
Abstract
Introduction
I
These findings highlight a promising new and safer therapeutic approach in the management of vascular inflammation that precedes or accompanies most cardiovascular disorders using two clinically available drugs. The synergistic anti-inflammatory effect was the consequence of increased RXRα, PPARα, and PPARγ expression as well as of RXRα/PPARα and RXRα/PPARγ interactions, leading to the inhibition of Nox5-mediated signaling pathways. Moreover, this new therapeutic regime results in minimal drug-associated side effects since low doses of both drugs are used, and rosuvastatin administration may beneficially counteract the dyslipidemic state linked to the chronic treatment with Bexarotene.
Ang-II is implicated in atherogenesis (9), and we demonstrated that 4 h of exposure to Ang-II in vivo caused arteriolar leukocyte adhesion in the mesenteric microcirculation of the rat (2). Notably, mononuclear leukocyte recruitment by Ang-II was found to be mediated largely by tumor necrosis factor-α (TNFα) and the subsequent increased endothelial expression of fractalkine (CX3CL1)(37, 54). Consequently, pharmacological modulation of inflammatory cell infiltration of the subendothelial space may impede the atherogenic process associated with different cardiovascular risk factors related to the rennin-angiotensin system.
Statins are a group of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors that were primarily used because of their lipid-lowering properties due to their capacity to block cholesterol biosynthesis (1). Thereafter, their ability to reduce cardiovascular mortality and stroke was found to go beyond their lipid-lowering attributes. Indeed, when rosuvastatin (Rosu) was administered to healthy subjects without hyperlipidemia but with elevated high-sensitivity C-reactive protein levels, the incidence of major cardiovascular events was reduced (52). These so-called pleiotropic effects of statins include modulation of inflammatory reactions and anti-oxidant properties, which may additionally play important roles in statin-mediated cardiovascular protection (38). Accordingly, there is evidence indicating that statins favorably influence endothelial cell function (38) and impair leukocyte trafficking at sites of inflammation (59). Furthermore, a growing body of experimental data suggests a potential interplay between statins and peroxisome proliferator-activated receptors (PPARs) (3, 6), and activation of PPARs can downregulate the expression of CAMs, proinflammatory genes, and leukocyte-endothelial cell interactions (40). Though statins in general are well tolerated, myopathy and acute renal events have been a significant concern with the use of high-potency statin drugs, in particular simvastatin and Rosu (8, 15, 23). Since these adverse effects are frequently dose related (8, 15, 23), there remains an overt need to identify agents that, when combined with statins, can provide the greatest benefit on cardiovascular disease with the least added risk.
Bexarotene (Bex) is a retinoid X receptor (RXR)-selective agonist currently used in the treatment of cutaneous T-cell lymphoma. We have previously shown that Bex can inhibit mononuclear leukocyte attachment to stimulated arterial endothelial cells through downregulation of redox-sensitive pathways and via RXR/PPARγ interaction (12, 55). Indeed, it is well established that PPARs form permissive RXR heterodimers that synergistically respond to agonists of RXR and the partner receptor (49). Unfortunately, Bex treatment is associated with unavoidable and dose-limiting side effects, in particular hypertriglyceridemia, hypercholesterolemia, and, to a lesser extent, hypothyroidism (7, 61). However, Rosu administration can reduce triglyceride levels (13) and the U.K. consensus statement on safe clinical prescribing of Bex recommend the use of this statin to prevent the dyslipidemic effects associated with administration of the RXR agonist (57).
In an attempt to identify more effective strategies to treat and prevent atherosclerosis, coronary heart disease, and co-morbid metabolic disorders characterized by endothelial dysfunction, we have evaluated the effect of combined treatment with Rosu and Bex. In this study, we provide both in vitro and in vivo evidence that the combination of Rosu+Bex at suboptimal concentration/doses provokes a clear impairment in Ang-II-induced mononuclear cell (MC) adhesion to the arterial endothelium. The underlying mechanisms involved in this beneficial synergism were also explored. We found that Nox5 downregulation and subsequent RhoA inactivation were involved in these responses. Furthermore, increased RXR/PPARα and RXR/PPARγ interactions were, in part, responsible for the observed anti-inflammatory activity.
Results
A combination of Rosu and Bex at suboptimal concentrations reduces Ang-II-induced human umbilical artery endothelial cell MC adhesion
Initially, we evaluated the effect of Ang-II challenge on mononuclear leukocyte-endothelial cell interactions in vitro using the dynamic flow chamber assay. Thus, freshly isolated human MCs were perfused across human umbilical artery endothelial cell (HUAEC) monolayers stimulated or not with Ang-II (1 μM) for 4 h. Ang-II caused a significant increase in mononuclear leukocyte adhesion to arterial endothelial cells (Fig. 1). To assess the effect of Rosu on Ang-II-induced MC recruitment, HUAECs were pretreated with the statin at two different concentrations (10 and 30 nM), 20 h before Ang-II stimulation. While 10 nM Rosu had no impact on Ang-II-induced MC recruitment, preincubation with 30 nM Rosu significantly reduced Ang-II-induced mononuclear leukocyte adhesion (Fig. 1A). A similar approach was followed to test the effect of Bex preincubation on Ang-II-induced responses, based on previous observations (55). As anticipated, 1 μM but not 0.3 μM Bex significantly inhibited MC adhesion to Ang-II-stimulated HUAECs (Fig. 1B). To evaluate the potential synergism of both drugs, different combinations of Rosu and Bex were assessed. All the combinations tested significantly impaired Ang-II-induced MC attachment to the arterial endothelium (Fig. 1C). Unexpectedly, the combination of Rosu at 10 nM and Bex at 0.3 μM, which displayed no significant effect when either treatment was tested alone, reduced adhesion by 51% (Fig. 1C). Consequently, in all subsequent in vitro experiments, this combination of Rosu and Bex (designated Rosu+Bex) was employed to explore the underlying mechanisms involved in this response.
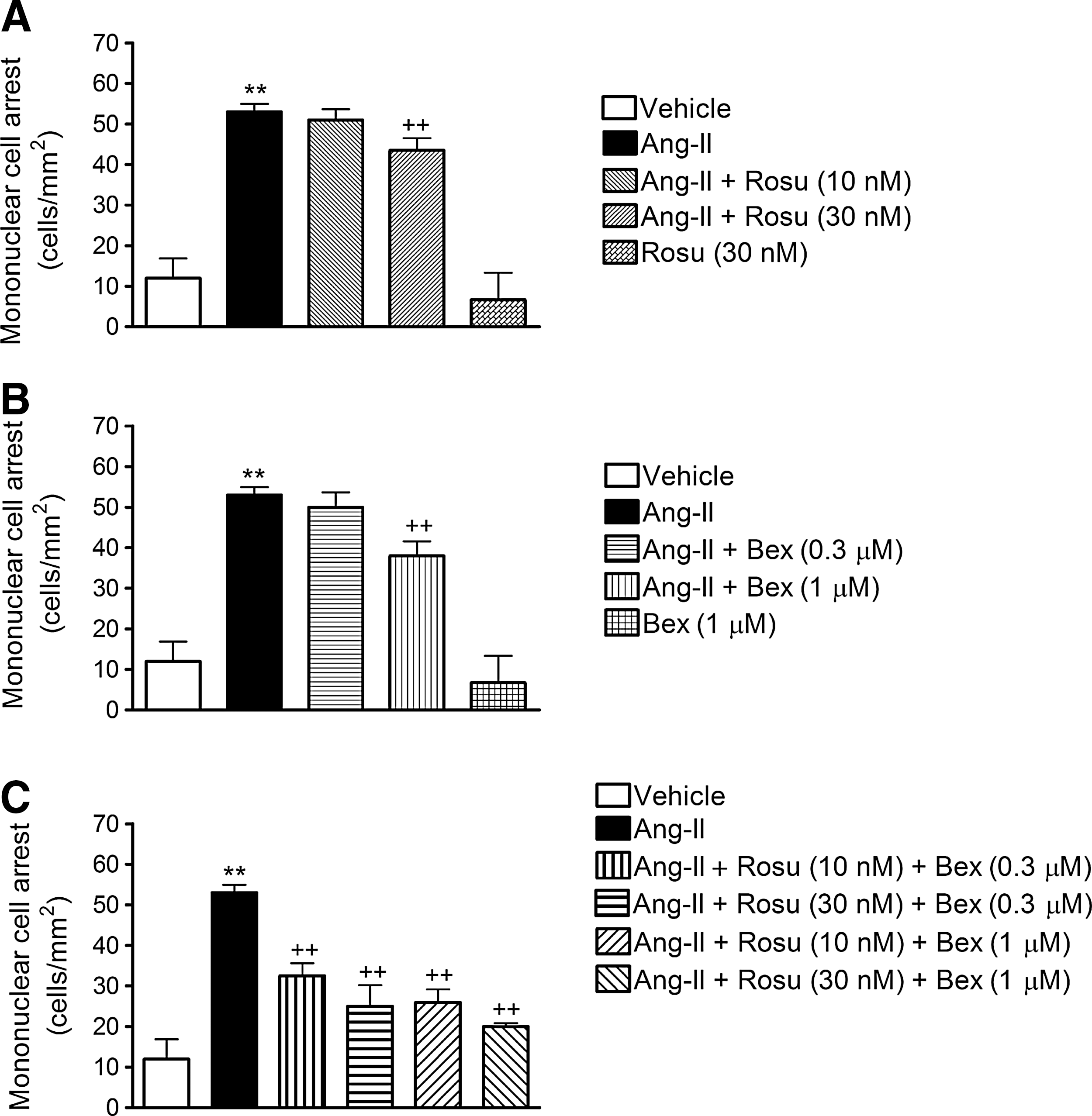
In addition, since there is evidence to support that Ang-II can decrease AT1 receptor expression, although in vascular smooth muscle cells (44), we evaluated this possibility. As illustrated in Supplementary Figure S1 (Supplementary Data are available online at
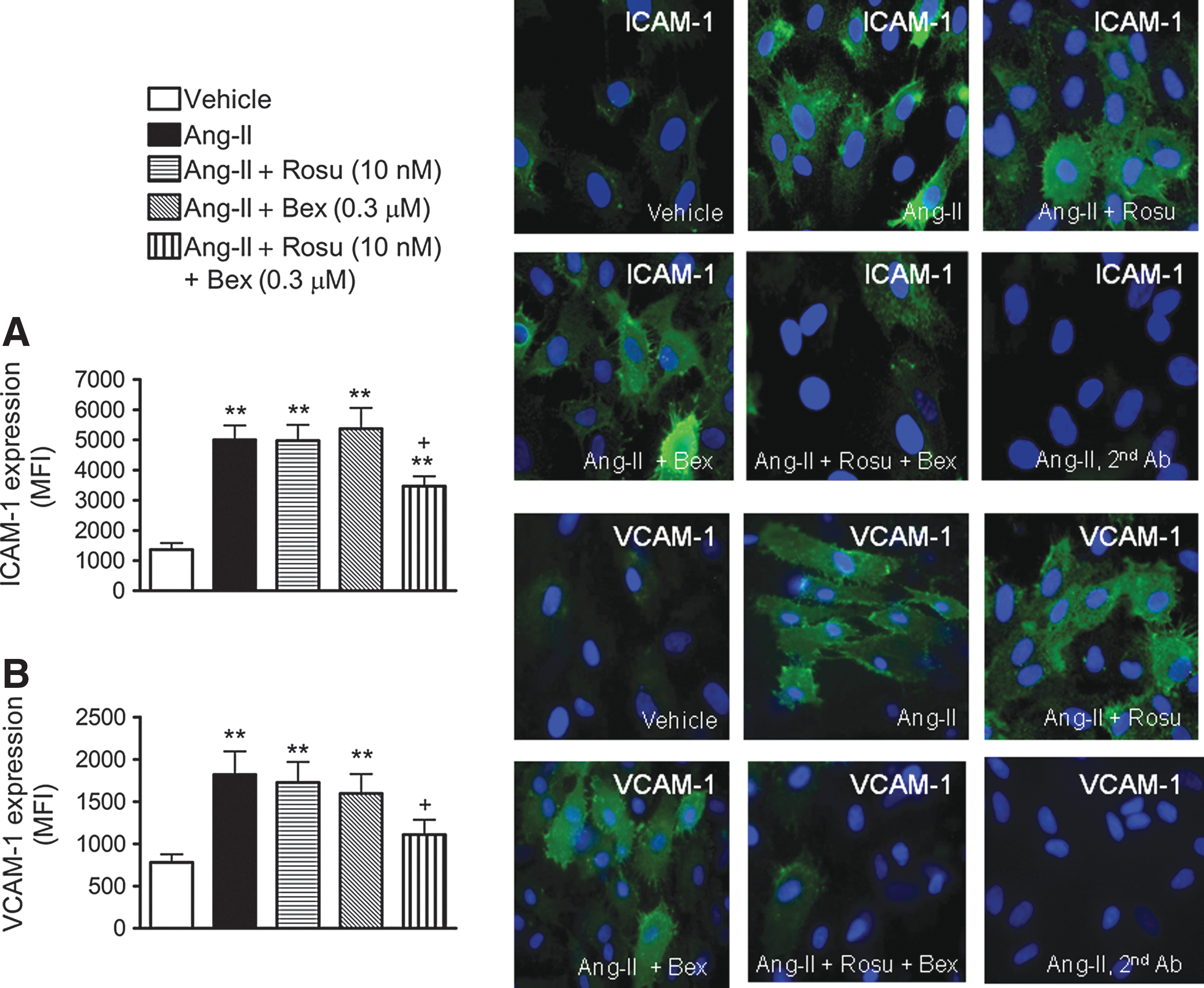
Decreased expression of ICAM-1 and VCAM-1 is involved in the reduced Ang-II-induced HUAEC-MC adhesion caused by suboptimal concentrations of Rosu+Bex
To investigate whether the inhibitory effects exerted by Rosu+Bex on Ang-II-induced mononuclear leukocyte adhesion were mediated through modulation of endothelial CAM expression, intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) protein expression were determined by flow cytometry and immunocytochemistry. Stimulation with Ang-II resulted in a significant upregulation of ICAM-1 and VCAM-1 expression when compared with unstimulated control HUAECs, as measured by both flow cytometry and immunofluorscence (Fig. 2A, B). Pretreatment of cells with Rosu or Bex did not modify the increased endothelial CAM expression induced by the peptide (Fig. 2A, B). In contrast, preincubation of HUAEC with Rosu+Bex resulted in a significant decrease in Ang-II-induced ICAM-1 and VCAM-1 expression, by 42% and 66%, respectively (Fig. 2A, B).
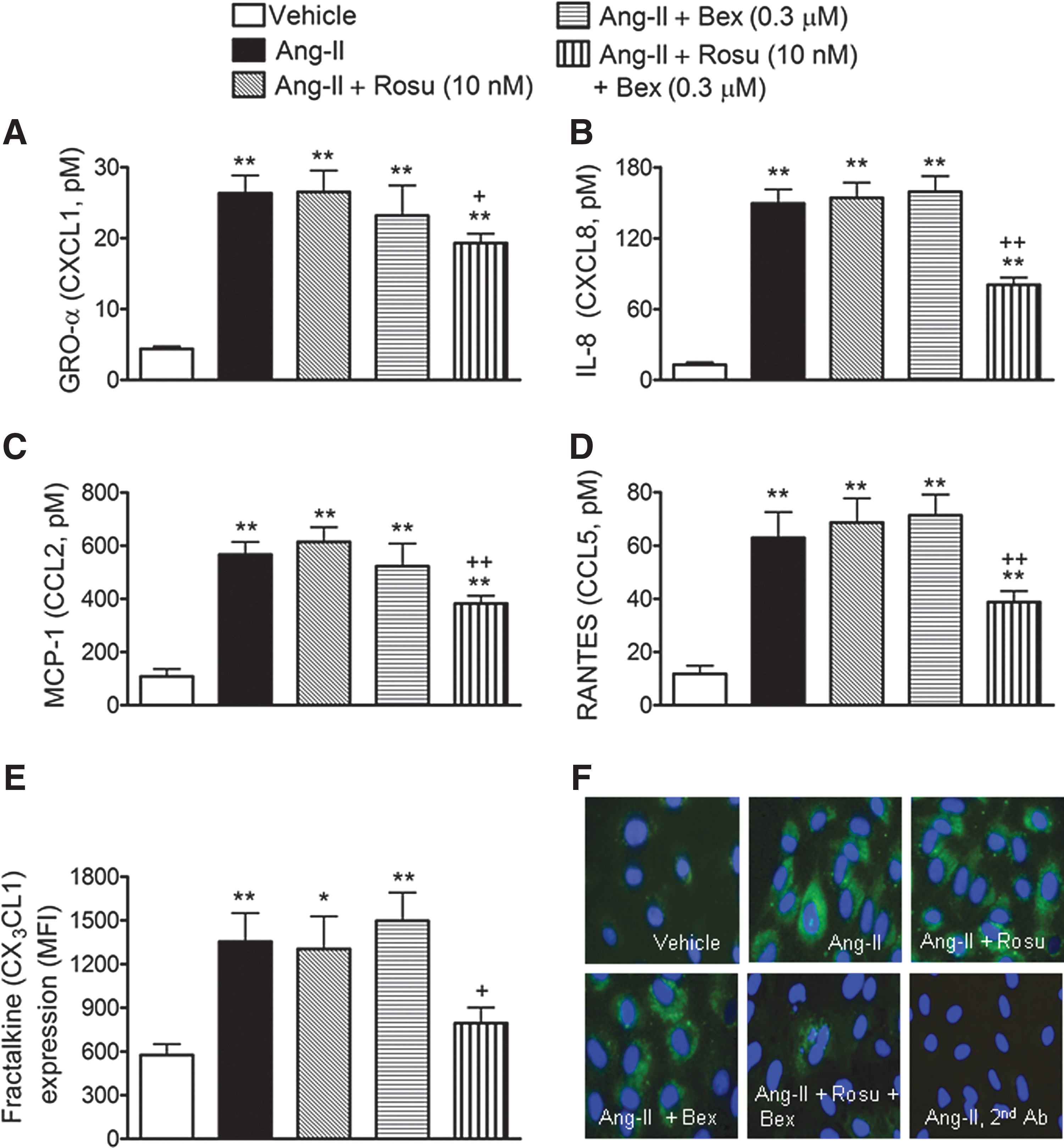
Inhibition of Ang-II-induced HUAEC chemokine synthesis by combination of Rosu+Bex at suboptimal concentrations
Since Ang-II stimulation of endothelial cells can induce the production and release of different CXC and CC chemokines, in addition to the expression of the membrane-bound chemokine fractalkine (36, 42, 54), we next evaluated the effect of Rosu+Bex on Ang-II-induced chemokine synthesis in human arterial endothelial cells. Significant increases in the levels of growth-regulated oncogene-α (GROα or CXCL1), interleukin-8 (IL-8 or CXCL8), monocyte chemotactic protein-1 (MCP-1 or CCL2), and regulated on activation, normal T cell expressed and secreted (RANTES or CCL5) were detected in the supernatant of HUAEC subjected to Ang-II stimulation for 4 h, as measured by enzyme-linked immunoassay (ELISA) (Fig. 3A–D). This increase was not inhibited when HUAECs were preincubated with either of the drugs alone, but was markedly diminished by pretreatment of cells with the combination of Rosu+Bex (Fig. 3A–D). Similarly, when HUAECs were stimulated with 1 μM Ang-II for 24 h, a significant increase in the expression of fractalkine was observed, by both flow cytometry and immunofluorescence (Fig. 3E, F). Again, preincubation of cells with Rosu+Bex caused a significant downregulation of the cell-membrane chemokine expression (Fig. 3E, F).
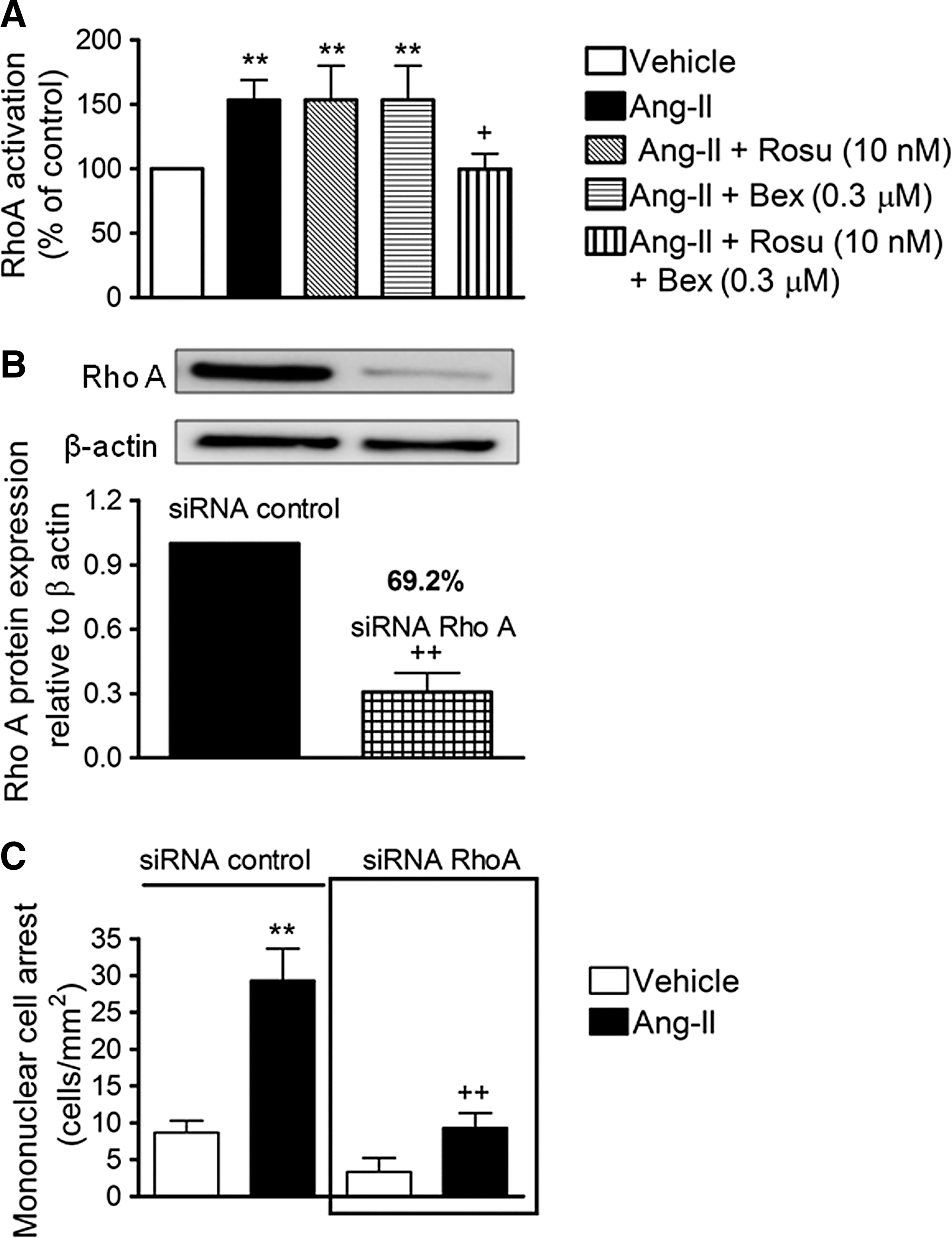
Combination of Rosu+Bex at suboptimal concentrations inhibits Ang-II-induced HUAEC RhoA activation and subsequent MC recruitment
It is well recognized that small GTP-binding proteins (G-proteins), such as Ras, Rho, and Rac, are activated by Ang-II through interaction with its AT1 receptor (22). To characterize the signaling mechanisms by which Rosu+Bex modulate arterial MC adhesion stimulated by Ang-II, we first established their effect on Ang-II-induced RhoA activation. As illustrated in Figure 4A, Ang-II stimulation of HUAEC for 1 h caused a significant increase in RhoA activation. Whereas pretreatment of cells with Rosu or Bex 20 h before Ang-II stimulation failed to modify RhoA activation, preincubation with the combination Rosu+Bex caused a significant inhibition of this response (Fig. 4A). To evaluate the impact of RhoA activation on Ang-II-induced MC recruitment, we silenced RhoA expression in endothelial cells with small interfering RNA (siRNA). Forty–eight hours after transfection with RhoA–specific siRNA, HUAEC exhibited a >69% reduction in RhoA protein compared with control siRNA-treated cells (Fig. 4B). Ang-II stimulation (1 μM, 4 h) produced a significant increase in MC arrest in control siRNA cells, which was abrogated in RhoA-silenced HUAECs (Fig. 4C). Furthermore, inhibition of the RhoA downstream target, Rho-associated protein kinase (ROCK), resulted in a significant impairment in Ang-II-induced arterial mononuclear leukocyte adhesion (Supplementary Fig. S2).
Combination of Rosu+Bex at suboptimal concentrations inhibits Ang-II-induced Nox5 expression in HUAECs
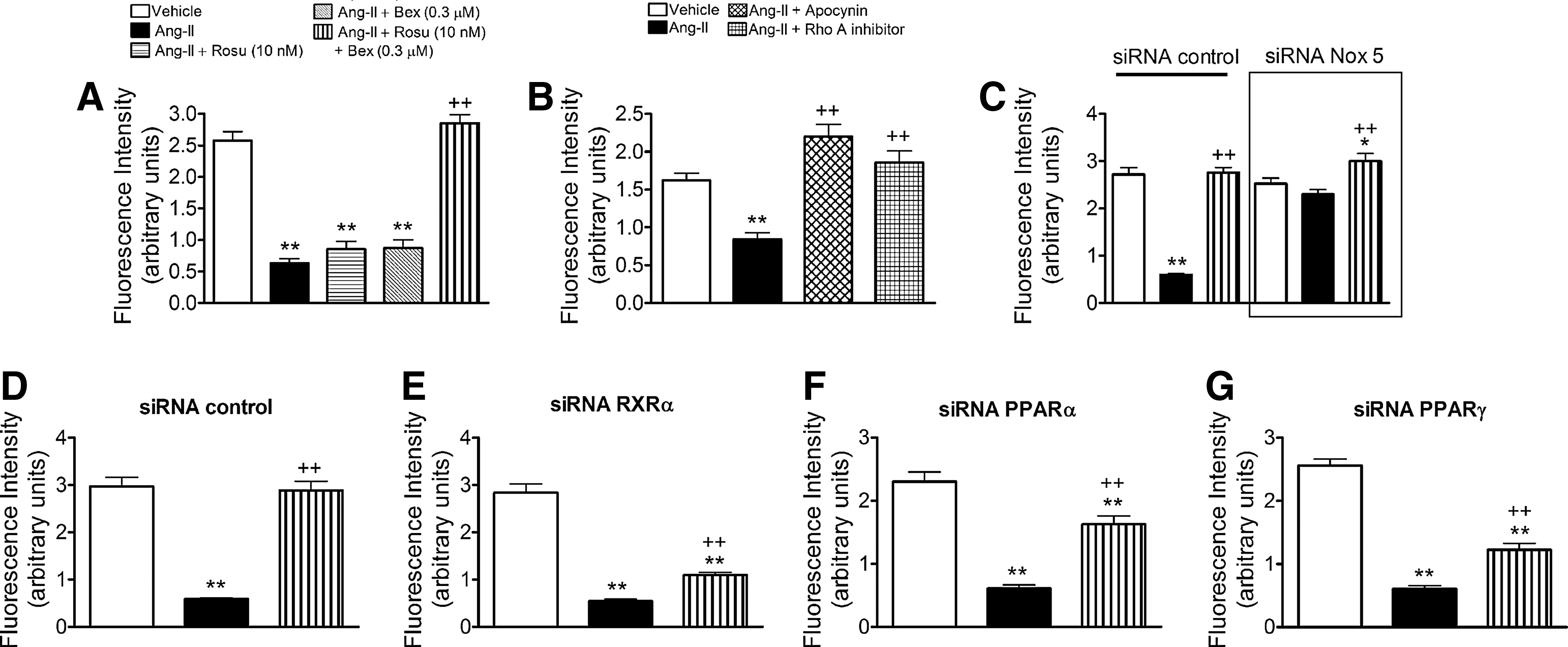
The production of ROS and subsequent activation of redox-sensitive signaling pathways mediates many of the inflammatory responses induced by Ang II (29, 30). Accordingly, endothelial Ang-II stimulation activated NADPH oxidase in HUAECs (Fig. 5A). Notably, this activation was diminished by cell pretreatment with Rosu+Bex, but not when either drug was used separately (Fig. 5A). Ang-II caused increased endothelial expression of Nox2, Nox4, and Nox5 (54) and Figure 5B–D. Interestingly, while the expression of these Nox isoforms induced by Ang-II were unaffected by Rosu or Bex pretreatment of the cells, Nox2 and Nox5 but not Nox4 expression was markedly reduced by preincubation with Rosu+Bex (Fig. 5B–D). Since MC arrest was found to be largely mediated via endothelial Nox5 expression (54) and ROS generation can activate RhoA (43), we next questioned whether the absence of endothelial Nox5 or antioxidant pretreatment resulted in a failure to activate this GTP-binding protein. To do this, we used both siRNA targeting Nox5 and the antioxidant apocynin. Nox5 protein expression was significantly reduced (62%) after 48 h of incubation with a specific siRNA (Supplementary Figs. S3 and S4). Further, knockdown of Nox5 or apocynin pretreatment resulted in the blockade of Ang-II-induced RhoA activation (Fig. 5E, F). Conversely, in RhoA-silenced HUAECs, or in HUAECs pretreated with an RhoA inhibitor (cell-permeable C3 transferase), the increased expression of Nox5 protein triggered by Ang-II remained unchanged (Fig. 5G, H). Moreover, while Ang-II-induced endothelial ROS generation was neutralized by preincubation with apocynin, it was unaffected by pretreatment of the cells with the RhoA inhibitor (Supplementary Fig. S5).
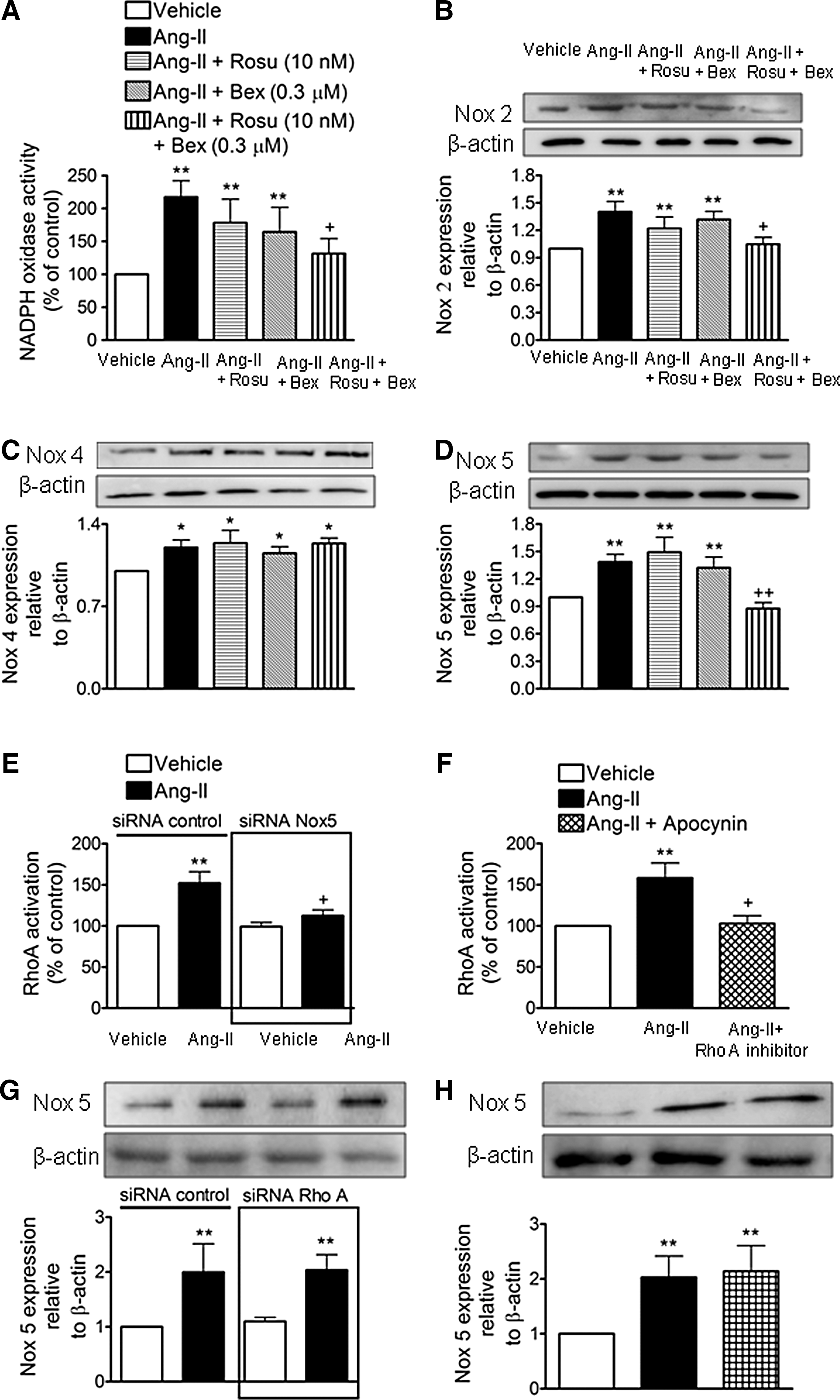
Suboptimal concentrations of Rosu+Bex provokes increased expression of endothelial RXRα, PPARα, and PPARγ
Given that statins and Bex can activate PPARs and RXRα nuclear receptors, respectively (3, 6, 55), we investigated whether the combination of Rosu+Bex exerted its inhibitory activity through changes in the endothelial expression of RXRα, PPARα, PPARβ/δ, or PPARγ. Immunoblot analysis showed that Ang-II-stimulated HUAECs, preincubated or not with Ros or Bex alone, expressed comparable levels of RXRα and PPAR nuclear receptors to vehicle controls (Fig. 6A–D). Importantly, when cells were treated with Rosu+Bex for 20 h followed by stimulation with Ang-II for 4 h, RXRα, PPARα, and PPARγ were significantly upregulated while PPARβ/δ expression was unchanged (Fig. 6A–D). To understand how increased expression of RXRα, PPARα, or PPARγ might contribute to the reduction of Ang-II-induced leukocyte-endothelial cell interactions elicited by pre-exposure of HUAEC to Rosu+Bex, we evaluated leukocyte adhesion to HUAECs individually silenced for expression of RXRα, PPARα, and PPARγ. Knockdown of RXRα, PPARα, and PPARγ resulted in a 57–66% decrease in receptor expression relative to control-siRNA cells (Supplementary Fig. S6). Notably, silencing of RXRα, PPARα, or PPARγ abolished the suppressive effects of Rosu+Bex on leukocyte adhesion to HUAEC stimulated with Ang-II (Fig. 6E–G). Furthermore, when HUAEC were immunoprecipitated with an anti-PPARα or an anti-PPARγ antibody, followed by immunoblotting with an anti-RXRα antibody, enhanced dimerization of RXRα with PPARα or PPARγ was detected with Rosu+Bex, relative to Ang-II-only stimulated cells (Fig. 6H), although the interaction seemed to be more pronounced with PPARγ.
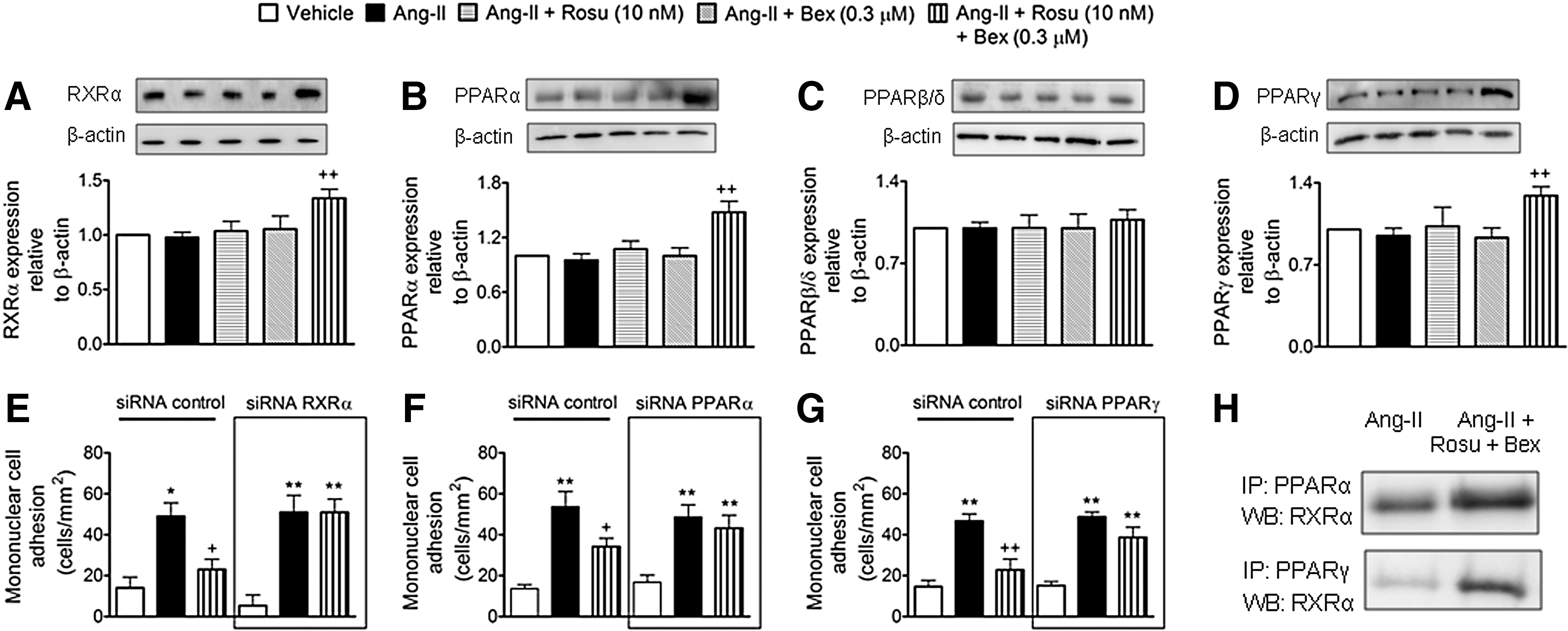
Reduction of RXRα, PPARα, or PPARγ expression blunted the inhibition of Ang-II-induced HUAEC RhoA activation and Nox5 expression by Rosu+Bex at suboptimal concentrations
To gain further insight into the inhibitory mechanisms involved in the Ang-II-induced MC arrest by Rosu+Bex, HUAEC were transfected with siRNAs against RXRα, PPARα, or PPARγ. Interestingly, the inhibitory effect of Rosu+Bex on RhoA activation or Nox5 downregulation in cells stimulated with Ang-II was abolished in cells silenced for these nuclear receptors (Fig. 7).
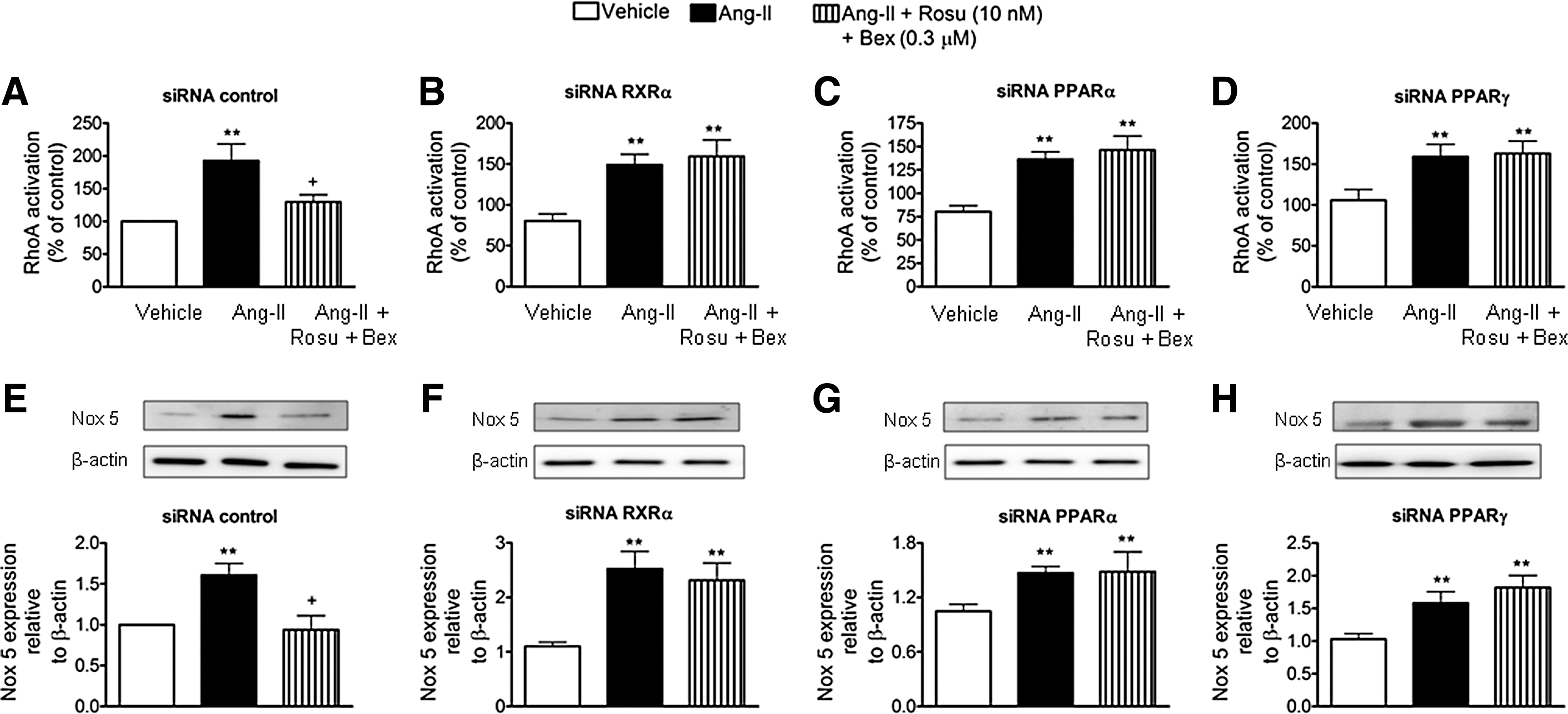
Combination of Rosu+Bex at suboptimal concentrations restored the inhibition in nitric oxide bioavailability induced by Ang-II in HUAECs
It is well known that Ang-II impairs endothelial function by decreasing nitric oxide (NO) bioavailability (32, 43). These effects can be mediated through superoxide anion generation and a reaction with NO, resulting in its quenching to form peroxynitrite, and endothelial NO synthase (eNOS) inhibition and uncoupling (32, 43). Therefore, we evaluated the effect of Rosu+Bex on the Ang-II-induced decrease in NO bioavailability in HUAEC. Ang-II stimulation of the cells reduced NO levels in HUAECs (Fig. 8A). Notably, this reduction was reversed by cell pretreatment with Rosu+Bex, but not when either drug was used separately (Fig. 8A). Pretreatment of the cells with the antioxidant apocynin or an RhoA inhibitor (cell permeable C3 transferase) reversed the reduction in NO availability induced by Ang-II (Fig. 8B). Similarly, silencing of Nox5 prevented the decrease in NO bioavailability provoked by Ang-II (Fig. 8C). More importantly, in the absence of RXRα, PPARα, or PPARγ, Rosu+Bex was unable to fully restore endothelial NO levels (Fig. 8D–G).
Suboptimal doses of Rosu+Bex impairs Ang-II-induced leukocyte-arteriolar adhesion in vivo
To connect our in vitro observations with what happens in vivo, we utilized a chronic model of Ang-II infusion. Animals were implanted with osmotic mini-pumps constantly releasing either Ang-II (500 ng/kg/min) (56) or vehicle, for 14 days. Consistent with the in vitro analysis, animals chronically infused with Ang-II showed a significant enhancement in arteriolar leukocyte adhesion compared with vehicle-infused mice (Fig. 9A). Co-infusion of Rosu at 1.25 mg/kg/day, or oral administration of Bex at 10 mg/kg/day, did not change arteriolar leukocyte adhesion caused by Ang-II systemic infusion (Fig. 9A). However, when the animals were co-infused with Ang-II plus the statin, and Bex was co-administered daily, leukocyte adhesion was significantly reduced by 65% (Fig. 9A). Moreover, Ang-II-induced expression of CD11b in circulating monocytes was also reduced by Rosu+Bex administration, whereas neutrophil CD11b expression was not affected by any treatment (Fig. 9B, C). In addition, immunohistochemistry analysis of cremasteric arterioles showed that the Ang-II-induced increase in ICAM-1, VCAM-1, and fractalkine expression was blunted by co-treatment with Rosu+Bex in mice (Fig. 9D). As expected, a significant reduction in circulating levels of chemokines, including CXCL1 and MCP-1, and a tendency for reduction in RANTES, were also observed in animals treated with Rosu+Bex and chronically subjected to Ang-II (Fig. 9E–G). In this experimental setting, glucose levels and lipid profile were unaffected by the different treatments (Supplementary Table S1). With regard to blood pressure, Ang-II infusion at this dose caused a small but significant increase in this parameter at day 14, which was more marked when the peptide was infused at 1000 ng/kg/min (Supplementary Table S2). However, none of the treatments applied affected the increase in blood pressure induced by Ang-II (Supplementary Table S2).
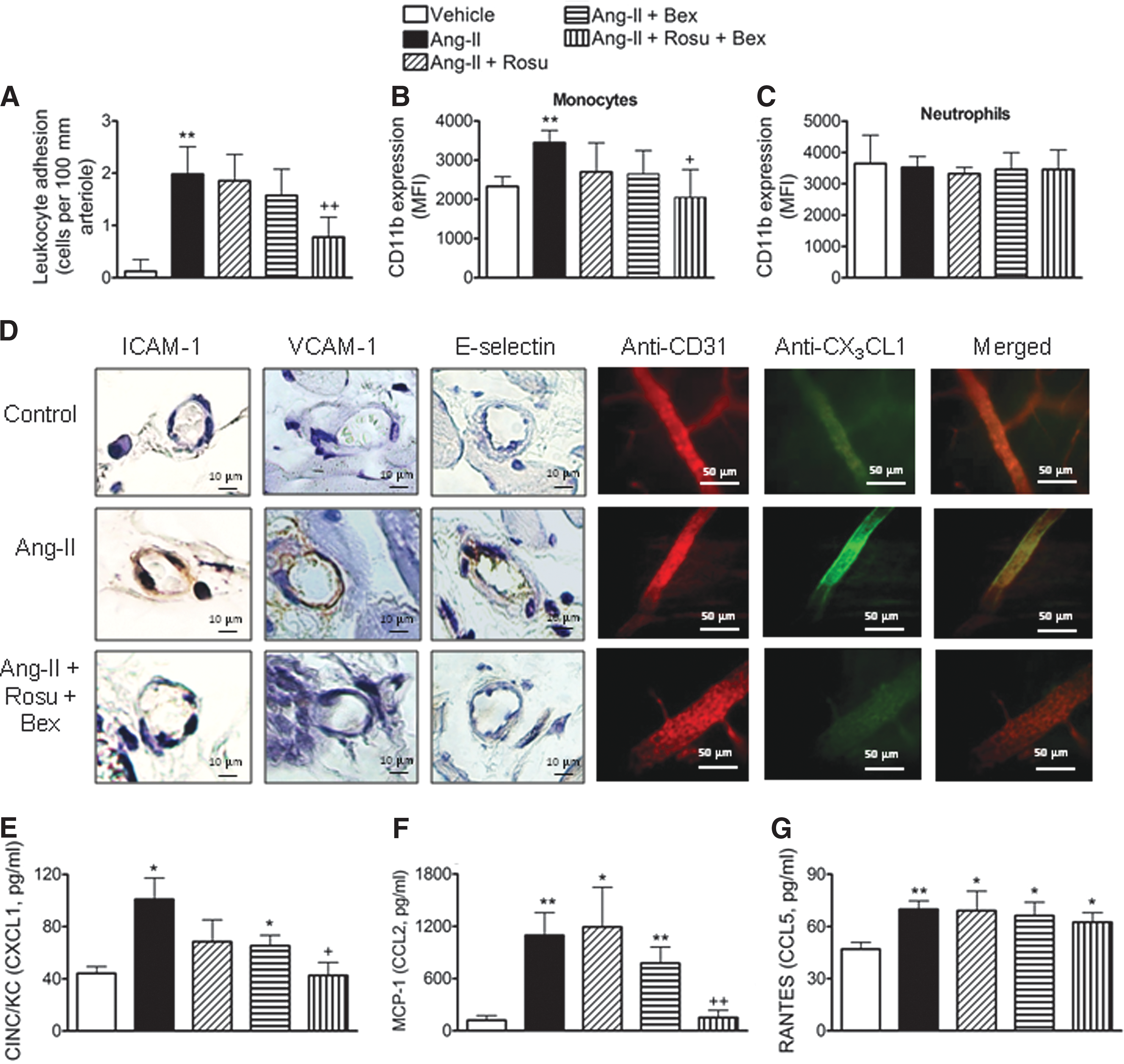
In an additional group of experiments using murine aortic endothelial cells, increased Nox2 and Nox4, but not dual oxidase 1 and 2 (Duox1 and Duox2), mRNA expression was detected in Ang-II-stimulated cells (Supplementary Fig. S7). Interestingly, whereas Nox2 expression was impaired by cell pretreatment with Rosu+Bex, Nox4 expression was unaffected by the combination treatment (Supplementary Fig. S7). In accordance with the results from human cells, neither drug affected Ang-II-induced Nox2 or Nox4 expression when evaluated separately (Supplementary Fig. S7).
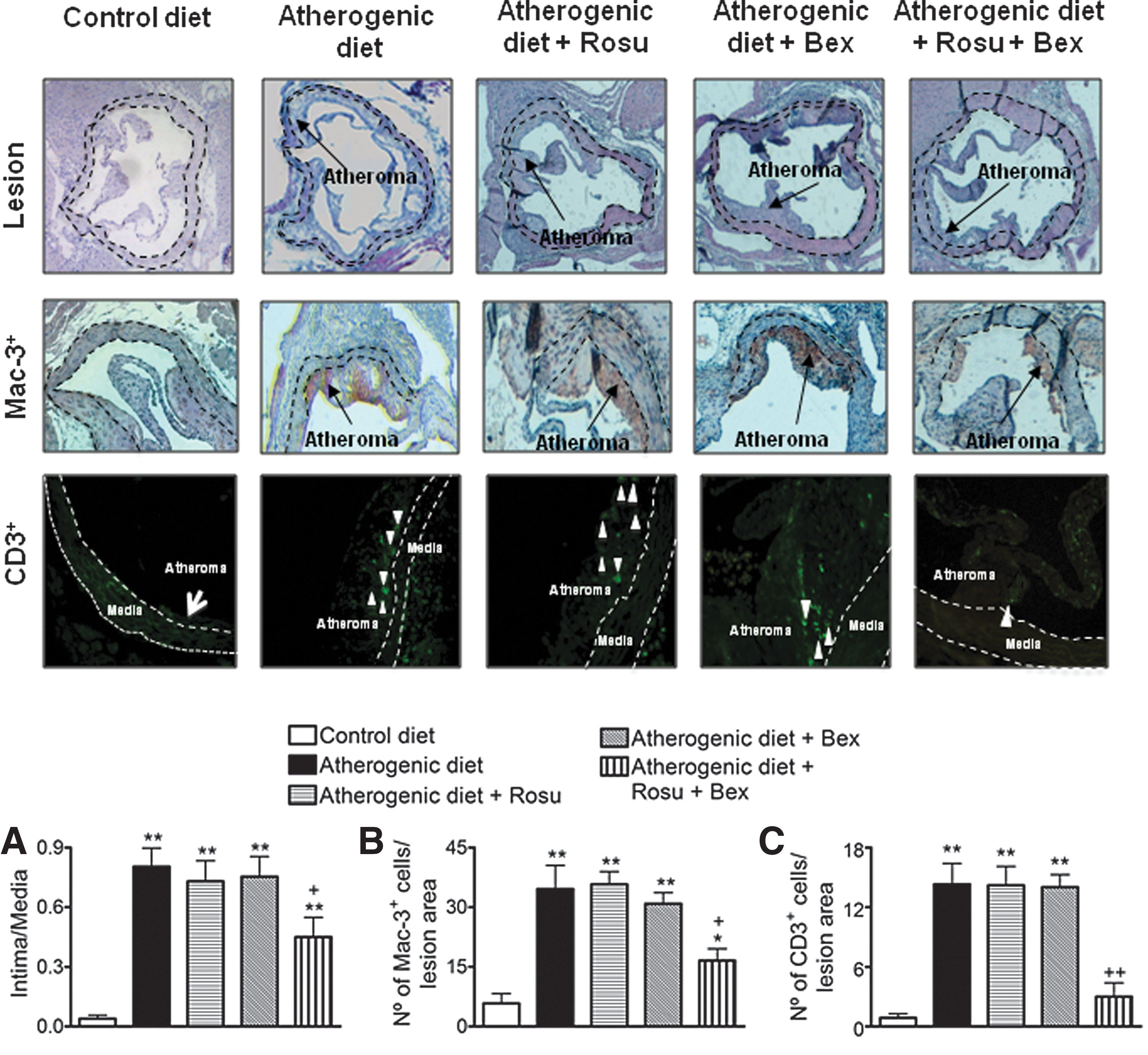
Suboptimal doses of Rosu+Bex reduce atherosclerosis development and cell composition in apoE−/− mice on atherogenic diet
To explore the potential relevance of these findings for atherosclerosis, 8-week-old apoE−/− mice were fed a high-fat atherogenic diet for 8 weeks. As expected, animals subjected to an atherogenic diet presented increased lesion formation and infiltration of macrophages and T cells within the lesion (Fig. 10). Co-infusion of Rosu at 1.25 mg/kg/day, or oral administration of Bex at 10 mg/kg/day, had no effect on these parameters (Fig. 10). Notably, chronic treatment with the statin plus Bex substantially decreased lesion formation and the associated MC infiltration (Fig. 10). Neither glucose level nor the lipid profile in atherogenic animals was significantly affected by the different treatments applied (Supplementary Table S3).
Discussion
Combination therapy that simultaneously addresses multiple mechanisms involved in the pathogenesis of atherosclerosis is an attractive emerging concept for preventing and/or slowing the progression of this disease. Using both in vitro and in vivo approaches, we provide the first demonstration of an anti-inflammatory effect of suboptimal concentrations/doses of two commercially available drugs, Rosu and Bex, in combination.
Mononuclear leukocyte infiltration into the subendothelial space is a key event in the atherogenic process (31) and treatment with Rosu or Bex has been shown to limit MC numbers in atherosclerotic plaques, or their interaction with the arterial endothelium (27, 55). In this study, stimulation of arterial endothelial cells with Ang-II, mimicking a dysfunctional endothelium, promoted the adhesion of mononuclear leukocytes, and this event was clearly reduced by different combined treatments of Rosu and Bex. Remarkably, this reduction was achieved with a suboptimal combination of Rosu (10 nM) and Bex (0.3 μM) since at these concentrations, neither the statin nor the RXR ligand displayed any significant efficacy when assessed individually. To further confirm this striking observation in vivo, daily Rosu administration at a dose four-fold-lower than that required to reduce atherosclerotic lesion formation (27), or Bex at a dose that was previously not found to inhibit acute TNFα-induced arteriolar leukocyte adhesion (55), was tested individually or in combination in mice subjected to chronic Ang-II infusion (56). In this model of systemic inflammation, arteriolar leukocyte adhesion remained unaltered in Rosu or Bex-treated animals, whereas combined administration of both drugs provoked a synergistic inhibitory effect. Furthermore, atheroma formation and MC infiltration in apoE−/− mice subjected to a high-fat diet was clearly diminished in animals treated with the drug combination, thus validating our in vitro observations in a relevant in vivo model.
Leukocyte/arterial interactions require both the expression of several types of CAMs in the endothelium and leukocytes as well as the presence of counter receptor molecules in the leukocyte/endothelial cell (14, 31). In addition to adhesion molecules, chemokines also have the potential to recruit specific cell types and are involved in the regulation of leukocyte trafficking (14). The ability of Ang-II to increase the endothelial expression of E-selectin, ICAM-1, and VCAM-1 and also the production of a wide array of leukocyte-recruiting chemokines is well known (2, 36, 42, 46, 54, 60). Statins are reported to reduce both the inflammation-induced expression of these endothelial CAMs and the synthesis of numerous chemokines (24, 38). More recently, we have demonstrated decreased expression of E-selectin, ICAM-1, and VCAM-1 and a decrease in the synthesis of CXCL1 or CCL2 by the endothelium with RXR agonists in an inflammatory environment (55). Therefore, it was perhaps not surprising that pretreatment of HUAECs with an Rosu+Bex combination reduced both the expression of CAMs and the synthesis of different chemokines involved in the MC arrest evoked by Ang-II. Furthermore, in the in vivo setting, the combined administration of suboptimal doses of Rosu+Bex, in addition to attenuating the increased expression of arteriolar E-selectin, ICAM-1, VCAM-1, and fractalkine, and the elevated circulating levels of CXCL1, CCL2, and CCL5 induced by chronic Ang-II exposure also decreased Ang-II-induced CD11b upregulation on circulating monocytes but not on neutrophils. This exciting observation may also suggest that this treatment may result in improved vascular function without compromising other neutrophilic responses required in host defense.
Having established the action of Rosu+Bex on the aforementioned components of the canonical leukocyte recruitment cascade, we sought to determine their effect on some intracellular signaling cascades triggered by activation of the AT1 Ang-II receptor. The RhoA-Rho kinase pathway has been implicated in leukocyte recruitment (4) and RhoA can be activated by Ang-II, as shown here and in a previous report (22). We show that targeted knockdown of RhoA in HUAECs diminished the adhesion of MCs triggered by Ang-II. Notably, Rosu+Bex abolished Ang-II-induced RhoA activation. Although blockade of the mevalonate pathway by statins can prevent the synthesis of isoprenoid intermediates, such as farnesyl pyrophosphate or geranylgeranyl pyrophosphate that are involved in the post-translational modification of numerous proteins including the γ subunit of heterotrimeric G proteins such as Rho (5, 19), the concentrations of statins required to inhibit Rho-kinase are much greater than those applied here (μM range vs. nM) (38, 51). Furthermore, to the best of our knowledge, the effect of Bex on RhoA activation has never been addressed. Therefore, additional studies were undertaken to address the inhibitory effect of Rosu+Bex on RhoA activation by Ang-II.
Guided by our initial observations, and given that RhoA is acknowledged as a primary target of oxidative stress on Ang-II endothelial stimulation (43), we explored the effect of the RXR agonist and statin combination on NADPH-oxidase-derived ROS production. Although Nox2, Nox4, and Nox5 are the most abundant NADPH oxidase isoforms in endothelial cells (29) and are induced by Ang-II (54), only Nox5 appears to play a major role in Ang-II-induced endothelial adhesiveness since both VCAM-1 and fractalkine upregulation and the subsequent MC arrest were found to be Nox5 mediated (39, 54). We found that Ang-II-induced production of ROS was attenuated by preincubation of HUAECs with Rosu+Bex. Moreover, Ang-II-dependent Nox2 and Nox5 expression was significantly reduced by the combined treatment and the former in murine aortic endothelial cells, which may account for the reduced arteriolar leukocyte adhesion encountered in animals subjected to chronic Ang-II infusion, as found in other animal models (11, 41). Furthermore, since mechanisms through which Ang-II-induced ROS production activate downstream signaling are elusive, we show here that RhoA activation is dependent on Nox5/NADPH oxidase activity. Conversely, knockdown of RhoA in HUAEC or its pharmacological inhibition affected neither Ang-II-induced ROS production nor the increase in Nox5 expression, indicating that ROS generation by Nox5 is an upstream regulator of RhoA activation in this experimental setting.
PPARs are ligand-activated nuclear hormone receptors that function as transcription factors. The three isoforms of PPAR (PPARα, PPARγ, and PPARβ/δ) are expressed in the endothelium and have been described to curb inflammation in atherosclerosis (3, 40). Ligand binding of PPAR promotes heterodimerization with the RXR receptor, inducing PPAR transactivation of target genes (40, 55). In this study, Rosu+Bex were found to enhance RXR, PPARα, and PPARγ protein expression in Ang-II-stimulated HUAECs. Importantly, specific knockdown of endothelial RXR, PPARα, or PPARγ reversed the inhibitory effects exerted by Rosu+Bex on Ang-II-induced mononuclear leukocyte adhesion. Finally, a clear interaction between RXR/PPARα and RXR/PPARγ was revealed on co-incubation with Rosu and Bex at suboptimal concentrations. Considering that PPARα or PPARγ agonists can inhibit RhoA activation (45, 50) and exert antioxidant properties (40), it seems likely that inhibition of Nox5 expression and prevention of ROS generation inhibit RhoA activation via RXR/PPARα and RXR/PPARγ. Indeed, knockdown of RXRα, PPARα, or PPARγ abolished the inhibitory action of the drug combination on Ang-II-induced RhoA activation and Nox5 expression. In addition, RhoA has also been reported to be an upstream regulator of mitogen-activated protein kinase (MAPK) family members, such as p38 MAPK (33), and the latter can regulate the transcription of many genes through its action on downstream targets such as NF-κB (20, 53); both are involved in inflammatory responses such as the MC recruitment induced by Ang-II (54). Therefore, the inhibition of Ang-II-induced Nox5 activation and expression by Rosu+Bex combination may inhibit the activation of RhoA and different MAPKs that lead to activation of several transcription factors, including NFκB, and the further regulation of genes encoding different CAMs and chemokines, which actively participate in the mononuclear leukocyte recruitment induced by Ang-II (Supplementary Fig. S8).
Finally, Ang-II impairs endothelial function through superoxide anion generation and peroxynitrite formation, and eNOS inhibition and uncoupling, reducing NO bioavailability (32, 43), and the anti-adhesive properties of NO are widely recognized (17, 21). Here, we report that Ang-II stimulation reduced NO availability in HUAEC and this reduction was reversed by pretreatment with Rosu+Bex. In addition, silencing of Nox5 prevented the effects caused by Ang-II and, in the absence of RXRα, PPARα, or PPARγ, the combination of Rosu+Bex was unable to fully restore endothelial NO bioavailability. These results suggest that the effects achieved by the drug combination are mainly due to their antioxidant properties.
In conclusion, this study provides the first evidence that combined therapy with Rosu and Bex at suboptimal concentrations/doses exerts synergistic beneficial effects on endothelial dysfunction produced by Ang-II. This anti-inflammatory activity was found to be mediated through an Nox5-RhoA-RXR/PPAR-associated mechanism, linking redox-signaling pathways to MC recruitment.
Materials and Methods
Human in vitro studies
All investigation with human samples carried out in this study conforms to the principles outlined in the Declaration of Helsinki and was approved by the institutional ethics committee at the University Clinic Hospital of Valencia, Spain. Written informed consent was obtained from all volunteers.
Cell culture
HUAECs were isolated by collagenase treatment (26) and maintained in human endothelial cell basal medium-2 (EBM-2) supplemented with endothelial growth medium-2 (EGM-2) and 10% FCS. Cells at passage 1 were grown to confluence on 24-well culture plates. Before every experiment, cells were incubated for 16 h in medium containing 1% FCS and then returned to 10% FCS-supplemented medium at the commencement of all experimental protocols.
Leukocyte-HUAEC interactions under flow conditions
HUAECs at passage 1 were grown to confluence and stimulated with Ang-II (1 μM) for 4 h. Rosu (10–30 nM), Bex (0.3–1 μM), or combinations of both were added to plates 20 h before Ang-II stimulation. In another set of experiments, cells were incubated with the ROCK inhibitor Y27632 (10 μM) 1 h before Ang-II stimulation (1 μM, 4 h). Human MCs were obtained from buffy coats of healthy donors by Ficoll–Hypaque density gradient centrifugation (37). The Glycotech flow chamber was assembled and placed onto an inverted microscope stage, and freshly isolated human MCs (1×106/mL) were then perfused across the endothelial monolayers (HUAECs or transfected HUAECs). In all experiments, leukocyte interactions were determined after 5 min at 0.5 dyn/cm2. Cells interacting on the surface of the endothelium were visualized and recorded (×20 objective, ×10 eyepiece) using phase-contrast microscopy (Axio Observer A1; Carl Zeiss microscope).
Flow cytometry
Confluent endothelial cells were stimulated with Ang-II (1 μM) for 4 h (ICAM-1 and VCAM-1 expression) or 24 h (CX3CL1 expression). In some experiments, cells were pretreated with Rosu (10 nM), Bex (0.3 μM), or a combination of Rosu (10 nM) plus Bex (0.3 μM) 20 h before Ang-II stimulation. Cells were detached, washed, and incubated at 2×106 cells/ml with a 1/100 dilution of primary antibodies against human VCAM-1 or ICAM-1 in PBS with 0.2% BSA and 0.05% NaN3 for 1 h on ice. Detection of primary antibodies was performed using the appropriate Alexa Fluor 488-secondary antibody (dilution 1/250). For fractalkine expression, cells were incubated with a PE-conjugated mAb against human CX3CL1 (1/25 dilution) for 1 h on ice in the same buffer. After two washes, cells were suspended in PBS containing 2% paraformaldehyde. The fluorescence signal of the labeled cells was then analyzed by flow cytometry (FACSVerse Flow Cytometer; BD Biosciences). The expression of ICAM-1, VCAM-1, and CX3CL1 was expressed as the mean of fluorescence intensity (MFI).
Real time-polymerase chain reaction
Confluent HUAEC or murine endothelial cells were stimulated with Ang-II (1 μM) for 4 h. In some experiments, cells were pretreated with Rosu (10 nM), Bex (0.3 μM), or a combination of Rosu (10 nM) plus Bex (0.3 μM) 20 h before Ang-II stimulation. RNA from endothelial cells was obtained using Trizol Reagent, and purity and concentration was determined by the A260/280 ratio. RNA (500 ng) was reverse transcribed with the Maxima First-Strand cDNA Synthesis kit and amplified with Luminar Color HiGreen HigBox qPCR Master Mix. Reactions were run on a 7900 Fast Real-Time PCR System, and results were analyzed with the software provided by the manufacturer (Applied Biosystems, Life Technologies). mRNA levels were determined following the 2−ΔΔCt method using mouse cyclophilin or human GAPDH as an endogenous control and normalizing with regard to the control group. The following primers were designed with Primer Express software (Forward: Fw; Reverse: Rv): Human AT1: Fw 5′ - ACGTGTCTCAGCATTGATCGAT-3′ and Rv 5′-TCGAAGGCGGGACTTCATT-3′ and human GAPDH: Fw 5′- ACCACAGTCCATGCCATCAC-3′ and Rv 5′-TCCACCACCCTGTTGCTGTA-3′. Mouse Cyclophilin Fw: 5′ TGGAGAGCACCAAGACAGACA-3′ and Rv 5′-TGCCGGAGTCGACAATGAT-3′; Mouse Nox 2: Fw 5′TCAAGACCATTGCAAGTGAACAC-3′ and Rv 5′-TCAGGGCCACACAGGAAAA-3′: Mouse Nox 4: Fw5′- AGCATCTGCATCTGTCCTGAAC-3′ and .Rv 5′-ACTGTCCGGCACATAGGTAAAAG-3′: Mouse Duox 1: Fw 5′-AGCTCTCCGGGTCTGCAA-3′ and Rv 5′-CACATCTTCAGCCCTTTGTAGCTT-3′; Mouse Duox 2: Fw 5′-GATCAGCATCAGACAGGGTTAGG-3′ and Rv 5′-TCTTCACGACGCGCTTTCT-3′.
Immunofluorescence
Confluent endothelial cells were grown on glass coverslips and stimulated with Ang-II (1 μM) for 4 h (ICAM-1 and VCAM-1 expression) or 24 h (CX3CL1 expression). In some experiments, cells were pretreated with Rosu (10 nM), Bex (0.3 μM), or a combination of Rosu (10 nM) plus Bex (0.3 μM) 20 h before Ang-II stimulation. The cells were fixed with 4% paraformaldehyde and blocked in a PBS solution containing 1% BSA. Next, they were incubated for 2 h with primary mouse antibodies against VCAM-1 or ICAM-1 (1/100 dilution), followed by a 45 min incubation at room temperature with an Alexa Fluor 488-conjugated goat anti-mouse secondary mAb (1/1000 dilution). For fractalkine detection, samples were incubated overnight with a primary mouse mAb against human CX3CL1 (1:200 dilution) in a 0.1% BSA/PBS solution at 4°C. Cell nuclei were stained with 4′-6-diamidino-2-phenylindole (DAPI). Images were captured with an immunofluorescence microscope (Axio Observer A1; Carl Zeiss microscope).
Chemokine detection
Confluent endothelial cells were stimulated with Ang-II (1 μM) for 4 h. In some experiments, cells were pretreated with Rosu (10 nM), Bex (0.3 μM), or a combination of Rosu (10 nM) plus Bex (0.3 μM) 20 h before Ang-II stimulation. Human chemokines GROα (CXCL1), IL-8 (CXCL8), MCP-1 (CCL2), and RANTES (CCL5) were measured in HUAEC culture supernatants by ELISA. After coating the plates overnight with the primary antibody, nonspecific binding sites were blocked with 3% BSA for 1 h. Supernatants and standards were added to PBS/0.5% BSA/0.05% NaN3 for 2 h. Biotinylated detector antibodies were added for 2 h, followed by neutravidin horseradish peroxidase for 1 h. All plate washes were carried out in four cycles in freshly prepared PBS/0.2% Tween 20. Enhanced K-Blue TMB substrate was added for 30 min, and the enzyme reaction was stopped by the addition of 0.19 M sulfuric acid. Absorbance was read at 450 nm, and the data were processed by GraphPad Prism software. Results are expressed as pM chemokine in the supernatant.
Gene knockdown by siRNA
Confluent HUAEC cultures were transfected with either control or Rho A, Nox 5, RXRα, PPARα, PPARβ, and PPARγ-specific siRNA using Lipofectamine RNAiMAX reagent. Protein expression was determined by immunoblot of cell lysates after 48 h and compared with the control siRNA to determine silencing efficiency.
Western blot and immunoprecipitation
After treatments, cells were washed, detached, collected, and centrifuged at 15,000 g at 4°C for 30 min. Protein content was determined according to Bradford's method. Samples were denatured, subjected to SDS-PAGE using a 10% running gel, and transferred to nitrocellulose membrane. Nonspecific binding sites were blocked with 3% BSA in TBS solution, and blots were incubated overnight with a mouse polyclonal antibody against human RhoA (dilution 1/250), a mouse polyclonal antibody against human Nox2 (0.2 μg/mL), a rabbit polyclonal antibody against human Nox4 (2 μg/ml), a rabbit polyclonal antibody against human Nox5 (dilution 1/500), a rabbit polyclonal antibody against human RXRα (dilution 1/500), a mouse polyclonal antibody against human PPARα (dilution 1/500), a rabbit polyclonal antibody against human PPARβ/δ (dilution 1/500), or a rabbit polyclonal antibody against human PPARγ (dilution 1/500). Subsequently, membranes were washed and incubated for 1 h with the corresponding secondary HRP-linked antibody and developed using the ECL procedure. Signals were detected using a luminescent reader (FujiFilm image Reader LAS1000; Fuji) and analyzed with ImageJ software (NIH, Windows free version).
For immunoprecipitation, cell extracts were prepared in 50 mM Tris-HCl (pH 8), 150 mM NaCl, 1% Nonidet P-40, with protease (1 mM PMSF, 40 μg/ml aprotinin, and 40 μg/ml leupeptin), and phosphatase (1 mM sodium ortovanadate and 1 mM NaF) inhibitors. Protein (∼200 μg) from cell extracts was incubated with 5 μg of the rabbit polyclonal antibody against human PPARα or PPARγ. Immunocomplexes were precipitated using anti-rabbit IgG beads following the manufacturer's instructions and suspended in sample buffer containing freshly added 50 mM dithiothreitol (DTT). Immunoblotting was performed with an antibody against RXRα. Membranes were developed using horseradish peroxidase-conjugated rabbit TrueBlot as secondary antibody. Chemiluminescent signals were developed with ECL.
RhoA activation assay
HUAECs were grown to 70% confluence in six-well plates. Then, cells were left untransfected or transfected with a control siRNA or specific siRNA against Nox5, RXRα, PPARα, or PPARγ using Lipofectamine RNAiMAX. Twenty-four hours after transfection, cells were pretreated for 20 h with Rosu (10 nM), Bex (0.3 μM), or a combination of Rosu (10 nM) plus Bex (0.3 μM) before Ang-II stimulation (1 μM, 1 h) since Ang-II-induced RhoA activation peaks within the first hour of challenge (58). In additional assays, untransfected HUAECs were pretreated with the antioxidant apocynin (30 μM, 1 h) and subsequently stimulated with Ang-II (1 μM, 1 h). Then, cells were lysed, protein content was determined, and the RhoA activity was quantified using a commercial Rho G-LISA™assay kit.
NADPH oxidase activity assay
The activity of NADPH oxidase was measured in HUAECs by lucigenin-derived chemiluminiscence. HUAECs were pretreated for 20 h with Rosu (10 nM), Bex (0.3 μM), or a combination of Rosu (10 nM) plus Bex (0.3 μM) before Ang-II stimulation (1 μM, 1 h) since superoxide release induced by Ang-II is detected after 1 h challenge (10). In some experiments, cells were pretreated with the antioxidant apocynin (30 μM, 1 h) or with an RhoA inhibitor (C3 transferase, 2 μg/ml, 4 h) before Ang-II stimulation (1 μM, 1 h). After treatments, HUAECs were washed with ice-cold PBS, scraped, and centrifuged at 13,000 rpm for 1 min at 4°C. The resulting cell pellet was homogenized in lysis buffer (pH 7.0) containing 50 mM KH2PO4, 1 mM EGTA, and 150 mM sucrose at 4°C, and protein content was determined. Cellular extracts were incubated in PBS containing 5 μM lucigenin and 100 μM NADPH. Thereafter, luminiscence was measured every 10 s for 5 min in an Optocomp luminometer (MGM Instruments). The enzymatic activity was expressed as relative light units (RLU)/μg of protein/min.
Measurement of NO production in HUAEC
Intracellular NO was monitored with 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-2-FM diacetate), a fluorescence indicator of NO that emits fluorescence in response to a reaction with NO, as previously described (47). To measure intracellular NO, HUAECs were seeded on 24-well plates. At 80% confluence, HUAECs were pretreated for 20 h with Rosu (10 nM), Bex (0.3 μM), or a combination of Rosu (10 nM) plus Bex (0.3 μM) and then stimulated with Ang-II (1 μM, 4 h). In another set of experiments, cells were pretreated with the antioxidant apocynin (30 μM, 1 h) or with an RhoA inhibitor (C3 transferase, 2 μg/ml, 4 h) before Ang-II stimulation (1 μM, 4 h). Finally, cells were transfected with a control siRNA or specific siRNAs against Nox5, RXRα, PPARα, or PPARγ. Twenty-four hours after transfection, HUAEC were pretreated for 20 h with or without Rosu (10 nM) plus Bex (0.3 μM) and then stimulated with Ang-II (1 μM, 4 h). After completion of the treatments, cells were loaded with 2.5 μM (DAF-2-FM diacetate) in EBM-2 supplemented with EGM-2 and 10% FCS for 30 min. After loading, cells were rinsed with PBS. To quantify the DAF-related fluorescence, cells were observed under an inverted fluorescence Nikon Eclipse Ti-S microscope. Fluorescence from five different fields per well was measured (excitation wavelength: 488 nm; emission wavelength: 515 nm). Fluorescence signals were quantified using NIS-Elements 3.2 software (Nikon Izasa S.A).
Animal studies
The animal protocol conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85–23, revised 1996) and was approved by the Ethics Review Board of the School of Medicine, University of Valencia. Colonies of C57BL/6 mice (22–30 g, Charles River) were bred and maintained under specific pathogen-free conditions. For the entire experimental period, the mice were fed with an autoclaved balanced diet with free access to water.
Ang-II was administered to randomly selected groups of mice via subcutaneous osmotic minipumps set to deliver saline or Ang-II (500 ng/kg/min) for 14 days. This dose was selected based on its mild effects on blood pressure and clear leukocyte infiltration as previously described (56). A second group of mice were infused with Ang-II and administered by gavage with Bex (10 mg/kg/day); a third group was infused with Ang-II and Rosu (1.25 mg/kg/day delivered by osmotic minipumps); and a final Ang-II infused group was treated with a combination of both drugs.
Intravital microscopy
Mice were anesthetized by i.p. injection with a mixture of xylazine hydrochloride (10 mg/kg) and ketamine hydrochloride (200 mg/kg). The mouse cremasteric preparation used in this study was similar to that previously described (48). The cremaster muscle was dissected from the tissues and exteriorized on an optical clear viewing pedestal. The muscle was cut longitudinally with a cautery and held flat against the pedestal by attaching silk sutures to the corners of the tissue. The muscle was then perfused continuously with warmed bicarbonate-buffered saline (pH 7.4) at a rate of 1 ml/min. The cremasteric microcirculation was observed using an intravital microscope (Nikon Optiphot-2, SMZ1; Badhoevedorp) equipped with a 50×objective lens (Nikon SLDW; Badhoevedorp) and a 10×eyepiece. A video camera (Sony SSC-C350P; Koeln) mounted on the microscope projected the image onto a color monitor, and the images were CCD recorded for playback analysis. Cremasteric arterioles (20–40 μm in diameter) were selected for the study, and the diameter was measured online using a video caliper (Microcirculation Research Institute, Texas A&M University, College Station, TX). Centerline blood cell velocity was also measured online using an optical Doppler velocimeter (Microcirculation Research Institute). Vessel blood flow was calculated from the product of mean RBC velocity (Vmean=centerline blood cell velocity/1.6) and cross-sectional area, assuming cylindrical geometry. Wall shear rate (γ) was calculated based on the Newtonian definition: γ=8 x (Vmean/Dv) s−1, in which Dv is vessel diameter (25).
The number of adherent leukocytes was determined offline during playback of videotaped images. A leukocyte was defined as adherent to arteriolar endothelium when it was stationary for at least 30 s. Leukocyte adhesion was expressed as the number per 100 μm length of vessel. In each animal, leukocyte responses were measured in three to five randomly selected arterioles. At the end of the experiments, animals were humanely euthanized by anesthetic overdose.
Whole mount immunohistochemistry
Once intravital microscopy determinations were performed, mice were sacrificed and the cremaster muscle was isolated and fixed in 4% paraformaldehyde for 10 minutes. The protocol followed was similar to that previously described (35); briefly, whole-mounted muscles were incubated for 2 h in 0.2% Triton X-100, 1% BSA, and 0.5% horse serum in PBS. They were then incubated overnight at 4°C with a primary antibody rabbit anti-mouse CX3CL1 (1/100 dilution) or eFluor 450-conjugated anti-mouse CD31 (PECAM-1) (1/100 dilution). Samples were subsequently washed with PBS and incubated for 1.5 h at room temperature with Alexa Fluor 488-conjugated donkey anti-rabbit secondary antibody (1/500 dilution). All antibodies were diluted in 0.1% PBS/BSA. The muscles were then mounted with Slowfade Gold Reagent. Images were acquired with a fluorescence microscope (Axio Observer A1) equipped with a 40× objective lens and a 10× eyepiece.
Immunohistochemistry
After completion of the intravital microscopy measurements, the cremaster muscle was isolated and fixed in 4% paraformaldehyde, dehydrated using a graded acetone series at 4°C, and embedded in paraffin wax for assessment of ICAM-1, VCAM-1, and E-selectin as previously described (55). Tissue sections (5 μm) were incubated against mouse ICAM-1, VCAM-1, and E-selectin (all dilutions 1:50) overnight at 4°C and then for a further 60 min at 37°C with a biotinylated anti-rabbit secondary antibody (1:500 dilution), streptavidin-HRP, and diaminobenzidine substrate. Slides were counterstained with hematoxylin. Positive staining was defined as an arteriole displaying a brown reaction product.
Determination of CD11b integrin expression by flow cytometry
Heparinized whole blood samples were stained for 30 min with saturated amounts (1:10 dilution) of PE-conjugated anti-CD115 antibody, VH-450-conjugated anti-Ly6G antibody, and APC-conjugated anti-CD11b antibody. Red blood cells were lysed using a commercial lysing solution, and samples were run in a flow cytometer (FACSVerse Flow Cytometer; BD Biosciences). The expression of CD11b (CFS fluorescence) in monocytes and neutrophils was measured according to the expression of CD115 and Ly6G, respectively, and expressed as the MFI.
Plasma chemokine detection
KC (CXCL1), MCP-1 (CCL2), and RANTES (CCL5) in mice plasma samples were measured using commercial ELISA kits. The OD values were recorded on a microplate reader at 450 nm. The concentrations of cytokines in samples were calculated from a standard curve generated using recombinant chemokines.
Measurement of glucose and lipid profile
Circulating glucose and lipid levels in plasma of mice fasted overnight were measured using enzymatic procedures (WAKO) and the Ascensia Elite glucometer (Bayer HealthCare).
Measurement of blood pressure
Systolic blood pressure was measured in conscious mice using a noninvasive tail cuff system with a photoelectric sensor (Niprem 645; Cibertec S.A) using a Niprem 1.8 software (Cibertec S.A.). During the procedure, animals were placed in the restrainer tube of a chamber that was kept at 36°C (LE5002 Pressure Meter; PANLAB) as previously described (62). Mice were acclimated to the instrument for 5 continuous days before baseline measurements and then daily for the remainder 14 days. Blood pressure measurements were determined every day. Each data are the average of 10 values recorded for each animal and used for analysis.
In an additional group of experiments, Ang-II 1000 ng/kg/min was administered via subcutaneous osmotic minipumps for 14 days. A second group of mice were infused with Ang-II and administered by gavage with Bex (10 mg/kg/day); a third group was infused with Ang-II and Rosu (1.25 mg/kg/day delivered by osmotic minipumps); and a final Ang-II-infused group was treated with a combination of both drugs.
Evaluation of diet-induced atherosclerosis
ApoE−/− (C57BL/6J) female mice were obtained from Charles River laboratories and kept on a low-fat standard diet (2.8% fat). At 8 weeks of age, mice were fed with a high-fat atherogenic diet (10.8% total fat, 0.75% cholesterol) for 8 weeks alone (atherogenic diet group) or treated with Bex (10 mg/kg/day) by gavage, Rosu (1.25 mg/kg/day delivered by osmotic minipumps), or a combination of both drugs. The control mouse group was maintained on a low-fat standard diet for 8 weeks. After treatments, hearts containing the aortic root were removed from mice, washed with PBS, fixed with 4% paraformaldehyde/PBS overnight, and paraffin embedded for sectioning and analysis, as described (54).
Atherosclerosis was evaluated in at least three to five aortic root cross-sections stained with hematoxylin/eosin (16). Lesion size was quantified as the intima-to-media ratio in cross-sections of paraffin-embedded aortic root. For macrophage quantification in lesions, a rat anti-Mac-3 monoclonal antibody (1/200 dilution) was used. After peroxidase inactivation (H2O2, 0.3%) and blockade with horse serum, samples were incubated overnight (4°C) with the primary antibody. Detection was performed with a biotin-conjugated goat anti-rat secondary antibody (1/300 dilution) followed by HRP-streptavidin and DAB substrate incubation. Slides were counterstained with hematoxylin and mounted with EUKITT. For T-cell detection within the lesion, aortic root cross-sections were blocked as described earlier, incubated overnight with an anti-CD3 antibody (1/75 dilution) followed by an incubation for 1 h at room temperature with an Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody, and mounted with Slow-Fade Gold antifade reagent. Preparations were analyzed by fluorescent microscopy with an inverted fluorescent microscope (Axio Observer A1).
Additional materials
Ang-II was purchased from Calbiochem. Bex, and Rosu were obtained from Axxora (BioVision). Endothelial basal medium-2 (EMB-2), EGM-2, and FCS were purchased from Lonza Iberica. Ketamine and xylazine hydrochloride were supplied by ORION Pharma. Apocynin, mouse anti-human β-actin mAb (clone AC-15), and anti-Nox5 C-terminal polyclonal antibody produced in rabbit that detects the 86 kDa band, hematoxylin, and Y27632 were purchased from Sigma-Aldrich. Nevertheless, there are other antibodies in the literature that can detect a 70 kDa band of Nox5 (39). The rabbit polyclonal against mouse CX3CL1, the PE-conjugated rat monoclonal against mouse CD31 (clone 390), and TrueBlot Anti-Rabbit Ig IP beads were provided by eBioscience. The PE-conjugated mouse monoclonal against human CX3CL1 (clone 51637), primary mouse monoclonal mAb against human CX3CL1 (clone 81506), primary chicken mAb against mouse E-selectin, human GRO-α, IL-8, MCP-1, and RANTES antibody pairs were purchased from R&D Systems. The mouse monoclonal anti-human RhoA Ab, rabbit polyclonal anti-human Nox4, the rabbit polyclonal anti-human PPARα Ab, the rabbit polyclonal anti-human PPARβ/δ Ab, and the rabbit polyclonal anti-human PPARγ Ab were supplied by Abcam. The primary mouse mAb against human VCAM-1, the primary mouse mAb against human ICAM-1, the mouse monoclonal anti-human Nox2 (clone NL7) Ab, and the DAB substrate was purchased from Serotec. Sodium heparin (5000 U/ml or 50 mg/ml) was supplied by Pharmaceutical Laboratories Rovi SA. Ficoll-Paque TM PLUS and ECL developer were purchased from GE Healthcare. DAPI, DAF-2-FM diacetate and Alexa Fluor 488-conjugated secondary antibodies were from Molecular Probes-Invitrogen. The secondary Abs, HRP-linked anti-goat, HRP-linked anti-rabbit, HRP-linked anti-mouse, and the anti-mouse CD3 antibody were purchased from Dako. HRP-Streptavidin was from LABVISION Corporation. The RhoA-specific siRNA and Ultra TMB-ELISA were purchased from Thermo Fisher Scientific, Inc. Nox5, RXRα, PPARα, PPARβ/δ, and PPARγ-specific siRNA were purchased from Dharmacon. The Alzet 2004 osmotic minipumps, C57BL/6 and ApoE−/− mice were from Charles River. The primary rabbit mAb against mouse VCAM-1, primary rabbit mAb against mouse ICAM-1, the PE-conjugated mAb against mouse CD115 (clone AF598), the VH-450-conjugated mAb against mouse Ly6G (clone 1A8) and APC-conjugated mAb against mouse CD11b (clone M1/70), and the lysing solution were from BD Pharmingen. The biotinylated anti-rabbit secondary Ab, rabbit polyclonal against PPARγ, rabbit polyclonal against RXRα, mouse monoclonal against PPARα, the anti-Mac-3 mAb (clone M3/84), and the biotin-conjugated goat anti-rat secondary Ab were purchased from Santa Cruz Biotechnology. The EUKITT was provided by Deltalab. The G-LISA RhoA activation assay Biochem kit and cell-permeable C3 transferase (RhoA inhibitor) were from Cytoskeleton, Inc. Slowfade Gold Reagent and lipofectamine RNAiMAX were from Invitrogen. The mouse RANTES ELISA kit was for RayBiotech. Recombinant human GRO-α, IL-8, MCP-1, and RANTES were acquired from Peprotech. Murine aortic endothelial cells were from Innoprot. Maxima First-Strand cDNA Synthesis kit and Luminar Color HiGreen HigBox qPCR Master Mix were from Fermentas, Thermo Fisher Scientific. The low-fat standard diet was from Panlab. The high-fat atherogenic diet (10.8% total fat, 0.75% cholesterol) was acquired from Sniff.
Statistical analysis
Values were expressed as mean±SEM. Differences between two groups were determined by paired or unpaired Student's t test, as appropriate. Data within multiple groups were compared using an analysis of variance (one-way ANOVA), including a Newman–Keuls post hoc test for multiple comparisons. Data were considered statistically significant when p<0.05.
Footnotes
Acknowledgments
This study was supported by grants SAF2011-23777, CPII13/00025, PI012/01271, CP10/00555, and PI13/00834 from the Spanish Ministry of Economy and Competiveness, Carlos III Health Institute, Spanish Ministry of Health, the European Regional Development Fund (FEDER), and research grants from Generalitat Valenciana (GVACOMP2014-006 and PROMETEO II/2013/014). PE is a recipient of a predoctoral grant (FPU) from the Spanish Ministry of Education. The technical assistance of Virginia López is greatly acknowledged. A part of these results were presented at the 19th World Congress on Heart Disease and the XXXV Congress of the Spanish Society of Pharmacology in 2014.
Author Disclosure Statement
The authors have no conflicting financial interests.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.