
Abstract
The studies described here identified the mechanisms of mitochondrial reactive oxygen species (ROS) release by Pseudomonas aeruginosa pyocyanin. The Asm/ceramide system is functional in mitochondria and is activated by ROS. Mitochondrial Asm mediates both cytochrome c release from mitochondria and cell death. Induction of cell death not only is a part of the method by which P. aeruginosa attack the host, but it also prevents an overshooting inflammatory reaction, which is observed in Asm-deficient cells. The results of these studies may be important for understanding sepsis and the often-lethal cytokine storm, as well as for defining molecular events involved in the interaction between pyocyanin and neutrophils in P. aeruginosa infections.
Introduction
P
We have previously shown that a deficiency in acid sphingomyelinase (human protein, ASM; EC 3.1.4.12, sphingomyelin phosphodiesterase; optimal pH 5.0; murine protein, Asm; gene symbol, Smpd1) overshoots the activation of the immune system after pulmonary infection with P. aeruginosa (9). At present, the mechanisms by which a deficiency in Asm disturbs the balance of innate immune activation are unknown. Asm is ubiquitously expressed and releases ceramide from sphingomyelin, predominantly not only in lysosomes but also in secretory lysosomes and on the plasma membrane (7, 13, 25). The ceramide molecules generated by Asm spontaneously associate to form ceramide-enriched membrane domains that serve to trap and cluster receptor and signaling molecules (8, 11, 12, 25). This spatial organization of receptors and signaling molecules seems to mediate many of the signaling effects of Asm and ceramide (11). This general function in the organization of cellular signaling explains the involvement of Asm in many forms of cellular stress.
ASM is also present in mitochondria (21). However, its physiological function and significance in these organelles are unknown.
P. aeruginosa can induce neutrophil death by a variety of factors, including exoenzyme U (Exo U), a phospholipase A2; pyocyanin, a phenazine; and rhamnolipids (17, 26). Bacteria that lack pyocyanin are less pathogenic in vivo (3), a finding indicating that this factor plays an important role in in vivo infection.
Pyocyanin is a redox-active compound capable of accepting and donating electrons. It has been detected at concentrations till 100 μM in the sputum of patients with cystic fibrosis (36). Because pyocyanin can cross biological membranes, it serves as a mobile electron carrier for P. aeruginosa (29). Under aerobic conditions, pyocyanin directly oxidizes reduced nicotinamide adenine dinucleotide phosphate (NADPH) in the host cell cytoplasm and donates accepted electrons to oxygen molecules to produce superoxide anions and reactive oxygen species (ROS) (29). Pyocyanin, however, has been also shown to decrease both cellular adenosine trisphosphate (ATP) levels and the mitochondrial membrane potential (MMP), a finding suggesting a direct interaction between pyocyanin and mitochondria (15, 22). Mitochondria are crucial for the production of ATP, the regulation of intracellular Ca2+ homeostasis, and the production of ROS. They are also key participants in the regulation of cell death.
Confocal microscopy studies indicate that extracellularly administered pyocyanin reaches mitochondria, where it may enhance the production of ROS and alter mitochondrial ultrastructure by mechanisms that are still poorly defined (22). Rho0 cells, which are devoid of the mitochondrial respiratory chain, have been reported to be almost entirely resistant to pyocyanin under aerobic conditions (1). Thus, elucidation of the mechanism by which pyocyanin affects mitochondrial functions in intact cells is an important but still largely unexplored topic.
The studies reported here focused on the molecular mechanisms that determine the balance between the proinflammatory and proapoptotic effects of pyocyanin in neutrophils. In particular, we investigated a possible effect of pyocyanin on mitochondria. Indeed, not only mitochondria are the checkpoint for the release of procaspases and caspase cofactors such as cytochrome c during cell death (6), but also they coordinate the inflammasome and inflammatory pathways (38).
We found that bacterial pyocyanin interferes with mitochondrial respiration and that this interference results in the formation of ROS and in the loss of MMP within a few minutes, even in intact cells. ROS then activate mitochondrial Asm, trigger the release of mitochondrial ceramide and the release of cytochrome c from mitochondria, and induce cell death. A deficiency in Asm prevents the pyocyanin-induced release of cytochrome c from isolated mitochondria, thereby preventing cell death. This reduced cell death, on the other hand, is associated with an increase in the release of interleukin-8 (IL-8) from pyocyanin-activated cystic fibrosis neutrophils but not from wild-type cells.
Results
Pseudomonas aeruginosa induces the death of neutrophils via acid sphingomyelinase
Neutrophils not only are key cells of the innate immune system but also play an important role in cystic fibrosis and its associated lung changes. To define the molecular mechanisms of the interaction between neutrophils and P. aeruginosa, we tested whether the P. aeruginosa strain ATCC 27853 induces the death of cultured and ex vivo neutrophils. We stimulated HL-60 cells with retinoic acid to induce their maturation to neutrophils and then determined cell death 8 h after infection with P. aeruginosa by staining the cells with Cy3-labeled annexin V and analyzing them by fluorescence-activated cell sorting (FACS). Furthermore, we isolated peritoneal neutrophils from wild-type and Asm-deficient mice and infected them with P. aeruginosa. The results showed that neutrophils lacking Asm are resistant to P. aeruginosa-induced death, whereas wild-type neutrophils rapidly undergo apoptosis after infection with P. aeruginosa strain ATCC 27853 (Fig. 1A). Similar findings were obtained with a second P. aeruginosa strain, 762 (not shown).
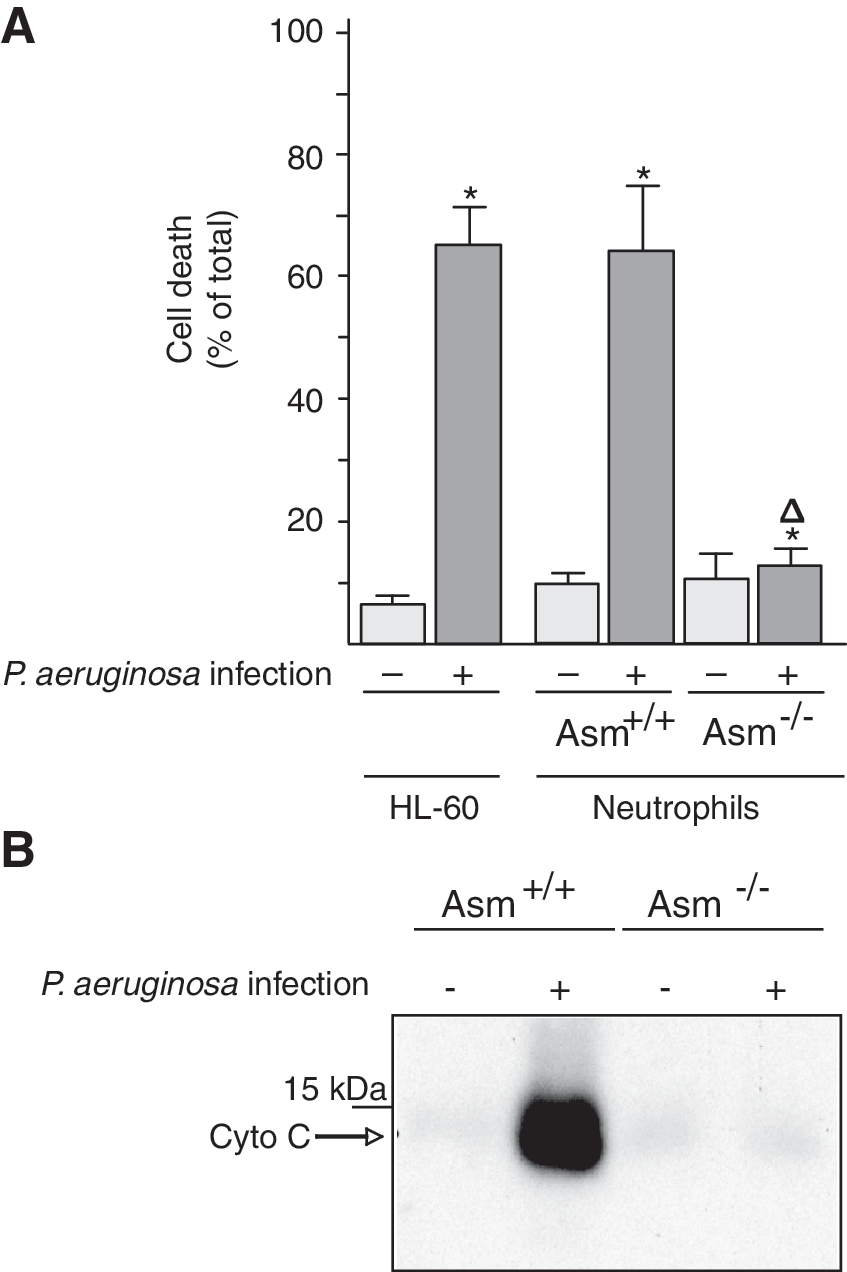
P. aeruginosa releases soluble toxins that are very important for the infection of mammalian hosts. To gain insight into the mechanism of P. aeruginosa-induced cell death, we incubated the pathogen directly with isolated mitochondria, key organelles in many forms of cell death. We determined cytochrome c release as a measurement for apoptotic changes in isolated mitochondria. Because it has been previously shown that Asm is present in the mitochondrial intermembrane space and that it associates with procaspase 3 and nitrous oxide (NO) synthases (21), we also tested the effect of P. aeruginosa on Asm-deficient mitochondria. The results showed that incubating isolated mitochondria from wild-type neutrophils with P. aeruginosa strains 762 or ATCC 27853 results in the release of cytochrome c, an effect that is abrogated by a deficiency of Asm in mitochondria isolated from Asm-deficient neutrophils (Fig. 1B). Control studies confirmed that the mitochondrial preparations did not contain lysosome-associated membrane protein 2 (LAMP-2) and cathepsin D but were very highly enriched in cytochrome c and in Tim and Tom proteins, findings that exclude contamination with lysosomes that also contain Asm (not shown).
To gain insight into the mechanisms by which P. aeruginosa kill neutrophils, we determined whether the bacterial product, pyocyanin, is involved in this process and whether pyocyanin acts in mitochondria by stimulating mitochondrial Asm and releasing ceramide.
Pyocyanin induces ROS in mitochondria
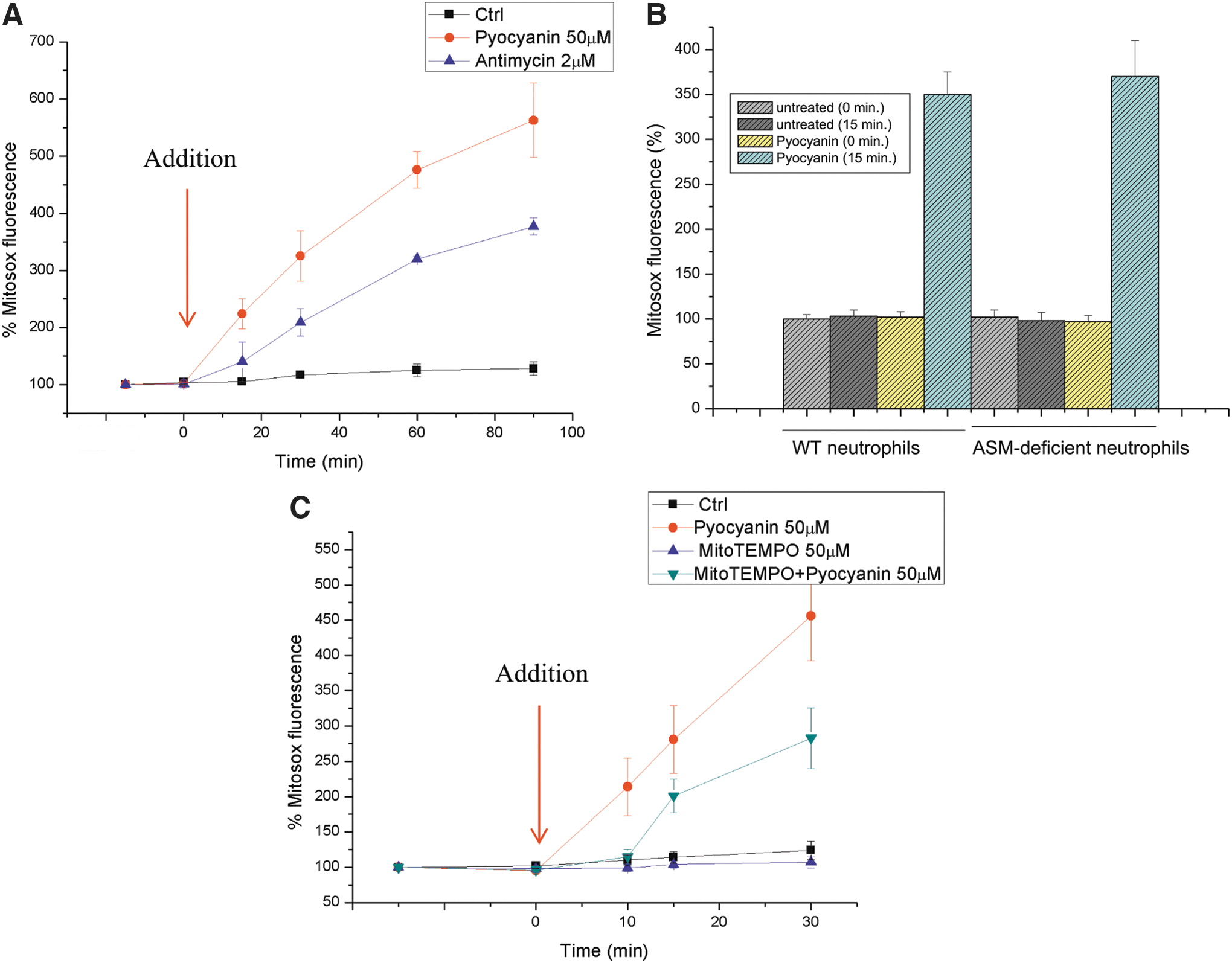
To gain in-depth insight into the possible interaction between pyocyanin and mitochondria, we first determined the short-term effects of pyocyanin on mitochondrial function in intact cells. The addition of 50 μM pyocyanin, a concentration that has been measured in the sputum of patients (36) and, thus, is pathophysiologically relevant, to Jurkat lymphocytes or neutrophils resulted in an immediate increase in superoxide anion production at the level of the mitochondria (Fig. 2A, B). This increase was assessed with MitoSOX Red, a mitochondria-targeted fluorogenic dye that can directly measure the superoxide generated in the mitochondria of live cells. Superoxide anion (O2 −•), the product of reducing oxygen by one electron (produced by various respiratory complexes that leak electrons to oxygen, producing primarily superoxide anions), is the precursor of most ROS. We used antimycin A, an inhibitor of complex III activity, as a positive control to force electrons to leak outside of the tight single-electron transfer mechanism, causing the production of superoxide anions. Dismutation of O2 −• produces hydrogen peroxide (H2O2), which, in turn, may be fully reduced to H2O or partially reduced to hydroxyl radical (OH•), one of the strongest oxidants. Although pyocyanin has been reported to directly interact with 2′,7′-dichlorodihydrofluorescein and dihydrorhodamine, which are commonly used to detect ROS (22), it did not oxidize MitoSOX (not shown). Incubation of cells with Mito-TEMPO, a mitochondria-targeted antioxidant with superoxide and alkyl-scavenging properties, significantly reduced pyocyanin-induced ROS production (Fig. 2C). These results suggest that a considerable portion of the pyocyanin-induced ROS production occurs at the level of mitochondria, although some pyocyanin-induced ROS production may also take place in the cytosol, mediated by the ability of pyocyanin to oxidize NADPH and to reduce cellular oxygen concentrations (29).
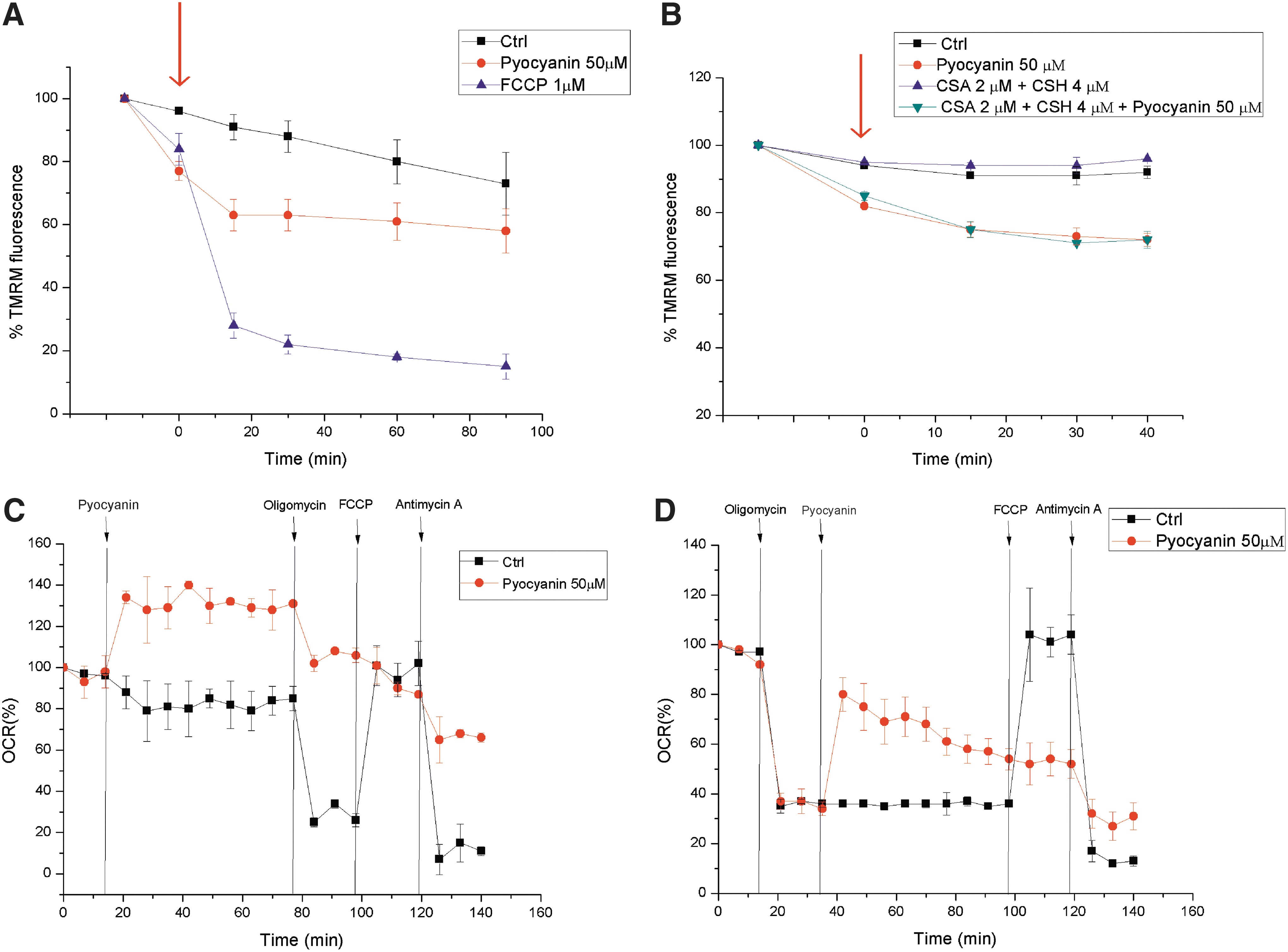
The addition of pyocyanin to intact cells not only led to ROS production but also resulted in a very rapid but incomplete dissipation of the MMP, Δψm (Fig. 3A). This change in Δψm was independent of the opening of the permeability transition pore (PTP) (34), because it also occurred in the presence of cyclosporin A, a potent PTP inhibitor (Fig. 3B). Next, we used an extracellular flux analyzer to determine the effects of pyocyanin on respiration in intact adherent mouse embryonic fibroblasts. The addition of pyocyanin to these cells immediately increased oxygen consumption (Fig. 3C). This apparent increase in respiration was only partially reversible by oligomycin, which blocks ATP synthase (Fig. 3C). The subsequent addition of the uncoupler carbonylcyanide-p-trifluoromethoxyphenyl-hydrazone (FCCP) did not restore respiration, whereas the addition of antimycin A, an inhibitor of complex III, further reduced but did not completely abolish oxygen consumption (Fig. 3C). Antimycin A, by binding to the Qi site of cytochrome c reductase, inhibits the oxidation of ubiquinol in the electron transport chain of oxidative phosphorylation. The inhibition of this reaction disrupts respiration and prevents the formation of the proton gradient across the inner membrane. Thus, pyocyanin drastically reduced the respiratory response to the subsequent addition of oligomycin and FCCP. A massive loss of cells because of death and detachment was excluded by direct microscopic observation of the cells at the end of the experiments. In contrast, if cellular respiration was depressed by oligomycin, the addition of pyocyanin induced a recovery of the respiratory rate (Fig. 3D). This recovery may be simply a reflection of the loss of Δψm induced by pyocyanin: According to the chemiosmotic model, depolarization with associated respiratory stimulation is likely to reflect the appearance of a Δψm-dissipating proton leak. Again, the subsequent addition of FCCP had no effect, whereas antimycin A reduced respiration (Fig. 3D). Similar effects were observed in both settings with 25 μM pyocyanin (not shown). These results suggest that the pyocyanin-induced increase in oxygen consumption is only partially caused by an increase in respiration, given the lack of full reversal by antimycin A. Alternatively, respiration may be increased bypassing complex III. Oxygen consumption is probably also enhanced by the involvement of molecular oxygen as an acceptor of electrons directly from pyocyanin, independent of the respiratory chain.
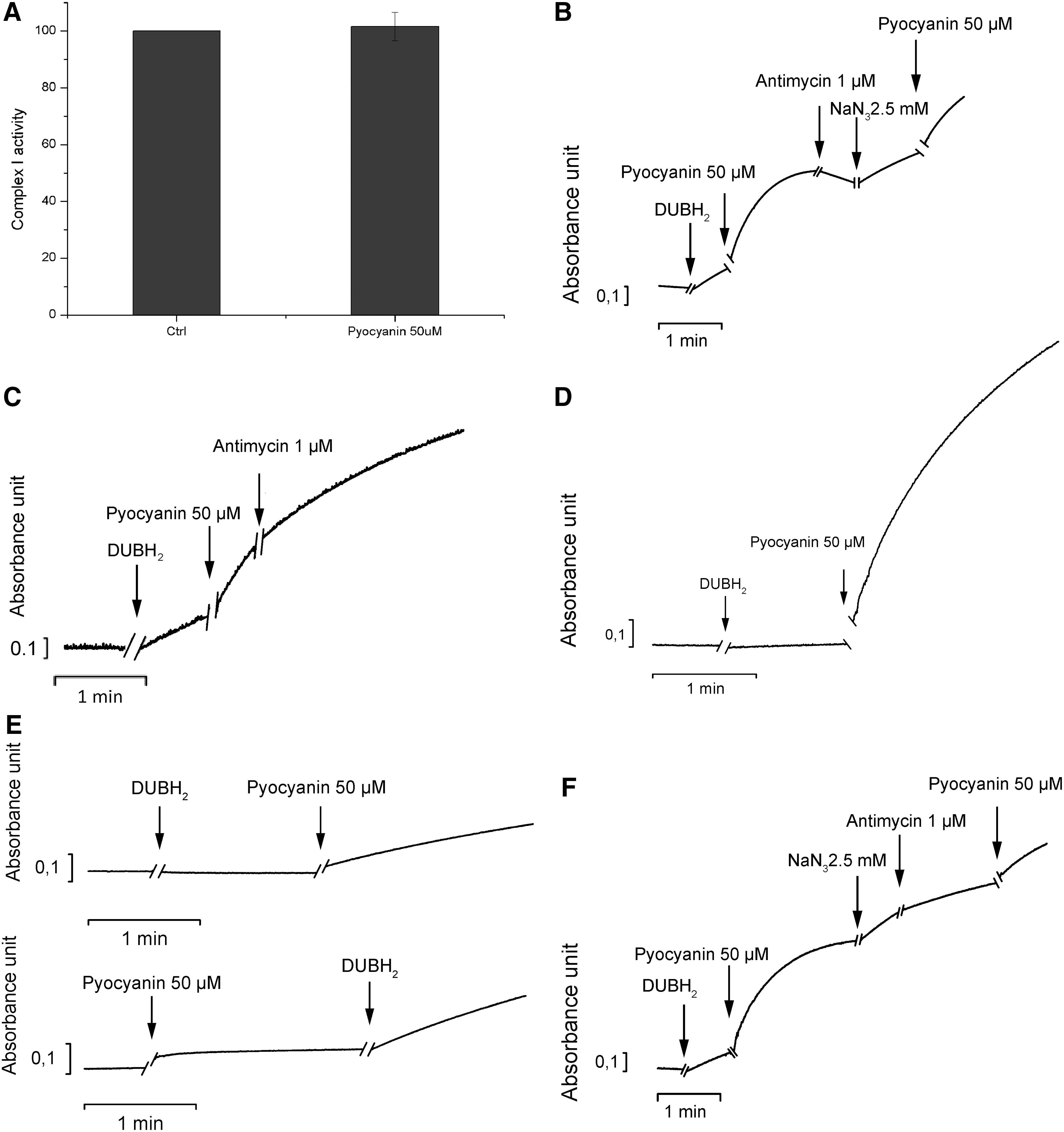
However, these experiments also point to a possible interaction between pyocyanin and the respiratory chain complexes. One possible explanation of the generation of mitochondrial superoxide by pyocyanin is the direct interference of the toxin with respiration, diverting a part of the electron flow to superoxide production. To test this hypothesis, we used the well-established method of alamethicin-permeabilized rat liver mitochondria (RLM). We addressed the function of complexes I and III, the main sites of ROS production, after the addition of pyocyanin. Pyocyanin did not alter the function of complex I (Fig. 4A). We assessed the function of complex III by measuring the reduction of cytochrome c concentrations in mitochondria in which the function of complex I was inhibited by rotenone. An increase in absorbance measured at 550 nm corresponds with the chemical reduction of this mobile electron carrier. On the addition of decylubiquinol, cytochrome c reduction occurred; this reduction was further stimulated by the addition of pyocyanin. Reduced cytochrome c is then reoxidized by complex IV. Adding the complex III inhibitor antimycin A during the re-oxidation phase did not drastically change the rate of reoxidation. The subsequent addition of sodium azide (NaN3), which inhibits complex IV activity, increased cytochrome c reduction (Fig. 4B). These results indicate that pyocyanin accepts electrons from decylubiquinol and reduces cytochrome c, thus replacing the function of complex III. Indeed, pyocyanin reduced cytochrome c even in the presence of the complex III inhibitor antimycin A (Fig. 4C). Reduction also occured when all complexes were inhibited (Fig. 4D) and even in the absence of mitochondria, provided an electron donor was present (Fig. 4E). The rate of cytochrome c reduction induced by pyocyanin was slightly lower when antimycin A was added after NaN3 than when NaN3 was used alone (Fig. 4F), a finding suggesting that maximal reduction of cytochrome c by pyocyanin is partially dependent on complex III activity.
ROS activate mitochondrial acid sphingomyelinase
Next, we tested whether the Asm/ceramide system functions as a downstream target of pyocyanin and ROS, because oxygen radicals have been shown to activate Asm (27), which, in turn, releases ceramide, a proapoptotic molecule. To this end, we purified mitochondria from freshly isolated peritoneal neutrophils and determined the activity of Asm after treatment with pyocyanin or H2O2. The results (Fig. 5A, B) demonstrate a rapid activation of mitochondrial Asm activity by pyocyanin or H2O2. The addition of Tiron, an antioxidant, to isolated mitochondria prevented the activation of Asm by pyocyanin (Fig. 5A), a finding demonstrating the crucial role of ROS in Asm stimulation by pyocyanin in mitochondria.
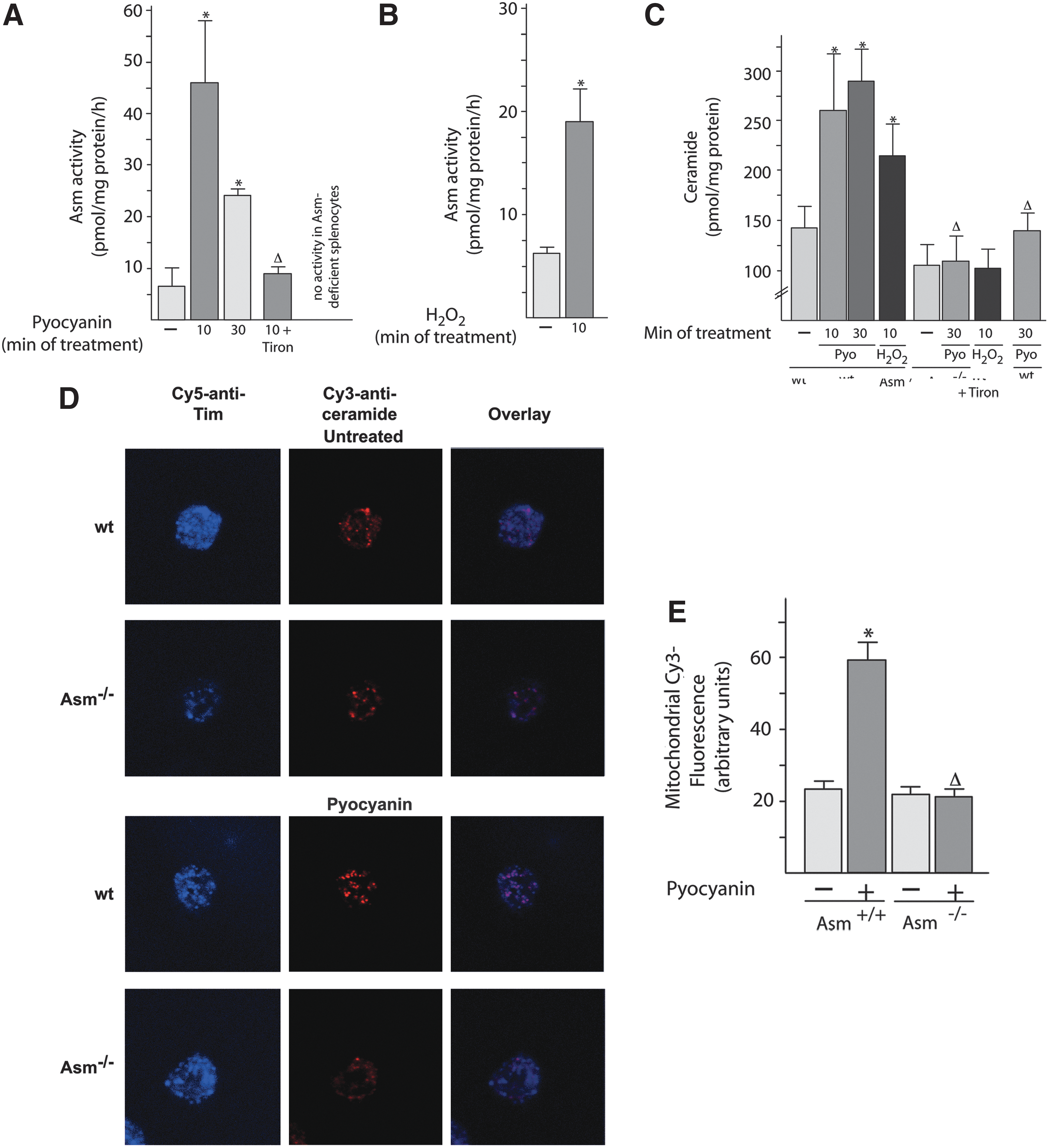
Activation of Asm in isolated mitochondria on treatment with pyocyanin or H2O2 resulted in a ceramide release that was abrogated in Asm-deficient peritoneal neutrophils (Fig. 5C). Confocal microscopy confirmed the increase in ceramide concentrations in wild-type mitochondria after treatment with pycoyanin; this increase did not occur in Asm-deficient cells (Fig. 5D, E). This finding indicates that Asm is necessary for the release of ceramide from isolated mitochondria after treatment with pyocyanin or H2O2.
Acid sphingomyelinase negatively regulates pyocyanin-induced production of IL-8
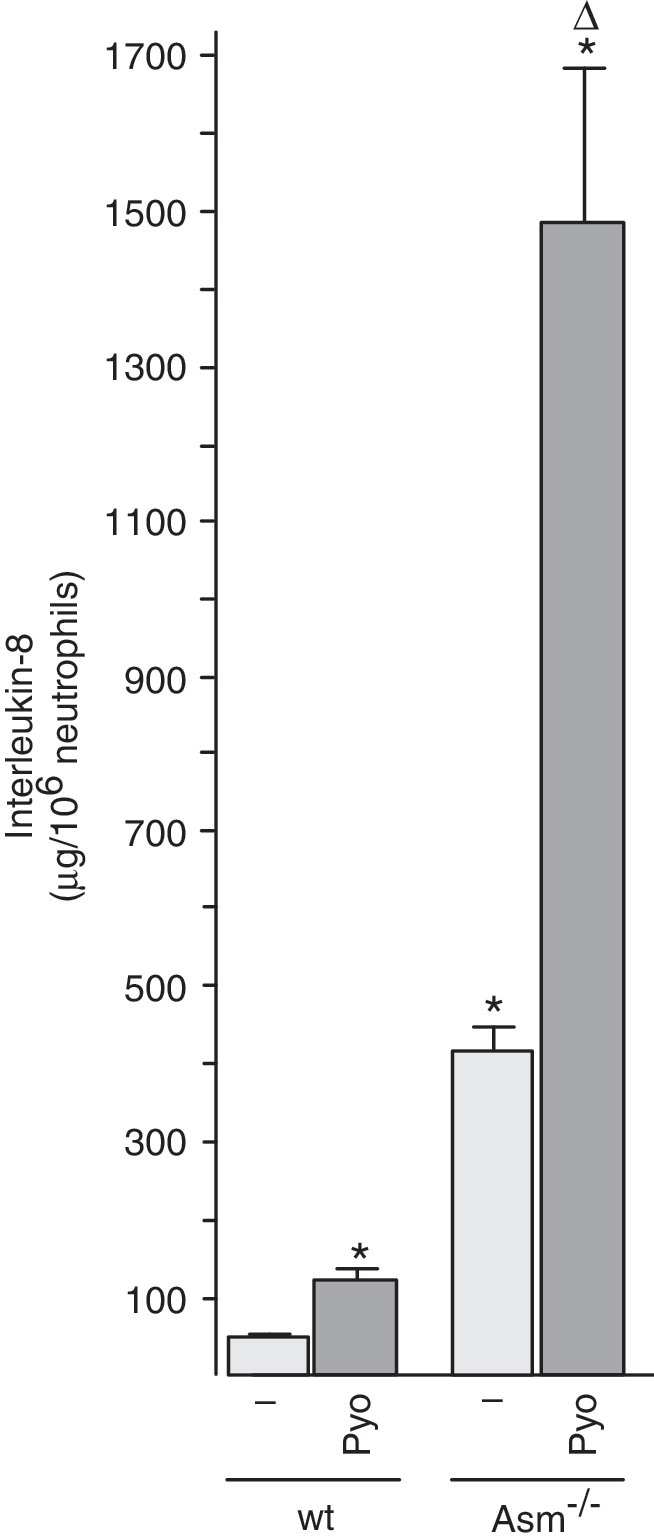
Mitochondria have been shown to be crucial in the activation of inflammasomes by recruiting and activating proinflammatory caspases; they are also crucial in the induction of cell death by the release of cytochrome c (6, 38). Therefore, we tested the effect of pyocyanin on the release of IL-8 and on the induction of death in freshly isolated neutrophils from wild-type and Asm-deficient neutrophils. The results demonstrate that pyocyanin induces the release of a higher amount of IL-8 from Asm-deficient neutrophils than from wild-type cells (Fig. 6). Basal levels of IL-8 were also higher in Asm-deficient neutrophils than in wild-type neutrophils. These findings indicate a negative regulation of IL-8 release from neutrophils by Asm and ceramide.
Pyocyanin induces the death of neutrophils via mitochondrial acid sphingomyelinase
Pyocyanin-induced cell death in wild-type neutrophils was determined by fluorescein isothiocyanate (FITC)-annexin V staining of intact peritoneal neutrophils. Asm-deficient neutrophils resisted the toxin and exhibited significantly less apoptosis than wild-type neutrophils after treatment with pyocyanin (Fig. 7A). In accordance, only isolated mitochondria from wild-type peritoneal neutrophils, not those from Asm neutrophils, released cytochrome c after treatment with pyocyanin (Fig. 7B). Similar results were obtained with neutrophils isolated from the spleen and with HL-60 cells (Fig. 7C).
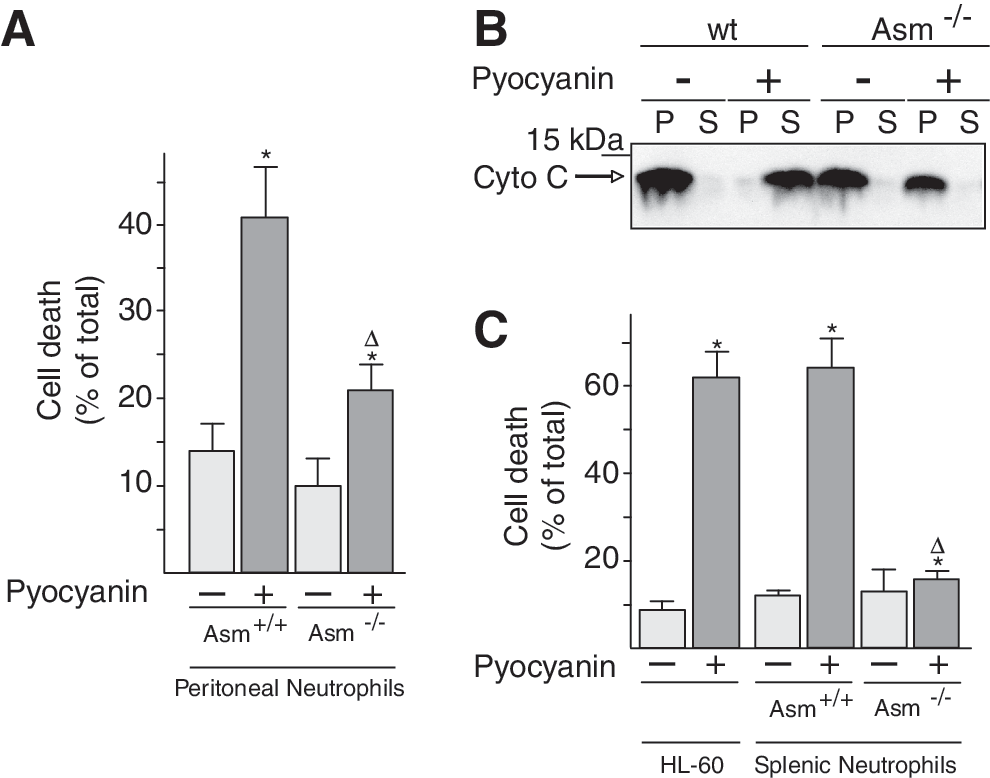
These results indicate that pyocyanin requires the expression of Asm if it is to mediate the mitochondrial release of cytochrome c and to trigger neutrophil death.
Discussion
Neutrophils are key elements of the host defense against infection with P. aeruginosa (16). Neutropenia greatly sensitizes mice to P. aeruginosa infections, with the result that neutropenic mice cannot eliminate the pathogens from the lung; they experience severe pneumonia and finally lethal sepsis (16). It is therefore of great clinical importance to understand the molecular mechanisms of the interaction between P. aeruginosa and neutrophils and, in particular, the mechanisms that allow the pathogen to escape the neutrophil attack.
Previous studies demonstrated that pyocyanin-induced death of neutrophils is an important element in the strategies used by P. aeruginosa as protection against the host defense (3). On the other hand, it is important for the host immune system to prevent overactivation, which may result in a very harmful destruction of parenchyma or even a lethal cytokine storm. Thus, the induction of neutrophil apoptosis may also contribute to the prevention of such an overactivation of the immune system and may balance the proinflammatory response. In this study, we tested this novel concept of a balance between immune activation and apoptosis.
We found that pyocyanin induces rapid death of wild-type neutrophils and that this death depends on the activation of Asm. We report a novel mechanism for this pyocyanin action, that is, activation of Asm in mitochondria on the generation of ROS induced by pyocyanin in mitochondria. At present, the function and regulation of mitochondrial Asm have not been elucidated, athough previous studies clearly showed mitochondrial expression of the enzyme and its association with pro-caspase 3, a protein involved in the induction of apoptosis on its release from mitochondria (21). Our studies of isolated mitochondria indicate that the release of cytochrome c as a prototypic event for the induction of mitochondrial changes during apoptosis is induced by pyocyanin via activation of Asm and is mediated by locally produced ROS. At present, it is unknown how ceramide mediates this proapoptotic effect. Several studies have shown that ceramide can form large pores that may allow proteins such as cytochrome c and proapoptotic caspases to be released from mitochondria (32). Ceramide may also interfere with the binding of cytochrome c to cardiolipin and may trigger the release of cytochrome c from this lipid (14, 24); such a release is an initial and necessary event in the induction of mitochondrial changes during apoptosis. Furthermore, ceramide has been shown to regulate several ion channels, including Ca2+ and K+ channels (2, 10, 20). C6 ceramide has been recently suggested to trigger PTP opening (28), and it may be possible that endogenous ceramide mediates death by a similar effect. Furthermore, ceramide has been shown to trigger the integration of Bax and Bak into mitochondrial membranes (19). Therefore, the generation of ceramide may allow these proteins, which are cytoplasmic or loosely associated with the outer mitochondrial membrane in viable cells, to change their conformation, integrate into the membrane, and cause apoptosis. It is certainly possible that ceramide activates several of these events that may act in concert. Finally, it is possible that ceramide is converted to proapoptotic sphingosine (for a recent review see Ref. 35), which may also permeabilize mitochondrial membranes, interfere with cytochrome c binding to cardiolipin, or both.
The molecular mechanisms of Asm activation have been only partially characterized. In vitro studies used purified Asm and demonstrated direct oxidation of Asm at cysteine residue 629, resulting in enzyme dimerization and activation (27). Our studies suggest a similar mechanism of mitochondrial Asm activation, because the effects of pyocyanin on Asm in isolated mitochondria were prevented by preincubation with the antioxidant Tiron. However, at present, it is unknown whether Asm dimerization occurs in mitochondria after stimulation. It is also possible that ROS regulate some yet unknown intermediates that transmit the signal to Asm.
In addition, Asm has been shown to be regulated by the lipid composition of membranes (23). Thus, anionic phospholipids, as well as diacylglycerol, ceramide itself, and free fatty acids, stimulate Asm activity (23). Whether pyocyanin induces a change in mitochondrial membrane lipids, for instance by oxidation, and thereby stimulates Asm has not yet been determined.
Finally, Asm is a pH-sensitive enzyme, and the activation of redox processes by pyocyanin may trigger a change in pH, in particular an acidification of the matrix that increases the activity of Asm and the generation of ceramide.
We found that pyocyanin increases the concentration of IL-8 in neutrophils from wild-type mice. To understand whether this increase depends on Asm, possibly by recruitment of inflammasome proteins to mitochondria (38), we tested whether an Asm deficiency impairs the release of IL-8 by neutrophils. Our findings demonstrate that Asm-deficient neutrophils respond to pyocyanin by releasing IL-8, the concentration of which is much higher in these neutrophils than in wild-type cells. These findings suggest that the release of IL-8 from neutrophils is negatively regulated by Asm. This conclusion is consistent with the findings of Yu et al. (37), who reported that a deletion of Asm results in an increase in IL-8 concentrations on infection with P. aeruginosa. A simple explanation for the enhanced production of IL-8 by cells lacking Asm on treatment with pyocyanin is the resistance of these cells to pyocyanin-induced death, whereas wild-type cells die on treatment with pyocyanin. However, it is also possible that mitochondrial ceramide serves to trap proinflammatory molecules, downregulating their activity or preventing them from their proper interactions with other molecules of the respective inflammasome. In this scenario, the absence of Asm and ceramide abrogates this inhibitory effect, resulting in a marked overactivation of neutrophils. Thus, the proinflammatory activity of neutrophils caused by pyocyanin is regulated by the balance between stimulation and ceramide-mediated death, downregulation of inflammation, or both. The observed form of death is most likely classical apoptosis brought about by changes in mitochondrial functions and not caused by pyroptosis, because Asm-deficient cells release massive amounts of cytokines but do not die.
Finally, because ROS activates death and it may also trigger the induction of cytokines, for instance by activation of an inflammasome, on treatment of neutrophils with pyocyanin, the absence of one pathway may shunt ROS to the other, resulting in an overactivation of this pathway.
These findings suggest that the induction of apoptosis in neutrophils is not only a part of the pathogen's attack against the host but also serves to balance the innate immune response and prevent an overshooting reaction.
Our studies further demonstrate that pyocyanin directly affects mitochondrial functions in intact cells. In particular, pyocyanin seems to have multiple effects within the cells and can provide an electron shunt pathway at various respiratory chain sites. Pyocyanin can accept electrons from both nicotinamide adenine dinucleotide (NADH) and ubiquinol; its measured electrochemical redox potential (0.045 mV; not shown) is higher than that of ubiquinol (−0.034 mV; see also results of experiments depicted in Fig. 4). In turn, pyocyanin can donate electrons not only to cytochrome c, as shown in this study, but also to molecular oxygen (29). As a redox cycler, pyocyanin can oxidize mitochondrial NADH, thereby decreasing proton export from the matrix through respiratory complexes and causing a significant depolarization and presumably a decrease in the pH of the matrix. Indeed, such a decrease in the MMP can be seen in intact cells within a few minutes of pyocyanin treatment (25% decrease within 30 min). Such a change is not related to the opening of the PTP, because this change also occurs in the presence of cyclosporin A, an inhibitor of the PTP. A recent study obtained a similar result with pancreatic acinar cells: 50 μM of pyocyanin decreased tetramethylrhodamine, methyl ester (TMRM) fluorescence by 18% within 40 min (4). The observed depolarization explains the ATP depletion of the cells, which has also been observed by several other groups (15, 22).
With regard to changes in the respiration of intact cells, pyocyanin increased their oxygen consumption, which was only partially sensitive to antimycin A, a finding suggesting that increases in these parameters are independent of complex III. Our interpretation is that this higher oxygen consumption occurs because of the conversion of molecular oxygen into superoxide radicals, a conversion that is not detected by the extracellular flux analyzer used in this study. Indeed, the addition of pyocyanin to intact cells immediately induces a substantially increased production of mitochondrial superoxide. The question arose as to whether mitochondrial ROS production is linked to a direct effect of pyocyanin on respiratory chain complexes. We found that pyocyanin does not affect complex I function but can “replace” complex III by draining electrons from ubiquinol and donating them to cytochrome c. These results suggest, first, that pyocyanin-induced superoxide production can be observed in mitochondria, primarily because NADH is the most concentrated in mitochondria and, second, that pyocyanin can provide an electron shunt pathway at various respiratory chain sites.
In summary, we found that the high concentrations of P. aeruginosa pyocyanin that are present in the lungs of patients infected with P. aeruginosa directly interfere with mitochondrial functions. This interference results in depolarization, release of ROS, activation of Asm, and formation of ceramide; this pathway triggers the release of cytochrome c and, finally, causes cell death, whereas it negatively regulates the induction of the proinflammatory cytokine IL-8. This pathway constitutes a novel pathway of host-pathogen interactions and serves to balance immune reactions.
Materials and Methods
Mice
Asm-deficient mice were in a C57BL/6 background, and littermates were used as controls. Mice were housed in isolated ventilated cages while providing a pathogen-free environment. The mice were routinely tested for microbial infections by bacterial culturing and serology; test results were negative before and during the study period. The hygienic status of the mice was repeatedly tested by a panel of common mouse pathogens according to the 2002 Federation of Laboratory Animal Science Associations (FELASA) recommendations.
Cells
To obtain mature primary neutrophils, we infected mice intraperitoneally for 3 h with 5×107 colony-forming units (CFU) of the clinical P. aeruginosa strain 762 (9). We then performed a lavage followed by Percoll purification of the cells. The identity of neutrophils was determined by staining with FITC-coupled anti-Gr1 antibodies followed by flow cytometric analysis. Isolated cells were cultured for 8 h in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS), 10 mM HEPES (pH 7.4), 2 mM
Jurkat T cells and HL-60 neutrophils were grown in RPMI 1640 supplemented as described earlier. HL-60 cells were differentiated for 36 h with 2 μM retinoic acid.
Mouse embryonic fibroblasts were grown in DMEM medium supplemented as previously described (18).
Bacterial cultures
P. aeruginosa strains 762 or ATCC 27853 were grown on tryptic soy agar plates (Becton Dickinson Biosciences). Bacteria were removed from the plate and suspended in 40 ml of prewarmed, sterile tryptic soy broth (Becton Dickinson Biosciences) in Erlenmeyer flasks. The optical density of the bacterial culture was adjusted to 0.225, and the bacteria were grown for 60 min at 37°C with shaking at 125 rpm. This procedure resulted in a culture in which the bacteria were in the early log phase. The bacteria were pelleted by centrifugation for 10 min at 1600 g and were then resuspended in prewarmed RPMI medium buffered with 10 mM HEPES. Aliquots were added to the cells or the mitochondria at a multiplicity of infection (MOI) of 10 bacteria per cell or mitochondrium.
Mice were infected by an intraperitoneal injection of 5×107 CFU P. aeruginosa strain 762 suspended in HEPES/saline (H/S; 132 mM NaCl, 20 mM HEPES, pH 7.4; 5 mM KCl, 1 mM CaCl2, 0.7 mM MgCl2, and 0.8 mM MgSO4) and were put to death after 3 h so that we could obtain mature peritoneal neutrophils.
Stimulations and infections
Cells or mitochondria were incubated with 50 μM pyocyanin or 10 μM H2O2 or were infected with P. aeruginosa at an MOI of 10 bacteria per cell for the indicated time.
Cell death
Cells were infected with P. aeruginosa at an MOI of 10 bacteria per cell or treated with 50 μM pyocyanin for 8 h. Cell death was determined by staining cells with FITC-coupled annexin V, followed by FACS analysis or by trypan blue staining and microscopic analysis of at least 200 cells per sample.
Isolation of mitochondria
Cells were incubated for 30 min at 4°C in buffer A, consisting of 0.3 M sucrose, 10 mM TES (pH 7.4), and 0.5 mM ethylene glycol tetraacetic acid (EGTA) (33). Cells were then disintegrated with a Dounce homogenizer with 40 strokes of the tight pestle. Nuclei and unbroken cells were pelleted by centrifugation for 5 min at 600 g and 4°C. Mitochondria in the supernatants were pelleted at 6000 g for 10 min at 4°C and resuspended in 50 mM PIPES-KOH (pH 7.4), 50 mM KCl, 2 mM MgCl2, 2 mM EGTA, 10 μg/ml aprotinin/leupeptin (A/L), 2 mM ATP, 10 mM phosphocreatine, 5 mM succinate, and 50 μg/ml creatine kinase. This buffer is slightly hyperosmotic to protect the mitochondria from swelling. Mitochondria were then incubated with the indicated doses of H2O2 or pyocyanin. The purity of the mitochondrial preparations was confirmed by Western blot analysis for the mitochondrial marker proteins cytochrome c, Tom, and Tim 23 and for proteins that are not present in mitochondria, in particular LAMP-1 and cathepsin D.
Cytochrome c release from isolated mitochondria
Isolated mitochondria were incubated with 50 μM pyocyanin for 15 min or with 10 μM H2O2 for 10 min. Stimulation was terminated by centrifugation at 4°C, after which supernatants were added to 5×sodium dodecyl sulfate (SDS) sample buffer. The pellets were resuspended in 1×SDS sample buffer. Samples were boiled, and proteins were separated on 12.5% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), eletrophoretically transferred onto nitrocellulose membranes, incubated with anti-cytochrome c antibodies (Becton Dickinson Biosciences), and developed with the Tropix electrochemoluminescence system.
Asm activity
Isolated mitochondria were stimulated with 50 μM pyocyanin for 10 or 30 min or with 10 μM H2O2 for 10 min. Samples were lysed in 250 mM sodium acetate (pH 5.0) and 1% NP40 for 10 min and homogenized by three rounds of sonication (10 s each) with a tip sonicator. The lysates were diluted to 250 mM sodium acetate (pH 5.0) and 0.1% NP40 and incubated with 50 nCi per sample [14C]sphingomyelin for 60 min at 37°C. The substrate was dried before use and resuspended in 250 mM sodium acetate (pH 5.0) and 0.1% NP40. Micelles were obtained by a 10-min bath sonication. The enzymatic reaction was stopped by the addition of 800 μl chloroform/methanol (2:1, v/v), phases were separated by centrifugation, and radioactivity of an aliquot of the aqueous phase was measured by liquid scintillation counting to determine the release of [14C]phosphorylcholine from [14C]sphingomyelin as a measure of acid sphingomyelinase activity.
Ceramide measurements
Mitochondria were stimulated as described earlier. Stimulation was terminated by lysis in 200 μl H2O and extraction in CHCl3:CH3OH:1 N HCl (100:100:1, v/v/v). The lower phase was collected, dried, and resuspended in 20 μl of a detergent solution consisting of 7.5% (w/v) n-octyl glucopyranoside and 5 mM cardiolipin in 1 mM diethylenetriamine-pentaacetic acid (DTPA). Samples were sonicated for 10 min, and the kinase reaction was performed for 45 min at room temperature by the addition of 70 μl of a reaction mixture containing 10 μl diacylglycerol kinase (GE Healthcare Europe), 0.1 M imidazole/HCl (pH 6.6), 0.2 mM DTPA (pH 6.6), 70 mM NaCl, 17 mM MgCl2, 1.4 mM EGTA, 2 mM dithiothreitol, 1 μM ATP, and 10 μCi [32P]γATP. The kinase reaction was terminated by extraction of the samples in 1 ml CHCl3:CH3OH:1 N HCl (100:100:1, v/v/v), 170 μl buffered saline solution (135 mM NaCl, 1.5 mM CaCl2, 0.5 mM MgCl2, 5.6 mM glucose, 10 mM HEPES [pH 7.2]), and 30 μl of a 100 mM EDTA solution. The lower phase was collected and dried, and the samples were spotted onto Silica G60 thin-layer chromatography (TLC) plates and developed with chloroform/acetone/methanol/acetic acid/H2O (50:20:15:10:5, v/v/v/v/v). The TLC plates were exposed to radiography films, the spots were removed from the plates, and the incorporation of [32P] into ceramide was measured with a phosphoimager (Fuji). Ceramide amounts were determined by comparison with a standard curve with C16 to C24 ceramides used as substrates.
Interleukin-8 measurements
IL-8 was measured by enzyme-linked immunosorbent assay according to the manufacturer's instructions (R&D).
Immunostaining of neutrophils
Neutrophils were cultured on glass cover slips and treated with pyocyanin for 6 h, fixed for 15 min in 1% paraformaldehyde, washed in phosphate-buffered saline (PBS), permeabilized for 10 min in 0.1% Triton X-100 in PBS, washed, blocked with 5% FCS and 0.025% Tween 20 in PBS, washed again in PBS, and stained with anti-ceramide (clone MAB 011, diluted 1:250; Glycobiotech) and anti-Tim 23 (#611223, diluted 1:200; Becton Dickinson Biosciences) for 45 min in H/S supplemented with 1% FCS. Cells were washed thrice for 5 min each with PBS plus 0.025% Tween 20 and once in PBS; they were then stained for 45 min at room temperature with Cy3-coupled anti-mouse immunoglobulin M (1:250 dilution) and DyLight 649 goat anti-mouse IgG antibodies (dilution, 1:500; all from Jackson ImmunoResearch). Cells were washed again thrice for 5 min each with PBS plus 0.025% Tween 20 and once in PBS; they were then embedded in Mowiol and analyzed by confocal microscopy. Cy3-fluorescence intensity, as a measurement for ceramide in mitochondria, was quantified with Photoshop.
Mitochondrial membrane potential and ROS production
MMP and ROS production were measured as reported by Leanza et al. (18). Briefly, MMP was monitored with TMRM (20 nM), whereas ROS production was measured with MitoSOX (1 μM). Jurkat cells were incubated for 20 min at 37°C in Hank's balanced saline solution. After incubation, the indicated compounds were added, and the decrease in TMRM fluorescence or the increase in MitoSOX fluorescence was measured by FACS.
Oxygen consumption assay
Oxygen consumption by adherent cells was measured with an XF24 Extracellular Flux Analyzer (Seahorse Bioscience). Wild-type mouse embryonic fibroblasts cells were seeded at 3×104 cells per well in 200 μl of supplemented culture medium (DMEM; Sigma-Aldrich). Oxygen consumption rate was measured at preset time intervals, and the instrument automatically carried out the preprogrammed additions of the various compounds (oligomycin, 1 μg/ml final concentration; FCCP, 400 nM; antimycin A, 1 μM), added as a solution in 70 μl of DMEM.
Isolation of RLM
After rats were sacrificied, the liver was removed and immediately immersed in an ice-cold isolation medium (250 mM sucrose, 5 mM HEPES, 2 mM EGTA; pH 7.5). The liver tissue was minced, thoroughly rinsed several times with ice-cold medium, and finally homogenized in the same solution with a glass Teflon Potter homogenizer. Mitochondria were then isolated by conventional differential centrifugation, as described by Schneider et al. (31). The protein content was measured by the biuret method, with bovine serum albumin (BSA) as a standard.
Activity of mitochondrial respiratory chain complexes
Complex activity was extrapolated from the slope of absorbance decrease; data are expressed as a percentage of the control (i.e., the activity without the addition of any derivative).
Complex I activity
To measure NADH-coenzyme Q (CoQ) oxidoreductase (complex I) activity, we incubated RLMs (50 μg of protein per ml) with 10 μM alamethicin, 3 mg/ml BSA, 10 mM Tris-HCl (pH 8.0), 2.5 mM NaN3, and 65 μM CoQ1. The reaction was started by the addition of 100 μM NADH. Changes in absorbance (340 nm) were monitored at 37°C with an Agilent Technologies Cary 100 UV-Vis spectrophotometer. Rotenone 2 μM was added to assess the rotenone-independent (and thus complex I-independent) activity to be subtracted.
Complex III activity
To measure CoQ cytochrome c oxidoreductase (complex III) activity, we added RLMs (10 μg protein per ml) to a cuvette containing 50 mM potassium phosphate buffer, pH 7.5, supplemented with 10 μM alamethicin, 3 mg/ml BSA, 2.5 mM NaN3, 2 μM rotenone, 0.025% Tween, and 75 μM oxidized cytochrome c. The reaction was started by the addition of 75 μM reduced decylubiquinol (produced by reduction of decylubiquinone in ethanol with NaBH4 shortly before use); changes in absorbance were monitored at 550 nm and 37°C. We added 2 μg/ml antimycin to assess the activity of the antimycin-dependent complex III enzyme.
Statistical analysis
All data are displayed as mean±standard deviation. All data were tested for normal distribution with the David–Pearson–Stephens test. Statistical analysis was performed with Student's t-test for single comparisons and ANOVA for multiple comparisons. The sample size planning was based on two-sided Wilcoxon–Mann–Whitney tests using the free software G*Power Version 3.1.7 of the University of Duesseldorf, Germany. Statistical significance was set at the level of p<0.05.
Footnotes
Acknowledgments
This study was supported by DFG grants GU 335/13-3 and GU 335/30-1 to E.G. and AIRC IG11814 to I.S. The authors thank Dr. Andrea Mattarei for measurement of the electrochemical redox potential of pyocyanin and Dr. Mario Zoratti for useful discussion.
Author Disclosure Statement
No competing financial interests exist.