
Abstract
Mitochondrial function and specifically its implication in cellular redox/oxidative balance is fundamental in controlling the life and death of cells, and has been implicated in a wide range of human pathologies. In this context, mitochondrial therapeutics, particularly those involving mitochondria-targeted antioxidants, have attracted increasing interest as potentially effective therapies for several human diseases. For the past 10 years, great progress has been made in the development and functional testing of molecules that specifically target mitochondria, and there has been special focus on compounds with antioxidant properties. In this review, we will discuss several such strategies, including molecules conjugated with lipophilic cations (e.g., triphenylphosphonium) or rhodamine, conjugates of plant alkaloids, amino-acid- and peptide-based compounds, and liposomes. This area has several major challenges that need to be confronted. Apart from antioxidants and other redox active molecules, current research aims at developing compounds that are capable of modulating other mitochondria-controlled processes, such as apoptosis and autophagy. Multiple chemically different molecular strategies have been developed as delivery tools that offer broad opportunities for mitochondrial manipulation. Additional studies, and particularly in vivo approaches under physiologically relevant conditions, are necessary to confirm the clinical usefulness of these molecules. Antioxid. Redox Signal. 22, 686–729.
I. Mitochondria
M
Within mitochondria, energy in the form of ATP is obtained in a reaction coupled with the reduction of O2 to form H2O. This process is mediated by the ETC in the IMM, which transfers electrons from the reduced co-factors (NADH and FADH2 using the tricarboxylic acid cycle and the β-oxidation of fatty acids) to the ultimate electron receptor O2. The transfer of electrons is coupled with the simultaneous transport of protons from the mitochondrial matrix across the IMM into the intermembrane space, thus generating a proton gradient between these two compartments, which is harnessed by the ATP synthase to produce ATP. Most of the O2 is completely consumed during this process; only a small part (1–2% in experiments with normal isolated mitochondria) leaks from complex I and III of the ETC in the form of superoxide anion (O2 •−) (30, 114).
A. Implication of mitochondria in cellular redox homeostasis
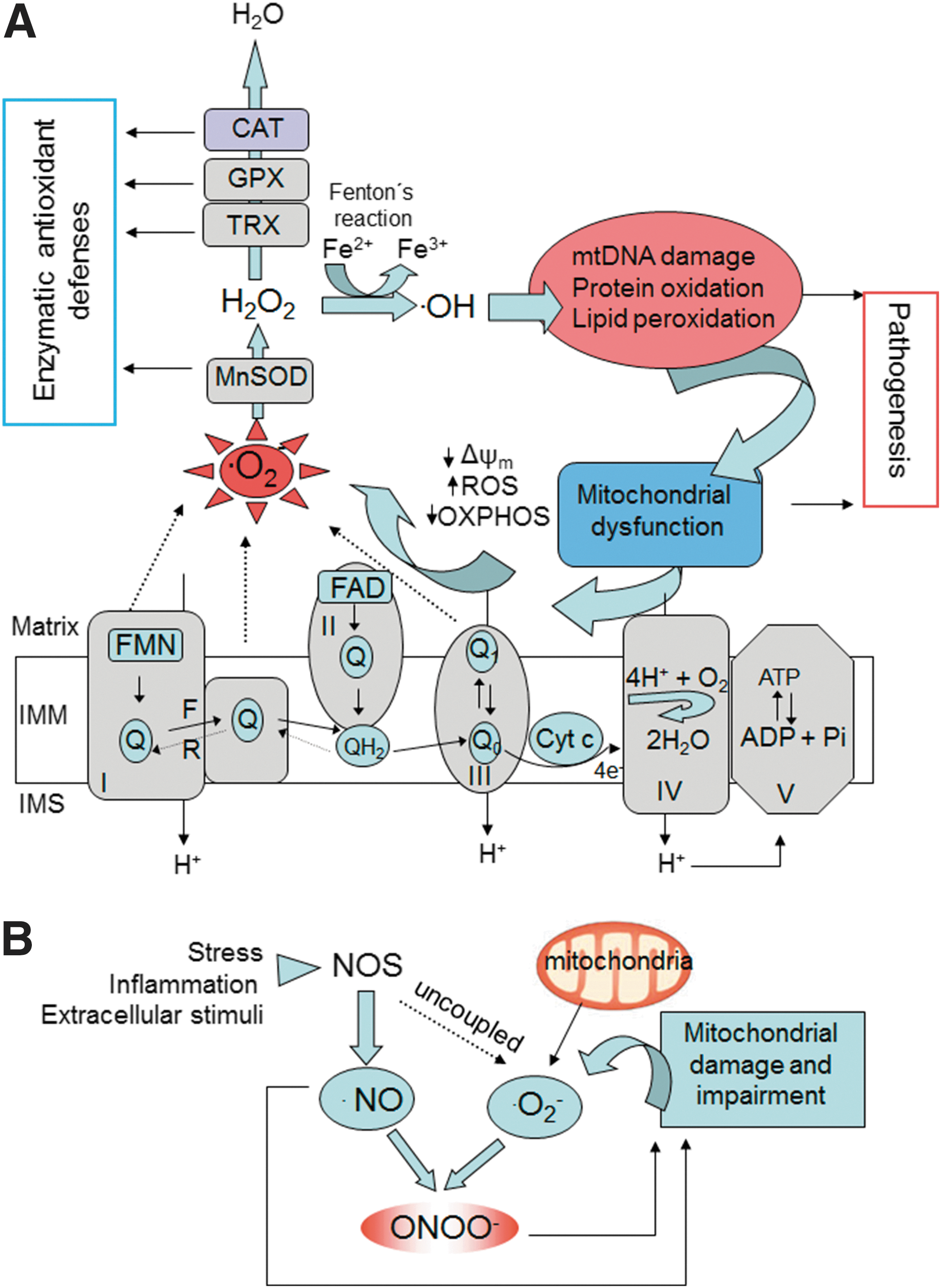
The mitochondrion is believed to be the major intracellular source of ROS, with specific sites at the ETC complexes constituting the foremost origin (Fig. 2A) (30, 78, 114). O2 •− seems to be the first radical to be generated, while other ROS are formed downstream, such as hydrogen peroxide (H2O2), which arises through the dismutation of O2 •− mediated by manganese superoxide dismutase (MnSOD), and hydroxyl radical (•OH), which is created through the reduction of H2O2 in the presence of reduced transition metals (78). This radical is highly reactive and, thus, very harmful to molecules and cellular membranes. Besides the activity of MnSOD, O2 •− can also be converted to H2O2 by other types of enzymes, including pyruvate dehydrogenase and α-ketoglutarate dehydrogenase, which generates both O2 •− and H2O2 via the oxidation pathway and can be considered a possible target for clinical research in the treatment of certain diseases that harbor mitochondrial dysfunction, such as neurodegenerative diseases (281). As a negatively charged molecule, O2 •− has limited mobility across biologic membranes and its diffusion depends on anion channels in the membrane. Thus, O2 •− is characterized by spatial specificity and can be confined within certain organelles, including mitochondria. Apart from complex I, O2 •− is also released into the intermembrane space from Qo of complex III, where it is mostly dismuted by SOD1. This has important implications for cytosolic signaling in many tissues, including smooth and cardiac muscle, as matrix and intermembrane space/cytosol ROS may change in the opposite direction under certain physiological conditions. For example, Waypa et al. demonstrated that increases in mitochondrial ROS can trigger hypoxia-induced Ca2+ responses in pulmonary artery smooth muscle cells (346) and that acute hypoxia induces O2 •− release from complex III of smooth muscle cells (347), constituting oxidant signals that diffuse into the cytosol and trigger increases in [Ca2+(i)] that cause acute hypoxic pulmonary vasoconstriction (348).
Other studies have indicated that in pulmonary vascular smooth muscle cells mitochondrial matrix oxidant signals generated during hyperoxia, specifically H2O2, activate phosphodiesterase type 5 in a reaction that involves cGMP-dependent protein kinase (86). In general, this action has important implications for the source of mitochondria-derived ROS and differential targeting of matrix, intermembrane space, and cytosolic antioxidants.
Although not a free radical, H2O2 is a very important biological marker of oxidative stress due to its ability to cross cellular membranes. In addition, it can act as an intracellular messenger in different redox pathways. H2O2 can be converted to H2O by several enzymes, including glutathione peroxidase (GPX) and catalase (CAT).
Another enzyme with antioxidant activity is heme oxygenase (HO-1), which metabolizes heme to biliverdin, iron, and carbon monoxide (27). HO-1 is activated under oxidative stress conditions (hyperthermia, hypoxia, heavy metals, etc.) and has been shown to be beneficial in several diseases, including cardiovascular pathologies, acute kidney injury, liver disease, and diabetes (112).
Under physiological conditions, ROS are released primarily from mitochondrial ETC, but there are other important sources that need to be taken into account, such as peroxidases, NADPH oxidase in the membrane of leukocytes, and certain types of non-phagocytic cells, myeloperoxidase, xanthine oxidase, cyclooxygenase, lipoxygenase, cytochrome P450 monooxygenase, or nitric oxide synthase (NOS). Therefore, other intracellular compartments that can contribute to the cellular production of ROS, besides mitochondria, involve the plasma membrane, peroxisomes, and the endoplasmic reticulum (ER). The reactive species generated from all these sources can damage many intracellular structures, which is related to the development of different pathologies (269). For example, NADPH oxidase, well known to be involved in the inflammatory process, catalyzes the production of O2 •− and is present mainly not only in leukocytes but also in other types of cells such as mesangial cells, epithelial cells, fibroblasts, chondrocytes, or endothelial cells, where it plays important regulatory functions (258). It has also been demonstrated that this enzyme plays a key role in the development of many diseases (90). In this sense, Wedgwood and Steinhorn have demonstrated that persistent pulmonary hypertension of the newborn (PPHN) increases p22 (phox) and Nox4 expression and activity, resulting in elevated H2O2 levels in the PPHN pulmonary artery. Increased H2O2 induces vasoconstriction via mechanisms involving SOD3 inactivation, and stimulates vascular remodeling via nuclear factor kappa B (NF-κB) activation and increased cyclin D1 expression (348). It is important to mention that the oxidants produced by immune cells have a dual function. On the one hand, they function as microbicidal agents by killing pathogens, and on the other hand, they can act as signaling molecules in different intracellular pathways (78, 90). In fact, ROS and reactive nitrogen species (RNS) can modulate several enzymes as well as membrane receptors, ion channels, lipid kinases and phosphatase transporters, and various transcription factors, including NF-κB, hypoxia-inducible factor alpha, and nuclear factor-E2-related factor 2 (Nrf-2) (78, 114). Furthermore, different proinflammatory cytokines such as interleukin (IL)-6 and tumor necrosis factor alpha (TNF-α) can be modulated by the levels of ROS, thus controlling the inflammatory response, and therefore the different leukocyte functions (adhesion, migration, and phagocytosis), and crucial cellular processes such as apoptosis and autophagy. Under oxidative stress conditions, the excessive production of ROS can damage vicinal cells, thereby contributing to tissue damage (270).
It should also be highlighted that redox signaling plays a role in the interplay between mitochondria and NADPH oxidases, and several reviews [for example, by Daiber (65)] have described different models of such signaling pathways: (i) NADPH oxidase activation can be triggered by angiotensin-II with subsequent opening of mitochondrial ATP-sensitive K channels in an ROS-dependent manner, leading to depolarization of mitochondrial membrane potential (Δψm) followed by mitochondrial ROS formation and respiratory dysfunction; (ii) Using pharmacological and genetic inhibitors, a role of mitochondrial ROS for the induction of NADPH oxidase via PKCɛ was demonstrated in a model of hypoxia-stimulated formation of mitochondrial ROS (mtROS); (iii) Cell death by serum withdrawal enhances ROS production in human 293T cells by stimulating both mitochondria and Nox1; and finally; (iv) Cross-talk between NADPH oxidases (serum-soluble gp91phox [Nox2]) and mitochondria has been observed in nitroglycerin-induced tolerance involving the mitochondrial permeability transition pore and ATP-sensitive K+ channels.
In addition to ROS, mitochondria produce RNS (Fig. 2B), which originate from nitric oxide (•NO), a gaseous molecule that can passively diffuse through mitochondrial membranes. One such RNS is peroxynitrite (ONOO−), a strong oxidant and nitrating agent formed by the reaction of O2 •− with •NO. Both ROS and RNS (collectively known as RONS) are produced through aerobic metabolism, and therefore act as “sensors” of intracellular alterations in O2 concentration. Through this action, RONS play an important role as signaling molecules involved in diverse cell functions in normal physiological conditions such as programmed cell death, regulation of stress responses, and cell proliferation (30, 79). •NO is an important intracellular signaling molecule that, unlike many other messengers, is both freely diffusible across membranes and (comparatively) highly reactive. Moreover, it is capable of reacting with a large number of intracellular components at considerable distances from its site of synthesis. •NO is released as a by-product by several isoforms of NOS, including a neuronal isoform (NOS1), an inducible isoform (NOS2) expressed in several types of cells, and the endothelial constitutive isoform (NOS3). In addition, a mitochondrially located •NO synthase has been described, which is responsive to changes in Ca2+ concentration in the mitochondrial matrix and plays a very important role in the modulation of mitochondrial O2 consumption (286). •NO can inhibit O2 consumption by inhibiting cytochrome c oxidase (complex IV of ETC), therefore inducing ROS production (55, 100, 192). In addition, •NO can inhibit mitochondrial complex I (269), an action that has important consequences regarding the OXPHOS process and the redox state of mitochondria. Furthermore, •NO can modulate mitochondrial biogenesis through its ability to regulate ROS production and mitochondrial O2 consumption (217, 254, 360). Besides mitochondria, RNS can be generated in mammalian cells by peroxisomes (94) or the plasma membrane.
It has been proposed that endogenous hydrogen sulfide (H2S), a naturally occurring gassotransmitter similar to •NO, plays a particularly important role in the process of acute O2 sensing in blood vessels (224) and the mammalian carotid body (225). Buckler has demonstrated that H2S can inhibit mitochondrial function over a similar concentration range than cyanide, suggesting that the effects of H2S on background K channels are a consequence of inhibition of OXPHOS (32).
Importantly, mitochondria should be viewed not only as a generator of toxic ROS but rather as a crucial regulator of ROS signaling and balance (78). Indeed, there is evidence of the role of mitochondria-generated ROS as signaling molecules and participants in cellular adaptative mechanisms, including the phenomenon of preconditioning. For instance, mitochondrial preconditioning induced by cyanide in cultured brain endothelial and neuronal cells triggers a protective response mediated by mtROS, which prevents apoptotic cell death and creates resistance against glucotoxicity (56). In a recent paper, Yee et al. postulate that, in nematodes, sensing of mtROS by the apoptotic pathway can elicit protective mechanisms that promote survival under stressful conditions, independently of apoptosis, which points to increased mtROS generation as a cause of extended longevity (368). Regarding ROS as signaling molecules, increasing evidence indicates that redox-dependent protein modification is an important mechanism in signal transduction involving redox modification of specific protein moities such as iron–sulfide (Fe–S) centers or cysteine residues (41). Inside mitochondria, several targets have been described; for example, the mitochondrial ETC, which contains the biggest multi-Fe–S protein, NADH dehydrogenase. Moreover, destabilization of the Fe–S cluster in aconitase by O2 •− inhibits this enzyme's activity, thereby limiting mitochondrial respiration. In addition, the peroxiredoxin (Prx) family of peroxidases, one of which (namely PrxIII) has been shown to be localized inside mitochondria, is believed to be involved in signaling loops through oxidation of a conserved cysteine residue present in the NH2-terminal region, the primary site of peroxide-induced oxidation. The importance of the maintenance of the cellular thiol/disulfide couples, which include thioredoxins (TRXs), Cys/CySS, and glutathione (GSH)/oxidized glutathione (GSSG), has led to a modification of the traditional concept of oxidative stress as an imbalance of prooxidants and antioxidants. Instead, the disruption of the existing thiol-dependent redox signaling and control mechanisms seems to be among the most sensitive and quantitatively relevant processes in oxidative stress (136). This “redox hypothesis,” an alternative to the “free radical hypothesis,” postulates that oxidizable thiols are common control elements for biologic processes and are functionally organized in redox circuits that are kinetically limited, insulated from each other, and highly responsive to redox conditions. It is the disruption of these circuits that causes oxidative stress. Since these redox mechanisms control a wide range of biologic processes that do not require macromolecular damage, including cell proliferation, apoptosis, or inflammation, it is conceivable that free radical scavenger trials have failed, because oxidative stress research has overemphasized the importance of free radical-induced damage to macromolecules as the underlying mechanism of pathogenesis (136). Moreover, the “redox hypothesis” is centered on redox compartmentalization and compartment-specific signaling, which can explain the characteristics of mitochondria versus those of other subcellular territories in the cellular redox metabolism. For instance, when HeLa cells are exposed to the cytokine TNF-α, compartmentalized, mitochondria-specific ROS generation occurs and results in TRX-2 oxidation, downstream signaling to cytoplasm with NF-κB activation and apoptosis; whereas TRX-1, the extra-mitochondrial TRX isoform, does not undergo oxidation (116).
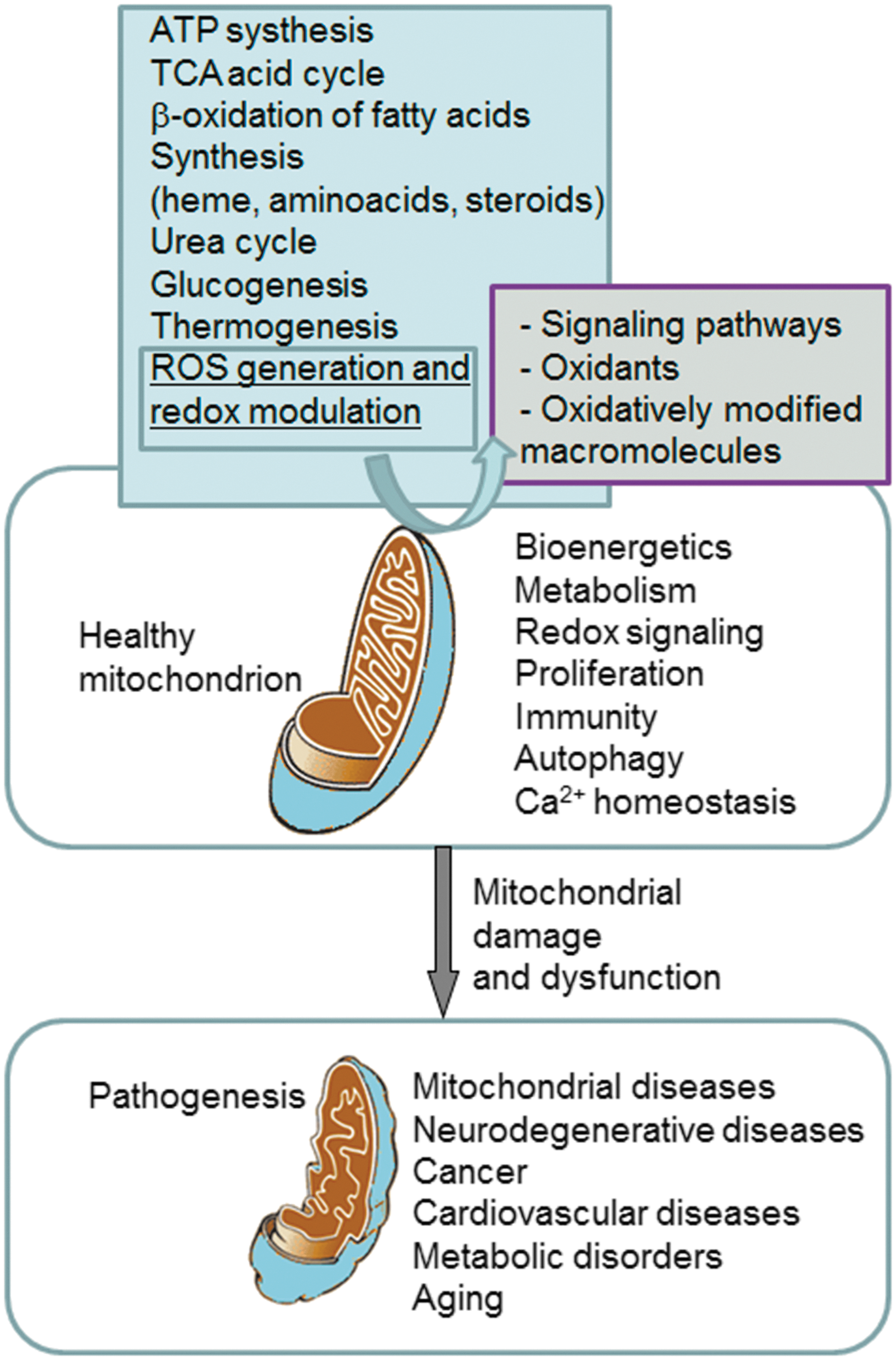
While low to moderate levels of ROS are physiological and can be beneficial or necessary for cell survival, high ROS levels are detrimental and associated with cell death. Clearly, mitochondrial dysfunction and oxidative stress are critical factors in the pathogenesis of several diseases. Nevertheless, the mechanisms of mitochondria-induced injury differ depending on the type of disease and the participation of ROS in the pathogenic processes varies largely (oxidative stress can be a cause, an aggravator, or a part of the outcome in a particular pathology). Diverse mechanisms of mitochondrial dysfunction have been described, including OXPHOS impairment, mtDNA depletion, membrane permeabilization, and alteration of fatty acid oxidation; therefore, specific drugs, including antioxidants, may have different effects in different pathological settings.
There is much evidence to show that mtROS can have an important impact on extra-mitochondrial structures. Increased mtROS production has been attributed an integral role in the acute inotropic response of cardiomyocytes to β-adrenergic stimulation. This is of clinical relevance, as chronically sustained adrenergic stress is associated with the development of heart failure and cardiac arrhythmias (9). Moreover, stimulation of β-adrenergic receptors causes apoptosis in adult rat ventricular myocytes through ROS/JNK-dependent activation of the mitochondrial death pathway (263). In a rat model of ischemia/reperfusion, caspase-8 activation has been shown to increase mtROS and •NO production, resulting in S-nitrosylation of ryanodine receptor RyR2 and depletion of calstabin2 from the channel complex, causing a diastolic sarcoplasmic reticulum (SR) Ca2+ leak that leads to acute pathological left ventricular remodeling (87).
1. ROS implication in specific aspects of mitochondrial function
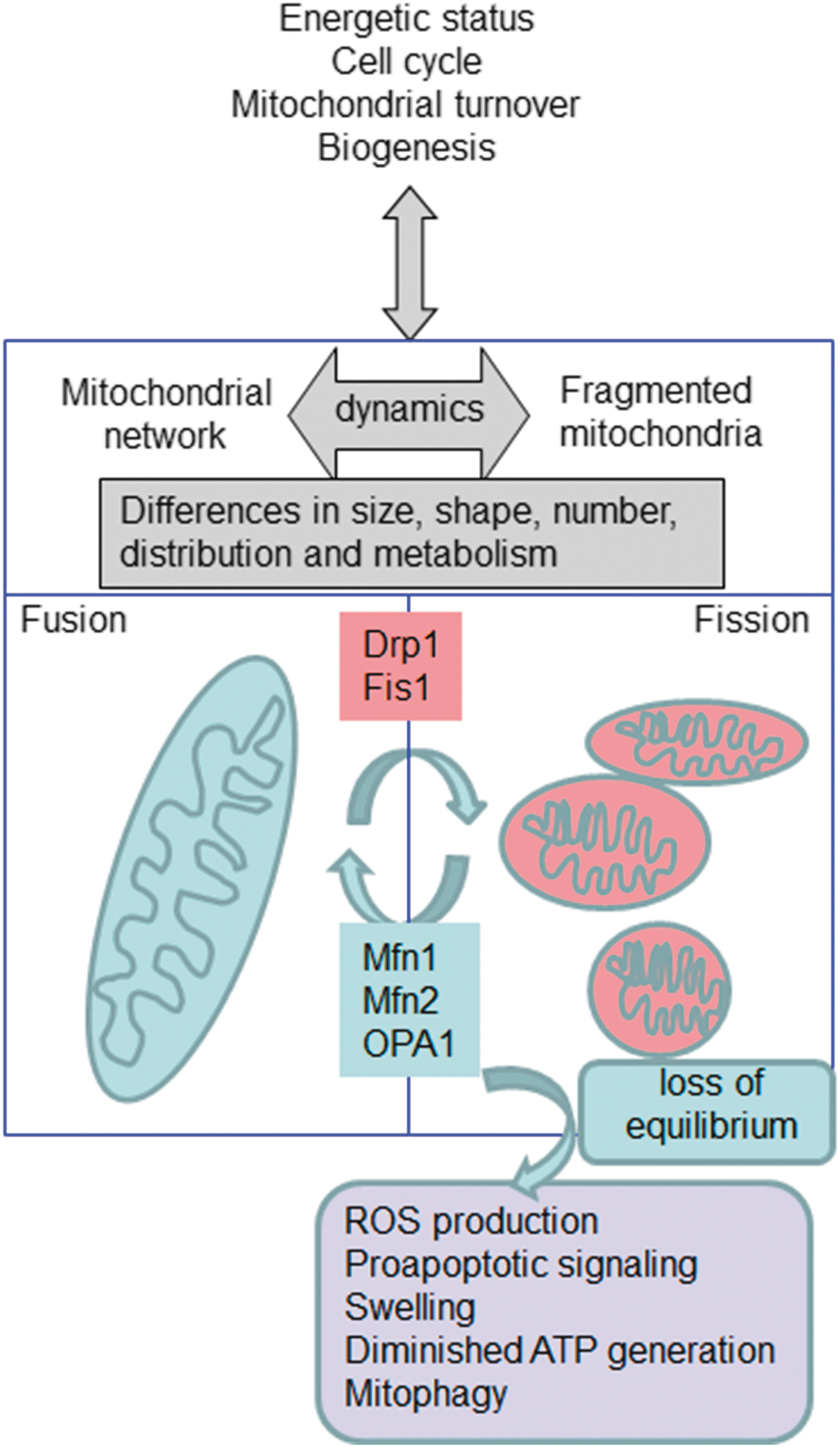
ROS are implicated in virtually all mitochondrial functions, from ATP generation, [Ca2+] buffering to induction of apoptosis. During recent years, the implication of specific mitochondrial functions such as mitochondrial dynamics (Fig. 3) and autophagy/mitophagy (Fig. 4) have been studied in different pathophysiological settings. In addition, both processes have been shown to be intimately connected to the redox balance in mitochondria through a complex and multi-way cause/effect relationship. For example, multiple reports describe that mitochondrial shape can influence cellular and physiological functions, such as ROS production, Ca2+ signaling, leukocyte migration, or lifespan (34, 35, 146). These actions can be explained by the specific mitochondrial localization and the relationship between mitochondrial shape and mitochondrial function. In fact, mitochondria are located where high amounts of ATP are required (34) and also where Ca2+ signaling needs tight regulation (126). Importantly, alterations in mitochondrial Ca2+ have been related to several pathologies, including neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS) (318), and aging (332). In recent years, it has been demonstrated that mitochondria actively interact with ER membranes through structure-denominated mitochondria-associated membranes, which enable direct and rapid exchange of lipids and Ca2+ between the two compartments. These complex ER–mitochondria interactions are emerging as a crucial hub for many cellular processes, such as Ca2+ signaling, mitochondrial dynamics, apoptosis, autophagy, and lipid biosynthesis/trafficking, with far-reaching implications for cell life and death (213). Mitochondrial localization and Ca2+ signaling in other areas of the cells have also been shown to be important and clinically relevant. In a recent elegantly performed study employing atomic force and electron microscopy, Dague et al. have shown that, after heart failure, cardiomyocytes exhibit overall sarcolemma disorganization with general loss of crests, accompanied by depletion of subsarcolemmal mitochondria (62).
a. Mitochondrial dynamics
The permanent and dynamic balance between fission and fusion states is very important to the cellular homeostasis. Alterations in the mitochondrial network have been associated with several human pathologies, including cardiovascular diseases (CVDs), diabetes, and genetic mitochondrial diseases. Several members of the fusion-regulating machinery have been described in detail (74) and comprise dynamin-homologous GTPases that participate in fission, fusion, and tabulation of membranes. In mammalian cells, it has been described that mitochondrial fission depends on dynamin-related protein (Drp1), a cytoplasmic dynamin GTPase involved in the fragmentation of peroxisomes, ER, and mitochondria (283). Drp1 is located in the cytoplasm, and several mechanisms facilitate its translocation to mitochondria, where it is located at the outer mitochondrial membrane (OMM). Drp1 translocation is triggered by mitochondrial dysfunction as an inducer of fragmentation (127). In fact, in mitochondrial dysfunction, there is an increase in the cytosolic Ca2+ that activates calcineurin and dephosphorylates Ser-637 residue of Drp1 (37), thereby inducing its translocation. This process is crucial in different conditions, such as ischemia-reperfusion damage of the heart (338), and can be counteracted by peptide inhibitors to inhibit Drp1-dependent cell death and mitochondrial damage (36). Its importance in Huntington's disease has also been underlined, where hyperactivation of calcineurin, which dephosphorylates Drp1 at Ser-637, induces an increase in Drp1 translocation to mitochondria, thereby increasing apoptosis (58).
It has also been described that Ser-637 can be phosphorylated by calmodulin-dependent protein kinase Iα; in this case, Drp1 phosphorylation is related with its mitochondrial location (115), with important homeostatic consequences. Mitochondrial Drp1 is also stabilized by other mechanisms, such as SUMOylation, a post-translational modification involved in various cellular processes (344), in this way gaining protection from degradation by the ubiquitin-proteasome system. This has been recognized as an important site for the regulation of mitochondrial function in several diseases models, such as cardiomyopathy and ischemia/reperfusion.
Mitochondrial fission 1 protein (Fis1), a mitochondrial protein anchored to the OMM, is also an important regulator of mitochondrial dynamics. Mitochondrial fragmentation can be caused by both Fis1 overexpression and the enhanced expression of a dominant negative mutant of Drp1 (131). Fis1 has also demonstrated additional functions, such as inducing apoptosis via the ER pathway (5), and can trigger autophagy to remove damaged mitochondria (102), thus playing an important physiological role.
Two mitofusins (Mfn1 and Mfn2) control OMM fusion. During mitochondrial fusion, Mfn1 is responsible for mitochondrial tethering and Mfn2 participates at the end of the process of fusion (155). Furthermore, it has been proposed that Mfn2 correlates with the proliferation of vascular smooth muscle cells (42) and with the oxidative metabolism in muscle (16). Mfn2 can also control the shape of ER and tethers it to mitochondria (67). In other types of cells, such as fibroblasts, Mfn1 is required for fusion triggered by optic atrophy 1 protein (OPA1) (49). OPA1 can be anchored to the IMM, the maintenance of which is another important function of OPA1 (221). At least eight splice variants of OPA1 have been described, and they are subject to a complex post-translational cleavage (222).
In general, mitochondrial dynamics represent the link between the shape of the organelle and pathophysiological processes, and can be considered a key target in the treatment of multiple diseases. Recently, mitochondrial dynamics has also been associated with cancer, as the migration of cells during cancer cell invasion, a phenomenon that is crucial for the invading and metastasizing capacity of the tumor, is related to specific intracellular localization of mitochondria. In faster moving cells, mitochondria are located in between the nucleus and the leading edge of the migrating cells, and this asymmetric distribution is governed by mitochondrial fusion (OPA1) and fission (Drp1) proteins (69).
Mitochondrial dynamics and ROS are closely related, and this relation may be crucial for many human diseases. There is abundant evidence that mitochondrial fragmentation and increased ROS generation coincide; however, it is unclear whether ROS is a cause, consequence, and/or exacerbating mechanism of mitochondrial fission. On the one hand, mitochondrial network formation is considered a regulator of mtROS generation. For example, mitochondrial fragmentation is necessary for high glucose-induced ROS overproduction and respiration increase in cultured rat hepatocytes (372), which supports results obtained in human aortic endothelial cells cultured under high glucose conditions where enhanced mtROS production, impaired endothelial nitric oxide synthase (eNOS) activation, and loss of •NO bioavailability are prevented by inhibiting mitochondrial fission (291). On the other hand, mtROS influence fission/fusion processes; for instance, Makino et al. provided evidence that in mouse endothelial cells, ROS scavenging prevents glucose-induced mitochondrial fragmentation, suggesting that an increase in ROS triggers fission under these conditions (186).
b. Autophagy
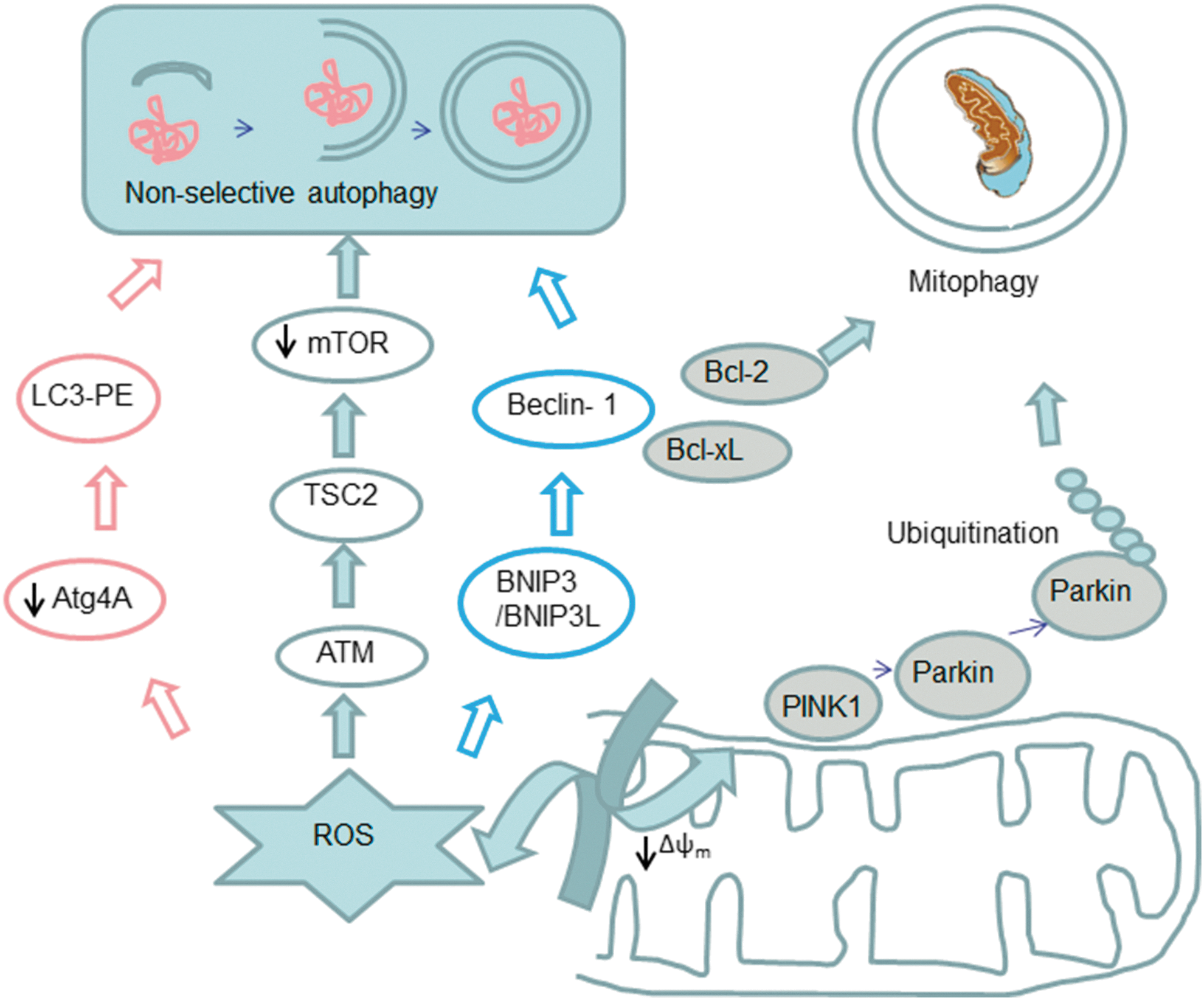
Another process that merits attention is autophagy (Fig. 4), which is vital for maintenance of cellular homeostasis and generation of energy, fatty acids, and amino acids for macromolecular synthesis and tissue remodeling. This process involves an intracellular, lysosomal degradation of cellular components such as macromolecules, protein aggregates, and organelles. Three different types of autophagy have been described so far (macro-, micro-, and chaperone-mediated autophagy), although the most evaluated process in mammalian cells is macro-autophagy (203). It begins with the sequestration and isolation of the cytoplasmic cargo by the autophagosome, followed by the fusion of these vesicles with the lysosome, which leads to the formation of autolysosomes, where cellular components are degraded by lysosomal enzymes, with the consequent release of ROS (146, 203, 204).
Autophagy has been related to the development of different diseases, including diabetes, neurodegenerative and myodegenerative disorders, Crohn's disease, various liver diseases, and inherited diseases such as Huntington's disease (57). Initially, autophagy was related to cell death, but mounting evidence has pinpointed the cell-protective role of this process (15, 22, 284). In fact, different studies have demonstrated the existence of a complex relationship between autophagy and cell death that determines whether a cell will live or die in response to anticancer therapies (22). ROS and autophagy are intimately connected (Fig. 4) (147). For example, nutrient starvation-induced ROS generation plays a key role in autophagy in cancer cell lines U87 and HeLa cells (44). Other studies have demonstrated that ROS scavenging or SOD overexpression can modulate autophagy and cell death in the presence of different inhibitors of the ETC (43,44). In this sense, mtROS can modulate autophagy by different pathways, including alteration of the proteins that are indirect participants in the autophagic process. For instance, it has been postulated that ROS in general, and specifically H2O2 released from mitochondria, modulate the activity of Atg4, which is a key cysteine protease in the autophagic process. Atg4 cleaves the C-terminus arginine residue of Atg8, which enables the conjugation of phosphatidylethanolamine with Atg8 and the subsequent recruitment of this protein on the autophagosomal membrane, a fundamental step in the final maturation of the autophagosome. H2O2 can also oxidize Atg4, thus enabling autophagosome completion. Therefore, mtROS can act as signaling molecules that trigger autophagy as a cell survival mechanism (284). Other studies have shown that, in the presence of ROS, there is an upregulation of beclin 1, a protein involved in the initiation of autophagy (287, 365). Moreover, as mentioned earlier, H2O2 and O2 •− can modulate the activity of different signaling pathways involved in autophagy. Several groups have demonstrated that ROS play a key role in the autophagy pathway in cancer cells. In fact, H2O2 can modulate Δψm, which leads to inhibition of the Akt/mTOR pathway that induces autophagy (375). Other studies have demonstrated that high levels of ROS induce autophagy that is dependent on p38 signaling in skeletal muscle or cardiomyocytes (193). In addition, Wong et al. have proposed that ROS and downstream activation of JNK and ERK pathways can be responsible for autophagy induction (355). Moreover, not only mtROS are involved in the autophagy process, as the release of ROS from NADPH oxidase has been shown to induce autophagy in immune cells.
Another kinase related to both autophagy and mitochondrial function is AMP-activated protein kinase (AMPK). Pharmacological AMPK activation has been shown to promote autophagy, and this effect is beneficial in several models of human diseases, such as a mouse model of Duchenne muscular dystrophy (234) or diabetic cardiomyopathy (120). The mechanism proposed for AMPK-mediated activation of autophagy involves direct phosphorylation of ULK1 (147).
Autophagy can regulate the mitochondrial network by eliminating damaged mitochondria. Kissova et al. have demonstrated that the Uth1p protein is responsible for the early selective elimination of mitochondria through autophagy under nutrient deprivation (149). This action has been defined as mitophagy, a term introduced by Lemasters to describe a type of macroautophagy where damaged mitochondria are joined by autophagosomes and removed to maintain mitochondrial homeostasis (165). In humans, the molecular mechanism that regulates mitophagy involves the parkin/PTEN-induced putative kinase protein 1 (PINK1) pathway. PINK1 is a mitochondrial kinase that is capable of identifying depolarized mitochondria. It undergoes voltage-dependent lysis and is removed from mitochondria in normal conditions; however, when mitochondria are impaired with the subsequent decrease in Δψm, PINK1 accumulates and recruits parkin (135, 214). Parkin can ubiquitinate proteins and recruit autophagic machinery to the damaged mitochondria for their removal (97). Many studies have pointed out that autophagy is a complex process which can be regulated by ROS at different points. Moderated levels of ROS can also induce mitophagy or autophagy to eliminate damaged mitochondria; on the contrary, high levels of ROS can modify signaling pathways and induce apoptosis (216). Importantly, there is evidence that mitochondria-targeted antioxidants can modulate autophagy, which will be discussed in detail.
In summary, mitochondria are a major source of ROS and RNS, species that can regulate mitochondrial activity through different mechanisms, including modulation of O2 consumption and OXPHOS, induction of mitochondrial membrane permeability transition, regulation of mitochondrial biogenesis, mitochondrial dynamics, and autophagy/mitophagy (23, 28, 218, 238). Another field of action involves the presence of oxidative stress and its consequences, such as lipid peroxidation, protein and DNA oxidation, which occur when there is a strong disruption of the cellular redox homeostasis (Fig. 2) (118).
2. Mitochondrial susceptibility to ROS/RNS damage
Mitochondria continuously generate ROS (7), and immediate and constant exposure to the latter renders mitochondrial components such as mtDNA, proteins, and membranes especially sensitive to such insult (Figs. 1 and 2). First, mtDNA accumulates much more oxidative damage than nuclear DNA due to its proximity to the source of ROS release, its less sophisticate repair systems, and the lack of histones that physically protect nuclear DNA from exogenous attacks (190). Currently, much research is focused on the proteins involved in the maintenance of the mitochondrial genome. One such protein is mitochondrial transcription factor A, a histone-like protein, member of a high mobility protein family and the first-identified mitochondrial transcription factor, which binds to mtDNA and is essential for its maintenance (139). Second, mitochondrial proteins are also particularly susceptible to oxidative damage because of the presence of Fe–S clusters in their structure that are susceptible to oxidant inactivation, and also of thiol residues that are vulnerable to S-nitrosation by RNS (63). In this sense, some mitochondrial proteins are known to be altered by redox modifications and ROS/RNS signaling, such as the enzyme aconitase (under aging conditions) (364) or α-synuclein and DJ-1 in Parkinson's disease (PD) (66). In addition, proteins integrating complexes of the ETC can be affected by ROS and RNS (63, 79). Damage to complex I, the most vulnerable ETC complex, increases ROS production, which leads to a vicious circle of further mitochondrial dysfunction. It is important to note that damage to complex I has a stronger impact on mitochondrial function than damage to other complexes, as mitochondria possess smaller amounts of complex I than other ETC complexes (212). Finally, it is known that phospholipids that integrate mitochondrial membranes are rich in unsaturated fatty acids, which are extremely vulnerable to lipid peroxidation by ROS. One of the lipids most affected by this oxidative damage is the IMM essential component cardiolipin, which is constituted exclusively by polyunsaturated fatty acids (PUFAs) and is also closely associated with mitochondrial ETC complexes (113). This ROS-induced damage to the IMM is particularly relevant, not only because this membrane contains ETC complexes but also because of a special feature, namely, elevated Δψm, which is necessary for maintaining its electrical properties. When IMM integrity and function are compromised by oxidative damage, respiratory energy is dissipated through the leakage of H+, which further compromises mitochondrial OXPHOS (85).
It is noteworthy that, in addition to mitochondria, other subcellular structures have been described as direct and specific targets of oxidative and nitrosative stress, including the ER, the plasma membrane, and the nucleus. For example, NAD(P)H oxidase has been associated with the SR of cardiac and skeletal muscle, and the O2 •− generated by this enzyme can oxidate RyR and therefore influence Ca2+ release during excitation–contraction coupling by the SR (129). RyR belongs to a class of intracellular Ca2+ channels and is an important mediator of Ca2+-induced Ca2+ release in excitable cells such as muscles and neurons. RyR2 (cardiac muscle) contains≈33 free thiol residues, rendering it highly sensitive to the cellular redox state. Cysteine oxidation and S-nitrosylation facilitate RyR opening and SR Ca2+ leak, which can provoke arrhythmia, skeletal muscle weakness, and muscle remodeling. Modifications of several sarcomeric proteins have also been identified and associated with defects in contractile function (306). Oxidative stress has also been shown to cause impairment of intracellular proteolysis via covalent binding of 4-hydroxy-2-nonenal, a major end product of lipid peroxidation, to proteasomes (220).
3. Cellular antioxidant defense systems
Excessive ROS production is usually compensated by the antioxidant defense system, thus maintaining the redox balance. Cellular antioxidants, including enzymes such as SOD and CAT and low-molecular-weight antioxidants such as vitamins C and E, and GSH, are the main orchestrators of this defensive barrier (Fig. 5) (78, 114).
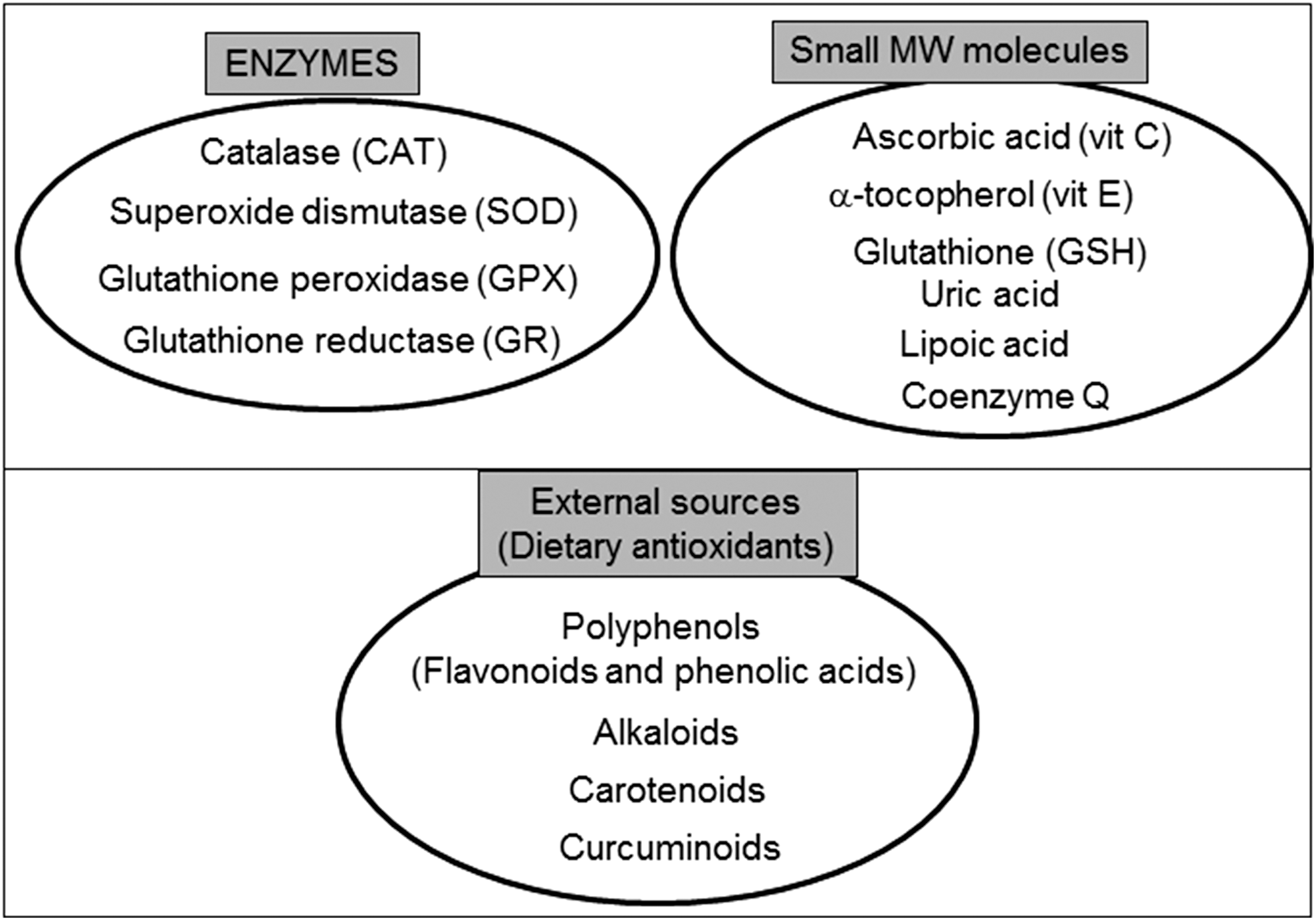
The tripeptide GSH (γ-glutamylcysteinylglycine) is the principle thiol antioxidant and redox buffer of the cell and is present in different intracellular locations, such as the nucleus, mitochondria, ER, or cytosol. Together with its oxidized form (GSSG), it maintains the redox balance, a function that is crucial to the fine-tuning of the cellular redox environment between control situations and oxidative stress conditions (336). GSH participates in the maintenance of an adequate redox state of the nuclear proteins through the presence of protein sulfhydryls that are vital for DNA expression and repairment. The protective effect of GSH against oxidative stress is due to its role as a (i) participant in amino-acid transport through the plasma membrane; (ii) cofactor of several detoxifying enzymes against oxidative stress (e.g., GPX); (iii) direct scavenger of singlet oxygen and •OH; (iv) detoxifier of H2O2 and lipid peroxides through the enzymatic action of GPX; and (v) regenerator of antioxidants such α-tocopherol or ascorbic acid back to their active forms. This capacity of GSH for regeneration of antioxidants is related with the redox state of the GSH disulfide–GSH couple (GSSG/2GSH). High levels of GSSG can induce oxidative stress and damage of multiple enzymes (333). In fact, one of the best methods to evaluate oxidative stress inside cells is the assessment of GSSG or the GSSG/GSH ratio (327).
Mitochondria are usually protected from oxidative insults and damage by a complex, multilayer network of mitochondrial antioxidant systems (Fig. 5). H2O2 can be readily converted to water by mitochondrial GPX, which oxidizes GSH to GSSG, and glutathione reductase then reduces GSSG back to GSH. GSH is synthesized in the cytosol, and its import into mitochondria is mediated by a transporter. In addition to GSH, mitochondria have two other small thiol-disulfide oxidoreductases—TRX and glutaredoxin—that play important roles in thiol redox control. Another detoxification enzyme, CAT, is present only in certain types of mitochondria [murine heart (265) or liver (280)]. In addition to these antioxidant enzymes, mitochondria possess several low-molecular-weight antioxidants, including α-tocopherol and ubiquinol. These molecules are particularly effective in scavenging lipid peroxyl radicals and preventing the free radical chain reaction of lipid peroxidation.
Another group of molecules implicated in redox control in the cell (and inside mitochondria) are metallothioneins (MT). These small cysteine-rich, metal-binding proteins play important roles in many biological processes such as metal ion homeostasis and detoxification, protection of cells from oxidative stress, cell proliferation, and cell survival. Their expression can be induced by many stimuli including hormones, cytokines, metal ions, oxidating agents, and specifically mitochondrial dysfunction (278). The mitochondrial distribution of these proteins is still not fully known, but it seems that some mitochondria do not contain them (e.g., heart mitochondria); however, their presence in the mitochondrial intermembrane space in the liver has been demonstrated (367). Abundant in vitro and in vivo evidence has pinpointed the antioxidant properties of MT based on their ability to act as important maintainers of the cellular zinc pool and as free radical scavengers. Using MT-overexpressing transgenic or MT-null mice, it has been shown that MT confer protection against the oxidative damage induced by a diversity of oxidative conditions, including doxorubicin cardiotoxicity, ischemia/reperfusion, diabetes, and alcohol administration (278).
II. Non-Targeted Antioxidant Treatments
Oxidative stress has been related to the pathophysiology of a wide variety of diseases (275, 276), including cardiometabolic diseases (hypertension, stroke, diabetes, metabolic syndrome), neurodegenerative disorders (Alzheimer's disease [AD], PD, and others), Duchenne muscular dystrophy, chronic obstructive pulmonary disease (COPD), and cancer.
It is not surprising that research over recent years has focused on antioxidants as a potential therapy with which to restore the normal physiology in these pro-oxidant conditions. Although many initial studies in cells or animal models have been successful, clinical trials have produced disappointing results. For example, in rat models of hypertension and renal dysfunction, treatment with vitamins C and E prevented renal inflammation and decreased arterial pressure, thus improving renal function (323). However, vitamin E treatment in hemodialysis patients with chronic kidney disease did not reduce oxidative protein modifications and lipid peroxidation (181), although a reduction in oxidative stress markers (60, 185) and improvements in endothelial function (356) have been reported. Regarding the antioxidant prevention of CVD, Papparella et al. showed that vitamin C prevented cardiac damage in rats with zidovudine-induced CVD, exerting its beneficial effects through inhibition of NADPH oxidase activity (230). In contrast, several clinical trials, such as the long-term trial performed by Sesso et al., have found that vitamins C and E have no preventive effect with regard to cardiovascular events, including coronary revascularization, non-lethal myocardial infarction, stroke, or CVD death (54, 178, 288). Another example of antioxidant therapy attempt is that used in AD, in which lipid peroxidation and β-amyloid deposition have been found to be reduced in the brain of mice treated with vitamin E in the early stage of the disease (312). In contrast to these results, this antioxidant did not improve cognitive defects in AD patients (176), thus calling into question its potential benefits in this neurodegenerative disorder (249). In addition to vitamins E and C, β-carotene has been presented as promising, but proved to have null or even detrimental effects on CVD when administered to high-risk subjects (54, 260).
The effects of antioxidants in cancer treatment are controversial. In fact, in the majority of past clinical trials, supplementation with antioxidants had no impact on the risk of developing a variety of cancers and in some studies, antioxidant supplementation was actually associated with an increased risk of cancer. The most notable of these studies were the β-carotene and Retinol Efficacy Trial (CARET) trial and the ATBC Study, in which daily supplementation with β-carotene or a combination of β-carotene and vitamin A increased incidence of lung cancer and all-cause mortality in smokers. The CARET evaluated the effect of daily retinyl palmitate (25,000 IU) and β-carotene (30 mg) administration on the incidence of lung cancer, other cancers, and death in 18,314 participants at a high risk of developing lung cancer because of a history of asbestos exposure or smoking. The CARET was aborted ahead of schedule in January 1996, because participants who had been randomly assigned the active intervention were found to have a higher rate of CVD mortality, and displayed a 28% increase in incidence of lung cancer and a 17% increase in incidence of death compared with participants in the placebo group (103). In another study, the effect of vitamin E and β-carotene (ATBC) on the incidence of lung cancer and other cancers was studied, and no reduction was found in the incidence of lung cancer among male smokers after 5–8 years of dietary supplementation with α-tocopherol or β-carotene. In fact, this trial pointed to the possibility that these supplements have harmful as well as beneficial effects (322).
The Selenium and Vitamin E Cancer Prevention Trial, which randomized 35,533 healthy >55 year-old men (>50 years if African American), revealed that neither selenium nor vitamin E, alone or together, prevented prostate cancer in this heterogeneous population (82). Other studies have demonstrated that N-acetylcysteine (NAC) and vitamin E markedly increase tumor progression and reduce survival in mouse models of B-RAF- and K-RAS-induced lung cancer, and accelerate tumor growth by disrupting the ROS-p53 axis. Given that somatic mutations in p53 occur late in tumor progression, antioxidants may accelerate the growth of early tumors or precancerous lesions in high-risk populations such as smokers and patients with COPD who receive NAC to relieve mucus production (282).
Exercise promotes longevity and ameliorates type 2 diabetes mellitus and insulin resistance (143, 229, 343). However, exercise also increases mtROS (248). Antioxidants are widely used as nutritional supplements, but whether they affect the health-promoting effects of exercise is not clear. In this sense, Ristow et al. (266) evaluated the effects of a combination of vitamin E (400 IU/day) and vitamin C (1000 mg/day) on insulin sensitivity in pretrained (n=20) and untrained (n=19) healthy young men. In both groups, exercise increased parameters of insulin sensitivity (glucose infusion rate and plasma adiponectin) only in the absence of antioxidants. This was paralleled by an increased expression of ROS-sensitive transcriptional regulators of insulin sensitivity and ROS defense capacity, peroxisome-proliferator-activated receptor gamma (PPARγ), and PPARγ coactivators PGC1α and PGC1β, again only in the absence of antioxidants. Exercise also increased the expression of SOD1, SOD2, and GPX, and this effect was inhibited by antioxidant supplementation. Consistent with the concept of mitohormesis, exercise-induced ROS generation reduces insulin resistance and causes an adaptive response by which the endogenous antioxidant defense capacity is enhanced. Thus, supplementation with antioxidants may preclude the health-promoting effects of exercise in humans.
Due to its essential role in mitochondrial respiration as an endogenous co-enzyme of ETC proteins and an ROS scavenger, Coenzyme Q10 (CoQ10), otherwise known as ubiquinone, exerts antioxidant effects in different pathological situations. Persson et al. demonstrated restored mitochondrial O2 consumption and an improvement in mitochondrial and renal functions in db/db diabetic mice fed a diet supplemented with the aforementioned coenzyme (236). Since CoQ10 is present at high concentrations in the heart, its potential benefits for cardiac dysfunction have been widely tested in humans with discrepant results (18, 345). Similar dicrepances have been reported in animal models; whereas CoQ10 attenuated amyloid β-peptide-induced mitochondrial dysfunction in brain mitochondria isolated from diabetic Goto-Kakizaki rats (207), and its administration was not fully advantageous for heart mitochondrial function in this model (223). More hydrosoluble molecules than ubiquinone have been developed and tested in different diseases; for example, the short-chain quinone idebenone, a synthetic derivative of CoQ10, has been shown to prevent cardiac hypertrophy in Friedreich's ataxia (119) and to improve measures of cognitive scores in patients with AD (351). Recently, idebenone has been awarded a temporary authorization and awaits marketing authorization for the treatment of Leber's Hereditary Optic Neuropathy, an inherited mitochondrial disease (Raxone®; Santhera Pharmaceuticals) (150).
Another strategy to preserve redox balance and an adequate mitochondrial function is the induction of endogenous antioxidants, and the Nrf-2 antioxidant signaling pathway is among the most eligible ones for this purpose. In patients with multiple sclerosis, the induction of Nrf-2 has been shown to be neuroprotective and anti-inflammatory (172), whereas sulforaphane induces enzymes downstream of Nrf-2 in airway disease, thus highlighting its potential antioxidant properties (264). However, bardoxolone methyl, a potent inducer of Nrf-2, has proved to have toxic effects in humans, leading to the interruption of an ongoing Phase III clinical trial (the BEACON study) (314).
Another potent antioxidant used in the treatment of diseases is lipoic acid (LA), which can be rapidly absorbed by cells and has a protective role against oxidative stress in different animal models (110, 111, 310). These studies demonstrated that LA can accumulate in different parts of the cell, but only in small concentrations within mitochondria (117). LA has powerful antioxidant properties and is therefore suitable for the treatment of diseases related to oxidative stress; for example, short-term and long-term supplementation of LA (200–1800 mg/day) in type 2 diabetic patients have beneficial effects in maintenance of glycemic control (200). LA can exert beneficial effects by several mechanisms, including improvement in vascular endothelial cell function (257), decrease in inflammation (104), amelioration of lipid abnormalities (376), and protection against myocardial ischemia/reperfusion injury (341) or antihypertensive effects (83). Defects in LA biosynthesis in human subjects can lead to the development of mitochondrial diseases, as has been shown in children with a mutation of NFU1, which is necessary for maturation of proteins such as succinate dehydrogenase or lipoic synthase (215). An in vitro study has shown that LA exerts a protective effect in fibroblasts obtained from patients with AD, as evidenced by decreases in oxidative stress and apoptotic markers, and this action is enhanced when LA is combined with NAC (206).
NAC is another important antioxidant with beneficial effects in different conditions. For instance, it might exert beneficial actions mediated by an increase in intracellular GSH levels, a major antioxidant, in different tissues (e.g., the lung) (255), and in particular respiratory diseases, including COPD, which is characterized, in part, by chronic mucus production, leading to an enhanced risk of infection. The benefits of short- and long-term administration of NAC for bronchial hypersecretion in chronic bronchitis have been studied since the early 1980s (320). Other research has focused specifically on NAC and has acknowledged the beneficial actions of oral administration during treatment of 3–6 months in reducing exacerbations and improving symptoms of chronic bronchitis (307). Moreover, administration of NAC is beneficial in acetaminophen poisoning, where it acts rapidly by increasing hepatic GSH synthesis, therefore protecting against oxidative stress (279). A number of clinical studies performed to investigate the potential of NAC as a therapeutic agent have been conducted in type 2 diabetes. This is of particular interest, as diabetic patients are a target group with a high risk of CVD in whom aspirin is ineffective in primary prevention of cardiovascular complications. For example, 6-month treatment with a combination of
There is abundant evidence in animal models of the utility of NAC for treatment or prevention of ROS-associated pathologies. Oral NAC treatment is beneficial against RNS-dependent ventricular tachycardia. In fact, Fauconnier et al. (87) demonstrated that diastolic SR Ca2+ leak via RyR2 due to S-nitrosylation of the channel and that calstabin2 depletion from the channel complex triggers cardiac arrhythmias, a situation in which treatment with NAC has beneficial effects. In addition, it has been demonstrated that NAC protects from ventricular arrhythmias by attenuating reduced connexin 43 expression and function via both PKA- and Epac-dependent pathways, which converge through the inactivation of glycogen synthase kinase-3β (164). Other studies have shown the beneficial effects of NAC in diabetes (140) in which diabetic C57BL/KsJ-db/db mice were exposed to antioxidant treatment (NAC, vitamin C plus E, or both). NAC increased glucose-stimulated insulin secretion and moderately decreased blood glucose levels, whereas vitamins C and E were not effective when used alone and only slightly effective when used in combination with NAC. Histologic analyses of the pancreas revealed that the β-cell mass was significantly larger in the diabetic mice treated with antioxidants than in the untreated animals. It was speculated that the antioxidant treatment suppressed apoptosis in β-cells without changing the rate of β-cell proliferation, supporting the hypothesis that, in chronic hyperglycemia, oxidative stress-induced apoptosis causes reduction of β-cell mass. The antioxidant treatment also preserved the amounts of insulin mRNA and peptide, making the extent of insulin degranulation less evident. Furthermore, expression of pancreatic and duodenal homeobox factor-1, a β-cell-specific transcription factor, was more evident in the nuclei of islet cells after the antioxidant treatment. In another study, Cuzzocrea et al. (61) demonstrated, using an animal model, that NAC (20 mg/kg; 30 min before reperfusion and 1, 2, and 6 h after reperfusion) reduced the formation of post-ischemic brain edema and attenuated the increase of brain levels of malondialdehyde (MDA) and myeloperoxidase in the hippocampus caused by cerebral ischemia. NAC treatment increased survival and reduced hyperactivity linked to neurodegeneration induced by cerebral ischemia and reperfusion. These results suggest that NAC alleviates damage induced by transient cerebral ischemia or brain injuries.
Trolox (6-hydroxy-2,5,7,8- tetramethylchroman-2-carboxylic acid) is a water-soluble analogue of the free radical scavenger α-tocopherol. Due to its enhanced water solubility, Trolox may function more rapidly during acute oxidative stress (357), while α-tocopherol requires several days of pretreatment to exhibit antioxidant benefits. Moreover, it seems that Trolox can scavenge peroxyl radicals better than α-tocopherol. For example, Wu et al. (358) demonstrated that Trolox protects human hepatocytes and myocytes against ROS. Furthermore, Trolox can reduce hypoxia/reoxygenation-induced hepatic injury in the isolated perfused rat liver (163). In cancer studies, it has been demonstrated that Trolox can inhibit breast cancer cell-induced osteoclast differentiation and the invasive behavior of cancer cells through PGE2-dependent and independent mechanisms, thereby suppressing osteolytic bone metastasis in breast cancer (162). Another example of the beneficial effects of Trolox has been described by Du et al. (81), who demonstrated that chitosan nanoparticles, when used as drug carriers for the delivery of Trolox, exert a protective effect against hypoxia-mediated oxidative stress and can block the mitochondria-dependent apoptotic pathway through upregulation of Bcl-2 expression and inhibition of Bax activation and Caspase-3 expression.
Another important antioxidant is the paraoxonase (PON) family of enzymes, which also contribute to vascular antioxidant defenses and protect against coronary artery disease (CAD), among other conditions (14). It has been described that PON1 and PON3 enzymes are synthesized in the liver and are associated with high-density lipoprotein (HDL) fraction. In fact, the capacity of HDL for controlling levels of HDL and low-density lipoprotein (LDL) lipid peroxidation depends on its PON levels (14). On the contrary, deletion of the PON1 gene can enhance oxidative stress in aorta and in mouse macrophages (277) and apoE knockout mice overexpressing PON1 have been shown to be protected against the atherosclerotic process (331). PON2 is expressed in various types of cells, and some polymorphisms have been associated with CVD (168). PON2 has demonstrated beneficial effects by scavenging ROS in fibroblasts, endothelial cells, and vascular smooth muscle cells (124). Some authors have demonstrated that overexpression of PON2 in apoE knockout mice protects against atherosclerosis. For these reasons, the increase in the levels of PON enzymes could be beneficial in oxidative stress-related conditions. In this sense, it has also been suggested that PON1 is a better atherosclerotic risk predictor than HDL in type 2 diabetes patients (232). The authors in question demonstrated that the levels of HDL and PON1 were negatively correlated with various atherogenic indexes but that the strength of negative correlation was always greater for PON1. In addition, and by using multiple linear regression analysis, they found that the regression coefficient (β) was always higher for PON1 than for HDL when taking the atherogenic indices such as atherogenic index of plasma=Log(triglicerides/HDL), atherogenic coefficient=(total cholesterol [TC]−HDL)/HDL, Castelli's risk index I (CRI I)=TC/HDL, and II (CRIII)=LDL/HDL as outcome variables (232). Several meta-analyses have been published about the importance of PON1 levels in human diseases. For example, a meta-analysis of 47 studies with 9853 CAD patients and 11,408 control subjects published in 2012 confirmed that lower plasma PON1 activity was related to an increased risk of CAD (339).
Importantly, some classical pharmacological agents have been attributed antioxidant properties. This is the case of statins (3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors) and β-adrenergic blockers. Several actions of statins have been related to their antioxidant capacity. First, they can suppress NADPH oxidase activity and expression (12, 239, 350). In fact, Pignatelli et al. (239) observed the effect of statins on 30 hypercholesterolaemic patients and 20 controls by assessing Nox2 and urinary isoprostane, a marker of oxidative stress. They demonstrated that the said patients had higher levels of Nox2 and urinary isoprostane. After atorvastatin treatment for 30 days, both soluble gp91phox and isoprostane were markedly reduced. The authors concluded that statins exerted an antioxidant action through the inhibition of serum gp91phox. Second, they can prevent eNOS uncoupling by upregulation of GTP cyclohydrolase 1 (GTPCH1) expression and increased tetrahydrobiopterin (BH4) biosynthesis (12, 350). Third, statins can induce antioxidant enzymes such as SOD1, SOD3, and GPX. Finally, they enhance eNOS expression and activity. For all these reasons, statins have beneficial effects in different conditions associated with oxidative stress and mitochondrial impairment. Some β-adrenergic blockers have demonstrated similar properties. One study evaluated the β-blockers atenolol, labetalol, metoprolol, pindolol, propranolol, sotalol, timolol, and carvedilol for their putative scavenging activity for ROS and RNS, and demonstrated that some were effective scavengers (101), which makes them useful in preventing oxidative damage in hypertension and other CVD that frequently emerge in association with oxidative stress. The beneficial effects of β-blockers associated with their antioxidant property have also been shown in arrhythmia (carvedilol improved intracellular Ca2+ handling and contractile dysfunction by correcting defective interdomain interaction within the RyR in the failing heart) (205) or nephrotoxicity (carvedilol protected rats from gentamicin-induced nephrotoxicity) (157).
Finally, the important antioxidant molecule resveratrol (3,5,4′-trihydroxy-stilbene) deserves a mention. This polyphenolic compound, present in red grapes and other plants including oriental herbal plants (170), exhibits ROS-scavenging activity and acts on several redox enzymes. Resveratrol has been shown to inhibit the activity and expression of NADPH oxidase in cardiovascular tissues (359), to accelerate mtROS detoxification by upregulating SOD2 (319), and to reduce mitochondrial radical production (250). Furthermore, it induces several antioxidant enzymes, such as CAT, GPX1, and SOD isoforms (170). Resveratrol can also prevent eNOS uncoupling by upregulating GTPCH1 and BH4 biosynthesis, and can enhance eNOS activity and expression (170). In addition, resveratrol supplementation can restore high-fat diet-induced insulin secretion dysfunction by increasing mitochondrial function in the islets (153), and preserve mitochondrial function by stimulating mitochondrial biogenesis together with attenuation of oxidative stress in regulatory T-cells of mice fed a high-fat diet (337). The potential effect of resveratrol has been evaluated in different clinical trials in humans with metabolic diseases (324, 325, 334). In one study, resveratrol showed beneficial effects in insulin resistance type 2 diabetic patients (29). It can also decrease insulin and glucose levels, enhance mitochondrial function, and suppress inflammation in healthy obese men (324). Its beneficial effects have also been demonstrated in type 2 diabetes by decreasing cholesterol, blood pressure, and HbA1c levels (20), while it promoted insulin sensitivity in the elderly (59). However, other studies have found no beneficial effects of resveratrol on the metabolism of obese men (250) or control non-obese women (373). Such studies provide discrepant evidence regarding the role of SIRT1 in resveratrol treatment (122), pointing to differences in the methodologies employed and the subjects studied, as well as the dosages, the routes of administration, and the bioavailability of resveratrol. A bioavailability study of resveratrol found that it can cross the blood–brain barrier (BBB) and reach the brain tissue rapidly after systemic administration (340), an effect related to the therapeutic potential of this compound in stroke and neurodegenerative diseases. Most clinical trials have been performed with high doses (ranging from 75 mg to 5 g per day) of resveratrol and during weeks or months (247). In general, it is unknown whether long-term consumption of resveratrol or low levels of resveratrol-rich foods can prevent or ameliorate the onset of type 2 diabetes. In addition, pharmacokinetic studies have detected nanogram levels of resveratrol in plasma, providing evidence that other resveratrol metabolites (e.g., resveratrol-3-sulfate, or resveratrol-3-O-glucuronide, which are more abundant in the blood) are responsible for the health-benefiting effects (247). Therefore, future studies taking into account these metabolites and focusing on systemic physiology in addition to tissue-specific effects may help understand the metabolic effect of resveratrol and its potential therapeutic actions in metabolic diseases or related pathologies.
Although some achievements have been made, in general terms, the experience with antioxidant therapies has not met the initial expectations. Multiple reasons could explain the lack of effectiveness of antioxidants: inadequate dose and/or timing of antioxidant supplementation, poor bioavailability, heterogeneous microenvironments in which antioxidants have to exert their action, antioxidant status of the study population, and untargeted delivery of the antioxidant compound. Regarding supplemental dosages, some studies may have administrated insufficient amounts of the antioxidant compound for assessing its beneficial effects (208), whereas excessive concentrations could have produced pro-oxidant effects in others (68, 227, 244), thus raising concerns about safety. The duration of antioxidant administration should also be taken into account, with acute pathological conditions requiring immediate and shorter intervals than chronic diseases (123). In terms of the microenvironment in which antioxidants are supposed to act, the same molecule may exert antioxidant or pro-oxidant effects depending on different factors; for example, the pressure of O2 in the case of β-carotene (33), or the amount of iron in the case of vitamin C (which induces the generation of •OH under iron overload) (121). In addition, it is important to note that redox signaling is a crucial part of many physiological processes, and that disruption of the redox equilibrium by excessive or inappropriate administration of antioxidants may have negative effects. This may be another contributing factor to the controversial results of clinical trials with antioxidant therapies, and it needs to be considered in the future.
In this context, some oxygenated metabolites are believed to have beneficial effects. Indeed, this is the case of n-3 PUFAs, eicosapentaenoic acid, and docosahexaenoic acid (DHA). DHA is very susceptible to spontaneous oxidation by non-enzymatic oxidation pathways. As the dogma regarding lipid peroxides has always dictated that they are undesirable and toxic, the possibility that they may be involved in mediating the beneficial effect of n-3 PUFAs is counterintuitive. However, mounting evidence suggests that in many contexts, spontaneously oxidized PUFAs can be beneficial, due largely to the fact that they are highly reactive agonists for certain receptors. For example, recent reports have demonstrated that oxidized DHA has a high affinity for the PPAR family of transcription factors, which regulate many cellular processes, including cellular development, differentiation, metabolism, and tumorigenesis. Moreover, oxidized DHA has a greater PPAR-activating effect than any other PPAR ligand tested (8, 128). These findings could have broad clinical implications, as they indicate that DHA peroxidation in vivo could greatly enhance its potency as PPAR agonist—a class of widely prescribed drugs that treat a variety of pathologies, from high cholesterol and arrhythmia to type 2 diabetes. In addition, it is possible that a large portion of the electrophysiological effects attributed to n-3 PUFAs are dependent on their oxidation (137).
Another explanation of the reported lack of benefits of antioxidants is the antioxidant status of the population selected for the study (309). This became evident in the clinical trial performed by Meagher et al., in which vitamin E supplementation did not affect lipid peroxidation in healthy subjects with normal endogenous levels of vitamin E due to its routine intake through the populations's Western diet (195). Finally, the most likely cause for these disappointing results is that the untargeted administration of general antioxidants leads to insufficient local amounts in the target tissue (123, 249) and, more specifically, in the primary source site of free radicals, that is, the mitochondrion. Many studies over the past decade have focused on the development of mitochondria-targeted antioxidants, with highly promising results (268) that will be discussed in detail.
III. Strategies for Mitochondrial Pharmacology with Special Focus on Antioxidants
Taking into account the important role of mitochondria in human pathophysiology (Fig. 1), these organelles are an obvious pharmacological target. Multiple compounds have been developed to alleviate their dysfunction in different pathological situations (295), with varying targets and mechanisms of action, including maintenance of Ca2+ homeostasis, regulation of mitochondrial dynamics, stimulation of mitochondrial biogenesis, or regulation of apoptosis and, particularly, oxidative stress (210).
A. Unspecific approaches
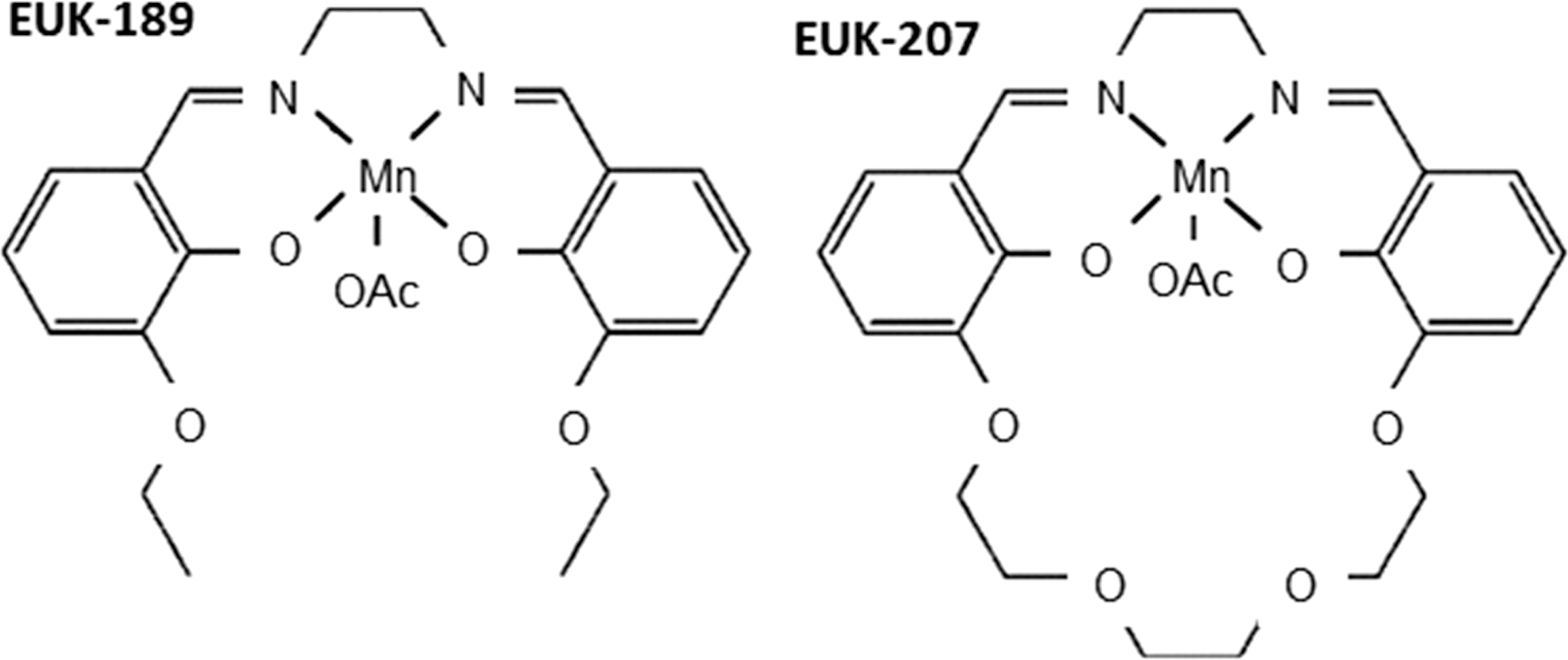
Although not designed to target mitochondria, several molecules display “mito-protective” activity; namely, an ability to attenuate mitochondrial injury (75). They are referred to as “synthetic catalytic scavengers or antioxidants” and were first investigated and developed by Eukarion (hence their code name EUK) (Fig. 6). These analogues of SOD-CAT mimetic have been shown to be beneficial in various models of oxidative stress; for example, salen Mn complexes were effective in a direct test of mammalian mitochondrial oxidative stress in SOD2 KO mice (196 –198), in which EUK-8, EUK-134, and EUK-189 significantly extended the lifespan of the animals (160, 197). Beneficial effects have been reported, not only in the brain but also in other tissues (198). Treatment with salen Mn mimetics has been shown to exert protective effects against oxidative stress in animal models of several diseases, such as PD (235), ALS (138), AD (198), stroke (17), and age-associated cognitive impairment (174). Specific animal models of ROS-related injury have revealed the potential of molecules such as EUK-207 to mitigate radiation damage in the lungs (161), or EUK-189 to prolong survival after a lethal dose of total body radiation (304). Some of these compounds are orally bioavailable, whereas others have been proposed for topic or parenteral administration. The compound EUK-134 has been used as an active ingredient in cosmetics for about 10 years, and another EUK compound is a clinical candidate for the prevention and treatment of certain neurodegenerative diseases.
Several studies have demonstrated the beneficial effect of antioxidant compounds on the vasculature through protection against oxidative damage and consequent restoration of endothelial function (285). Novel approaches have been developed in this aspect, with certain compounds having been shown to render protection against these CVD and other related diseases by acting specifically on mitochondria. For example, diazoxide, an FDA-approved drug for the management of symptomatic hypoglycemia, has been identified as cardioprotective. Diazoxide is generally believed to inhibit succinate dehydrogenase, a mitochondrial complex II protein (51). This inhibition, which also takes place in the heart, can occur at concentrations that are often used to study cardioprotection, and therefore may constitute a mechanism involving partial uncoupling of OXPHOS and/or modulation of ROS production. However, there is contrasting evidence regarding its targets and mechanisms of action. It seems that diazoxide exerts an ambivalent effect on mtROS production, as shown in a mechanistic study using submitochondrial particles and intact rat heart mitochondria, in which, depending on the metabolic state and Δψm, diazoxide-mediated inhibition of complex II either promoted transient generation of signaling ROS at complex III (during preconditioning) or attenuated the production of deleterious ROS at complex I (during ischemia and reperfusion) (80). Whatever the underlying molecular mechanism(s) may be, it is clear that improved mitochondrial function is a principal cardioprotective effect of diazoxide (52).
B. Targeted mitochondrial delivery
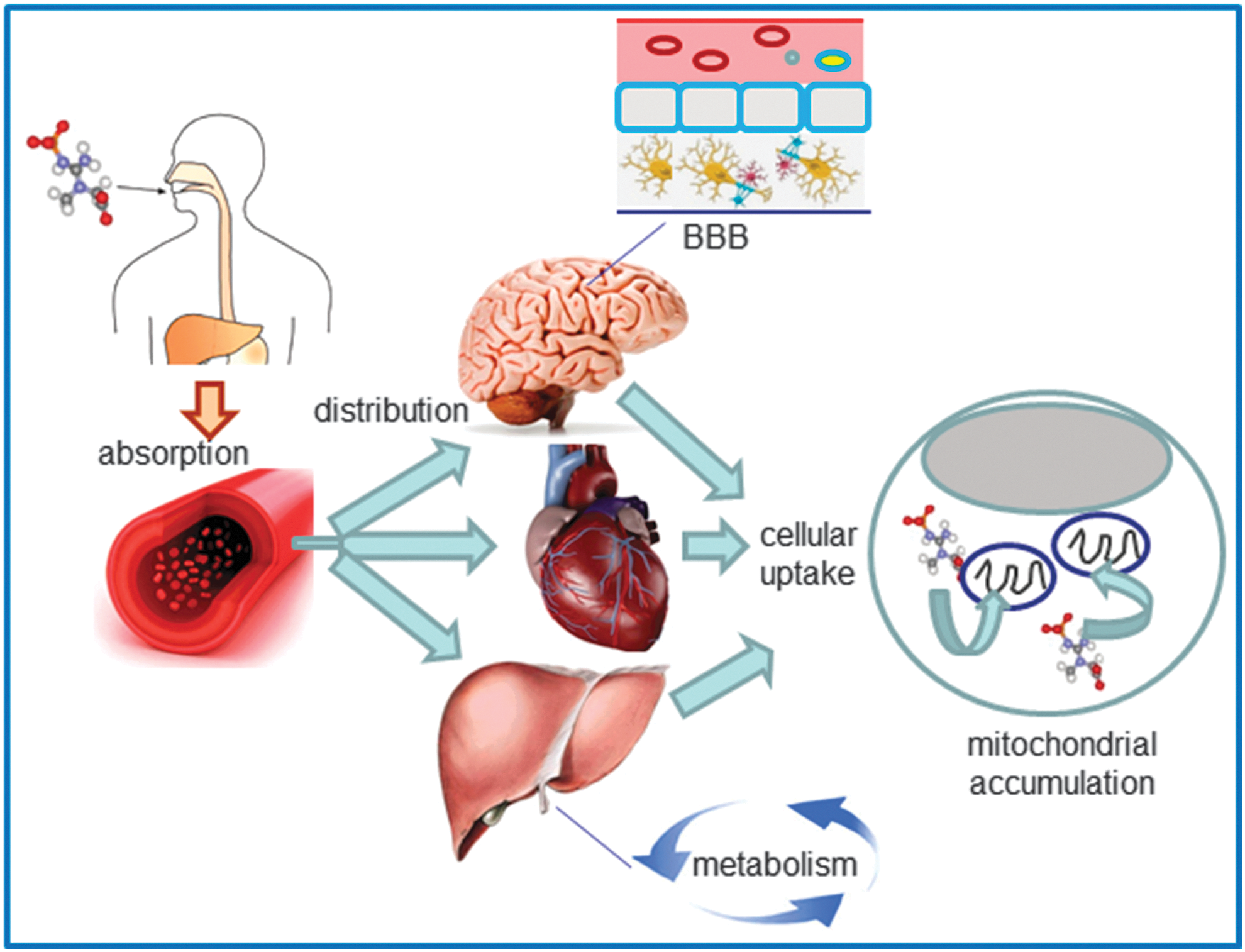
The delivery of drugs to specific subcellular territories is a promising avenue for therapeutics in many human diseases. It improves the therapeutic efficacy of compounds through accumulation of the drug near the specific target and reduction of the deleterious consequences of its off-target subcellular localization. Moreover, targeted delivery systems may improve certain characteristics of the drug and thus overcome the classical limitations of conventional drug administration, such as low bioavailability, insolubility, and drug resistance. Due to the crucial implication of mitochondria in human pathophysiology, the elaboration of methods and construction of vehicles for efficient selective drug delivery to these organelles is currently the focus of molecular pharmacological research. Even when the compound has the adequate physicochemical properties as to overcome the anatomical, immunological, and biochemical barriers to reach the target tissue and cells, it still has to cross several membranes to reach its final destination inside the mitochondrion. This greatly limits its action at the specific site inside this organelle; therefore, selective targeting of the drug to the mitochondrial localization is necessary. Mitochondrial drug targeting is a complex process that depends on the particular nature of these cellular compartments, including the presence of specific transporters located on the mitochondrial membrane. Diffusion through the IMM is difficult; therefore, mitochondria-targeted compounds need to be encapsulated inside a drug carrier. Moreover, this encapsulation and the subsequent release of the compound inside the mitochondrion must preserve the drug's pharmacological stability/activity. In recent years, great efforts have been made to find solutions for the development of efficient drug carriers. These molecules display several features: (i) ability for binding to the pharmacologically active form of the drug or a prodrug; (ii) an efficient transport system that carries the drug to the site of its action; (iii) specific and selective targeting to the mitochondrial compartment; and (iv) release of the drug inside the mitochondrion (Fig. 7). Current strategies for mitochondrial drug delivery include passive and active targeting (287). In the case of active targeting, specific interactions that take place at mitochondrial sites, including ligand-receptor associations and antigen-antibody binding, are exploited and take advantage of the compatibility between the physicochemical properties of the carrier molecule (electric charge, hydrophilicity, size, and mass) and those of the mitochondrial compartment. Due to mitochondrial morphological properties, passive targeting to these organelles is difficult; hence, active targeting strategies are being developed. Small molecules have been efficiently targeted to mitochondria in vivo by several targeting strategies; namely, through enclosure inside liposomes (287), conjugation to lipophilic cations (210), and incorporation into mitochondria-targeted peptides (315) (Table 1). So far, the molecules employed in these approaches involve relevant antioxidants as well as substrates and coenzyme components of the ETC, such as cytochrome c, B2, succinate, and vitamin B1, and proapoptotic proteins including those of the Bax/Bcl2 family and p53 (287).
Major characteristics are detailed, together with examples of compounds and settings in which they have been reported to have beneficial effects.
AD, Alzheimer's disease; ALS, amyotrophic lateral sclerosis; AMPK, AMP-activated protein kinase; CAD, coronary artery disease; CNS, central nervous system; CVD, cardiovascular disease; ETC, electron transport chain; H2O2, hydrogen peroxide; HCV, human hepatitis C virus; HSVEC, human saphenous vein endothelial cells; MitoQ, mitoquinone; MnSOD, manganese superoxide dismutase; mtDNA, mitochondrial DNA; mtROS, mitochondrial ROS; O2 •−, superoxide anion; ONOO−, peroxynitrite; PD, Parkinson's disease; SkQ1, plastoquinonyl decyltriphenylphosphonium; SS, Szeto-Schiller; UVA, ultraviolet A; Δψm, mitochondrial membrane potential.
1. Lipophilic cations
Lipophilic cations take advantage of mitochondrial ΔΨm to facilitate their selective targeting and accumulation within the mitochondrial matrix (273). This process can be expressed by the Nernst equation, by which the uptake of these molecules increases ∼10-fold for every ∼60 mV of membrane potential, leading to significant uptake within mitochondria in vivo (246, 271) (Figs. 8 and 9). Massive mitochondrial targeting is enabled by the electrochemical gradient from the plasma membrane potential (−30 to −60 mV) to the IMM potential (Δψm), (−150 to −180 mV), which provides a potent force for the selective targeting of large lipophilic cations and their concentration inside the mitochondria (211, 295). This high negative membrane potential present in the mitochondria is not found in any other subcellular compartment, which offers a very selective molecule delivery to these organelles. Several lipophilic cations, including triphenylphosphonium (TPP+), rhodamine 123, flupirtine, MKT-077, and anthracyclins, exhibit selective mitochondrial accumulation on cellular entry (95).
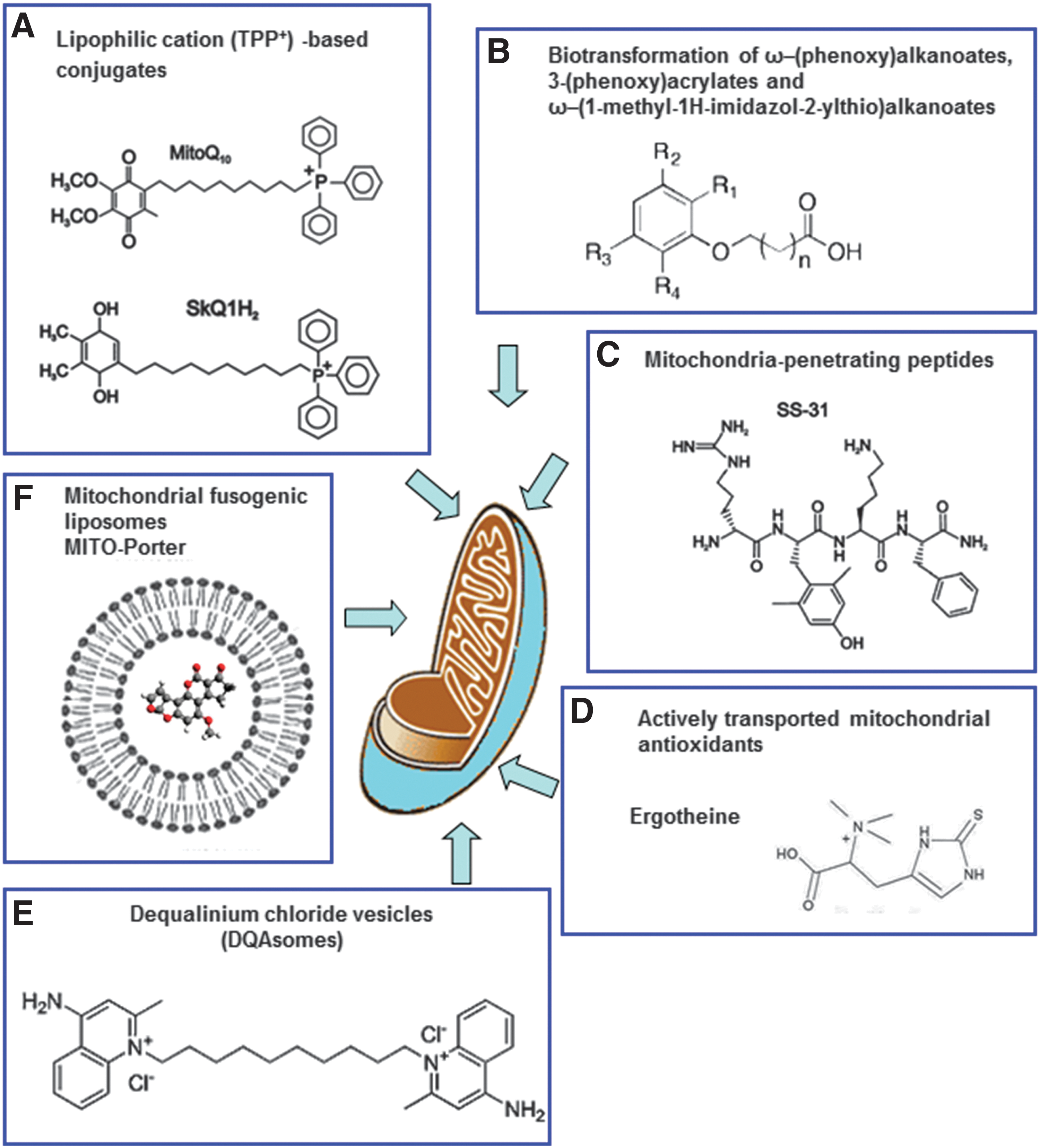
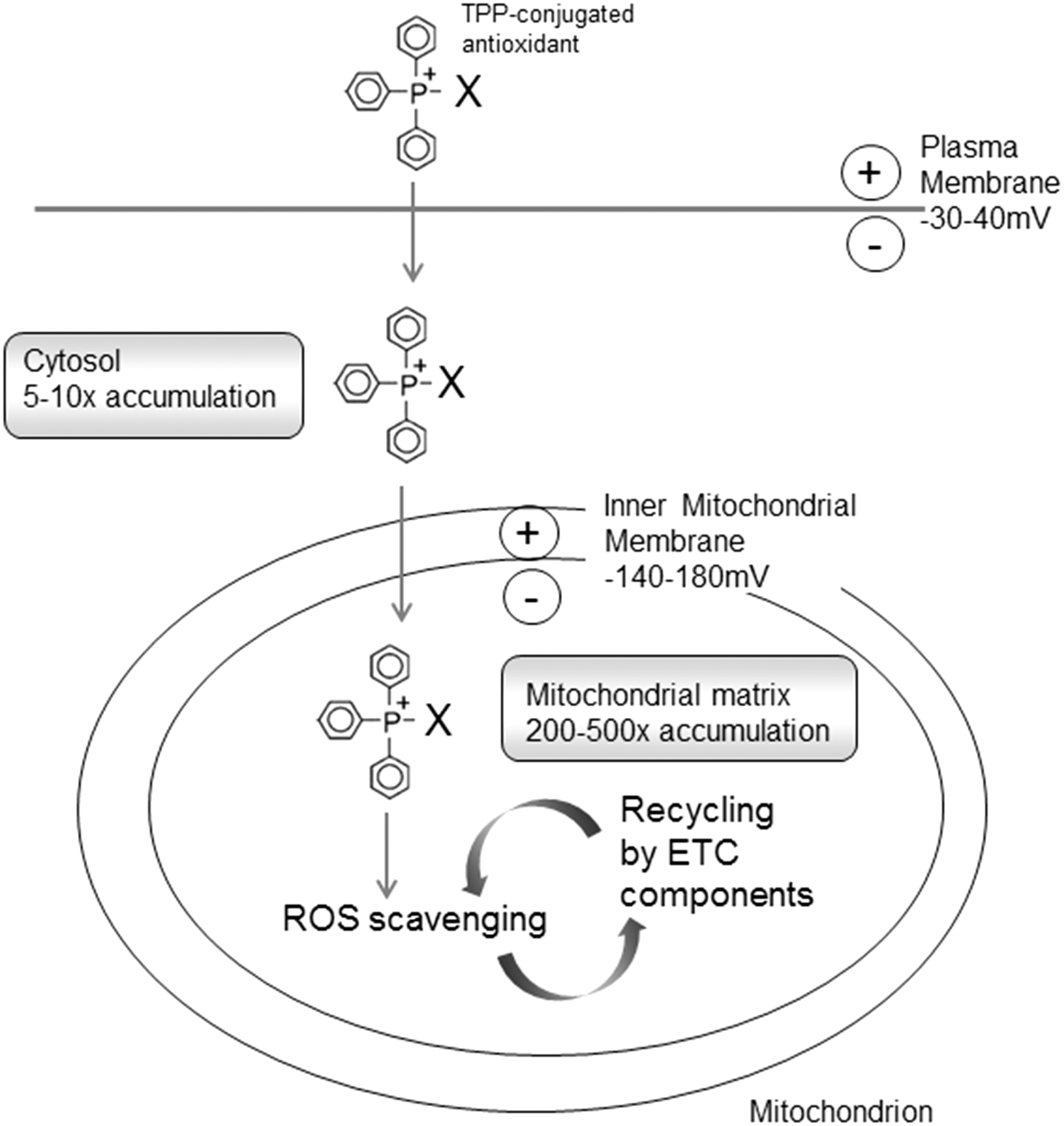
The delivery of pharmacological agents into the cell through the use of lipophilic cations was first shown with the lipophilic cation rhodamine 123 in combination with the anticancer drug cisplatin (321). TPP+ and TPMP+ (its methylated form) are the most widely employed lipophilic cations for mitochondrial targeting of antioxidants (210, 211). An alternative lower-molecular weight spin trap bearing an N-arylpyridinium ion (267) has been used and positive results are obtained, though its efficacy needs further confirmation. The TPP moiety is driven by the plasma membrane potential, which allows rapid cellular uptake of bioactive molecules, followed by specific mitochondrial matrix accumulation. Once inside the mitochondrion, TPP+ molecules position themselves primarily on the mitochondrial matrix-facing surface of the phospholipid bilayer, with the functional group and the linker positioned within the IMM (211). The extent to which TPP+ molecules anchor themselves to the IMM depends on the hydrophobicity of the molecule and on the length of both the linker and the functional group. A large number of compounds have been generated through conjugation to the TPP moiety (Fig. 10), of which several are antioxidants, such as ubiquinone (13), tocopherol (298), LA and LA spin traps, (31), ebselen (91), resveratrol (21), nitrones (209), plastoquinone (293), and TEMPOL (330). These Mito-derived redox modulators are reported to diminish the level of a great variety of reactive species (e.g., H2O2, •NO, ONOO−, lipid peroxyl, and alkoxyl radicals). The accumulation of antioxidants within mitochondria though the IMM anchoring of their TPP+-conjugates has been proposed to control mitochondrial redox signaling and prevent membrane lipid peroxidation. Since lipid peroxidation is a crucial feature of mitochondrial oxidative damage, TPP+-conjugates that are effective against lipid peroxidation are the focus of current research. Lipophilic cations show great potential for the successful delivery of antioxidants to mitochondria; however, they present several disadvantages: (i) limited capacity (only low-molecular-weight molecules and electrically neutral chemicals can be successfully transferred); (ii) sublocalization (these compounds tend to localize in the mitochondrial matrix and the matrix-facing surface of IMM; thus, the targeting of important processes that occur on the outer leaflet of the IMM, the OMM, or the intermembrane space is largely limited or impossible); and (3) toxicity (at high concentrations, they can depolarize ΔΨm and compromise cell viability).
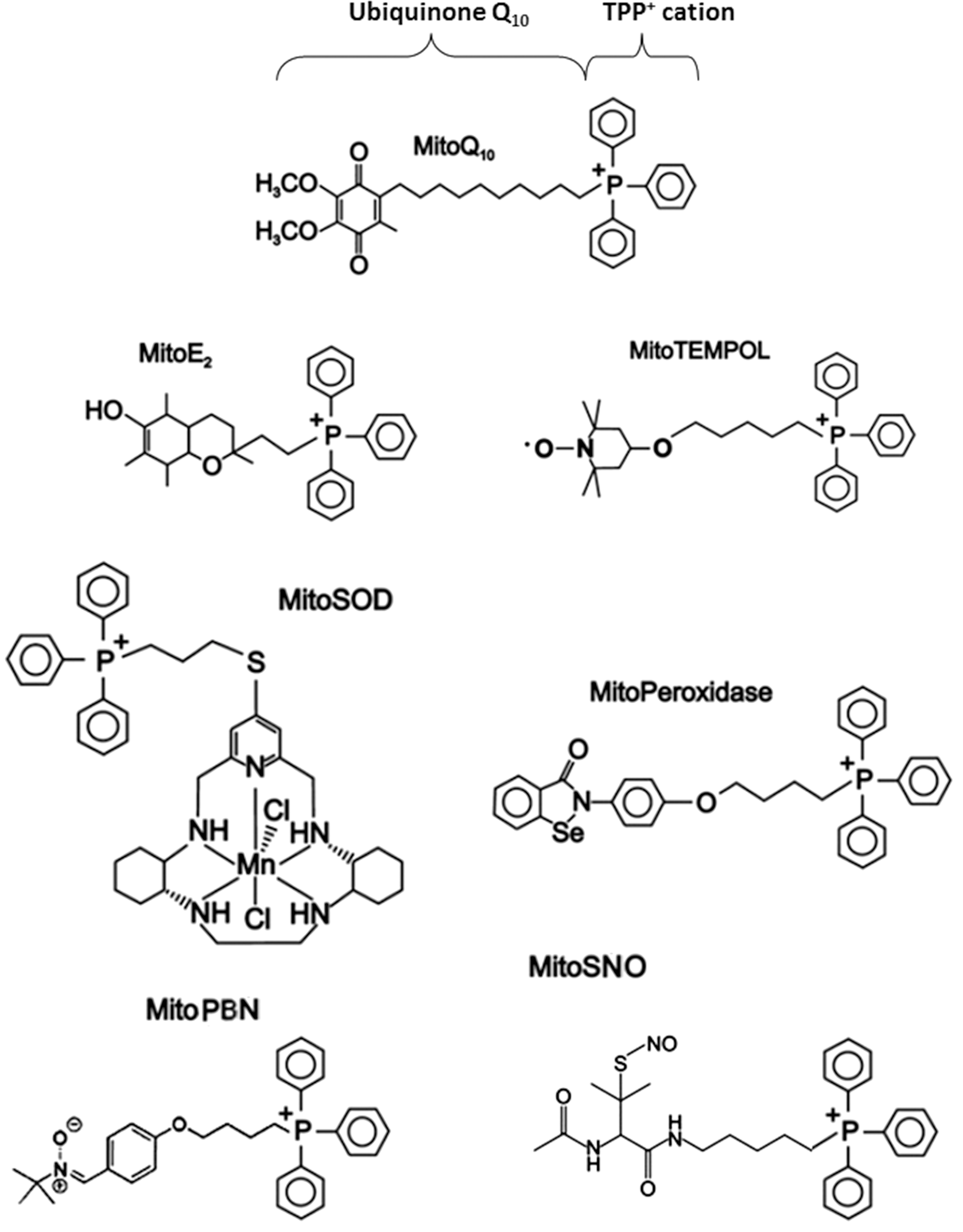
The TPP-bound version of ubiquinol (mitoquinone [MitoQ]) is the most extensively evaluated and best understood mitochondria-targeted antioxidant (50, 210, 211, 298). The cellular uptake of MitoQ is far more rapid than that of methylTPP, presumably because its higher hydrophobicity reduces the activation energy available for passage across the plasma membrane. The longer a molecule's alkyl chain is, the greater the mitochondrial accumulation of MitoQ; in this context, MitoQ10, which has a 10-carbon atom-alkyl chain, shows optimum performance (Fig. 10). MitoQ uptake occurs primarily in mitochondria rather than in other cell compartments, as supported by the fact that its accumulation in cells is largely blocked when Δψm is disrupted by the uncoupler carbonylcyanide-p-trifluoromethoxyphenylhydrazone (FCCP). A further indication that the mitochondrial concentration of TPP-containing molecules within cells is membrane potential—dependent is the FCCP-sensitive protection afforded by MitoQ in a cell model of Friedreich's ataxia, in which FCCP does not alter the efficiency of the non-targeted compounds decylQ and idebenone (132). Within mitochondria, MitoQ is accumulated at the matrix-facing surface of the IMM, where complex II of the ETC recycles it into the active ubiquinol form (MitoQH2), which remains stable during long-term incubation (13, 210). This reduction is crucial for the compound to function as a reducing agent, as the totality of the effects of MitoQ can probably be attributed to accumulation of the antioxidant ubiquinol form. This form has been shown to be a highly effective antioxidant by reacting with ROS and also inhibiting ONOO− formation. The antioxidant property of the ubiquinol form of MitoQ is based on its oxidation into the ubiquinone form, which is then rapidly re-reduced by complex II, thus regaining its antioxidant efficacy. Given that MitoQ is generally adsorbed to the IMM, and the fact that its linker chain enables the penetration of the active antioxidant form, ubiquinol, deep into the membrane core, this molecule is believed to be effective as an antioxidant against mitochondrial lipid peroxidation (144, 294). The reactivity of MitoQ10 toward O2 •− and other O2 radicals has been studied in detail (188). The ubiquinone form of MitoQ directly reacts with other ROS such as O2 •−, although, as occurs with other ubiquinols, its reaction with H2O2 can be considered negligible. MitoQ10 reacts with O2 •− in water and methanol with rate constants of 2.0×108 M −1 s−1 and 4.2×108 M −1 s−1, respectively, forming the semiquinone radical MitoQH, which then dismutes to MitoQ10 and the quinol MitoQH2. In addition, the reverse reaction of MitoQH with O2 displays rate constants of 2.9×107 M −1 s−1 and 7.3×106 M −1 s−1 in water and methanol, respectively. MitoQH2 rapidly reacts with ONOO− (whereby MitoQH is produced and undergoes dismutation), an action that is particularly effective against lipid peroxidation, which accounts for the protection it is reported to afford against ONOO−-induced damage (294). In short, in isolated mitochondria, MitoQ seems to fulfill most of the requirements of a successful mitochondria-targeted antioxidant.
Stable-free radical nitroxides with antioxidant properties (TEMPO derivatives) are another group of mitochondria-targeted antioxidants that have been described. The action of these compounds to metabolize ROS is atributed primarily to cyclic one- or two-electron transfer in three oxidation states: the oxammonium cation, the nitroxide, and the hydroxylamine (353). Nitroxides undergo a very rapid, one-electron reaction to the corresponding hydroxylamine, which has antioxidant activity and can be converted to nitroxides by H2O2 or other oxidants, such as transition metals. The nitroxide moiety is considered essential for full antioxidant activity, whereas substitution at the four-position affects potency. One-electron redox cycling of six-member ring nitroxides such as TEMPOL is enhanced by their ability to undergo reversible “boat-and-chair” conformational change; this is not possible with five-member ring nitroxides, which may be responsible for their lesser biological activity (353). MitoTEMPO, which possesses O2 •− and alkyl radical scavenging properties, is a combination of the pleiotropic intracellular antioxidant piperidine nitroxide TEMPO (2,2,6,6-tetramethylpiperidin-1-yloxy) and the TPP cation. A related molecule is MitoTEMPOL, in which the conjugate molecule is 4-hydroxy-TEMPO (TEMPOL) and, therefore, less hydrophobic. TEMPOL is an amphilite nitroxide that concentrates on hydrophobic microdomains of cell membranes.
Antioxidants targeted to mitochondria have been extensively used in isolated preparations of these organelles and cells, with toxicity being one of the main issues addressed. The massive accumulation of lipophilic cations within isolated mitochondria has been shown to disrupt mitochondrial membrane integrity and uncouple OXPHOS, which occurs as a consequence of the adsorption of cations to the matrix surface of the IMM (262). In accordance with this idea, the disruption of the mitochondrial function occurs at lower concentrations when more hydrophobic TPP cations are employed, and the degree of this interference shows a positive correlation with the quantity of the compound adsorbed to the IMM (262). The effects of MitoQ on mitochondria that occur as a result of non-specific interference are assessed using decylTPP as a control compound, which has a similar hydrophobicity to that of MitoQ, but lacks the ubiquinol moiety and therefore does not display antioxidant properties. Mitochondrial function is disrupted by MitoQ and decylTPP at similar concentrations, which implies that some of the bioenergetic and redox effects detected in cell culture systems and ascribed to mitochondria-targeted antioxidants are, in fact, produced by the linker group and are independent of the antioxidant functional group (262). Another study has reported impairment of the mitochondrial efflux of Ca2+ in cell culture systems exposed to TPP+, an effect attributed to direct inhibition of Na+ (or H+)/Ca2+ exchanger (167). MitoQ can also act as a pro-oxidant by generating O2 •− in accordance with the capacity of all ubiquinols, including that derived from MitoQ, to deprotonate in water and form ubiquinolate, which triggers O2 •− formation from O2 (130). This pro-oxidant activity is limited if the ubiquinol moiety is maintained in the lipid phase, and may also largely depend on the concentration employed (50). In this sense, MitoQ has been shown to induce O2 •− production, redox cycling at mitochondrial complex I, and apoptosis in isolated mitochondria from bovine aortic endothelial cells (77). More recent data with the same cell type have shown that MitoQ alters mitochondrial respiration in a concentration-dependent manner (100–300 nM) and that this effect is not primarily due to a protonophoric action but rather to a pro-oxidant action of MitoQ (92). Importantly, the cationic component, methylTPP did not modify the bioenergetic profile of the cells in question. In light of all this evidence, the effect of TPP compounds is likely to be dependent on cell type, as the bioenergetics of cells show a wide range of responses depending on their number of mitochondria and regulation. Obviously, such deleterious effects would limit the amounts of TPP+-conjugated targeted antioxidants that can be used; in this sense, when comparing cells to preparations of isolated mitochondria, it is imperative to keep in mind that, as a result of the presence of the plasma membrane potential, targeted antioxidants accumulate within cells at a concentration that is 5- to 10-fold greater than that in the extracellular environment (262). For cultured mammalian cells, this concentration generally ranges from 0.1 to 0.5 μM, but can vary considerably due to its dependence on cell density, cell type, and incubation conditions. However, inhibition of OXPHOS in cell culture as a result of the use of TPP+-conjugated compounds is obtained only at concentrations of these agents that are 10–100 times greater than those generated through oral therapy in animal models and human subjects (271).
a. In vitro results
MitoQ has been employed in a large range of both mitochondrial and cell models (211) (Table 1), and abundant evidence proves it is effective against mitochondrial oxidative damage. For example, detailed studies of the interaction of MitoQ with mtROS have been performed in rotenone-treated fibroblasts (154). MitoQ did not decrease O2 •− production when this process was analyzed by dihydroethidium oxidation, but it prevented lipid peroxidation when it was detected by the fluorescent marker C11-BODIPY. This finding is in agreement with the model of MitoQ action described by studies with isolated mitochondria in which the main antioxidant action of MitoQ was the prevention of lipid peroxidation. The protective action of MitoQ against oxidative stress has also been shown with regard to another target of ROS-induced damage: mtDNA. In a more recent study involving human dermal fibroblasts, Oyewole et al. demonstrated that MitoQ alleviated ultraviolet A- and H2O2-induced mtDNA damage. Interestingly, tiron, an unrelated compound with ROS-scavenging properties, was shown to be more efficient than MitoQ, despite it being a non-selective antioxidant (228). Tiron permeabilizes the mitochondrial membrane and localizes to mitochondria; however, it is not a targeted drug, and its action is not limited to mitochondria. The authors of the study in question speculated that the fact that tiron rendered greater protection than MitoQ in this model may have been a result of the dual action of tiron as a radical scavenger and a metal chelator.
In another study, mitochondria-targeted antioxidants were employed in β-cells such as RINm5F and HIT-T15 under conditions of glucotoxic and glucolipotoxic stress typical of type 2 diabetes (171). β-cells under oxidative stress conditions manifested increased levels of mitochondrial antioxidant enzymes (such as MnSOD), enhanced expression of mitochondrial ETC complex subunits and lipogenic enzymes (such as fatty acid synthase, ATP-binding cassette transporter A1, and acetyl-CoA carboxylase), induction of apoptosis, incremented intracellular lipid droplet accumulation, ER stress, mitochondrial membrane depolarization, presence of oxidative stress, sterol regulatory element binding protein 1c, and NF-κB expression, in parallel with a reduction in citrate synthase activity, ATP concentration, and insulin release. These manifestations were related with mitochondrial oxidative stress and were prevented by administration of the mitochondria-targeted antioxidants MitoQ and MitoTEMPOL, thereby improving insulin secretion and promoting the survival of these cells. The anti-inflammatory effect of MitoQ has been investigated in an endothelial cell model of sepsis in which it was shown to reduce oxidative stress and protect against mitochondrial damage, as indicated by a lower rate of ROS formation and maintenance of Δψm. These effects were accompanied by suppressed proinflammatory cytokine release from cells, while production of the anti-inflammatory cytokine IL-10 was enhanced (180).
A recent report by Mackenzie et al. (184) indicated a novel, mtROS-mediated activation of AMPK in the endothelium of patients with type 2 diabetes and CAD. Human saphenous vein endothelial cell (HSVEC) cultures obtained from these patients revealed an increase in AMPK activity and enhanced mitochondrial H2O2 production with regard to that in healthy volunteers. Short-term treatment with MitoQ10 of HSVEC reduced AMPK activation, but without modifying basal mitochondrial H2O2 production (184).
A potential therapeutic effect of mitochondria-targeted antioxidants has also been demonstrated in models of tolerance to nitroglycerine (GTN), in which the deleterious effects of GTN on mitochondrial O2 consumption, aldehyde dehydrogenase 2 activity and of ROS on the mitochondria were prevented by MitoQ (84, 96). Nevertheless, whether this constitutes the primary mechanism by which MitoQ is protective in all cell types and forms of oxidative stress remains to be explored. A large body of evidence demonstrates that antioxidants are capable of specific modulation of the cell survival/cell death pathways, and specifically those related to mitochondrial function, including apoptosis, mitophagy, mitoptosis, and necrosis. Such processes reveal the special attention that mitochondria-targeted antioxidants merit. Moreover, in HeLa cells and fibroblasts, MitoQ has been shown to efficiently inhibit mitochondrial fission induced by application of specific chemical inhibitors of the mitochondrial ETC (myxothiazol and piericidin), which endorses the potential of this mitochondria-targeted agent in other mitochondrial processes such as mitochondrial dynamics (240).
Regarding autophagy, 0.5 mM MitoTEMPOL has been shown to diminish both basal and induced (pharmacological induction with rapamycin or induction by nutrient deprivation) autophagy in murine C2C12 myoblasts, thus pinpointing the role of mtROS in muscle autophagy (256). Moreover, the study in question also reported that MitoTEMPOL had a greater ability than the untargeted version of TEMPOL to alleviate the increase in ROS (mitochondrial O2 •−) associated with autophagy.
It is important to bear in mind that the protective effects of antioxidants depend on cell type, number of mitochondria in the cells, and the extent to which cells depend on OXPHOS. Moreover, caution need to be taken for in vitro experimental conditions that often do not reflect the physiological redox environment. In this sense, there is evidence of a lack of beneficial effects of Mito-compounds in certain models. For instance, in a study evaluating the effect of palmitate on cell viability and mitochondrial respiratory function of murine C2C12 myoblasts, neither MitoQ (0.5 μM) nor MitoTEMPOL (10 μM) was capable of preventing cell death, whereas only MitoTEMPOL blocked palmitate-induced mtDNA damage. Indeed, MitoQ increased H2O2 production, and both antioxidants had a marked negative effect on cellular respiration (231).
The usefulness of Mito-compounds as anticancer drugs merits detailed analysis. Recent findings have highlighted the complex role of antioxidants in survival and cell death signaling and have shown that, though protective for normal tissue, Mito-compounds may exert cytotoxic effects in cancer cells. MitoQ was shown to be cytotoxic, inducing both autophagy and apoptosis in human breast cancer cells, an effect not observed with the untargeted ubiquinone (259). The mechanism suggested for this action involved the pro-oxidant capacity of MitoQ. Importantly, MitoQ is much more cytotoxic for cancer cells than for normal cells. In this latter publication, the GI50 (growth inhibition) value for MitoQ in the breat cancer cell lines MDA-MB-231 and MCF-7 was 296 and 113 nM, respectively, whereas it was >10 μM in normal mammary epithelial cells. There may be several reasons for the cytotoxic specificity of Mito-compounds in cancer versus healthy cells. First, cancer cells have a greater Δψm than healthy cells, which enhances mitochondrial accumulation of these agents. Second, cancer cells have higher levels of ROS and may be more susceptible to further tilting of the redox balance.
b. In vivo results
Mitochondria-targeted antioxidants have also been employed in in vivo studies (Table 1). In the case of MitoQ, the role it plays in in vitro settings of mitochondrial oxidative stress has been shown to be beneficial in many conditions; moreover, its protective actions have also been observed in animal models of cardiometabolic diseases, including ischemia/reperfusion, diabetes, and sepsis, and also in patients (297). Notably, it has been demonstrated that MitoQ is uptaken rapidly from the blood into cells (246). To be of acceptable therapeutic relevance (among other necessary properties), mitochondria-targeted antioxidants must reach and accumulate inside the mitochondria within the cells of patients, ideally after oral administration (Fig. 7). Taking into account that TPP cations can easily cross phospholipid bilayers, they should pass efficiently from the digestive system to the bloodstream and then from the plasma to most tissues. It has been demonstrated that, when simple alkylTPP compounds are injected into mice intravenously, they undergo rapid clearance from the plasma and accumulate significantly in different organs, including the heart, skeletal muscle, brain, liver, and kidney (299). Importantly, TPP-derived compounds in the form of tritiated methylTPP, MitoE2, or MitoQ were made bioavailable orally to mice through their drinking water, after which they were uptaken into the plasma and delivered to mitochondria-rich tissues such as the heart, brain, liver, kidney, and muscle (299). MethylTPP has been shown to be cleared from all organs at a similar rate by a first-order process with a half-life of about 1.5 days (299). The study in question confirmed the distribution of orally administered alkylTPP compounds to all organs due to their facile permeation through biological membranes. The non-specific toxicity of alkylTPP cations found in mitochondria and cells also occurs in vivo, and this is probably the major limiting factor regarding the amounts of the compound that can be safely administered. In crude toxicity assessments (299), intravenously administered methylTPP and MitoE2 showed no toxicity at 300 nmol (∼4–6 mg/kg), but were toxic at 500 nmol (∼6–10 mg/kg). MitoQ was tolerated marginally better, with no toxicity at 750 nmol (∼20 mg/kg), but with evident toxicity at 1000 nmol (∼27 mg/kg). When mice were administered 500 μM TPMP, MitoE2, or MitoQ via their drinking water, no toxic effects were observed over the course of the experiment, which lasted 43, 14, and 14 days for TPMP, MitoE2, and MitoQ10, respectively (299). These similar levels of toxicity are consistent with the hypothesis that the deleterious effects of these compounds are largely attributable to non-specific disruption of the mitochondria due to the accumulation of large amounts of the lipophilic cation. To summarize, it is possible to administer alkyltTPP compounds to animals orally, as they are uptaken from the gut into the circulation with reasonable bioavailability. They are then rapidly cleared from the plasma as they reach target tissues, where they accumulate in the mitochondria of cells. Having confirmed the viability of the long-term exposure to mitochondria-targeted antioxidants, the following step is to assess whether the amount of accumulated compound is sufficient to act as an antioxidant in vivo. MitoQ has proved effective in several animal models of oxidative stress, such as various CVD settings, including endotoxin-induced cardiac dysfunction (313), age-related arterial endothelial dysfunction (99), cardiac ischemia/reperfusion (2), and doxorubicin-induced cardiac toxicity (40) and was shown to confer protection against an increase in blood pressure in rats that spontaneously develop hypertension (105). In one study, rats were administered 500 μM MitoQ for 2 weeks via their drinking water, after which their hearts were isolated and exposed to ischemia–reperfusion injury in a Langendorff perfusion system. In that study, MitoQ provided greater protection against tissue damage, loss of heart function, and mitochondrial dysfunction than the control compounds that employed methylTPP or short-chain quinol (2). MitoQ-mediated prevention of the lipid peroxidation in the IMM seems to be the most likely cause for the protection observed in this experimental model (2). In another work, MitoQ protected against oxidative damage caused by nitroglycerin in the rat aorta in a model of nitrate tolerance (84). MitoQ also reversed age-related endothelial dysfunction in mice. Its acute (ex vivo) administration or chronic supplementation through the animals' drinking water completely restored carotid artery endothelium-dependent dilation in older mice by improving •NO bioavailability. The improvements in endothelial function were associated with the normalization of age-related increases in total and mitochondria-derived arterial O2 •− production and oxidative stress (99). MitoQ has also been reported to prevent endotoxin-induced reductions in cardiac mitochondrial and contractile function in rodents (313). Drug-induced cardiotoxicity has also been assessed; for example, in a rat model of cardiomyopathy induced by doxorubicin, MitoQ significantly reversed the effects of this antineoplasic drug, allowing the left ventricular function and several mitochondrial parameters, such as cytochrome c activity and expression, to be recovered (40). The cardioprotective effect of Mito-compounds toward doxorubicin has also been shown through administration of MitoTEMPOL to immune-competent, spontaneously hypertensive rats. The underlying mechanism involved an increase of the pro-survival autophagy marker LC3-II and a decrease of the apoptosis marker caspase-3 in the heart (70). In the study in question, doxorubicin-treated rats were implanted with the breast tumor cell line SST-2 and of note, MitoTEMPOL caused significant tumor reduction and appeared to increase the antitumor effect of doxorubicin (70), thus reproducing the anticancer properties of Mito-compounds observed in vitro. Similarly, MitoQ prevented mitochondrial impairment in a rat model of cocaine-induced cardiac dysfunction (335). In addition, administration of MitoQ10 to young, stroke-prone, spontaneously hypertensive rats was shown to improve endothelial function, reduce cardiac hypertrophy, and protect against the development of hypertension (105). In vivo treatment with the mitochondria-targeted antioxidant MitoTEMPO has been found to partially correct endothelial dysfunction and prevent vascular inflammation in Ang II-treated PGC-1α KO mice (156). Similarly, MitoTEMPO has proved to be effective in a mouse model (ACE8/8) of cardiac renin–angiotensin system activation with elevated cardiac ROS, a high rate of spontaneous ventricular tachycardia, and sudden cardiac death secondary to a reduction in connexin 43 level. 2-week treatment with MitoTEMPO reduced sudden cardiac death, decreased spontaneous ventricular premature beats and ventricular tachycardia inducibility, reduced elevated mtROS to control levels, and prevented structural damage to mitochondria. Of note, general antioxidants, including sepiapterin (the precursor of BH4), TEMPOL, apocynin (nicotinamide adenine dinucleotide phosphate oxidase inhibitor), and allopurinol (a xanthine oxidase inhibitor), did not prevent ventricular tachycardia and sudden cardiac death, which highlights the importance of mitochondrial targeting versus non-targeting therapies (301). In addition, it has been demonstrated that apocynin does not inhibit all Nox isoforms (e.g., Nox4, which is inducible and may be involved in the pathology of chronic cardiac disease, among others) and must be metabolised intracellularly to be effective, and all cell types do not contain the relevant mechanisms. A very recent study by McLachlan et al. has assessed the combined effect of MitoQ10 and a low dose of the angiotensin receptor blocker losartan on stroke-prone, spontaneously hypertensive rats. The authors report an additive therapeutic benefit, with a significant attenuation of left ventricular hypertrophy and hypertension. In addition, MitoQ10 was shown to mediate a direct antihypertrophic effect on rat cardiomyocytes in vitro. These results endorse MitoQ10's potential as a novel therapeutic intervention in conjunction with current antihypertensive drugs (194).
MitoQ has also been shown to be beneficial in a rat model of sepsis, a pathological process characterized by a systemic dysregulated inflammatory response and oxidative stress, often leading to organ dysfunction, organ failure, and death. In lipopolysaccharide-peptidoglycan treated rats, treatment with MitoQ resulted in lower levels of biochemical markers of acute liver and renal dysfunction and an increase in Δψm in most organs (179). MitoE, another mitochondria-targeted antioxidant, has also demonstrated its utility in a rat model of pneumonia-related sepsis (369). The study in question confirmed the in vivo distribution of MitoE in the heart and liver at 24 h after its oral administration, and provided evidence that MitoE improves mitochondria-specific antioxidant defense and mitochondrial function (respiration and mitochondrial membrane integrity) in the heart, provides cardiac protection from sepsis, and suppresses sepsis-induced inflammation. Importantly, untargeted vitamin E also exhibited anti-inflammatory and protective effects, but to a lesser extent than the mitochondria-targeted molecule.
There is also in vivo evidence of the beneficial effects of MitoQ in metabolic pathologies, including diabetes and obesity. Chacko et al. demonstrated that MitoQ improved glomerular and tubular function in a Ins2(+/−) (AkitaJ) mouse model of type 1 diabetes when administered orally over a 12-week period (38). Creatinine levels were not significantly changed by MitoQ, but it reduced urinary albumin to levels similar to those exhibited by non-diabetic animals. In addition, it prevented the increase in the nuclear accumulation of the pro-fibrotic transcription factors β-catenin and phospho-Smad2/3. The same authors have also reported the efficacy of MitoQ in decreasing ethanol-induced micro- and macrohepatosteatosis in rats (39). A recently published study has demonstrated a protective role of mitochondria-targeted antioxidants in obesity-related comorbidities. Zucker obese fatty (ZOF) rats displayed higher levels of ROS in the smooth muscle cells and the aortic endothelium as well as increased UCP-2 and antioxidant enzyme activity in comparison with Zucker control rats. In ZOF rats, MitoQ reduced lipid peroxides to levels similar to those present in lean rats and also improved their metabolic profile, thereby restoring coronary collateral growth in response to ischemia to control levels (252). MitoQ has also been shown to render protective effects in animal models of metabolic syndrome and atherosclerosis (fat-fed ApoE−/−and ATM+/−/ApoE−/−mice, which are haploinsufficient for ataxia telangiectasia-mutated protein kinase, ATM). In one study, MitoQ prevented hypercholesterolemia (an increase in hypertriglyceridemia and adiposity related to metabolic syndrome) when administered orally for 14 weeks. It also reduced hepatic steatosis, hyperglycemia, and lipid and DNA oxidative damage (8-oxo-G) in different organs. Furthermore, a lower macrophage content together with diminished cell proliferation was observed within the plaques of fat-fed ATM+/−/ApoE−/−and ATM+/+/ApoE−/−mice after administration of MitoQ (199).
It is noteworthy that, in recent years, MitoQ has also been shown to be efficient in central nervous system (CNS) pathologies. In the case of AD, Ma et al. have reported that amyloid β-induced impairments in mice with hippocampal synaptic plasticity were reversed by decreasing mitochondrial O2·− through administration of MitoQ (182). MitoQ was shown to significantly reduce neuronal apoptosis and alleviate oxidative stress in rats exposed to Dichlorvos, a synthetic insecticide related to organophosphate pesticides known to induce neurodegeneration with clear signs of mitochondrial dysfunction, manifested as inhibition of mitochondrial complex I, cytochrome oxidase generation of ROS, and apoptotic cell death (342).
In conclusion, extensive evidence suggests that Mito-compounds are a promising approach to therapeutic intervention in numerous aspects of human pathophysiology.
c. MitoQ as a pharmacological agent
The design of MitoQ as a pharmacological agent has differed somewhat from that of most other drugs. In medicinal chemistry, compounds are typically developed to interact with a specific target in the biological system, such as a receptor-binding site. For this purpose, performing a preliminary screening is often necessary to confirm solubility, bioavailability, and capacity of a drug to cross phospholipid bilayers (173). Mitochondria-targeted antioxidants based on TPP lipophilic cations have several unusual properties. They are targeted to an organelle to modify a general, rather than a specific, process (namely oxidative damage), and have the peculiar pharmacological feature of being both relatively water soluble and membrane permeant. MitoQ, for example, is readily bioavailable and passes easily through biological membranes, even though its molar mass is relatively large for a pharmaceutical agent and has a high octanol—phosphate buffered saline partition coefficient. Therefore, if lipophilic cations such as MitoQ fulfil their potential as effective drugs, a new and different approach to the traditional view of medicinal chemistry and drug discovery would be endorsed. MitoQ is now being developed as a pharmaceutical agent by Antipodean Pharmaceuticals, Inc. In the search for a commercially satisfactory stable formulation, the creation of the compound has been based on the methane sulfonate counter anion and its decomposition is inhibited by complexation with β-cyclodextrin (50). This preparation is easily manufactured in tablet form and has passed conventional animal toxicity tests with no observable adverse effects at 10.6 mg/kg. In human studies, MitoQ has shown a positive pharmacokinetic behavior when administered orally at 80 mg (1 mg/kg), resulting in a plasma Cmax of 33.15 ng/ml and Tmax at ∼1 h, and 10% oral bioavailability. This formulation has good pharmaceutical characteristics and has been tested in two phase II clinical trials. In the first, which evaluated MitoQ10's ability to slow down the progression of PD, no difference was observed between placebo and experimental groups. In hepatitis C virus-infected patients who also had chronic liver hepatitis, MitoQ was found to decrease serum alanine transaminase, but had no effect on viral load (296).
d. Mito-compounds and nitrosative stress
•NO and RNS play a crucial role in oxidative stress-related disorders. A wide variety of •NO donors and S-nitrosating agents have been shown to protect the ischemic myocardium from infarction. Recently, mitochondria-selective S-nitrosating agents have been developed; MitoSNO, the best known, is the product of a covalent linking of an S-nitrosothiol to the lipophilic TPP cation (Fig. 10), which enables extensive and rapid accumulation within mitochondria (driven by Δψm), where it generates •NO and S-nitrosate thiol proteins. MitoSNO has been shown to provide protection in an in vivo rodent model of ischemia reperfusion, in which it reversed S-nitrosation of complex I by slowing down the reactivation of mitochondria during the crucial first minutes of reperfusion of ischemic tissue, thereby decreasing ROS production, oxidative damage, and tissue necrosis (48).
2. Cationic plastoquinone derivatives
Another group of synthetic cationic mitochondria-targeted antioxidants are SkQs-cationic plastoquinone derivatives containing positively charged phosphonium or rhodamine moieties connected to plastoquinone by decane or pentane linkers (Fig. 11). The prototypical example of these molecules is the plastoquinonyl decyltriphenylphosphonium (SkQ1). When SkQ1 electrophoretically enters the cell (assuming a plasma membrane potential of −60 mV) as a monovalent penetrating cation, it accumulates in the cytosol and the intermembrane space of mitochondria by a factor of 10, and then by factor 1000 when it enters, again electrophoretically, the mitochondrial matrix, which is 180 mV more negative than the intermembrane space. The membrane/water distribution coefficient, which is about 104 for SkQ1, also has a bearing. Hence, the total magnification of SkQ1 concentration in the inner leaflet of the IMM with regard to the extracellular space is expected to be 108. The relative accumulation may even be higher, as the SkQ1 cation forms a complex with cardiolipin anion, which seems to be the prime target of the antioxidant activity of SkQs (11). In a similar fashion to MitoQ, SkQs are rechargeable antioxidants, as their inactive oxidized forms are reduced (i.e., re-activated), in the case of SkQ by complex III of ETC (11). The efficacy of SkQs has been demonstrated in many in vitro and in vivo models (Table 1).
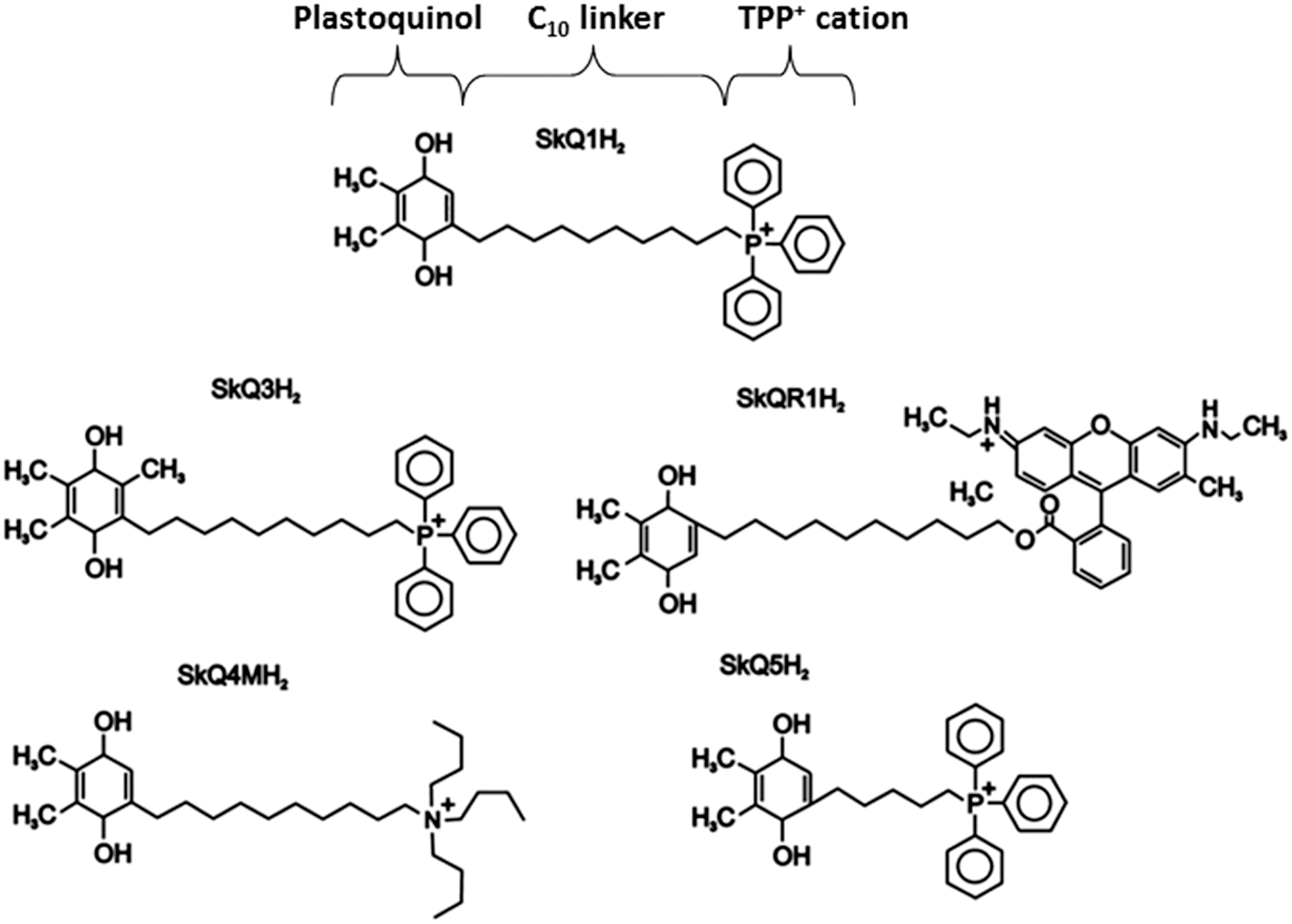
a. In vitro results
SkQ1 has been shown to be protective in in vitro models of hemolysis; it protected erythrocyte membrane lipids from peroxidation induced by the lipophilic free radical initiator 2,2-azobis-(2,4-dimethylvaleronitrile) and avoided hemolysis induced by the water-soluble free radical initiator 2,2-azobis-(2-methylpropionamidine)-dihydrochloride, but it had no antioxidant effect when the process was assessed by lipid peroxidation (226). The concentrations at which this occurred were approximately two to three orders of magnitude higher than those that have been proved active in a system of isolated mitochondria, which reflects the low ability of this compound to accumulate in erythrocytes due the low potential across the erythrocyte membrane (−10±3 mV) and emphasizes SkQ1 accumulation in mitochondria as a result of the electrophoretic transport. In addition, higher concentrations of SkQ1 displayed hemolytic abilities, likely due to its detergent-like effects that can cause disruption of the lipid membranes and not due to pro-oxidant activity, as Trolox was not capable of reversing it.
b. In vivo results and clinical studies
Several pathophysiological settings have been explored to test the utility of SkQ compounds in vivo. In a rat model, SkQR1 rescued kidney function from the deleterious effects of myoglobinuria (rhabdomyolysis) or ischemia/reperfusion (241), and the report suggested that SkQR1 exerted protective effects not only through direct scavenging of ROS but also by providing the kidney with elements of ischemic tolerance signaling mechanisms. Similarly, SkQR1 was shown to be beneficial in a rat model of acute pyelonephritis, where it inhibited leukocyte infiltration in the kidney and the spreading of the inflammatory process (242). SkQR1 recovered normal ROS levels in the leukocytes and expression of Bcl-2 in the kidney of pyelonephritic rats. Remarkably, treatment with SkQR1 during the first 2 days after infection resulted in significantly greater animal survival; therefore, the antioxidant protected the organisms from the development of sepsis. (SkQR1). Another study reported the nephroprotective action of SkQR1in two animal models of kidney failure (ischemia, rhabdomyolysis) and in an in vitro setting of pyelonephritis (243), as well as a neuroprotective role in a rat model of focal brain ischemia (243). Importantly, C12R, a conjugate of rhodamine with a linker chain of 12 carbon atoms, did not exhibit neuroprotective effects in this model of stroke, thereby showing that this biological activity could not be attributed to the part of the molecule not carrying the quinone residue. Moreover, SkQ1 has been reported to prolong the lifespan of many organisms, including mammals (292). It has proved to be particularly effective in animals suffering from eye diseases, retarding the development of (and in certain cases even reversing) symptoms of cataract, glaucoma, macular degeneration, uveitis, and dry eye syndrome (292). In the case of dry eye syndrome, clinical trials have shown that 3-week therapy with SkQ1 eliminates symptoms in 60% of patients (292). Indeed, SkQ1-containing eye drops (in the form of “Visomitin”) are commercially available in Russia since 2012, and clinical trials of SkQ1 as a medicine against age-related cataract and glaucoma are close to completion (292). Several recent studies have provided evidence of SkQ1 as a promising candidate for the treatment or prophylaxis of age-related and neurodegenerative disorders, particularly AD. The beneficial prophylactic effect of long-term treatment with SkQ1 has been shown in OXYS rats, senescence-accelerated rats that also develop AD-like pathology. SkQ1 reduced age-related alterations of behavior and spatial memory deficit and slowed down pathological accumulation of AβPP, Aβ and hyperphosphorylation of tau-protein (305). Another study revealed that an in vivo and in vitro injection of SkQ1 can compensate Aβ-induced oxidative damage of long-term synaptic plasticity (LTP) in the rat hippocampus. The neuroprotective activity of the analog of SkQ1 lacking plastoquinone (C12TPP) was found to be much lower than SkQ1, thus identifying the plastoquinone part of the SkQ1 molecule as being responsible for the LTP rescue (141).
A significant feature of SkQs is that they can be administered effectively in extremely low doses in in vivo treatments, thus significantly diminishing the probability of adverse side effects. However, similar to the majority of antioxidants, when their concentration is increased, which can represent overdose, SkQs become pro-oxidants. This has been shown in isolated yeast mitochondria, where lipophilic cations including SkQ1, SkQ3, and MitoQ acted as antioxidants, uncouplers, pro-oxidants, and detergents depending on the concentrations used (311). Fortunately, the window between the anti- and pro-oxidant activities of plastoquinone and its derivatives is large (30–1000 times), and is likely to be even wider than that for MitoQ.
c. Other derivatives
A similar strategy for mitochondria-targeted antioxidant therapy also involves the use of a series of novel compounds composed of natural constituents, such as conjugates of the plant alkaloids berberine and palmatine with the antioxidant plastoquinone
3. Peptide-based targeting
Szeto-Schiller (or SS)-peptides (378) and mitochondria-penetrating peptides (MPPs) (370) are peptide-based targeting agents for delivering antioxidants to mitochondria (Fig. 8). SS-peptides were developed by Szeto and Schiller and constitute a series of 4 small, cell-permeable antioxidant compounds (SS-19 H-Tyr-
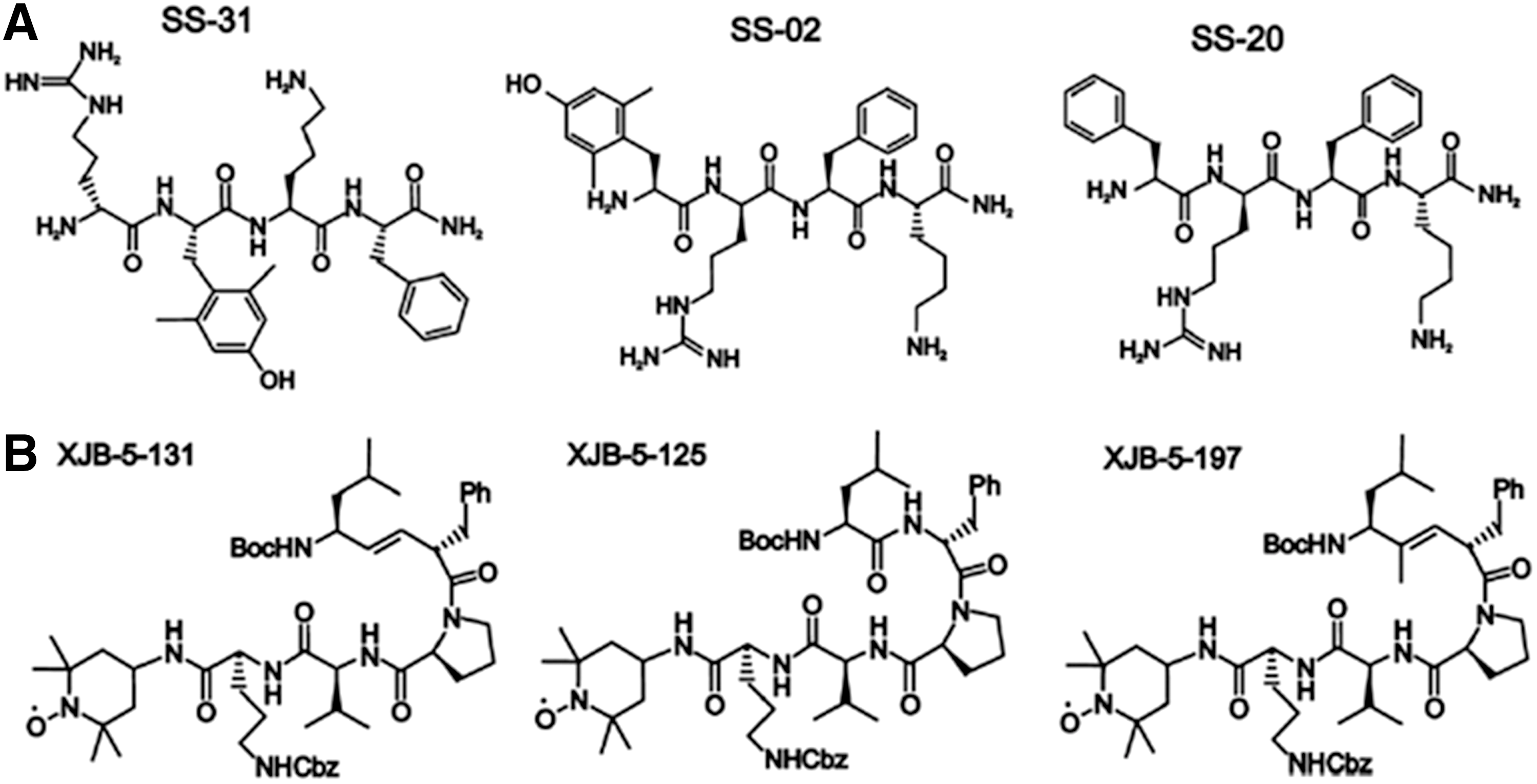
SS-peptides can scavenge H2O2 and ONOO− and inhibit lipid peroxidation, antioxidant actions attributed to a tyrosine or dimethyltyrosine residue in their structure, the latter of the two being more effective with regard to ROS scavenging. This moiety is believed to scavenge •OH and ONOO−, and possibly HOCl and peroxyl radicals, but a direct reaction with O2 •− or H2O2 seems unlikely. The specific location of these residues does not appear to be significant, as SS-31 is as effective as SS-02 in scavenging H2O2 and inhibiting LDL oxidation (261). SS-peptides have been reported to be beneficial against the conditions of oxidative stress both in cellular models of disease and in isolated mitochondria, with SS-31 proving to be the most efficient (187, 352, 378).
a. In vitro studies
The protective effects of these mitochondria-targeted therapies have been demonstrated in major comorbidities associated with diabetes, such as some vasculopathies, namely nephropathy and retinopathy (Table 1). Li et al. described a protective effect of peptide SS-31 on hyperglycemia-induced damage in human retinal endothelial cells (HRECs) (169). They reported that SS-31 decreased ROS production (especially in mitochondria), impaired the translocation of cytochrome c from the mitochondrion to the cytosol, controlled Δψm, decreased the expression of caspase-3, and enhanced the expression of TRX-2 in HRECs. In addition, cultured C2C12 myoblasts under basal condition or subjected to triggers of autophagy (pharmacological induction with rapamycin or induction by nutrient deprivation) display diminished autophagic flux and decreased rate of proteolysis when exposed to 1 μM SS-31 (256).
b. In vivo studies
SS-31 has been found to protect cells from mitochondrial toxicity in rodent models of human diseases, including ischemic brain injury (47), myocardial infarction (46), and ALS (237). Other animal studies have confirmed the efficacy of SS peptides aimed at PD (366), AD (187), obesity (11), and unilateral urethral obstruction (202) and those employed for cell and organ transplantation (316), although their exact mechanism of action and whether it unequivocally includes an antioxidant activity is not always clear. This antioxidant tetrapeptide has also proved to be beneficial in a mouse model of oxaliplatin-induced neuropathy (328). In a recent study, concomitant SS-31 administration (chronic treatment) with repeated oxaliplatin administration attenuated both cold and mechanical hypersensitivity, whereas acute SS-31 administration after symptoms manifested themselves only reversed cold hypersensitivity (328). The authors concluded that SS-31 has the potential to prevent acute and chronic neuropathy but is helpful only in the treatment of acute neuropathy. SS-31 utility has also been shown in a rat model of microvascular rarefaction or loss of microvascular density, a phenomenon implicated in the progression from acute ischemic kidney injury to chronic kidney disease. Microvascular dropout is thought to begin with ischemic damage to endothelial mitochondria due to cardiolipin peroxidation, resulting in loss of cristae and the failure to regenerate ATP on reperfusion. In a recent study, Liu et al. showed that SS-31 prevented swelling of mitochondria and protected mitochondrial cristae in both endothelial and epithelial cells. This was associated with a significantly reduced loss of peritubular capillaries and cortical arterioles, interstitial inflammation, and fibrosis 4 weeks after ischemia (175). The mechanism proposed for this action of SS-31 is its selective binding to cardiolipin and inhibition of cardiolipin peroxidation by cytochrome c peroxidase activity. Actually, a recent report has explored in detail the interaction of SS-31 and cardiolipin (23). Using as a model liposomes and bicelles containing phosphatidylcholine alone or with cardiolipin, Birk et al. (23) proved that SS-31 selectively targeted cardiolipin and that this interaction occured only with liposomes and bicelles containing cardiolipin in a ratio of 1:1. Moreover, SS-31 modulated the interaction of cardiolipin with cytochrome c, inhibiting cytochrome c/cardiolipin complex peroxidase activity while protecting the ability of cyttochrome c to serve as an electron carrier. In fresh mitochondria, SS-31 increased state 3 respiration and ATP generation efficiency. Another mechanism responsible for the beneficial effects of SS-31 in vascular diseases has been related to its ability to reduce the expression of CD36, a multifunctional class B scavenger receptor that mediates free radical production and tissue injury in cerebral ischemia. In the work in question, C57BL/6 mice were subjected to transient middle cerebral artery occlusion and treatment with SS-31 immediately after reperfusion significantly attenuated ischemia-induced GSH depletion in the cortex and reduced infarct size. Importantly, the protective effect of SS-31 was absent in CD36 KO mice, indicating that SS-31 was acting through inhibition of CD36 (47).
In a mouse model of pressure overload (by transverse aortic constriction)-induced heart failure, SS-31 ameliorated cardiac malfunction and the proteomic remodeling associated with it (64). SS-20 was also tested, and its protective effect was found to be more modest and possibly related to the fact that SS-20, unlike SS-31, is not a direct ROS scavenger. Importantly, SS-31 administration largely prevented mitochondrial oxidative damage and the process of autophagy in the heart induced by chronic pressure overload. Several studies using neurons treated with Aβ25–35 peptide and primary neurons from Tg2576 mice (AβPP transgenic) have highlighted the potential of SS-31 for treatment of AD (261). Primary neurons from AβPP mice present decreased anterograde mitochondrial movement, increased mitochondrial fission and decreased fusion, abnormal mitochondrial and synaptic proteins, and defective mitochondrial function when compared with wild-type neurons. SS-31 protects neurons from Aβ toxicity by reducing Aβ-induced mitochondrial toxicity, increasing axonal transport of mitochondria and enhancing synaptic viability. It also decreased levels of mitochondrial fission proteins (Drp1, Fis1) and matrix protein CypD in neurons affected by AD. Moreover, it enhanced the number of healthy and intact mitochondria and increased synaptic outgrowth and neuronal branching. These are very promising findings that encourage the evaluation of SS-31 in clinical trials with AD patients.
c. Mitochondria-penetrating peptides
MPPs consist of between four and eight alternating positively charged and hydrophobic partly unnatural aminoacids. They are believed to be a promising targeting approach for the mitochondrial delivery of small molecules (biotin and trolox have already been tested), though it is yet to be determined whether MPPs are effective in the transport and targeting of larger cargos such as DNA or nano-carriers (125, 371). In terms of the accumulation of MPPs in the mitochondrial matrix, specific chemical properties, such as electric charge and hydrophobicity, are clearly considered relevant (371), although the exact mechanisms that enable MPP transport through the phospholipid bilayer, including the role of ΔΨm and other mitochondrial factors in this process, are yet to be determined.
d. XJB peptides
Novel mitochondria-targeted molecules also include XJB peptides, a series of hemigramicidin-TEMPO compounds (Fig. 12B). These molecules are composed of 4-NH2-TEMPO, a stable nitroxide radical that exhibits antioxidant properties in vitro, conjugated to a pentapeptide fragment from gramicidin S (Leu-
4. Nanotechnology
Nano-preparations, or nano-particles (NP), are nano-sized, uniformly dispersed particles composed of biodegradable and biocompatible materials such as polymers, self-assembled lipids, or inorganic (metallic and magnetic) materials that are capable of carrying molecules with pharmacological activities such as peptides, proteins, nucleic acids, or small-molecular-weight drugs and prodrugs. There are several reasons that drug delivery systems based on NP are advantageous when compared with conventional drugs: (i) increased drug concentration at a specific site through passive and active targeting, thus reducing concentration-dependent side effects; (ii) better pharmacokinetics and pharmacodynamics properties; and (iii) improved cellular internalization and organelle-specific delivery. Recently, great interest has been focused on the development of specific delivery systems based on nano-materials designed to carry antioxidant agents. Selective targeting has been proposed in different strategies of mitochondrial medicine, including liposome-based vehicles (described in detail in section “Liposomal carriers”) and polymer-based, noble metal oxide-based, or carbon-based nano-antioxidants (107).
a. Colloidal dequalinium vesicles
One of the most extensively studied compounds among mitochondriotropic nano-carriers is the so called dequalinium-based liposome-like vesicle, or DQAsome (107, 361, 374). The antimicrobial and antineoplastic agent dequalinium (1,1′-(1,10-decamethylene-bis-[aminoquinaldinium])-chloride) is a dicationic amphiphilic molecule; it possesses two symmetrical cationic charge centers separated by a hydrophobic carbon chain (Fig. 8). DQAsome is generated through the self-assembly of dequalinium chloride into vesicle-like aggregates during sonication in aqueous suspension. DQAsomes penetrate cells through the process of endocytosis and, once inside cells, escape endosomal compartments through destabilization of endosomal membranes. It has been postulated that these delivery particles are attracted to mitochondria by electrostatic interactions, after which they fuse with the OMM. Several studies have demonstrated that DQAsomes can render efficient and selective delivery of nucleic acids to mitochondria (71, 349, 374), which opens an avenue for mitochondria-targeted gene therapy. Apart from nucleic acid delivery, DQAsomes have also been investigated for mitochondrial targeting of drugs (72, 333). However, this approach for delivery of either drugs or DNA into mitochondria needs further analysis, as liposomes designed from dequalinium analogues may have cytotoxic effects (333). Very recently, the mitochondrial delivery of DQAsome loaded with the antioxidant curcumin has been reported in Caco-2 cells (380). The same study did not detect any significant cytotoxicity in human epithelial A549 cells exposed for 72 h to DQAsomes at concentrations below 3 μM and postulated that curcumin-loaded DQAsomes may represent a promising inhalation formulation as a novel approach to the treatment of acute lung injury.
b. Liposomal carriers
Liposomal carriers (liposomes) are nano-carrier delivery systems that are capable of carrying hydrosoluble drugs in their core or lipid-soluble compounds in their outer membrane layers. An advantage of this strategy compared with chemically modified drugs is that active molecules are encapsulated without modifying their molecular structure, which preserves the drug's pharmacodynamics profile. When employed in antioxidant therapy, lisposomes are constituted by phosphatidylcholine, phosphatidylglycerol, and cholesterol, and further include molecular antioxidants, antioxidant enzymes, or a combination of various agents with antioxidant activities (308). It appears that antioxidants such as quercetin, the liposomally encapsulated NAC (4), and Siliphos (a complex formed by silybin and phospholipids) have therapeutic effects in a model of sepsis. Siliphos has been shown to be hepatoprotective in a rat model of steatosis (73) and to ameliorate liver enzyme levels in patients with non-alcoholic fatty liver disease (177) by enhancing mitochondrial function and through an insulin-sensitizing action.
In recent years, a Japanese group developed MITO-Porter (Fig. 8), a liposome-based nano-carrier that delivers cargo to mitochondria through a membrane fusion mechanism based on the multifunctional envelope-type nano-device, which consists of a condensed plasmidic DNA core and a lipid envelope that mimics envelope-type viruses (152). This mechanism seems to be able to transport proteins, functional nucleic acids, and small bioactive molecules (362). These delivery systems are useful in that they can transport encapsulated molecules of varying physicochemical characteristics or size. Mitochondrial delivery using MITO-Porter takes place over three steps: (i) delivery of the carrier from the extracellular space to the cytosol; (ii) intracellular trafficking of the carrier, including mitochondrial targeting; and (iii) mitochondrial delivery via membrane fusion (Fig. 13). This system of delivery allows large cargos to be delivered to mitochondria, provided they can be encapsulated in the MITO-Porter. Recently, the same group created a conjugate nano-carrier that targets mitochondria; it contains a mitochondria-targeting signal peptide and MITO-Porter to ensure selectivity for mitochondrial delivery (363). A dual function MITO-Porter (DF-MITO-Porter) that integrates the octaarginine (R8)-modified liposome for cytoplasmic delivery has also been created (361). High-density R8-modified liposomes are internalized primarily via macropinocytosis and are efficiently delivered to the cytosol in a similar fashion to an adenoviral vector (145). This exploits the characteristics of R8, a synthetic peptide that mimics the trans-activating transcriptional activator derived from the human immunodeficiency virus. DF-MITO-Porter includes a complexed particle of cargo coated with two mitochondria-fusogenic inner membranes and two endosome-fusogenic outer membranes. Modification of the outer envelope surface with a high density of R8 enables efficient internalization of the carrier into cells (first step). Once inside the cell, the carrier escapes from the endosome into the cytosol via membrane fusion, a process mediated by the outer endosome-fusogenic lipid membranes (second step). Indeed, a major limiting factor of nano-carrier systems for intracellular delivery is their escape from endosomes after endocytosis; only a small fraction of the endosome degrades spontaneously, which severely limits the possibility of the endocytosed material to reach the cytosol and, subsequently, the mitochondria. To overcome this limitation, nano-carriers are chemically designed to provoke the rupture of the endosome and to facilitate their release into the cytosol. After this step of intracellular trafficking, a third step consists of the binding of the carrier to mitochondria via R8, which is followed by fusion with the mitochondrial membrane (fourth step). This nano-carrier system, which can accomplish both efficient cytoplasmic delivery and mitochondrial macromolecule targeting, could open up new avenues of mitochondrial disease therapies. The efficacy of DF-MITO-Porter for the delivery of DNase I to the mitochondrial matrix has been demonstrated (361). The report in question described the process of construction of this particle, for which it was first necessary to determine the optimal conditions for complexation between DNase I protein and stearyl R8 through a process of three steps: (i) construction of the NP to contain an adequate amount of DNase I; (ii) coating of the NP obtained in step 1 with a mitochondria-fusogenic envelope; and (iii) further coating of the endosome-fusogenic envelope in a step-wise fashion. When DF-MITO-Porter was tested for cytotoxicity in HeLa cells, it was found that mitochondrial membrane fusion activity did not universally correlate with cytotoxicity (361). Rather, cytotoxicity was related to the composition of the inner envelope; among several lipid components tested, including sphingomyelin, cholesterol, cholesteryl hemisuccinate, cardiolipin, phosphatidic acid, phosphatidyl glycerol, phosphatidyl inositol, and phosphatidyl serine, sphingomyelin showed the best performance in terms of cell viability.
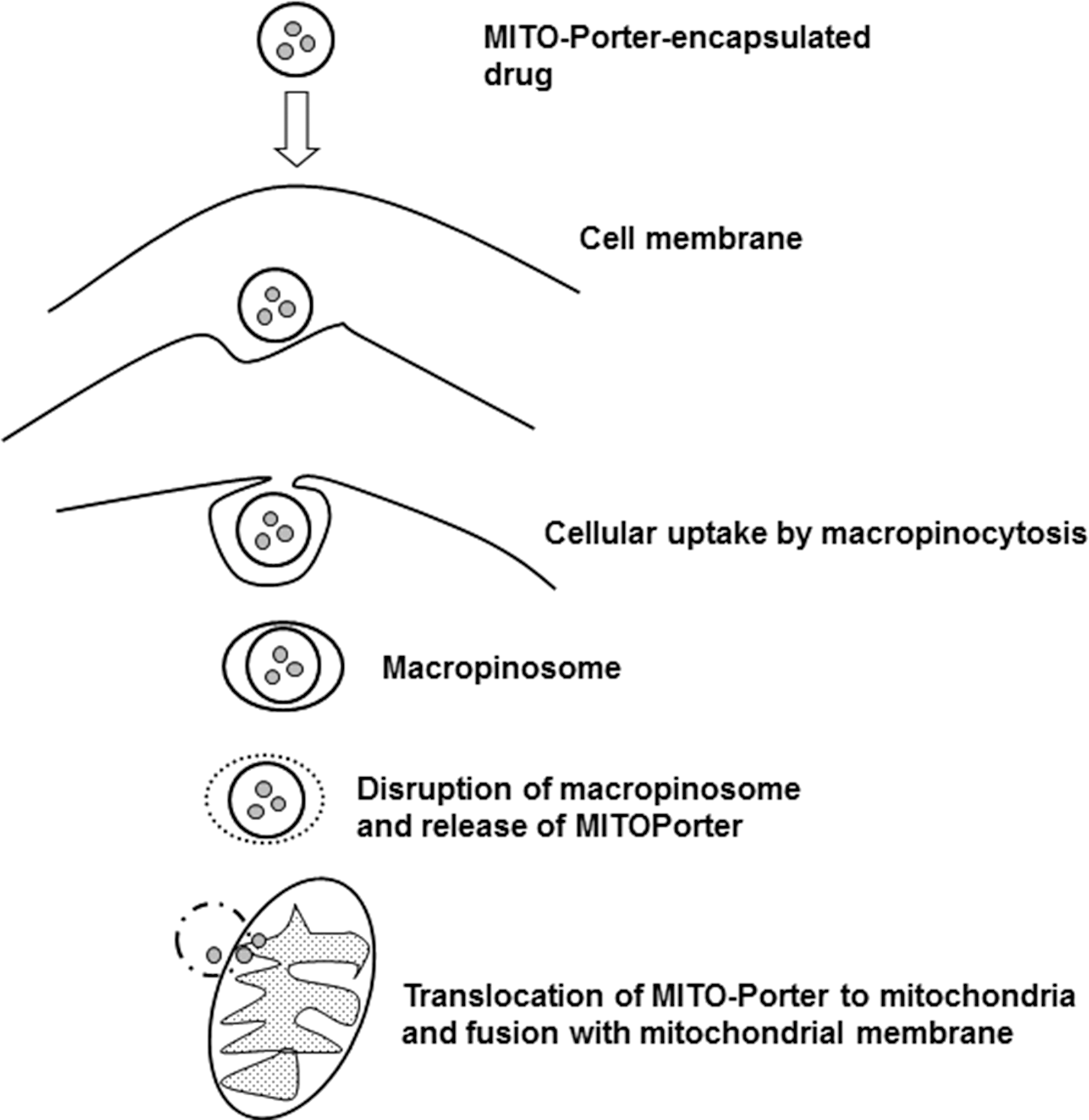
Mixed complex molecules containing both liposomes and lipophilic cations have also been designed. Effective delivery into mitochondria has been achieved with antitumor drug-loaded liposomes surface-functionalized with TPP (24, 26).
5. Mild mitochondrial uncoupling
Modulation of mitochondrial function to diminish the production of ROS by this organelle is another possibility. It is known that high mtROS generation is positively correlated with high Δψm. Mitochondrial uncoupling is a physiological process by which proton leak across the IMM allows protons to bypass complex V (ATP synthase), thereby disabling ATP synthesis as a result of the ETC function. In the mammalian cell, this process is regulated by the activity of the specialized proteins located at the IMM known as uncoupling proteins (UCP). Mitochondrial uncoupling can also be induced chemically by addition of protonophores such as 2,4-dinitrophenol (DNP). There is evidence that dissipation of Δψm by means of mild uncoupling can significantly reduce mtROS production and therefore protect macromolecules, such as nucleic acids, lipid, and proteins, from oxidative damage. This is the basis of the “uncoupling to survive theory,” which associates mild mitochondrial uncoupling with the prolonging of the lifespan, an effect already shown in mice (303) and Caenorhabditis elegans (166). Therefore, mild mitochondrial uncoupling by activation of the physiological regulators of the uncoupling process or by addition of chemical uncouplers may represent a plausible strategy for selective intervention in mtROS generation and function. There is abundant evidence that genetic modulation of innate UCP activity may be a promising avenue for treatment; for example, overexpression of hUCP2 was found to partially rescue the phenotype of PD in mice (10, 53). However, findings with UCP transgenics are contradictory in general and difficult to extrapolate to human therapeutics. With this in mind, direct mitochondrial uncoupling using chemical uncouplers currently seems a more rational approach, and compounds already studied include DNP, FCCP, and carbonyl cyanide m-chlorophenyl hydrazone (CCCP). Leaving to one side the discrepancies in the reported data, these compounds have been shown to prolong lifespan in some animal models (107). However, we need to bear in mind that although DNP, CCCP, and FCCP may have foreseeable clinical applications, their use is largely limited by the narrow therapeutic range they exhibit. For instance, despite being banned as a legal weight loss drug, DNP is still sold for this purpose and has produced more than 60 recorded deaths, of which 12 have occurred over the past decade. Classic symptoms include tachycardia, tachypnoea, and hyperthermia, culminating with deadly cardiovascular collapse and cardiac arrest (109). In this sense, a safer profile is displayed by the uncoupler butylated hydroxytoluene (BHT), which has been reported to partially uncouple isolated rat mitochondria and rat thymocytes in vitro, even at very low concentrations (2×10−3 nM) (179). Although BHT showed antioxidant properties in vitro, mice treated with this compound did not have uncoupled mitochondria and did not present a significant decrease in ROS production or oxidative damage of proteins, which calls into question the assumption that in vitro results will be observed in in vivo settings (114). With the aim of improving mitochondrial selectivity and, thus, therapeutic safety, conventional chemical uncouplers such as BHT and DNP have been conjugated with the TPP cation to generate Mito-BHT (179) and Mito-DNP (20) (Fig. 14). MitoBHT was shown to be effective in terms of mitochondrial uncoupling in isolated rat mitochondria and cultured rat thymocytes, an effect that was believed to be due to enhancement of the proton leak through the adenine nucleotide translocator (179). In contrast, MitoDNP was ineffective in uncoupling isolated rat mitochondria, probably as a result of the absence of efflux of the deprotonated MitoDNP from the mitochondrial matrix back to the mitochondrial intermembrane space (25). Importantly, the mitochondria-targeted TPP-conjugated plastoquinone SkQ1, previously described in section “Cationic plastoquinone derivatives,” not only displays antioxidant properties but can also act as an uncoupler by facilitating fatty acid cycling in IMM, thus dissipating Δψm, an effect observed in both isolated rat and yeast mitochondria (289). Bringing mild uncoupling one step further, Quin et al. synthetized a series of novel caged mitochondrial uncouplers designed to sense redox state (specifically H2O2 levels) and to respond only when there is an excess of this oxidant in the mitochondria. When levels of H2O2 are high, these molecules release the chemical uncouplers DNP or FCCP, thus dissipating Δψm and, consequently, lowering mtROS production (253). The trigger mechanism for this release exploits the reaction of arylboronate esters with H2O2 in mildly alkaline media, which converts them into phenols (e.g., at pH 8.3, which is the approximate pH of the matrix of active mitochondria), an effect already employed to make a range of fluorescent sensors that are sensitive to and selective for H2O2 (300). After this conversion, the molecule is fragmented and the mitochondrial uncoupler is released. Although H2O2-activated uncouplers may be useful, their beneficial effect and practical utility need to be further assessed both in vitro and in vivo.
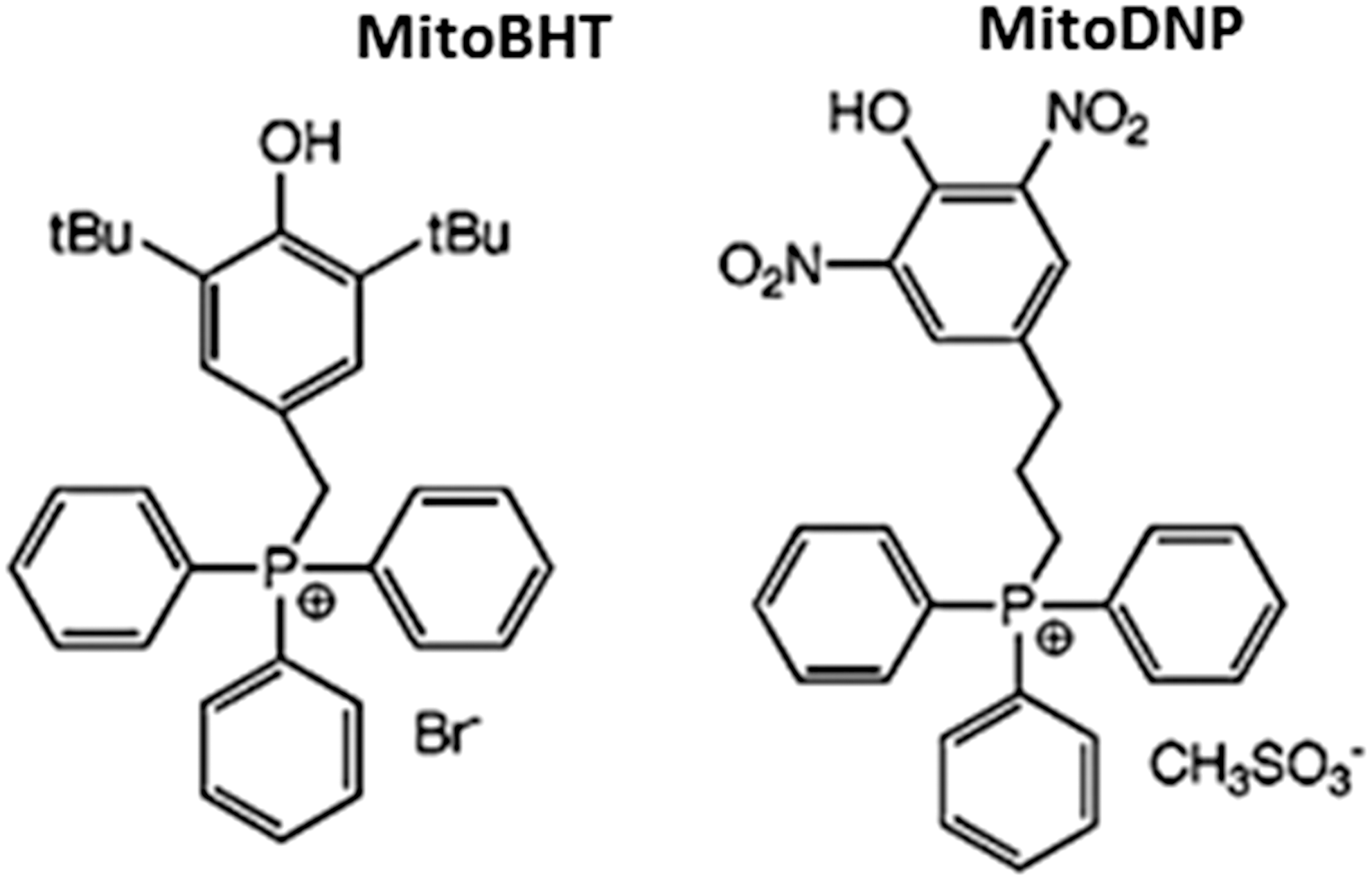
6. Other strategies
a. Organelle-specific bioactivation reactions
Another promising strategy for selective mitochondrial delivery of compounds is through an organelle-specific bioactivation reaction (6) (Fig. 8). This approach is based on the presence of bioactivating enzymes in subcellular territories such as mitochondria, which can be exploited to catalyze the conversion of a prodrug to a drug. Mitochondrial enzymes that are potential candidates for controlled xenobiotic transformations include mitochondrial monoamine oxidase B, cytochrome P450, and mitochondrial medium-chain acyl-CoA dehydrogenase (6).
One strategy uses the enzymes of mitochondrial β-oxidation, which catalyze the biotransformation of dietary and endogenously generated fatty acids along with a long list of xenobiotic alkanoic acids, including tianeptine, chlorophenoxybutyric acid, and 5-hydroxydecanoic acid. Mitochondrial targeting of antioxidants using mitochondrial β-oxidation has been studied in the case of alkanoate-based prodrugs (6). The transporters and enzymes that make up the fatty acid β-oxidation pathway for short and medium chain fatty acids are found only in mitochondria, which make the biotransformation of the aforementioned molecules selective for mitochondria. The potential of thia- and oxaalkanoate-based prodrugs has been assessed through studies of the biotransformation of ω-(phenoxy)alkanoates, 3-(phenoxy)acrylates and ω-(1-methyl-1H-imidazol-2-ylthio)alkanoates (Fig. 8). Importantly, 3- and 5-(1-methyl-1H-imidazol-2-ylthio)alkanoates have been shown to undergo a rapid biotransformation into the antioxidant methimazole and to exert a cytoprotective effect in a hypoxia-reoxygenation model of rat cardiomyocytes (272). In the same model, 3-(2,6-dimethylphenoxy)propanoic acid and 3-(2,6-dimethylphenoxy)acrylic acid also afforded cytoprotection. The protective effect of these compounds was blocked by etomoxir, an inhibitor of carnitine palmitoyl transferase I, the mitochondrial enzyme that enables their delivery into mitochondria. Very importantly, despite being reported as an efficient and versatile ROS scavenger (148), methimazole failed to provide cytoprotection for cardiomyocytes under conditions of hypoxia-reoxygenation when administered alone. This evidence supports the idea that antioxidants need to be specifically delivered into mitochondria so that they can reach the necessary “in situ” concentration to be active. Further studies are required to confirm the therapeutic potential of these and similar molecules in vivo.
b. Mn porphyrin-based cellular redox modulators
Mn porphyrin (MnP)-based cellular redox modulators also merit attention. These compounds have been designed to mimic mitochondrial SOD (MnSOD), an enzyme with a fundamental role in mitochondrial redox homeostasis. Such compounds belong to the class of cationic Mn(III) N-substituted pyridyl- and N,N′-disubstituted imidazolylporphyrins. Some of them have rate constants k cat (O2 •−) that are almost identical in magnitude to those of SOD enzymes. The first potent porphyrin-based SOD mimics, MnTM-2-PyP5+ and MnTE-2-PyP5+, were developed about 20 years ago. Since then, several other molecules have been generated based on the same principle; namely MnTDE-2-ImP5+ and, more recently, MnTnHex-2-PyP5+ and MnTnOct-2-PyP5+, which are produced by lengthening the alkyl chains of meso pyridyl groups while maintaining their total cationic charge and, therefore, their redox activity. The structure of the cationic MnP molecule combines the following critical features that allow it to mimic both the action and site of endogenous MnSOD: (i) the redox active Mn site; (ii) a multiple cationic charge; and (iii) four alkyl chains (the longer the alkyl chains, the higher the accumulation of MnP in mitochondria) (201). Available data suggest that MnP accumulates inside mitochondria, specifically in the mitochondrial matrix. The reason for this selective accumulation may be their cationic charge that is attracted by the negative Δψm and the anionic phosphate groups of cellular membranes. Porphyrin accumulation was investigated in heart mitochondria of mice after intraperitoneal injections over 7 days; the lipophilic compound MnTnHex-2-PyP5+ accumulated several-fold more in mitochondria than in cytosol compared with the hydrophilic analog MnTE-2-PyP5+ (326). The complex reactivities of MnPs within the cells make further mechanistic studies necessary; however, it seems that apart from acting as scavengers of O2 •−, synthetic SOD mimics have an impact on levels of O2 •−-derived or related molecules such as •OH, ONOO−, NO2, CO3 −, and lipid peroxyl radicals. Reduction of CO3 − by MnPs and reactivity toward •NO has also been reported. Ferrer-Sueta et al. showed that ≥3 μM MnTE-2-PyP5+ protects submitochondrial particles against oxidative stress imposed by ONOO− (88). In another work, accumulation of MnTE-2-PyP5+ in mouse heart mitochondria was reported to be 5.1 μM, a level high enough to render protection of mitochondria against oxidative damage (302).
The reaction in vivo will ultimately depend on the levels of MnPs, ROS, and RONS, cellular reductants, including GSH, ascorbate, BH4, and flavoprotein components of the mitochondrial respiration, and their co-localization with MnPs. MnP-based SOD mimics have proved to be effective in many in vitro and in vivo models, including tumorigenesis (particularly in combination with radiation and chemotherapy), renal ischemia reperfusion, diabetes, and CNS injuries and disorders (stroke, cerebral palsy, spinal cord injury, subarachnoid hemorrhage, AD, and chronic morphine tolerance) (as thoroughly reviewed by Tovmasyan et al.). Some studies have produced relevant data for the pathogenesis and/or management of CVD and related states, such as cardiometabolic pathologies, including diabetes (326).
c. Technology of biodegradable polymers
The use of platform technology, specifically the technology of biodegradable polymers, constitutes another promising approach for targeting bioactive molecules to the mitochondrial matrix (189). This method involves a rationally designed system of polymeric NP and contains the combination of a targeted poly(
d. Actively transported mitochondrial antioxidants
Exploiting actively transported mitochondrial antioxidants is another encouraging approach for specific targeting of these organelles (Fig. 8). Although natural antioxidants generally have a good safety profile, their use is limited because of their incapacity to penetrate mitochondria. However, there are several exceptions, and the antioxidant best understood in this regard is ergothioneine (EGT) (Fig. 8), a naturally occurring sulfur-containing amino acid that animals acquire through their diet. In humans, EGT has been shown to accumulate in specific tissues at millimolar concentrations (107), and rat studies have shown the accumulation of EGT in the mitochondrial fraction of hepatic cells (142). It has been postulated that the active transport of EGT through the plasma membrane is mediated by an organic cation transporter (OCTN1), is compound specific, and occurs in an Na+-dependent way (108). The mitochondrial localization of this transporter has also been confirmed (159). Several studies have shown the antioxidant properties of EGT in vitro, where it scavenges several ROS, including •OH, ONOO−, and hypochlorous acid (107), and can deactivate singlet oxygen (kΔ=2.3×107 M −1 s−1) at a rate higher than that achieved by other natural thiols, including GSH (274). In vitro studies have analyzed the potential of EGT to protect DNA against damage induced by various reactive species, and this has been related to its cytoprotective role, such as in the case of PC12 cells exposed to H2O2 (52). It seems that EGT acts by conferring protection to mitochondrial components that are vulnerable to ROS generated by the mitochondrial ETC. This is supported by the fact that H2O2 treatment of HeLa cells in which OCTN1 had been silenced provoked increased mtDNA damage (233). Furthermore, addition of EGT potentiated the protective effect of CoQ10, an antioxidant primarily found in mitochondria, in a study that assessed alloxan-induced lipid peroxidation of phosphatidylcholine liposomes (76). EGT also protects against metal-induced oxidative damage in accordance with its ability to react with bivalent metal cations such as Cu2+ and Fe2+ forming redox-inactive complexes (107). Of note, and unlike GSH, which generates ROS in the presence of Cu2+ through the formation of a redox-active Cu (I)-(GSH)2 complex, the association of EGT and Cu2+ is relatively stable and does not generate free radicals (379). In summary, EGT seems to possess a protective capacity against mtROS in vitro, though its actions in vivo await further assessment.
IV. Conclusions
Oxidative stress is implicated in a wide range of pathologies. Mitochondria generate most of the cell's energy by OXPHOS and are considered the main source of ROS in many cell types. Mitochondria-generated ROS are crucial signaling molecules and participants in many cellular adaptative mechanisms. However, when the redox balance is disrupted as a consequence of excessive ROS generation or insufficient scavenging, mtROS become harmful. The continuous generation of ROS inside mitochondria and the specific molecular/morphological characteristics of these organelles versus other parts of the cell (both mtDNA and proteins are particularly susceptible to oxidative damage) make this cellular compartment highly vulnerable to the impact of ROS. Oxidative stress leads to mitochondrial impairment, both of which are features of the pathophysiology of a wide variety of diseases, including cardiometabolic diseases, neurodegenerative disorders, cancer, and other age-related diseases.
For decades now, numerous studies have focused on the potential of non-targeted antioxidant therapy for restoring normal physiology in oxidative stress conditions. In general, studies in cells or animal models have been successful, but clinical trials have led to disappointing and contradictory results.
Taking into account the importance of their role in human pathophysiology, mitochondria stand out as a key pharmacological target. Multiple compounds have been developed to treat mitochondrial dysfunction in different pathological situations with varying targets and mechanisms of action. In the present review, we have explored the characteristics and the use of different mitochondria-targeting strategies, including lipophilic cations, cationic plastoquinone derivatives, liposomal carriers, peptide-based targeting, uncouplers, and other recently described compounds. The majority of these compounds have demonstrated their beneficial effects in different models of oxidative stress, and some of them have proved to be effective in clinical trials.
Moreover, multiple novel delivery tools have recently been developed and are currently under evaluation, which offers broad opportunities for mitochondrial modulation. Many of these approaches are promising according to the results obtained in vitro or in cell-free systems; however, additional in-depth studies, particularly in vivo, are lacking to ensure translation of findings to more clinically relevant settings. Specific animal disease models are necessary to address the ability of a given compound to prevent and/or treat oxidative stress-related dysfunction. Finally, human clinical trials under physiologically relevant conditions are crucial to assess the clinical relevance of these molecules.
Funding
This study was financed by grants PI13/1025, PI11/00327, CIBER CB06/04/0071, PROMETEOII/2014/035, and GV/2014/118 and by the European Regional Development Fund (ERDF). V.M.V. is recipient of a contract from the Ministry of Health of the Valencian Regional Government (CES10/030).