
Abstract
Introduction
T
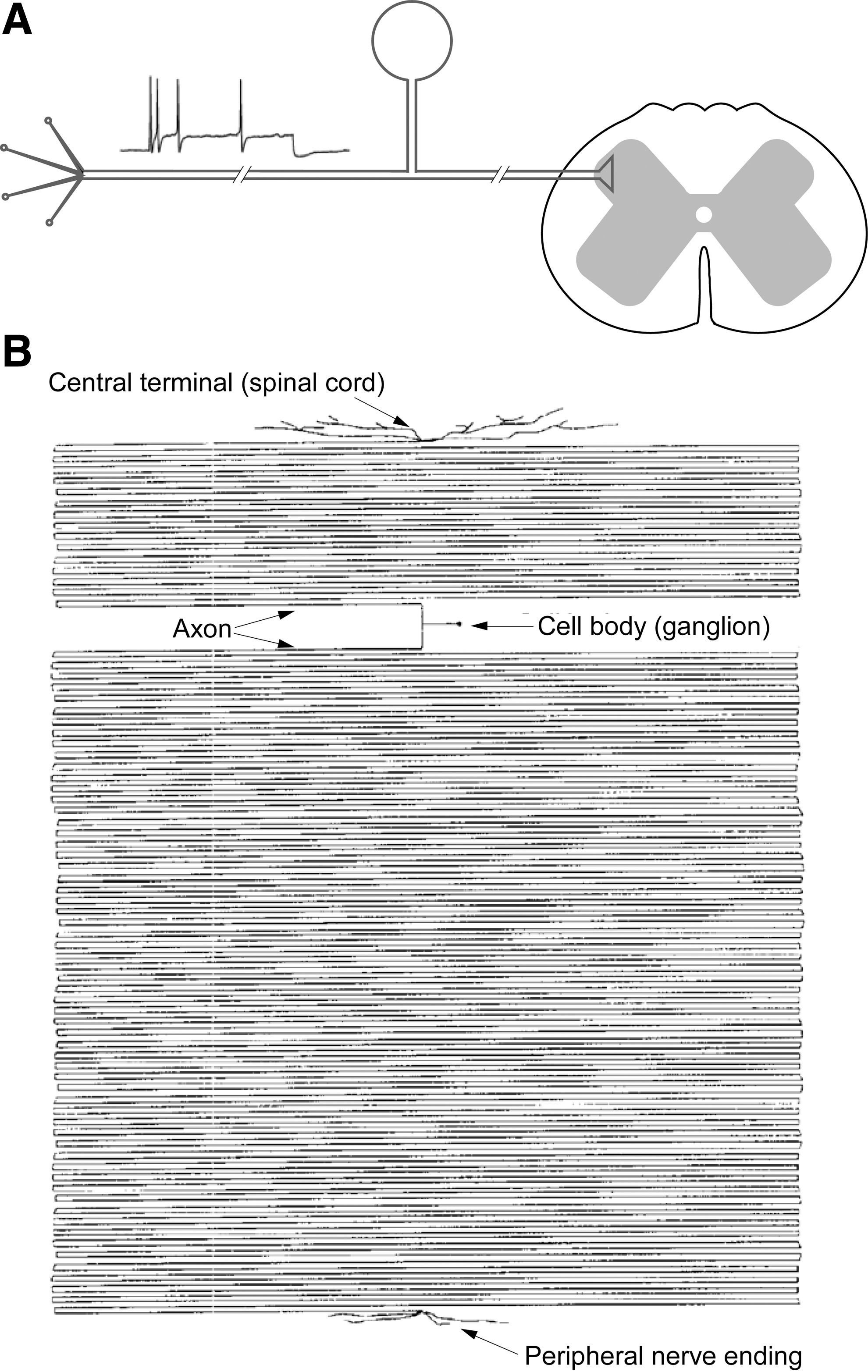
Mechanisms of ROS Generation
Molecular mechanisms of ROS metabolism are well studied in the brain with much less literature focused specifically on the peripheral nervous system. Although such mechanisms may differ between neuron types, the key steps of ROS metabolism are similar. One of the major ROS species in cells is the superoxide anion (O2 •−), which is generated by one-electron reduction of O2. In mitochondria, this mainly happens at electron transport chain (ETC) complexes I and III [Fig. 2; reviewed in Murphy (118)], α-ketoglutarate dehydrogenase (175, 199), α-glycerophosphate dehydrogenase (200), and some other ETC sites. Outside of the mitochondria, O2 •− can be produced by xanthine oxidase-catalyzed hypoxanthine oxidation (81), plasma membrane NADPH oxidase (87), as well as by the phospholipase A2-dependent cyclo-oxygenases and lipoxygenases (33). The O2 •− is highly reactive and can oxidize many moieties; however, it is usually rapidly converted to hydrogen peroxide (H2O2) in a dismutation reaction catalyzed by the enzyme superoxide dismutase [i.e., mitochondrial manganese SOD (MnSOD) or cytosolic copper/zinc SOD (Cu/ZnSOD)] (Fig. 2) (143). H2O2 is also a potent oxidizer and it can be involved in further reactions and produce several other ROS, including the highly reactive hydroxyl radical (•OH) (in a Fenton reaction with Fe2+) (140).
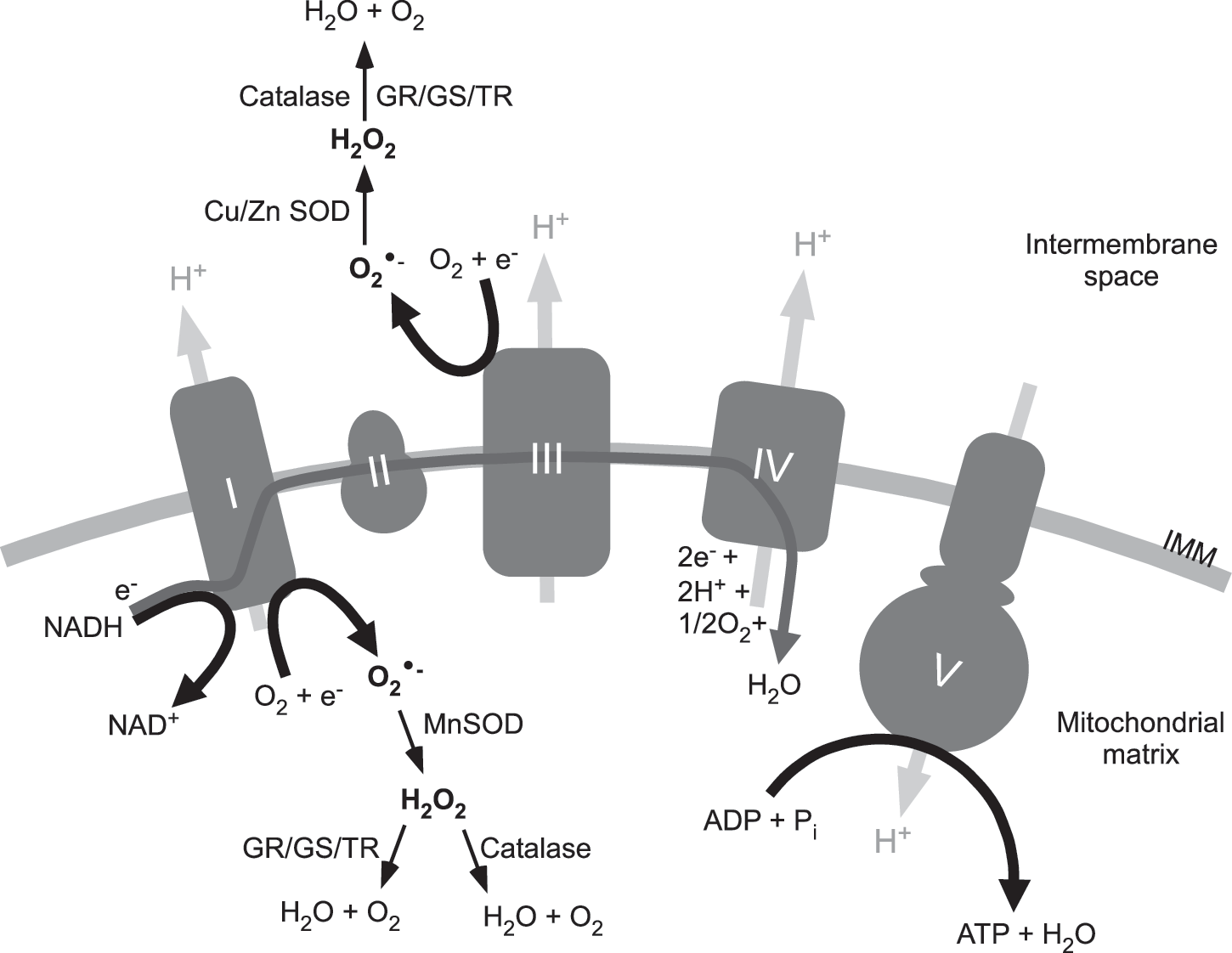
Mechanisms of NO Generation
The metabolism of nitrosyl species is tightly linked with oxygen and ROS. Synthesis of NO and
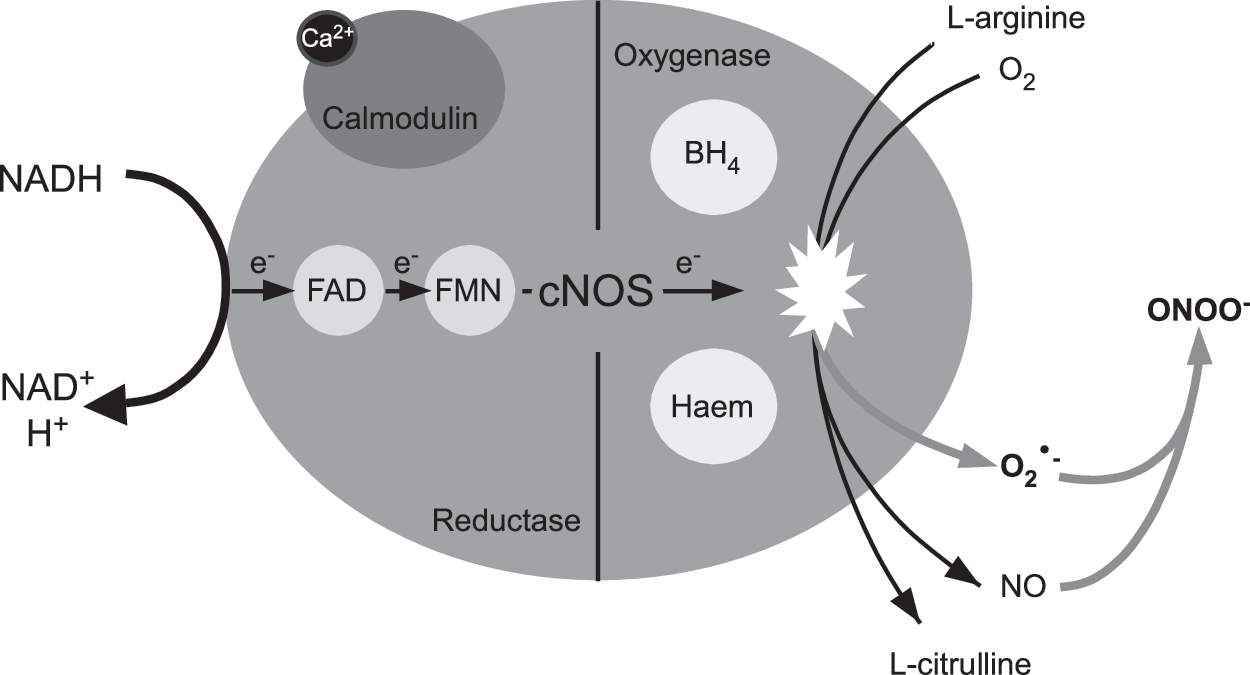
Interplay Between NO and ROS
The reversible binding of Ca2+ to calmodulin is the primary controller of cNOS activity, with increases in free cytosolic Ca2+ initiating the production of NO [reviewed in Schmidt et al. (162)]. Inhibition of cNOS is triggered through negative feedback of NO on the enzyme in a concentration-dependent manner. For cNOS to exclusively produce NO and
ROS and the Peripheral Somatosensory System
Increased ROS generation is implicated in several pathological conditions related to pain; these include inflammation [i.e., chronic pancreatitis (190)] and diabetes mellitus. Diabetes is often accompanied by the peripheral neuropathies (diabetic neuropathy), and oxidative stress is a widely accepted key factor in its development (53). The incidence of neuropathy in diabetic patients can be very high (up to 50%); the major feature of this syndrome is sensory and motor fiber degeneration, with longer axons being the most affected. Diabetic neuropathy is an incapacitating condition accompanied by pain, foot ulceration, digestive abnormalities, sensory loss, erectile dysfunction, and heart arrhythmias among other conditions. In both type 1 and type 2 diabetes, there is significant accumulation of extracellular glucose, which is transported into the cytosol of cells faster than endogenous metabolic pathways can accommodate it (167). Under these conditions, glucose enters an alternative metabolic pathway where it is converted to sorbitol by the enzyme aldose reductase (112). This reaction requires NADPH, which, in turn, becomes less available for the production of the intracellular antioxidant glutathione from glutathione disulfide by the NADPH-dependent enzyme glutathione reductase. Reduced glutathione levels, in turn, make cells highly vulnerable to ROS (112, 201). Hyperglycemia may cause redox disturbances in a number of additional ways. Glucose can undergo auto-oxidation in the presence of trace amounts of free transition metals, yielding α-ketoaldehydes, H2O2 and ROS (213). Thus, reduced antioxidant capacity in combination with increased ROS production creates a cellular background for oxidative stress. As extensively described in excellent recent reviews [i.e., Refs. (112, 201)], oxidative stress and ROS are largely responsible for the neurodegeneration underlying diabetic neuropathy. Importantly, it has long been noted that the longer axons suffer the most (which is probably to do with the metabolic challenges associated with long axons).
NO and the Peripheral Somatosensory System
NO is a molecular mediator with diverse physiological roles, including vasodilation, blood clotting, and nociception. It is released from arteries, following mechanical stimuli or from dural mast cells or nerve fibers, after inflammation, contributing to pain (17). In the central and peripheral nervous systems, NO displays many properties of a neurotransmitter (55). As a gasotransmitter, NO is diffusible across membranes and its physiological effects are relatively short lived, being oxidized to nitrite in the time frame of seconds (110). NO-mediated signaling pathways include the activation of soluble guanylate cyclase (GC) and the cyclic guanosine monophosphate (cGMP)/protein kinase G (PKG) pathway (25), direct S-nitrosylation of protein thiols (addition of a nitrosyl ion NO− to generate a nitrosothiol, RS-N=O) (95) and protein tyrosine nitration (addition of NO2 to generate 3-nitrotyrosine) (16, 124). Each of these pathways has been linked to pathology of one form or another and diversion toward one of these pathways over another depends on a multitude of factors in the system, including the presence of transition metal complexes and redox status (195). With such varied and wide-ranging effects, understanding the individual signaling pathways of NO is of extreme importance, though relatively little is known about the molecular players involved in these processes. NO has been implicated in migraine, as it was reported to induce CGRP release from trigeminal afferents and CGRP levels are increased during a migraine attack in humans (51, 58). In animal models, migraines can be induced by NO donors and reduced with CGRP antagonists (85, 210) or trigeminal denervation (210). Trigeminal models show that NO increases the release of CGRP from afferent fibers (113), which is not caused by increases in CGRP mRNA (50) but which could be the result of increased neuronal activity. An injection of NO donors into the periphery is also painful and in rat paw causes hyperaemia via release of the vasodilatory CGRP from afferent nerve fibers and up-regulation of prostaglandin production (68). However, antinociceptive effects of NO have also been suggested; see “K
The contribution of the individual NOS isoforms to inflammatory pain has been elucidated using isoform-specific knockout mice. In mice deficient for nNOS, thermal hyperalgesia, after an injection of complete Freund's adjuvant (CFA) was halved, mechanical hypersensitivity was absent and CFA-induced increases in CGRP in DRG neurons were reduced (21). These data suggest that nNOS plays a significant role in sensitization of DRG neurons in inflammatory pain. However, both iNOS and eNOS also contribute (21). In DRG neurons, nNOS is expressed in early developmental stages but is down-regulated with age (141, 194); both nNOS and iNOS expression was reported to be up-regulated in DRG after injury and inflammation (1, 60, 103, 104). Increased NO synthesis in microglia and astrocytes in combination with the properties that enable retrograde messenger activity has implicated astrocytes and microglia as an external source of pathogenic NO (179). Tissue inflammation thus likely leads to increases in all three isoforms (nNOS, iNOS, and eNOS) with vasodilation also increasing blood flow. Candidate mediators of this effect are the P2 purinergic receptors, as the P2 receptor antagonist pyridoxalphosphate-6-azophenyl-2′,4′-disulphonic acid (PPADS) decreased face-rubbing activity, NOS isoform expression in TG, and Fos expression in the spinal trigeminal nucleus after a subcutaneous injection of formalin (22).
An important aspect of sensory signaling by NO has recently been identified in a study that focused on the production of NO in the skin. TRPV3, a heat-sensitive ion channel (169), is expressed in keratinocytes and on activation, induces NO production (115). This is an interesting finding, as a number of the NO-modulated ion channels described next are also expressed in skin cells, including Kv7 (146), TRPV1 (20), and TRPA1 (11) channels. Characterization of the precise details of the signaling pathways between the skin and the peripheral nervous system will provide important contributions to our understanding of the role of NO and ROS in sensory processing.
While our understanding of NO signaling in the peripheral somatosensory system is still “work in progress,” roles of NO in the CNS are much better understood; several recent reviews cover these roles in processes as diverse as synaptic plasticity (26, 64, 67), excitotoxicity (29), or neurodegeneration (179) and we direct readers interested in such effects to these fine papers.
Physiological Relevance and Considerations for Future Studies
The cellular functions of NO are complex and are highly dependent on NO concentration. Broadly speaking, high concentrations of NO are cytotoxic, while low concentrations are cytoprotective. The complexities of measuring NO levels, coupled with the fact that NO freely diffuses across membranes in three-dimensional space, has made it difficult to quantify the levels that accumulate during physiological signalling in vivo. In fact, the use of the word “accumulate” may well be misleading in this context, as the presence of NO as a signaling molecule is likely highly transient. This has led to controversy and difficulties within the field, as astoundingly different physiological concentrations of NO have been reported. We also need to consider the precise biological context, with regard to both normal and pathological conditions. In this way, levels measured via various means (using electrode measurements, fluorescent probes, biosensors, and selectivity of NO for GC-coupled NO receptors) and by different groups have been reported from micromolar to picomolar concentrations [reviewed in Hall and Garthwaite (63)]. Certainly high concentrations of NO (potentially micromolar levels) can be released from inflammatory cells after their activation, while levels released from endothelial cells and neurons have been harder to measure accurately. New findings suggest that picomolar concentrations of NO can now be measured. In fact, it would appear that cells are able to detect exceedingly low concentrations of NO, even down to the picomolar level and even when the exposure is brief (e.g., subsecond range) (214).
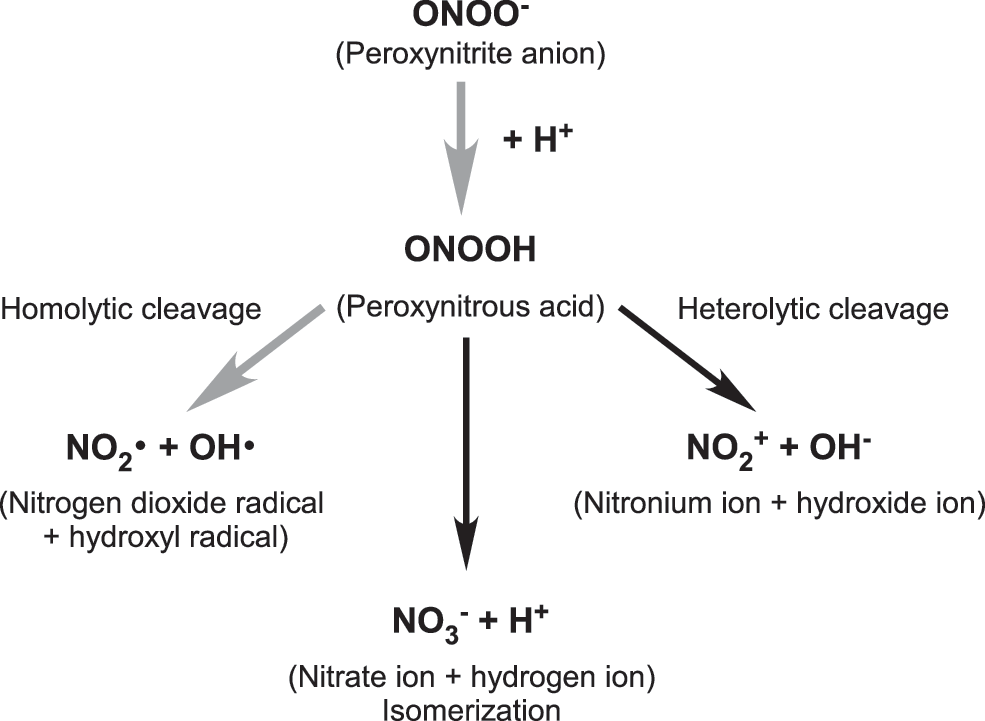
In addition, the function of NO is tightly coupled to oxidative stress (see, e.g., Figs. 3 and 4), which makes it often difficult to separate pure NO effects from these that are more complex and require secondary mechanisms (e.g., ROS). Thus, it is still unclear whether NO is able to bind Cys directly at detectable levels or whether Cys nitrosylation requires an additional oxidative mechanism to occur (54). Certainly the biochemical tests (the “biotin switch assay” being the most prominently used method) suggest that in many proteins, cysteines are indeed nitrosylated but we will need to turn to more precise techniques in the future to confirm this.
Many of the same considerations are also true for ROS, whose precise concentrations in cells are difficult to measure, and thus the biological relevance of many of the proposed ROS mechanisms remains in question (181). Consequently, tonic intracellular H2O2 concentrations in mammalian cells were reported to vary in the range of 1–700 nM (181) and H2O2 production can increase in ischemic brain by more than 10-fold (174), suggesting that an even greater range of concentrations may potentially be achievable in vivo. Clearly, caution has to be executed when designing (and interpreting) in vitro experiments with extracellular sources of highly concentrated ROS or NO donors.
Regulation of Sensory Neuron Ion Channels by ROS and NO
M channels
Among a plethora of ion channels expressed in somatosensory (and particularly nociceptive) neurons, one subfamily is gaining increased recognition for its central role in setting and controlling background excitability, a subfamily of voltage-gated K+ channels called KCNQ (Kv7 or “M channels”). These K+ channels conduct slow, non-inactivating “delayed-rectifier” K+ currents with a threshold for activation well below −60 mV (can be as low as −80 mV) (70). These properties enable some M channels to remain active at the resting membrane potential of a sensory neuron (∼−60 mV) (6, 91, 92, 160). Since M channels have strong outwardly rectifying voltage dependence, their “background” activity represents an “intrinsic voltage-clamp” mechanism that controls the resting membrane potential, threshold for action potential (AP) firing, and accommodation within trains of AP [reviewed in Refs. (41, 49)]. Functional M channels are expressed in a variety of locations, including DRG neuron cell bodies (80, 130, 228), nerve fibers (42, 80, 129, 154), dorsal roots/central terminals (149), and nociceptive nerve endings (129). As expected from their biophysical properties, M channel activity strongly affects afferent fiber excitability both in vitro and in vivo. Thus, M channel inhibition increases and M channel enhancers decrease action potential firing in vitro [e.g., in cultured DRG neurons (91, 92, 98, 117, 130) or in skin-nerve preparations (129)]. Accordingly, a hind paw injection of the M channel blocker XE991 in rats induces moderate pain (94, 98), while peripheral injections of M channel pharmacological enhancers, such as retigabine and flupirtine, produce analgesic effects (98, 152).
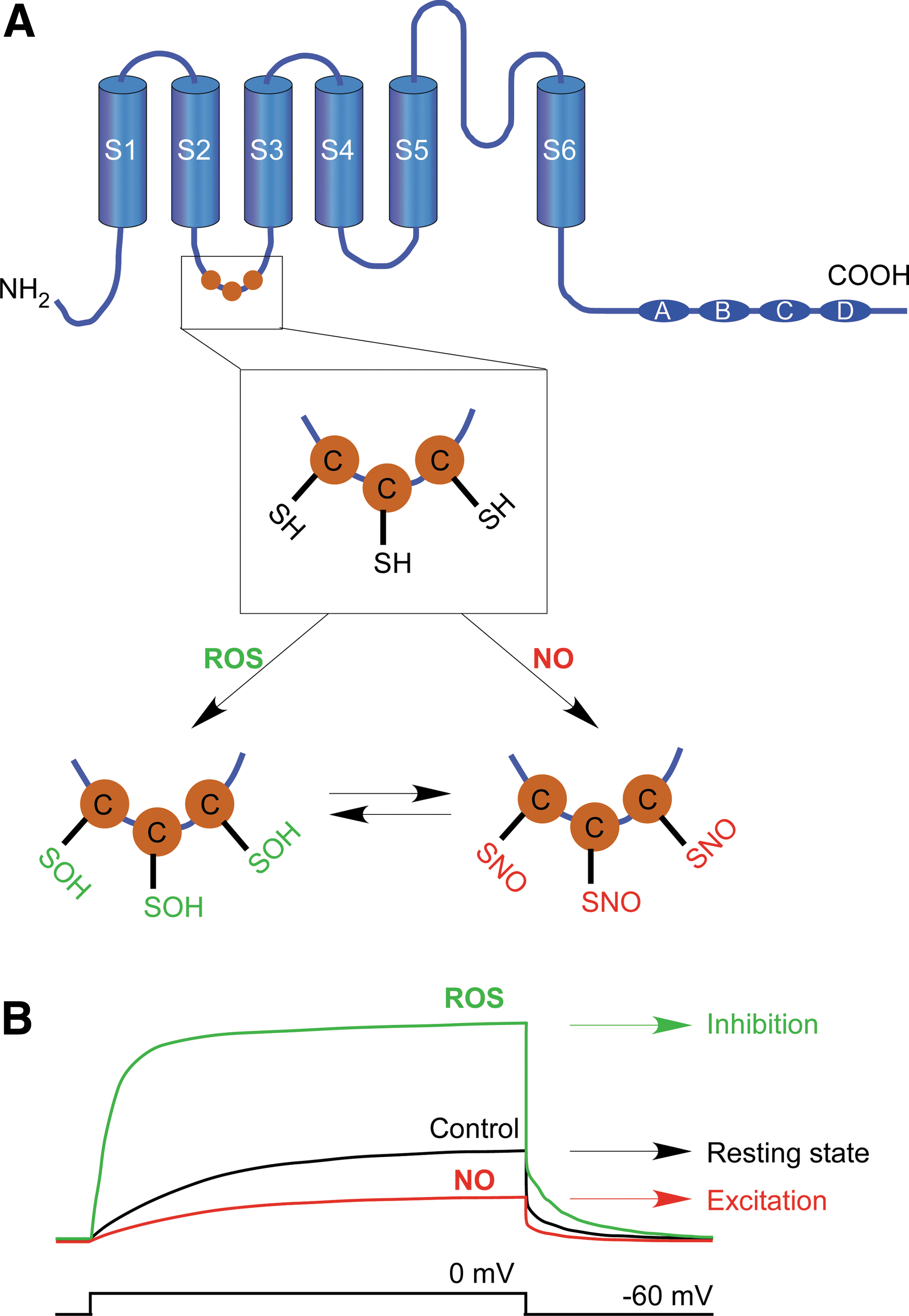
In recent studies, we have found that four (Kv7.2–Kv7.5) out of five M channel subunits possess a reactive cluster of three successive cysteines in the S2–S3 linker (equivalent to the positions C156, C157, and C158 in Kv7.4; Fig. 5), which can be oxidized by physiologically relevant concentrations of H2O2 (56, 91) or S-nitrosylated by NO donors (126). Oxidation results in potent augmentation of M channel activity, while nitrosylation results in channel inhibition (Fig. 5). With regard to the H2O2 effect, we found that Kv7.2, 7.4, and 7.5 are potently augmented by extracellular H2O2 concentration as low as 5 μM. Kv7.1 and Kv7.3 are largely insensitive to H2O2 despite the fact that Kv7.3 (but not Kv7.1) possesses the conserved triple-cysteine cluster which is necessary for H2O2-induced augmentation of Kv7.2, Kv7.4, and Kv7.5 currents. The H2O2 induced a leftward shift in channel voltage dependence and induced a prominent increase in channel open probability (P o) at all voltages. Kv7 channels have highly divergent tonic maximal P o values (that is P o at saturating voltages under “control” conditions); thus, Kv7.2, 7.4 and Kv7.5 have a rather low P o in the range of 0.1–0.2, whereas Kv7.3 has a maximal P o near unity (90, 165), which is likely to explain the fact that H2O2 did not augment Kv7.3 current (as measured at saturating voltage of 0 mV). Kv7.1 differs from the rest of the family in that it has only one cysteine residue within the “triple C” region; however, reconstitution of the complete CCC sequence in Kv7.1 by substitution of R86 and S87 with cysteines bestowed some H2O2 sensitivity to this mutant, further supporting the conclusion that this triplet of cysteines mediates the redox modulation of Kv7 channels. Interestingly, it seems that oxidation of individual cysteines within the triple-C cluster has a cumulative effect on M channel activity; thus, substitution of all three cysteines was required to completely abolish channel sensitivity to H2O2, while mutation of one or even two cysteines resulted in partial reduction (but never a complete loss) of H2O2 sensitivity (56). Cysteine residues can be reversibly oxidized to cysteine sulfenic acid (Cys-SOH); this derivative can be further (irreversibly) oxidized to sulfinic acid (Cys-SO2H) and sulfonic acid (Cys-SO3H); alternatively two juxtaposed Cys-SOH can react and form intra- or intermolecular disulfide bonds with another protein or low-molecular-weight thiol such as glutathione (128). We were unable to detect the formation of intermolecular disulfide bonds specific to the triple cysteine region under non-reducing PAGE. Given the fact that the H2O2 effect on Kv7 channel activity is completely reversed by the reducing agent dithiothreitol (DTT) (56), while Cys-SO2H and Cys-SO3H modifications cannot be reduced by DTT (135), our working hypothesis is that cumulative oxidation of cysteines to cysteine sulfenic acids within the triple-C cluster mediates oxidative augmentation of M-current (Fig. 5A). Interestingly, in further experiments using patch-clamp electrophysiology and the biotin-switch technique, we found that the same triplet of cysteines can be directly S-nitrosylated in the presence of the NO donor S-nitroso-N-acetyl-DL-penicillamine (SNAP), an effect that leads to a marked decrease of M channel activity (126). This finding raises an interesting possibility that M channels, in fact, may serve a role of cellular sensors conveying intracellular ROS and NO levels into the electrical excitability of neuronal membranes. Indeed, small-diameter DRG and TG neurons respond to NO donors with (i) M channel inhibition; (ii) increased excitability; and (iii) increased CGRP release (126). Conversely, H2O2 and endogenous ROS mediate the opposite effect, that is, (i) enhancement of M channel activity; (ii) moderate hyperpolarization in small DRG neurons; and (iii) inhibition of CGRP release (91). Moreover, these anti-excitatory effects were also observed in response to endogenous O2 •− release from mitochondria stimulated in small DRG neurons by substance P (91). Thus, redox/NO-sensitivity of M channels may indeed play vital regulatory and signaling roles within the somatosensory system and particularly within nociceptive fibers. However, this role of a membrane integrator of redox/NO status of the cytosol is extremely complex. NO is generated by eNOS and nNOS in the trigeminovascular system (142) and by immune cells during an inflammatory response, via cytokine-induced activation of iNOS (38), while many pathways involved in inflammatory signaling also inhibit M-current (61, 74, 75, 93, 94, 98, 224, 225). Gq-protein-coupled receptors, such as the bradykinin B2 receptor, evoke an intracellular signaling cascade that stimulates cleavage of phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3), triggering Ca2+ release from IP3-sensitive stores. Both PIP2 depletion and Ca2+ release inhibit M-current directly. As previously shown, NO synthases are calmodulin dependent and elevated Ca2+ triggers NO production from nNOS by activating calmodulin (2); while NOS activity is inhibited by PIP2 (218). In this way, Gq-PCR inflammatory signaling would be able to increase NO levels by two means, the removal of PIP2-mediated inhibition of NOS and increases in Ca2+ activating NOS. This suggests that inflammatory Gq-PCR signaling and NO may work in concert to ensure robust inhibition of M-current. In addition to these effects, NO can react rapidly with endogenous O2 •− and form a strong neurotoxic oxidant, ONOO−, a major cytotoxic agent produced during inflammation, sepsis, and ischemia/reperfusion (15, 95). Formation of ONOO− by O2 •− and scavenging of NO leads to the inhibition of mitochondrial respiration and SOD activity, potentially establishing a feed-forward loop for ONOO− formation and oxidative modification of M channels (although the direct effect of ONOO− on M channel activity has not been demonstrated thus far). This provides for a potential difference in the cellular effects of NO, depending on redox state, by which ONOO− formation augments M-current, decreasing excitability but NO inhibits M-current, increasing excitability. As a gasotransmitter, NO and its effects are short lived and highly regionally specific, features that underlie a possibility for location- and temporal-specific regulation of ion channels. Thus, at precise loci, the redox environment will determine the ultimate effect of NO release on M-current.
KATP channels
ATP-sensitive K+ channels (KATP) are heteromeric channels formed by Kir6.1 or Kir6.2 subunits of inward-rectifier K+ channels and regulatory sulfonylurea receptor subunits SUR1 or SUR2 (65). Physiologically, KATP channels are inhibited by intracellular ATP and activated by cytosolic ATP depletion, thus serving the role of cellular metabolic sensors. KATP channels have been suggested to play a neuroprotective role in parts of the CNS in some pathological conditions, such as ischemia (216). In the peripheral nervous system, KATP channels (i.e., Kir6.2, SUR1, and SUR2) are expressed in a subpopulation of DRG neurons of various sizes (229). KATP currents in small DRG neurons are relatively small, but KATP channel enhancers (diazoxide and pinacidil) hyperpolarize nociceptive neurons and reduce bradykinin-induced pain (48). Similar to M channels, KATP channels are augmented by endogenous H2O2, as has been shown in dorsal striatal neurons (12), and the resulting hyperpolarization of neurons has been suggested as a mechanism of neuroprotective silencing in hypoxia (198). The molecular mechanism of the KATP channel sensitivity to ROS remains to be established; thus far, it has been reported that the effect is probably direct and is mediated by decreasing channel sensitivity to ATP (71); SUR1-containing KATP channels are more sensitive to H2O2 (147). It is not yet tested whether ROS sensitivity of KATP channels plays any role in sensory neurons.
KATP channels are also sensitive to NO; however, in contrast to M channels, NO, similar to ROS, activate KATP channels in DRG neurons (as shown using excised-patch electrophysiology); the effect is possibly mediated via the direct S-nitrosylation of the SUR1 subunit of the channel complex (77). However, an indirect activation of KATP channels in DRG neurons by NO acting via the classical NO/sGC/cGMP/PKG pathway has also been suggested in other studies (170, 171) (Fig. 6). After peripheral nerve injury, the nociceptive effects of NO are exacerbated, potentially due to inflammation and associated increases in neuronal excitability. The NO donor SNAP caused activation of KATP channels, resulting in decreased neuronal excitation of large DRG neurons after axotomy (77). Peripheral injections of NO donors showed antihyperalgesic effects in some studies (although pro-algesic effects were also demonstrated in others and these were suggested to be mediated by KATP channel opening via the NO/sGC/cGMP/PKG pathway (156).
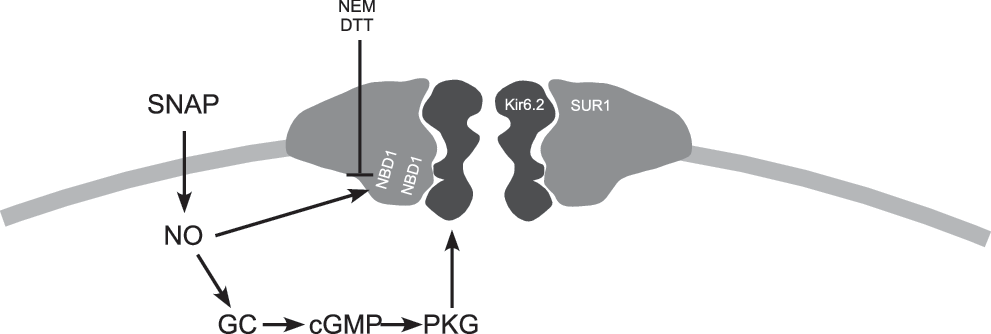
As per examples given earlier, it appears that peripheral somatosensory actions of NO can be either pro-nociceptive or antinociceptive depending on the experimental conditions and models used [for review see Refs. (40, 134)]. For instance, a dual action of locally applied NO donors on tactile allodynia induced by surgical incision in rats has been noted (136); lower concentrations of SNAP reduced allodynia in a GC-dependent manner, while higher concentrations enhanced allodynia, independently of GC. Since NO has opposing effects on M channels and KATP channels, it is conceivable that at least some pro-algesic effects can be mediated by M channel inhibition, while analgesic effects can potentially be mediated by KATP activation. In such a scenario, the exact outcome of the NO effect would depend on the relative expression and activity of M channels and KATP channels in the affected neurons and the spatial correlation of the NO release events with the locality of these channels within the neuronal plasma membrane. It should be noted that KATP channels appear to be slightly more sensitive to NO as compared with M channels; the activating effect of SNAP on KATP channels reached saturation at 100 μM (77), while for M channels the SNAP IC50 has been determined at 370 μM (126).
BK channels
Large-conductance Ca2+-activated K+ channels (Slo1, BK, and Maxi-K) are expressed in DRG neurons of various sizes as demonstrated with correlative patch clamp analysis and RT-PCR (89, 163); the auxiliary subunit β2 was suggested to participate in the BK channel complex in DRG (89, 163). The major functions of BK channels in sensory neurons were suggested to include shortening of the action potential, acceleration of repolarization, and fast after-hyperpolarization (89, 163). These effects may both limit neuronal excitability/output or, somewhat paradoxically, increase it as fast repolarization and more prominent after hyperpolarization may result in an increased firing rate due to improved recovery of voltage-gated Na+ channels (VGNC) from the inactivation (82). The BK channel protein contains multiple cysteine and methionine residues that potentially can be oxidized to modulate channel function (i.e., human Slo1 protein AAB65837 has 29 cysteines and 30 methionines) and BK channels are well established as O2 sensors, responsive to gasotransmitter modulation (78, 157). The general mechanisms of redox modulation of BK channels were recently reviewed in depth in an excellent review (157); therefore, here, we only briefly consider some key facts. The presence of multiple cysteines and methionines within Slo1 proteins results in a complex response to oxidizers and cysteine-modifying reagents. Thus, heterologousely expressed Slo1 channels are inhibited by H2O2 and activated by DTT (45), presumably via cysteine-oxidation mechanisms. The application of various cysteine-modifying agents such as 2-(trimethylammonium)ethyl methanethiosulfonate (MTSET), (2-sulfonatoethyl)methanethiosulfonate (MTSES), or N-ethylmaleimide (NEM) to Slo1 changed channel voltage- and Ca2+ dependence of steady-state activation. Moreover these agents, while acting on the same cysteines, had different effects on channel function. For instance, MTSET increased Slo1 K+ conductance and shifted voltage dependence to more negative voltages, while MTSES and NEM had the opposite effects (226). There is also evidence that methionine oxidation and cysteine oxidation may have opposing effects on channel activity (191). Thus, redox modulation of BK channels is incredibly complex and the outcome of such modulation is likely to depend on the nature of the redox changes and local environment. The significance of redox modulation of BK channels for sensory neurons has not yet been elucidated.
Literature on the regulation of BK channels by NO is more coherent, as there is a broad agreement that in neurons and smooth muscle cells, BK channel activity can be augmented via the classical NO/sGC/cGMP/PKG pathway (10, 82, 102, 164, 217), by which the activation of PKG by cGMP leads to phosphorylation of the Slo1 channel protein at three specific PKG phosphorylation sites, an effect that results in enhancement of channel activity (86).
While redox/NO modulation of BK channels can be complex, there is possibly a pattern for reciprocal effects; however, in contrast to M channels, ROS, such as H2O2, are rather inhibitory, while NO enhances channel activity. It would be interesting to investigate how (and if ) the redox/NO modulation of BK channels affects peripheral somatosensory transmission.
A-type K+ channels
Somatosensory neurons express robust A currents (IKA, inactivating voltage-gated K+ current) that can be further pharmacologically and electrophysiologically separated into two or three subtypes which are distinguishable by different inactivation kinetics (i.e., slow- and fast-inactivating IKA) and sensitivity to tetraethylammonium (TEA) (3, 52, 59, 111, 173, 219); see Du and Gamper (49) for review. The IKA with slower inactivation kinetics is predominantly found in small-diameter nociceptors that also express TTX-resistant Na+channels and TRPV1 (59, 223). IKA is conducted by Kvs, several members of which are expressed in sensory neurons. Thus, the expression of several members of the Kv1.1 channel family has been reported with Kv1.1, Kv1.2, and Kv1.4 being the most abundantly expressed subunits (79, 145, 204, 219). Kv1.1 and Kv1.2 are predominantly found in non-nociceptive, large-diameter DRG neurons. In small-diameter nociceptive DRG neurons, the predominant Kv1 α-subunit is Kv1.4 (145, 204), a rapidly inactivating A-type K+ channel (37). Inactivating subunits of Kv3 (Kv3.4) and Kv4 (Kv4.3) channel families are also expressed in sensory neurons and contribute to IKA, particularly in nociceptors (32).
Several A-type forming Kv subunits have been found to be sensitive to ROS. Thus, inactivation of Kv1.4 and Kv3.4 is slowed by oxidation, an effect reversed by reducing agents (155). Cysteine C13 was found to be responsible for this effect in Kv1.4 (155) [a detailed discussion on the regulation of Kv channel inactivation by ROS can be found in Sahoo et al. (157)]. Interestingly, the modulation of native IKA by ROS has been reported in small-diameter DRG neurons (69), suggesting that the redox sensitivity of Kv channels may have a physiological role in peripheral somatosensory processing. Thus, the oxidizing agents DTBNP and chloramine-T reduced fast inactivation of IKA in cultured small DRG neurons, while the reducing agent DTT reversed this effect (69). If the fast inactivation of IKA being reduced by oxidation, the underlying Kv channels would become less susceptible for cumulative inactivation during repetitive firing, which, in turn, may affect firing rate and thus modulate the sensitivity of nociceptive fibers. It, however, should be noted that in the study (69), the IKA has been pharmacologically isolated in DRG neurons by the addition of 5 mM TEA to inhibit the delayed-rectifier K+ channel current. However, at this concentration, TEA would only partially block M channels; only Kv7.2 is sensitive to TEA in a low millimolar range, while other M channel subunits expressed in DRG, Kv7.3 and Kv7.5 are poorly TEA insensitive (62, 166, 221). The classical M channel, which is a Kv7.2/Kv7.3 heteromer, has a TEA IC50 in the range of 5 mM (62, 166, 208, 221) and, thus, will only be 50% blocked at this concentration. M channels have slow kinetics and as discussed earlier are potently augmented by oxidizing agents [and these effects are reversed by DTT (56, 91, 92)]. Therefore, the increase in steady-state K+ current on application of oxidizing agents that was interpreted as reduction of inactivation (69) is also consistent with an increased contribution of M-current to the total K+ current recorded. Clearly, further research is needed to elucidate the exact molecular mechanisms of sensory neuron K+ current modulation by redox signaling.
T-type Ca2+ channels
T-type Ca2+ channels (encoded by the CACNA1G, CACNA1H, and CACNA1I genes, which code for Cav3.1–3.3 channel α-subunits, respectively) are expressed in a subpopulation of DRG neurons of various types [from small/medium-diameter nociceptors to some large-diameter, mechanosensitive neurons (39, 73, 153)]. These channels differ from other voltage-gated Ca2+ channels (VGCCs) in several important features; thus, they are activated at voltages below −60 mV (hence, they are also termed “low-voltage activated” Ca2+ channels [LVA]); T-type channels activate and inactivate rapidly and have a smaller single-channel conductance than high-voltage-activated VGCCs (30, 72, 133). In the CNS, T-type channels are responsible for pacemaker activity and low threshold spikes; they also may display a significant window current (i.e., they can be tonically active) at membrane potentials close to the resting membrane potential and consequently can contribute to tonic Ca2+ influx (30, 72, 133). In DRG neurons, Cav3.2 is the predominantly expressed T-type channel subunit (168, 189). It is hypothesized that in sensory fibers, T-type channels can participate in setting the threshold for action potential firing and control burst firing (119, 121, 211). It has been shown that in nociceptive neurons, T-type current amplitudes can be augmented by reducing agents such as DTT or
Carbon monoxide (CO) is increasingly being recognized as a potent intracellular second messenger or gasotransmitter [for review see Peers and Steele (131)]. A recent study reported CO-mediated inhibition of Cav3.2 in DRG neurons, an effect which was based on a redox mechanism distinct from that involving the MCO of His191 (23). CO is produced in cells by the heme oxygenase (HO) enzyme, which degrades heme and produces biliverdin, free iron (Fe2+), and CO. Growing evidence suggests that HOs can modulate nociception; thus, HO-1 is induced in DRG neurons in response to heat stress (132) or axonal damage (107). Accordingly, local endogenous CO has been suggested to suppress inflammatory hyperalgesia in the carrageenan model of pain (178). Moreover, the chemical induction of HO-1 inhibited the nociceptive response to formalin, while the transcriptional inhibition of HO-1 production prevented this effect (151). Boycott et al. (23) have shown that heterologousely expressed Cav3.1, Cav3.2, and Cav3.3 as well as native T-type current in cultured DRG neurons is potently inhibited by the CO donor CORM-2, an effect that was significantly reversed by DTT. CO sensitivity of Cav3.2 (but not the other two subunits) was mediated by an extracellular redox-sensitive site, which was also highly sensitive to thioredoxin (TR) but was distinct from His191. It has been suggested that TR acts as a tonic, endogenous regulator of Cav3.2 channels; while HO-1-derived CO disrupts this regulation, causing channel inhibition. Taken together, these studies emphasize the importance of T-type channels and their redox sensitivity to peripheral somatosensory (and particularly nociceptive) transmission.
TRP channels
Transient receptor potential (TRP) channels are universal cellular sensors that are capable of responding to versatile external stimuli such as temperature, pH, and changes in the chemical milieu. Several TRP channels are preferentially expressed in peripheral somatosensory neurons where these mediate neuronal responses to the environment. Probably the best studied sensory TRP channels within peripheral sensory nerves are TRPV1 and TRPA1 [for a review, see e.g., Vay et al. (202)]. TRPV1 is a member of the vanilloid subfamily of TRP channels; it is expressed in subpopulations of all the major types of nociceptive neurons (nociceptive Aδ fibers, peptidergic and non-peptidergic C-fibers). TRPV1 is a versatile sensor that is activated by a wide range of stimuli such as capsaicin, noxious heat, protons (acidification), endocannabinoids, ethanol, camphor, allicin, 2-APB, lidocaine, ω-3 fatty acids, and many others. The mechanisms of TRPV1 activation and its role in thermosensation and pain have been extensively reviewed [e.g., Refs. (31, 44, 202)]; therefore, here, we will only consider mechanisms relevant to the topic of this article. TRPV1 (together with several other TRP channels, for example, TRPC1, TRPC4, TRPC5, TRPV3, and TRPV4) is activated by NO in a process that is likely mediated by direct S-nitrosylation of cysteines (222). Likewise, TRPV1 is also activated by H2O2. In TRPC5, the action of these cysteine-modifying agents was localized to C553 and C558 within the extracellular S5–S6 linker (Fig. 7A); TRPV1 also has two cysteines in this region (C616 and C621), accordingly it was suggested that TRPV1 is regulated by the same mechanism as the substitution of these two residues reduced sensitivity to H2O2 and the cysteine-modifying reagent 5-nitro-2-PDS (222). Surprisingly, the sensitization of heat- and capsaicin sensitivity of TRPV1 by both oxidizing (diamide, H2O2 and chloramine-T) and reducing (DTT and reduced glutathione) agents has also been reported (186, 205). Interestingly, in the latter study, the effect of the reducing agents was linked to the same extracellular cysteines C616 and C621 (with the additional C634 that was also considered important for the effect in this study); while the sensitizing effect of the oxidizing compounds was found to be dependent on intracellularly facing residues. Chuang and Lin have identified the C-terminal cysteines C772 and C783 as being necessary for the sensitizing action of oxidizing agents (36). TRPV1 can also be activated by pungent compounds from onion and garlic, such as allicin (106). The action of allicin has been localized to a single cysteine C157 within the TRPV1 N-terminus (159). This is a physiologically relevant mechanism, as it underlies the responses of sensory neurons that express TRPV1 (and TRPA1, which is also sensitive to allicin via a similar mechanism) to these pungent stimuli. While the precise mechanics of redox/NO regulation of TRPV1 are still emerging, it is clear that the channel is very sensitive to such modulation and that multiple mechanisms exist. Since tissue damage and inflammation are linked to ROS and RNS production, it is likely that redox/NO modulation of TRPV1 has a prominent role in inflammatory pain mechanisms.
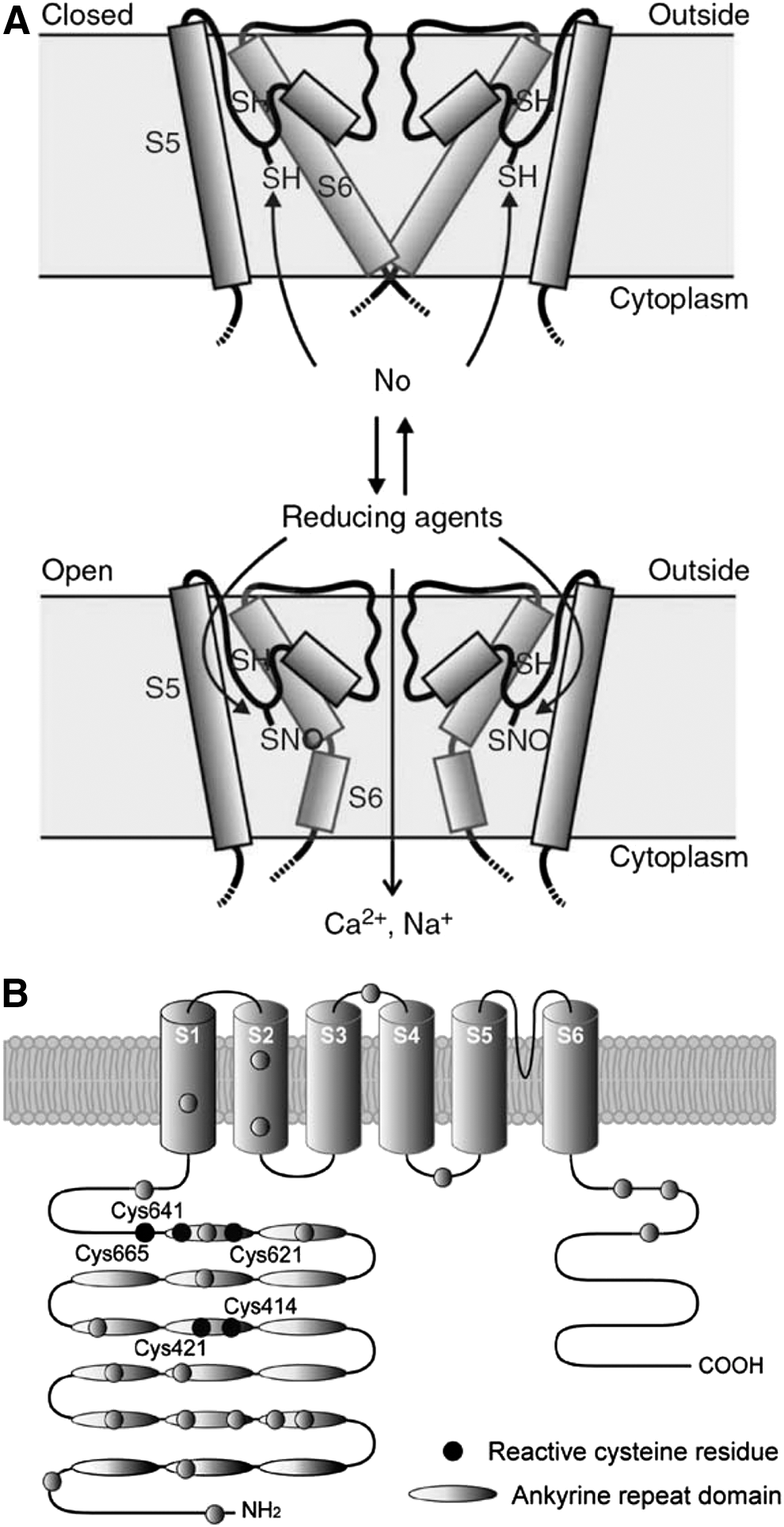
TRPA1 is the only member of the ankyrin subfamily of TRP channels in mammals; similar to TRPV1, TRPA1 is also highly expressed in peripheral somatosensory fibers, particularly in those that are activated by both noxious cold and a wide range of environmental irritants (13, 19, 182). Similar to TRPV1, TRPA1 is a “promiscuous” sensor that is activated by a variety of stimuli, including temperatures below 15°C, cinnamaldehyde, endocannabinoids, acrolein, allicin, icilin, gingerol chlorine, ROS, nicotine, and many others [for review see Refs. (14, 123, 202)]. Some of the compounds that activate TRPV1 can also activate TRPA1; moreover, both channels are often co-expressed in the same sensory neurons and possibly interact physically (4, 158, 176). Similar to the case of TRPV1, both NO donors (SNAP, NOR3) and ROS (H2O2) activate TRPA1 (both the native channel in sensory neurons and heterologously overexpressed TRPA1) (7, 18, 114, 161, 187). Again, similar to TRPV1, a common theme in the action of ROS, NO, and irritants is multiple modifications of channel cysteines. Thus, two groups have identified five cysteines (all in the channel N-terminus) to be responsible for TRPA1 activation by irritants but only one of them (C622 in mouse TRPA1) was identified by both groups (66, 105). Covalent modification of lysine 708 (in human TRPA1) by isothiocyanates was also reported to contribute to the activation of TRPA1 (66). In another study, C421, C641, and C665 of human TRPA1 (all within the N-terminus) were found to be mostly responsible for channel TRPA1 activation by NO donors and H2O2 (187); positions of the reactive cysteines in the human TRPA1 N-terminus are indicated in Figure 7B. Thus, while having similar functional outcomes, the sensitivity of the TRPA1 channel to redox/NO modulation may be underwritten by somewhat different structural mechanisms as compared with TRPV1 in which both the N-terminus and the pore region were implicated. Modulation of TRPA1 via modification of reactive cysteines has obvious physiological importance, as it underlies sensory responses to airborne irritants (13, 18) and is likely to contribute to disorders such as asthma (27) and chronic itch (97, 212).
Other channels
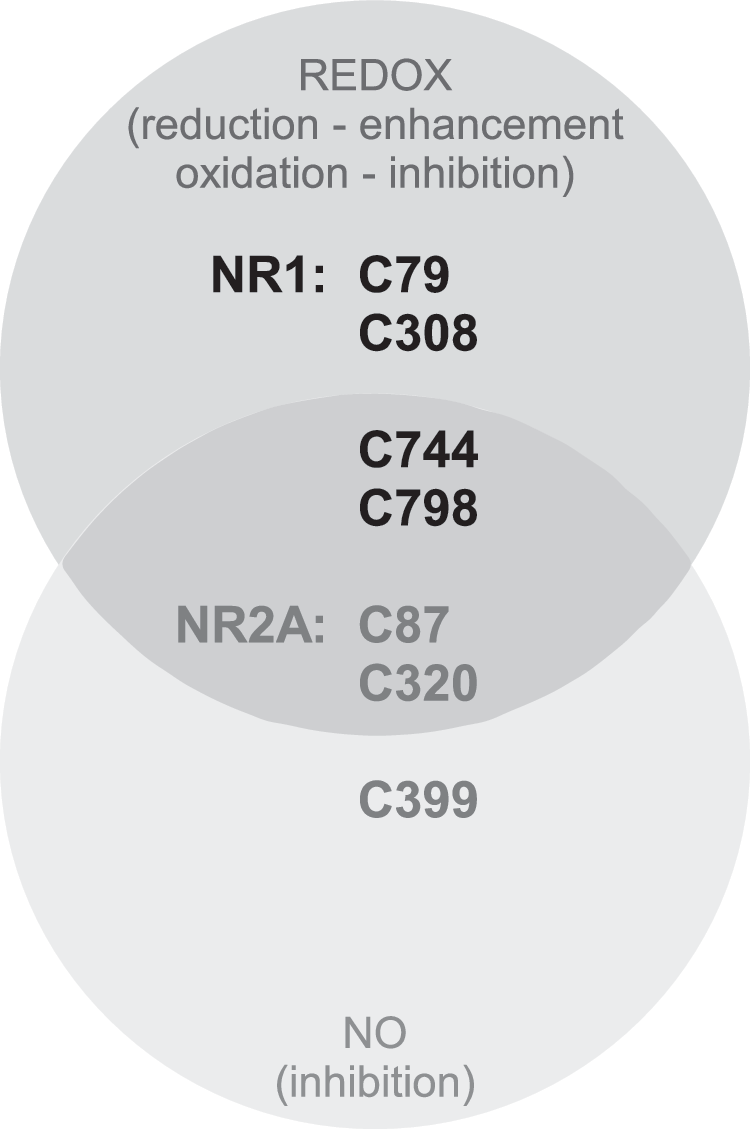
The number of sensory ion channels being identified as NO- and ROS-regulated is growing rapidly, indicating the importance of these signaling molecules in controlling excitability and pain processing. In DRG neurons, acid-sensing ion channels are potentiated by NO, and NO donors increased the pain of acid injections into the skin in humans (28). Potentiation of VGNC by endogenous ROS in small- and medium-sized DRG neurons has been recently suggested, as VGNC current density decreased after application of ROS scavengers (207). In addition, another study reported that in vitro hyperglycemia produced both increased ROS production and augmented VGNC currents in cultured DRG neurons. These are, however, indirect evidence and the sensitivity of VGNC to ROS needs to be tested further. Peripheral sensory neurons use glutamate as their main neurotransmitter; interestingly, DRG neurons also abundantly express somatic N-methyl-
Role for Lipids in Redox and NO Modulatory Pathways
When considering the regulation of channel proteins by ROS and NO, the contribution of modifications to the lipid membrane in which these channels are located should not be overlooked. Many of the channels mentioned earlier display a complex relationship not only with ROS and NO but also with the molecular composition of the lipid bilayer. Oxidative stress is the physiological state in which the formation of ROS is favored and oxidative damage to a cell or tissue can subsequently result. A large proportion of ROS are free radicals that possess one unpaired electron, giving them a high degree of reactivity. These species are produced during normal cellular metabolism, particularly cellular respiration that occurs in the mitochondria. Cells have a protective defence system made up of enzymatic and non-enzymatic antioxidants that decrease levels of ROS on their generation (see, e.g., Fig. 2). Consequently cells exist in a state of equilibrium with a fine balance between the production and sequestration of ROS (47). Neurons have an inherent susceptibility to oxidative stress for a number of reasons, including a deficiency in antioxidants compared with other tissues of the body, meaning a reduced ability to defend against ROS attack (9). In addition, there is a greater production of ROS in neurons due to their high rate of metabolic activity and a high content of transition metals that are actively redistributed within the cell (46). Finally, the brain is composed of a high proportion of polyunsaturated fatty acids (PUFA), which are susceptible to free radical attack, causing changes in membrane permeability, fluidity, and function (148).
Higher concentrations of cellular ROS arise due to dysregulation of cellular H2O2, NO, and •OH processing. The increase in cellular ROS is a self-perpetuating mechanism, as ROS damage lipids, which, in turn, also become radical species. Lipid peroxidation occurs via a ROS mediated bis-allylic reaction. The ROS removes a hydrogen atom from the carbon adjacent to the double bond of PUFA, generating species that are highly reactive electrophilic aldehydes (206). This lipid peroxidation impairs the normal function of the cell as lipid radicals and ROS are highly reactive and thus induce lipid–lipid and lipid–protein cross-linking, which leads to an increase in membrane rigidity, a decrease in membrane enzyme activity, as well as alterations to membrane permeability and ion channel activity (8, 185, 220).
Phospholipids, sphingolipids, and sterols are known to regulate a number of channels, including M channels (192), inwardly rectifying Kir channels (150), TRPV1 (100, 138), and TRPM8 (99). These lipid species are particularly susceptible to peroxidative damage due to higher levels of PUFAs, which are thought to be targeted by oxidative radicals in disease (137). Lipid rafts are membrane microdomains that are rich in cholesterol and sphingolipids, which further control ion channel distribution and function. These lipid rafts control the function of a number of ion channels in sensory neurons, including M channels (125), Ca2+-activated Cl− channel ANO1/TMEM16A (76) purinergic P2 receptors (57), and voltage-gated sodium channels (139). These papers show that the manipulation of cholesterol levels within the membrane has profound effects on ion channel function. Therefore, we can consider lipids as playing an important role in signaling by ROS and NO. However, due to the inherent difficulties in manipulating levels of individual lipid species in sensory neurons, their precise contribution to changes in ion channel activity by ROS and RNS remains to be determined.
Concluding Remarks
The picture being assembled in the regulation of ion channels by ROS and RNS is one of extraordinary complexity. These species are able to regulate channels by direct modification of amino acid residues, affecting signaling pathways and/or altering redox state. ROS and RNS have interconnected modes of action and important physiological roles. However, the exact mechanisms of ROS- and RNS-mediated regulation of peripheral sensory neuron excitability are, at best, fragmented and incomplete. We know that many ion channels which are expressed in sensory fibers are sensitive to ROS and RNS, and we also know that these reactive species are being generated at increased levels in many pain states (i.e., in inflammation, injury, etc.); nevertheless, a consistent and inclusive picture is yet to be produced. Obviously, intensive future research is needed; moreover, these future studies will need to consider the system, as a whole, rather than individual reactions and responses, in order to decipher the most important effects. Evidently, a focus on ROS and NO signaling in sensory processing is crucial not only for understanding such processing (particularly in the disease states where these transmitters are implicated) but also for the development of future therapeutics.
Footnotes
Acknowledgments
The work that has led up to this review has been supported by MRC, BBSRC, and the Wellcome Trust.