
Abstract
Introduction
N
The Proteasome: An Intracellular Protein Processing Plant
The proteasome is an essential protease of the ubiquitin-proteasome pathway (UPP) that degrades the bulk of intracellular proteins, including vital regulatory factors and damaged polypeptides (21). Twenty years have passed since the physiological significance of the UPP, and, in particular, since the proteasome first started emerging (40, 104, 105, 112). As the broad involvement of the proteasome in the regulation of cell cycle, apoptosis, antigen processing, or stress response became evident, disrupting this ubiquitous protease function with specific inhibitors seemed to provide a viable pro-apoptotic or anti-inflammatory strategy. Indeed, proteasome inhibition has proved to be a very effective anti-cancer treatment (see Soriano et al., this Forum). Up to now, two small-molecule drugs, bortezomib (Velcade™) and PR-171 (Kyprolis™), are FDA approved to treat blood cancers, and several other similar-acting compounds are in human trials (1, 26, 66). All these drugs specifically target orthosteric sites of the proteasome. Before we can begin to discuss the usefulness of allosteric sites, we need to briefly present the structure of the proteasome, revealed by X-ray crystallography (50, 63, 85, 130) and cryo-electron microscopy (cryoEM) combined with molecular modeling studies (30, 79, 80).
The proteasome is a complex and diverse enzymatic factory that is often compared with a molecular organelle (see Soriano et al., this Forum). The name “proteasome” encompasses several assemblies with a common catalytic core and additional regulatory proteins that are capable of attaching to the core and modulating its function (49). All “greater proteasome” assemblies contain a tube-shaped 20S (S denotes Svedberg sedimentation units) catalytic core of 700 kDa (core particle [CP]) that is constructed from four hetero heptameric rings of pseudo seven-fold symmetry. The two α and two β rings are stacked in an α−β−β−α manner and are built from tightly arranged homologous subunits, respectively, designated α1–α7 and β1–β7. The external α rings form the α face: a nearly flat surface that binds additional protein modules regulating the proteasome function (Fig. 1A). The center of the α ring is occupied by “the gate,” which provides passage to the central channel leading to the antechamber and then into the spacious catalytic chamber, formed inside β-rings. The gate is tightly closed in the canonical crystal structures of wild-type Eukaryotic 20S. Substrate access to the catalytic chamber is enabled when the gate is opened (50, 63, 130). In humans, as in other Eukaryotes, there are three pairs of active centers that cleave peptide bonds on the carboxyl side of hydrophobic (chymotrypsin-like peptidase activity [ChT-L]), basic (trypsin-like activity [T-L]), and acidic (post-glutamyl peptide hydrolyzing activity [PGPH]; caspase-like) amino acids. The ChT-L site can additionally process branched amino acids (branched amino-acid processing activity), and the PGPH site is capable of also cleaving after small neutral amino acids (small neutral amino-acid processing activity) (97). The ChT-L, T-L and PGPH activities are provided by active centers that are harbored by β5, β2, and β1 subunits, respectively (5, 27). All active sites are of the rare N-terminal threonine type (29, 121). The constitutive, or “housekeeping” β5, β2, and β1 subunits of higher vertebrates can be replaced by interferon-γ inducible β5i, β2i, and β1i “immunosubunits.” Here, the active centers are modified to accommodate more efficient production of peptides serving as antigens for major histocompatibility complex class I antigen processing (40, 63, 113). In addition to housekeeping and immunoproteasomes, there are thymus-specific “thymo-proteasomes” that feature a specific β5t subunit accompanied by β2i and β1i. This plays a role in the positive selection and maturation of CD8+ T cells (129).
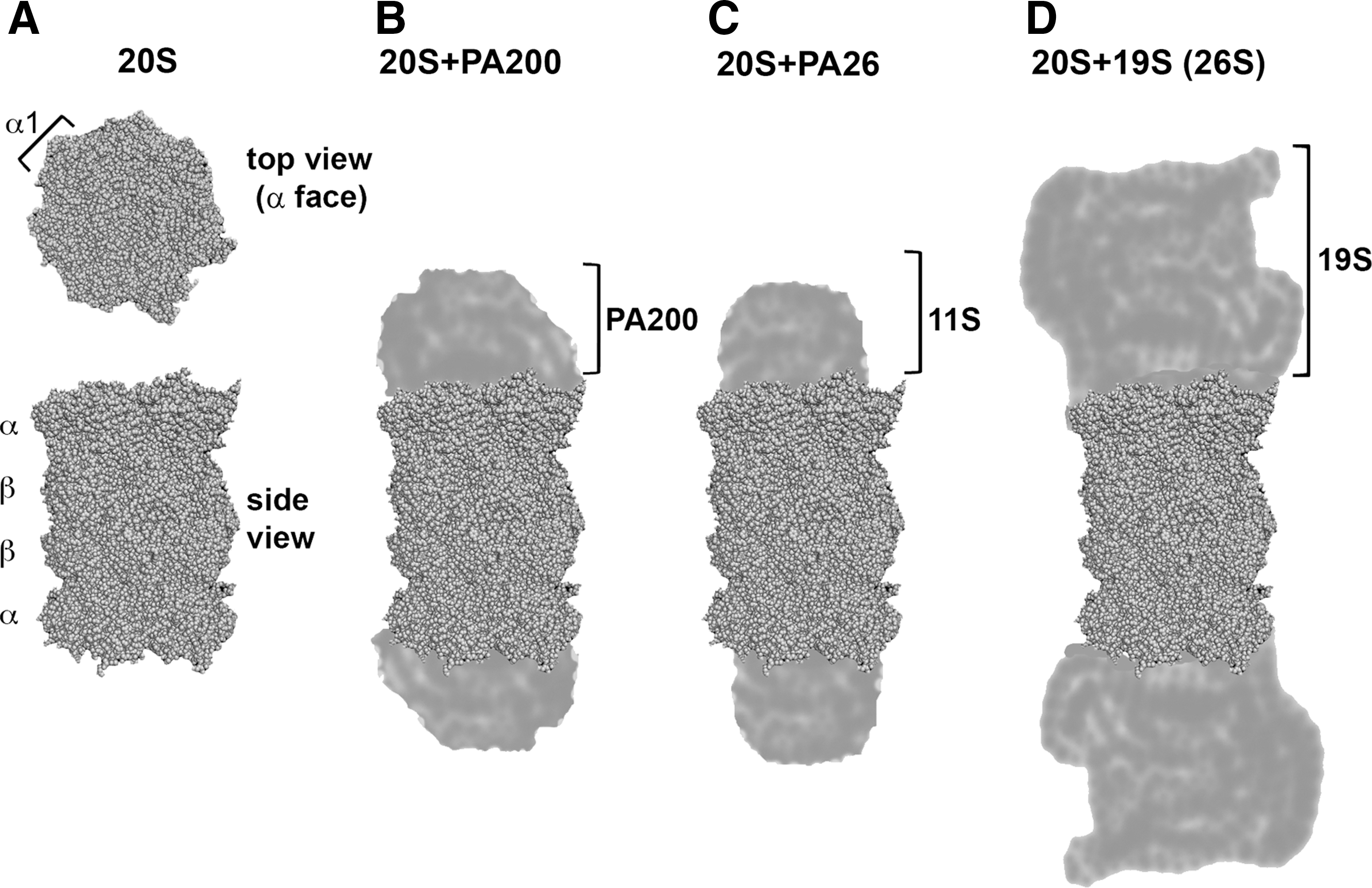
The 20S core alone is capable of degrading peptides, denatured proteins, and proteins with unorganized domains (84, 94). The activity of the core can be modulated by the attachment of additional proteins or protein complexes to one or both α rings. The two α faces may be occupied by the same or distinct modules (49, 59). The proteasome inhibitor PI31 is perhaps the smallest attachment, of molecular weight, comparable to the core subunits (20, 87). PI31 most likely regulates protein–protein interactions within the “greater proteasome” (7, 18, 69). Another single protein module is proteasome activator PA200 (Blm10), a large, 200 kDa protein of HEAT repeat-driven circular structure, that is predominantly found in mixed assemblies PA200-20S-19S. This assembly is primarily involved in DNA repair and maintaining mitochondrial function (Fig. 1B) (115, 118, 131). Next in the line of special attachments to the 20S core are 11S PAs/regulators with 28 kDa subunits (PA28/regulator [REG]; Fig. 1C) (32, 134). They come in two homologous forms, as either heptamers built from α and β subunits or from γ subunits (110). The interferon-γ inducible PA28αβ facilitates the production of antigenic peptides and the degradation of oxidatively damaged proteins (47, 107). The PA28γ is often found in the nucleus and assists in the degradation of specific substrates (110). Direct structural evidence and biochemical data demonstrate that the activators open or disorganize the gate, thus enabling the substrates to enter and the cleaved products to leave the catalytic chamber (110, 115, 134). While not diminishing the role of the above modules, the 19S cap, or regulatory particle (RP) is the most broadly involved in cellular physiology. It is also the largest and most complex of the core-binding assemblies. The 19S-20S-19S assembly is known as the 26S proteasome, a giant enzyme complex of 2.5 MDa (Fig. 1D). The core needs to be decorated by at least one cap to recognize, bind, and process the UPP substrates marked for degradation by polyubiquitin chains. The 19S resists crystallization attempts; however, detailed models based on cryoEM and molecular modeling have been recently proposed (30, 79, 80). The 19S cap is built from 18 constitutive subunits, forming the base and lid subcomplexes (45). The eight “Rpn” (RP non-ATPases) subunits of the lid, Rpn3, Rpn5-Rpn9, Rpn11, and Rpn12, are responsible for binding the substrates and removing the polyubiquitin chains. The base subcomplex consists of six AAA ATPases termed Rpt1-Rpt6 (RP ATPases), and four non-ATPase subunits Rpn1, Rpn2, Rpn10, and Rpn13 (30, 36, 79). The ATPases form a ring or a “spiral staircase” structure, work as reverse chaperones, unfold the protein substrates, open the gate, and direct the substrates to the core for proteolytic processing (9). In addition to the constitutive subunits, several other proteins, among them being substrate-bringing vehicles, deubiquitinylating enzyme or ubiquitin ligase, can transiently bind to the 19S (23, 28, 44, 57). These additional constitutive or transient protein ligands can supplement the enzymatic capabilities of the proteasome by extra ATPase, protease, or ligase. Clearly, the proteasome is an extraordinary complex enzyme assembly, in both structural and functional aspects. It is, thus, not only an attractive but also a challenging subject for studies on allostery.
The Proteasome As an Allosteric and Dynamic Protein Assembly
The structural organization of the core proteasome as a compartmentalized enzyme assembly with multiple active centers lends itself to widespread involvement of allosteric interactions, which are unequivocally connected to structural dynamics. This is in stark contrast to the manner in which the proteasome has been presented for many years as a static and non-allosteric molecule (11). Moreover, the proteasome has been contrasted with chaperonins of similar size and shape, but with robust molecular dynamics, such as GroEL (22, 116, 136). Nevertheless, evidence of allosteric regulation of the proteasome abounds is provided by several diverse methods, including biochemical activity tests, atomic force microscopy (AFM), nuclear magnetic resonance (NMR) spectroscopy, and EM. Starting with the classical biochemistry, analysis of enzyme kinetics for three proteasome peptidases indicates that the active centers are bound in a web of interactions, from positive cooperativity of the two ChT-L sites to complex activation/inhibition schemes possibly involving noncatalytic substrate binding sites (70, 91, 98). Putative allosteric sites are also involved in actions of a leading anti-AIDS drug, ritonavir that is designed to block essential peptidase of the human immunodeficiency virus (HIV) impairing its ability to replicate. Apparently, ritonavir inhibits the core 20S with a unique noncompetitive-competitive, allostery-involving mechanism of action (119).
Activation of the 20S core on binding the PA28/REG is also an allosteric process. The increase in activity may be simply explained by gate opening and indiscriminate, increased influx of substrates. The very diverse effects of PA28αβ and PA28γ on the three peptidase activities of the core, however, are better explained by allosteric interactions (109, 110).
In fact, the gate opening provides an excellent example of allosteric signaling and molecular dynamics. The first crystal structure of the core proteasome was solved for the archaebacterial 20S, homologous but simpler than the eukaryotic 20S particle (85). The general architecture of the Thermoplasma acidophilum proteasome is very similar to the eukaryotic 20S core; however, the former is built from only one kind of α and one kind of β subunits. The homoheptameric α ring apparently does not support a full protection of the central channel: The structural model presents an always-open entrance to the inner compartment (85). Analysis of the primary structure of eukaryotic α subunits reveals that they evolved to include “extensions” of N-termini. Indeed, the center of the α face is sealed by tightly interwoven N-terminal “tails” of α subunits in the crystal structure models of both yeast and mammalian CPs (50, 63, 130). This finding at first created serious confusion, as it was not apparent if and how the tails may loosen up, nor where the true entrance used by the substrates was. The widely used in vitro method of “activation” of the core by treatment with a low concentration of detergent sodium dodecyl sulfate was hardly transferable to the natural conditions. However, subsequent crystal structures of the yeast 20S core activated by gate-disrupting mutation (48) or by the attachment of PA26, a homolog of PA28 (134), revealed the open gate and unobstructed entrance to the central channel. Simultaneously, AFM imaging demonstrated not only that the open-gate conformation exists, but also that the gate is dynamic with every single proteasome molecule able to switch constantly between the closed and open conformations (Fig. 2) (99).
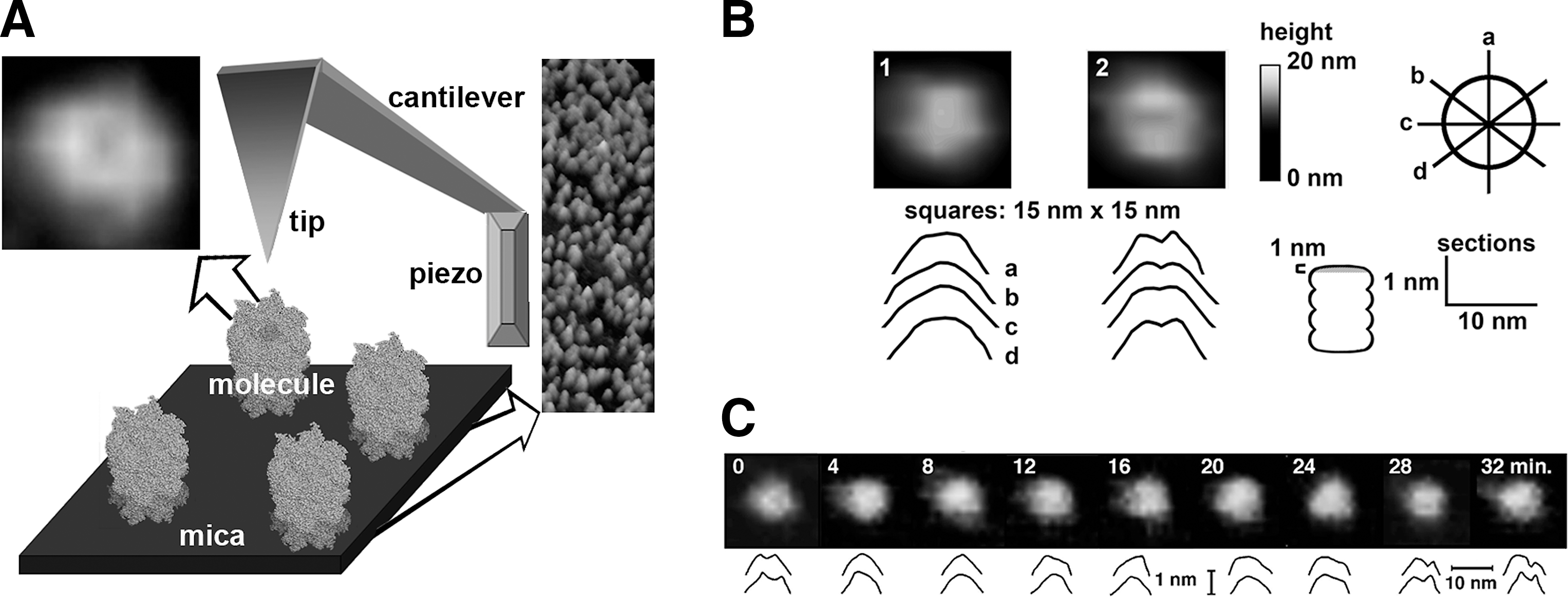
AFM Is an Excellent Technique to Study Protein Dynamics
AFM belongs to the group of scanning probe microscopies (SPM) where an image is created based on interactions of the scanning probe with the surface on an imaged object. AFM is the most popular among the SPM methods used for biological applications (34, 38). In the noninvasive oscillation (tapping) mode AFM, a very small and sharp probe (“tip”) mounted on the cantilever oscillates above the object (for example, a protein molecule), scanning it in a raster motion (Fig. 2A). The object to be scanned needs to be immobilized; however, this can be achieved in a very gentle way by simply depositing the molecules on a weakly charged, flat, and clean surface such as freshly cleaved mica (39). The probe is positioned close enough to communicate with the surface atoms of the molecule, mostly by van der Waals interactions. The probe oscillations are modified as a result of variations in the distance between the tip and the scanned sample. The feedback loop between a sample/tip positioning system and a computer-controlled piezo element constantly corrects the set distance. The image of the sample's surface has three-dimensional topographical information and is generated by plotting the z (vertical direction) correction signal from the feedback loop against the x and y plane (34). There is no electromagnetic wave-dependent limitation of resolution of the imaging. The practical resolution for biomolecules depends on the size and shape of the tip, the quality of electronics, and the dynamics of the object, and reaches 0.1 nm in vertical and 1 nm or less in lateral directions (101). Nanometer-scale allosteric motions, especially in large molecules, are perfectly within the range of detection by AFM. The advantages of the method include the opportunity to monitor in real time changes of the topography of live molecules in liquid. Detecting the spontaneous opening and closing motions of the gate demonstrates the conformational diversity of the proteasome core (Fig. 2B, C). Moreover, the gate area is not the only dynamic part of the molecule. Analysis of lateral dimensions of the particles reveals that they assumed either barrel-like (short and wide) or cylinder-like (long and narrow) shapes, correlating with the gate closing and opening (100). Apparently two forms of particles with well-distinguishable structural features remain in equilibrium with the prevailing closed gate, barrel shaped, and less populated open gate, cylinder shaped (100). The partition of conformers, about 75% (closed) to 25% (open), indicates a relatively small difference in the free energy between the two conformers, as even the less populated state is readily detectable (100). Still, the crystal structure of the free, wild-type core captures the more stable and thus more populated, closed-gate conformer (50). Importantly, the dynamic nature of the entrance to the central channel has been confirmed by NMR spectroscopy of the archaebacterial core (111). NMR is usually used to study dynamics of much smaller proteins; however, the homooligomeric architecture of archaebacterial proteasome rings enabled to overcome the size limitations (125). Even if the archaebacterial gate is rudimentary, it still provides a movable barrier sequestering larger substrates (124), a startling example of the long evolutionary history of the concept of dynamic gating. Coming back to the elegant NMR studies, the most recent data demonstrated the conformational diversity of the archaebacterial proteasomes, which is in full agreement with the AFM imaging (114).
The significance of dynamic gating and conformational diversity for catalysis is obvious: The inactive (latent; closed-gate) proteasome core periodically opens and accepts substrates. Moreover, AFM imaging combined with biochemical and mutational studies reveals a positive feedback loop between catalysis and gate opening. Apparently, a tetrahedral transition state in a catalytic center sends an allosteric signal to the gate, promoting its opening (103). Thus, while supplies last, the next substrate molecule can enter the proteasome inner compartments, proceed to degradation, and promote gate opening for the subsequent substrate molecule. The process is mediated by the amine group of the catalytic N-terminal threonine, and it can be provoked not only by substrates but also by inhibitors mimicking the tetrahedral transition state, such as bortezomib, and by certain active site mutations (103). Here, the orthosteric site performing its catalytic duties acts at the same time as an allosteric site, sending signals to a distant gate. Binding of a substrate or a certain inhibitor shifts conformational equilibrium of the proteasome population toward more of the open conformers (103). We may speculate that random molecular reorganizations in the active site area are responsible for the closed-open switches in the absence of a specific ligand. It is reasonable to speculate that the same or similar allosteric route, from the active sites to the α face, is used in another phenomenon, which is the stabilization of 26S proteasome observed on bortezomib treatment (72).
The structural integrity of 26S assembly is controlled at several levels, with most, if not all of them, clearly involving allostery. Biochemical methods and EM have been used to propose a model of allosterically driven disassembly of the 26S, controlled by ATP hydrolysis and correlating with the catalytic cycle (6). This model remains controversial, and results of a distinct set of experiments under different conditions failed to support it (74). However, the idea of periodic lowering of the affinity between the core and the cap may still be viable. The reverse-chaperone and polypeptide-translocase activities of the 19S base ATPases are expected to involve robust mechanical interactions between the substrate, the base, and the 20S core (61, 88, 124). Focusing on the base-core interface, the most striking effect of the base attachment is gate opening (123, 124).
Earlier, we discussed the allosteric gate opening “from the inside” responsible for conformational diversity as well as effective catalysis by the 20S core. Attachment of regulatory modules to the α face opens the gate “from the outside” (115, 123, 134). All the modules dock in the grooves positioned between α subunits, halfway from the gate to the rim of α ring (Fig. 1) (115, 123, 134, 137). The 2–3 nm long distance between the binding grooves and the effector site (the pivot residues of the gate forming tails of α subunits) invokes allosteric signaling as the cause of gate opening “from the outside.” One more class of ligands proposed to induce gate opening are hydrophobic peptides (71). These peptides may bind to several new allosteric sites (71). Alternatively, they may utilize the canonical grooves on the α face, the substrate binding pockets in the inner compartments of the core, or serve as substrates, triggering gate opening “from the inside.”
The movements related to opening and closing of the gate are not restricted to the gate-forming N-terminal tails of α subunits. An analysis of cryoEM images of the 26S proteasomes reveals radial displacements of α subunits, postulated to open the gate (31). Interestingly, we observe a similar phenomenon, manifested by a significantly increased diameter of the top of α ring, in the AFM images of open-gate conformation of the 20S core (Gaczynska and Osmulski, Unpublished observations). Apparently, multisubunit rings of the 20S core and the 19S base are conformationally diverse. The dynamic changes in the α ring and ring of ATPases may stand behind the first observed case of multiple conformational forms of the proteasome assembly, the “wagging” of 19S cap in 26S. The “wagging” was reported after analysis of three-dimensional EM images, clearly showing distinct positioning of the 19S respective to the 20S core (80). The different poses might have been artifacts forced by fixing of the molecules for EM imaging. However, even more robust bends of the 26S structure were observed by AFM, with unfixed, live molecules, lightly immobilized and submerged in a buffer (38). It is tempting to speculate that “wagging” is a manifestation of conformational diversity of the 26S proteasome, and that substrate processing is tied to conformational changes. Indeed, a recent report presents an elegant model of the 19S cap dynamics, involving twisting motions of the ring of ATPases, periodic widening of the ring's pore, and exposure of the Rpn11 deubiuquitinylase, all of which facilitate the processing of polyubiquitinylated proteins (86).
Summarizing, the proteasome assemblies appear dynamic and highly allosteric. The so-far described cases of molecular dynamics and allosteric regulation follow modern views on conformational diversity, population shifts, and dynamics driven catalysis. For now, we can distinguish several allosteric routes between distant parts of the assemblies. Some of the routes can be utilized by small-molecule ligands. We will discuss these next.
Proteasome Inhibitors: The Blockers of Catalytic Centers
Development of inhibitors specific to the proteasome played a pivotal role in establishing the physiological significance of the UPP (see Soriano et al., this Forum). Some of the common inhibitors targeting active sites of multiple serine or cysteine proteases; for example, leupeptin (N-acetyl-Leu-Leu-Arg-aldehyde) (135) or 3,4-dichloroisocoumarin (98) affect proteasome activities. Their value, however, in in cellulo or in vivo studies is obviously limited. Since the first reports in the early 1990s, that small-molecule inhibitors show reasonable specificity toward the proteasome, seven drugs have been already approved or tested in humans. All of these drugs are covalent, competitive inhibitors that primarily target the β5/β5i active sites which are responsible for the “workhorse” post-hydrophobic (ChT-L) cleavages (14). These new drug developments are based on the serendipitous observation that hematological cancer cells, for example in multiple myeloma and lymphoma, are exceptionally sensitive to blocking the UPP and respond by the induction of apoptosis (1, 33). Several proteasome-specific competitive inhibitors that differ in their chemical mechanism of action to block the catalytic centers are currently used in research (Fig. 3A). These are also under scrutiny as possible treatments for human cancers and other diseases. Two proteasome-targeting drugs are currently approved by the FDA. The first, bortezomib (Velcade, MLN-314; Millenium Pharmaceuticals, Inc.) is a dipeptidyl boronic acid (pyrazylcarbonyl-PheLeu-boronate; Fig. 3B) (2); whereas the second FDA-approved drug is carfilzomib (Kyprolis; Onyx Pharmaceuticals), an epoxyketone derivative. Two other boronates, ixazomib and delanzomib (Millenium Pharmaceuticals, Inc. and Cephalon, respectively), as well as two other epoxyketone derivatives (epoxomicin and oprozomib) (Fig. 3C) (76, 106), and a lactone derivative, marizomib (NPI-0052; Nereus Pharmaceuticals), are currently undergoing clinical trials (Fig. 3D) (16).
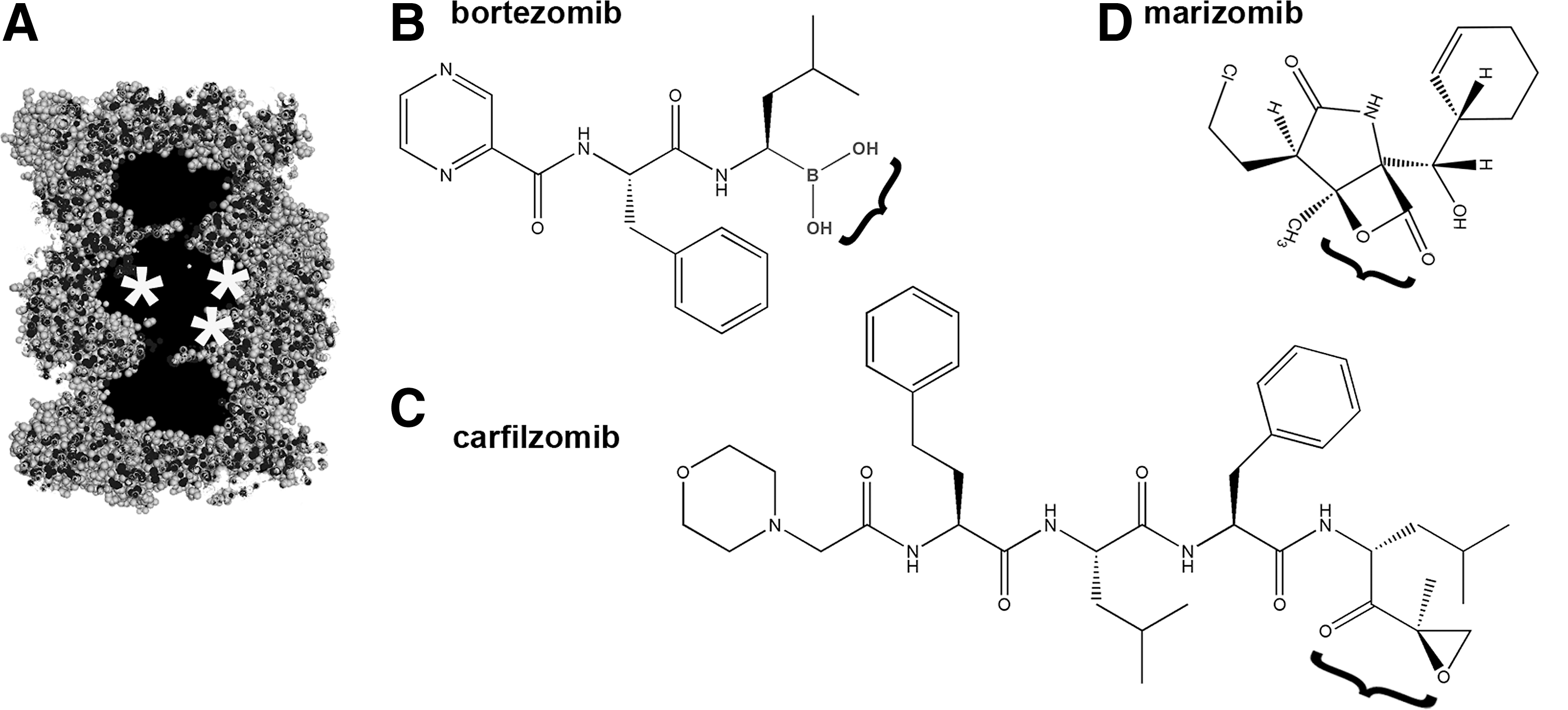
Many of these inhibitory compounds are using “warheads” developed for serine or cysteine proteases. In such cases, the proteasome specificity is bestowed by the peptide or peptidomimetic moieties that are designed to fit in the substrate-binding pockets (13). Peptide aldehydes, vinyl sulfones (VSs), and boronates belong to this group. Examples of such inhibitors include carbobenzoxy-Leu-Leu-Leucinal (MG132) (112), carbobenzoxy-Leu-Leu-Leu-VS (12), or carbobenzoxy-Leu-Leu-Leu-boronate (MG262) (72), all of which have the same peptide moiety but distinct pharmacophores, and all primarily target the β5/β5i active sites. Changing the moiety attached to the pharmacophore alters the affinity of the inhibitor to the distinct active centers. A notable example is provided by a derivative of the familiar Leu-Leu-Leu-VS sequence that is extended by aminohexanoic acids (Ahx) and blocked with adamantanyl acetate (Ada): Ada(Ahx)3LLL-VS and binds to all three kinds of active sites with a similar affinity (67). Inhibitors targeting primarily post-acidic or post-basic cleavages are less frequently used than those binding to the ChT-L active sites, and they are mostly represented by VS and epoxyketone derivatives (15, 53, 91, 93, 132). The use of α′,β′-epoxyketone derivatives as proteasome inhibitors started with the natural antibiotics eponemycin and epoxomicin (78) and later evolved into an important drug development enterprise (76, 106). The exceptional specificity of epoxyketones as inhibitors of proteasome function exploits the rare N-terminal nucleophile catalytic mechanism of the proteasome: The compounds engage both amino and hydroxyl groups of the active site Thr1, whereas most of the other inhibitors target the Thr1 hydroxyl (51). The greater specificity of carfilzomib and other epoxyketone-based drugs renders their use to be more advantageous than that of bortezomib. The boronate warhead of bortezomib is highly reactive and unfortunately also reacts with nonproteasomal targets, contributing to several adverse side effects (4) (see also Drews and Taegtmeyer, this Forum). Interestingly, the exquisite specificity of epoxyketones can go as far as strongly preferring the immunosubunit β1i (low molecular weight protein 2) to the housekeeping β1 (Y) (60).
The lactone-derivative class of competitive proteasome inhibitors warrants special attention. The first extensively studied compound from this group is derived from a natural fungal antibiotic lactacystin (omuralide). This, when in aqueous environments, rearranges its structure into clasto-lactacystin β-lactone, which is capable of blocking Thr1 hydroxyl. Lactacystin was pivotal in the discovery of the catalytic mechanism of the proteasome (29). Interestingly, prodrugs for statins, used in the treatment of hypercholesterolemia, weakly target the proteasome with their lactone rings (90). Lactone rings are common in many natural products and these may work as unsuspected proteasome inhibitors. Similarly, plant polyphenols, which are abundant in numerous nutraceuticals and in green tea, may block proteasome in a manner resembling lactones (92). Relatively recent additions to the family of proteasome inhibitors are syrbactins, which are bacterial products. These cyclic peptides are highly specific ligands of the β5 active site (54, 73) and extremely potent proteasome inhibitors. The proteasome inhibiting compounds mentioned earlier directly engage catalytic threonine, whereas several inhibitors are known to occlude the active sites without binding to Thr1. These include fungal product gliotoxin (75), and cyclic peptide fungal metabolites from the TMC series, such as TMC-95A and others (52). The end product of lipid peroxidation, 4-hydroxy-2-nonenal (HNE), also strongly, albeit nonspecifically, modifies the proteasome, with deleterious effects on catalytic activity (96). Gliotoxin, TMC peptides, or HNE likely do not directly involve the allosteric effects.
This brief overview of the best-known proteasome inhibitors will not be complete without mentioning the synergistic effects displayed in vivo by their combination. This phenomenon was noted for bortezomib and marizomib (17), and also for bortezomib and delanzomib (117). Moreover, carfilzomib and other new generation proteasome targeting drugs are capable of overcoming the common phenomenon of bortezomib resistance in cancer cells (76, 106). The effect of synergy may stem from slightly distinct specificities of the different drugs. All of them target primarily the ChT-L activity; however, these also target other catalytic sites, although their second-choice active site preferences are different. These secondary actions apparently translate into induction of separate apoptotic pathways (17). The structural diversity of the various proteasome inhibitors is likely pivotal in overcoming resistance to bortezomib, especially if the resistance is caused by a mutation in the active site (35, 128). The occurrence of such mutations brings into the spotlight the well-known deficiency of all drugs binding to active centers of enzymes: Sooner or later, a mutation may occur, rendering a drug unusable.
Proteasome Regulators: Beyond the Catalytic Centers
Given the undisputed clinical successes and number of known proteasome-inhibiting compounds as well as the level of structural and functional knowledge of their mechanism of action, it is not surprising that drug development has focused, to a large extent, on mechanisms that block the active centers of the proteasome. However, the diversity of such inhibitors pales in comparison with potential allosteric regulators. The term “regulators” is broadly used to indicate non-straightforward functional effects of these ligands. For example, they may activate latent 20S proteasomes and inhibit the activated 20S cores. The three major peptidase activities may respond in distinct ways to allosteric regulators: One peptidase may be activated and another may be inhibited by the same compound. In addition to or instead of influencing peptidase activities, the ligands may interfere with interactions between the CP and regulatory modules. The variety of in vitro detectable actions translates into intriguing physiological outcomes in cultured cells or in animal studies; for example, a substrate-specific inhibition. Substrate-specific activation may be of high interest to prevent accumulation and aggregation of polypeptides causing cell damage in neurodegenerative and other aggregation diseases (see McKinnon and Tabrizi, this Forum). On one hand, the expanded arsenal of effects promises precise tailoring of pharmacological interventions within the UPP. On the other hand, the limited knowledge about proteasome allostery complicates the task of designing allosteric drugs. Fortunately, recent years brought a significant advance in mapping the allosteric routes in the 20S core proteasomes and challenging them with small molecules, usually binding in a non-covalent, fully reversible manner. Next, we present the better traveled allosteric routes and the respective ligands.
Allosteric Route: From the Catalytic Centers to the Grooves on α Face
As outlined earlier, substrates and certain competitive inhibitors binding to orthosteric sites may double as allosteric ligands, promoting opening of the gate (99, 100, 103). Gate opening can be detected by AFM imaging of the 20S core and confirmed by functional studies of the latent or allosterically open core decorated with 11S activator (103). Interestingly, inhibitors promoting gate opening such as bortezomib and epoxomicin support stability of the 26S assembly, and serve as an elegant example of long-distance signaling between the active centers and the grooves used to anchor the 19S module (72). It is tempting to speculate that the same molecular route from the active sites is used to reach the grooves and the gate, both of which are positioned on the α face. The significance of this route for drug design is yet to be evaluated. By now, it appears to be safe to assume that blocking the workhorse ChT-L catalytic site by boronates or epoxyketones results in much more dramatic effects in cellular physiology than stabilization of the 26S assembly. However, mapping the relevant allosteric route may enable the design of specific small-molecule regulators targeting yet-undiscovered allosteric sites between the catalytic centers and the grooves. Such regulators would affect the stability of higher-order proteasome assemblies without blocking the active sites, and, thus, would provide very precise means to intervene into the UPP performance.
Allosteric Route: From the Grooves on α Face to the Gate
The surface of the α ring is not only conveniently flat to avoid steric hindrance with large protein ligands, but also equipped with anchor sites for securing the protein–protein interactions. As mentioned earlier, the sites are in the form of grooves positioned on the interfaces of α subunits, about halfway between the gate and the rim of α ring (Fig. 4A). The grooves are similar but not identical and may display certain levels of specificity or redundancy, depending on the ligand. Most of the protein ligands, the Rpt2, Rpt3, and Rpt5 ATPases of the 19S base; PAN ATPases in Archaebacteria; the PA200 (Blm10) activator; and, most likely, the PI31 inhibitor utilize the C-terminal hydrophobic amino-acid residue (Hb)YX motifs (hydrophobic amino acid–Tyr–any amino acid) for secure docking in the grooves (Fig. 4B) (115, 123). The 11S activator is the sole exception to the HbYX rule. Subunits of PA28/REG and homologous PA26 insert their C-terminal fragments to the grooves; however, the HbYX motifs are not present and the binding is stabilized by interactions between the activation loops of each 11S subunit and the α face, near the respective grooves (Fig. 4B) (32, 134). Remarkably, short fragments of the original “anchors” can mimic some functional effects of the activators. These observations strongly suggest that allosteric switches are positioned in the grooves and are flipped on binding of the HbYX motif. Indeed, as little as ten C-terminal amino-acid residues of Rpt2, Rpt5, PAN ATPase, PA200, or PI31 are sufficient to enhance at least some peptidase activities (Fig. 4B) (43, 123) (Madabhushi, Osmulski, and Gaczynska, Unpublished observations). Moreover, treatment of the proteasome with the C-terminal peptides derived from Rpt5 or PI31 shifts the conformational equilibrium of the 20S core toward more of the open particles, as revealed with AFM imaging (Osmulski and Gaczynska, Unpublished observations). The presence of HbYX motif does not guarantee modulation of peptidase activities. The Rpt3 ATPase possesses the same motif and binds to the α face; however; it is implicated in stability of base-core interactions rather than activation of the core (77, 123). Interestingly, the HbYX motif is not unconditionally required for the peptide-induced, groove-related effects on the peptidase activities. Treatment of the human 20S proteasome with a combination of 12-residue peptide fragments derived from the C-terminus and from the activation loop of PA28 subunit (Fig. 4B) resulted in more than 50% activation of the post-hydrophobic cleavages, even if the peptides were present at as low concentrations as 0.5 μM each (65). These results suggest that both the stable binding of the 11S and its allosteric effects require both the C-terminal and activation loop components.
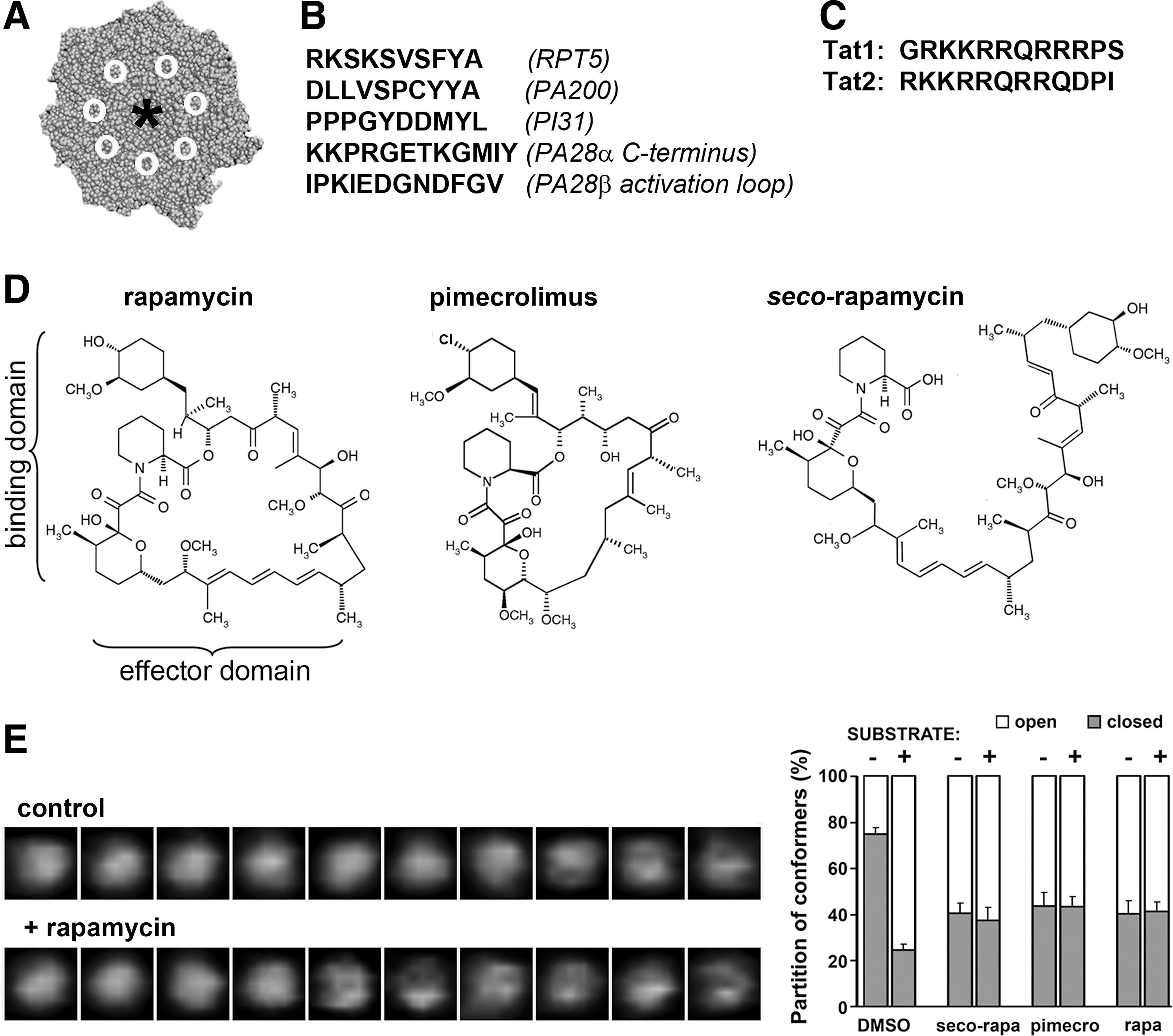
The C-terminal “tail” peptides of regulatory modules have not been modified to enhance their performance, with the exception of a brief attempt to mix and match their HbYX-containing fragments (77) (Tokmina-Lukaszewska, Gaczynska, Osmulski, and DeMartino, Unpublished observations). Moreover, the structures of C-terminal tails of PA28 subunits do not seem to determine their functional specificity, hampering the design of peptides that modulate the proteasome peptidase activities (81). Most importantly from the practical point of view, the groove-binding peptides in their present form would be likely rapidly degraded in the cytosol before even reaching the proteasome. Fortunately, the high propensity to degradation is not of particular concern in the case of another family of small ligands of the proteasome, the transactivator (Tat) peptides (Fig. 4C). Tat peptides are fragments of human immune deficiency virus type 1 transactivator (HIV-1 Tat), an 86-residue RNA-binding protein that is responsible for transactivation of viral and certain host genes. The poorly structured HIV-1 Tat forms a homoheptameric complex, affects angiogenesis and the immune response, and inhibits ChT-L activity of the proteasome (10, 42, 64, 120). HIV-1 Tat can reportedly interact with multiple α and β subunits of the 20S core (3); however, it is reasonable to assume that the high-affinity binding is related to the REG/Tat-proteasome binding (RTP) site present in the HIV-1 Tat (62). The functional RTP site is shared by HIV-1 Tat and PA28α subunit and, consequently, the Tat protein competes with 11S for binding to the α face (62, 120). Even if the direct structural identification of the docking place of HIV-1 Tat is missing, we may hypothesize that it is positioned in or in the close vicinity of α face grooves. The RPT site is located near the C-terminus in PA28α, whereas it encompasses the essential basic domain of the viral protein in HIV-1 Tat (65). Highly basic peptides derived from the Tat protein are used for a variety of tasks, including the transport of cargo molecules through the membrane (68); however, here, we focus on their proteasome-targeting ability. The two 12-residue fragments (Fig. 4C) containing the RTP site and carved out of the basic domain (Tat1), as well as the basic and Gln-rich domains (Tat2) of HIV-1Tat noncompetitively inhibit the ChT-L and PGPH peptidases of the activated human 20S core at high nanomolar concentrations, and also efficiently compete with the core activation by PA28αβ complex (65). The Tat1 peptide is better characterized to date than Tat2. Tat1 induces a shift toward the higher abundance of the open conformer in AFM studies. It also inhibits the ChT-L peptidase of the latent 20S, and strongly affects the pattern of degradation products of poorly structured proteins (65) (Karpowicz, Gaczynska, and Osmulski, Unpublished observations). The affinity of Tat1 to the proteasome can be improved by modifying its structure with non-natural amino acids (Karpowicz, Jankowska, Gaczynska, and Osmulski, Unpublished observations). The intracellular effects of Tat1 and its derivatives are currently under vigorous investigation.
The small peptide ligands of the α face grooves described earlier are derived from natural protein ligands of the 20S core. The ligand described next breaks this rule: It is not a peptide and is not based on a natural proteasome ligand. This compound rapamycin (sirolimus) is a canonical allosteric drug, inducing dimerization of a pivotal regulator of intracellular metabolism mammalian target of rapamycin (mTOR) and FK-binding protein 12 (Fig. 4D). This natural macrocyclin inhibits mTOR by binding to its allosteric site, located near the kinase domain containing the orthosteric site (8). The binding domain of the macrocyclin ring interacts with mTOR, whereas the effector domain recruits FKBB12 (8). In vivo treatment with rapamycin induces numerous pleiotropic effects, including immunosuppression, pro-apoptotic and anti-cancer actions, as well as prolonging longevity (55, 58, 133). In vitro, at low micromolar concentrations rapamycin inhibits the degradation of casein, a poorly structured protein, by the latent human 20S core (102). The various peptidase activities of the 20S core respond to varying concentrations of rapamycin differently. At high nanomolar concentrations of rapamycin, post-acidic cleavages are inhibited by a noncompetitive mechanism. At low micromolar concentrations, the ChT-L peptidase is strongly inhibited, while the T-L peptidase activity is increased two-fold (102). Interestingly, rapamycin abolishes activation of the 20S core by 19S complex or by peptides containing the HbYX motif. The most straightforward explanation of the finding would be competition of rapamycin and the peptides for binding to the α face (102). Indeed, molecular modeling strongly suggests that rapamycin may fit very well into selected grooves on the α face (Bohmann, Gaczynska, and Osmulski, Unpublished observations). Moreover, treatment with the drug affects conformational dynamics of the α face. AFM imaging of the human 20S proteasomes revealed that rapamycin shifts the conformational equilibrium toward a higher abundance of the open particles (Fig. 4E) (102). We speculate that on docking to the α face, rapamycin exploits the allosteric route used by gate-opening regulatory modules. There is one more intriguing aspect of the conformational dynamics related to effects of rapamycin. Treatment of the 20S core with the saturating concentration of a model peptide substrate induces maximum 75% of the open particles (99, 100, 103). A saturating concentration of rapamycin pushes the equilibrium to about 60% of the open proteasomes. When the rapamycin-treated 20S proteasomes are subsequently treated with a substrate, the partition of open and closed forms stays at 60% and 40%, respectively, and does not shift to 75% and 25%, even if the active sites of the core are not directly blocked and, thus, ought to be capable of engaging substrates (Fig. 4E) (102). The nature of the link between apparent rapamycin-induced perturbation in conformational dynamics and the effects on proteasome catalytic activity is purely speculative at this point. There may be a direct allosteric communication between the binding groove and the catalytic sites, perhaps independent from gate opening. On the other hand, there may be an allosteric link between the gate and the catalytic sites, involving the dynamics of gate movements. Such a link could be far more complex than a non-allosteric effect of increased flux of substrates through a longer open gate. Dissecting the structural and functional effects of rapamycin–proteasome interactions is of great value for learning about the role of allostery in proteasome function. The difference in in vitro affinities of rapamycin to mTOR and also to the proteasome definitely favors the former as the primary, in practice the sole, in vivo target of the drug. It is likely that close homologs of rapamycin, rapalogs, and single-domain derivatives of rapamycin, such as pimecrolimus (Fig. 4D), will affect proteasome function using similar pathways to those outlined earlier. However, there is a great opportunity to design “rapamycin–inspired” ligands specific to the proteasome. Apparently, the first in vivo metabolite of rapamycin, the open-ring seco-rapamycin (Fig. 4D), exerts similar effects to those of the parent drug on the proteasome while not inhibiting mTOR activity (102). Pilot tests indicate that seco-rapamycin is cytotoxic to those cells which are resistant to bortezomib treatment (Osmulski and Gaczynska, Unpublished observations). Studies are in progress to identify the proteasome targeting pharmacophore of rapamycin and seco-rapamycin, and to utilize this in the design of a new class of allosteric proteasome regulators.
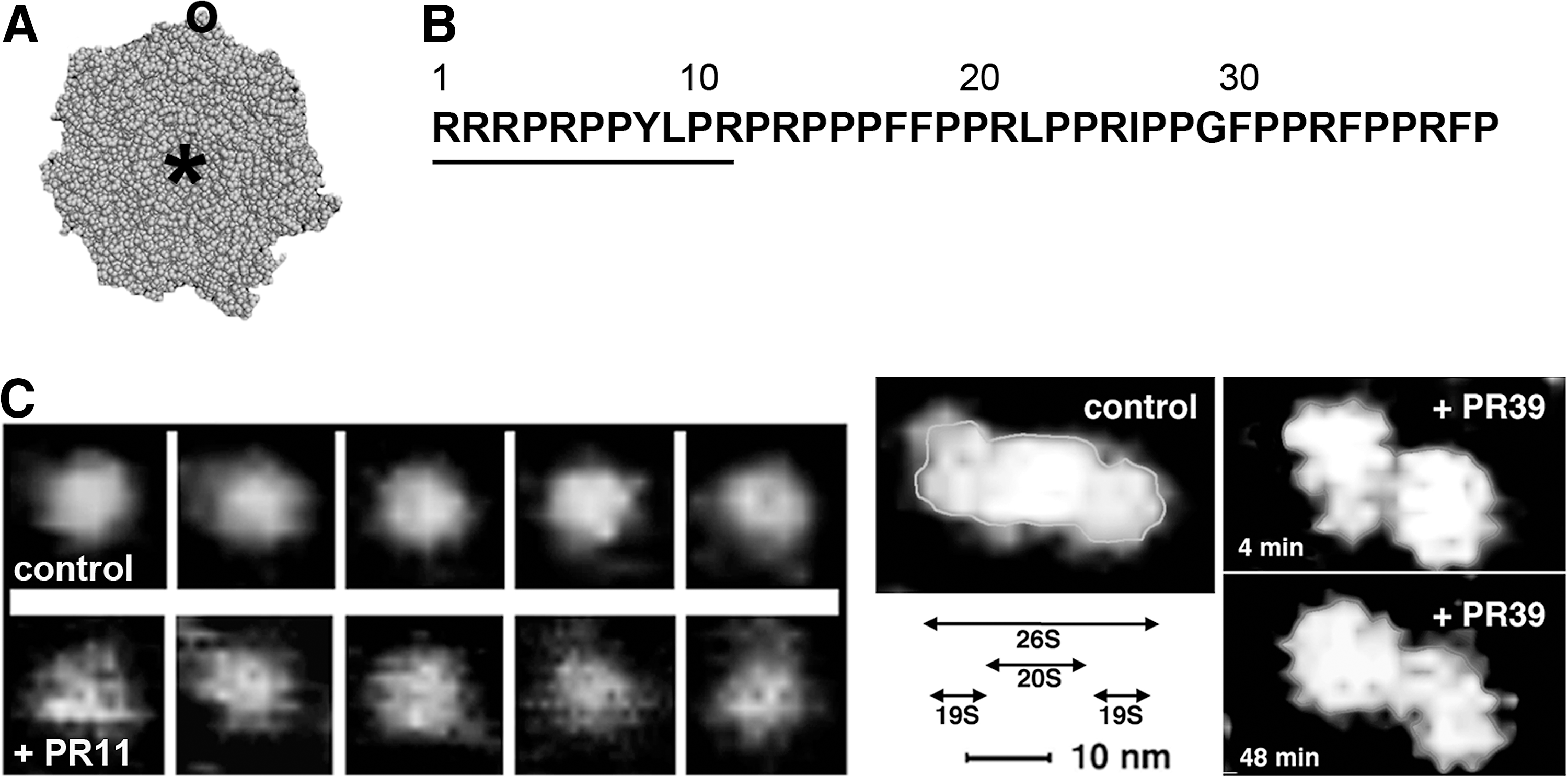
Allosteric Route: From the Outer Rim of α Ring to the α Face
The grooves on the mid-region of α face are universally used by protein regulators of the proteasome (115, 123, 134). There are no known physiological ligands of the outer rim of α ring; however, the rim may bear a binding site for proline- and arginine-rich (PR) peptides. PR peptides are allosteric ligands that are derived from an antibacterial peptide from the cathelicidin family (Fig. 5A). Cathelicidins are a part of innate immune response of mammals, killing bacteria by a non-pore forming mechanism (19). The effects of PR peptides on the proteasome were first reported with PR39, a 39-residue-long peptide from a porcine intestine with no known homologue among human cathelicidins (Fig. 5B). In vitro, PR39 inhibits the ChT-L and PGPH activities of purified activated 20S proteasome in a noncompetitive manner. The N-terminal arginines of PR39 are essential for these biological effects (37). PR39 also competes with the 19S cap binding to the 20S core and inhibits the already assembled 26S proteasome (37). Importantly, PR39 can be shortened to only 11 N-terminal residues (Fig. 5B) without any loss of function, although some loss of affinity is evident (37). Yeast two-hybrid screen, molecular modeling, and cross-linking/mass spectrometry studies indicate that the binding site for PR peptides is the C-terminal part of the α7 subunit, positioned on the outer rim of α face (41) (Osmulski, Gaczynska, Tokmina-Lukaszewska, Slusarz, and Slusarz, Unpublished observations). Interestingly, AFM studies revealed that PR peptides structurally destabilize the α face. Instead of the well-distinguished conformers, images of the 20S particles treated with PR39 or PR11 show a “shaky” α face with a partially open gate (Fig. 5C) (37). Structural destabilization was not restricted to the core, but was apparent for the 26S proteasome treated with PR peptides as well (Fig. 5C) (37). We hypothesize that there is an allosteric route extending from the α7 C-terminus into the whole α face. The PR peptides disrupt this route, affecting dynamics of open–close switches. Such destabilization of the α face is likely a culprit in a lowered affinity between the 20S and 19S subassemblies, and in the striking disruption of the 26S super-assembly (Fig. 5C). The drastic changes in molecular dynamics of the α face may account for all the observed effects of PR peptides on catalytic activity. However, the existence of an additional allosteric route between the rim of α ring and the catalytic chamber is still possible. These intriguing molecular effects may be accompanied by in vivo actions of considerable pharmacological interest. The strongly basic PR peptides readily traverse cell membrane. Higher nanomolar to lower micromolar concentrations of the compounds are required both in vitro and in cellulo to demonstrate effects of treatments (41). PR peptides have likely additional intracellular targets besides proteasomes (108); however, their directly proteasome-related actions include blocking the degradation of two key transcription factors: hypoxia-inducible factor (HIF)-1α and inhibitor of κB, where NF-κB is nuclear factor kappa-light-chain-enhancer of activated B cells; IκB, without detectable attenuation of the whole UPP (41, 82). HIF-1α is a regulator of intracellular oxygen distribution, and its accumulation induces angiogenesis (82). Stabilizing IκB, in turn, has anti-inflammatory effects, as IκB blocks nuclear factor-κB, the master regulator of inflammation (41). This seemingly substrate-specific inhibition remains unexplained; however, the potential clinical value of the phenomenon is immense. The beneficial effects of PR peptides in animal studies include improved wound healing and protection of the brain and heart from inflammation-driven deleterious effects of ischemia–reperfusion injury after stroke or heart attack (41, 82) (see also Drews and Taegtmeyer,
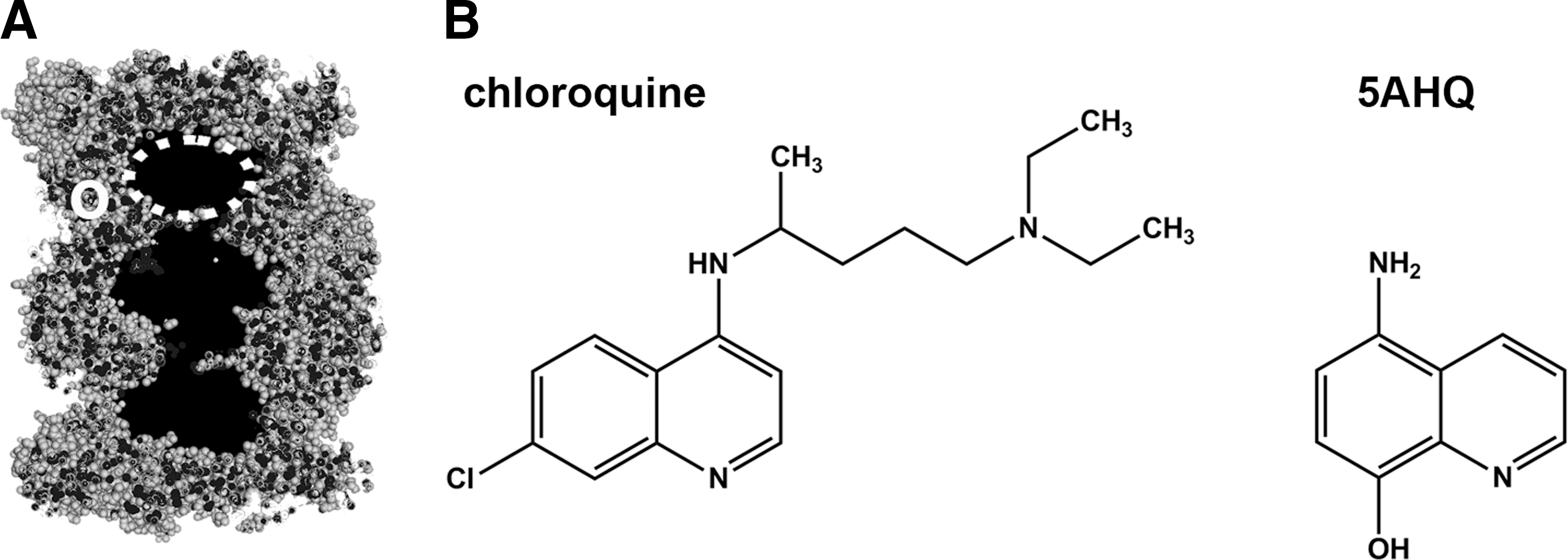
Allosteric Route: From the Interface of α and β Rings to the Antechamber
As in the case of the allosteric site on the C-terminus of α7 subunit, the interface of α and β rings have no known physiological ligands. However, NMR studies on the allostery of Thermoplasma proteasome performed on the complete core complex, the “half proteasome” dimer of α rings, or a single homoheptameric α ring demonstrated that chloroquine, a well-known anti-malaria drug, binds in this region (Fig. 6A, B) (126). A relatively large negatively charged area in the vicinity of α Ile157 was identified as the site of drug-proteasome interactions, possibly stabilized by hydrogen bonds. Interestingly, the lining of the antechamber is affected by the binding of chloroquine, clearly indicating a novel allosteric route (Fig. 6A) (126). In vitro, chloroquine at millimolar concentrations inhibits the degradation of model peptide substrates and disordered proteins (114, 126). The affinity of chloroquine to the eukaryotic or archaebacterial 20S cores is rather low; however, a screen of chemical library of compounds with a quinoline pharmacophore turned in 5-amino-8-hydroxyquinoline (5AHQ) as a viable candidate for proteasome targeting allosteric drug (Fig. 6B) (83). NMR studies indicated that 5AHQ binds to the archaebacterial proteasome in the region identical or very similar to that identified for chloroquine, and that antechamber is affected by the binding, as was the case for chloroquine (83). Since the architecture of the α–β interface is well conserved between the archaebacterial and eukaryotic proteasomes, we may assume that the very similar allosteric routes constitute the molecular basis for functional effects of the drugs on the proteasome (83, 114, 126). As in the case of PR peptides described earlier, the presence of additional allosteric routes besides the established interface-antechamber link should be taken into account, especially that a part of NMR studies was performed without the presence of the β ring. For example, binding of the drugs may destabilize the α−β ring interactions and propagate allosteric signals directly to the catalytic chamber. Speculations about the molecular mechanism aside, the functional effects of 5AHQ are worth attention. In vitro, 5AHQ noncompetitively inhibits eukaryotic proteasome ChT-L and PGPH activities in micromolar concentrations, and displays synergistic effects when combined with bortezomib (83). 5AHQ is cytotoxic for established blood cancer cell lines, for primary myeloma and leukemia cells, and, importantly, also suppresses tumor growth in the mouse model of leukemia (83). The drug synergizes with bortezomib not only in vitro but also in cell culture, which is a phenomenon of unknown molecular mechanisms (83). Moreover, 5AHQ is cytotoxic to myeloma and non-small cell lung cancer cells, which displayed intrinsic or acquired resistance to bortezomib due to mutations in the β5 inhibitor-binding pocket (24, 83). Obviously, the phenomenon of overcoming resistance to established drugs is of considerable interest for the prospective use of noncompetitive proteasome inhibitors in clinics. The intracellular specificity of 5AHQ remains to be established, and most likely, the proteasome will not be the sole target of this compound. However, both the affinity and the specificity of 5AHQ toward the proteasome are much better than in the case of chloroquine. Clearly, the anti-cancer preclinical effects of 5AHQ noted so far are very promising.
Concluding Remarks
Perhaps it is not surprising for such a giant enzyme complex with so many active sites and protein-binding domains that there are so many types of small-molecule compounds which are known to affect its catalytic performance. Here, we described only those compounds that have been well studied, with reasonable well-delineated molecular mechanisms of action. We restricted this discussion to the catalytic core 20S particle, central for all assemblies and obligatory for the complex to bear the “proteasome” name. The described regulators engaging allosteric routes are likely to constitute a tip of the iceberg of potentially useful compounds, and are considered highly desirable due to the great diversity of their functional effects. There are two major approaches to search for more allosteric regulators. First, the already-identified allosteric routes can be targeted in rational drug design. The designer compounds may be destined to target any point within the allosteric path. The most straightforward and likely successful approach would be the targeting of the known receptor site. However, disrupting signaling between the receptor and effector sites may have the advantage of the lowest evolutionary pressure for drug-binding spots and a greater level of diversity of functional effects. Second, a broad high-throughput screening, virtual or with a library of compounds using a carefully targeted readout should fish out a variety of proteasome regulators. Some of the regulators may affect yet-unknown allosteric routes. The lead compounds may not necessarily be proteasome specific; however, they can be further optimized for improved in vivo performance. The critical issue in both the approaches is the application of an appropriate in vitro readout. Testing the workhorse ChT-L peptidase activity with a fluorogenic model peptide substrate, so popular in screens for competitive inhibitors, is not sufficient here. Allosteric ligands may perform poorly with the single-peptidase test yet display desirable in vivo effects, including interactions with regulatory modules, substrate specific inhibition or activation, or changes in a product's pattern. Using an array of model substrates and selected protein substrates should help pinpoint shifts in specificity. Fluorescent probes that are sensitive to structural changes in drug-treated proteasome particles would also come handy. Summarizing, searching for, and characterizing allosteric regulators of the proteasome are not trivial tasks. However, allosteric drugs may provide a truly comprehensive targeting of the UPP in general and the proteasome in particular, which is beneficial for the treatment of a variety of clinically relevant conditions.
Footnotes
Acknowledgments
M.G. and P.A.O. are grateful to the William and Ella Owens Foundation for Medical Research; to the Mike Hogg Foundation and Institute for Integration of Medicine and Science for their support. They thank Dr. Rochelle Buffenstein for a critical reading of this article and invaluable expert advice.