
Abstract
Introduction
C
Novel findings of this study include that the demonstration of (i) small ubiquitin-like modifier type 1 (SUMO-1) expression blocks hypertrophic responses in isolated cardiomyocytes stimulated by phenylephrine; (ii) SUMO-1 gene transfer abrogates the hypertrophic response to pressure overload in vivo; and (iii) SUMO-1 expression protects SERCA2a from oxidative stress. These observations suggest that post-translational modifications of SERCA2a caused by the toxic environment of the hypertrophied and failing myocardium can be prevented by the expression of SUMO-1.
Impaired cardiac isoform of sarcoplasmic reticulum calcium ATPase (SERCA2a) function underlies many of the effects of pressure overload-induced hypertrophy on cardiomyocyte performance, and it has been shown to be critical in the progression of compensatory hypertrophy to heart failure (28, 49, 51). Our group along with other investigators has shown that increasing the expression of SERCA2a by gene transfer in various animal models of heart failure improves cardiac function. Following a long line of investigation, SERCA2a gene therapy is now undergoing testing in human clinical trials (19, 29, 70). SERCA2a protein is also sensitive to oxidative stress. Hydrogen peroxide and hydroxyl radicals directly inhibit ATPase activity by interfering with ATP binding, thereby impairing SR calcium pump rate (67). Furthermore, oxidative modifications of SERCA2 have also been identified. Increased tyrosine nitration of SERCA2a is positively correlated with aging (68). In addition, cystein oxidation of SERCA2 contributes to the decrease in its activity that leads to impaired calcium handling and relaxation in the aging heart (55).
We have recently found that the levels and activity of SERCA2a in cardiomyocytes are modulated in parallel with the levels of a cytoplasmic protein, small ubiquitin-like modifier type 1 (SUMO-1). We also found that SERCA2a is SUMOylated at lysine residues 480 and 585 and that this post-translational modification is responsible for stabilizing SERCA2a as well as for enhancing its activity. Furthermore, we showed that increasing SUMO-1 levels led to restoration of SERCA2a levels, improved hemodynamic performance, and reduced mortality in a murine model of heart failure (34). More recently, we demonstrated that SUMO-1 gene transfer and its combination with SERCA2a led to a reversal of heart failure in a porcine model of ischemia-induced heart failure (63).
It is well known that global increases in protein SUMOylation occur in response to cellular stress. Recent studies have also shown an intimate relationship between SUMO and oxidative stress. Oxidative stress can affect the conjugation profile of SUMO to a target protein in a concentration- and time-dependent fashion (5, 47). In human neuronal cells, overexpression of SUMO increases cell survival in the presence of oxygen/glucose deprivation, suggesting that SUMO is a component of ischemic tolerance (40). However, it is unknown what happens to the expression of SUMO-1 in the setting of compensated hypertrophy and the transition from hypertrophy to heart failure and whether modulating SUMO-1 in this setting alters the phenotype. In this study, we examined the effects of overexpressing SUMO-1 on the hypertrophic response both in vitro and in vivo. Since oxidative stress is a critical component of cardiac hypertrophy and heart failure, the effects of SUMO-1 gene transfer on oxidative stress are also examined.
Results
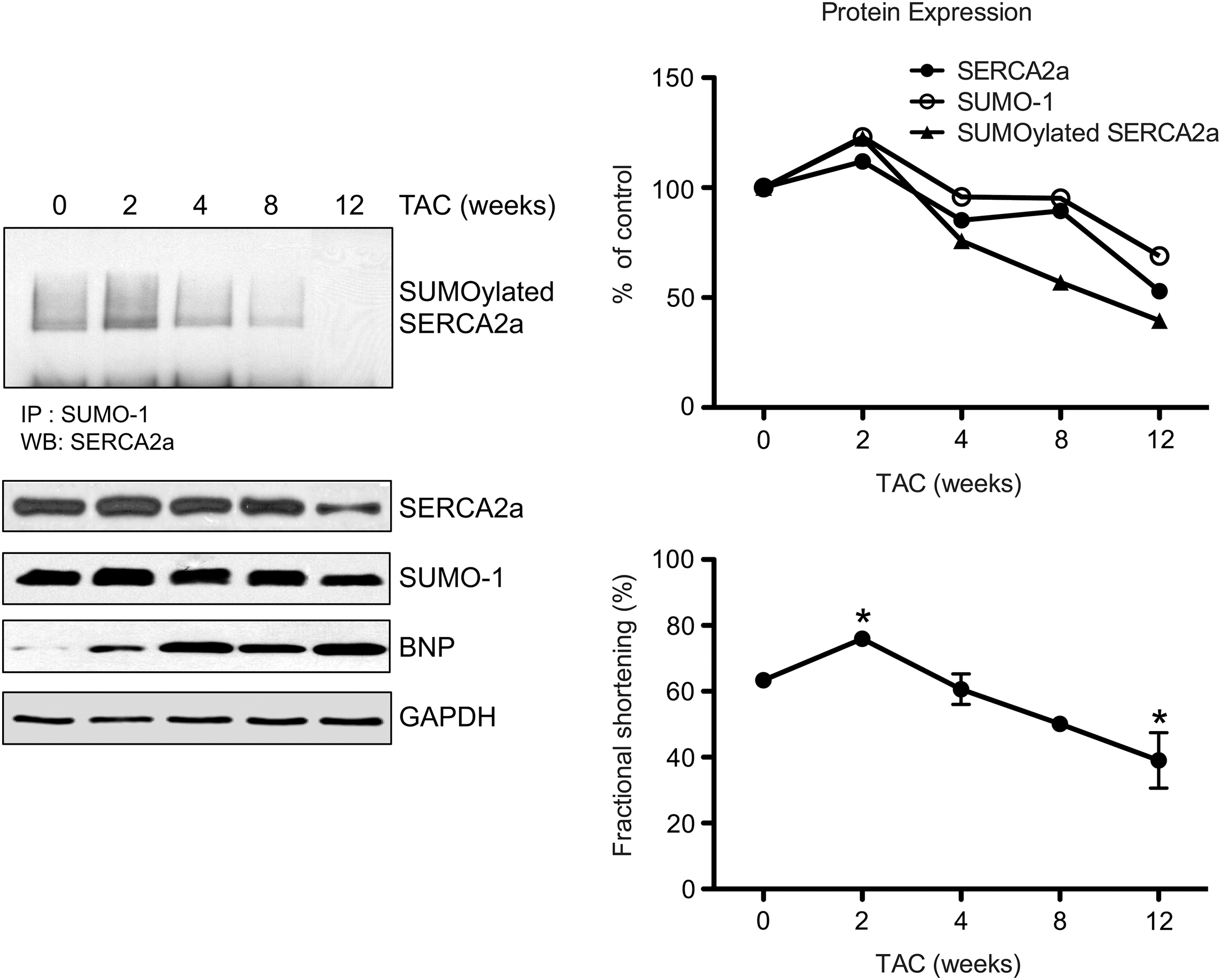
SUMO-1 is differentially expressed throughout the progress of heart failure
To gain insights into a possible role of SUMOylation in the stress response during the progression to heart failure, SUMO-1 expression levels were quantified in transverse aortic constriction (TAC) mice at different time points and related to cardiac function. Western blot analysis showed that protein levels of both SUMO-1 and SERCA2a were altered by TAC-induced pressure overload (Fig. 1). Both SUMO-1 and SERCA2a protein levels were increased in mice at 2 weeks of TAC (23% in SUMO-1 level and 12% in SERCA2a level, respectively, vs. no TAC control), when brain natriuretic peptide (BNP) level was significantly increased (1.9-fold, vs. no TAC control) and systolic function was significantly increased during compensatory hypertrophy (fractional shortening [FS]; 75.9% in 2 weeks TAC vs. 63.3% in baseline), and declined thereafter. At 12 weeks of TAC when LV function was severely decreased (FS; 39% vs. 63.3% in baseline, p<0.05), SUMO-1 and SERCA2a protein expression was decreased (32% decreased in SUMO-1 level and 48% decreased in SERCA2a level compared with baseline). TAC also altered SUMOylated SERCA2a levels in a time-dependent manner. SUMOylation status of SERCA2a was increased at 2 weeks of TAC but greatly decreased at 12 weeks of TAC. These data suggested that loss of SUMO-1 levels contributes to the decreased SUMOylation of SERCA2a during the transition to heart failure.
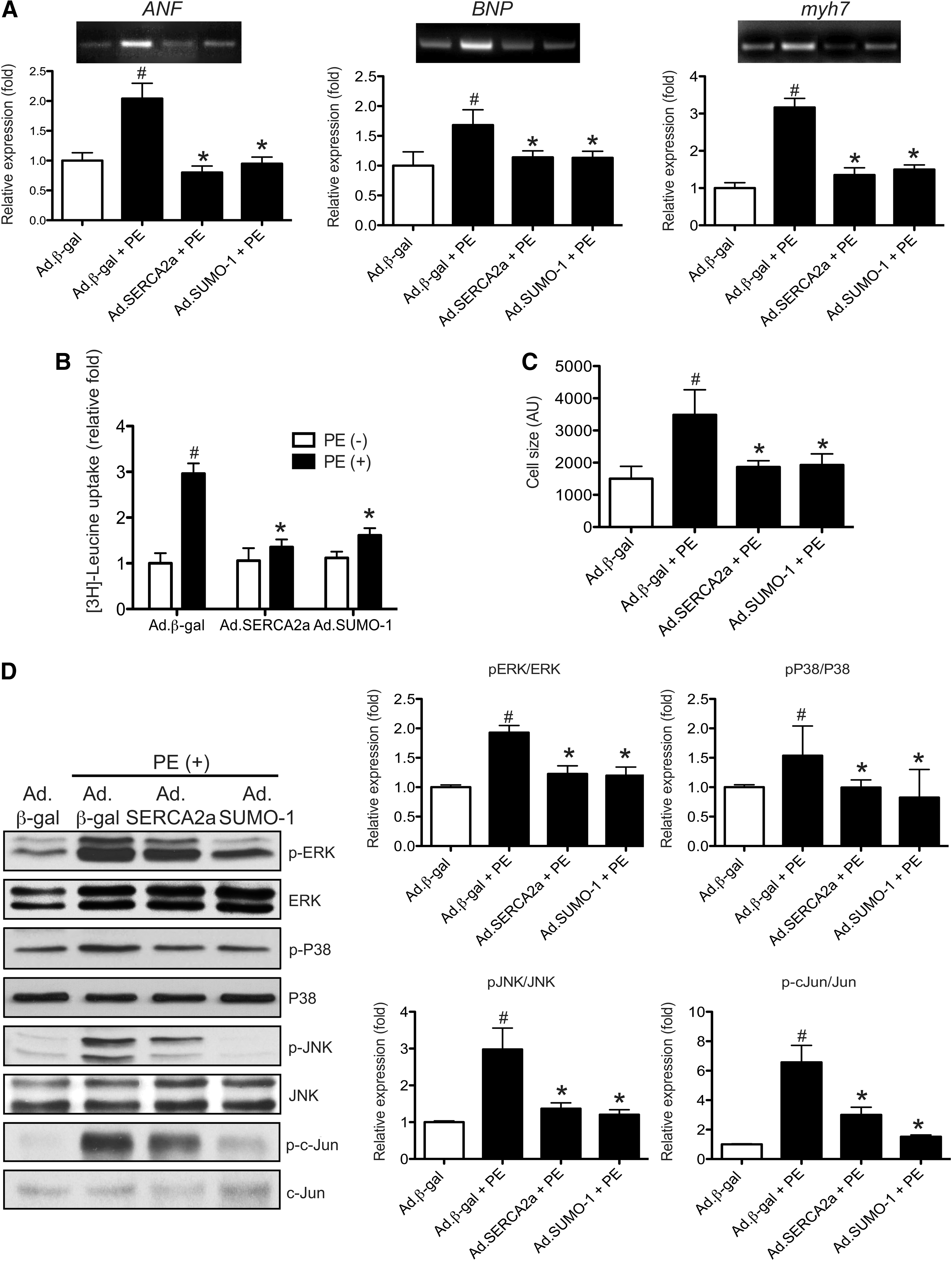
SUMO-1 overexpression inhibits the phenylephrine-induced hypertrophic response
First, the ability of SUMO-1 to inhibit cardiac hypertrophy was evaluated in vitro. The cultured neonatal rat cardiomyocytes were infected with either Ad.SUMO-1 or Ad.SERCA2a or Ad.β-gal. At 24 h after infection, the cardiomyocytes were further stimulated with 100 μM phenylephrine (PE), an agonist for cardiac hypertrophy, for 24 h. PE-treated cardiomyocytes showed a significantly (p<0.05) increased in mRNA level of fetal genes such as ANF (2-fold, vs. no PE), BNP (1.6-fold, vs. no PE), and myh7 (3.1-fold, vs. no PE) in the Ad.β-gal-infected cardiomyocytes. In contrast, no significant PE-induced increases in these typical markers for cardiac hypertrophy were observed with either Ad.SUMO-1- or Ad.SERCA2a-infected cardiomyocytes (Fig. 2A). Another feature of the hypertrophic response of cardiomyocytes is a pronounced increase in protein synthesis. The increased protein synthesis induced by PE treatment (2.9-fold, vs. no PE), as determined by 3H-leucine incorporation, was significantly (p<0.05) suppressed by SUMO-1 overexpression (0.54-fold, vs. Ad.β-gal control) and SERCA2a overexpression (0.45-fold, vs. Ad.β-gal control) (Fig. 2B). Stimulation with PE results in an increase in cell size of cardiomyocytes infected with Ad.β-gal. In contrast, no significant increase in cell size was observed in cardiomyocytes infected with either Ad.SUMO-1 or Ad.SERCA2a after PE treatment (Fig. 2C). Interestingly, PE treatment caused a 57% decrease in SERCA2a protein expression, whereas SERCA2a protein level was maintained in SUMO-1 overexpressing cells compared with Ad.β-gal overexpressing cells (Supplementary Fig. S1; Supplementary Data are available online at
SUMO-1 pretreatment prevents pressure overload-induced cardiac hypertrophy
To determine whether SUMO-1 overexpression inhibits hypertrophic response in vivo, SUMO-1 was pretreated by adeno-associated vector type 9 (AAV9)-mediated gene delivery. AAV9 is known to be a cardiotropic vector. AAV9.SUMO-1 was injected into B6C3/F1 male mice through the tail vein at a dose of 5×1010 viral genomes per mouse (vg/mice). Mice injected with same doses of either AAV9.SERCA2a or AAV9.GFP viruses were used as positive and negative control groups, respectively. Six weeks after the injection, mice were subjected to TAC operation. Cardiac function was accessed by echocardiography at day 0 (baseline), 2, and 12 weeks after TAC operation (Fig. 3A).
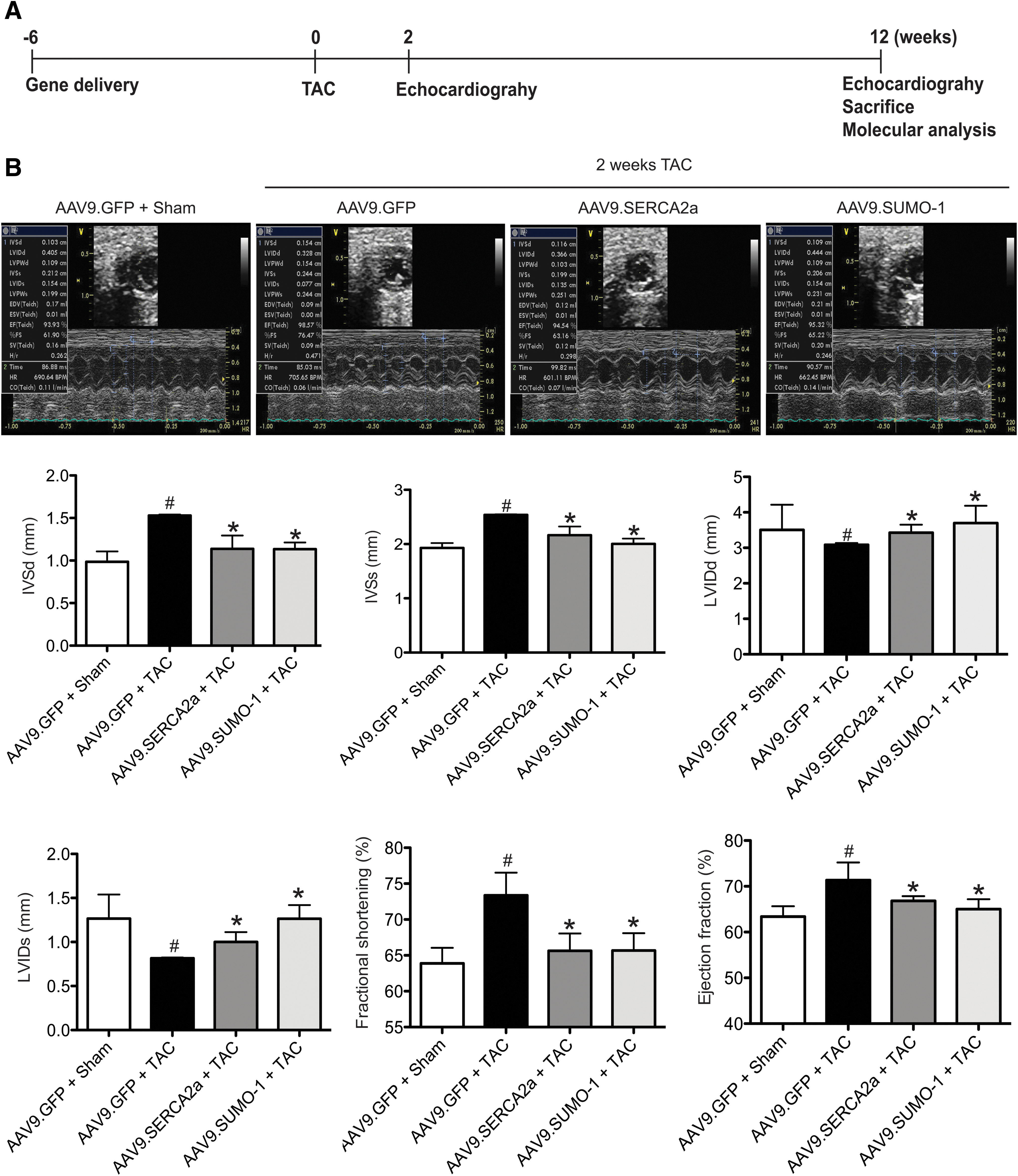
There were no significant differences in LV function and LV dimension between the gene-delivered groups before TAC (Table 1). By contrast, at 2 weeks of TAC, AAV9.GFP preinjected mice exhibited hypertrophic phenotypes with significant increases in interventricular septal thickness in diastole (IVSd; p<0.05, vs. sham), interventricular septal thickness in systole (IVSs; p<0.05, vs. sham), left ventricular posterior wall thickness in diastole (LVPWd; p<0.05, vs. sham), left ventricular posterior wall thickness in systole (LVPWs, p<0.05 vs. sham), and LV FS (p<0.05, vs. sham) and significant decreases in left ventricular internal diameter in diastole (LVIDd; p<0.05, vs. sham) and left ventricular internal diameter in systole (LVIDs; p<0.05, vs. sham). SUMO-1 pretreatment, however, significantly suppressed these hypertrophic phenotypes. The increase in LV end-diastolic and systolic wall thickness and ventricular posterior wall thickness in diastole caused by TAC was significantly preserved in AAV9.SUMO-1 preinjected mice compared with AAV9.GFP preinjected control (p<0.05). Consistently, LV FS and ejection fraction (EF) was maintained in SUMO-1 pretreated mice (FS; 65.58% in 2 weeks' TAC, vs. 64.23% in sham and EF; 95.52% in TAC, vs. 95% in 2 weeks' sham) (Fig. 3B and Table 2). Similar results were obtained in the AAV9.SERCA2a preinjected group. TAC-induced hypertrophy was strongly prevented by the pretreatment with SERCA2a. These data indicated that SUMO-1 pretreatment abrogated the development of left ventricular hypertrophy induced by pressure overload in vivo.
All data represent the mean value±the SD of cardiac functional parameters.
EF, ejection fraction; FS, fractional shortening; HR, heart rate; IVSd, interventricular septal thickness in diastole; IVSs, interventricular septal thickness in systole; LVIDd, left ventricular internal diameter in diastole; LVIDs, left ventricular internal diameter in systole; LVPWd, left ventricular posterior wall thickness in diastole; LVPWs, left ventricular posterior wall thickness in systole.
All data represent the mean value±the SD of cardiac functional parameters.
p<0.05 compared with the sham groups.
p<0.05 compared with the AAV9.GFP control; significant effect of TAC.
TAC, transverse aortic constriction.
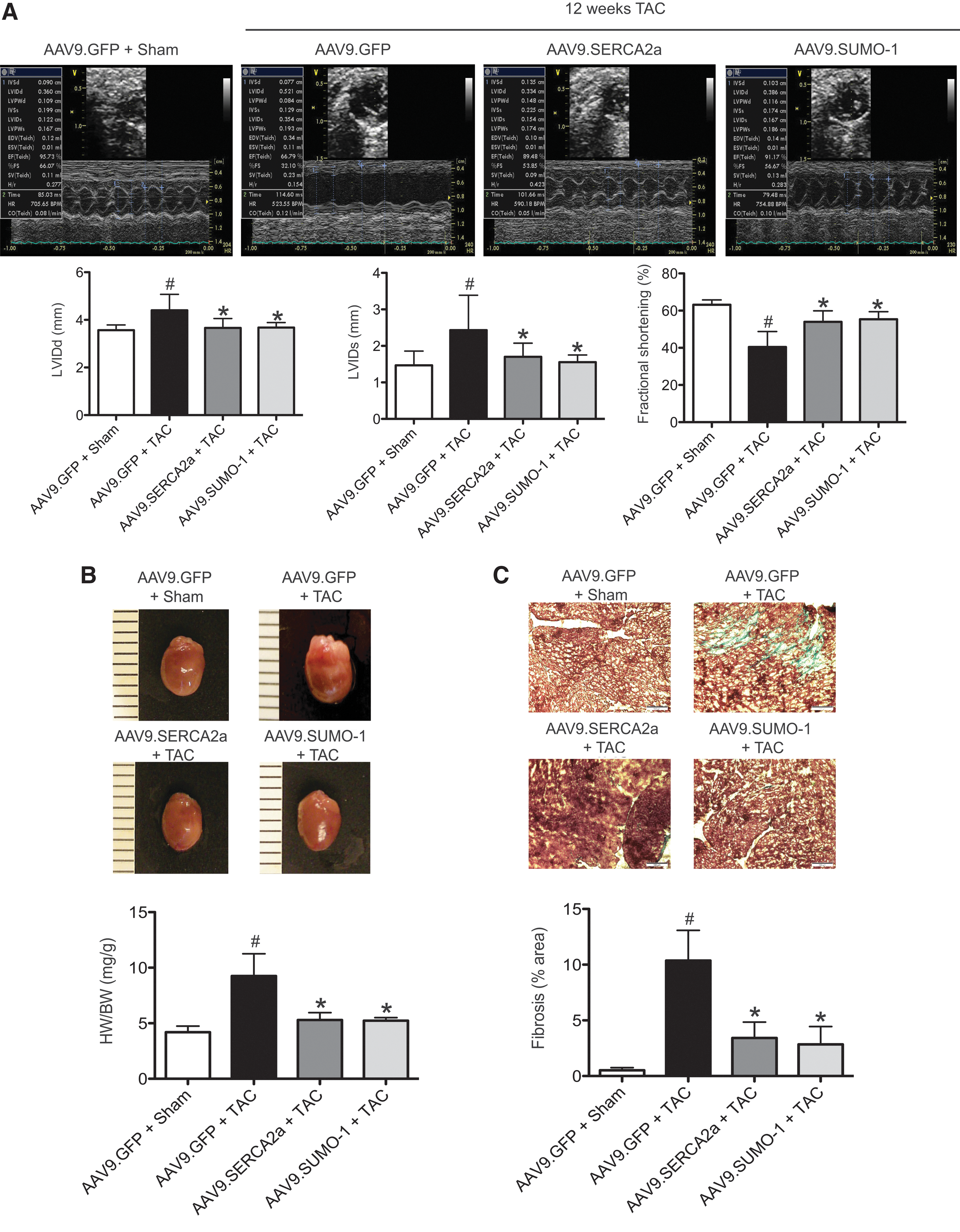
SUMO-1 pretreatment prevents development of heart failure
The beneficial effects of SUMO-1 pretreatment were also observed in the transition from hypertrophy to failure. All the remaining viable mice were subjected to echocardiography to assess the progression of heart failure. As expected, the prolonged pressure overload induced the typical phenotypes of heart failure. Increased LV wall thickness was significantly reduced at 12 weeks of TAC (p<0.05, vs. 2 weeks of TAC) (Table 3). The degree of LV dilation assessed as LV end-diastolic and systolic diameters was significantly increased in AAV9.GFP preinjected mice at 12 weeks of TAC (p<0.05, vs. 2 weeks of TAC and p<0.05, vs. sham). Systolic function was significantly decreased in AAV9.GFP preinjected mice as demonstrated by decreases of LV FS and EF after TAC, as compared with the sham (p<0.05) and 2 weeks of TAC (p<0.05) (Fig. 4A and Table 3). However, development of heart failure was significantly ameliorated in AAV9.SUMO-1 preinjected mice compared with the AAV9.GFP preinjected control group (p<0.05). The LV chamber cavity was significantly maintained in the AAV9.SUMO-1 group (p<0.05). LV FS and EF were significantly enhanced in AAV9.SUMO-1 pretreated mice compared with the green fluorescent protein (GFP) preinjected control animals (p<0.05) (Fig. 4A and Table 3). At 12 weeks of TAC, the heart weight-to-body weight ratio was significantly higher in the AAV9.GFP preinjected group (ratio 9.25±2.0) than both AAV9.SUMO-1 (ratio 5.29±0.67) and AAV9.SERCA2a (ratio 5.23±0.27) pretreated groups (Fig. 4B). All animals pretreated with AAV9.SUMO-1 retained cardiac function to the same degree as the mice injected with AAV9.SERCA2a. No statistically significant changes in echocardiographical parameters were observed in sham-operated groups (Table 3). The TAC operation significantly increased fibrosis in the AAV9.GFP control group compared with sham (2.1%±1.2% vs. 11.6%±3.2%), but this increase was reduced in the TAC mice receiving either AAV9.SUMO-1 or AAV9.SERCA2a (Fig. 4C). As expected, the anti-hypertrophic effects of SUMO-1 prolong survival in animals with TAC-induced heart failure (Supplementary Fig. S3).
All data represent the mean value±the SD of cardiac functional parameters.
p<0.05 compared with the 2 weeks' TAC.
p<0.05, compared with the AAV9.GFP control; Significant effect of TAC.
p<0.05 compared with the sham groups.
SUMO-1 overexpression protects SERCA2a function from oxidative stress
Oxidative stress plays an important role in the transition of cardiac hypertrophy to heart failure. SUMO-1 has also been demonstrated to play a role in stress responses, as its expression increases during hypoxia (59). We postulated that oxidative stress could inhibit SERCA2a function and SERCA2a SUMOylation status. To investigate the effects of SUMO-1 on oxidative stress-induced SERCA2a dysfunction, SERCA2a SUMOylation status, SERCA2a oxidation, and SERCA2a activity were determined in cardiomyocytes stimulated with H2O2. Isolated neonatal cardiomyocytes were infected with adenoviruses carrying SUMO-1 or β-gal genes for 24 h and then, the cells were exposed to H2O2 for 6 h. H2O2 reduced SERCA2a SUMOylation as well as SERCA2a's ATPase activity (Fig. 5A), but endogenous levels of SERCA2a protein were not significantly changed. Furthermore, nitrosylated tyrosine residues of SERCA2a, which serve as a maker of oxidative post-translational modification, were significantly increased in H2O2-treated cardiomyocytes compared with no stimulation. However, pretreatment of SUMO-1 showed enhanced SERCA2a SUMOylation, reduced nitrosylation, and a 25% inhibited H2O2-mediated reduction of SERCA2a enzymatic activity (Fig. 5A).
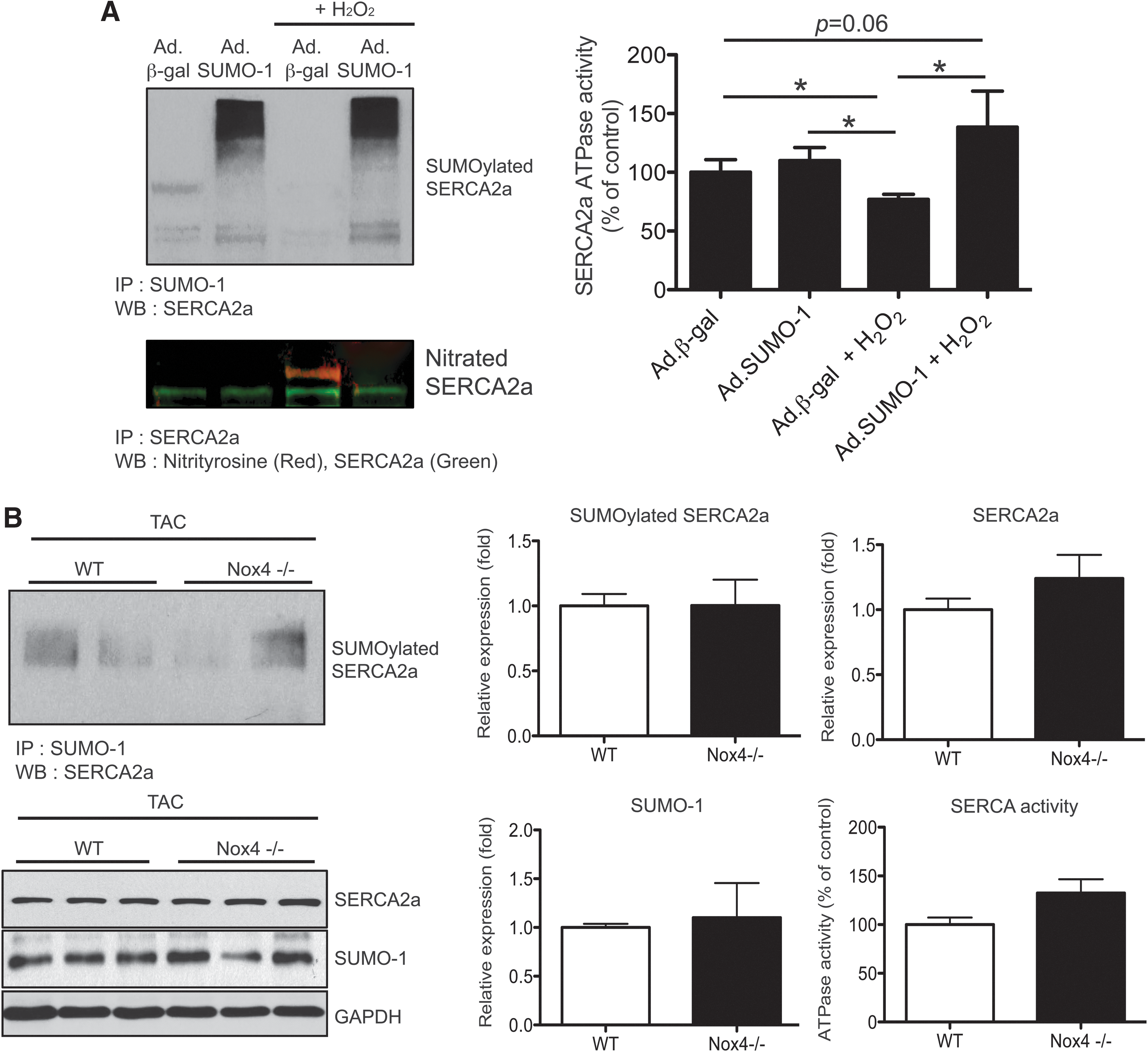
To further define the mechanism underlying the enhanced antioxidant effects associated with SUMO-1, we next examined the effect of nicotinamide adenine dinucleotide phosphate oxidase 4 (Nox4) on SUMO-1 protein and SERCA2a SUMOylation during hypertrophy. Nox4 is a major enzyme that produces H2O2 in cardiomyocytes. Moreover, cardiac Nox4 expression level is increased in response to the PE and pressure overload is known to result in an increase oxidative stress and apoptosis. Interestingly, we observed that PE-induced upregulation of Nox4 expression was inhibited in SUMO-1 overexpressing cardiomyocytes (Supplementary Fig. S4). On the other hand, Western blot analyses showed that SUMO-1 expression, SERCA2a SUMOylation, and SERCA2a expression were preserved in the hearts isolated from cardiac-specific Nox4 knockout mice with TAC compared with wild-type mice with TAC (Fig. 5B). Furthermore, SERCA2a activity protected from TAC operation in Nox4 is deficient in mice (Fig. 5C). Taken together, these data suggest a protective role of SUMO-1 in oxidative stress-induced SERCA2a dysfunction and Nox4-derived oxidative stress may influence SERCA2a SUMOylation and protein expression as well as its activity.
SUMO-1 pretreatment reduces the TAC-induced increase in oxidative stress and reduction of SERCA2a level
In an attempt to gain insights into the mechanisms by which overexpression of SUMO-1 confers resistance to oxidative stress, we examined whether TAC induces generation of ROS. ROS levels in myocardial tissues were measured by dichlorofluorescein fluorescence (Fig. 6A). There was a 1.8-fold increase in ROS production in TAC hearts preinjected with AAV9.GFP compared with sham control. However, SUMO-1 overexpression significantly prevented TAC-induced ROS generation. To further examine the extent of oxidative stress in the heart, the level of 4-hydroxynonenal (HNE)-modified protein was assessed in cardiac tissues using immunohistochemical staining with anti-4-HNE antibody. 4-HNE-modified protein adducts in the hearts of TAC mice were significantly increased compared with sham-operated controls. However, TAC-induced HNE formation was significantly decreased in the hearts overexpressing SUMO-1 (Fig. 6B). Furthermore, TAC animals exhibited significant increases in the level of nitrotyrosine on SERCA2a and decreased SERCA2a SUMOylation. In contrast, SUMO-1 overexpressing hearts showed a decrease in SERCA2a oxidation and increases in the level of SUMOylated SERCA2a (Fig. 6C). SUMO-1 pretreatment also resulted in a strong restoration of SERCA2a protein levels at 12 weeks of TAC mice (Supplementary Fig. S5). These results point to the possibility that SUMO-1 overexpression contributes to diminished oxidative stress by reducing ROS levels.
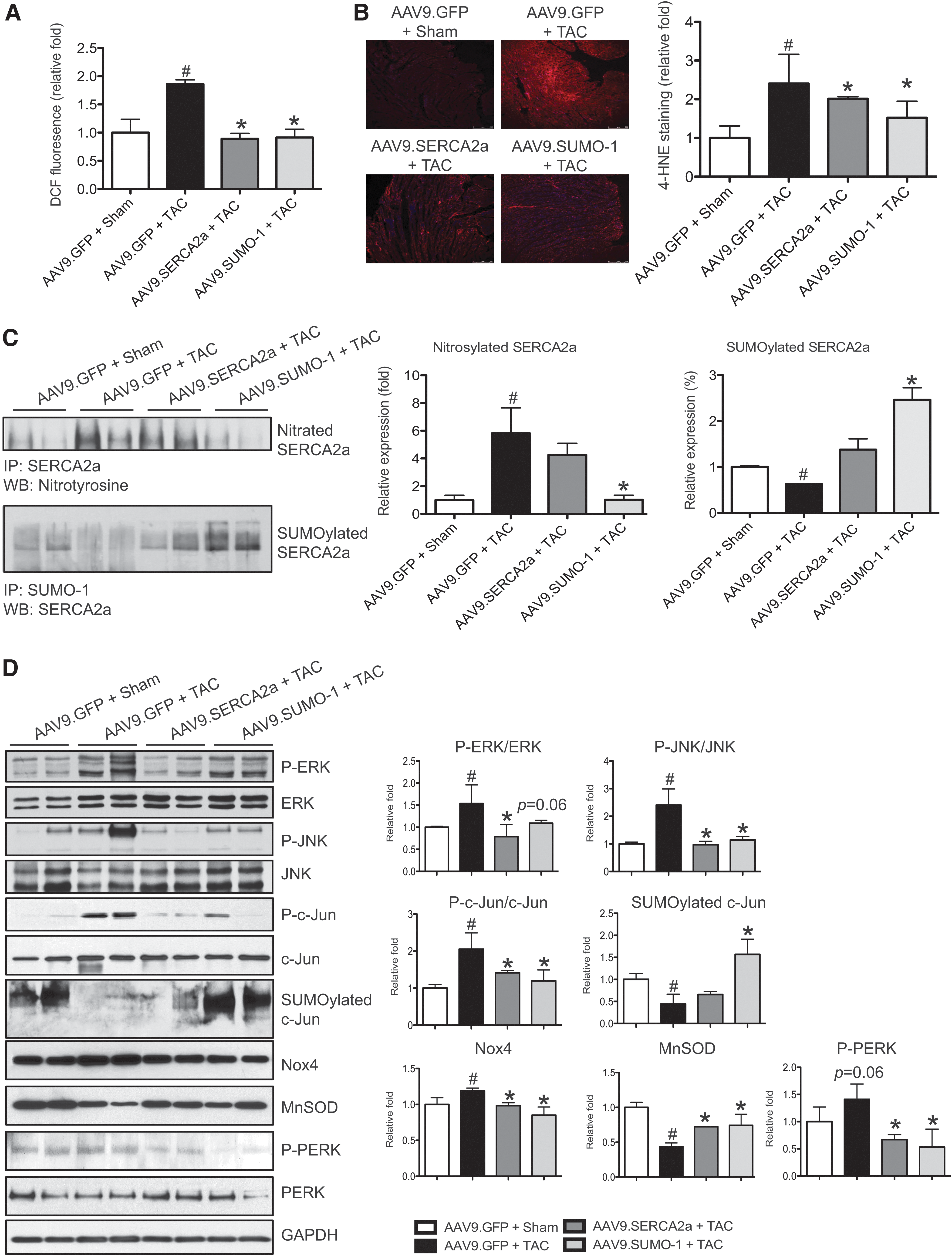
SUMO-1 overexpression contributes toward regulating redox signaling-mediated cardiac stress
We next examined whether the anti-oxidant effect of SUMO-1 overexpression is correlated with redox-sensitive signaling molecules in pressure-overload-induced heart failure mice. Similar to the observation in vitro cardiomyocytes, TAC-induced activation of MAPK declined in SUMO-1 overexpressing hearts. TAC significantly elevated phosphorylation of ERK and JNK, and this activation subsequently affected increased phosphorylation of c-Jun (Fig. 6D). However, we did not observe a significant induction of MAPKs and c-Jun phosphorylation in TAC hearts that received AAV9.SUMO-1. We further tested the relationship between MAPK signaling and SUMO-1 in the pathogenic progression. In isolated neonatal cardiomyocytes, inhibition of the MEK/ERK pathway by U0126 prevented PE-induced downregulation of SERCA2a expression; whereas the JNK inhibitor SP600125 had no such effect (Supplementary Fig. S6A). In the presence of PE, SUMO-1 expression was increased in the cells pretreated with JNK inhibitor (SP600125) compared with the cells without reagent (Supplementary Fig. S6B).
In mice preinjected with AAV9.GFP and subjected to 12 weeks of TAC, Nox4 protein level was significantly increased compared with sham-operated mice preinjected with AAV9.GFP (1.7-fold increase, p<0.05). Interestingly, Nox4 level was greatly reduced in SUMO-1 overexpressing hearts with TAC. To further understand how the SUMO-1 overexpression leads to downregulation of Nox4, we checked the SUMOylation status of c-Jun, which is an important transcriptional regulator of Nox4 expression. SUMOylation of c-Jun was significantly increased in TAC mice pretreated with AAV9.SUMO-1 compared with any other groups, and this suggests an involvement of SUMO-1 in Nox regulation in vivo. In contrast, the level of mitochondrial anti-oxidant, MnSOD was decreased in the TAC mice injected with AAV9.GFP (38% decrease) compared with sham control (p<0.05). However, MnSOD expression was normalized in AAV9.SUMO-1 pretreated groups. Furthermore, SUMO-1 overexpression reduced the TAC-induced phosphorylation of PERK, a marker of endoplasmic reticulum (ER) stress (Fig. 6D). Almost molecular profiles were identical between SUMO-1 and SERCA2a pretreatment groups. These data indicate that pretreatment of SUMO-1 decreased oxidative stress and partially contributed to TAC-induced SERCA2a dysfunction and a decrease of LV function.
Discussion
SUMO belongs to a large family of ubiquitin (Ub)-related proteins. The overall sequence identity with Ub is small (around 18%); however, the C-terminus of the protein, which confers most of its activity, is almost super-imposable to the equivalent region of Ub (3, 50). Both SUMOylation and ubiquitination induce reversible covalent post-translational modification of lysine amino-acid residues in their substrate proteins, leading to changes in stability, activity and/or sub-cellular localization of the modified proteins they target (15, 18, 20, 21). Unlike ubiquitination, SUMOylation does not tag substrate proteins for degradation. In fact, SUMOylation and ubiquitination may even counteract one another by competing for the same lysine residues to define the fate of substrate proteins (7, 12, 23, 26). SUMOylation has been found to be highly relevant in signal transduction, particularly in response to cellular stress and disease (57, 61). We have previously reported the beneficial effects of SUMOylation on SERCA2a's biological properties using a murine model of heart failure (34). More recently, the positive effects of SUMO-1 gene transfer on SERCA2a and cardiac function were further confirmed in a porcine model of ischemia (63), suggesting a huge therapeutic potential of SUMO-1 for heart failure.
In this study, we found that SUMO-1 overexpression via gene transfer prevents the development of cardiac hypertrophy both in vitro and in vivo. In addition, we found that SUMO-1 gene transfer blocks the transition from compensated hypertrophy to heart failure. We also found that SUMO-1 abrogates the damage of ROS formation in the setting of hypertrophy and failure of SERCA2a.
At the molecular level, we found that PE-induced MAPK activation was inhibited by either SUMO-1 or SERCA2a overexpression in cardiomyocytes. In TAC mice, SERCA2a overexpression seems more efficient in inhibiting MAPK activation (e.g., ERK) than SUMO-1 overexpression, suggesting that Ca2+ is an important upstream activator of MAPKs. Previously, it was shown that thapsigargin administration, an inhibitor of SERCA2a enzyme, induced ERK activity in human foreskin fibroblasts, underscoring the importance of SERCA2a in Ca2+-mediated MAPK activation (10). MAPK regulation by SUMO-1 seems to influence downstream signaling cascades. JNK activation was found to be in parallel with phosphorylation of c-Jun. Previous studies have reported that c-Jun activation has pro-hypertrophic and pro-apoptotic effects (16, 32, 52). In this study, an increase in JNK-dependent phosphorylation of c-Jun in pressure-overload-induced heart failure was significantly decreased by SUMO-1 overexpression. We tested the effects of MAPK inhibition on SUMO-1 and SERCA2a expression. PE-mediated MEK-ERK activation might induce SERCA2a downregulation, whereas JNK activation did not affect SERCA2a expression. In contrast, JNK activation during cardiac hypertrophy appears to contribute to SUMO-1 downregulation. These data strongly suggest that an association exists between MAPK and SUMO-1 in cardiomyocytes. The ERK and JNK are activated in response to hypertrophic stimuli in cultured cardiomyocytes (4, 11); however, the importance of this pathway and regulatory mechanisms in the cardiac hypertrophy remains controversial. Further studies, therefore, will be needed to investigate the detailed mechanisms underlying the regulation of each of the MAPKs vis a vis SUMO-1 and SERCA2a is to produce ROS; while all other major oxidases produce ROS as a secondary process either to catalyze another oxidative reaction or dysfunctional abnormal forms of enzymes generate ROS. The Nox family consists of seven members (Nox1–5, Duox1, and Duox2) and they are differentially expressed in various tissues. Nox1, Nox2, and Nox5 mainly produce superoxide and Nox4 mainly produce hydrogen peroxide. Unlike other Nox family members, Nox4 activity is not controlled by binding regulatory proteins. The regulatory mechanism of Nox4 in the heart, however, remains poorly understood. Previous studies have shown that Nox4 is elevated in pathological conditions (8, 38, 42), and it directly impairs SERCA function (41). Induction of Nox4-derived ROS is known to coordinate the activity of ERK and JNK signaling by modulating ER stress signaling (65). In our study, we also observed an elevation of Nox4 expression when the MAPKs were activated in response to the hypertrophic stimuli such as PE in vitro and TAC in vivo. Recently, negative effects of SUMO-1 on Nox enzymes have been reported (53) by which Pandey et al. (53) showed that endogenous SUMO-1 regulates ROS output and inhibits Nox activity, although they did not provide concrete evidence that Nox is a direct target of SUMO-1. These previous studies and our findings would suggest that there is a direct relationship between Nox4 and SUMO-1. It is interesting to note that Nox4 expression was decreased by SUMO-1 gene transfer. Increased SUMOylation of c-Jun by SUMO-1 overexpression may contribute to downregulation of Nox4 in pressure-overloaded hearts. A c-Jun/c-fos-dependent transcriptional activation of Nox has been reported in previous studies (46). SUMOylation inhibits c-Jun/c-fos activity, which may turn off the Nox4 transcription. In SERCA2a overexpressing hearts, the level of Nox4 was also normalized. c-Jun SUMOylation seems to be increased (not statistically significant) compared with the control TAC hearts. Thus, c-Jun activation on phosphorylation by JNK, which is inhibited by either SUMO-1 or SERCA2a overexpression, may be involved in the regulation of Nox4. c-Jun activity is also closely associated with MnSOD transcripts and its activity. Endogenous c-Jun represses ROS-reducing genes such as MnSOD and catalase via mitochondrial complexes and Nox enzyme (32).
At a cellular level, elevated levels of ROS impair cardiomyocyte function by damaging multiple channels and transporters (24, 43, 69). ROS disrupts the structural integrity of ion channels via membrane lipid peroxidation (17, 37). ROS also decreases expression and activity of SERCA through post-translational modifications (1, 31, 58, 60, 68). Both tyrosine nitration and cystein oxidation of SERCA are known as important mechanisms that damage SERCA and lead to its decreased activity. Both these processes increase during aging. In this study, we observed the inhibitory effects of SUMO-1 pretreatment on SERCA2a oxidation, suggesting the potential role of SUMO-1 in the aging heart. Concomitant with these adverse effects on cardiomyocyte function, ROS stimulates a number of responses associated with the ventricular remodeling processes. These include ROS-mediated activation of matrix metalloproteinases to alter the architecture of the extracellular matrix, modulation of signal transduction pathways that initiate cardiomyocyte hypertrophy, and apoptosis or cell death (9, 35, 39). Clearly, SUMO-1 can affect multiple critical proteins beyond SERCA2a that can lead to decreased oxidative stress.
Taken together, these observations suggest that one mechanism by which to halt deleterious ventricular remodeling and abnormal cardiomyocyte functional responses is by increasing SUMO-1. Concomitant with the protective effects of SUMO-1, we found a decrease in oxidative stress and interdependence between the Nox and SUMO pathways.
Clinical implications
More than 5.8 million Americans are suffering from heart failure, and there is an urgent unmet need for novel strategies to treat heart failure. In previous studies, we identified SUMO-1 as a new regulatory mechanism of SERCA2a showing that it enhanced its function and improved its protein stability and activity. We showed that in rodent and large animal models of heart failure, SUMO-1 gene transfer results in improved cardiac function and survival. In fact, co-expression of SUMO-1 and SERCA2a led to synergetic benefits. In this study, when we delivered SUMO-1 by gene transfer, we found that it protected SERCA2a from oxidative stresses that are prevalent in heart failure. We further found that SUMO-1 modulated important redox regulating molecules, which have been implicated in age-related heart diseases. Taken together, our results indicate that SUMO-1 may be a new therapeutic target not only in advanced heart failure but also during the early phases of transition from cardiac hypertrophy to failure.
Materials and Methods
Animals and gene delivery
All procedures were approved by the local Institutional Animal Care and Use Committee (IACUC). Male B6C3/F1 mice were purchased from Charles River Laboratories. Six week-old male mice received AAV9 vectors carrying SUMO-1, SERCA2a, and GFP via tail-vein injection. After 6 weeks of gene delivery, cardiac function was measured and randomized.
Surgical interventions
Mice underwent TAC using a supraclavicular construction model. TAC or sham surgery was performed at 6 weeks postgene delivery. Mice were anesthetized with intraperitoneal ketamine and placed on a ventilator. A 2- to 3-mm longitudinal cut was made in the proximal portion of the sternum, enabling visualization of the aortic arch. The transverse aortic arch was ligated between the innominate and left common carotid arteries with an overlaying 27-gauge needle. The needle was immediately removed, leaving a discrete region of constriction. The animal sham group underwent a similar procedure without ligation.
Echocardiography
Transthoracic echocardiography was performed using a Vivid 7000 GE machine equipped (GE Healthcare) with an H13L transducer (14 MHz). Two-dimensional and M-mode images were obtained in the short-axis view. The heart rate, left ventricular internal diameter in diastole (LVIDd), and left ventricular internal diameter in systole (LVIDs) were measured over the course of at least three repeated cardiac cycles. The EF and FS were then calculated.
Cell culture and hypertrophic stimulation
Primary cultures of cardiomyocytes from 2-day-old Sprague–Dawley rats were used. Briefly, ventricular tissue was enzymatically dissociated, and the resulting cardiomyocytes was isolated using Neonatal Cardiomyocyte isolation kit (Worthington Biochemical). Cells were plated onto collagen-coated culture dishes with a specific culture medium (DMEM supplemented with 10% fetal bovine serum and 2 mM
Quantitative RT-PCR
Total RNA from NRVMs was isolated using RNeasy isolation kit (Qiagen) according to the manufacturer's instructions. cDNA was generated from total RNA using the Superscript II (Invitrogen). PCR was performed using an ABI Prism 7500 Real-Time PCR System. Results were expressed as target gene/18S ribosomal RNA. The following primers were used for qPCR:
ANF, sense 5′-CAGACCGATGAAGCGGGGGC-3′, antisense 5′-CTTCGCAGGCTCCGAGGGC-3′; BNP, sense 5′-AGACAAGAGAGAGCAGGACACC-3′, antisense 5′-CTTGAACTATGTGCCATCTTGG-3′; myh7, 5′-GAGGGCGGACATTGCCGAGT-3′, 5′-AAGGCTCCAGGTCTCAGGGCTTC-3′; 18S, sense 5′-TCAAGAACGAAAGTCGGAGG-3′, antisense 5′-GGACATCTAAGGGCATCAC-3′.
Measurement of protein synthesis rate
Isolated cardiomyocytes were cultured in serum-free DMEM medium for 12 h. The cells then were stimulated with PE for 12 h and incubated in the same medium containing 1.5 μCi/ml [3H]leucine (NEN Research Products) for an additional 20 h. Total proteins were precipitated by the addition of 10% trichloroacetic acid for 45 min on ice, re-solubilized in 0.4 N sodium hydroxide for 1 h at 37°C, and subjected to liquid scintillation counting.
Molecular analysis
The frozen tissues were crushed and homogenized in lysis buffer [50 mM Tris-Cl pH 8.0, 150 mM NaCl, 0.1% Triton X-100, 10 mM EDTA, complete protease inhibitor (one tablet per 10 ml; Roche), and a protein phosphatase inhibitor cocktail (Sigma-Aldrich)] using the MP homogenate system (FastPrep homogenizer). The insoluble portion was removed by centrifugation at 30,000 g for 20 min. SERCA2a SUMOylation assay was performed as previously described (21). For in vivo SUMOylation assays, cardiac tissue extracts were immunoprecipitated with a SUMO-1 agarose resin (Santa Cruz Biotechnology) overnight at 4°C. The resins were washed thrice with cold lysis buffer. The immunocomplexes were then resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), and Western blot analysis was performed using a SERCA2a-specific antibody. For SERCA2a oxidation analysis, SERCA2a was immunoprecipitated from cardiac tissue extracts with anti-SERCA2a antibody. Nitrosylation of SERCA2a was then probed with the anti-nitrotyrosine antibody. For general Western blotting, cardiac proteins (10–50 μg) were separated by SDS-PAGE, transferred to a nitrocellulose membrane (Bio-Rad), and probed with the following antibodies that were specific for SERCA2a (produced by 21st Century Biochemicals): glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Sigma-Aldrich), SUMO-1 (Santa Cruz Biotechnology), p-ERK, ERK, p-p38, p38, p-JNK, JNK, p-c-Jun, c-Jun, 3-nitrotyrosine (Cell Signaling), BNP (Santa Cruz Biotechnology), anti-rabbit-HRP, and anti-mouse-HRP secondary antibodies (Sigma-Aldrich). Blots were developed with Super Signal West Pico (Pierce). Protein band densities were quantified by using quantity ImageJ software (NIH). Inhibitors of ERK (U0126) and JNK (SP600125) were purchased from Calbiochem.
Immunostaining
Myocardial fibrosis was determined by Masson's trichrome staining (Sigma-Aldrich) of paraffin-embedded sections and semi-quantitatively rated. For 4-HNE staining, heart tissues were fixed and embedded. The sections were subjected to immunostaining with anti-HNE antibody (Alpha Diagnostics).
Statistical analysis
Results are presented as mean±SD. Data were analyzed using an unpaired t-test for comparisons between two means or a one-way ANOVA with the Bonferonni correction for comparisons between>2 means. A data comparison between the AAV9.SUMO-1 (or AAV9.SERCA2a) and AAV9.GFP groups was analyzed by using a post hoc Tukey test. p<0.05 was considered significant. Statistical analysis was performed using GraphPad Prism software (GraphPad Software, Inc.).
Footnotes
Acknowledgments
This work is supported by NIH K99 HL116645, R01 HL093183, HL117505 01A1, HL088434, P20HL100396, an NHLBI Program of Excellence in Nanotechnology (PEN) Award, Contract No. HHSN268201000045C, and P50 HL112324.
Author Disclosure Statement
Dr Hajjar is the scientific cofounder of Celladon Corporation, which is developing AAV1.SERCA2a for the treatment of heart failure. No competing financial interests exist for the other authors.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.