
Abstract
Introduction
T
Our data support a novel view that the pathogenic role of nucleotide oligomerization domain-like receptor protein with pyrin domain containing 3 inflammasome activation in endothelial dysfunction in response to various danger factors is not only due to instigation of canonical sterile inflammatory response but also due to its direct actions on endothelial cells beyond canonical inflammation. Our findings also provide new insights that therapeutic strategies mainly targeting inflammation may only have partial effects when the intracellular inflammatory machinery, the inflammasome, is triggered in the vasculature. This is because non-canonical actions occur before or concurrently with classical inflammatory responses, which, ultimately, may initiate or exacerbate vascular injury during atherogenesis.
Endothelial dysfunction is developed in the very early stages of cardiovascular diseases and/or with various coronary risk factors such as dyslipidemia, obesity, diabetes mellitus, hypertension, or hyperhomocysteinemia (31). Recent studies have indicated that Nlrp3 inflammasome activation is critical for the development of atherosclerosis on endogenous danger factors such as cholesterol crystals (10, 29). Cholesterol crystals can be found in all stages of atherogenesis and is present in early atherosclerotic lesions (10, 36) and in obese mice (4). Engulfment of cholesterol crystals by macrophage leads to lysosomal destablization and cathepsin B release (10), which results in Nlrp3 inflammasome activation and canonical inflammatory effects, including recruitment and infiltration of inflammatory cells into vasculature and subsequent vascular inflammation and injury (10). Thus, cholesterol crystals have been considered one important endogenous danger signal-associated dyslipidemia to trigger sterile inflammation that initiates atherogensis (10, 45). Despite these pilot studies in the immune cells, it remains unknown whether activation of inflammasomes in endothelial cells (ECs) by endogenous danger factors, including cholesterol crystals, could cause endothelial dysfunction in coronary circulation.
To test these possibilities, this study first determined whether Nlrp3 gene deletion or inhibition of inflammasome activity could ameliorate endothelial dysfunction in coronary arteries in a typical mouse model of acute hypercholesterolemia. Then, we examined whether such a role of inflammasome in endothelial dysfunction was associated with inflammasome-dependent production of cytokines or DAMPs. We further determined the dependence of cholesterol crystal-induced endothelial dysfunction on Nlrp3 inflammasome and its associated detrimental effects, including reactive oxygen species (ROS) and pyroptosis. Our findings suggest a direct link between endothelial Nlrp3 inflammasome activation and endothelial dysfunction, which is distinct from canonical inflammasome-secreted cytokine (IL-1β/IL-18)-induced inflammatory responses such as inflammatory cell recruitment and infiltration in the vasculature. This novel action of Nlrp3 inflammasomes in ECs may initiate or exacerbate vascular injury in coronary circulation during hypercholesterolemia.
Results
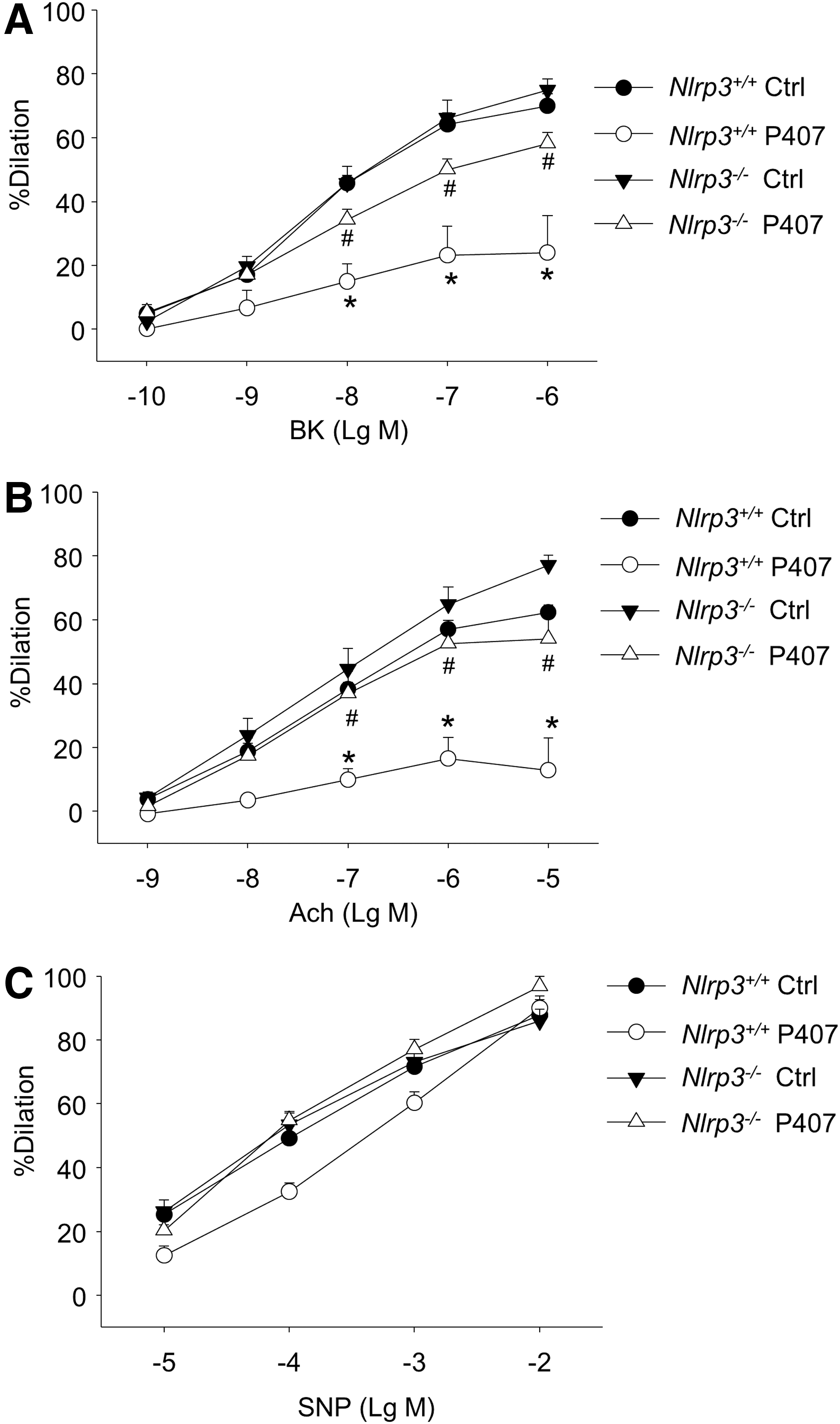
Nlrp3 deficiency restored endothelium-dependent vasodilation in coronary arteries of mice with hypercholesterolemia
We first investigated the role of Nlrp3 inflammasome in endothelial dysfunction associated with hypercholesterolemia by analyzing endothelium-dependent or -independent vasodilation responses in isolated, perfused, and pressurized coronary arteries from Nlrp3+/+
or Nlrp3−/−
mice. Mice were injected with poloxamer 407 (P407), a hydrophilic tri-block copolymer comprising polyoxyethylene and polyoxypropylene units, which induces acute hypercholesterolemia via upregulation of both the protein expression and activity of HMG-CoA reductase in the liver (20). As shown in Figure 1A, bradykinin (BK) produced a concentration-dependent vasorelaxation in coronary arteries from control Nlrp3+/+
mice with a maximal response of 70±3.7%. Such a BK-induced dose-dependent vasodilator response was significantly attenuated in coronary arteries from Nlrp3+/+
mice with P407-induced hypercholesterolemia, with maximal inhibition of 67.2%. BK produced a similar concentration-dependent vasorelaxation in coronary arteries from control Nlrp3−/−
mice with a maximal response of 74.9±3.5%. However, the hypercholesterolemia-induced impairment on endothelium-dependent vasodilation was markedly ameliorated in coronary arteries from Nlrp3−/−
mice (maximal inhibition of 22.7%). Similarly, Nlrp3 gene deletion restored endothelium-dependent vasodilation response to acetylcholine (Ach) (Fig. 1B). In contrast, hypercholesterolemia did not impair endothelium-independent vasodilation responses to sodium nitroprusside (SNP) (Fig. 1C). Nlrp3 gene deletion had no effect on P407-induced increases in plasma cholesterol levels (Supplementary Fig. S1; Supplementary Data are available online at
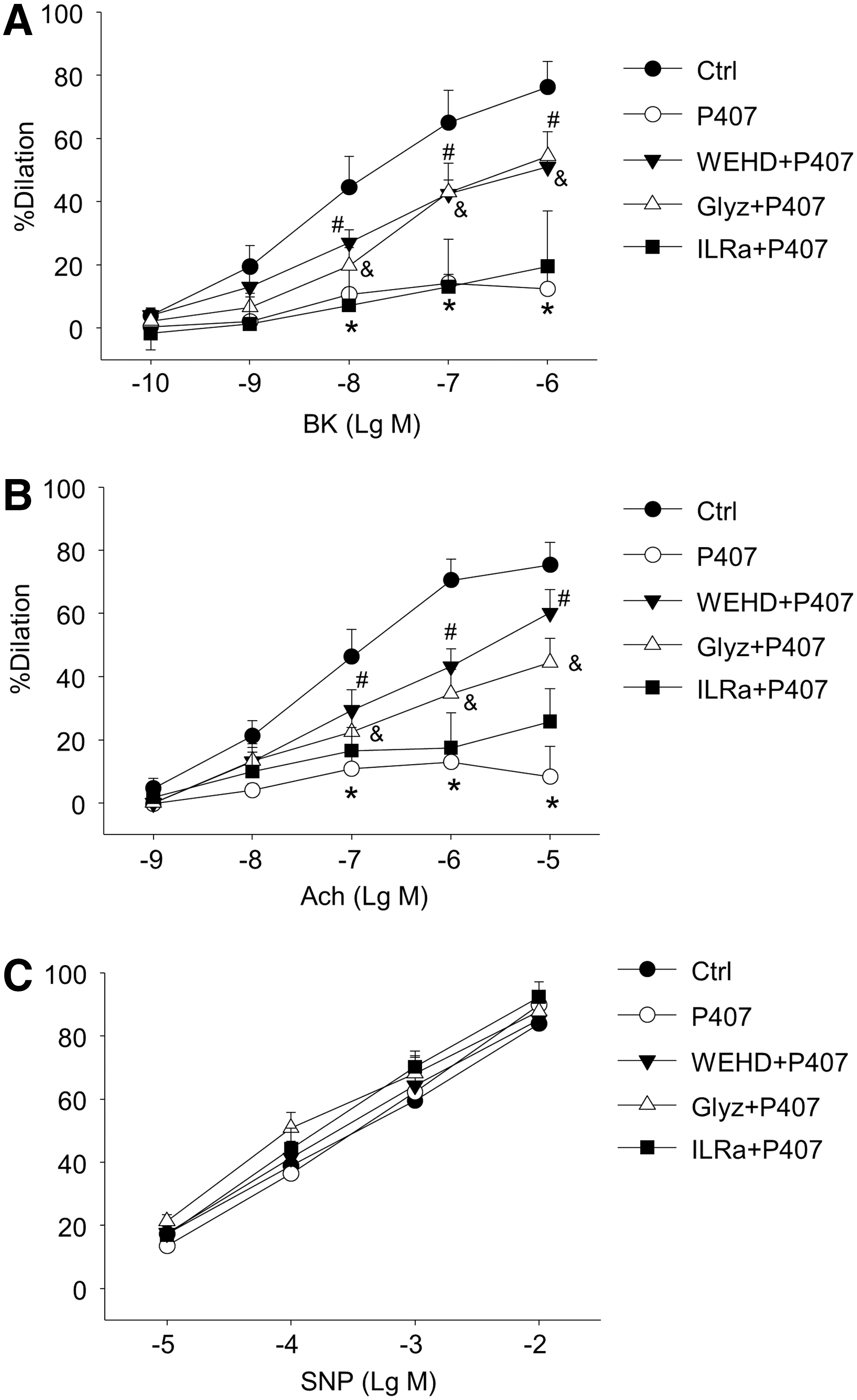
Inhibition of caspase-1 or HMGB1 improves endothelium-dependent vasodilation in mice with hypercholesterolemia
Nlrp3 inflammasome is activated to produce a number of inflammasome products, including the prototype cytokine IL-1β as well as a recently reported danger factor HMGB1 (33, 35). Thus, we examined the role of Nlrp3 inflammasome or its products in endothelial dysfunction in vivo in mice with acute hypercholesterolemia. As shown in Figure 2A and B, previous treatment of Nlrp3+/+ mice with caspase-1 inhibitor WEHD or HMGB1 inhibitor glycyrrhizin markedly attenuated hypercholesterolemia-induced impairment on endothelium-dependent vasodilation to BK or Ach. In contrast, previous treatment of mice with IL1 receptor antagonist (IL1Ra) did not improve endothelium-dependent vasodilation to either BK or Ach. As shown in Figure 2C, previous treatment of mice with WEHD, glycyrrhizin, or IL1Ra did not change endothelium-independent vasodilation responses to SNP in coronary arteries from hypercholesterolemic mice. We also demonstrated that WEHD and glycyrrhizin had no effect on endothelium-dependent or–independent vasodilation in coronary arteries from Nlrp3+/+ mice without hypercholesterolemia (Supplementary Fig. S3) or from Nlrp3−/− mice with hypercholesterolemia (Supplementary Fig. S4). These data suggest that caspase1/HMGB1 signaling contributes to endothelial dysfunction in vivo in hypercholesterolemic mice.
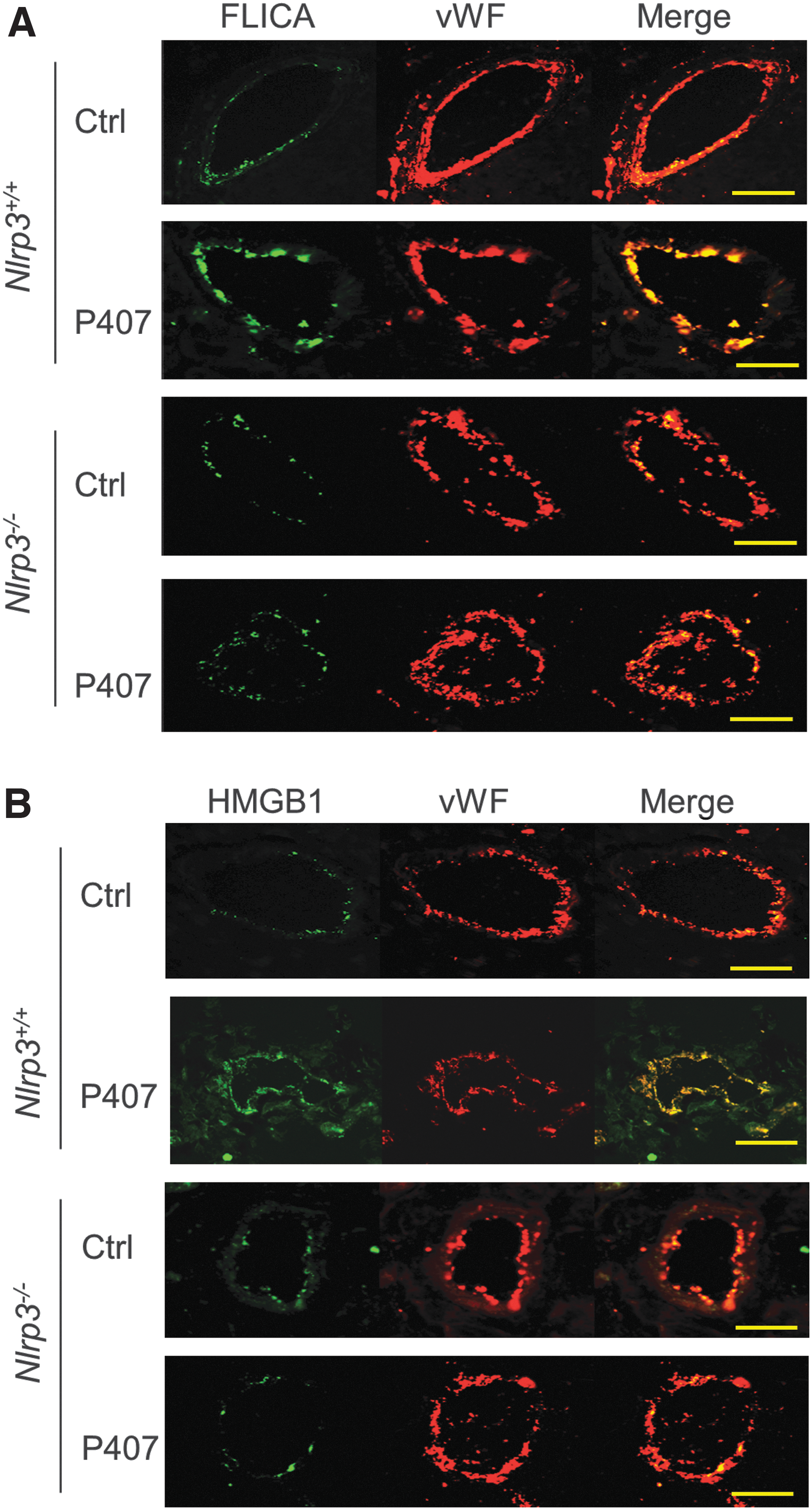
Activation of caspase-1 and increased expression of HMGB1 in coronary arterial intima of hypercholesterolemic mice
We further examined whether hypercholesterolemia increases Nlrp3-dependent caspase-1 activation and HMGB1 expression in the coronary arterial endothelium in mice. To this end, fluorescent labeled inhibitor of caspases (FLICA), a specific green fluorescent probe for binding the active form of caspase-1, was used to detect increased caspase-1 activity in coronary arteries of frozen heart sections. As shown in Figure 3A, confocal microscopic studies demonstrated that caspase-1 activity was indeed increased (green FLICA fluorescence) in the coronary arterial endothelium [EC marker von Willebrand factor (vWF), red color] from hypercholesterolemic mice compared with normal mice, as shown by increased colocalization of FLICA with vWF (yellow color), which was blocked by knocking out Nlrp3 gene. As shown in Figure 3B, correlated with increased caspase-1 activation, hypercholesterolemia markedly increased HMGB1 in coronary arterial intima in wild-type mice as shown by increased yellow color intensity, while such hypercholesterolemia-induced HMGB1 increases were not observed in the arterial intima of Nlrp3−/− mice.
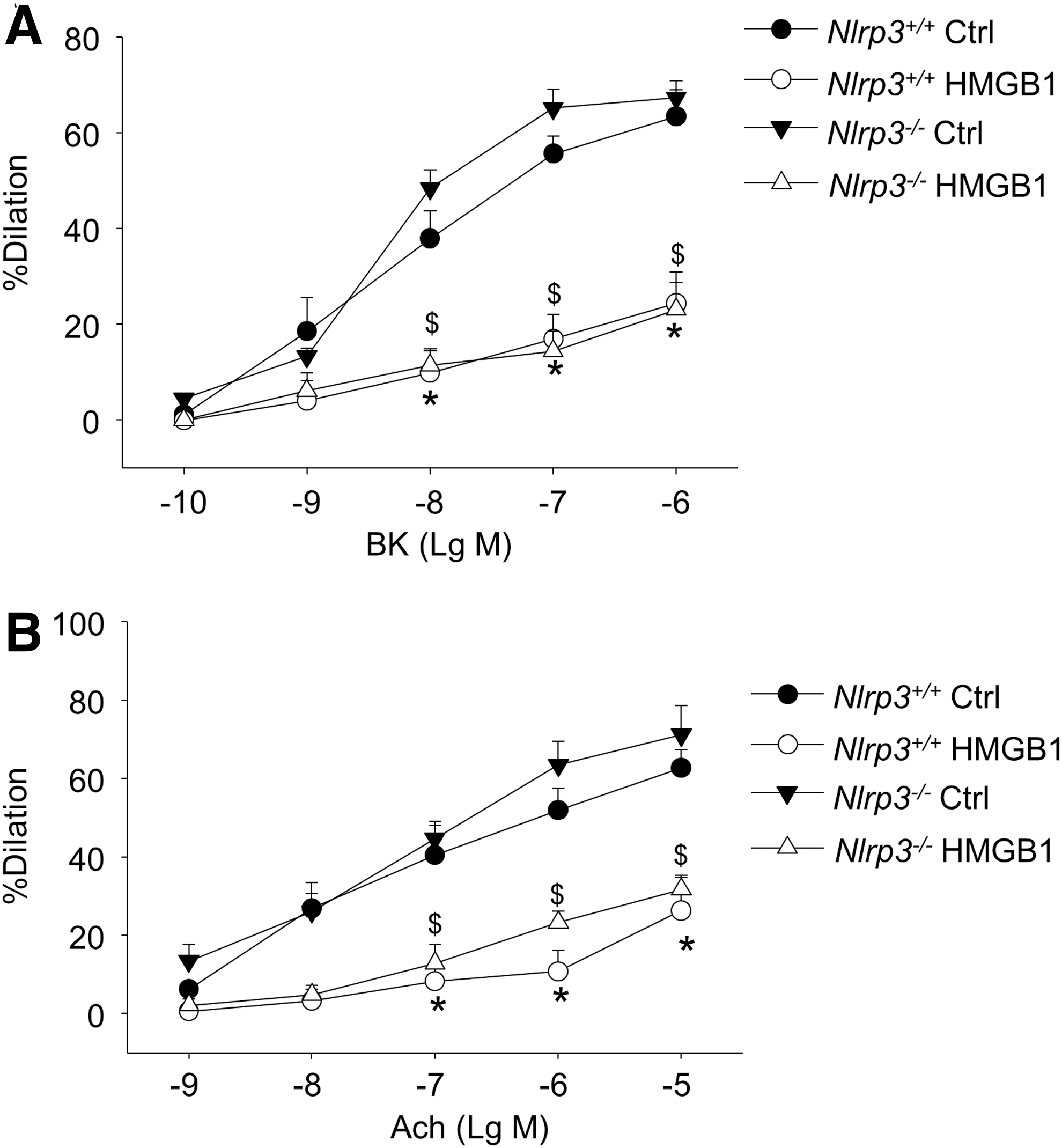
Exogenous HMGB1 impairs endothelium-dependent vasodilation in coronary arteries
We further tested whether luminal incubation of the recombinant HMGB1 can cause impairments on endothelium-dependent vasodilatory responses in coronary arteries from normal control Nlrp3+/+ or Nlrp3−/− mice. As shown in Figure 4A and B, HMGB1 treatment significantly decreased the endothelium-dependent vasodilation of normal Nlrp3+/+ arteries to BK or Ach. HMGB1 had similar inhibitory effects on BK or Ach-induced endothelium-dependent vasodilation in normal Nlrp3−/− arteries.
Cholesterol crystal induces formation and activation of Nlrp3 inflammasomes in cultured ECs
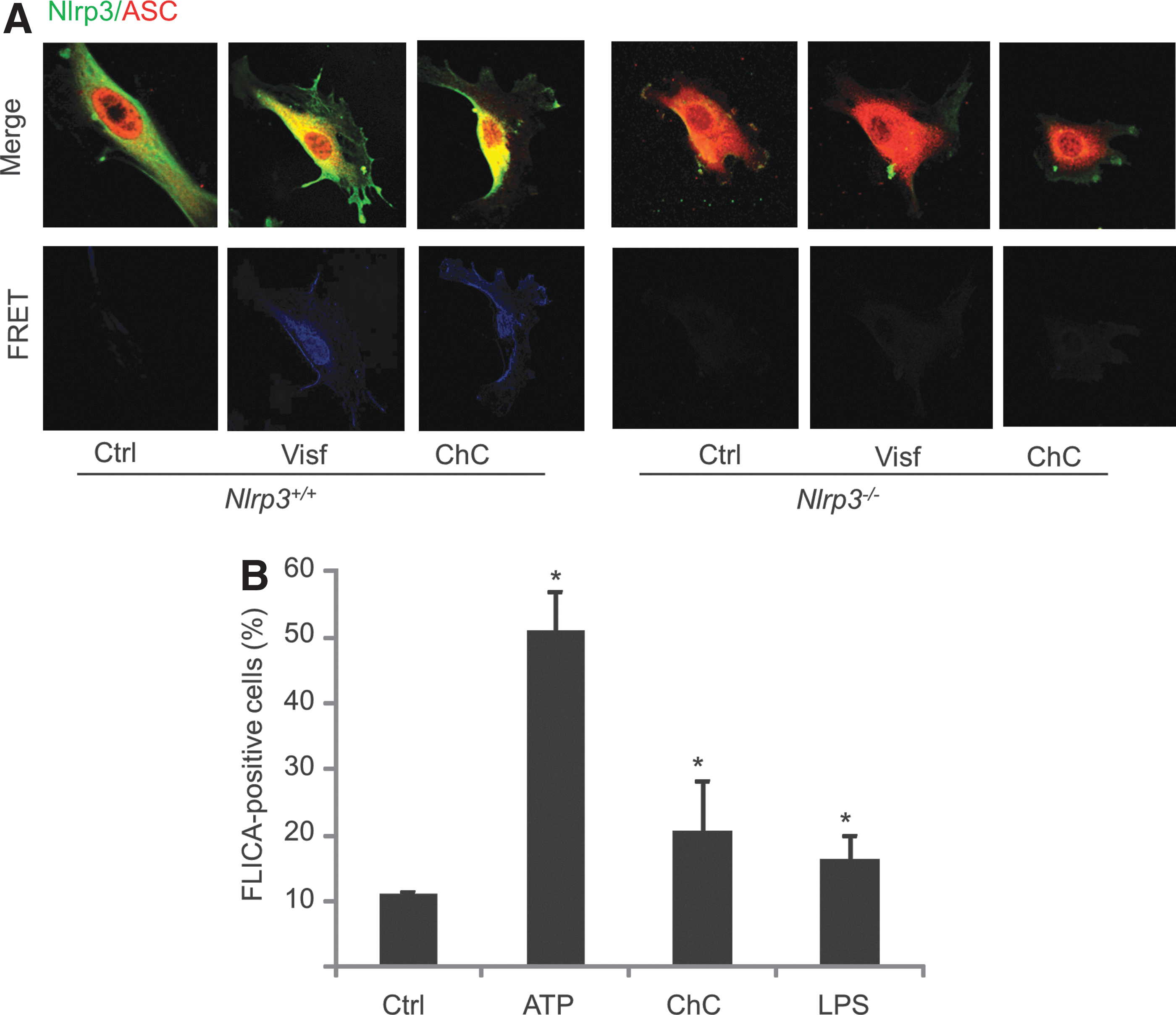
We next examined whether endothelial inflammasomes can be activated by danger signals associated with hypercholesterolemia. The crystal form of cholesterols has recently been considered an endogenous danger signal to trigger Nlrp3 inflammasomes in macrophages (10, 45), but it is unknown whether cholesterol crystals can also activate endothelial inflammasomes. Using primary cultured coronary arterial ECs from mice, we first analyzed the colocalization of inflammasome components by confocal microscopy. As shown in Figure 5A, in wild-type cells, the colocalization of Nlrp3 (Alexa-488 labeled) with Asc (Alexa555 labeled) was markedly increased in response to cholesterol crystal treatment, indicating increased interaction or assembly of these inflammasome molecules. Such an increased interaction was further confirmed by observing increased fluorescence resonance energy transfer (FRET) between Alexa488 and Alexa555 on cholesterol crystal stimulation. As expected, such cholesterol crystal-induced formation of Nlrp3 inflammasomes was abolished in Nlrp3−/− cells. Consistently, as shown in Figure 5B, FLICA analysis demonstrated that caspase-1 activity was elevated by cholesterol crystals in cultured ECs, which is comparable to classic Nlrp3 inflammasome activator ATP or lipopolysaccharide.
Activation of endothelial Nlrp3 inflammasomes by cholesterol crystals impairs endothelium-dependent vasodilation in coronary arteries
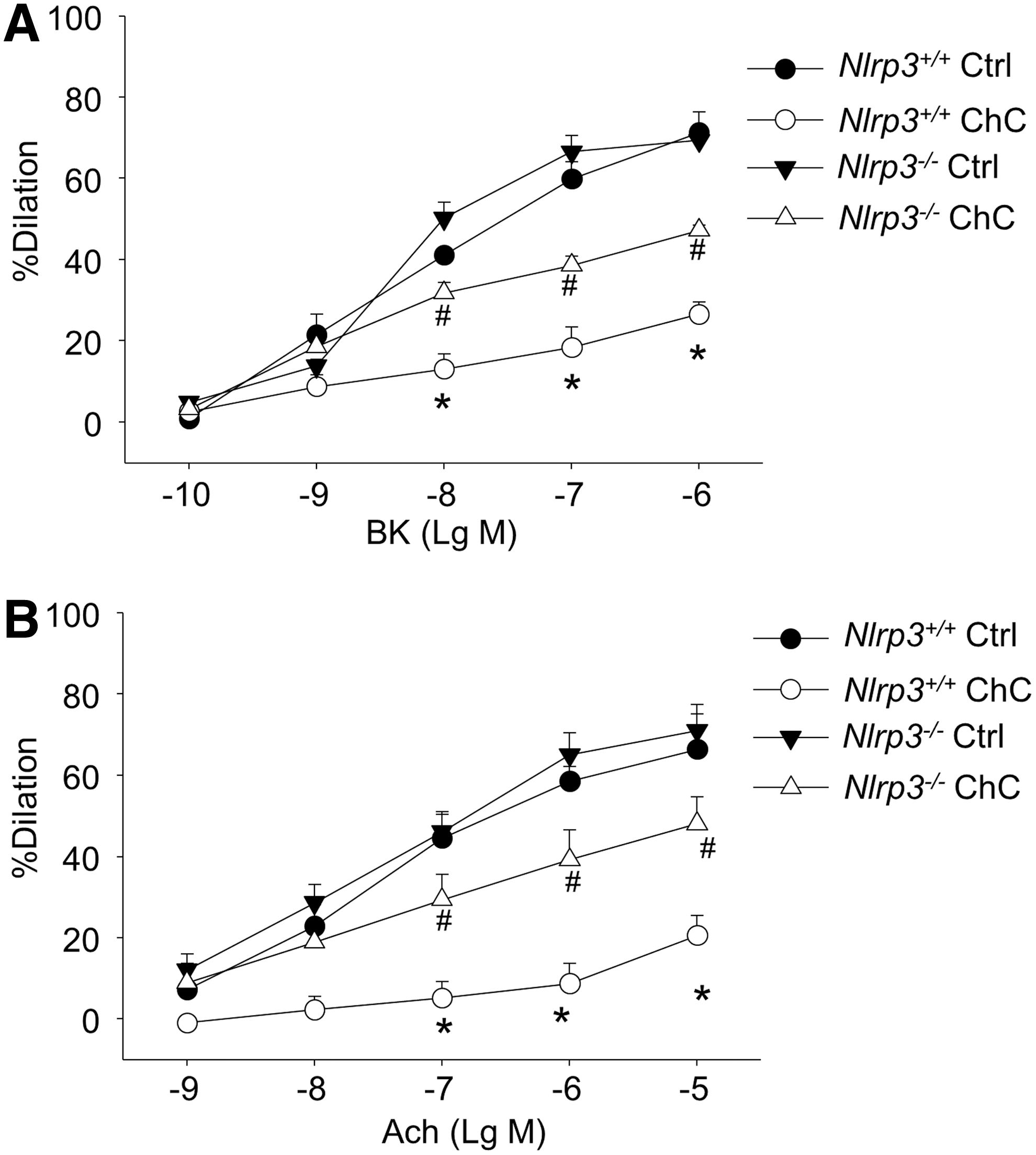
We also investigated whether luminal incubation of cholesterol crystals directly attenuates BK- or Ach-induced endothelium-dependent vasodilatory responses in isolated, perfused, and pressurized coronary arteries from normal Nlrp3+/+ or Nlrp3−/− mice. As shown in Figure 6A, cholesterol crystals markedly attenuated BK-induced dose-dependent vasorelaxation in coronary arteries from Nlrp3+/+ mice, while such a detrimental effect of cholesterol crystals was significantly reversed when Nlrp3 gene was deleted. Similarly, Nlrp3 gene deletion markedly restored endothelium-dependent vasodilation response to Ach (Fig. 6B).
Effects of Nlrp3 inflammasome activation by cholesterol crystals on superoxide and nitric oxide production and cell survival in ECs
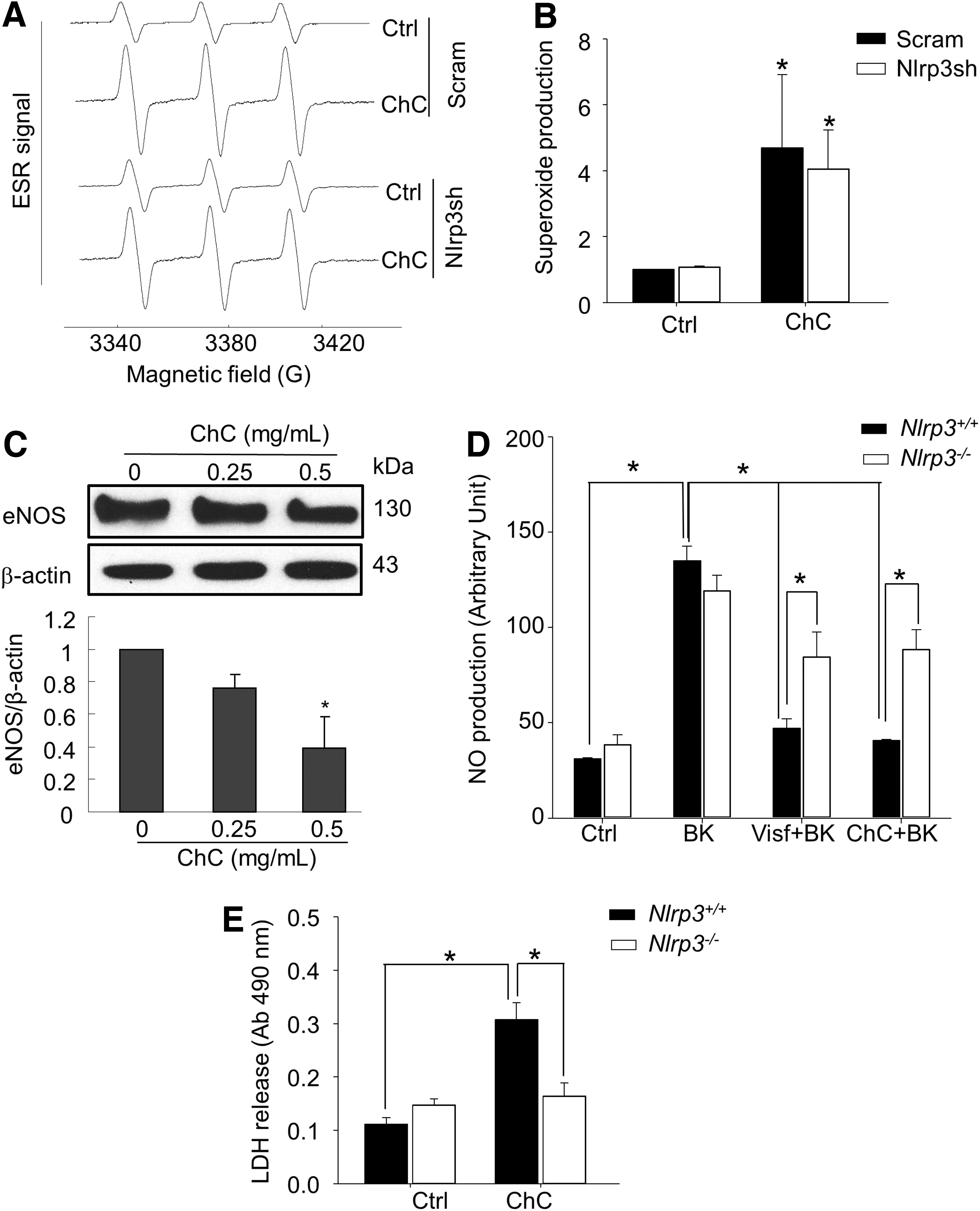
We then examined whether the detrimental action of activated Nlrp3 inflammasomes on endothelial function is associated with superoxide (O2 •−) production or cell death in ECs. We first used electron spin resonance (ESR) spectrometric analysis to determine the production of O2 •− in ECs with or without Nlrp3 shRNA transfection. Figure 7A depicts representative ESR spectrographs of O2 •− production as trapped by a spin trap probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH) under different treatments. As shown in summarized data in Figure 7B, cholesterol crystal treatment significantly increased O2 •− production in ECs. However, such cholesterol crystal-induced O2 •− production in ECs was not altered by Nlrp3 gene silencing, suggesting that O2 •− production may be an upstream event of inflammasome activation (Nlrp3 silencing efficiency was shown in Supplementary Fig. S5). On the other hand, we demonstrated that incubation of ECs with cholesterol crystals decreased the protein expression of endothelial nitric oxide synthase (eNOS), the nitric oxide (NO)-producing enzyme in ECs (Fig. 7C). Consistently, we found that cholesterol crystal inhibited BK-induced NO production in coronary arterial ECs from Nlrp3+/+ mice, whereas Nlrp3 gene deletion significantly reversed such inhibition of NO production by cholesterol crystal (Fig. 7D). This inhibitory action of cholesterol crystals is similar to that of visfatin, an injurious adipokine (Fig. 7D). In addition, we also examined the cell viability of ECs by lactate dehydrogenase (LDH) release assay. Cholesterol crystal treatment markedly increased the release of the cytosolic protein LDH into the culture medium in primary cultured coronary arterial ECs from Nlrp3+/+ mice, which was completely abolished by Nlrp3 deficiency (Fig. 7E). Similarly, inhibition of caspase-1 also abolished cholesterol crystal-induced LDH release in ECs (Supplementary Fig. S6). Thus, cholesterol crystal treatment of ECs increases their pryroptosis, a form of cell death that depends on Nrlp3 inflammasome and caspase-1 activity.
Discussion
The primary goal of this study is to reveal whether Nlrp3 inflammasome activation in ECs contributes to endothelial dysfunction associated with dyslipidemia in the early stage of atherogenesis. An in vivo animal model of acute hypercholesterolemia and ex vivo vascular reactivity and in vitro cell studies demonstrated that activation of Nlrp3 inflammasome under hypercholesterolemia condition or by cholesterol crystal stimulation causes endothelial dysfunction in mouse coronary arteries. Such detrimental action of Nlrp3 inflammasome is associated with increased pyroptosis and the HMGB1 but not with IL-1β signaling in the ECs. These results for the first time demonstrate a novel action of endothelial inflammasomes in endothelial dysfunction beyond canonical inflammation.
It have been well established that arterial ECs are vulnerable to the atherogenic effects of hypercholesterolemia (15). A hallmark of endothelial dysfunction or injury associated with hypercholesterolemia is a reduced capacity to release endothelium-derived relaxing factor, primarily NO produced by eNOS (15). Consistently, this study demonstrated that endothelium-dependent vasodilation responses to typical dilators, including BK and Ach, are impaired in a mouse model of acute hypercholesterolemia. We further revealed that Nlrp3 gene deletion markedly ameliorates such hypercholesterolemia-induced impairment in coronary arteries. In contrast, endothelium-independent vasodilator responses of coronary arteries to SNP were similar in both wild-type and Nlrp3-deficient mice. To our knowledge, our findings for the first time reveal a crucial role of Nlrp3 inflammasome in promoting endothelial dysfunction, possibly via inhibiting eNOS-derived NO signaling.
It has been indicated that the NLRP3 inflammasome/IL-1β pathway promotes atherogenesis in humans and mice (11, 12). Caspase-1 was also demonstrated to promotes atherosclerosis in apolipoprotein E-null [Apoe(−/−)] mice by enhancing the inflammatory status of the lesion through a mechanism likely involving activation of lesion-associated immune cells and expression of cytokines, including interferon-γ and IL-1β (4, 44). In addition to Nlrp3, Nlrp1 has also been implicated in the regulation of immune-inflammatory processes in arterial ECs, contributing to endothelial activation and vessel remodeling (3). All these previous studies have emphasized the role of Nlrp3 inflammasome/caspase-1/IL-1β signaling in immune cells. However, in this study, we demonstrated that inhibition of caspase-1 but not antagonism of IL-1β receptor in vivo protected endothelial dysfunction in coronary arteries from mice with acute hypercholesterolemia. These findings suggest that other caspase-1-depenent signaling is involved. The production of DAMPs is another important feature of intracellular inflammatory machinery activation (33, 35). HMGB1 is a prototype DAMP that has been shown to participate in the regulation of a variety of cell functions and activities, including cell survival and endothelial progenitor cell homing. In this study, we found that hypercholesterolemia increased the caspase-1 activity and enhanced production of HMGB1 in the coronary arterial endothelium, while Nlrp3 gene deficiency abolished such hypercholesterolemia-induced increases (Fig. 3). We also demonstrated that recombinant HMGB1 proteins directly impaired endothelium-dependent vasodilation regardless of Nlrp3 expression, suggesting that HMGB1 signaling is downstream of Nlrp3 inflammasome activation (Fig. 4). There is a concern that recombinant HMGB1 may directly act on smooth muscle cells to inhibit smooth muscle relaxation. Given the fact that impairment of endothelium-dependent but not endothelium-independent dilation is ameliorated by Nlrp3 gene deletion (Fig. 1), it is plausible that Nlrp3/caspase-1/HMGB1 signaling is directly involved in endothelial dysfunction during hypercholesterolemia. One possible mechanism for HMGB1 in hypercholesterolemia-induced endothelial dysfunction in coronary arteries may be due to the activation of receptor for advanced glycation end products, which has been identified as an HMGB1 interacting receptor and involved in diabetes-associated endothelial dysfunction (36). Together, our data implicate that the role of Nlrp3 inflammasome in atherogenesis may depend on cell types and disease stages: In immune cells, Nlrp3 inflammasome activation is likely to be linked with IL1 receptor-mediated signaling, while in vascular cells Nlrp3 inflammasome may be associated with caspase-1 HMGB1 signaling, which may represent a very early signaling event during atherogenesis.
Our findings further demonstrated that activation of endothelial Nlrp3 inflammasome is sufficient to trigger endothelial dysfunction. Cholesterol crystals have been considered an endogenous danger signal to trigger sterile inflammation that initiates atherogenesis (10, 45). In this study, we further demonstrated that cholesterol crystals could induce formation and activation in cultured ECs. Cholesterol crystals also induced impairment in endothelium-dependent vasodilation. Our finding is consistent with a previous report that cholesterol crystals impairs endothelial barrier function in cultured human aortic ECs, possibly via cAMP and RhoA activation (1). Together, these data strongly suggest that activation of Nlrp3 inflammasome in ECs is able to cause endothelial dysfunction in the absence of immune cells. It should be noticed that Nlrp3 deficiency only partially reversed the detrimental effect of luminal incubation of cholesterol crystals in coronary arteries, whereas it almost completely reversed P407-induced endothelial dysfunction (Fig. 1). This may be due to the difference in the experimental settings (in vivo hypercholesterolemia model vs. ex vivo luminal perfusion of cholesterol crystals). In addition to hypercholesterolemia, P407 administration also induces hypertriglyceridemia (20), which may also trigger Nlrp3 inflammasome activation and its subsequent detrimental effects on endothelial functions. Nonetheless, these data suggest that cholesterol crystals induce both Nlrp3-dependent and Nlrp3-independent impairments in EC functions. We further explored the molecular mechanisms mediating the production of these novel effects of Nlrp3 inflammasome in ECs. The production of ROS was shown to be one of the major early factors mediating death factor-induced endothelial injury (5, 6, 40, 49, 50). NADPH oxidase-derived ROS associated with ceramide-enriched membrane raft clustering (21, 47, 52) were found to primarily contribute to the death factor or adipokine visfatin-mediated endothelial dysfunction (13, 17, 46, 48, 52, 53). We recently demonstrated that ROS is able to activate Nlrp3 inflammasomes in ECs (44). In this study, cholesterol crystals markedly increased endothelial O2 •− production; however, such O2 •− production was not affected by Nlrp3 gene silencing. Together, these data suggest that O2 •− production is an upstream triggering mechanism for activation of endothelial Nlrp3 inflammasomes. The Nlrp3-independent impairment may be associated with cholesterol crystal-induced production O2 •− production, which results in direct trapping of NO decreasing NO bioavailability. On the other hand, our data for the first time suggest that cholesterol crystal-induced O2 •− production also causes impairment beyond its trapping action by activating endothelial Nlrp3 inflammasomes, leading to Nlrp3-dependent inhibition of endothelium-dependent dilatory responses. In this study, we observed that cholesterol crystal treatment markedly decreased eNOS expression in ECs (Fig. 7C), which suggests that activation of Nlrp3 inflammasome could lead to a downregulation of expression of this NO-producing enzyme. Consistently, cholesterol crystal pretreatment blunted agonist (BK)-induced NO production in ECs (Fig. 7D). However, Nlrp3 gene deletion significantly reversed the cholesterol crystal-induced inhibition of NO production. This inhibitory action of cholesterol crystals is similar to that of visfatin, a known adipokine that produces ROS-dependent impairment on endothelial function and Nlrp3 inflammasome activation (44, 46). Together, these data suggest that Nlrp3-dependent downregulation of eNOS activity is another contributing mechanism that is responsible for cholesterol crystal-induced decreases in NO bioavailability and subsequent endothelial dysfunction. Activation of Nlrp3 can induce cell death via caspase-dependent mechanisms (27, 34). Previous studies have shown that activation of Nlrp3 inflammasome has also been shown to induce cell death in human retinal ECs (28). Nlrp3 deficiency prevented caspase 3-mediated apoptosis by upregulating autophagy, specifically mitophagy in lungs and lung ECs during hyperoxia (56). Here, we further demonstrated that cholesterol crystal markedly induced Nlrp3-dependent pyroptosis in ECs. However, it remains whether Nlrp3-dependent downregulation of eNOS is directly associated with enhanced pyroptosis in ECs. It should be noted that more mechanistic studies are needed to further dissect the pathogenic mechanisms for Nlrp3-dependent actions in endothelial dysfunction both in vitro and in vivo. To achieve such goals, endothelial-specific Nlrp3 knockout mice will be implemented in future studies. Nonetheless, our data support the view that at least Nlrp3/caspase-1-dependent downregulation of eNOS activity or EC pyroptosis may initiate or contribute to the development of endothelial dysfunction in coronary arteries.
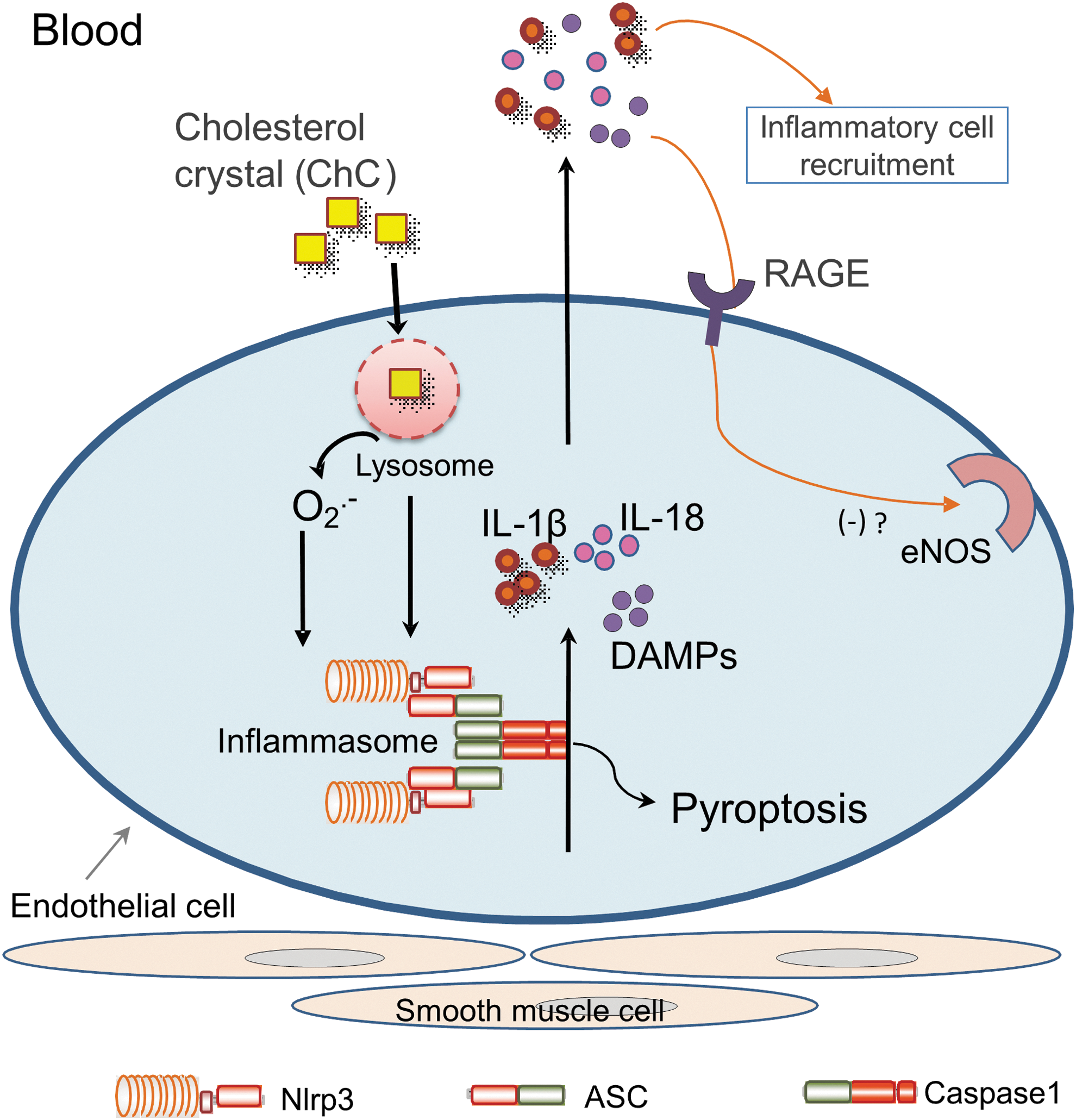
In summary, as described in a diagram in Figure 8, assembly and activation of endothelial Nlrp3 inflammasome induced by hypercholesterolemia leads to caspase-1 activation and increased expression and release of DAMPs such as HMGB1 in ECs, which may directly induce endothelial dysfunction. Our data support a novel view that the pathogenic role of Nlrp3 inflammasome activation in endothelial dysfunction and vascular injury in response to various danger factors is not only due to instigation of canonical sterile inflammatory response but also due to its direct actions on ECs beyond canonical inflammation. These non-canonical actions may include pryoptosis, release of DAMPs, and interference with endothelium-dependent vasodilation. Our findings provide new insights that therapeutic strategies mainly targeting inflammation may only have partial effects when the intracellular inflammatory machinery, the inflammasome, is triggered in the vasculature. This is because non-canonical actions occur before or concurrently with classical inflammatory responses, and, ultimately, may initiate or exacerbate vascular injury during atherogenesis under coronary artery disease risk factors such as hypercholesterolemia.
Materials and Methods
Mice
Nlrp3-deficient (Nlrp3−/− ) and wild-type (Nlrp3+/+ ) mice (C57BL6 background) were obtained from Jackson Laboratories and used in this study. The mouse genotype was confirmed by reverse transcription–polymerase chain reaction. All experimental protocols were reviewed and approved by the Animal Care Committee of Virginia Commonwealth University. All animals were provided standard rodent chow and water ad libitum in a temperature-controlled room.
Acute hypercholesterolemia in mice
Poloxamer 407 (P407; Sigma-Aldrich) was used to induce acute hypercholesterolemia in 6–8 week-old male mice as previously described (19). In brief, Nlrp3−/− or Nlrp3+/+ mice were intraperitoneally injected with P407 (0.5 g/kg), while control groups of mice were administrated with the same volume of sterile saline. Some groups of Nlrp3+/+ mice were also intraperitoneally injected with indicated inhibitors or antagonists 30 min before P407 administration. Twenty-four hours after P407 or normal saline treatment, blood samples of mice were obtained by periorbital sampling when mice were under ether anesthesia. Then, the plasma samples were isolated by centrifugation and frozen at −80°C until analysis. After blood collection, the animals under ether anesthesia were sacrificed by cervical dislocation. The hearts with coronary artery were obtained for assaying endothelial function or preparation of frozen sections.
Vascular reactivity in in vitro perfused and pressurized mouse coronary arteries
Mouse septal or left descending coronary arteries (∼100 μm) were dissected in ice-cold physiological saline solution (PSS) containing the following composition (in mM): NaCl, 119; KCl, 4.7; CaCl2, 1.6; MgSO4, 1.17; NaH2PO4, 1.18; NaHCO3, 24; ethylenediamintetraaceticacid (EDTA), 0.026; and glucose, 5.5, pH 7.4 and carefully cleaned of fat and connective tissues under a dissection microscope. Dissected arteries were immediately transferred to a water-jacketed perfusion chamber and cannulated with two glass micropipettes at their in situ length as previously described (54). Any branches of coronary arteries were carefully tied with sutures. The outflow cannula was clamped, and the arteries were pressurized to 60 mm Hg and equilibrated in PSS at 37°C. PSS in the bath was continuously bubbled with a gas mixture of 95% O2 and 5% CO2 throughout the experiment. After a 1 h equilibration period, the arteries were precontracted with U46619 (10–100 nM) until a ∼50% of decrease in resting diameter was reached. Once steady-state contraction was obtained, cumulative dose–response curves to the endothelium-dependent vasodilator BK (10−10–10−6 M) or Ach (10−9–10−5 M) were determined by measuring changes in internal diameter. The vasodilator response was expressed as the percent relaxation of U46619-induced pre-contraction based on changes in arterial internal diameter. Internal arterial diameter was measured with a video system composed of a stereomicroscope (Leica MZ8), a charge-coupled device camera (KP-MI AU; Hitachi), a video monitor (VM-1220U; Hitachi), a video measuring apparatus (VIA-170; Boeckeler Instrument), and a video printer (UP890 MD; Sony). The arterial images were recorded continuously with a videocassette recorder (M-674; Toshiba).
Immunofluorescent staining of coronary arteries
The hearts with intact coronary arteries were obtained for coronary dissection or frozen in liquid nitrogen for preparation of frozen section slides. The expression of indicated proteins was detected in mouse heart frozen slides using corresponding immunofluorescence labeled antibodies as previously described (2, 23, 41). Briefly, the tissues of hearts with coronary arteries were frozen in Tissue-Tek OCT, cut by cryostat into 10 μm sections, and mounted on Superfrost/Plus slides for immunofluorescent staining. After fixation with acetone, the slides were incubated with indicated antibodies (1:50) overnight at 4°C. After incubation with primary antibodies, the slides were washed and labeled with corresponding Alexa488 or Alexa555 conjugated secondary antibodies (Invitrogen) (200) or labeled with FLICA probes from caspase-1 FLICA™ kit (Immunochemistry). The slides were then washed, mounted, and subjected to confocal microscopic analysis (Fluoview FV1000; Olympus).
Cell culture
Isolation and characterization of mouse coronary arterial ECs were performed as previously described (22, 39). Briefly, the hearts was excised from six-week-old male Nlrp3−/−
mice or their wild-type littermates with an intact aortic arch and immersed in a petri-dish filled with ice-cold Krebs-Henseleit (KH) solution (mM): 118 NaCl, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 2.5 CaCl2, and 11 glucose. Surrounding fat and connective tissue were removed from the heart. The cleaned heart with intact aorta was transferred to another petri-dish with fresh KH solution. A 25-gauge needle filled with Hank's balanced salt solution (HBSS; in mM: 5.0 KCl, 0.3 KH2PO4, 138 NaCl, 4.0 NaHCO3, 0.3 Na2HPO4·7H2O, 5.6
The mouse microvascular EC line was purchased from ATCC (also known as EOMA cells). This cell line was originally isolated from mouse hemangioendothelioma. These vascular ECs were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco), supplemented with 10% FBS (Gibco) and 1% penicillin–streptomycin (Gibco) in humidified 95% air and 5% CO2 mixture at 37°C. Cells were passaged by trypsinization (Trypsin/EDTA; Sigma), followed by dilution in DMEM medium containing 10% FBS. Cells were used for experiments between passages 6 and 13.
Confocal microscopic and FRET analysis
For confocal analysis, cells were grown on glass coverslips, stimulated or unstimulated, fixed in 4% paraformaldehyde in phosphate-buffer saline (PBS) for 15 min as described (55). After being permeabilized with 0.1% Triton X-100/PBS and rinsed with PBS, the cells were incubated overnight at 4°C with indicated primary antibodies: goat anti-Nlrp3 (1:200; Abcam) and rabbit anti-Asc (1:200; Abcam). After washing, these slides were incubated with either Alexa-488- or Alexa-555-labeled secondary antibodies for 1 h at room temperature. The slides were mounted and subjected to examinations using a confocal laser scanning microscope (Fluoview FV1000; Olympus). An acceptor bleaching protocol was used to measure the FRET efficiency between Alexa488-Nlrp3/Alexa555-Asc as previously described (18, 22). Briefly, after the pre-bleaching image was normally taken, the laser intensity at the excitement wavelength of the acceptor (Alexa555) was increased from 50% to 98% and continued to excite the cell sample for 2 min to bleach the acceptor fluorescence. After the intensity of the excitement laser for acceptor was adjusted back to 50%, the post-bleaching image was taken for Alexa488. A FRET image was obtained by subtraction of the pre-bleaching images from the post-bleaching images and given a dark blue color.
Preparation of cholesterol crystals
Cholesterol (Sigma; cat. C8667) dissolved in 95% ethanol (12.5 g/l) was heated to 60°C, filtered through Whatman filter paper while still warm, and left at room temperature to allow crystallization to proceed. Then, relatively large cholesterol crystals were formed and collected by filtering, autoclaved, ground using a sterile mortar and a pestle to yield a size range of 1–10 μm, and stored at −20°C until use.
FLICA assay of caspase-1 activity in cell culture
Cells were labeled with FAM-YVAD-fmk caspase-1 FLICA™ kit (Immunochemistry), which binds the active form of caspase-1. Flow cytometric analysis was performed according to the manufacturer's manual. In brief, cells were incubated with FLICA and propidium iodide (PI) at room temperature for 1 h. After two washes of cells with PBS, FLICA and PI fluorescence were analyzed by flow cytometry with a Guava EasyCyte (Guava Technologies). The percentage for FLICA-positive cells was used to represent the relative active caspase-1 level as previously described (36).
Nucleofection
Transfection of Nlrp3 shRNA plasmids (Origene) was performed using a 4D Nucleofector X-Unit (Lonza) according to the manufacturer's instructions as previously described (42). Briefly, cells were trypsinized and centrifuged at 90 g for 10 min. The cell pellet was resuspended in 100 μl SF Nucleofection solution (Lonza) (with the program code DS198). The program was chosen based on the fact that Nucleofection efficiency was more than 80% as analyzed by flow cytometry using control GFP plasmids. For each Nucleofection sample, 2 μg plasmid DNA was added in 20 μl SF Nucleofection solution. After Nucleofection, cells were cultured in DMEM medium for 24 h and then cells were ready for the treatments.
ESR spectrometric detection of O2 •−
ESR detection of O2 •− was performed as previously described (51). In brief, protein homogenates was gently isolated and resuspended with modified Krebs–4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer containing deferoximine (100 μM; Sigma) and diethyldithiocarbamate (5 μM; Sigma). A spin trap, CMH (Noxygen) (1 mM final concentration), was then added to the mixture in the presence or absence of manganese-dependent superoxide dismutase (SOD, 200 U/ml; Sigma). The cell mixture loaded in glass capillaries was immediately analyzed for O2 •− production at each minute for 10 min using a Miniscope MS200 ESR spectrometer (Magnettech, Germany). The ESR settings were as follows: biofield, 3350; field sweep, 60G; microwave frequency, 9.78 GHz; microwave power, 20 mW; modulation amplitude, 3G; 4096 points of resolution; receiver gain, 100; and kinetic time, 10 min. The SOD-inhibitable signals were normalized by protein concentration and compared among different experimental groups.
Western blot analysis
Western blot analysis was performed as previously described (23). In brief, proteins from the ECs were extracted using sucrose buffer (20 mM HEPES, 1 mM EDTA, 255 mM sucrose, cocktail o protease inhibitors [Roche], pH 7.4). After boiling for 5 min at 95°C in a 5× loading buffer, 30 μg of total proteins were separated by a 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The proteins of these samples were then electrophoretically transferred at 100 V for 1 h onto a polyvinylidene fluoride membrane (Bio-Rad). The membrane was blocked with 5% nonfat milk in Tris-buffered saline-Tween 20. After washing, the membrane was probed with 1:1000 dilution of primary mouse or rabbit antibodies against eNOS (Cell Signaling) or β-actin (Santa Cruz) overnight at 4°C followed by incubation with horseradish peroxidase-labeled immunoglobulin G (1:5000). The immuno-reactive bands were detected by chemiluminescence methods and visualized on Kodak Omat X-ray films. Densitometric analysis of the images obtained from X-ray films was performed using the Image J software (NIH).
Determination of NO production
Cells were cultured in 12-well plates (105 cells/well) and incubated with 10 μM nonfluorescent dye 4-amino-5-methylamino-2′,7′-difluorescein (Molecular Probes) for 30 min at 37°C. Cells were scraped down, and the green fluorescence of cells was immediately analyzed by flow cytometry using a flow cytometer (GUAVA) (55).
LDH assay of cell viability
For analysis of cell survival, cells were cultured in 96-well tissue culture plates. After stimulation, plasma membrane integrity was assayed by measuring the release of cytosolic protein LDH. The LDH released into the supernatant was analyzed by using a LDH assaying kit (Promega). The absorbance (490) nm was determined by using a microplate reader (BioTek FLx800).
Statistical analysis
Data are presented as means with standard error of the means. Significant differences between and within multiple groups were examined using one-way analysis of variance test followed by Duncan's multiple-range test. A Students' t test was used to detect significant differences between two groups. The statistical analysis was performed by SigmaStat 3.5 software (Systat Software). p<0.05 was considered statistically significant.
Footnotes
Acknowledgments
This study was supported by National Institutes of Health R01 grants (HL057244, HL075316, and HL091464 to P.-L.L.; HL122937 and HL122769 to Y.Z.), National Institutes of Health CTSA grant UL1TR000058, and VCU CCTR Endowment Fund to Y.Z.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.