzed with peptides mapping regions in the σ1R loop and cd (GenScript Co.). The purity of these peptides was higher than 95%.
PKCγ-mediated phosphorylation of the HINT1-RGSZ2 assembly. The Gα subunits were previously incubated with 10 μM GTPγS in 50 μl of 10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA buffer (Gi2GTPγS) and the unbound GTPγS was removed (centrifugal filter devices; 10 kDa nominal MW limit, Amicon Microcon YM-10#42407; Merck-Millipore, Barcelona, Spain). After 20 min, the HINT1 and RGSZ2 proteins were incorporated into HINT1-RGSZ2 complexes, then Gαi2GTPγS subunits were added to a final 100 nM, and incubation was continued for an additional period of 15 min. Subsequently, 30 nM PKCγ in 100 μl of kinase buffer (60 mM HEPES-NaOH [pH 7.5]; 3 mM MgCl2; 3 mM MnCl2; 3 μM Na-orthovanadate; 1 mM DTT and 250 μM ATP) was added to the mixture; the reaction was conducted at room temperature, and it was terminated after 20 min by the addition of the PKC inhibitor Gö7874 (Calbiochem; #365252) at a concentration of 5 μM.
The influence of SUMO1 on σ1R's association with the NR1 C0-C1-C2 subunits was determined through the preincubation of recombinant σ1R (100 nM) with agarose-SUMO1 for 30 min with rotation at room temperature in 150 μl of 50 mM Tris-HCl, pH 7.5, 2.5 mM CaCl2, and 0.2% CHAPS. After the removal of free σ1R, NR1 C0-C1-C2 (100 nM) was added to these protein mixtures and incubated for an additional 30 min. In a set of assays, free SUMO1 was added to preformed agarose-NR1-σ1R complexes (100 nM) for 30 min at RT. Agarose-SUMO1 pellets containing the bound proteins were obtained by centrifugation, washed thrice, solubilized in 2×Laemmli buffer, and analyzed by Western blotting.
Animals and evaluation of antinociception
Wild-type and homozygous (σ1R−/−) male sigma receptor knockout mice, backcrossed (N10 generation) onto a CD1 albino genetic background (Harlan Iberica, Barcelona, Spain), and homozygous (HINT1−/−), generously supplied by I.B. Weinstein/J.B. Wang, were used in this study (32, 51). The mice were housed and used in strict accordance with the European Community guidelines for the Care and Use of Laboratory Animals (Council Directive 86/609/EEC). All the procedures for handling and sacrificing the animals were approved by the Committee on Animal Care at CSIC. The animals were housed at 22°C under a 12 h light/dark cycle (lights on from 8 a.m. to 8 p.m.). Food and water were provided ad libitum. The response of the animals to nociceptive stimuli was assessed using the warm water (52°C) tail-flick test. The tail-flick analgesic test applies a thermal noxious stimulus to promote flicking of the mouse's tail, and opioids given by icv route increase the time elapsed between application of the stimulus and the flick. This response comprises a spinal reflex that is under facilitator drive by the brain stem nociceptive modulating network. After icv administration of opioids, the MORs in the ventral region of PAG play an important role in the supraspinal pathways that modulate spinal nociceptive processing (66, 67). Thus, icv morphine modulates descending serotoninergic and adrenergic systems and inhibits responses to nociceptive stimuli, including nociceptive withdrawal reflexes that are organized segmentally, such as the hind limb withdrawal and tail flick reflexes (18). In this analgesic test, the baseline latencies ranged from 1.7 to 2.0 s, and this parameter was not significantly affected by the σ1R ligands or the solvent used ethanol/Cremophor EL/physiological saline (1:1:18), 1.9±0.2 s (n=10). A cut-off time of 10 s was used to minimize the risk of tissue damage. Antinociception is expressed as a percentage of the maximum possible effect (MPE=100×[test latency−baseline latency]/[cut−off time-baseline latency]). Groups of 8 to 10 mice received a dose of morphine, and antinociception was assessed at different time intervals thereafter.
Production of acute and chronic tolerance to the antinociceptive effect of morphine
The development of morphine-induced acute opioid tolerance was monitored as described (15). Briefly, the animals received an icv priming dose of 10 nmol morphine in the right lateral ventricle. A 10 nmol dose produced 70%–80% of the MPE in the “tail-flick” test for analgesia. Controls were injected only with the opioid priming dose, whereas the experimental groups received the drug under study before the morphine priming dose or 24 h later for a few minutes (typically 30 min) before the morphine test dose of 10 nmol morphine. At this point, the analgesic effect of the 10 nmol priming dose had dissipated as evidenced by the restoration of baseline latencies in the tail-flick test. In some assays, the desensitizing effect of icv administration of a priming dose of 10 nmol morphine was addressed by an identical test dose of the opioid administered 24 and 48 h later.
For the study of the time interval required to recover analgesic response from acute tolerance, all the mice were icv-injected with 10 nmol morphine and divided into sub-groups of eight mice each. At increasing intervals postopioid priming dose, a different group was icv-injected with the morphine test dose of 10 nmol morphine. The antinociceptive effect of morphine was evaluated at 30 min postinjection, allowing time for the compound to reach its peak analgesic effect. Development of acute tolerance was ascertained through the comparison of the effects promoted by the morphine priming and test doses. Thus, data are expressed as the mean±SEM from groups of eight mice.
Mice were also pretreated with a schedule of chronic morphine administration (29). The mice were anesthetized by isoflurane inhalation before stereotaxically implanting a sterile cannula in the lateral ventricle (coordinates: 0.3 mm caudal, 1 mm lateral from bregma, and depth 2.3 mm) and fixing it to the skull with dental cement. The animals were allowed to recover for 4 days before the experiments commenced, and icv injections of 10 nmol morphine per mouse were administered daily for 7 consecutive days. The placement of the cannula was verified for each mouse, and only the data obtained from mice with a correctly inserted cannula were included in the statistical analysis.
Immunohistochemistry
A rabbit IgG labeling kit (Zenon Tricolor Rabbit IgG labeling Kit #1; Molecular Probes, Eugene, OR) was used for triple-labeling with the rabbit polyclonal IgGs against: σ1R (internal region 139–157, Invitrogen, Madrid, Spain, 42-3300; Alexa Fluor 488), NMDAR NR1 subunit (C-terminus, Merck-Millipore, Chemicon AB9864; Alexa Fluor 555), and MOR (C-terminal region, Abcam, Cambridge, United Kingdom, ab134054; Alexa Fluor 647). Mouse coronal brain sections with the PAG were incubated with the labeling complex overnight and examined by confocal microscopy (Leica TCS SP-5/LAS AF Lite Software; Microsystems, GmbH, Hohenstein-Ernstthal, Germany). Controls for immunohistochemistry were performed according to standard protocols (for further see details in Supplementary Materials and Methods section and Supplementary Fig. S3).
Immunoprecipitation and Western blotting
After icv morphine, groups of eight mice were killed at various intervals postopioid; PAG were obtained and processed to obtain the synaptosomal pellet as previously described (49), and used for MOR and NR1 immunoprecipitation and co-precipitation of HINT1, NR1, and MOR. This procedure has been described elsewhere (13, 17). Further details are provided in Supplementary Materials and Methods section.
Statistical analysis
ANOVA, followed by the Student–Newman–Keuls test (SigmaStat; SPSS Science Software, Erkrath, Germany) was performed, and significance was defined as p<0.05.
Footnotes
Acknowledgments
The authors would like to thank Concha Bailón and Gabriela de Alba for their excellent technical assistance. Cajal drawings were provided by the Legado Cajal, Cajal Institute, CSIC, Madrid, Spain. This research was supported by the “Ministerio de Sanidad y Consumo-Plan de Drogas 2011–2014” and the “Ministerio de Economía y Competividad (MINECO), SAF 2012-34991.”
Author Disclosure Statement
The authors declare that, excluding income received from their primary employer “Ministerio de Economía y Competitividad, MINECO,” no financial support or compensation has been received from any individual or corporate entity over the past 3 years for research or professional services and that there are no personal financial holdings that could be perceived as constituting a potential conflict of interest. M.M. and J.M.V. are full-time employees at Esteve.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.