
Abstract
Introduction
L
The present study provides evidence that hypoxia-responsive microRNA (miR)-101 downregulates cullin 3 by targeting its 3′untranslated region and activates the nuclear factor erythroid-derived 2-related factor 2 (Nrf2)/heme oxygenase-1 (HO-1)/vascular endothelial growth factor (VEGF)/endothelial nitric oxide synthase (eNOS)-derived nitric oxide (NO) production pathway. Elevated NO further amplifies HO-1-mediated, but oxygen-independent, VEGF production through S-nitrosylation of Kelch-like ECH-associated protein 1, leading to a positive feedback circuit between the Nrf2/HO-1 and VEGF/eNOS axes. Moreover, miR-101 overexpression promotes angiogenesis and improves perfusion recovery in a mouse hindlimb ischemia model. These data highlight the importance of miR-101 as a novel therapeutic angiomiR to regulate angiogenesis and vascular remodeling.
VEGF, a known pro-angiogenic factor, is mainly upregulated by the oxygen-sensitive transcription factor hypoxia-inducible factor-1α (HIF-1α), a master factor of cellular response to hypoxia (37). Under normoxic conditions, HIF-1α is hydroxylated at proline residues 402 and/or 564 by prolyl hydroxylase domain protein (PHD) and increases interaction with the von Hippel–Lindau (VHL) protein, leading to ubiquitination-dependent proteasomal degradation (51). However, hypoxia inhibits hydroxylase activity, subsequently leading to HIF-1α stabilization. Conversely, this transcription factor is also regulated in an O2-independent manner via an interaction with heat shock protein 90 (HSP90) and translational activation (14, 51). We and others have shown that degradation products, such as bilirubin and carbon monoxide (CO), of heme by heme oxygenase-1 (HO-1) stimulate angiogenesis by increasing VEGF expression (13, 30), implicating HO-1 as a potent angiogenic modulator.
HO-1, a phase II enzyme, catalyzes the rate-limiting step in heme degradation, resulting in CO liberation, iron, and biliverdin, in which biliverdin is converted to bilirubin by biliverdin reductase (44). HO-1 is induced by protecting the transcription factor nuclear factor erythroid-derived 2-related factor 2 (Nrf2) from Kelch-like ECH-associated protein 1 (Keap1)/cullin (Cul)3/E3 ligase complex-mediated degradation after exposure to various stimulants, including heme, hypoxia, cytokines, and growth factors (52). Recent studies demonstrated that either Cul3 mutation or siRNA-based Cul3 knockdown confers Nrf2 activation and upregulates phase II genes (43, 47). However, Cul3 overexpression negatively regulates the biological function of Nrf2 (43). These evidence suggest a possibility that functional loss of Cul3 may increase Nrf2 stability, which then leads to induction of HO-1 expression and an angiogenic switch.
MicroRNAs (miRNAs) are small non-protein-coding single-stranded RNA molecules of 21–23 nucleotides that negatively regulate protein expression in various organisms, mainly by promoting mRNA degradation or inhibiting mRNA translation via binding to the 3′untranslated region (3′UTR) of their targeted mRNAs (3, 4). It is now evident that miRNAs are major players involved in many aspects of vascular homeostasis and the pathogenesis of cardiovascular diseases, including angiogenesis, vascular remodeling, and myocardial infarction (4, 5, 9, 24). Since Poliseno et al. first demonstrated that miRNAs (miRs)-221/222 modulate the angiogenic activity of stem cell factor by targeting its receptor c-Kit (49), many miRNAs have emerged as critical players in the regulation of angiogenesis and vascular function. For example, miR-34a is known to regulate cardiac aging and function via inhibition of senescence-associated PNUTS expression (6). miRs-199a/590 is also suggested to stimulate cardiac regeneration via proliferation of adult cardiomyocytes (22). In addition, the miR-132 and miR-17–92 cluster, which is expressed in human tumors, promotes tumor angiogenesis by suppressing p120RasGAP expression and the thrombospondin family, respectively (2, 19). Moreover, miR-132 secreted by human pericyte progenitor cells has been shown to improve cardiac function via induction of angiogenesis in a paracrine mode of action after myocardial infarction in the mouse (35). Although some miRNAs, including miRs-10/30 family/126, are shown to facilitate angiogenesis by promoting VEGF signaling (7, 29, 46), miRs-492/195/16/15a function as anti-angiogenic molecules via VEGF expression inhibition (48, 53, 57). Therefore, identifying the function of new pro-angiogenic miRNAs may be valuable to further understand the molecular mechanisms of neovascularization and establish new therapeutic strategies for ischemic diseases.
Some miRNAs are identified to be regulated under hypoxic conditions, and a subset of hypoxic responsive miRNAs, including Let-7 and miRs-93/103/107/424, is considered pro-angiogenic (13, 27, 30). Although miR-101 is downregulated in some human prostate tumors during tumor progression (56), this molecule is upregulated in endothelial cells (ECs) exposed to shear stress and can modulate endothelial homeostasis (12). In this study, we found that miR-101 is increased under hypoxic conditions and stimulates angiogenesis to improve blood perfusion in mouse ischemic hindlimbs via the HO-1/VEGF axis. This effect is achieved by targeting Cul3, a key modulator of Nrf2-mediated HO-1 induction. Our study thus concludes that hypoxia-responsive miR-101 plays an important physiological role in post-ischemic neovascularization and vascular remodeling.
Results
Hypoxia-induced miR-101 regulates Cul3 expression in human umbilical vein endothelial cells
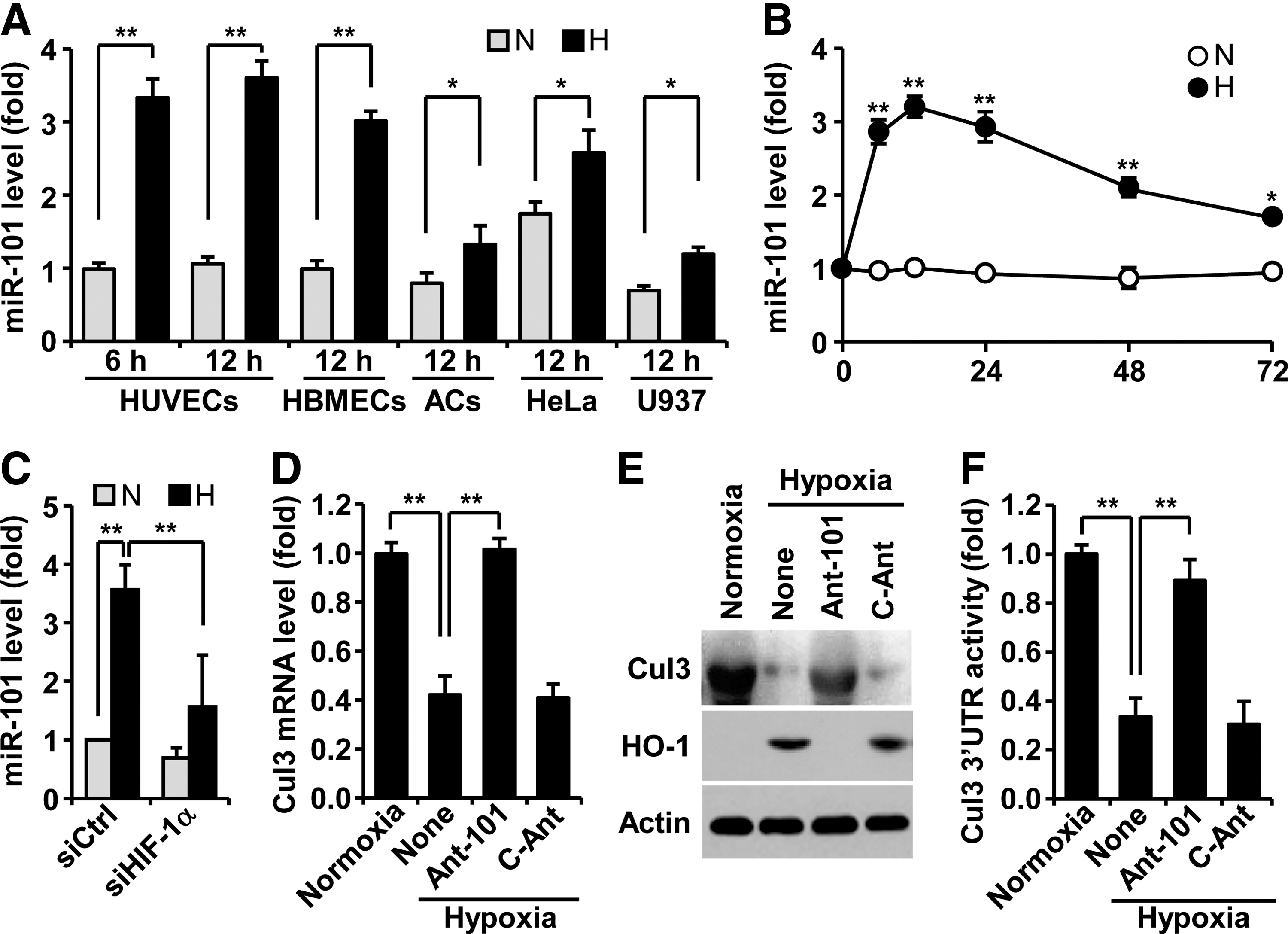
To assess whether hypoxia regulates miR-101 biogenesis, human umbilical vein endothelial cells (HUVECs) were exposed to normoxia and hypoxia, and expression levels of miR-101 were determined by real-time polymerase chain reaction (PCR). Hypoxia induced about a 3.5-fold increase in miR-101 expression at 6 h, as compared with normoxia, and no further increase at 12 h (Fig. 1A). Human brain microvascular ECs revealed a similar response to hypoxia-induced miR-101 expression, and additional cell types, including astrocytes, HeLa, and U937, also increased, but to a lesser extent (Fig. 1A). Interestingly, basal levels of miR-101 in normoxic HeLa cells were significantly higher than those of other normoxic cells. The level of miR-101 rapidly increased in ECs 6 h after exposure to hypoxia and remained almost unchanged until 24 h. Thereafter, a gradual decrease in its steady-state level was observed at 72 h (Fig. 1B). Elevated miR-101 levels in hypoxic HUVECs were blocked by HIF-1α knockdown (Fig. 1C), suggesting that miR-101 is upregulated in an HIF-1α-dependent manner under hypoxic conditions. We next predicted highly reliable targets of miR-101 using TargetScan, microRNA.org, and miRDB. Among 31 target genes, Cul3 was selected as a new target of miR-101 (Supplementary Fig. S1A; Supplementary Data are available online at
miR-101 upregulates Nrf2-dependent HO-1 expression by directly targeting Cul3
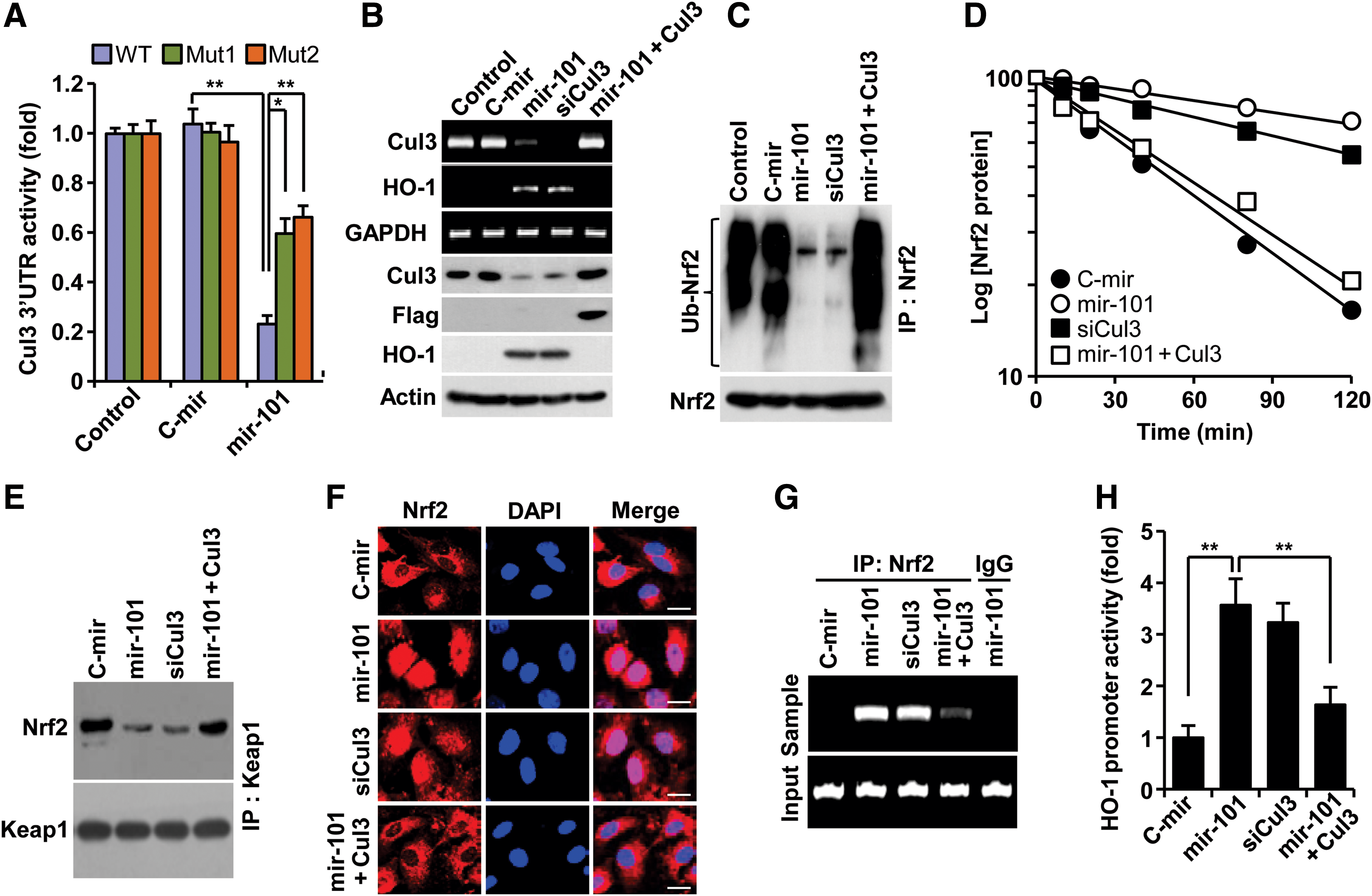
Since Cul3 is a scaffold protein in the E3 ligase complex, which negatively regulates HO-1 expression via ubiqutination-mediated proteasomal degradation of Nrf2 from Nrf2-Keap1 system (38), we determined a functional role of miR-101 in Nrf2-dependent HO-1 expression via Cul3 downregulation. Transfection with a precursor miR-101 (pre-mir-101) expression vector increased mature miR-101 levels (Supplementary Fig. S2A). Overexpression of mir-101 effectively inhibited Cul3 3′UTR-WT reporter activity, as compared with the control. This inhibition was markedly, but not completely, reversed by each single mutation of two putative miR-101-binding seed sites (Fig. 2A; Supplementary Fig. S1D). These data indicate that miR-101 binds to both sites. As expected, mir-101 overexpression resulted in a decrease of Cul3 mRNA and protein levels and a subsequent increase in HO-1 expression (Fig. 2B), leading to elevated HO-1 enzyme activity (Supplementary Fig. S2B). We further examined whether miR-101 elicits Nrf2-dependent HO-1 expression via regulation of Cul3-based E3 ligase activity using several biochemical methods. We found that miR-101 significantly inhibited Nrf2 ubiquitination compared with the negative control (Fig. 2C), which resulted in about a fivefold increase in Nrf2 half life (46 vs. 228 min) (Fig. 2D). Furthermore, mir-101 overexpression markedly inhibited the interaction between Keap1 and Nrf2 (Fig. 2E). These effects of miR-101 were nearly abolished by Cul3 overexpression. Further, Cul3 knockdown effectively increased the half life of Nrf2 protein similar to miR-101 (Fig. 2B–E). As a result of this inhibition, miR-101 increased Nrf2 nuclear translocation as confirmed by confocal microscopy (Fig. 2F). We next examined a functional role of nuclear Nrf2 accumulated by miR-101 in HO-1 promoter activation. Chromatin immunoprecipitation (ChIP) assay showed an increase in functional binding to the HO-1 promoter in HUVECs overexpressing mir-101 (Fig. 2G). Moreover, luciferase reporter assay indicated that miR-101 significantly increased HO-1 promoter transcriptional activity as compared with the control (Fig. 2H). Expectedly, the promotive effects of miR-101 on Nrf2-dependent HO-1 promoter activity were abolished by Cul3 overexpression. Moreover, Cul3 knockdown showed a similar outcome to Nrf2-mediated HO-1 promoter activity induced by miR-101 (Fig. 2F–H). We further examined the functional role of Nrf2 in hypoxia- and miR-101-mediated HO-1 expresion using siRNA technology. Nrf2 knockdown suppressed the induction of HO-1 expression in ECs by hypoxia and miR-101 (Supplementary Fig. S3A, B). These results indicate that miR-101 increases Nrf2-dependent HO-1 expression through direct targeting of Cul3 3′UTR.
miR-101 promotes HIF-1α-mediated VEGF expression via HO-1 induction
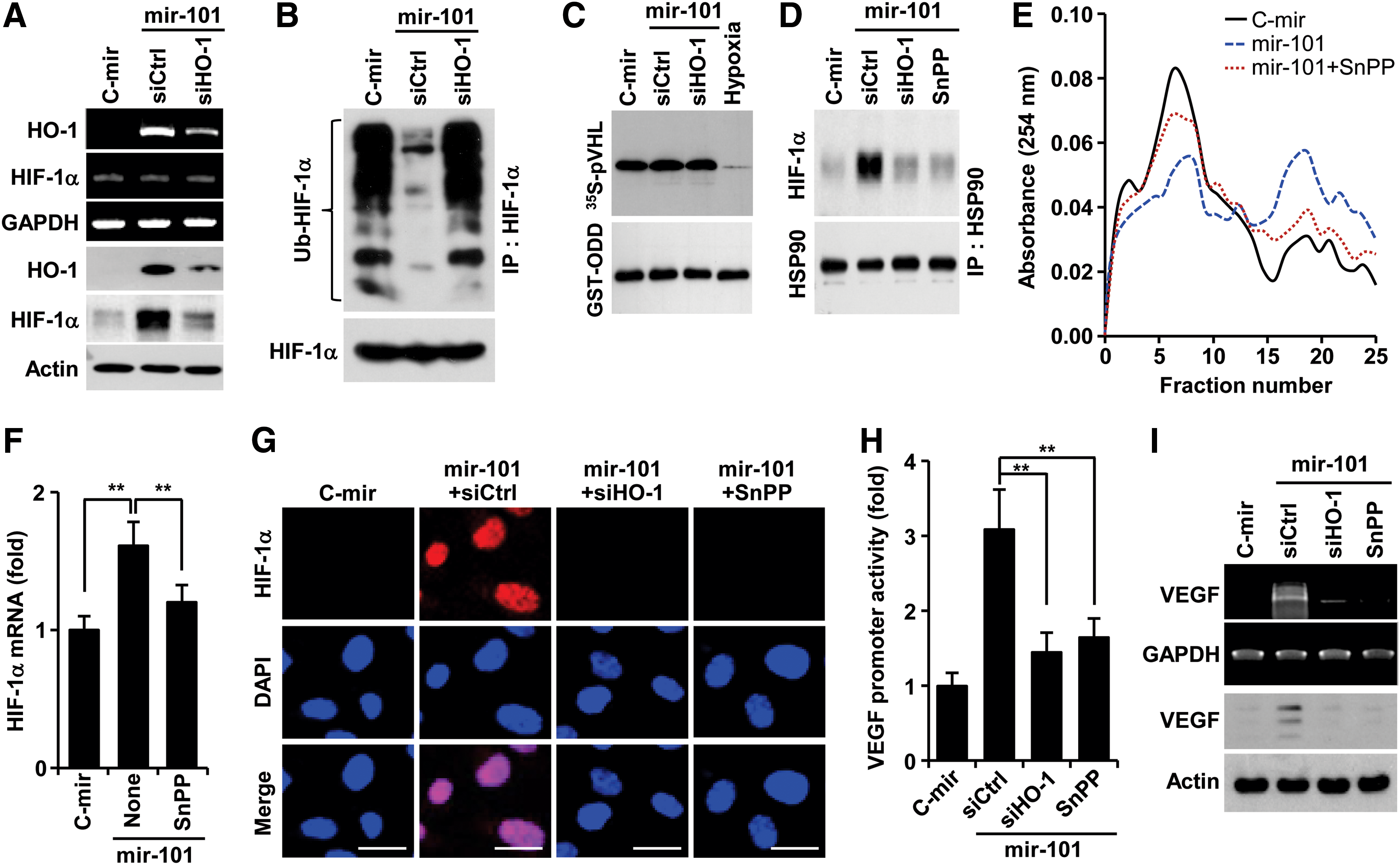
The reaction product of HO-1, CO elevates VEGF expression by increasing HIF-1α protein levels via two distinct mechanisms, translational activation and stabilization of HIF-1α protein (14). We examined whether miR-101 promotes HO-1-mediated stabilization of HIF-1α, which plays an important role in VEGF expression. HUVECs overexpressing mir-101 upregulated HO-1 expression and subsequently stabilized HIF-1α protein, and these increases were inhibited by transfection with HO-1 siRNA (Fig. 3A). These results suggest that miR-101 regulates post-translational increases in HIF-1α protein level in an HO-1 dependent manner. However, miR-101 inhibited ubiquitination of HIF-1α, which was reversed by HO-1 knockdown (Fig. 3B), without affecting biological activity of PHD, the key enzyme responsible for degrading HIF-1α (34), as compared with its activity under hypoxic condition as a positive control (Fig. 3C). This indicates that miR-101 enhances HIF-1α protein levels in an HO-1-dependent, but not in a PHD-dependent manner. Since HSP90 inhibits HIF-1α ubiquitination by physically interacting with HIF-1α, leading to protection of HIF-1α from PHD-independent degradation (14), we further examined the effect of miR-101 on the interaction between HSP90 and HIF-1α. Overexpression of mir-101 enhanced the interaction between HIF-1α and HSP90, and this interaction was blocked by HO-1 knockdown and the HO-1 inhibitor tin-protoporphyrin IX (SnPP) (Fig. 3D). Interestingly, miR-101 increased the formation of a high-molecular-weight polysome complex, which was associated with a high level of HIF-1α mRNA in pooled polysome fractions from 16 to 22. These effects were attenuated by co-treatment with SnPP (Fig. 3E, F), reflecting an increase in HIF-1α mRNA's translational activity. Consequently, miR-101 increased nuclear accumulation of HIF-1α, and this event was suppressed by treatment with HO-1 siRNA and SnPP (Fig. 3G). In addition, miRNA-101 significantly increased VEGF promoter activity and expression, and both effects were reduced by HO-1 siRNA and SnPP (Fig. 3H, I). The HO-1 byproducts, CO, biliverdin, and bilirubin, but not Fe2+, increased the protein levels of HIF-1α and VEGF as well as tube formation (Supplementary Fig. S4A, B). These results indicate that miR-101 stabilizes HIF-1α protein and induces VEGF expression and angiogenesis by increasing HO-1 expression and its reaction products.
miR-101 amplifies VEGF expression via a positive feedback circuit between HO-1 activity and VEGF/endothelial nitric oxide synthase axis by S-nitrosylating Keap1
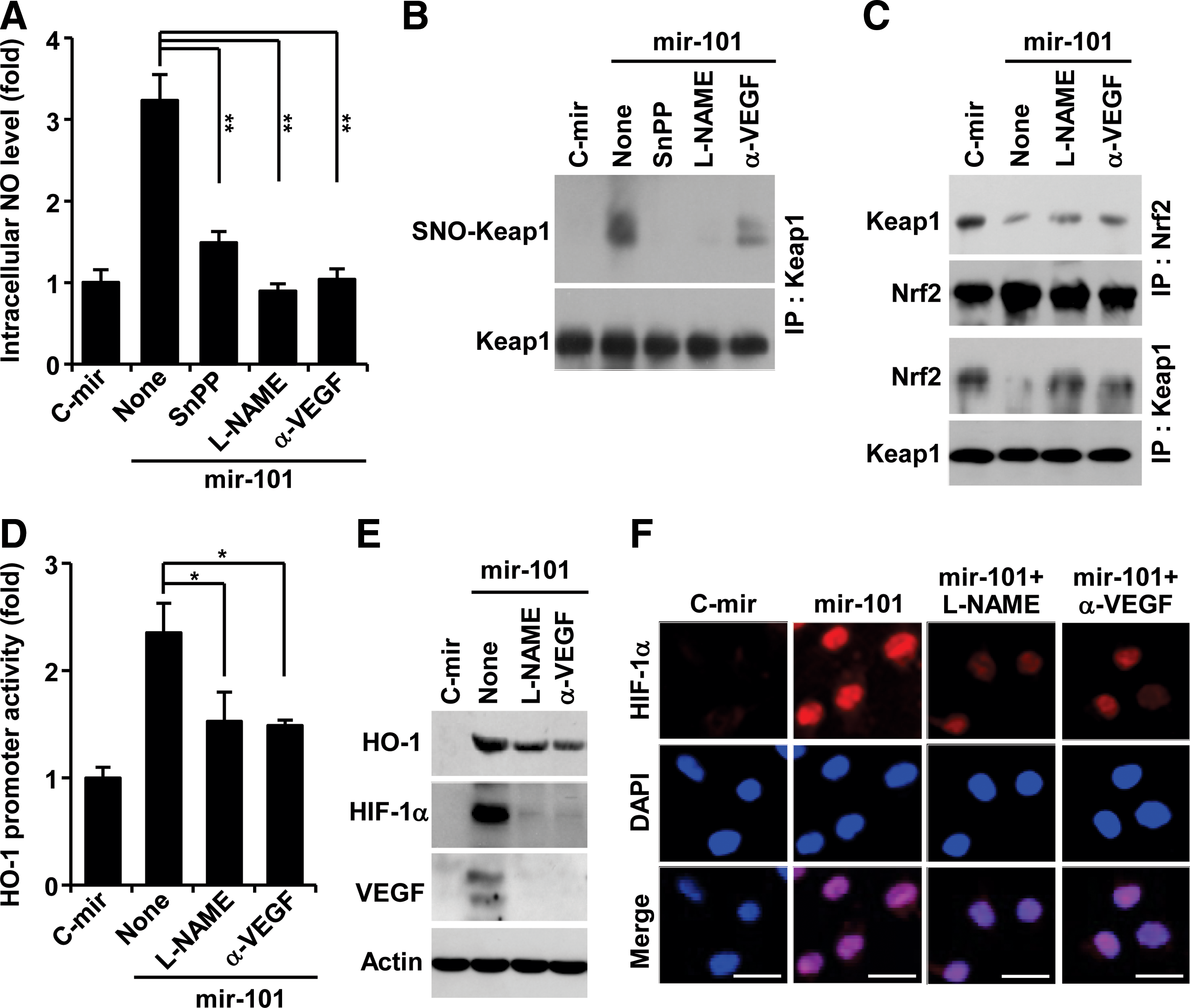
VEGF promotes endothelial nitric oxide synthase (eNOS)-derived nitric oxide (NO) production in ECs, and NO plays important roles in a variety of physiological systems via modification of protein thiols (26, 50). We examined the effects of miRNA-101 on the intracellular NO level in ECs. HUVECs expressing mir-101 exhibited significant increases in NO production, which was inhibited by treatment with SnPP, the NOS inhibitor N
G-nitro-
miR-101 stimulates angiogenesis in vitro and in vivo
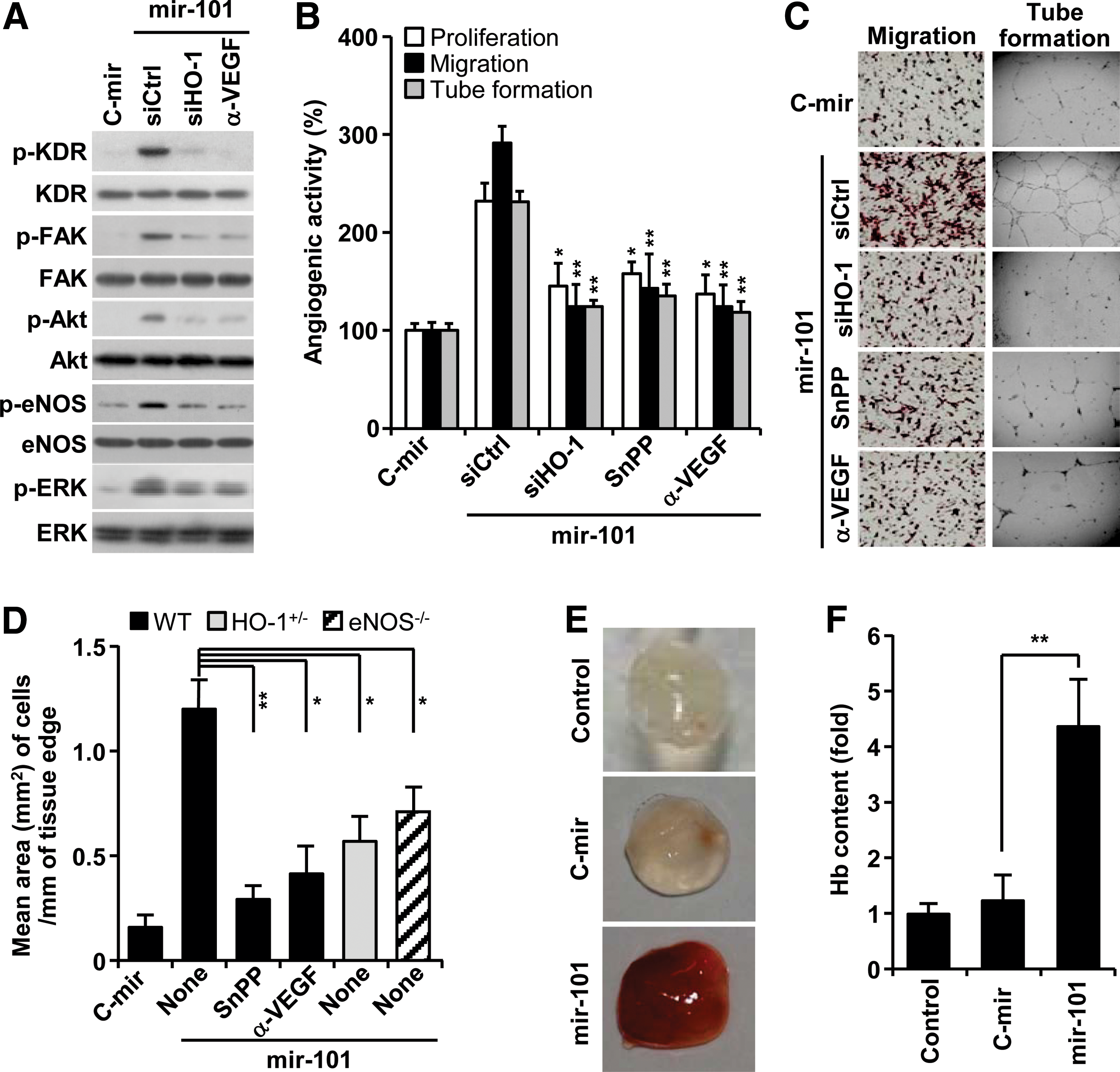
Based on our data that miRNA-101 increased VEGF production via HO-1 induction (Fig. 3, 4), we further examined the effects of miR-101 on the activation of angiogenic signal mediators (14). Overexpression of mir-101 increased VEGF receptor-2 (KDR) phosphorylation and its downstream effectors, including FAK, Akt, eNOS, and ERK, and these effects were blocked by HO-1 siRNA and an anti-VEGF antibody (Fig. 5A). As expected, miRNA-101 increased proliferation, migration, and tube formation of HUVECs. These effects were significantly suppressed by HO-1 siRNA, SnPP, and an anti-VEGF antibody (Fig. 5B, C). Mouse aortic rings transfected with mir-101 lentivirus, which was confirmed to express mature miR-101 and activate Cul3-mediated HO-1/VEGF axis in HUVECs (Supplementary Fig. S5A, B), led to longer and more abundant sprouts compared with control aortic rings. Importantly, this effect was abolished by the addition of SnPP and an anti-VEGF antibody (Fig. 5D; Supplementary Fig. S6). Moreover, the angiogenic sprouting activity of miR-101 was significantly attenuated in aortic rings from HO-1+/− and eNOS−/− mice (Fig. 5D; Supplementary Fig. S6). These data suggest that miRNA-101 stimulates angiogenesis by amplifying VEGF production via a cross-interaction between HO-1 and eNOS pathways. We further examined the role of miR-101 in the development of functional vasculature in nude mice by Matrigel plug assay. The plug containing mir-101-overexpressing HUVECs appeared dark red (Fig. 5E) and abundantly filled with intact red blood cells (RBCs), as determined by measuring hemoglobin contents (Fig. 5F). However, the plug containing control mir-expressed HUVECs was pale in color and minimally influxed with RBCs, comparable to Matrigel alone (Fig. 5E, F). These results indicate that miR-101 is capable of promoting a functional vasculature via in vivo angiogenesis.
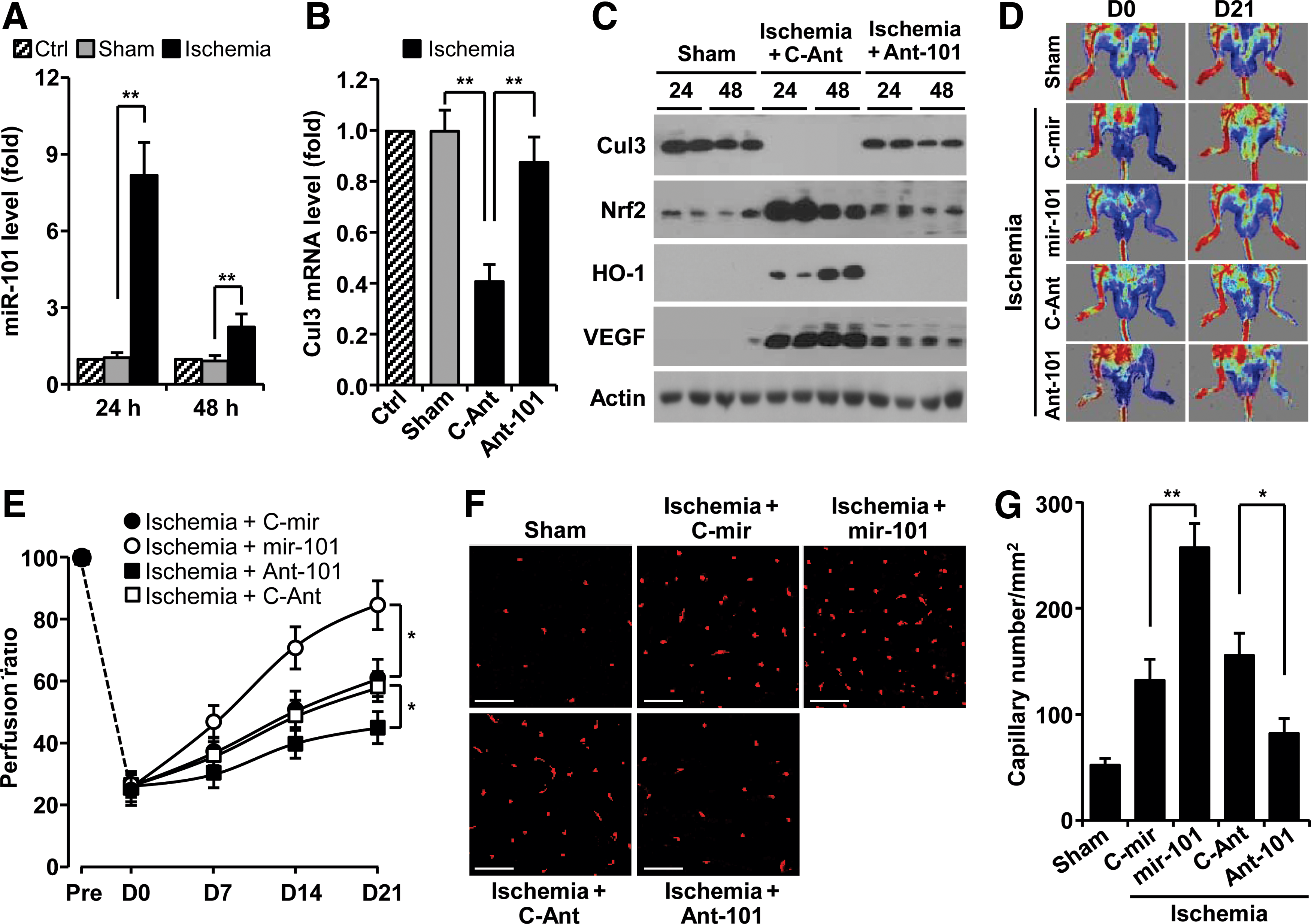
Hypoxia-responsive miR-101 promotes neovascularization
We next examined whether miR-101 expression is regulated in a mouse hindlimb ischemia model. The levels of miR-101 were increased about eight and twofold in gastrocnemius muscles harvested at 24 and 48 h after femoral artery ligation, compared with those of sham-operated muscles (Fig. 6A). As expected, the levels of Cul3 mRNA and protein were effectively decreased in ligated hindlimb muscles compared with sham tissues (Fig. 6B, C). However, the protein levels of Nrf2, HO-1, and VEGF were dramatically increased in ligated tissues (Fig. 6C). These expressional changes were restored by treatment with antagomiR-101 (Fig 6B, C). We further examined the in vivo role of hypoxia-responsive miR-101 in neovascularization and blood flow after hindlimb ischemia. Mice treated with mir-101-expressing lentivirus demonstrated significantly improved blood flow in ischemic hindlimbs compared with mir-control lentivirus-transfected mice (Fig. 6D, E). In contrast, mice treated with antagomiR-101 showed impaired perfusion recovery in the ischemic hindlimb compared with control animals treated with control antagomiR (Fig. 6D, E). Consistent with improved perfusion recovery, ischemic muscle from miR-101-treated mice showed higher capillary density than mock-treated mice (Fig. 6F, G). Moreover, treatment with antagomiR-101 reduced the number of capillaries in the ischemic hindlimb. Taken together, our results demonstrate that miR-101 promoted functional angiogenesis and was associated with significantly improved blood perfusion after hindlimb ischemia.
Discussion
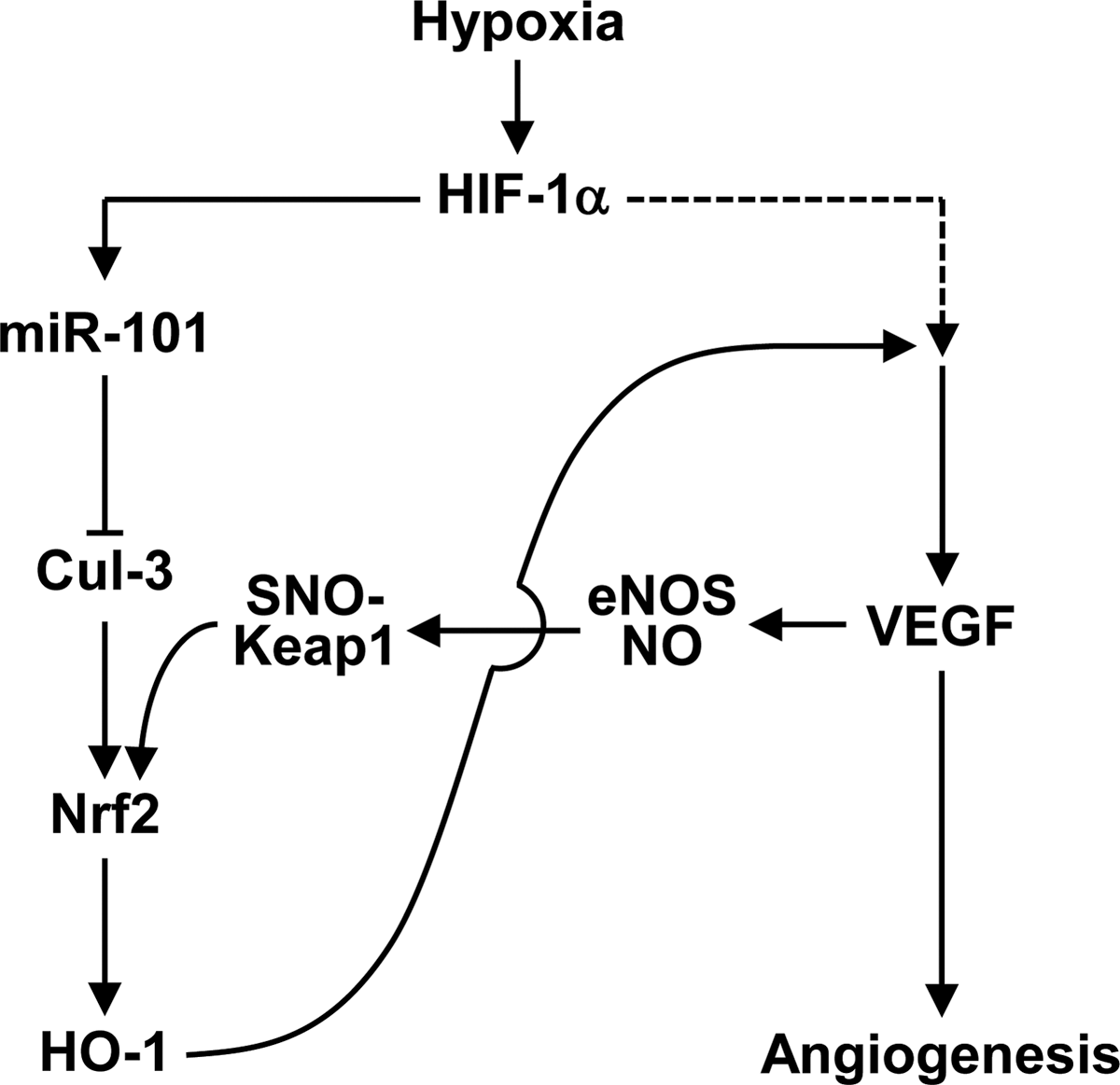
Several hypoxia- or ischemia-responsive miRNAs, including Let-7 and miRs-93/103/107/210/424 (13, 27, 30, 39), are known to play an important physiological role in post-ischemic vascular remodeling and angiogenesis. Unfortunately, there is still limited information on the role of miRNAs in ischemia-associated vascular homeostasis and diseases. A previous study provided the possibility that hypoxia can increase miR-101 expression in ECs compared with normoxic cells using array analysis (27). However, its expressional regulation and function have not been elucidated. In this study, we identified an important role of hypoxia-responsive miR-101 in promoting angiogenesis and blood perfusion. The conclusion of a novel role of miR-101 is supported by several lines of evidence. First, we provide data that miR-101 is upregulated both in vitro and in vivo in response to hypoxia and increases VEGF expression. The expression of VEGF is associated with HO-1 upregulation via activation of the Keap1/Nrf2 pathway by targeting the 3′UTR of Cul3. Second, our data show that an miR-101-mediated increase in VEGF expression is amplified via a positive feedback loop, where VEGF-induced S-nitrosylation of Keap1 promotes Nrf2-dependent HO-1 induction. Finally, our in vitro experiments show that miR-101 improves angiogenic behavior of ECs via activation of VEGF-mediated angiogenic signal cascades. In vivo, we confirmed the role of miR-101 in angiogenesis and hemodynamic recovery in a mouse hindlimb ischemia model. Based on these findings, we identify miR-101 as a novel angiomiR, which promotes neovascularization via a positive circuit between HO-1/CO and VEGF/eNOS/NO axis by targeting Cul3 (Fig. 7).
Although increasing evidence indicates that several miRNAs play an important role in vascular homeostasis and angiogenesis, the molecular targets and biological functions of only a few angiomiRs are identified in ECs and non-ECs (11, 58). One main hurdle that has limited the determination of a specific miRNA function is the difficulty of identifying determinant target genes. The first identified target of miR-101 is EZH2, a tumor suppressor that functions as a negative regulator of a mammalian histone methyltransferase. The miR-101 locus is somatically lost in some solid tumors, leading to overexpression of EZH2 and concomitant dysregulation of epigenetic pathways to promote tumor progression (54, 56). Recently, miR-101 induced in human breast tumor MCF-7 cells by starvation and etoposide was shown to specifically downregulate several autophagic genes, including STMN1, ATG4D, and RAB5A, leading to an efficient decrease of tumor cell autophagy (25). Interestingly, contradictory reports show that under hypoxic conditions, miR-101 can be increased in ECs (27) and downregulated in squamous cell carcinoma (39). However, our data demonstrate that miR-101 was effectively induced in response to hypoxia, particularly in an HIF-1α-dependent manner, in ECs compared with other cell types. Thus, the mechanism for miR-101 function and expression is mediated by a diverse range of targets and transcriptional machinery that likely vary depending on the cell type and environmental setting.
Here, we have found new potential target genes, including Cul3, Cul4B, and Cul5, for hypoxia-responsive miR-101 by performing in silico analysis using algorithm of TargetScan (Supplementary Fig. S1B). Cullin family proteins consisting of seven members are molecular scaffolds for E3 ligase, which play diverse and essential roles in many biological processes via mediating ubiquitination of target proteins. Crucial substrates of Cul3, Cul4B, and Cul5 are identified as Nrf2, CDT1, and EPAS1, which are involved in phase II gene expression, chromatin regulation, and cartilage destruction, respectively (21, 59). In fact, functional inhibition of Cul3 effectively stabilizes the transcription factor Nrf2 (21). Importantly, a wide range of evidence shows that Nrf2 is an important transcription factor for phase II genes, including HO-1, which promotes angiogenesis and survival of ECs by increasing VEGF expression (32, 33). Here, we provide evidence that hypoxia-inducible miR-101 inhibits Cul3 expression by directly targeting its 3′UTR, leading to upregulation of Nrf2-mediated HO-1 expression and concomitant promotion of angiogenesis. Thus, this study demonstrates a signaling pathway that links miR-101 to Nrf2-dependent HO-1 expression via direct targeting of Cul3.
It is well established that expression of HO-1 is regulated by the Nrf2/Keap1/Cul3/Rbx1 E3 ligase complex. As mentioned earlier, Cul3 binds the RING domain protein Rbx1 that, in turn, recruits an E3 ligase, which facilitates Nrf2 ubiquitination and degradation. Dominant-negative Cul3 is unable to recruit E3 ligase and stabilize Nrf2, a transcription factor required for phase II genes (21, 38). Moreover, siRNA-mediated knockdown of Cul3 significantly increased Nrf2 protein levels and induced phase II enzymes (43). Of these enzymes, HO-1 catalyzes heme to produce CO, biliverdin, and bilirubin, which play important roles in preventing EC death and promoting angiogenesis (8). In this study, we identify hypoxia-responsive miR-101 as a new member of the angiomiR family. In fact, this miRNA promotes both in vitro and in vivo angiogenesis by activating the HO-1/HIF-α/VEGF axis via Cul3 targeting. In keeping with these findings, several lines of evidence also show that hypoxia and hemin (an inducer of HO-1) induce HO-1 expression and concomitant augmentation of VEGF production, which enhances EC proliferation and in vitro capillary formation (18, 32). In addition, hemin-induced VEGF production and angiogenesis were not elicited in normal ECs by co-treatment with the HO-1 inhibitor (20) or in ECs from HO-1−/− mice (16), strongly implicating a role of HO-1 byproducts in angiogenesis. The HO-1 reaction product, CO, promoted VEGF expression via translational activation and stabilization of the HIF-1α protein (14), and expression of VEGF by hypoxia was significantly blocked by inhibition of HO-1 activity (20, 32). These findings indicate that HO-1 is critically involved in hypoxia-induced VEGF production. In accordance with this view, the present study demonstrates that hypoxia-responsive miR-101 increases HIF-1α and VEGF protein levels, which were suppressed by HO-1 siRNA and an HO-1 inhibitor. In addition, our data show that miR-101 increased HIF-1α protein levels via HSP90-mediated HIF-1α stabilization and translational synthesis of HIF-1α, whose effects were associated with HO-1 induction and activity. Moreover, these results indicate that miR-101 is a typical angiomiR, which functions as a positive regulator of VEGF expression via HIF-1α activation by modulating the Cul3/Nrf2/HO-1 pathway.
The HO-1/CO pathway positively modulates HIF-1α-mediated VEGF synthesis and promotes angiogenesis via activation of several intracellular signal pathways, including PI3K/eNOS-derived NO production. Conversely, VEGF activates HO-1 via activation of Nrf2 (40). Since both CO and NO, produced from HO-1 and eNOS, respectively, share similar biological properties, such as the ability to regulate angiogenesis and vasodilation, it is now evident that both gaseous molecules do not always work independently, but rather can cross-modulate each other's biological activity in the vasculature. Although both molecules readily react with ferrous heme-iron, only NO modifies free cysteine residues to disulfide linkage or S-nitrosylation and also interacts with iron–sulfur clusters. NO can induce S-nitrosylation of critical cysteine residues in Keap1, which coincides with disruption of Nrf2 from the Keap1-Cul3 ubiquitination system (55). Although several mechanisms for VEGF-mediated Nfr2 activation have been proposed (23, 40), our data show that miR-101 elicits Keap1 S-nitrosylation, a crucial step of Nrf2-dependent HO-1 induction, which is associated with activation of the VEGF/eNOS axis (Fig. 7). These evidence suggest that miR-101 stimulates the angiogenic processes via a positive feedback loop between Nrf2/HO-1 and VEGF/eNOS/NO axes after initially targeting the 3′UTR of Cul3.
Angiogenesis is a physiological process that may also occur as a natural repair mechanism against ischemic diseases, such as ischemic stroke, myocardial infarction, and peripheral arterial disease. Although several pro-angiomiRs have been identified (31, 58), there is still limited application to therapeutic angiogenesis. Previous reports point to the HO-1/VEGF/eNOS axis as one of the determinants of angiogenesis and improved blood flow in the ischemic hindlimb (40, 45). These data suggest that miRNAs activating this signaling axis can be used as potential therapeutic agents in patients with vascular obstructions. We confirmed the angiogenic function of miR-101 in a murine hindlimb ischemia model by gain- and loss-of-function experiments using exogenous miR-101 and its antagomiR. Indeed, mir-101 overexpression enhanced perfusion recovery via an increase in capillary density in the ischemic hindlimb. However, antagomiR-101 impaired angiogenic responses to ischemia. These data suggest that miR-101 promotes angiogenesis via activation of the HO-1/HIF-1α/VEGF signaling axis by directly targeting Cul3. However, we cannot exclude the possibility that a pro-angiogenic function of miR-101 can be associated with the regulation of other targets, because more than 30 genes can be regulated as determined by computational algorithms for miRNA target perfection. One of miR-101 target genes, Cul5 can regulate the post-translational stability of EPAS, which promotes VEGF expression. This possibility is currently under investigation.
In summary, our present findings demonstrate that miR-101 induced by hypoxia is an important regulator in vascular remodeling and angiogenesis. Increased miRNA-101 expression in hypoxic ECs and hindlimb muscles was identified as a novel mechanism contributing to angiogenesis by triggering HO-1/HIF-1α/VEGF signaling axis via negative expression of Cul3, a critical scaffold protein in the E3 ligase complex. Moreover, miR-101 improved neovascularization and blood flow recovery in the ischemic hindlimb. Therefore, hypoxia-responsive miR-101 may provide a potential therapeutic avenue for the treatment of ischemia-related diseases. Importantly, this angiogenic modulation is a potential novel mechanism independent of canonical (oxygen-dependent) HIF-1α-mediated angiogenic pathway.
Materials and Methods
Plasmid construction
To generate the pre-miR-101 or control pre-miR expression vectors, oligonucleotides of human pre-miR-101 and control pre-miR were purchased from Bioneer and cloned into BamHI and HindIII sites of pSilencer 2.1-U6 (Invitrogen). The 3′UTR (∼0.5 kb) of Cul3 was prepared using human genomic DNA by PCR using the following primers, 5′-CGGCCTCGAGATTCTTGTCAGATATCCTG-3′ (forward) and 5′-AATGCGGCCGCACACCTGACTTAGGTACTA-3′ (reverse). The PCR product was ligated at the XhoI and NotI sites of psiCHECK™-2 vector (Promega). The luciferase reporter vectors, pGL3-HO-1-Luc and pGL3-VEGF-Luc, were used for promoter activity assay (14, 36).
Cell culture and treatment
HUVECs were cultured as previously described (36). Cells were transfected with 1 μg/ml of expression vectors (pSilencer 2.1-U6/pre-miR-101, pSilencer 2.1-U6/control pre-miR, psiCHECK™-2/Cul3 3′UTR, psiCHECK™-2/Cul3 3′UTR mutant 1, psiCHECK™-2/Cul3 3′UTR mutant 2, pGL3-HO-1-Luc, pGL3-VEGF-Luc, and p3xFLAG-CMV10-Cul3) and 100 nM of siRNA targeted to HO-1 or Nrf2 or Cul3 (Santa Cruz Biotechnology), miR-101 (Qiagen), control miRNA (Qiagen), or antagomiR-101 (Bioneer) using a microporator (NanoEnTek) or Lipofectamine™ 2000 (Invitrogen) for 24 h and treated with or without 20 μM SnPP (Frontier Scientific), 1 mM L-NAME (Sigma-Aldrich), or 0.5 μg/ml VEGF-neutralizing antibody (R&D Systems) to VEGF for 12 h. The cells were used for further analyses.
Hypoxia treatment
Cells were maintained in a hypoxic chamber (Coy Laboratory Products, Inc.) flushed with a gas mixture containing 94% N2 and 5% CO2. Under these conditions, O2 levels in the medium were determined to be 2±1%.
In vitro angiogenic assays
HUVECs were transfected with pSilencer 2.1-U6/mir-101 or in combination with HO-1 siRNA, followed by treatment with SnPP and a VEGF neutralizing antibody. Proliferation, migration, and tube formation of HUVECs were carried out as previously described (41).
Ex vivo aortic ring and Matrigel plug assays
All animal studies were approved by the Institutional Animal Care and Use Committee of the Kangwon National University. Aortic rings from 7-week-old female C57BL/6J wild-type, eNOS−/−, and HO-1+/− mice (Jackson Laboratory) were transduced with lentivirus carrying either mir-101 or control mir (Thermo Fisher Scientific, Inc.) and incubated at 37°C for 18 days. Aortic ring sprouts were photographed using a microscope that was equipped with a digital camera. For a quantitative assessment of sprouting, the area of sprouting per millimeter of tissue was assessed using Image J software (NIH;
PCR analysis
miRNAs were isolated using an miRNeasy mini kit (Qiagen) according to the manufacturer's protocols. cDNA for determining miRNAs were obtained from 1 μg of miRNAs using a miScript II RT kit. Quantitative real-time PCR (qRT-PCR) was performed with miScript SYBR Green PCR kit according to the manufacturer's instructions. The miR-101 level was analyzed by miScript primer assay using a target miR-101-specific primer and universal primer (Qiagen). In addition, the Cul3 mRNA expression levels were determined by iTaqTM SYBR Green Supermix with ROX (BioRad) with ABI PRISM 7000 Sequence Detection System (Applied Biosystem). The following sets of primers were used: 5′-GTGGTAAACCAACACAGCGG-3′ (forward) and 5′-GGGTCGGATTCACCTTGTTT-3′ (reverse) for Cul3; 5′-CGCCACAGTTTCCCGGAGGG-3′ (forward) and 5′-CCCTCCAAAATCAAGTGGGG-3′ (reverse) for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The fold change of miR-101 and Cul3 mRNA was calculated using the 2−ΔΔCt method as previously described (42). For reverse transcription PCR analysis, total RNAs were isolated from the indicated cells using Trizol reagent (Invitrogen). RT-PCR analysis was performed as previously described (14). The following sets of primers were used: 5′-AGTCGGACAGCCTCAC-3′ (forward) and 5′-TGCTGCCTTGTATAGGA-3′ (reverse) for human HIF-1α; 5′-CAGGCAGAGAATGCTGAG-3′ (forward) and 5′-GCTTCACATAGCGCTGCA-3′ (reverse) for human HO-1; 5′-GAGAATTCGGCCTCCGAAACCATGAACTTTCTGT-3′ (forward) and 5′-GAGCATGCCCTCCTGCCCGGCTCACCGC-3′ (reverse) for VEGF; 5′-CAGGGCTGCTTTTAACTCTG-3′ (forward) and 5′-TAGAGGCAGGGATGATGTTC-3′ (reverse) for GAPDH. PCR products were analyzed on 1.2% agarose gels.
Luciferase assay
HUVECs were transfected with psiCHECK™-2 vector containing the Cul3 3′UTR region as well as antagomiR-101 or negative control antagomiR for 24 h and were maintained under normoxia or hypoxia for 12 h. Promoter activity was assayed using a dual-luciferase report assay kit (Promega). Cells were also transfected with pGL3-HO-1-Luc (or pGL3-VEGF-Luc) or in combination with pSilencer 2.1-U6/pre-miR-101 (mir-101), pSilencer 2.1-U6/control pre-miR (C-mir), p3xFLAG-CMV10-Cul3, Cul3 siRNA, HO-1 siRNA, or control siRNA for 24 h, followed by treatment with SnPP, L-NAME, and a VEGF neutralizing antibody for 12 h. Luciferase activity was assayed using a luciferase assay system.
ChIP assay
ChIP assay was performed as previously described (36). Briefly, DNA/protein cross-linking was obtained in HUVECs by incubating cells for 20 min at 37°C in 1% formaldehyde. After sonication, chromatin was immunoprecipitated overnight with 10 μl of anti-Nrf2 antibody (Santa Cruz Biotechnology). Targeted promoter sequences of HO-1 were identified by PCR (150 bp, 30 cycles at 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s) using primer pairs spanning HO-1-specific promoter regions containing the antioxidant response element-binding sequence. The products (150 bp) were identified on a 2% agarose gel. The primer sequences are as follows: 5′-GGGATTAAACCTGGAGCAGC-3′ (forward) and 5′-TTTTTCCTGCTGAGTCACGG-3′ (reverse) for HO-1.
Immunoprecipitation
Cellular proteins from HUVECs were incubated with an antibody for Keap1 or Nrf2 in RIPA buffer with constant rotation overnight at 4°C. Cell lysates were incubated with antibodies against Keap1 (Santa Cruz Biotechnology), Nrf2, HIF-1α (Novus Biologicals), and HSP90 (Santa Cruz Biotechnology), and immune complexes were collected by centrifugation after incubation with an antibody against protein G-Sepharose (Millipore). Immunoprecipitates were analyzed by sodium dodecyl sulphate polyacryl amide gel electrophoresis, followed by Western blot analysis using the indicated antibodies.
Biochemical analyses
Western blot analysis for target proteins in whole-cell lysates as well as cytosolic and nuclear fractions was performed as previously described (36). S-nitrosylation of Keap1 was measured by an S-nitrosylated protein detection kit (Cayman) according to the manufacturer's protocols. The intracellular NO level was measured by using 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (Molecular Probes) according to a previous protocol (15). PHD activity was assayed by determing the interaction between [35S]methionine-labeled VHL protein and purified glutathione S-transferase-oxygen-dependent degradation domain (amino acids 401–603 of human HIF-1α) that was preincubated with cell lysates as previously described (14). Hemoglobin was measured using the Drabkin reagent kit 525 (Sigma-Aldrich) to quantify blood vessel formation in Matrigel plugs.
Immunohistochemistry and vascular density
HUVECs were fixed in 3.7% formaldehyde for 15 min at room temperature, washed gently, and permeabilized with 0.1% saponin. Cells were incubated with antibodies (1:100) against Nrf2 and HIF-1α for 2 h and then incubated with Alexa Fluor antibody (1:200; Invitrogen). For nuclear staining, cells were further incubated for 30 min with 4′,6-diamidino-2-phenylindole (1 μg/ml; Sigma-Aldrich). After mounting, nuclear translocation of transcription factors was observed by confocal microscopy. For measurement of vascular density, the gastrocnemius muscles were dissected out from ischemic mouse hindlimbs, and coronal sections (10 μm) were fixed with formalin, processed, and stained for blood vessels with Texas Red-conjugated mouse CD31 antibody (DB Pharmingen).
Polysome assay
Polysome analysis was performed as previously described (14). HUVECs were lysed in 400 μl lysis buffer. After centrifugation at 2000 g for 5 min, the supernatant was added to heparin (a broad-range RNase inhibitor) at a final concentration of 200 μg/ml. After removing debris by centrifugation at 10,000 g for 5 min, the cytoxolic supernatants were laid on a 20–50% sucrose gradient (total volume: 5 ml), and centrifuged at 39,000 rpm for 120 min in Beckman SW-55Ti rotor at 4°C. Fractions (0.2 ml) were collected and monitored for absorbance at 254 nm. RNA was isolated from pooled polysome fractions from 16 to 22, and HIF-1α mRNA levels were analyzed by qRT-PCR.
Hindlimb ischemia
Lentivirus carrying either control pre-miR or pre-miR-101 and antagomiR-101 were delivered into the right gastrocnemius muscle of 8-week-old female C57Bl/6J mice by three injections (total 2.5×106 virus particles in 50 μl/mouse and total 8 mg/kg of antagomiR-101 or control antagomiR) under anesthesia with ketamine (100 mg/kg) und xylazine (2 mg/kg). After 30 min, unilateral femoral artery occlusion was performed by double ligation of the superficial femoral artery proximal to the deep femoral artery and distal femoral artery. Mice were also i.v. injected with 8 mg/kg of antagomiR-101 at days 3 and 7 after hindlimb ischemia. Sham-operated control animals were subjected to the same surgical protocol, but the femoral artery was not ligated. Blood flow in both hindlimbs was determined by laser-Doppler perfusion imaging (Moor Instruments). Flow ratios of the occluded/nonoccluded leg were compared between experimental groups. The gastrocnemius muscles were surgically removed for miR-101 analysis, target gene expression, and immunohistochemistry.
Statistical analysis
Quantitative data are expressed as the mean±standard deviation of at least three separate experiments. Statistical significance was determined using either one-way analysis of variance or unpaired Student's t test, depending on the number of experimental groups analyzed. Significance was established at a p-value<0.05.
Footnotes
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (NRF-2011-0028790). The authors thank Dr. Elaine Por for helpful comments and a critical reading of this article.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.