
Abstract
Introduction
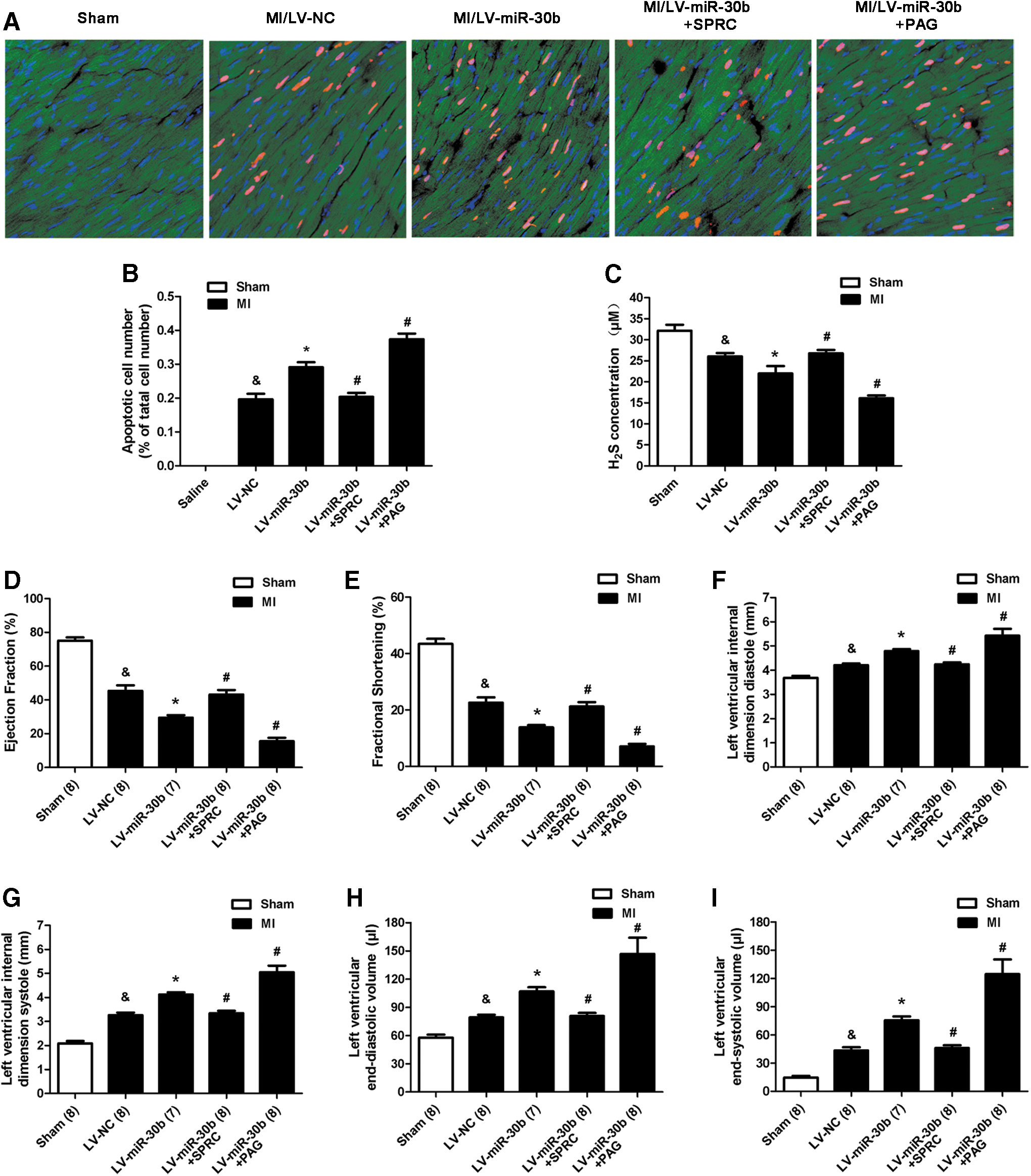
A
MicroRNAs (miRNAs) are endogenous small noncoding RNA acting as negative regulators of gene expression by mRNA degradation or translational repression (1, 8). Growing evidence shows that miRNAs are extensively involved in the pathogenesis of heart diseases (15), including cardiac hypertrophy (30), cardiac fibrosis (36), arrhythmia (43), heart failure (33), and MI (16, 27). The miR-30 family, including miR-30a, miR-30b, miR-30c, miR-30d, and miR-30e, has been shown to alter cell proliferation, differentiation (37), migration, and invasion (6) in various carcinomas. The details of miR-30 involvement in cardiovascular pathophysiology have recently emerged. A study has shown that downregulated miR-30a aggravates angiotensin II-induced myocardial hypertrophy by targeting beclin-1 (26). Wei et al. (42) recently demonstrated that NF-κB positively regulated miR-30b expression in Ang II-induced cardiomyocytes apoptosis, and Bcl-2 was a direct target for miR-30b. Duisters et al. (13) showed that miR-30c is predominantly expressed in fibroblasts and that it can regulate connective tissue growth factors in myocardial matrix remodeling. MiR-30c and miR-30d overexpression has also been proved to induce cardiomyocyte hypertrophy (20). Studies also showed that miR-30 family plays an important role during sprouting angiogenesis by targeted delta-like 4 (DLL4) (3). Furthermore, the upregulation of circulating miR-30a may be used as a potential biomarker for acute myocardial infarction (AMI) (23). Dong et al. (11) also studied AMI and observed that miR-30b-5p and miR-30c expressions were upregulated in the noninfarcted areas of the infarcted heart. These results suggest that miR-30 may be a new pathological factor and therapeutic target for cardiovascular diseases.
We demonstrated for the first time that the miR-30 family was able to regulate hydrogen sulfide (H2S) production in the heart by directly targeting cystathionine-γ-lyase (CSE). This revealed a new molecular control mechanism for endogenous H2S production in the heart at the microRNA level. We also found that miR-30–CSE–H2S axis contributes to protection against cardiomyocyte ischemic injury both in vitro and in vivo by regulating H2S production. This may provide a new potential therapeutic target for ischemic heart disease.
Hydrogen sulfide (H2S), which is the third gaseous mediator alongside carbon monoxide (CO) and nitric oxide (NO) in mammals, has been reported to provide cardioprotection in various models of cardiac injury, including MI (28). Physiologically, H2S is generated from L-cysteine and catalyzed by cystathionine-β-synthase, cystathionine-γ-lyase (CSE), or 3-mercaptopyruvate sulfurtransferase (32, 40). CSE is the predominant H2S-generating enzyme in the cardiovascular system (25).
A growing body of evidence shows that H2S derived from CSE modulates cardioprotection in myocardial ischemic injury. Transgenic mice with cardiac-restricted overexpression of CSE result in increased H2S production and cardioprotection in response to both acute myocardial ischemia/reperfusion (I/R) injury (14) and ischemia-induced heart failure (5). In contrast, deficiency of CSE significantly attenuates H2S and results in exacerbating myocardial I/R injury (4). Moreover, our previous studies (38, 39, 46) have shown that CSE expression in the rat heart is significantly reduced at 48 h after MI induction. Qipshidze et al. (28) also found that induction of MI for 4 weeks decreases expression of CSE in mice heart. These findings indicate that lower endogenous levels of H2S that are attributable to the suppression of CSE expression are pathogenic in cardiac ischemic injury. However, the mechanisms for the alterations in CSE expression in these pathological conditions remain unclear. In addition, the molecular mechanism for the regulation of endogenous H2S production in the heart is not yet known.
In this study, we provide a better understanding of the relationship between miR-30 family and H2S production and reveal the mechanism for endogenous H2S production at miRNA level. Furthermore, we clarify the protective effect of miR-30 family inhibition on myocardial ischemic injury by regulating the production of H2S both in vitro and in vivo.
Results
The expressions of CSE and miR-30 family are changed in response to MI
To explore whether miRNAs regulated CSE, we first undertook a bioinformatic approach with targetscan software (
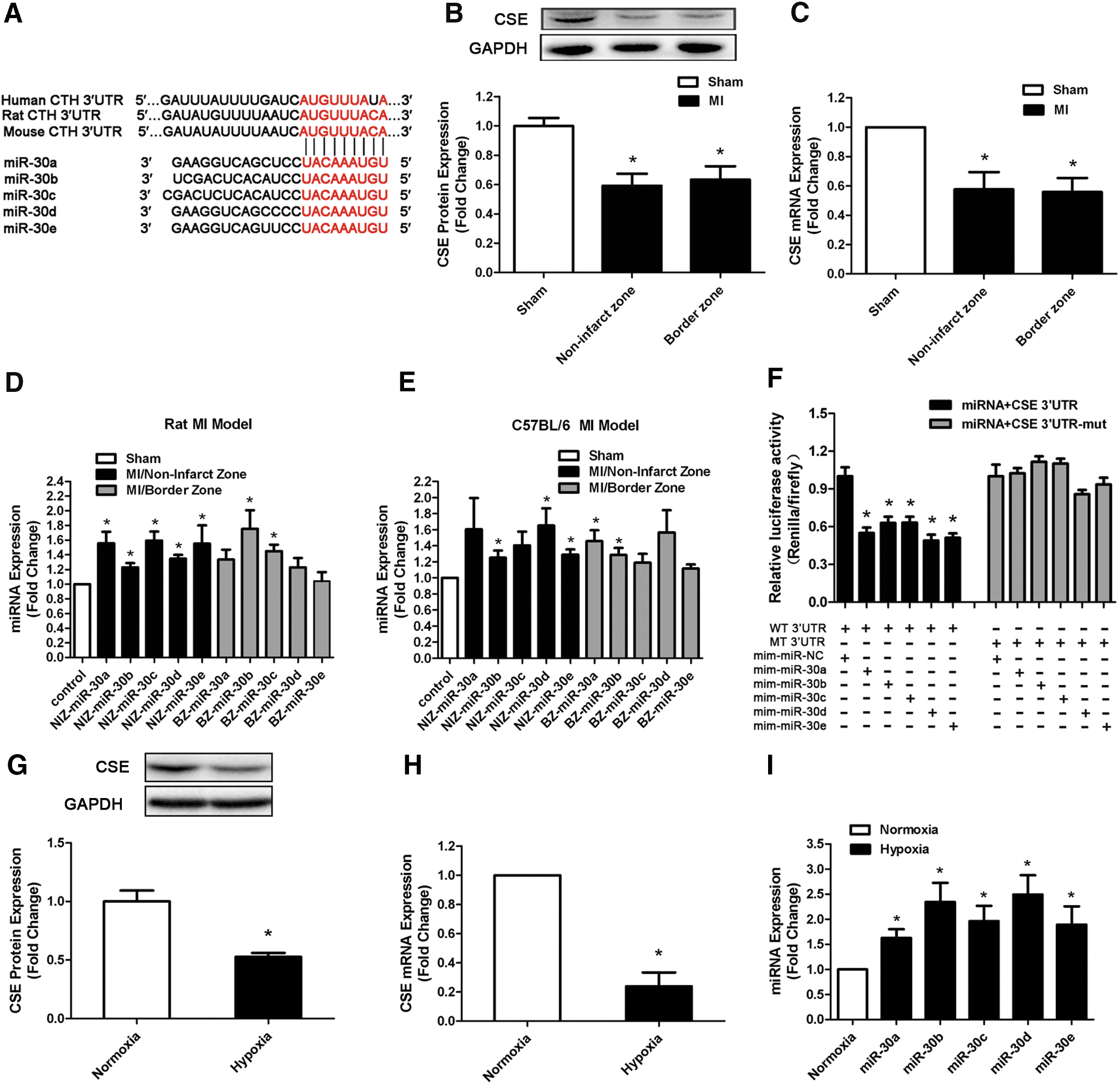
Then, we examined CSE expression in different heart regions in a rat MI model at 48 h after MI induction. Real-time polymerase chain reaction (PCR) and Western blot revealed that CSE levels decreased in the remote and border zone regions after MI compared with control tissue from sham-operated animals (Fig. 1B, C). However, all members of the miR-30 family were observed to be upregulated in these two regions of both rat and mouse MI models (Fig. 1D, E). Furthermore, in hypoxic myocardial cell model, we also found that CSE expression was decreased (Fig. 1G, H) and miR-30 family was increased (Fig. 1I) when compared with normoxia group. These results indicated a correlation between increased miR-30 levels and the decrease in CSE expression.
CSE is a direct target of the miR-30 family
To further validate that the miR-30 family is able to directly bind to CSE and inhibit CSE expression, we inserted the full-length 3′-UTR of the CSE transcript into a dual-luciferase expression plasmid (pMir-report) and cotransfected them into HEK293 cells with miR-30 family member mimics or negative control (NC) miRNA. As expected, the cotransfection of miR-30 family members with the reporter construct containing the 3′-UTR segment of the CSE resulted in a significant decrease in luciferase activity, but this was not observed with control miRNA. No effects were observed with a construct containing a mutated segment of the CSE 3′-UTR (seed sequence TGTTTACA was mutated to AGATAAGT) (Fig. 1F). This indicated that CSE transcripts represented a genuine target of the miR-30 family.
MiR-30 family regulates CSE mRNA and protein levels
To determine the role of the miR-30 family in CSE regulation, gain-of-function and loss-of-function approaches were performed in primary neonatal rat myocardial cells. First of all, we transfected cultured myocardial cells with miR-30 family member mimics (50 nM) or an NC miRNA. Forty-eight hours after transfection, we observed a significant negative correlation between all of the miR-30 family member mimics and CSE expression in primary cardiomyocytes. The real-time PCR and Western blot results showed that the overexpression of each miR-30 family member clearly decreased the CSE expression at both the mRNA and protein levels (Fig. 2A, B).
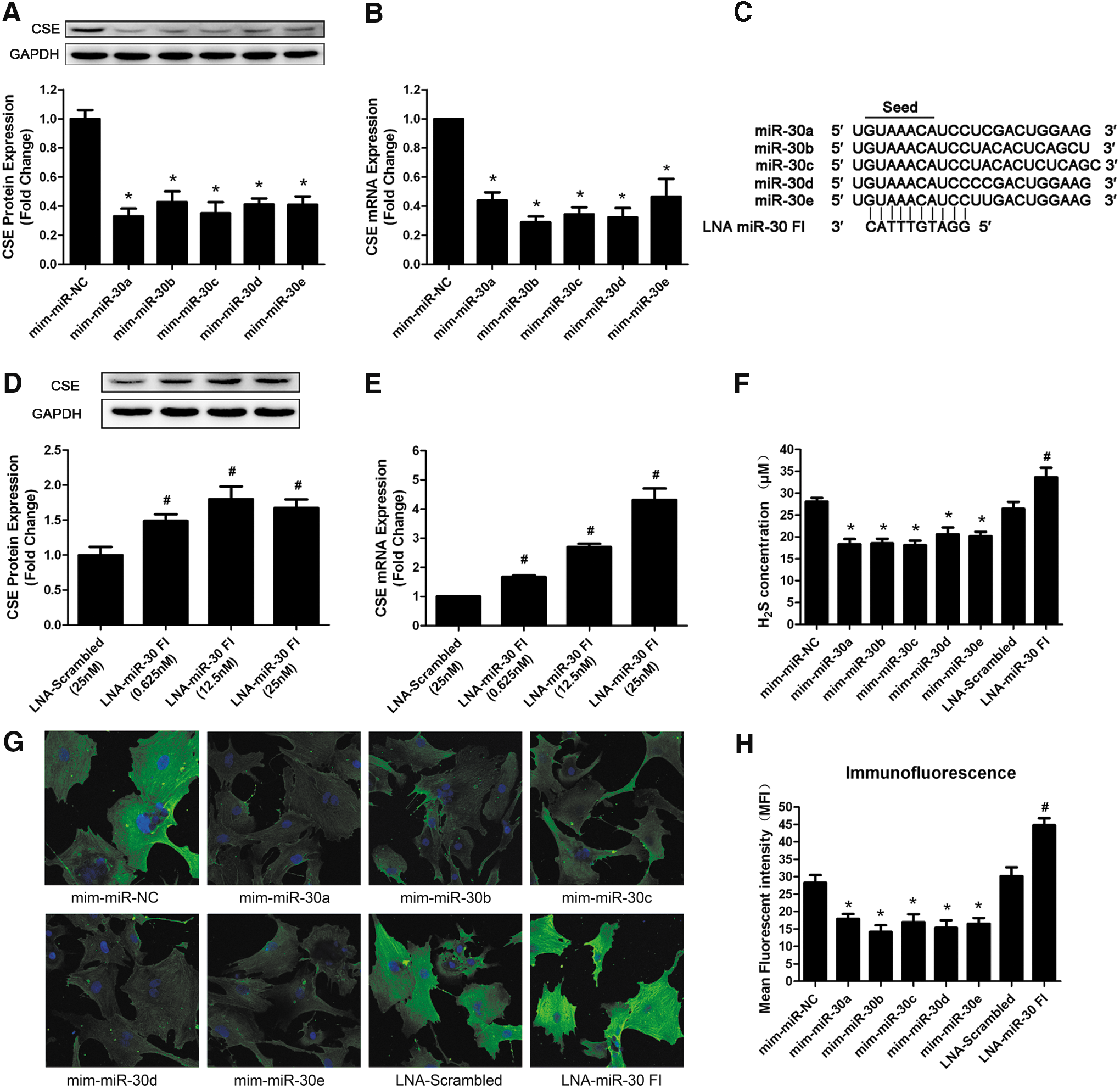
Subsequently, we tested whether knockdown of the whole miR-30 family induced an upregulation of CSE levels. Therefore, we designed a 10-mer locked nucleic acid (LNA)-miR-30 family inhibitor (LNA-miR-30 FI) that was complementary to the seed region of the miR-30 family, as described by Obad et al. (24), which was predicted to target all miR-30 family members (Fig. 2C). The successful knockdown of miR-30 family members was confirmed by real-time PCR. At 25 nM, the LNA-miR-30 FI reduced the expression of the miR-30 family members by ∼70% compared with that in LNA-scrambled control-transfected cells (Supplementary Fig. S1; Supplementary Data are available online at
The effects of miR-30 mimics and miR-30 FI were also confirmed by immunohistochemistry. As shown in Figure 2G and H, transfection of miR-30 family member mimics resulted in weaker green fluorescence intensity compared with the NC group. However, the green fluorescence intensity in the LNA-miR-30 FI group was stronger than in the LNA-scrambled group.
The H2S concentration in the supernatant after transfection was also determined. It was found that miR-30 family mimics reduced H2S production, whereas LNA-miR-30 FI increased H2S level at 48 h after transfection (Fig. 2F).
The miR-30 family affects hypoxia-induced cardiomyocyte injury
CSE is the main enzyme and plays a major role in catalyzing the production of H2S in the cardiovascular system. We aimed at determining the effects of the miR-30 family on cardiomyocytes under hypoxia condition by regulating CSE expression. Eight hours of hypoxia, the cell viability decreased 35.85%±5.75%, and the lactate dehydrogenase (LDH) release significantly increased 86.61%±19.55% when compared with normoxic group (Fig. 3A, B), indicating that the hypoxic conditions were sufficient to induce cardiomyocyte injury in vitro.
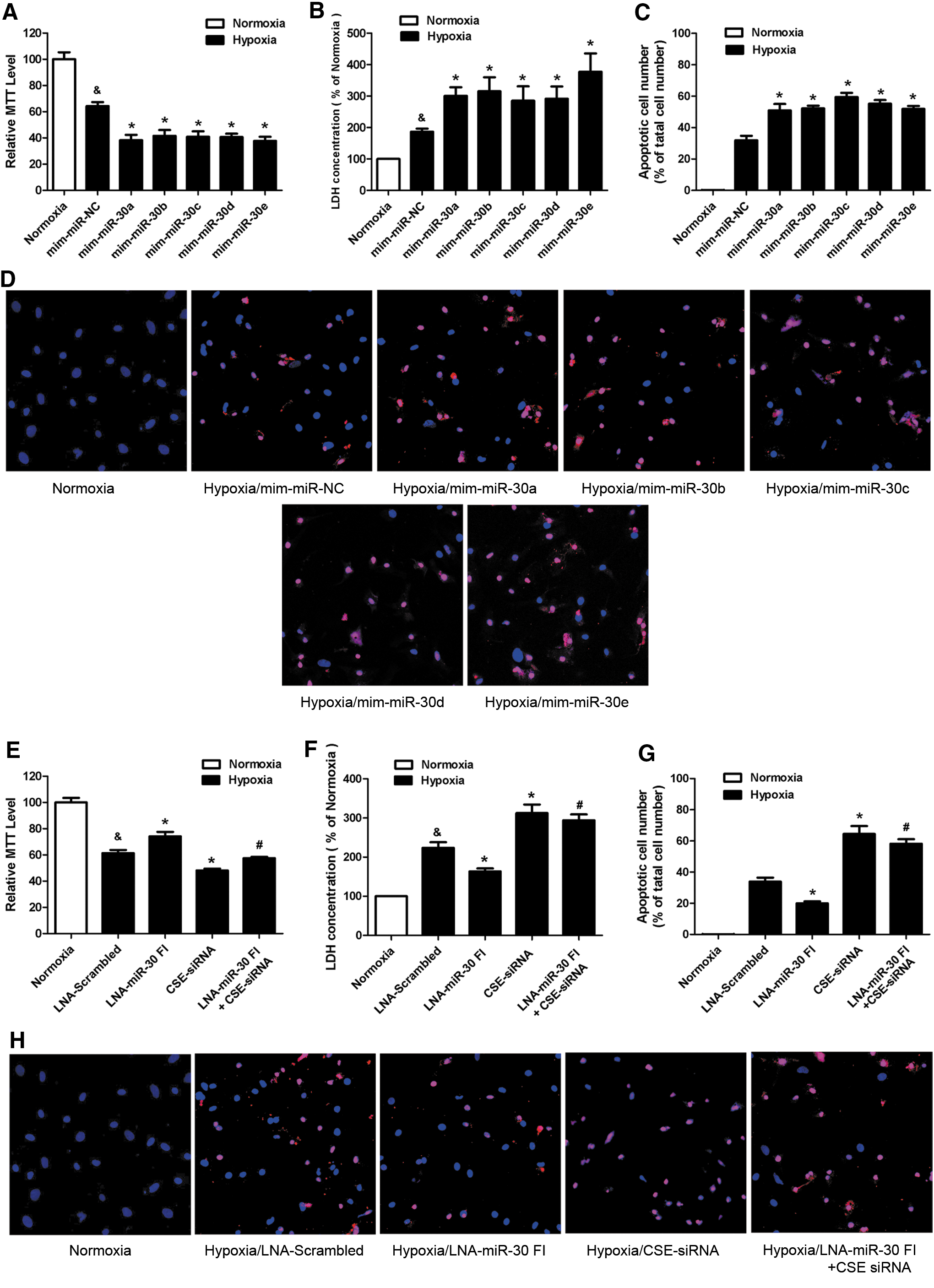
Subsequently, using this hypoxic model, we confirmed that upregulation of CSE by an overexpression plasmid (pcDNA3.1-CSE) markedly increased the CSE and H2S level and protected cardiomyocyte against hypoxia injury, determined by 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetra-zolium (MTT) and LDH assay (Supplementary Fig. S2). On the other hand, blockade of CSE with siRNA significantly reduced the CSE and H2S level and aggravated hypoxia cardiomyocyte injury (Supplementary Fig. S2). In miR-30 transfection groups, we found that the overexpression of each miR-30 family member decreased cell viability and increased LDH release when compared with NC under hypoxic conditions (Fig. 3A, B). On the other hand, the knockdown of the whole miR-30 family with the LNA-miR-30 FI increased cell viability and reduced LDH release (Fig. 3E, F). However, this protective effect was absent after cotransfecting LNA-miR-30 FI with CSE siRNA (Fig. 3E, F).
We also used a TUNEL assay to determine the effects of the miR-30 family in hypoxia-induced apoptosis. The results showed that the number of apoptotic cells significantly increased after transfecting miR-30 family mimics (Fig. 3C, D), but reduced after transfecting the miR-30 FI (Fig. 3G, H) when compared with NC. Nevertheless, the apoptotic cells' number was increased again after cotransfecting miR-30 FI with CSE siRNA (Fig. 3G, H).
Taken together, these data showed that miR-30 overexpression aggravated cardiomyocyte injury during hypoxia and that the LNA-miR-30 FI protected cardiomyocytes against hypoxia-induced injury. Notably, CSE-siRNA abolished the protective effect of LNA-miR-30 FI.
Silencing the miR-30 family protects against MI injury in mice
To investigate whether the beneficial effects existed in in vivo conditions, we knocked down the miR-30 family with a tail vein injection of LNA-miR-30 FI. Scrambled oligo and saline were used as controls. With an LNA-miR-30 FI injection (5 mg/kg) for 3 successive days, real-time PCR analysis revealed a dramatic reduction in miR-30 family expression levels in heart tissue compared with the LNA-scrambled control (Fig. 4A). This indicated that LNA-miR-30 FI efficiently downregulated miR-30 family expression levels in the heart. In parallel, the administration of the LNA-miR-30 FI and not the LNA-scrambled control was associated with greatly increased CSE expression levels in the left ventricle, and the data showed that LNA-miR-30 FI increased the CSE expression in a dose-dependent manner at both the mRNA and protein levels (Fig. 4B, C). In our experiment, 2.5 mg/kg of LNA-miR-30 FI showed the highest efficiency in increasing CSE protein expression in vivo, and was, therefore, chosen in subsequent experiments.
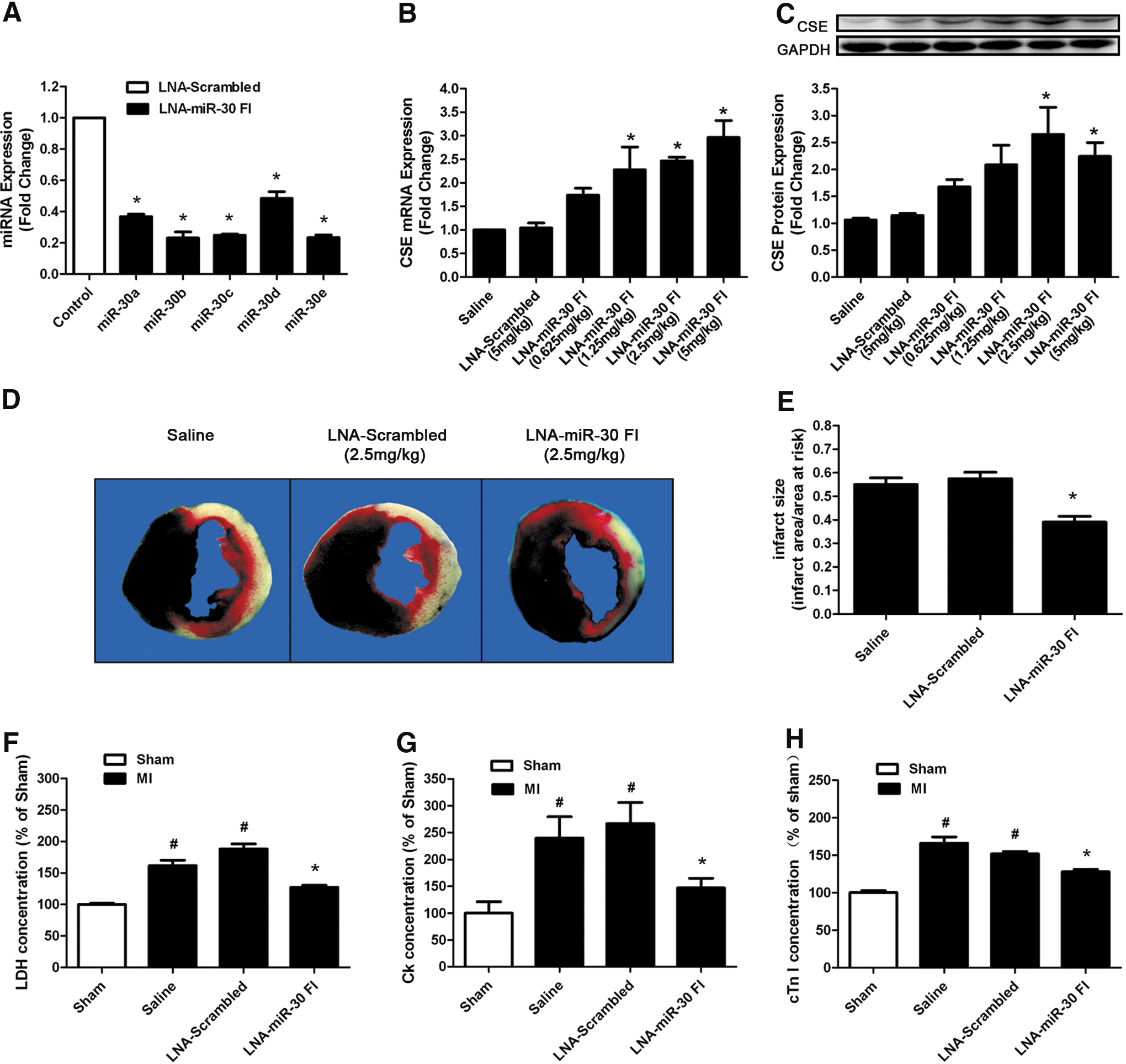
Next, the hearts treated with LNA-miR-30 FI for 3 days were subjected to coronary occlusion to induce an MI model. The extent of MI was determined at 48 h after surgery, and the results indicated that treatment with LNA-miR-30 FI resulted in a significant decrease in infarct size compared with treatment with either saline or LNA-scrambled control (Fig. 4D, E). The area at risk (AAR) was nearly the same among groups (data not shown).
LDH, creatine kinase (CK), and cardiac troponin-I (cTn-I) are three reliable biomarkers of cardiomyocyte injury. To confirm the degree of cardiomyocyte injury in ischemia, we determined LDH, CK, and cTn-I concentrations in plasma. Consistent with the results of cardiac infarct size, our data showed that LDH, CK, and cTn-I concentrations in plasma were markedly increased in the MI group compared with the sham group, whereas the knockdown of the miR-30 family by LNA-miR-30 FI inhibited the increases in serum LDH, CK, and cTn-I levels that were caused by MI injury (Fig. 4F–H).
Cardiac function was examined using echocardiography at 5 days after MI surgery. The echocardiography revealed that the left ventricular ejection fraction (EF) and fractional shortening (FS) were significantly decreased (Fig. 5A, B), whereas the left ventricular internal dimension systole (LVIDs), left ventricular internal dimension diastole (LVIDd), left ventricular end-systolic volume (LVESV), and left ventricular end-diastolic volume (LVEDV) were increased in the MI models (Fig. 5C–F), indicating impaired cardiac function. The downregulation of the miR-30 family attenuated the deterioration in left ventricular performance, as indicated by the increased EF and FS, which corresponded with the decreased LVIDd, LVIDs, LVEDV, and LVESV. In contrast, the LNA-scrambled control and vehicle control produced no such effects. The improvement in ischemia injury was in parallel with the increase in CSE expression and H2S concentration in response to LNA-miR-30 FI at 48 h after injury (Fig. 5G, H).
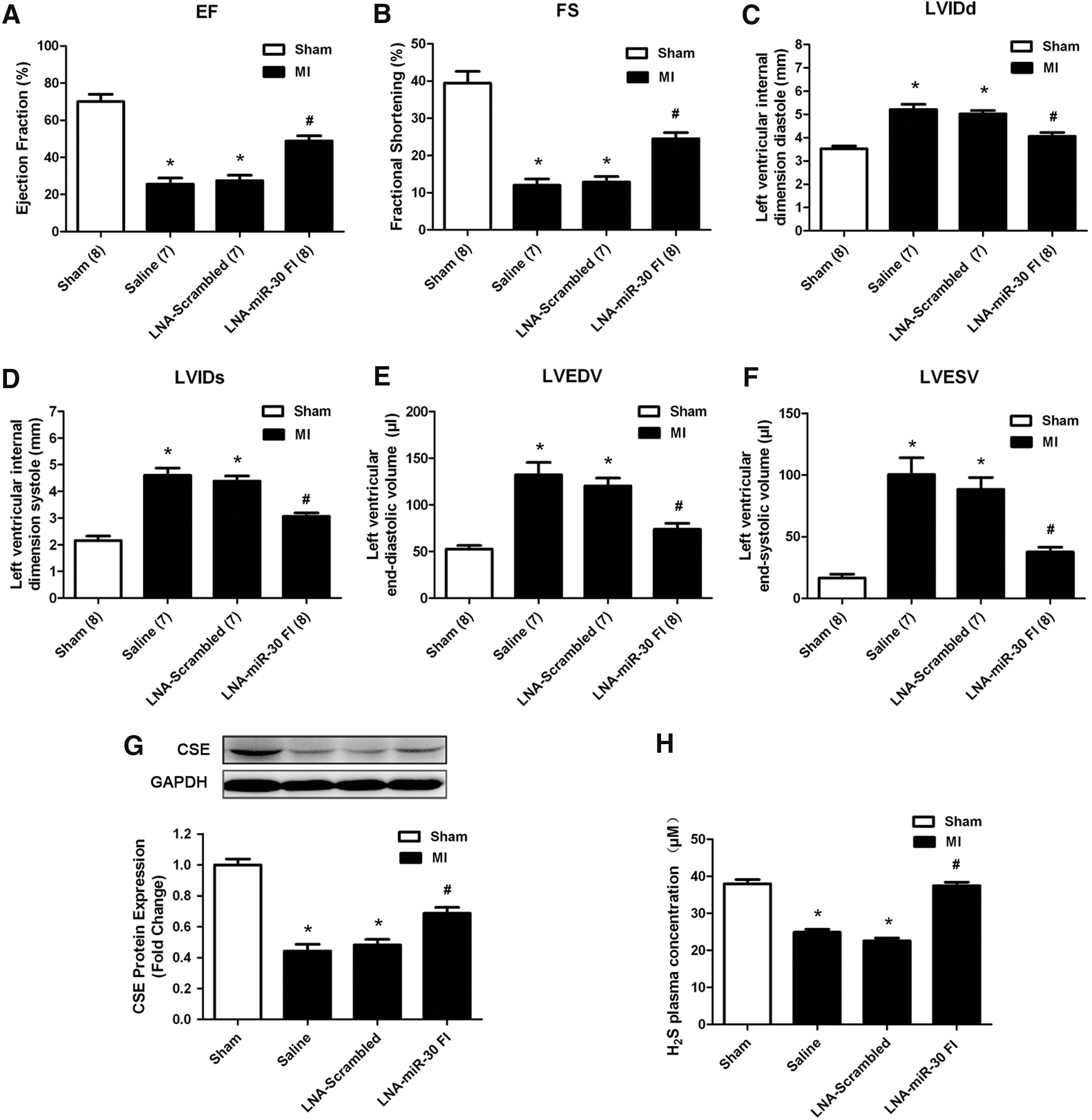
We also examined the effects of LNA-miR-30 FI on Bcl-2 and Bax expression in the left ventricle of the ischemic heart. We observed that downregulation of the whole miR-30 family enhanced Bcl-2 levels and reduced Bax levels after MI injury (Fig. 6A, B). We further examined apoptosis of cardiomyocytes that were subjected to ischemia injury by TUNEL staining in the peri-infarct area of the infarcted heart. Compared with the saline control, LNA-miR-30 FI markedly decreased the apoptosis of cardiomyocytes, while the LNA-scrambled control produced no effects (Fig. 6C, D). In addition, we also found the activities of antioxidant defensive enzymes such as superoxide dismutase (SOD) and catalase (CAT) were decreased in left ventricular tissue of MI control group, but increased after using LNA-miR-30 FI (Fig. 6E, F). An additional biomarker of oxidative stress named malonaldehyde (MDA) was increased in the MI control group, but reduced after treatment with LNA-miR-30 FI (Fig. 6G).
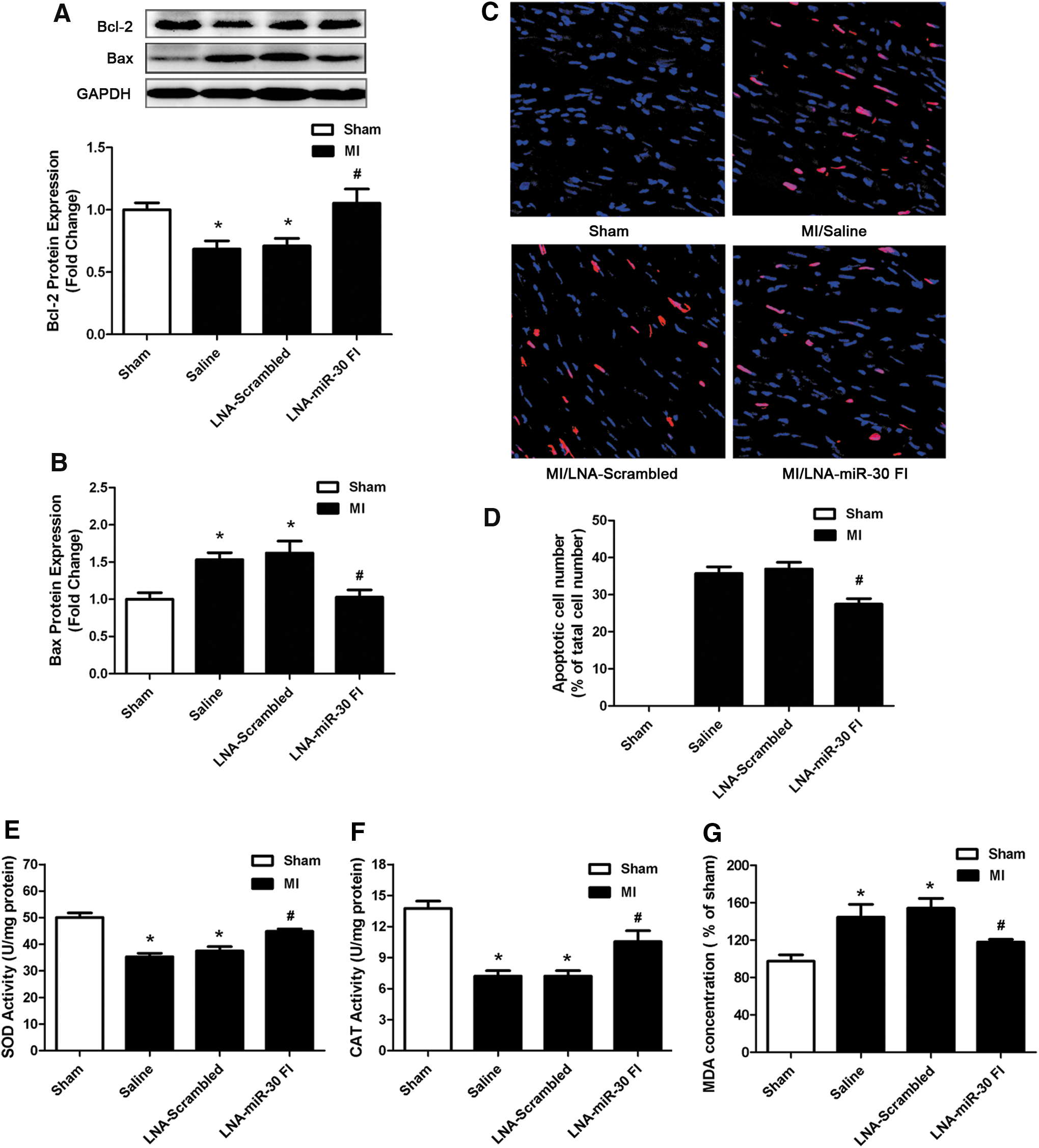
Taken together, these data indicated that the systemically applied LNA-miR-30 FI reduced infarct size and improved cardiac function in response to ischemia injury. This corresponds with the increase in H2S production that occurred in response to miR-30 inhibition.
The cardioprotective effect of LNA-miR-30 FI was absent in CSE knockout mice
Furthermore, in order to ascertain whether the protective effect of LNA-miR-30 FI is, indeed, dependent on CSE, CSE knockout mice (CSE−/−) were used to check whether the protective effect is still present after administration of LNA-miR-30 FI. The results showed that CSE protein was absent in heart of CSE−/− mice, endogenous H2S levels in plasma were decreased by about 50% when compared with wild-type (WT) mice. However, there was no significant difference between CSE−/− mice and CSE−/− mice treated with LNA-miR-30 FI (Supplementary Fig. S3).
Deficiency of CSE exacerbates myocardial ischemia injury. Myocardial infarct size was significantly greater in CSE−/− mice MI model as compared with WT control hearts (Fig. 7A, B). In parallel, circulating plasma levels of the LDH, CK, and cTnI evaluated as an additional marker of myocardial injury were significantly increased in CSE−/− mice MI model (Fig. 7C–E). However, administration of LNA-miR-30 FI showed no improvement. There was no difference between CSE−/− mice group and CSE−/− mice administered with LNA-miR-30 FI group (Fig. 7 A–E).
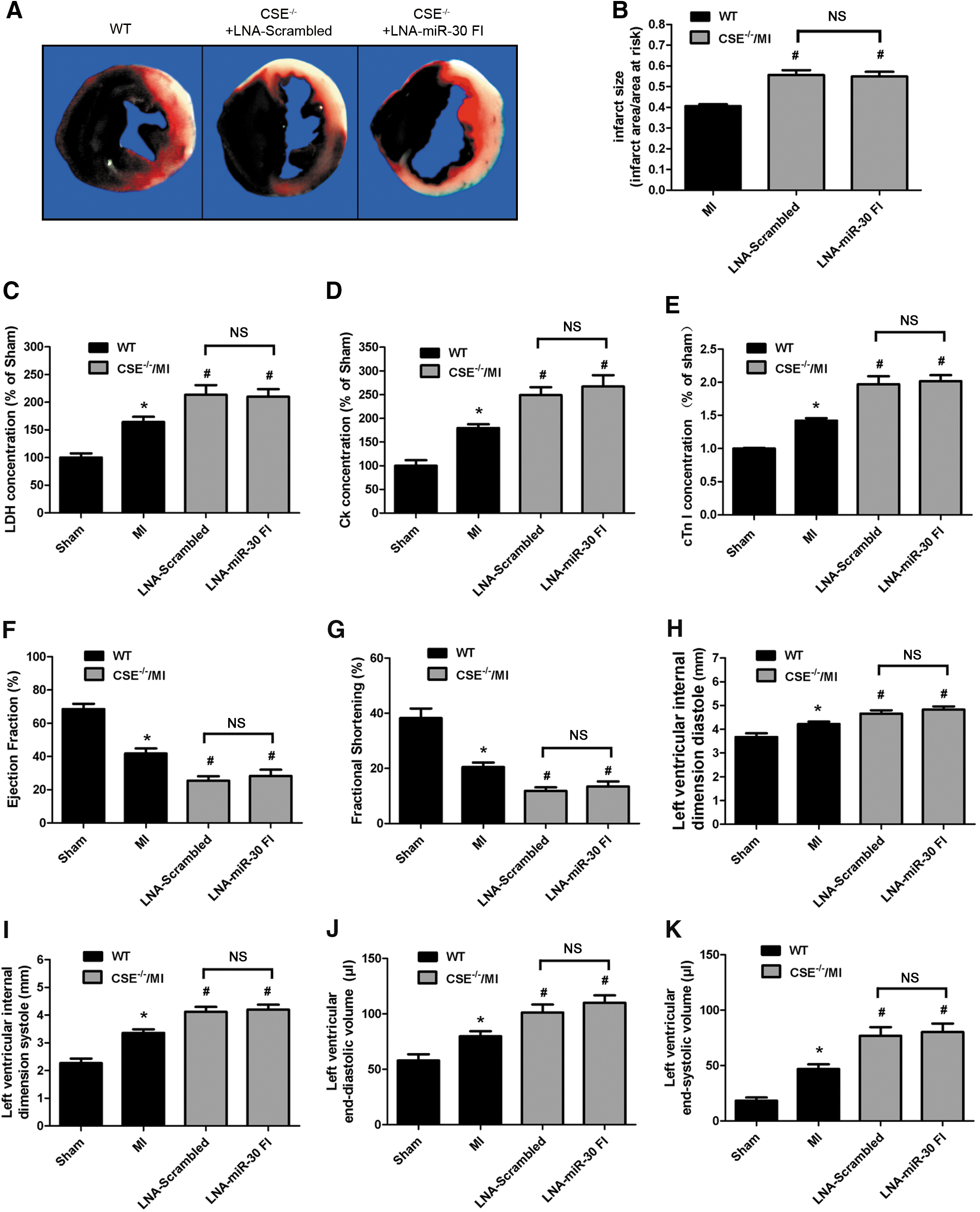
Moreover, LNA-miR-30 FI-treated CSE−/− mice displayed no significant increase in EF, FS as compared with LNA-scrambled treated CSE−/− mice, which displayed significantly decreased EF and FS after MI (Fig. 7F, G). These findings provide direct evidence that LNA-miR-30 FI plays its protective role by regulating the expression of CSE and the production of H2S.
MiR-30b overexpression aggravates MI injury in mice
Since the change of miR-30b expression is the most obvious in both murine MI model and cardiomyocyte hypoxic model (Fig. 1D, E, I), we hypothesize that the increased miR-30b expression levels, occurring in response to ischemic injury, may have the most contribution in the MI model. Thus, to investigate the functional significance of miR-30b in ischemic heart, we tested whether miR-30b overexpression could exacerbate cardiac dysfunction by reducing H2S production. Furthermore, we used S-propargyl-cysteine (SPRC) and Propargylglycine (PAG) to confirm the possible role of CSE-mediated mechanism in the effect of miR-30b on ischemic injury. SPRC is a novel modulator of CSE that has been proved to be able to protect against myocardial ischemia by increasing CSE expression. PAG is an inhibitor that can reduce endogenous H2S production by inhibiting CSE (38, 39).
First, we verified that a single administration of SPRC could increase the expression of CSE and the level of H2S (Supplementary Fig. S4A, B) in plasma, and then protect the cardiac ischemia injury by reducing infarct size (Supplementary Fig. S4C, D) and improving heart function (Supplementary Fig. S5). However, administration of PAG alone could decrease the level of H2S in plasma and aggravate cardiac injury (Supplementary Figs S4 and S5).
Subsequently, we used lentivirus (LV) carrying the precursor of miR-30b to overexpress miR-30b in vivo, at 8 days after infection with 2.5×107 TU lentiviral particles; the expression of miR-30b was significantly increased (Fig. 8A). Correspondingly, CSE expression was decreased at both the mRNA and protein levels (Fig. 8B, C).
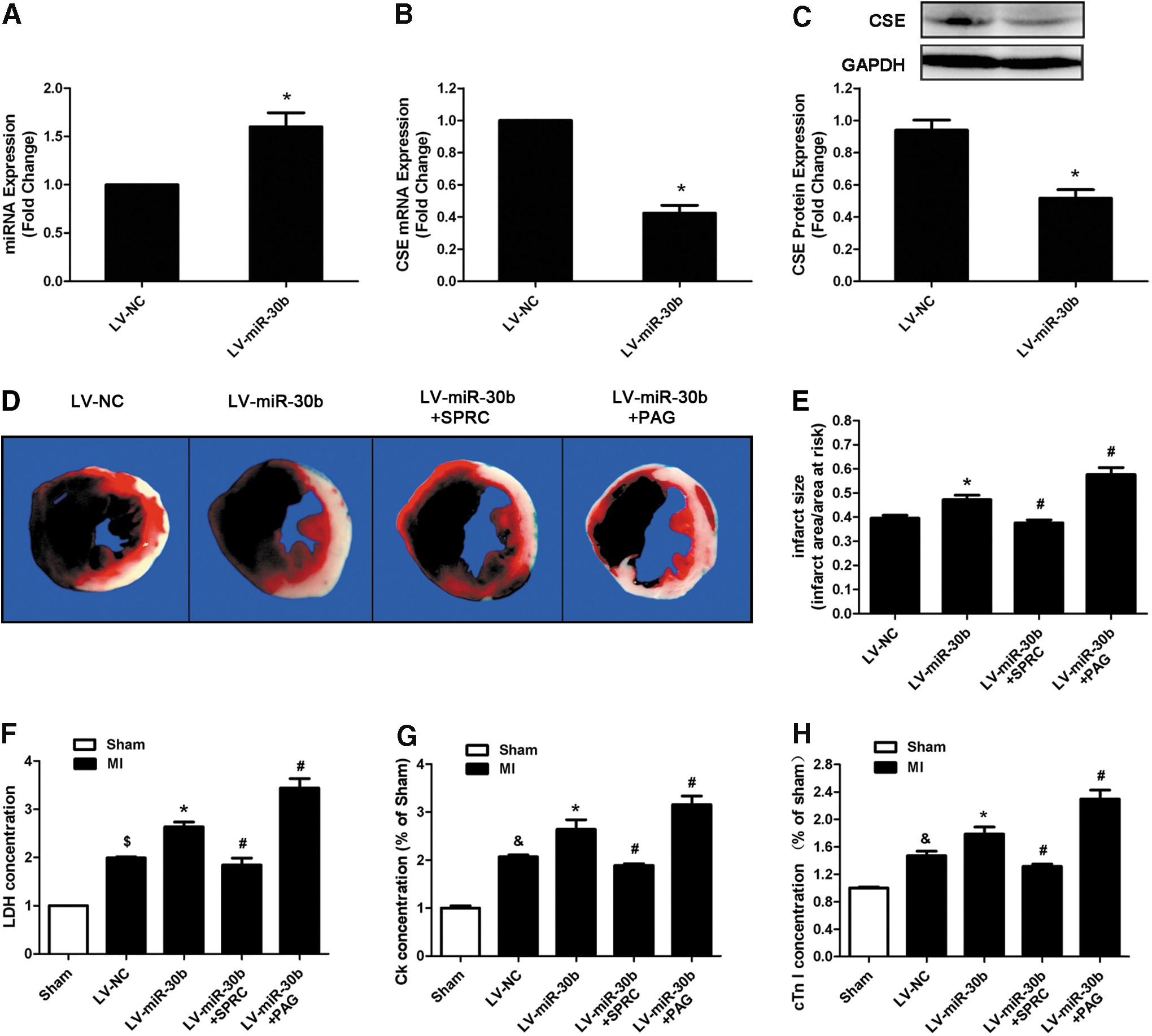
Then, we used MI model combined with SPRC and PAG to investigate the effect and mechanism of LV-miR-30b on ischemia injury. Two days after MI induction, we observed that miR-30b overexpression increased the infarct area (Fig. 8D, E), the LDH, CK, and cTn-I concentrations (Fig. 8F–H), and also increased the apoptotic cardiomyocyte number in the peri-infarct area compared with the LV-NC group after MI (Fig. 9A, B). Treatment of LV-miR-30b plus SPRC abolished the negative effects of LV-miR-30b on augmentation of infarct size (Fig. 8D, E) and apoptotic cardiomyocyte number (Fig. 9A, B). However, treatment of LV-miR-30b plus PAG further increased infarct size (Fig. 8D, E) and apoptotic cell number (Fig. 9A, B). Moreover, miR-30b overexpression destroyed cardiac function (Fig. 9D–I). LV-miR-30b plus SPRC also rescued the cardiac damage caused by a single administration of LV-miR-30b, but LV-miR-30b plus PAG further exacerbated the cardiac damage (Fig. 9D–I). These results corresponded with the H2S concentration in plasma after administration with LV-miR-30b, SPRC, or PAG. As Shown in Figure 9C, LV-miR-30b decreased the H2S level, whereas administration of LV-miR-30b plus SPRC abrogated the negative effect of LV-miR-30b on H2S level. However, LV-miR-30b plus PAG further decreased the H2S concentration in plasma.
Discussion
Taken together, our results identified the miR-30 family as potentially important regulators of CSE gene expression. The overexpression of the miR-30 family members decreased CSE expression and H2S production and subsequently aggravated the hypoxia-induced cardiomyocyte injury in vitro. In contrast, downregulation of the entire miR-30 family increased CSE expression and H2S production and protected hypoxia injury in cardiomyocytes. In the in vivo study, we observed that the downregulation of the miR-30 family reduced the infarct size of the heart, which was subjected to coronary artery ligation, and improved the impaired cardiac function by increasing CSE expression and H2S production. The overexpression of miR-30b would aggravate MI injury because of H2S reduction. Our data demonstrated for the first time that the miR-30 family was able to regulate H2S production by directly targeting CSE. These findings revealed a new molecular control mechanism for endogenous H2S production in the heart at the miRNA level. Furthermore, the therapeutic potential of miR-30 family inhibition may open a new avenue for the treatment of ischemic heart diseases.
MiR-30 family members included miR-30a, −30b, −30c, −30d, and −30e, which are encoded by six genes located on human chromosomes 1, 6, and 8. All of them share a common 8-mer conserved seed sequence at their 5′ terminals, but the flanking sequences between the members are substantially different from each other. The miR-30 family is abundantly expressed in the heart under physiological conditions (19). The levels of redundancy suggest a critical functional role for this miRNA family. As shown in Figure 2C, all five members of the miR-30 family have one 8-bp conserved target site in the 3′-UTR of CSE. It has been reported that binding sites with as few as 7 bp of complementarity (seed sequence) to the 5′-end of miRNA are sufficient for regulation in vivo (2, 9). This strongly led us to believe that the 3′-UTR of CSE could be a target of miR-30 family. In addition, we observed that the miR-30 family was upregulated and the CSE expression was downregulated in the remote and border zone of murine MI model as well as in the cardiomyocyte hypoxic model. This result also implied that CSE may be regulated by the miR-30 family, because miRNA is a negative regulator of gene expression (8). As expected, the notion was verified by the results of luciferase assay, gain-of-function and loss-of-function assay.
CSE, which is a predominant H2S-producing enzyme in the cardiovascular system, is responsible for major H2S production in the heart (22, 25). Thus far, there have been only a few publications that have delineated the relationship between miRNAs and H2S production. Yang et al. (44) have observed that miR-21 represses human SP1 protein expression by targeting the 3′-UTRs of the SP1 gene in SMCs, which, in turn, downregulates CSE mRNA expression and indirectly reduces H2S production; this was confirmed by Cindrova-Davies et al. (7) and Toldo et al. (34). In this work, we provided novel evidence that the miR-30 family was able to regulate H2S production by directly targeting CSE. MiR-30 suppresses CSE expression at both mRNA and protein levels, indicating that miR-30 regulates CSE expression by promoting mRNA degradation. On the other hand, Shen et al. (31) and Liu et al. (21) found that H2S participate in different pathological and physiological processes by regulating miRNA expression. These demonstrate that the regulation between H2S and miRNA is mutual.
A growing body of experimental literature has indicated that increased H2S levels exogenously or endogenously protect against MI injury (17, 28), but that decreased H2S levels produced by inhibiting CSE result in aggravated MI injury (1, 38, 46). In this study, we observed that the overexpression of miR-30 family members reduced CSE expression and H2S production. In contrast, the downregulation of the miR-30 family increased CSE expression and H2S production. These findings predicted that miR-30 may participate in MI injury by regulating H2S levels. To confirm this presumption, we used an in vitro model of hypoxic injury and an in vivo MI model and observed that the overexpression of the miR-30 family aggravated hypoxia-induced injury. However, the downregulation of the miR-30 family by LNA-miR-30 FI protected cardiomyocytes in vitro, but this effect was abolished by cotransfecting with CSE-siRNA, indicating that the effect of miR-30 depends on the regulation of CSE. This pointed out that the additional increase in CSE level caused by LNA-miR-30 FI was required to establish the protective effects on cardiomyocytes during hypoxia. Similarly, the downregulation of the miR-30 family by LNA-miR-30 FI in vivo protected mouse heart against MI injury, but this cardioprotective effect was absent in CSE−/− mice model. This indicated that the protective effect of LNA-miR-30 FI, indeed, depends on CSE. Furthermore, the overexpression of miR-30b exacerbated MI injury, and this role of miR-30b can be rescued by SPRC, which is a novel modulator of CSE, or be further aggravated by PAG, which is a selective inhibitor of CSE. These effects were associated with the changes in H2S concentration in plasma, indicating that miR-30 participates in the protection of ischemic injury by regulating H2S production.
The effect of LNA-miR-30 FI on improvement of cardiac function may be attributable to the anti-oxidant action and anti-apoptotic effect of H2S. We previously reported (39) that a modulator of endogenous hydrogen sulfide-SPRC has cardioprotective effects in MI by reducing the deleterious effects of oxidative stress through the modulation of the endogenous H2S level. Similarly, Geng et al. (17) also proposed that H2S protects the heart from isoproterenol-induced ischemic injury, at least in part by scavenging oxygen-free radicals and attenuating lipid peroxidation. Besides, studies also show that H2S has anti-apoptotic properties and is known to increase Bcl-2 and reduce Bax expression (10, 41). Our results showed that LNA-miR-30 FI increased the activities of SOD and CAT, but decreased the MDA levels. Moreover, inhibition of the miR-30 family reduced apoptotic cell numbers in hypoxia cell model and in the peri-infracted region of MI model. In addition, we observed that miR-30 FI administration in vivo increased Bcl-2 and decreased Bax expression. Taken together, we propose that the effects of miR-30 on cardiac function were a consequence of the increase in H2S production induced by miR-30 FI.
Finally, we have shown that the LNA-modified miR-30 FI is efficient in the potent silencing of miR-30 family members both in vitro and in vivo. This indicated that LNA-modified oligos targeting seed region is a good method to inhibit a whole miRNA family simultaneously. Obad et al. (24) have found that 7- to 8-mer fully modified LNA oligonucleotides which target the miRNA seed sequence can inhibit entire miRNA families that share the same seed sequence. Hullinger et al. (18) also observed that using an 8-mer LNA-anti-miR complementary to the miR-15 family seed region dose dependently represses all miR-15 family members in both murine and porcine cardiac tissue. LNA-modified oligonucleotides targeting seed region have become an increasingly and widely used experimental approach to inhibit miRNAs family simultaneously. A practical problem in administering LNA-miRNA inhibitor systemically is the effect on nontarget tissues, in that most miRNAs are expressed in many different tissue types. This could be remedied by oligonucleotide packaging such as lipid formulations and conjugation with high-affinity molecules using nanotechnology for guidance to the target tissues (35). The ability of the LNA-miR-30 FI in protecting cardiomyocytes against ischemic injury suggests the potential of miR-30 as a target for molecular therapy. Normalization of the miR-30 level to the normal range may well be a novel strategy for protecting cardiac ischemic injury.
In conclusion, our study is the first to identify and validate CSE as a target of the miR-30 family and to demonstrate the role of the miR-30 family in MI injury. We propose that the miR-30–CSE–H2S axis contributes to protection against cardiac ischemic injury. The miR-30 family functions during MI may provide additional insights into the role of miRNAs in these processes and establish miR-30 as potential therapeutic targets for ischemic heart diseases.
Materials and Methods
Animals
Healthy male Sprague–Dawley rats (weight, 200–250 g) and C57BL/6 (8–10 weeks old) mice were used for this experiment. The CSE knockout mice were a gift from Shanghai Research Center for Model Organisms, Shanghai, China. CSE-KO and WT control animals used in this study were littermates obtained via heterozygous breeding. Genotype was determined by a single PCR with forward primer (5′-CCTGGATATAAGCGCCAAAG-3′) and reverse primer (5′-AGGAACCAGGGCGTATCTCT-3′). This reaction yields a 309 bp product from a transgenic allele. The forward primer with another reverse prime (5′-GAGAATTCCATTGCTCAGG-3′) produced a 167 bp product from a WT allele. All animal experiments were performed in accordance with the Animal Management Rules of the local authorities and approved by the Animal Research Ethics Committee of the School of Pharmacy of Fudan University.
MI models
Left descending coronary artery ligation was performed to induce MI model as previously reported (46). Detailed description can be found in the online supplementary materials.
Real-time PCR
The total RNA that was enriched with small RNAs was isolated from cultured cardiomyocytes or heart tissue with a mirVanamiRNA isolation kit (Ambion, Austen, TX) according to the manufacturer's instructions. For the quantitative detection of CSE mRNA, the SYBR Green I (Takara, Dalian, China) incorporation method was used with GAPDH as an internal control. The levels of miR-30 family were detected with the TaqMan MicroRNA Assay Kit (Applied Biosystems, Foster City, CA) with U6 as an internal control. More details can be found in the online supplementary materials.
Cell culture
Primary cardiomyocytes were obtained from the ventricles of neonatal Sprague–Dawley rats (1–2 days old) according to the method described by Wang et al. (38). The isolated primary cardiomyocytes were seeded at a density of 1×106 cells/ml in D/F12 that was supplemented with 10% FBS (Gibco, Grand Island, NY), 100 U/ml penicillin, 100 mg/ml streptomycin, and 100 μM 5-bromodeoxyuridine (Sigma, St. Louis, MO). HEK293 cells were cultured in high-glucose Dulbecco's modified Eagle's medium (Gibco) that was supplemented with 10% fetal bovine serum. All cells were cultured in a humidified incubator at 37°C with 95% O2 and 5% CO2.
Transfection of miRNA mimics or LNA-miRNA Inhibitor
MiR-30a, miR-30b, miR-30c, miR-30d, and miR-30e mimics and the miRNA mimic Negative Control #1 were obtained from Ambion. The LNA-modified miRNA FI and a scrambled NC were obtained from Exiqon (Vedbaek, Denmark). For details, see the online supplementary materials.
Plasmid construction and transfections
The open reading frame (ORF) of CSE (GenBank ID: NM_017074) was amplified and cloned into the expression vector pcDNA3.1 (+) to obtain pcDNA3.1-CSE plasmid, which was transfected into primary cardiomyocytes with Lipofectamine 3000 (Invitrogen, Carlsbad, CA). More details can be found in the online supplementary materials.
Hypoxia cell model
Hypoxia cell model was induced in accordance with the technique described by Rakhit et al. (29). Cardiomyocytes were first placed in a hypoxic solution (NaCl, 116 mM; KCl, 50 mM; CaCl2, 1.8 mM; MgCl2·6H2O, 2 mM; NaHCO3, 26 mM; and NaH2PO4·2H2O, 1 mM) and subsequently incubated in a humidified atmosphere at 37°C in a 3-gas hypoxic chamber that was maintained at 5% CO2, 1% O2, and 94% N2 for 8 h.
Cardiomyocyte survival assays
After exposure to normoxia or hypoxia for 8 h, MTT and TUNEL assay were used to determine cell survival and apopotosis. Detailed description can be found in the online supplementary materials.
LDH, CK, and cTn-I levels
LDH, CK, and cTn-I were detected to evaluate the severity of cardiomyocyte injury. More details can be found in the online supplementary materials.
Antioxidant enzyme analysis
For the determination of antioxidant enzyme activities, 0.1 g left ventricular tissues were homogenized as 1:100 in 50 mM ice-cold potassium phosphate buffers (pH 6.8). MDA, CAT, and SOD were detected by using commercially available kits according to the manufacturer's instructions (Jiancheng Bioengineering Institute, Nanjing, China).
Measurement of H2S concentration
H2S concentration was measured as previously described (38). More details can be found in the online supplementary materials.
Luciferase reporter constructs and luciferase assay
The WT 3′-UTR fragment of CSE that contained the putative binding site for the miR-30 family was obtained by PCR and inserted into a pmiR-RB-REPORT dual luciferase reporter vector (Ribobio, Guangzhou, China). The seed region mutant plasmid was also constructed as a control. Luciferase assays were performed according to the manufacturer's instructions (Promega, Madison, WI). For details, see the online supplementary materials.
Western blot analysis
Total protein samples were extracted from the cultured primary cardiomyocytes and heart tissue for protein immunoblotting as previously described (38). For details, see the online supplementary materials.
Immunofluorescence staining
Forty-eight hours after transfection, cardiomyocytes were fixed with 4% paraformaldehyde (Sigma), which was followed by blocking with 3% BSA (Amresco, Solon, OH). The monoclonal mouse anti-CSE primary antibody (Abcam, Cambridge, United Kingdom) was then added, and the cells were incubated for 1 h at room temperature. Then, the cells were incubated with Alexa Fluor 488-conjugated secondary antibody (Molecular Probe, Grand Island, NY) in the dark for 1 h. For nuclear staining, the cells were stained with 4,6′-diamidino-2-phenylindole (DAPI) for 5 min before examinations for visualization with a laser scanning confocal microscope (Zeiss, Oberkochen, Germany).
Delivery of the miRNA modulator and drugs
The indicated doses of in vivo LNA-miR-30 FI obtained from Exiqon were intravenously injected into C57BL/6 mice (8–10 weeks old) for 3 consecutive days before MI surgery; LNA-scrambled control or a comparable volume of saline was also injected as a control.
GFP-tagged lentivirus particles carrying the mmu-mir-30b precursor and its control were purchased from GeneChem (Shanghai, China). The lentiviral transduction was conducted according to advice from GeneChem. In brief, 2.5×107 TU of lentivirus particles were diluted in 200 μl saline and administered into the tail vein at 8 days before MI surgery. Meanwhile, 20 mg/kg of SPRC or 15 mg/kg PAG (Sigma) was injected intraperitoneally on a daily basis for 8 days in different groups before surgery.
Infarct size determination
Forty-eight hours after the MI surgery, the mice were anesthetized and a median sternotomy was performed. Evans Blue dye (0.3 ml of a 1.0% solution; Sigma) was injected into the heart from the cardiac apex to delineate the ischemic zone from the nonischemic zone. 0.1% triphenyltetrazolium chloride (TTC, w/v) solution was used to demarcate the viable and nonviable myocardium within the AAR. More details can be found in online supplementary materials.
Echocardiography
Five days after the MI surgery, two-dimensional echocardiography was performed on mice with a Vevo770 ultrasound machine (VisualSonics, Inc., Toronto, Canada) that was equipped with a 10-MHz phased-array transducer to test left ventricular function. A detailed description can be found in online supplementary materials.
Statistical analysis
All data analyses were performed using GraphPad Prism 5 software. One-way analysis of variance was used to examine the statistical comparisons among multiple groups. Two-tailed Student's t-tests were performed to examine the statistical significance of the differences between two groups. Differences were considered significant with p<0.05.
Footnotes
Acknowledgments
This work was supported in part by the National Nature Science Foundation of China (No.: 81402919), the National Basic Research Program of China (973 Program, No.: 2010CB912600), the National Science and Technology Major Project (No.: 2012ZX09501001-003), the key program of National Nature Science Foundation of China (No.: 81330080), and the Key Program of Shanghai Committee of Science and Technology in China (No. 10431900100).
Author Disclosure Statement
No competing financial interest exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.