
Abstract
Introduction
A
Nuclear hormone receptors are a family of transcription factors involved in diverse physiological functions. Several members of this superfamily are expressed in the cardiovascular system, pivotally regulating cardiovascular function (8, 12, 52, 54). The vitamin D receptor (VDR), also known as NR1I1 (nuclear receptor subfamily 1, group I, member 1), is a member of the nuclear receptor superfamily (7). Upon activation by natural (i.e., vitamin D) or synthetic VDR agonists, VDR forms a heterodimer with the retinoid-X-receptor and binds to VDR response elements on DNA, resulting in expression or transrepression of target genes. In addition to its classic function in the regulation of skeletogenesis and mineral homeostasis through increasing the intestinal absorption of calcium, VDR has also been reported to regulate a variety of other metabolic pathways, such as those involved in the kidney disease, immune response, and cancer (7). Moreover, recent evidence has demonstrated the presence of VDR in the vasculature and heart, with important roles in maintaining the optimal function of cardiovascular system (10, 13, 44, 58, 60). In cardiomyocytes, although the importance of VDR in attenuating cardiac hypertrophy and preventing heart failure after cardiac remodeling has been reported (3, 4, 6, 9, 11), little information is available regarding the roles of VDR in acute stress in myocardium. Most importantly, the roles of VDR in the pathophysiology of acute myocardial ischemia/reperfusion (MI/R) injury have not been investigated.
To the best of our knowledge, this study investigated for the first time the roles of vitamin D receptor (VDR) in the pathophysiology of acute ischemia/reperfusion-induced myocardial injury. We have found that endogenous VDR was upregulated after acute myocardial ischemia/reperfusion (MI/R), and that VDR acted as a novel endogenous self-defensive and cardioprotective receptor against MI/R injury. We have further demonstrated that VDR activation reduced oxidative stress through metallothionein-dependent mechanism, and inhibited apoptosis and autophagy dysfunction-mediated cell death, providing new insights into the cardioprotective mechanisms and pleiotropic functions of VDR in the myocardium.
Therefore, the aims of this study were to investigate whether VDR activation protects against or mediates MI/R-induced myocardial injury and clarify the effects of VDR on cardiomyocyte apoptotic and autophagic pathways in the pathophysiology of MI/R injury.
Results
VDR is expressed in mouse heart tissue and upregulated by MI/R
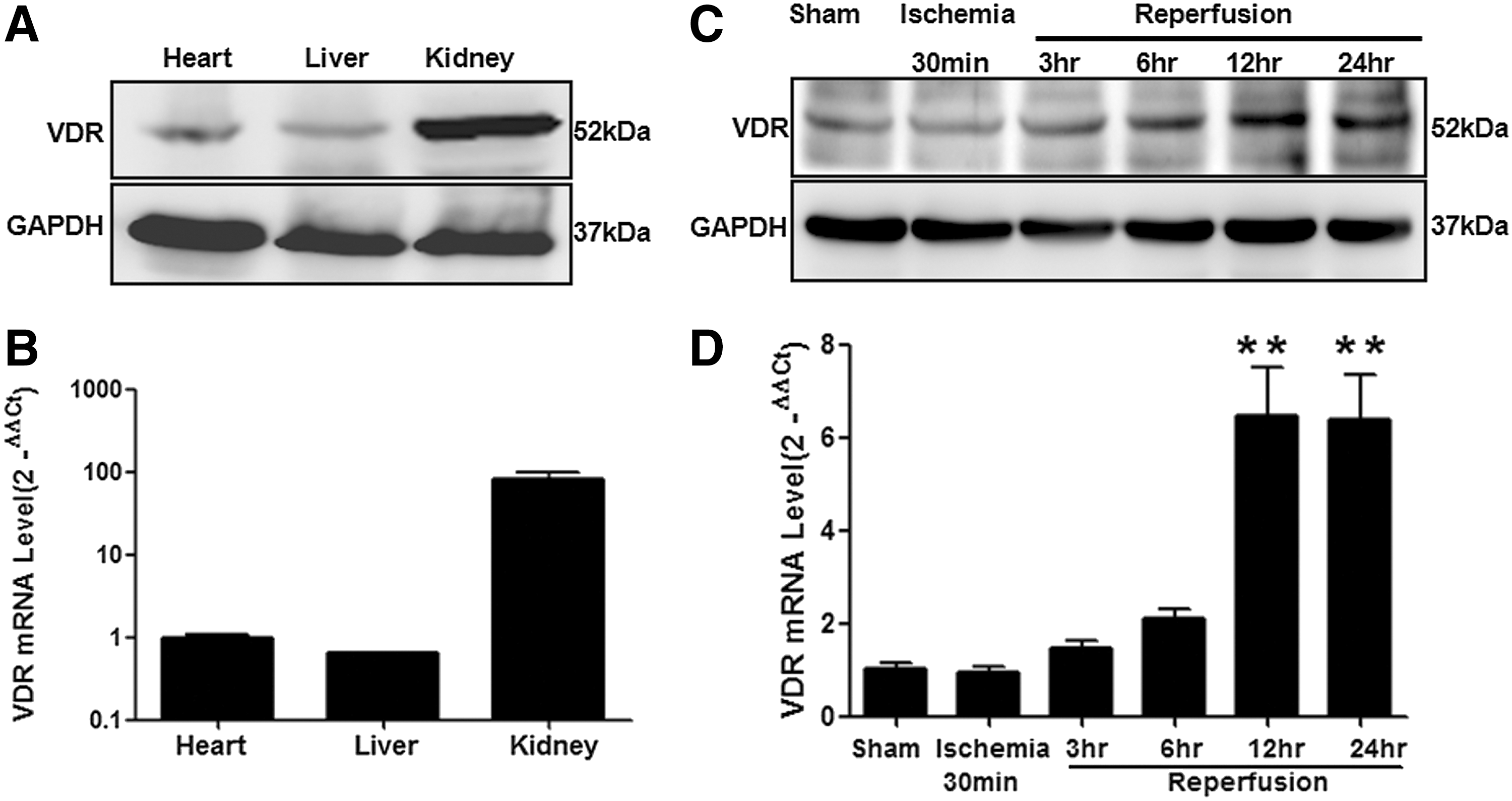
To confirm the presence of VDR in cardiac tissue, we detected endogenous VDR expression in the heart samples obtained from left ventricular (LV) segments of adult mice using both Western blot (Fig. 1A) and real-time quantitative polymerase chain reaction (RT q-PCR) analyses (Fig. 1B). VDR was expressed in mouse heart tissue at a similar level to the liver but at a relatively lower level to the kidney. Importantly, a significant increase in VDR expression was observed in cardiac tissue obtained from ischemic/reperfused myocardium (Fig. 1C, D). Time-course studies of VDR expression suggested that VDR levels were not significantly changed during ischemia, but markedly increased in the ischemic area at risk (AAR) after reperfusion (Fig. 1C, D).
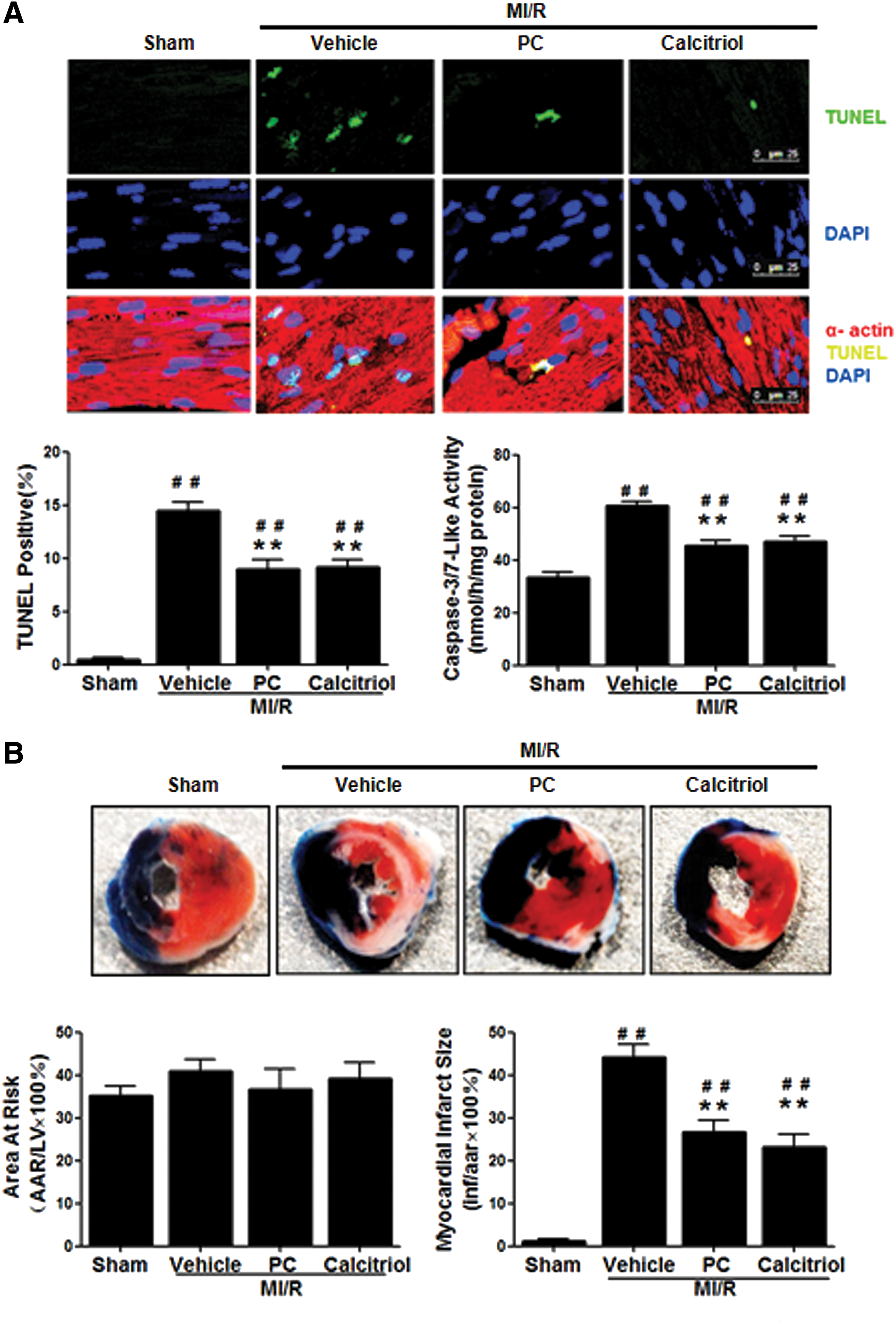
Activation of VDR reduces MI/R-induced myocardial apoptosis, infarct size, and cardiac dysfunction
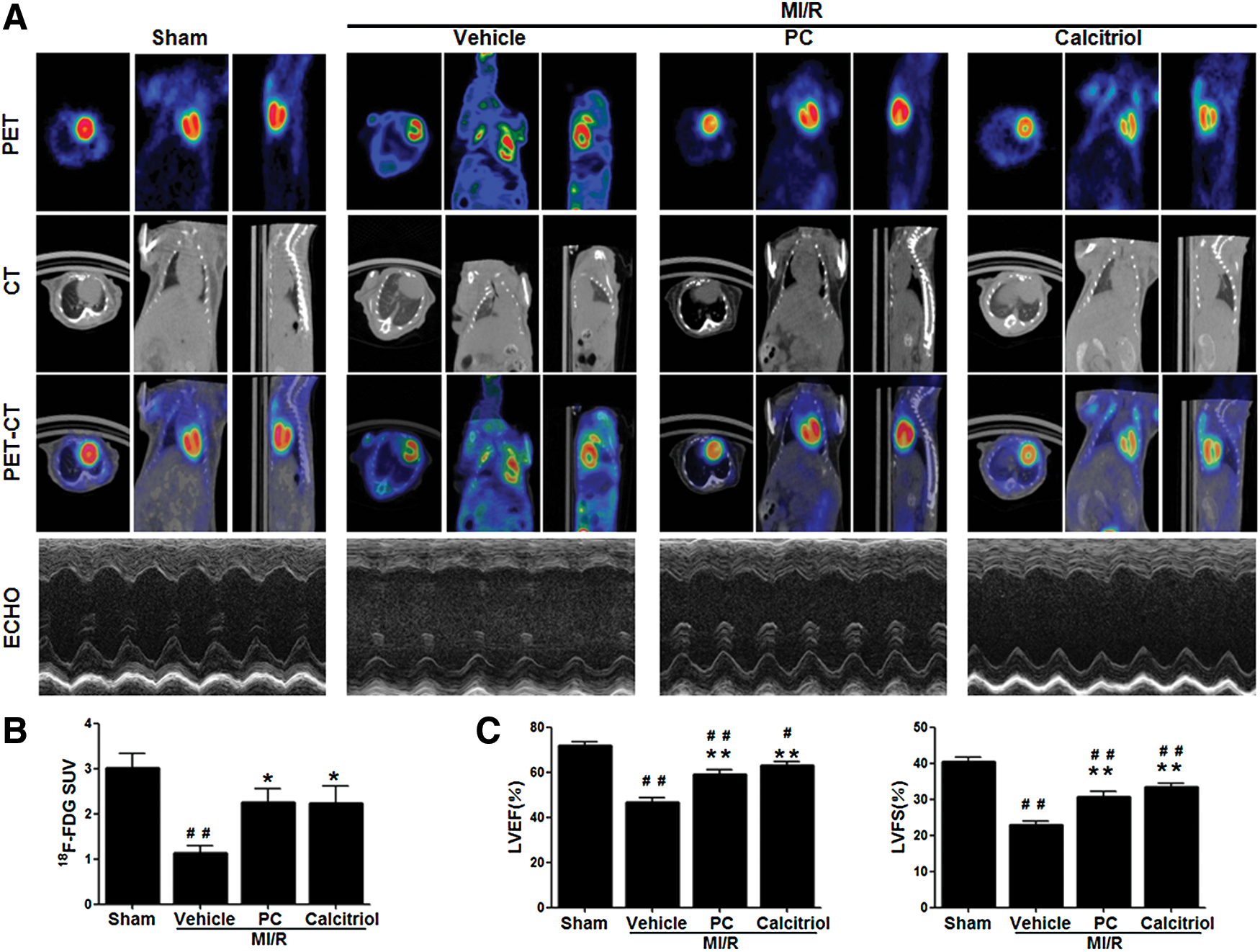
To determine whether post MI/R upregulation of VDR mediates myocardial reperfusion injury or acts as a self-defensive pro-survival signal, mice were treated with vehicle, calcitriol (a natural VDR agonist), or paricalcitol (PC, a synthetic VDR agonist), and MI/R injury determinants were assessed. Treatment with VDR agonists had no significant effects on heart rate and mean arterial blood pressure (Supplementary Fig. S1; Supplementary Data are available online at
Activation of VDR inhibits MI/R-induced endoplasmic reticulum stress and mitochondrial dysfunction
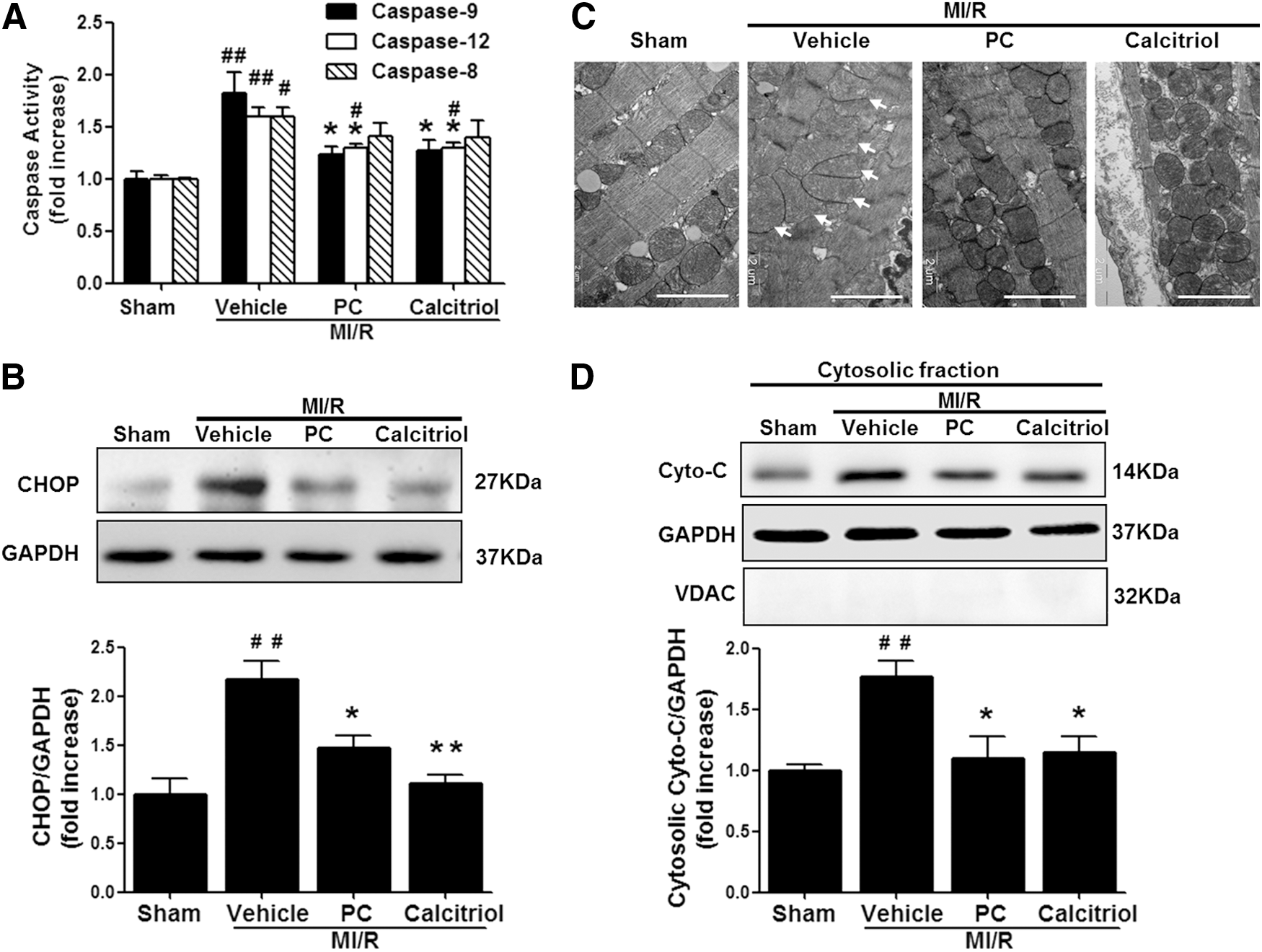
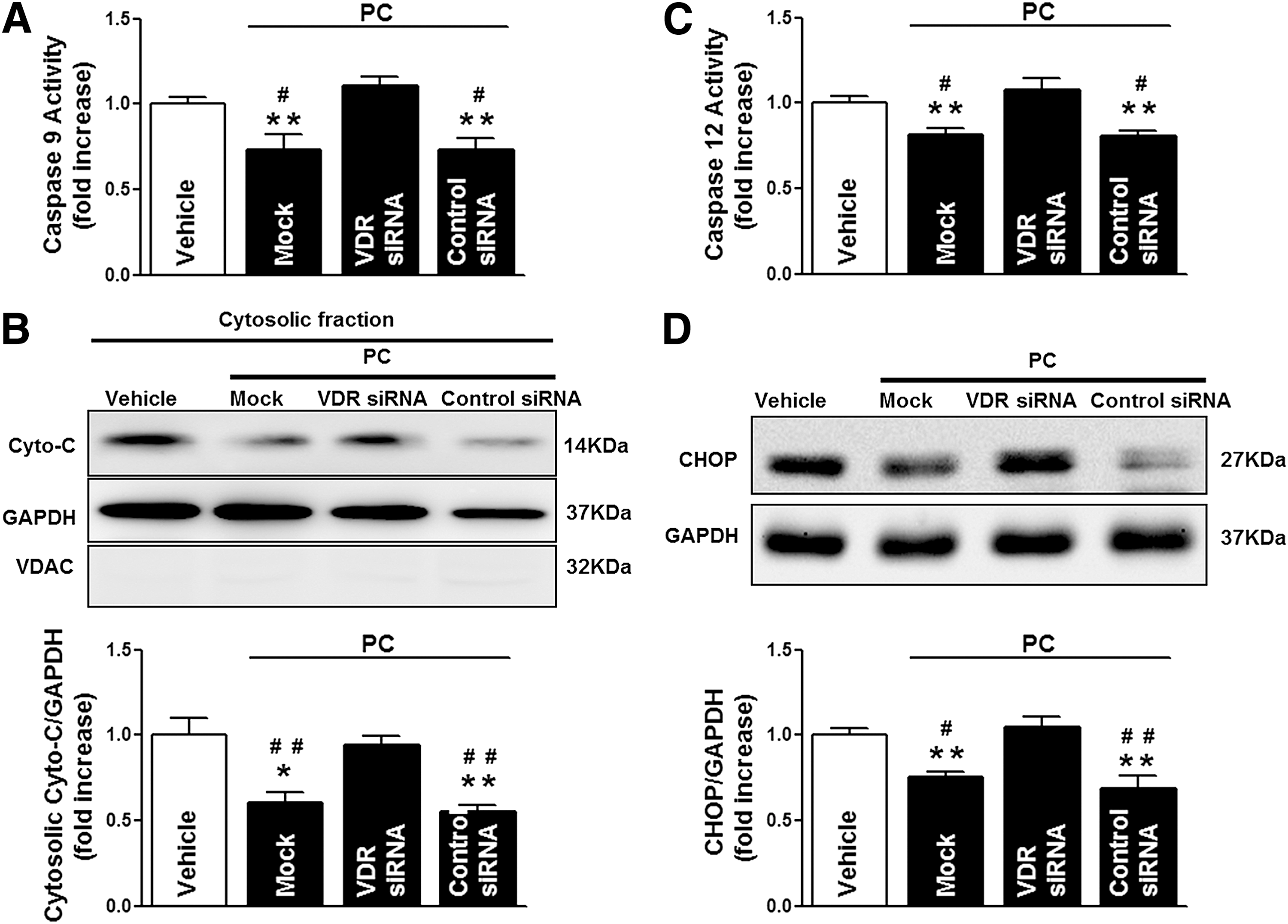
To examine the potential mechanisms behind VDR activation-elicited cardioprotection against apoptosis and MI/R injury, the effects of VDR activation on caspase-12 (an endoplasmic reticulum [ER] stress-activated caspase), caspase-9 (a mitochondrial pathway-activated caspase), and caspase-8 (an initiator caspase of the extrinsic death receptor pathway) activities were analyzed. MI/R activated all three caspases, while VDR agonists significantly reduced MI/R-induced caspase-12 and caspase-9 activities without altering caspase-8 activity (Fig. 4A). Moreover, MI/R stimulated CCAAT/enhancer-binding protein homologous protein (CHOP) overexpression and induced mitochondria swollen and cytochrome c (Cyto-C) release to the cytoplasm, all of which were significantly inhibited by VDR agonist administration (Fig. 4B–D). Overall, these results suggest that VDR activation reduces MI/R-induced ER stress and mitochondrial dysfunction.
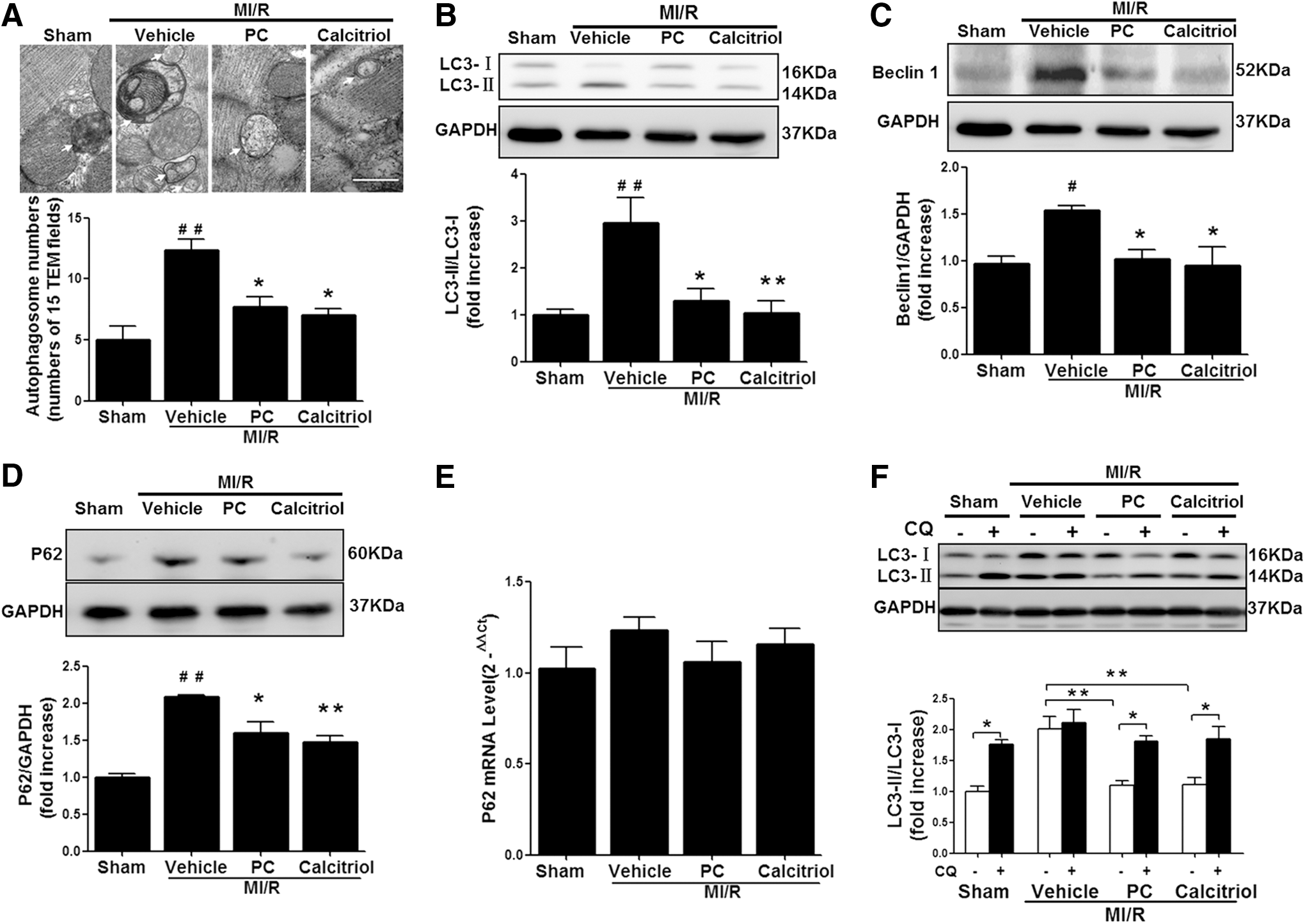
Activation of VDR inhibits MI/R-induced autophagy dysfunction
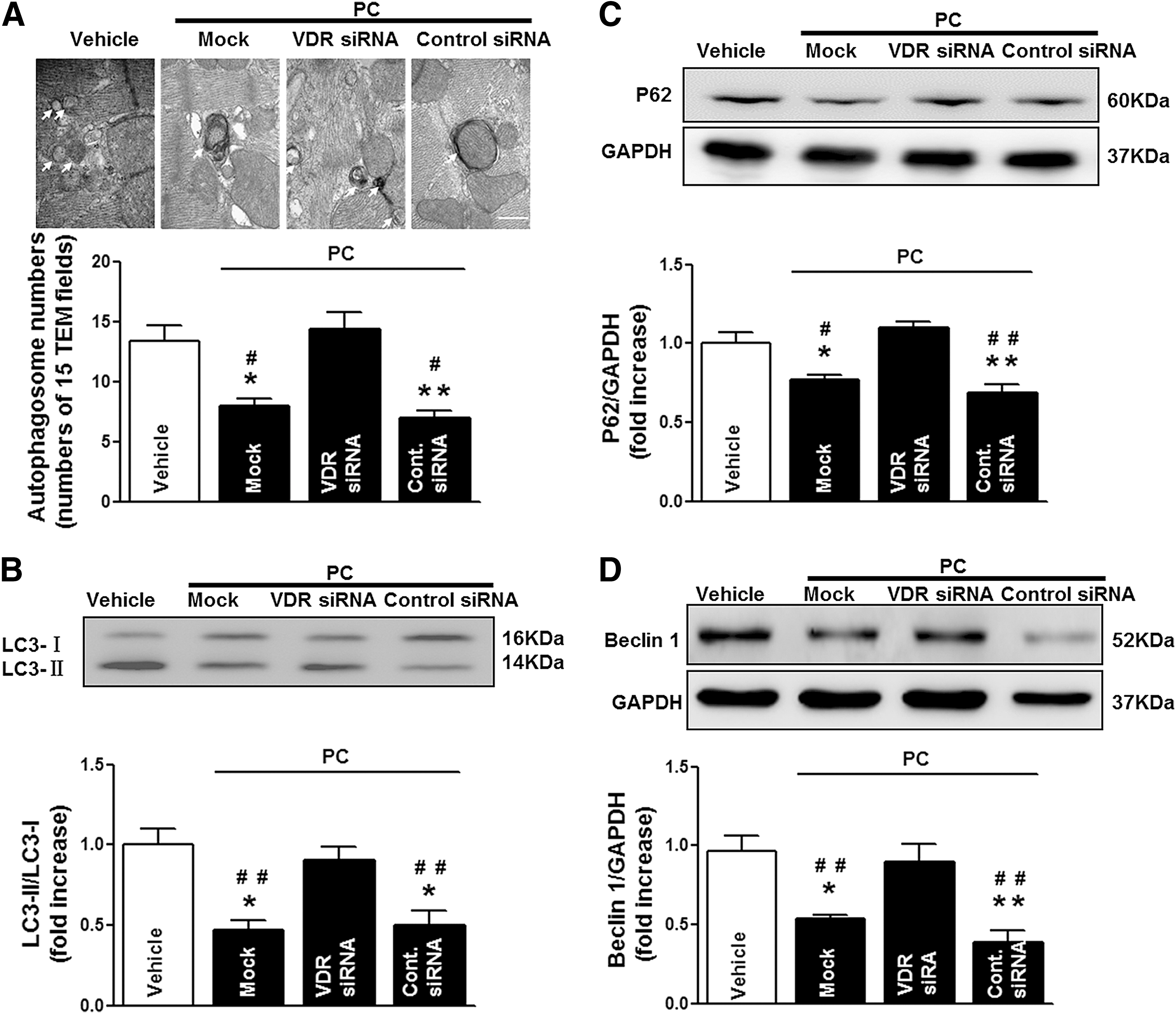
In addition to apoptosis, increasing lines of evidence suggest that autophagy dysfunction is involved in nonapoptotic cell death by MI/R (43). Therefore, we further investigated the effects of VDR activation on autophagy in the ischemic/reperfused myocardium. As shown in Figure 5A, transmission electron microscopy (TEM) revealed a large number of autophagosomes in the vehicle-administrated mouse heart, while calcitriol or PC treatment reduced autophagosome abundance. To further confirm that the activation of VDR was involved in the regulation of autophagic activity, we measured the conversion of the soluble form of microtubule-associated protein light chain 3 (LC3-I) to the lipidated and autophagosome-associated form (LC3-II) and the activation of Beclin 1, an autophagy-related protein with an essential role in the reperfusion-mediated autophagy (35 –37). As shown in Figure 5B and C, the ratio of LC3-II/LC3-I and the expression of Beclin 1 were dramatically increased in the ischemic/reperfused heart, indicating stimulated autophagy and increased autophagosome abundance. Interestingly, the MI/R-induced increase of LC3-II/LC3-I ratio was not accompanied by the decrease of p62, a specific autophagic substrate protein and the hallmark representing autophagic flux (Fig. 5D, E). In addition, preventing autophagosome-lysosome fusion with chloroquine (CQ) did not induce additional increase of LC3-II/LC3-I ratio after MI/R in vehicle-treated mice (Fig. 5F). These results suggested that MI/R induced the dysfunction of autophagy and the impairment of autophagosome clearance (35, 36). Compared with vehicle, calcitriol or PC treatment significantly reduced the ratio of LC3-II/LC3-I and the expression of Beclin 1 accompanied by the decrease of p62 protein (Fig. 5B–E). However, pretreatment with CQ significantly increased the LC3-II/LC3-I ratio in VDR-agonist-treated mice (Fig. 5F), indicating preserved “intact” autophagy flux by VDR activation. Given the distinct role of autophagy in the different phase of MI/R (33, 36, 37), we further investigated the effect of VDR agonists on autophagy at 30 min after ischemia and at 3 or 24 h after reperfusion, respectively. As shown in Supplementary Figure S2, autophagy flux was stimulated by 30 min of ischemia, but impaired by 30 min of ischemia followed by either 3 or 24 h of reperfusion. Compared with vehicle, VDR agonist administration did not alter the autophagic activity and autophagy flux at 30 min after ischemia, but restored the impaired autophagy flux at both 3 and 24 h after reperfusion. Taken together, these results suggest that VDR activation inhibits autophagy dysfunction-mediated cell death through restoration of autophagy flux.
VDR activation reduces MI/R-induced oxidative stress
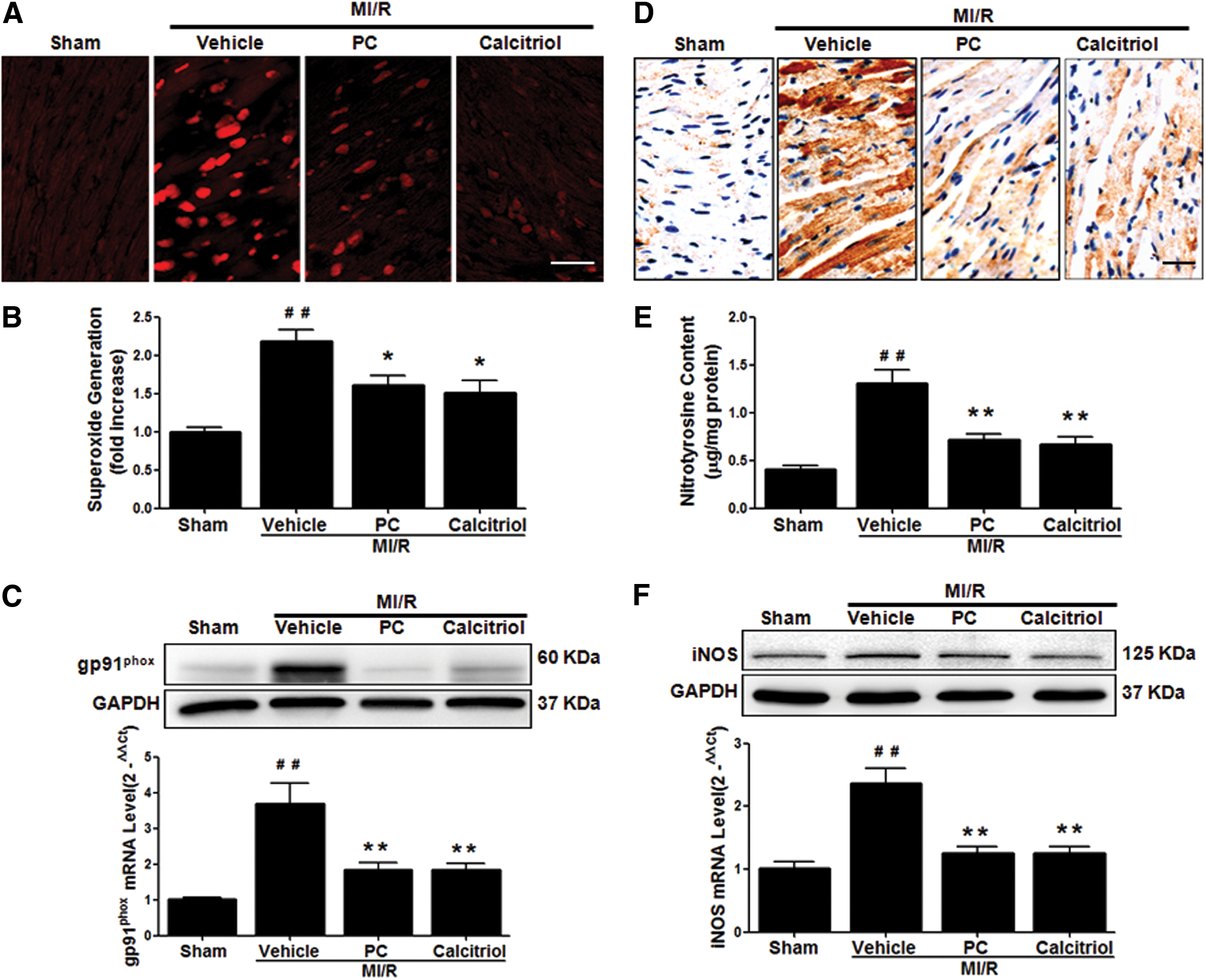
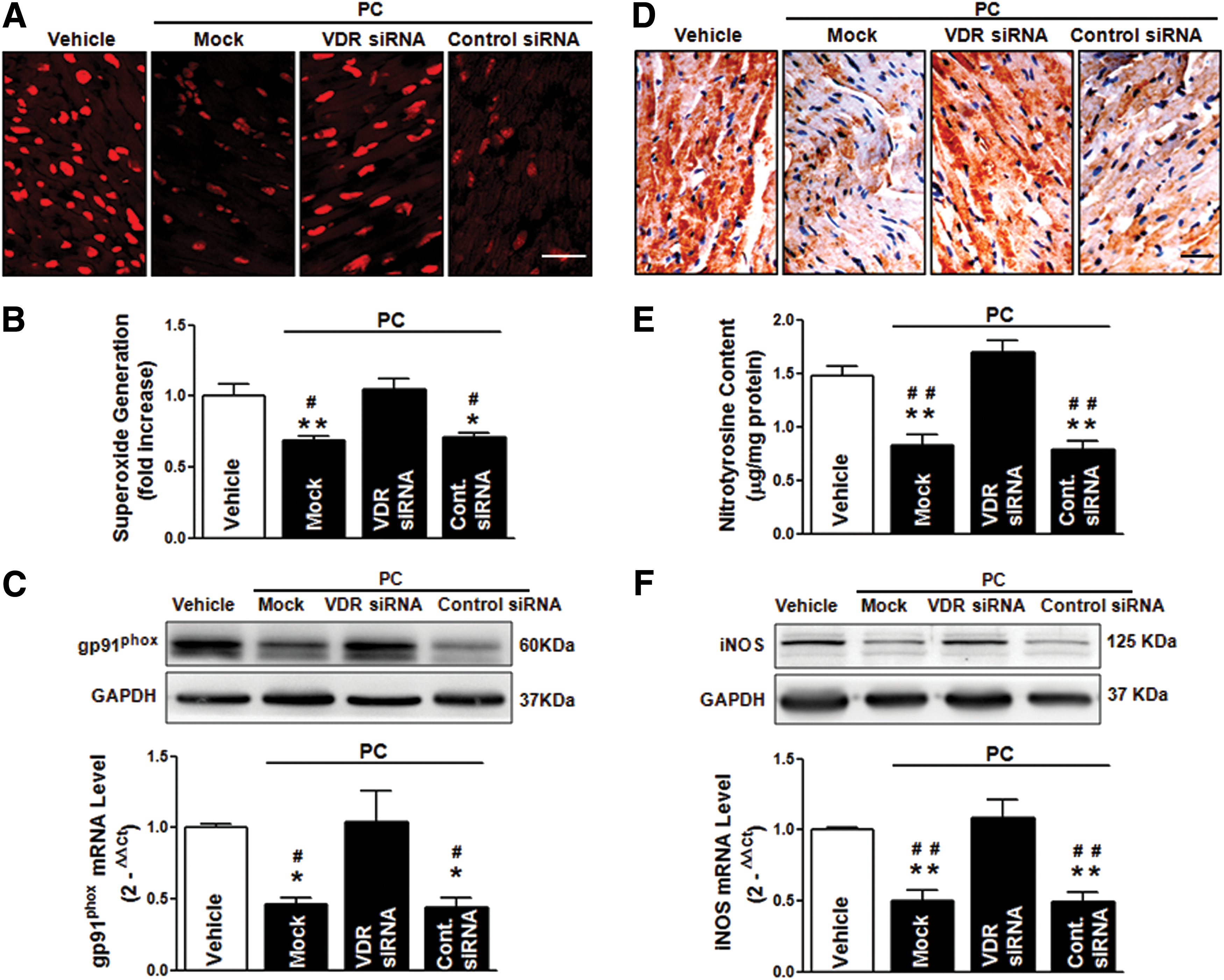
To further determine the molecular mechanisms underlying the protective actions of VDR agonists against apoptosis and autophagy dysfunction, we assessed the roles of VDR in regulating oxidative stress, a crucial upstream mediator of apoptosis and autophagy. Both calcitriol and PC treatment significantly reduced MI/R-induced ROS steady-state levels (Fig. 6A), superoxide generation (Fig. 6B), and gp91phox NADPH oxidase subunit overexpression (Fig. 6C). Moreover, VDR agonists significantly reduced tissue nitrotyrosine content (Fig. 6D–E), and inhibited inducible nitric oxide synthase (iNOS) expression (Fig. 6F). Collectively, these results demonstrate that the cardioprotection of VDR activation during MI/R involves the reduction of oxidative stress.
Cardioprotective effects of VDR agonists are dependent on the specific VDR signaling
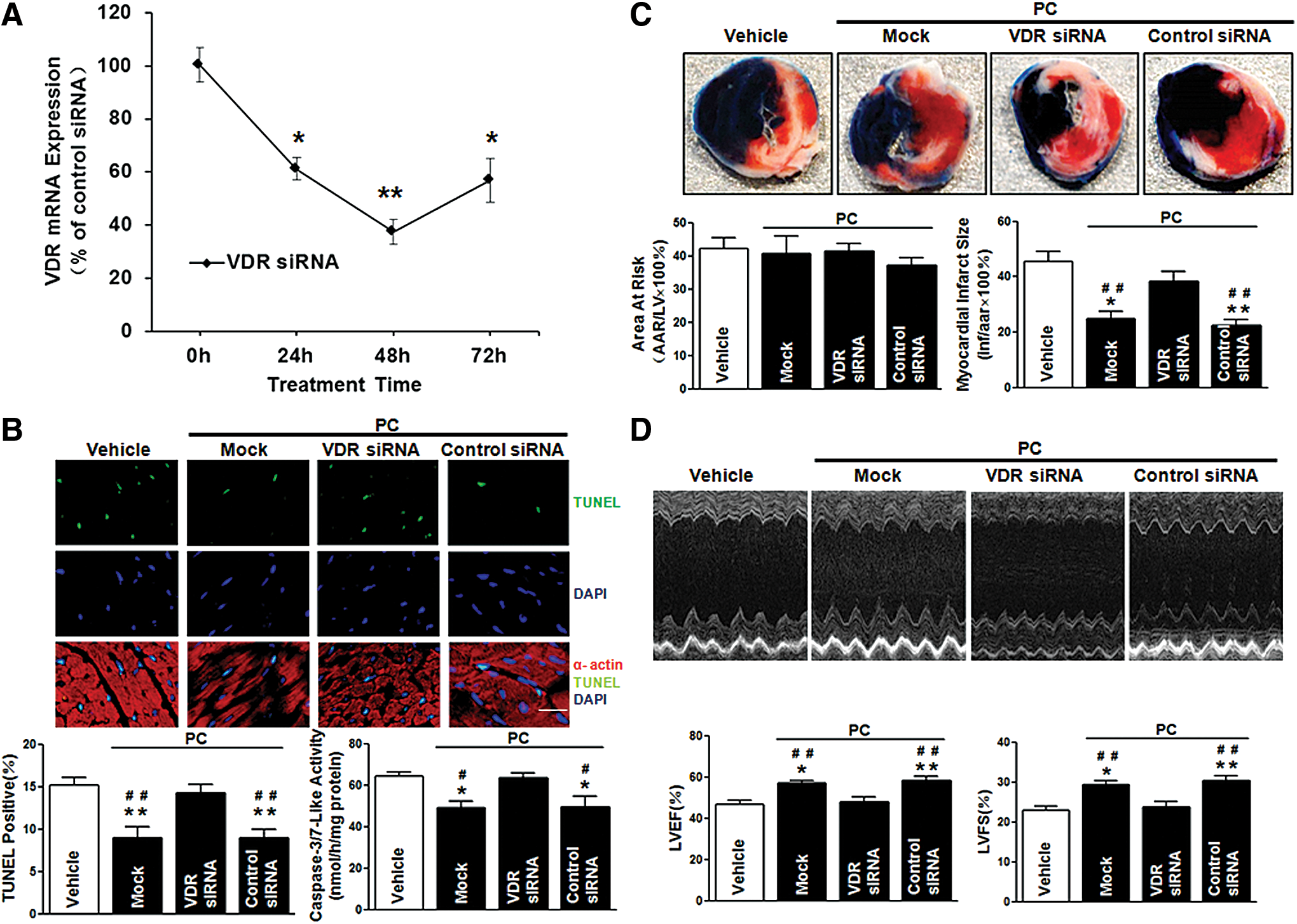
To further confirm the VDR-dependent protective role of VDR agonists in MI/R injury, we utilized in vivo siRNA gene silencing technique through intramyocardial delivery with either VDR siRNA or control siRNA, as previously described (52). Our pilot experiments showed that cardiac VDR expression reached nadir (∼40% of the control levels) at 48 h after siRNA injection (Fig. 7A). Thus, the MI/R protocol was performed at 48 h after intramyocardial siRNA delivery. No difference was observed in the AAR among all groups, indicating that the experimental ischemia/reperfusion-induced myocardial injury was comparable. Notably, VDR agonist administration in control siRNA-treated mice remained effective in reducing MI/R-induced myocardial apoptosis, decreasing infarct size, and enhancing LVEF and LVFS. However, these cardioprotective effects of VDR agonists were abrogated when endogenous cardiac VDR was knocked down (Fig. 7B–D). Mechanistically, VDR knockdown (KD) abrogated the beneficial effects of VDR agonists on preserving the function of mitochondria and ER (Fig. 8A–D), restoring autophagy flux (Fig. 9A–D), and inhibiting oxidative stress (Fig. 10A–F). Collectively, these results demonstrate that the cardioprotective effects of VDR agonists are dependent on the specific VDR signaling.
Adenovirus-mediated cardiac VDR overexpression protects heart against MI/R injury
To obtain more direct evidence supporting VDR activation being cardioprotective against MI/R injury, cardiac-specific gene overexpression of VDR by in vivo intramyocardial adenovirus-encoded VDR (Adv-VDR) transfection was performed before MI/R. Intramyocardial Adv-VDR transfection led to a ∼2.5-fold increase in VDR expression (Supplementary Fig. S3A). Compared with vehicle or adenovirus control (Adv-EGFP), Adv-VDR significantly reduced the number of apoptotic nuclei (TUNEL-positive nuclei) and caspase-3/7-like activity (Supplementary Fig. S3B), decreased infarct size (Supplementary Fig. S3C), and improved echocardiographic measurements of LVEF and LVFS (Supplementary Fig. S3D). Adv-EGFP did not alter MI/R-induced caspase-9 and caspase-12 activation, while Adv-VDR significantly reduced MI/R-induced caspase-9 and caspase-12 activation (Supplementary Fig. S4A, B). In addition, compared with vehicle or Adv-EGFP control, Adv-VDR reduced autophagosome abundance and the LC3-II/LC3-I ratio (Supplementary Fig. S4C, D). Furthermore, Adv-VDR inhibited MI/R-induced gp91phox expression and resultant superoxide generation, and attenuated MI/R-stimulated iNOS expression and tissue nitrotyrosine production (Supplementary Fig. S5A–D). Collectively, these data suggest that VDR is a pro-survival protein, and genetic augmentation of VDR contributes to the cardioprotection against MI/R injury.
Activation of VDR protects cardiomyocytes against hypoxia-reoxygenation injury in vitro
Having demonstrated that activation of VDR protected heart against MI/R injury in vivo, we further examined whether VDR agonists exerted these beneficial effects directly on cardiomyocytes. As illustrated in Supplementary Figure S6A and B, we detected the expression of VDR in both neonatal rat ventricular myocytes (NRVMs) and neonatal rat cardiac fibroblasts (NRCFs). Hypoxia insult alone did not significantly affect VDR expression in myocytes; however, the levels of VDR increased significantly after 3–6 h of reoxygenation (Supplementary Fig. S6A). Pretreatment of NRVMs with VDR agonists reduced hypoxia/reoxygenation (H/R)-induced ROS, attenuated caspase-3/7-like activity, restored the H/R-impaired autophagy flux, and inhibited lactate dehydrogenase (LDH) release (Supplementary Fig. S7A–F). Interestingly, when NRCFs obtained from the same cultures were subjected to the same H/R protocol, we did not observe any significant changes of VDR expression in cardiac fibroblasts (Supplementary Fig. S6B). Thus, although both myocytes and fibroblasts express VDR, only myocytes exhibit a significant response to H/R with an increase in VDR (Supplementary Fig. S6A, B). In the H/R experiments performed on a co-culture of NRVMs/NRCFs (5: 3) separated by cell culture transwells, VDR activation reduced the H/R-induced LDH release and caspase-3/7-like activity in the Mock or control siRNA group. However, these beneficial effects of VDR activation were abrogated when the expression of VDR in NRVMs was selectively knocked down before co-culture (Supplementary Fig. S6C–E). These results suggest that the cardioprotection of VDR activation is mainly dependent on cardiomyocyte-expressed VDR.
Mechanism of VDR-mediated cardioprotection during MI/R-essential role of metallothionein-dependent anti-oxidative action
Recently, it has been suggested that metallothionein (MT) and glucose-6-phosphate dehydrogenase (G6PD) were the direct target genes of VDR, and might be involved in the anti-oxidative and cytoprotective effects of VDR (5, 17, 56). Therefore, we hypothesized that VDR-mediated anti-oxidative and cardioprotective effects may depend on MT and/or G6PD. To test this hypothesis, we first pretreated NRVMs with VDR agonists for 24 h, then detected the expression of MT, G6PD, and other cardioprotective antioxidant enzymes such as catalase (CAT), thioredoxin (Trx), superoxide dismutase 1 (SOD1), glutathione peroxidase 1 (GPx1), and peroxiredoxin 6 (Prx6). Pretreatment of NRVMs with VDR agonists upregulated MT expression without altering G6PD expression (Supplementary Fig. S8A–D). In addition, VDR activation did not alter the expression of CAT, Trx, SOD1, GPx1, and Prx6 in NRVMs (Supplementary Fig. S8A). Moreover, by utilizing in vivo gain- and loss-of-function approaches, we observed that enhanced activation of VDR via adv-mediated gene overexpression upregulated MT expression; while VDR-knockdown (KD) downregulated MT expression in mouse heart tissue. In contrast, the expression of G6PD was not altered in both gain- and loss-of-function experiments (Supplementary Fig. S9A–C). Moreover, in vivo KD of MT abrogated not only the anti-oxidative effect but also the anti-apoptotic and autophagy-modulating effects of VDR activation (Supplementary Fig. S10A–D). Importantly, the cardioprotection of VDR activation was lost in the MT-KD mice (Supplementary Fig. S10E). Taken together, these results suggest that MT-dependent anti-oxidative mechanism is crucial for the VDR activation-elicited cardioprotection.
Discussion
In this work, we provided new insights into understanding the novel roles for VDR in the heart. The novel contributions include the following: First, we demonstrated that the endogenous VDR was expressed in adult cardiac tissue and markedly upregulated by MI/R. Second, we proved that the activation of VDR significantly reduced infarct size and improved cardiac function in the setting of MI/R injury. Third, mechanistic studies demonstrated that VDR activation inhibited apoptosis and autophagy dysfunction-mediated cell death, and reduced oxidative stress via MT-dependent mechanism. Finally, utilizing in vivo siRNA gene silencing and adenovirus vector-mediated gene overexpression, we demonstrated that cardiac-specific VDR signaling mediated the cardioprotection against MI/R injury. Taken together, we have obtained the first direct evidence that VDR acts as a novel endogenous cardioprotective receptor against MI/R injury.
VDR, a unique member of the nuclear receptor superfamily, classically functions as a mineral metabolism sensor with well-recognized roles in regulating calcium and phosphorus uptake and transport (7). It is most highly expressed not only in small intestine, colon, kidney, bone, and skin but also in other tissues and cell types such as the endocrine organs, immune system, brain, and muscle (7). Importantly, a growing body of evidence suggests that VDR is also expressed in cardiovascular system and plays an essential role in the regulation of cardiovascular physiology/pathology. In vessel system, it has been reported that VDR activation sustained proper endothelial cell function (13), suppressed vascular smooth muscle cell proliferation (10), and inhibited cholesterol sequestration in macrophages collected from patients with diabetes mellitus (44). In myocardium, VDR activation attenuated cardiac hypertrophy, and prevented heart failure after cardiac remodeling (3, 4, 6, 9, 11). However, little information is available regarding the roles of VDR in acute stress in myocardium (e.g., acute MI/R injury). To the best of our knowledge, our data demonstrated for the first time that the endogenous VDR levels were markedly increased after acute MI/R stress. Enhanced activation of VDR by pharmacological agonists dramatically decreased apoptosis and infarct size, and improved echocardiographic LV function and Micro-PET/CT18F-FDG uptake. Furthermore, siRNA-mediated silencing of endogenous VDR blocked the cardioprotective effects of VDR agonists, demonstrating a direct receptor-dependent mechanism. Most importantly, adenovirus-mediated cardiac-specific VDR overexpression significantly mitigated MI/R injury, further supporting that VDR acted as a self-defensive protein to overcome the acute pathological stress in MI/R.
Apoptosis, the type I programmed cell death, is the major form of cell death after a short period of ischemia followed by reperfusion (40). Our study demonstrated that VDR activation downregulated the marker of ER stress (caspase-12 activation and CHOP expression) and mitochondrial stress (caspase-9 activation and Cyto-C release) without altering caspase-8 activity (the extrinsic apoptotic marker). These observations suggested that VDR activation achieved anti-apoptotic action mainly through inhibition of ER stress and attenuation of mitochondrial dysfunction. The involvement of VDR in regulating apoptosis has also been reported in several cell and tissue types. Interestingly, VDR activation was shown to be anti-apoptotic in several cell types and pro-apoptotic in others. For example, VDR activation inhibited apoptosis in keratinocytes, islet cells, and tubular cells, but induced apoptosis in a number of tumor models, such as carcinomas of the breast, colon, and prostate (7, 47). Hence, the role of VDR in inhibiting or promoting apoptosis may depend on the cell and tissue types (7).
Autophagy, an important intracellular bulk degradation process of lysosome-dependent turnover of damaged cytosolic proteins and organelles, is critical for maintenance of normal cell phenotype and function (41, 50). However, the autophagic machinery might also be used for self-destruction and contribute to cell death when pathological stress induced autophagy dysfunction (27, 36, 43). Recently, autophagy has been reported as a novel regulatory target to limit MI/R injury (33, 35 –37). In this study, we observed that MI/R dramatically induced autophagosome formation (as evidenced by the upregulation of Beclin 1 and the increase of the LC3-II/LC3-I ratio), and impaired autophagosome clearance (characterized by the accumulation of p62 autophagic substrate protein and the absence of CQ-induced additional increase of LC3-II/LC3-I ratio), suggesting that MI/R induced autophagy dysfunction with autophagosome accumulation. These findings are in line with the recent report that MI/R impaired autophagic flux with reduced clearance of autophagosomes (35, 36). We further showed that VDR activation inhibited the MI/R-induced autophagy dysfunction, as evidenced by the reduction of Beclin 1 expression, the decrease of the LC3-II/LC3-I ratio and p62 protein abundance, and the restoration of autophagy flux. This study provided a novel cardioprotective mechanism for VDR by decrease of autophagy dysfunction-mediated cell death in the setting of MI/R.
Oxidative stress has been recognized as a major mediator of MI/R injury, and is causatively related to ER stress, mitochondrial impairment, and autophagy dysfunction (19, 24, 39, 48). Thus, we further examined the effects of VDR activation on oxidative stress. We showed that VDR activation attenuated oxidative stress in both in vivo MI/R and in vitro H/R experiments. Although the anti-oxidative action of VDR has been increasingly recognized in various types of cells (13, 49, 53, 55), little is known on the molecular mechanism by which VDR decreases oxidative stress. Recently, it has been suggested that G6PD and MT were the direct target genes of VDR and might be involved in the anti-oxidative and cytoprotective effects of VDR (5, 17). Thus, we further examined the role of G6PD and MT in the VDR activation-elicited cardioprotection. We demonstrated that VDR activation upregulated the expression of MT (but not G6PD) in cultured NRVMs in vitro and in mouse heart tissue in vivo. Furthermore, MT-KD abrogated not only the anti-oxidative effect but also the anti-apoptotic and autophagy-modulating effects of VDR activation, suggesting that MT-dependent anti-oxidative mechanism was crucial for the VDR activation-elicited cardioprotection.
The current experimental findings are scientifically and clinically important for the following reasons. First, the identification of VDR as a novel endogenous cardioprotective factor against MI/R injury, in conjunction with the recent findings of other investigators regarding the role of VDR in cardiovascular tissues (3, 4, 6, 9 –11, 13, 44), broadens our understanding of the multiple biological functions of VDR beyond being a mineral metabolism sensor. Interestingly, recent clinical studies have demonstrated that functional genetic variation in VDR was related to ischemic heart disease risk (45), and the inhibition of VDR signaling due to vitamin D deficiency was associated with incident major adverse cardiac events after AMI (42). Second, the novel regulatory roles that VDR plays in oxidative stress and apoptotic/autophagic pathway in cardiomyocytes suggest that pharmacologic manipulation of VDR activity may beneficially treat cardiac dysfunction related to dysregulation of oxidative stress or apoptosis/autophagy. Our observations provide new support for VDR as an important regulator and direct target of cardiovascular biology.
Several members of the nuclear hormone receptor superfamily have been reported to be involved in MI/R injury pathophysiology. The peroxisome proliferator-activated receptors (PPARα, β, and γ) (8, 18, 26), liver X receptors (22, 29), thyroid hormone receptor (46), estrogen receptors (30), androgen receptors (57), and glucocorticoid receptor (25) have been proposed as the endogenous cardioprotective receptors against MI/R injury, while farnesoid-X-receptor (52), mineralocorticoid receptors (32, 54), and Nur77 (12) exacerbate MI/R-induced myocardial injury. Our study adds novel evidence that VDR acts as an endogenous cardioprotective nuclear receptor against MI/R injury. Given the distinct regulatory roles of nuclear receptors in MI/R injury, it is conceivable that potential regulatory cross-talk among VDR and other nuclear receptors might maintain the delicate homeostatic balance between cellular death and survival in the heart.
In conclusion, VDR is a novel endogenous self-defensive and cardioprotective receptor against MI/R injury, via mechanisms (at least in part) reducing oxidative stress, and inhibiting apoptosis and autophagy dysfunction-mediated cell death. VDR, therefore, represents a potentially attractive molecular target for the treatment of ischemic heart diseases.
Materials and Methods
Reagents
Calcitriol, CQ, 5-bromo-2′-deoxyuridine (BrdU), 2,3,5-triphenyltetrazolium chloride (TTC), and 4′-6-diamidino-2-phenylindole (DAPI) were obtained from Sigma-Aldrich. PC was purchased from ALP Pharm Beijing Co., Ltd. Dihydroethidium (DHE) and TRIzol Reagent were from Life Technologies. In vivo-jet Polyethylenimine (PEI) was obtained from Polyplus (Polyplus-transfection SA).
Animals and experimental protocols
Adult male C57BL/6 mice (22–26 g) were obtained from Shanghai Jiao Tong University and housed at 25°C±5°C under a 12-h light/dark cycle. All animal experiments were carried out according to the National Institutes of Health Guidelines on the Use of Laboratory Animals, and was approved by the Institute's Animal Ethics Committee of Shanghai Jiao Tong University. The mice were randomly assigned to the following groups: sham, vehicle, calcitriol, and PC. Calcitriol or PC was dissolved in a 95% propylene glycol and 5% ethyl alcohol solution and was administered through an intraperitoneal injection at 1 μg/kg 15 min before reperfusion. The vehicle group received vehicle injections using the same schedule. The pharmacological dosages were chosen based on previous reports (4, 6, 15). The surgical procedures were performed as described in previous studies (16, 52). Briefly, the heart was manually exposed without intubation through a small incision, and a slipknot was made around the left anterior descending coronary artery at 2–3 mm from its origin using a 6-0 silk suture. The sham-operated animals were subjected to the same surgical procedure, but the ligation remained untied. After 30 min of ischemia, the slipknot was released, and the myocardium was reperfused for the indicated time. Mice that fully recovered from the surgical procedure were returned to standard animal housing conditions.
Hemodynamic measurements
Blood pressure and heart rate in conscious mice were measured by a tail-cuff system (BP-2010; Softron), as previously described (22, 23). Before study initiation, the mice were adapted to the apparatus for at least 5 days. Multiple measurements generated an average value for each mice.
In vivo siRNA-mediated cardiac-specific gene silencing
Stealth siRNAs targeting mouse VDR (Cat. No. MSS238646, MSS238647, and MSS278682), ambion in vivo siRNAs targeting mouse MT (Cat. No. s201712, s70097, and s70098), and control nonspecific siRNA oligos (Negative Control siRNA) were obtained from Life Technologies. To knock down cardiac-specific VDR or MT expression, in vivo siRNA gene silencing through intramyocardial delivery was used as previously described (52). Briefly, mouse-specific siRNAs were complexed with in vivo jet-PEI delivery reagent in 5% glucose according to the manufacturer's instructions. The mice were anesthetized with 2% isoflurane, and the heart was exposed via a left thoracotomy at the fifth intercostal space. Mouse-specific siRNAs (20 μl; 1 μg/g) were delivered via three separate intramyocardial injections (32.5-gauge needle) to temporarily blanch the LV free wall.
In vivo adenovirus-mediated cardiac-specific gene overexpression
Adenoviral shuttle vector pDC316-mCMV-EGFP was obtained from Biowit Technologies, and adenoviral backbone plasmid pBHGloxdeltaE13Cre was obtained from Microbix biosystems, Inc. The mouse cDNA for VDR was cloned into the adenoviral shuttle vector to generate pDC316-mCMV-VDR-EGFP vector. VDR adenoviruses were generated following the instructions of AdMax™ Adenoviral Vector Creation System, and resulting viral titers were determined using the Adeno-X-Rapid Titer-Kit (BD Biosciences Clontech) (31, 34). The control virus (Adv-EGFP) carried the coding sequence for EGFP with an empty coding sequence for VDR. The mice were anesthetized with 2% isoflurane, and the heart was exposed via a left thoracotomy at the fifth intercostal space. Adenovirus (4×109IFU/ml) was administered by a direct injection in the left ventricular free wall (three sites, 10 μl/site, 32.5-gauge needle). Myocardial target gene expression was analyzed 4 days after virus injection as previously described (2).
Tissue harvest and determination of myocardial infarct size
The mice were anesthetized using 2% isoflurane gas. The chest was opened, and the heart was removed and immediately placed in ice-cold phosphate-buffered saline. Only hearts demonstrating infarctions involving the anterior and apical regions were snap frozen and stored in liquid nitrogen (N2) for biochemical assays. In some cases, whole transverse sections were fixed with 4% formaldehyde polymerization solution or placed in an optimal cutting temperature compound for immunohistochemistry. The myocardial infarct size was determined using Evans blue-TTC double staining methods as previously described (52). Briefly, at the end of a 24-h reperfusion period, the ligature around the coronary artery was re-tied and 0.2 ml 2% Evans blue dye was injected into the left ventricular cavity. The dye was circulated and uniformly distributed, except in the area of the heart previously perfused through coronary artery occlusion in the AAR. The heart was quickly excised, frozen at −70°C, and sliced into 1-mm sections perpendicular to the long axis of the heart. The slices were incubated separately using a 24-well culture plate in 1% TTC solution, pH 7.4, at 37°C for 10 min, and photographed using a digital camera. The Evan's blue-stained blue area (area not at risk, ANAR), TTC-stained red area, and TTC-negative staining white area (infarcted myocardium) were measured using the computer-based image analyzer SigmaScan Pro 5.0 (Systat Software). The myocardial infarct size was expressed as a percentage of the infarct area (Inf) over the AAR (Inf/AAR), and the size of the AAR was expressed as the percentage of the AAR over the total LV area (AAR/AAR+ANAR) as previously described (52).
Determination of myocardial apoptosis
Myocardial apoptosis was determined by TUNEL technique via a Fluorescein In Situ Cell Death Detection Kit (Roche Diagnostics) as previously described (52). Apoptotic nuclei were detected by green fluorescein staining, total cardiomyocyte nuclei were DAPI labeled, and cardiomyocytes were identified by anti-α-actin antibody. For overexpression procedure, kit for immunohistochemical detection and quantification of apoptosis (Roche Diagnostics) was used and apoptotic nuclei were detected by brown staining. Cardiac caspase-3/7-like activity was measured via Colorimetric Assay Kit (Millipore). Briefly, 200 μg of total protein from tissues was loaded to each well of the 96-well plate and incubated with 25 μg Ac-DEVD-pNA at 37°C for 120 min. The pNA absorbance was quantified by a microplate reader (BioTek) at 405 nm. Cardiac activation of caspase-8, caspase-9, and caspase-12 was evaluated utilizing respective caspase Fluorometric Assay Kits (BioVision). The procedure was performed as per the manufacturer's instructions. Briefly, 200 μg of total protein from tissues per assay and a final concentration of 50 μM of AFC-conjugated with substrates specific for caspase-8, −9, and −12 (IEDT, LEHD, and ATAD), respectively, was loaded. Samples were read by a fluorimeter equipped with a 400-nm excitation and a 505-nm emission filter. Caspase-8, −9, and −12 activities were calculated against the mean value of caspase activities from corresponding control.
Western blot analysis
The proteins from the ischemic/reperfused heart tissues were prepared using standard protocols, and the protein concentrations in the lysates were determined using the Pierce BCA Protein Assay Kit (Thermo Scientific). For quantification of Cyto-C release, cytosolic and mitochondrial fractions were separated as previously described (14). Equal quantities of proteins (30–50 μg/lane) were subjected to 8%–12% SDS-PAGE, depending on the target proteins, electrotransferred onto nitrocellulose membranes, and incubated with primary antibodies against VDR (Santa Cruz Biotechnology; 1:200), CHOP [Cell Signaling Technology(CST); 1:1000], Cyto-C (CST, 1:1000), VDAC (CST, 1:1000), gp91phox (Santa Cruz; 1:200), iNOS (Millipore; 1:2000), LC3 (CST, 1:1000), p62 (CST, 1:1000), Beclin 1 (CST, 1:1000), G6PD (CST, 1:1000), MT (Santa Cruz; 1:200), and GAPDH (CST, 1:1000). After incubation with the corresponding secondary antibodies, the protein bands were detected using enhanced chemiluminescence (Millipore), and the quantitation was performed using Quantity One 4.4.0 software (Bio-Rad).
Real-time quantitative PCR
Total RNA was isolated from tissues or cells using TRIzol Reagent (Invitrogen) and purified using the RNeasy Total RNA Isolation Kit (Qiagen). Real-time quantitative PCR was performed using the ABI 7500 Real-Time PCR System (Applied Biosystems) and the SYBR®Premix Ex Taq™ Perfect Real-Time Kit. For the mouse gene expression, the following SYBR Green real-time PCR primers were used: VDR, forward 5′-GTGGACATTGGCATGATGAAGG-3′ and reverse 5′-CATCGAGCAGGATGGCGATA-3′; gp91phox, forward 5′-TGATCCTGCTGCCAGTGTGTC-3′ and reverse 5′-GTGAGGTTCCTGTCCAGTTGTCTTC-3′; Nos2, forward 5′- CAAGCTGAACTTGAGCGAGGA-3′ and reverse 5′-TTTACTCAGTGCCAGAAGCTGGA-3′; p62, forward 5′-GATGTGGAACATGGAGGGAAGAG-3′ and reverse 5′- AGTCATCGTCTCCTCCTGAGCA-3′; GAPDH, forward 5′- TGTGTCCGTCGTGGATCTGA-3′ and reverse 5′- TTGCTGTTGAAGTCGCAGGAG-3′. For the rat gene expression, the following SYBR Green real-time PCR primers were used: VDR, forward 5′-TGACCCCACCTACGCTGACT-3′ and reverse 5′-CCTTGGAGAATAGCTCCCTGTACT-3′; CAT, forward 5′-ATCAGGGATGCCATGTTGTT-3′ and reverse 5′-GGGTCCTTCAGGTGAGTTTG-3′; SOD1, forward 5′-TCACTTCGAGCAGAAGGCAA-3′ and reverse 5′-TTGTTTCTCGTGGACCACCA-3′; GPx1, forward 5′-CGACATCGAACCCGATATAGA-3′ and reverse 5′-ATGCCTTAGGGGTTGCTAGG-3′; Trx, forward 5′-TCCAATGTGGTGTTCCTTGA-3′ and reverse 5′-ACCAGAGAACTCCCCAACCT-3′; Prx6, forward 5′-CACATCCGCTTCCACGATTTCCTA-3′ and reverse 5′-CCAGGCAAAATGGTCCTCAACAGA-3′; MT, forward 5′-TGGACCCCAACTGCTCCTG-3′ and reverse 5′-TCAGGCGCAGCAGCTGCAC-3′; G6PD, forward 5′-GCCTTCTACCCGAAGACACCTT-3′ and reverse 5′-CTGTTTGCGGATGTCATCCA-3′; GAPDH, forward 5′-TCCATGACAACTTTGGCATC-3′ and reverse 5′-CATGTCAGATCCACCACGGA-3′.
Measurement of ROS generation
DHE was used to detect ROS steady-state levels within the myocardium in-situ or NRVMs (38). Unfixed frozen cross-sections (5 μm) were incubated with DHE (5 μM) at 37°C for 30 min in a humidified chamber protected from light, followed by 5 min of washing in PBS to remove the nonintercalated EB molecules. The images were obtained with a Leica laser scanning confocal microscope (Leica TCS SP5 II). Superoxide production in ischemic/reperfused heart tissues or hypoxic/reoxygenated NRVMs was measured by lucigenin-enhanced chemiluminescence as previously described (59). Superoxide production was expressed as relative light units (RLU) per second per mg heart weight (RLU·mg−1·s−1) and was calculated against the mean value of RLU from corresponding control.
Determination of nitrotyrosine content in cardiac tissue
Paraffin-embedded slices were stained with primary antibody against nitrotyrosine (Upstate, Millipore; 1:100). The immunostaining was performed using the Vectastain ABC kit (Vector Laboratories; 1:200), and the slides were analyzed under light microscopy. The quantification of the nitrotyrosine content in cardiac tissue was determined using the Nitrotyrosine ELISA Kit (Abnova). The results are presented as microgram of nitrotyrosine per milligram of protein.
Transmission electron microscopy
Hearts demonstrating infarctions were fixed with 2% glutaraldehyde for 2 h, then postfixed in 1% OsO4 for 2 h, and embedded as monolayers in LX-112 (Ladd Research). Sections were stained in uranyl acetate and lead citrate, and observed with an electron microscope (Philips CM-120; Philips Electronic Instruments). Random sections were taken by an electron microscopy technician blinded to the treatments.
Echocardiographic measurements
We assessed the cardiac function after 24 h of reperfusion using an echocardiographic imaging system (Vevo 770; VisualSonic). The mice were anesthetized using 1.5% isoflurane, and two-dimensional echocardiographic views of the mid-ventricular short axis were obtained at the papillary muscle tips below the mitral valve. The LV wall thickness and internal dimensions were measured, and the LVFS and LVEF were calculated.
18F-FDG micro-PET/CT scanning and analysis
Micro-PET/CT scanning for detection of viable myocardium was performed as previously described (22, 28). After 24 h of reperfusion, the animals were anesthetized with 2% isoflurane in O2 gas for the 18F-FDG injection [a single injection of 0.1 ml FDG with an activity of 10 MBq intravenously in the tail vein as previously described (22, 28)]. Subsequently, the mice were immediately awakened and placed back in the anesthesia cage. Two hours after the administration of the tracer injection, the animals were anesthetized with isoflurane, placed prone on the PET scanner bed near the central field of view, and maintained under continuous anesthesia for the duration of the study. The Inveon Acquisition Workplace (IAW) was used for scanning. Ten-minute static PET scans were acquired, and the images were reconstructed using a three-Dimensional Ordered Subsets Expectation Maximum (OSEM3D) algorithm, followed by Maximization/Maximum a Posteriori (MAP) or FastMAP through IAW. The 3D regions of interest (ROIs) were drawn over the heart based on the CT images, and the tracer uptake was measured using the Inveon Research Workplace software 3.0. The individual quantification of 18F-FDG uptake was calculated in each mouse. The mean SUV were determined by dividing the relevant ROI concentration by the ratio of the injected activity to the body weight.
Cell culture and in vitro experimental protocols
Primary cultures of NRVMs were prepared as previously described (52). Briefly, cells were isolated from hearts of 1–2-day-old Sprague–Dawley rats by trypsin digestion, purified by differential preplating, and maintained in DMEM with 10% (v/v) fetal bovine serum in a humidified incubator (37°C; 95% air+5% CO2). To reduce the background of nonmyocytes, BrdU (100 μM) was added during the first 48 h. NRCFs were isolated by enzyme digestion as described earlier for myocytes, with removal of myocytes by selective attachment of nonmyocytes to tissue culture dishes during the preplating procedure. NRCFs were grown in DMEM supplemented with 10% (v/v) fetal bovine serum, and used for experiments within three passages.
In all experiments, cells were rendered quiescent by serum starvation for 24 h before treatment. For VDR agonism experiments, cultures were treated with calcitriol (20 μM) or PC (20 μM) for 24 h. To dissolve the VDR agonists, ethanol (EtOH, final concentration <0.1%) was present in the buffer of all groups. The pharmacological dosages were chosen based on previous reports (3). For H/R experiments, the hypoxic condition was created by incubating the cells in an anaerobic chamber equilibrated with 95% N2 plus 5% CO2 at 37°C for 6 h. The cells were then reoxygenated under normoxic conditions in a 95% air/5% CO2 humidified atmosphere at 37°C for the indicated time. Normoxic control cells were incubated at 37°C in a 95% air/5% CO2 only. Co-culture of NRVMs/NRCFs (5: 3) separated by cell culture transwells (transwell, 0.3 μm pore size, Falcon, Becton Dickinson) was performed as described in a previous study (1). NRCFs were seeded in cell culture transwells and positioned above NRVMs culture dishes at 24 h before H/R, thereby allowing a paracrine interaction with NRVMs during H/R. In some experiments, the expression of VDR in NRVMs was selectively knocked down using VDR siRNA at 48 h before applying the H/R protocol.
Measurement of LDH activity
LDH activity in the culture supernatant was measured by LDH cytotoxicity assay kit (Beyotime). The procedure was performed according to the manufacturer's instruction. Briefly, 120 μl of the culture supernatant was loaded to each well of the 96-well plate and incubated with the reaction buffer containing 20 μl lactate solution, 20 μl 2-p-iodophenyl-3-nitrophenyl tetrazolium chloride, and 20 μl diaphorase at 37°C for 30 min. Then, the absorbance was quantified at 490 nm.
Measurement of fluorescent LC3 puncta
Imaging studies for mRFP-GFP-LC3 in NRVMs were performed as previously described (21). Briefly, NRVMs cultured on coverslips were transduced with Adv-mRFP-GFP-LC3 (Hanbio Co. LTD.) at 20 MOI. Two hours later, the culture media containing virus were replaced with fresh media. H/R experiments were performed at 24 h after transduction. After the treatment, the cells were washed with PBS, fixed with 4% paraformaldehyde, and viewed under a Leica laser scanning confocal microscope (Leica TCS SP5 II). The number of GFP and mRFP dots was determined by manual counting of fluorescent puncta in five fields from three different myocytes.
In vitro gene silencing with siRNA
Predesigned siRNA targeting rat VDR (Cat.No. SI02053695, SI02053702, and SI02053709) were purchased from Qiagen GmbH. AllStars Negative Control siRNA (Qiagen; Cat. No. SI03650318) was used as a negative control. This validated nonsilencing control has no homology to any known mammalian gene and has been validated using Affymetrix GeneChips (Qiagen). Cells were transfected for 24 h with VDR or control siRNA by using RNAiFect Transfection Reagent (Qiagen) according to the manufacturer's instructions. Preliminary experiments were carried out to establish optimal transfection conditions. Mock-transfected cells were treated identically except for the omission of siRNA. Experiments were performed at 48 h after siRNA transfection.
Statistical analysis
The data are presented as the means±SEM. Statistical analysis was performed with Mann–Whitney test for two-group comparisons. Statistical differences of multiple treatments were calculated with one-way analysis of variance followed by the Bonferroni post hoc test when appropriate. Western blot densities were analyzed with the Kruskal–Wallis test followed by Dunn's post hoc test. Probabilities of 0.05 or less were considered statistically significant.
Footnotes
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81330006, 81170192, 81470389, 81270282, 81070176, and 81200163 to B.H., J.P., and Q.H.), Key Basic Research Program of Shanghai Committee of Science and Technology (14JC1404500 to J.P.), Program for New Century Excellent Talents from Ministry of Education of China (NCET-12-0352 to J.P.), Shanghai Shuguang Program (12SG22 to J.P.), International Cooperation Program of Shanghai Committee of Science and Technology (12410708300 to J.P.), and Foundation of Shanghai Jiao Tong University (YG2013MS42 and YG2012MS07 to J.P. and A.Y.).The authors thank Professor Jianguo Jia, Doctor Jian Wu, and Doctor Yong Ye (Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital, Fudan University, Shanghai, China) for echocardiographic measurements.
Author Disclosure Statement
No conflicts of interest exist for any of the authors.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.