
Abstract
Introduction
O
Several enzymes in the body are capable of producing ROS. Among them are xanthine oxidase (104), cytochrome P450 oxidases (50), lipoxygenases (192), uncoupled nitric oxide synthase (NOS) (174), NADPH oxidases (catalytic subunit of NADPH oxidases [NOX]) (13), monoamino oxidases (48), and the mitochondrial electron transport chain (163). The majority of these enzymes only produce ROS after they have been damaged by ROS, as, for example, is the case for uncoupled endothelial nitric oxide synthase (eNOS) (174) and xanthine oxidase (104). In contrast, NADPH oxidases produce ROS as their primary and sole function. They are widely distributed throughout different tissues and organs and were suggested to play important roles in multiple diseases associated with oxidative stress [reviewed in (13)]. Therefore, NADPH oxidases are considered prime target candidates for the treatment of these diseases. In that setting, various compounds have been postulated as NADPH oxidase inhibitors.
Here, we give an overview of the most important NADPH oxidase inhibitor candidates and critically review their use in in vivo proof-of-concept studies of NADPH oxidase inhibition in various diseases.
The NADPH Oxidase Enzyme Family
The NADPH oxidase enzyme family contains a homologous catalytic subunit, NOX. Seven NOX members exist that are characterized by at least six trans-membrane helices containing two iron-heme prosthetic groups, as well as a flavin adenine dinucleotide (FAD) and an NAPDH-binding domain in the cytosolic c-terminus (Fig. 1). The NOX isoforms dual oxidase 1 (DUOX1) and 2 (DUOX2) have an additional trans-membrane domain and an extracellular N-terminus that contains a peroxidase-like domain. Therefore, they were termed DUOX1 and DUOX2. However, according to current knowledge, human DUOX enzymes do not display any peroxidase activity (105 –107). Therefore, others and we suggest that the field considers terming these isoforms NOX6 and NOX7 rather than DUOX1 and DUOX2.
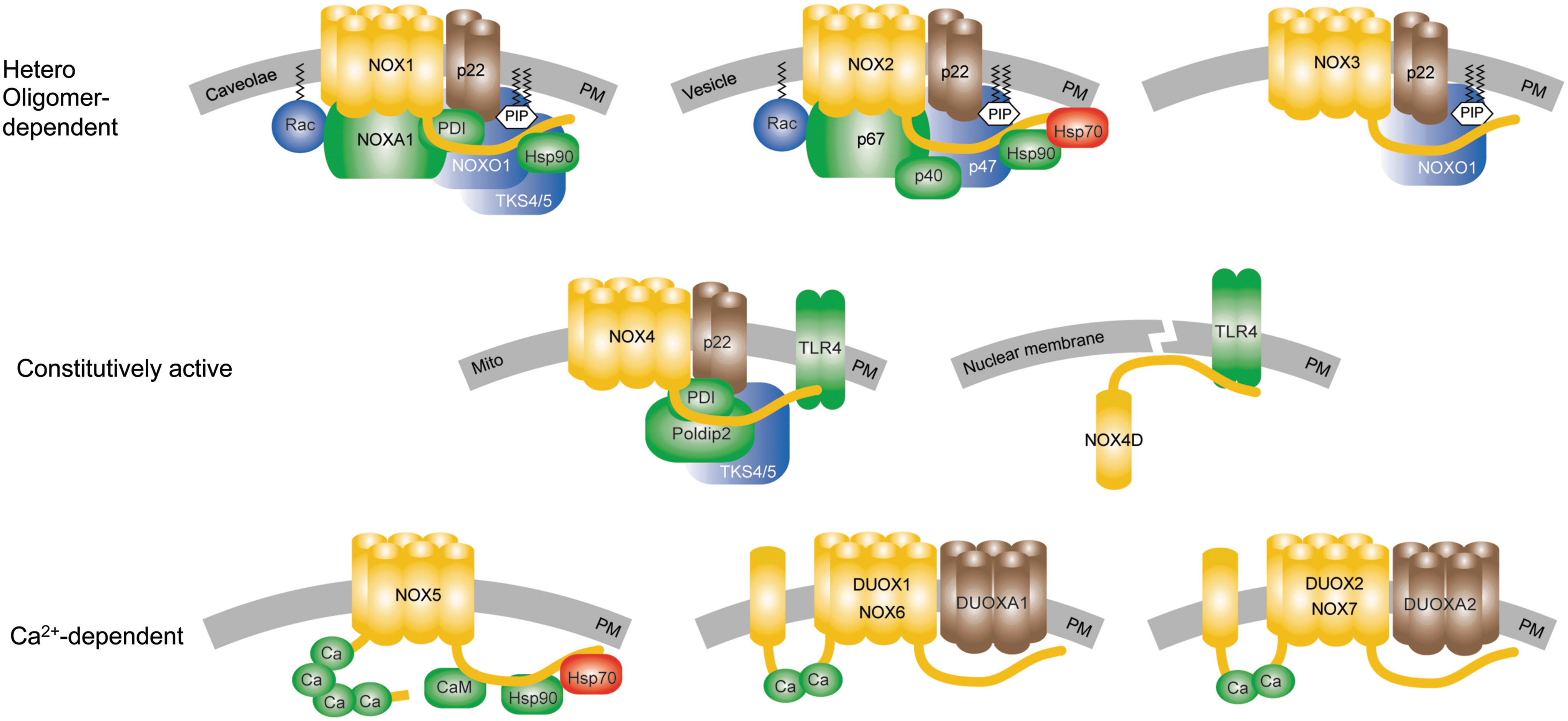
NOX enzymes differ in enzymatic complex composition, modes of activation, and the products of their enzymatic reaction. Most NOX isoforms require at least one cytosolic or membrane-bound binding partner for activity (Fig. 1). NOX1–4 associate with the stabilizing membrane protein, p22phox. DUOX1 and DUOX2 associate with their respective maturation factors, the membrane proteins DUOXA1 and DUOXA2. All other associated proteins can be classified as activators or organizers (126). The activating proteins activate or increase the enzymatic ROS production. These are p67phox and p40phox for NOX2 and its analogue NOXA1 for NOX1. The organizer proteins set the structural requirements for the activation or binding of the activator proteins. p47phox stabilizes the complex formation for NOX2. NOXO1 enables the active complex formation for NOX1 and NOX3. In addition, NOX1-3 seems to require the small GTPase, Rac, for activity, although the role of Rac for NOX3 in vivo is still under debate (171, 172).
Recently, additional NOX-interacting partners have been suggested to bind to and enhance the activity of NOX4, such as polymerase (DNA-directed) delta-interacting protein 2 in focal adhesions (102), and activated Toll-like receptor-4 (130, 168). Additional factors, which might bind and activate both NOX1 and NOX4, such as protein disulphide isomerase (75) and tyrosine kinase substrate with 4/5 SH3 domain (43, 57), have been identified. Furthermore, heat shock protein 90 (Hsp90) has been suggested to bind to and regulate the stabilities of NOX1, NOX2, and NOX5; while Hsp70 most likely facilitates the degradation of NOX2 and NOX5 (28, 29). However, heterologous expression in cells may influence the expression of other proteins and interfere with protein processing and folding. In addition, in vitro systems may lack interacting partners that are important in vivo. Thus, there may be even yet undiscovered NOX binding partners. Accordingly, the physiological roles and tissue specificities of these recently reported protein–protein interactions and stabilization-destabilization mechanisms by chaperones for NOX activity regulation need to be further analyzed in vivo.
NOX isoforms differ not only in their subunit requirements, but also in their modes of activation. NOX1–3 seem to be dynamically activated or deactivated by complex formation with the NOX regulatory proteins (NOXA1 and NOXO1 for NOX1; p67phox, p47phox, and p40phox for NOX2; and NOXO1 for NOX3). NOX5 and DUOX 1/2 are activated intra-molecularly by the binding of calcium to their intracellular EF-hand motifs (8, 111, 146). The calcium sensitivity of NOX5 can be further enhanced by the binding of calmodulin or Hsp90 to the C-terminus of the EF-hand motifs (28, 170) and by phosphorylation (159). DUOX1 and DUOX2 bind their respective maturation factors, DUOXA1 and DUOXA2, enabling translocation from the endoplasmic reticulum to the plasma membrane before they can be activated (63).
NOX4 is an exceptional member of the NOX family. Although binding proteins have been suggested to enhance NOX4 activity (see earlier), these do not seem to be required for NOX4's basal activity, at least in vitro. According to the current knowledge, NOX4 is constitutively active and is mainly regulated via regulation of its expression.
All NOX isoforms catalyze the transfer of two electrons from NAPDH via their FAD domain and two iron-heme prosthetic groups to molecular oxygen. While NOX1, NOX2, NOX3, and NOX5 generate superoxide, NOX4, DUOX1, and DUOX2 mainly release hydrogen peroxide. In case of NOX4, the produced superoxide seems to be trapped and dismutated to hydrogen peroxide in an extracellular loop of NOX4 (169). For DUOX1 and DUOX2, the maturation factors DUOXA1 and DUOXA2 seem to be responsible for the type of ROS produced and released (72, 112). However, since iron-heme groups generally perform single-electron transfers, it is likely that DUOX1 and DUOX2 release hydrogen peroxide in a similar mechanism as proposed for NOX4.
NADPH oxidases are widely distributed through different tissues and cell types. For a full list, we refer the reader to the review of Bedard and Krause (13). Briefly, NOX1 is mainly expressed in the colon, NOX2 in phagocytes and B lymphocytes, NOX3 in the inner ear and some fetal tissues, NOX4 in the kidney and the blood vessels, and NOX5 in lymphoid tissue and testis. Notably, NOX5 is not expressed in rats and mice. Therefore, the information on NOX5 localization and function is limited. DUOX1 and DUOX-2 are most abundantly expressed in thyroid and lung tissue.
Often, suggested roles for NAPDH oxidases evolved mainly from tissue expression studies. For instance, the potential roles of NOX1, NOX2, NOX4, and NOX5 were based on their expression in cardiovascular tissues (93), of NOX2 and NOX4 in brain tissue (23, 140), of NOX2, NOX4, and DUOX2 in the lung (26, 67, 173), as well as of NOX1, NOX2, NOX4, and NOX5 in cancer cells and tumors (17). However, whether or which role NOX isoforms might play in health and disease in these tissues is still the subject of ongoing research.
NADPH Oxidase Inhibitors
Historical small-molecule NADPH oxidase inhibitors
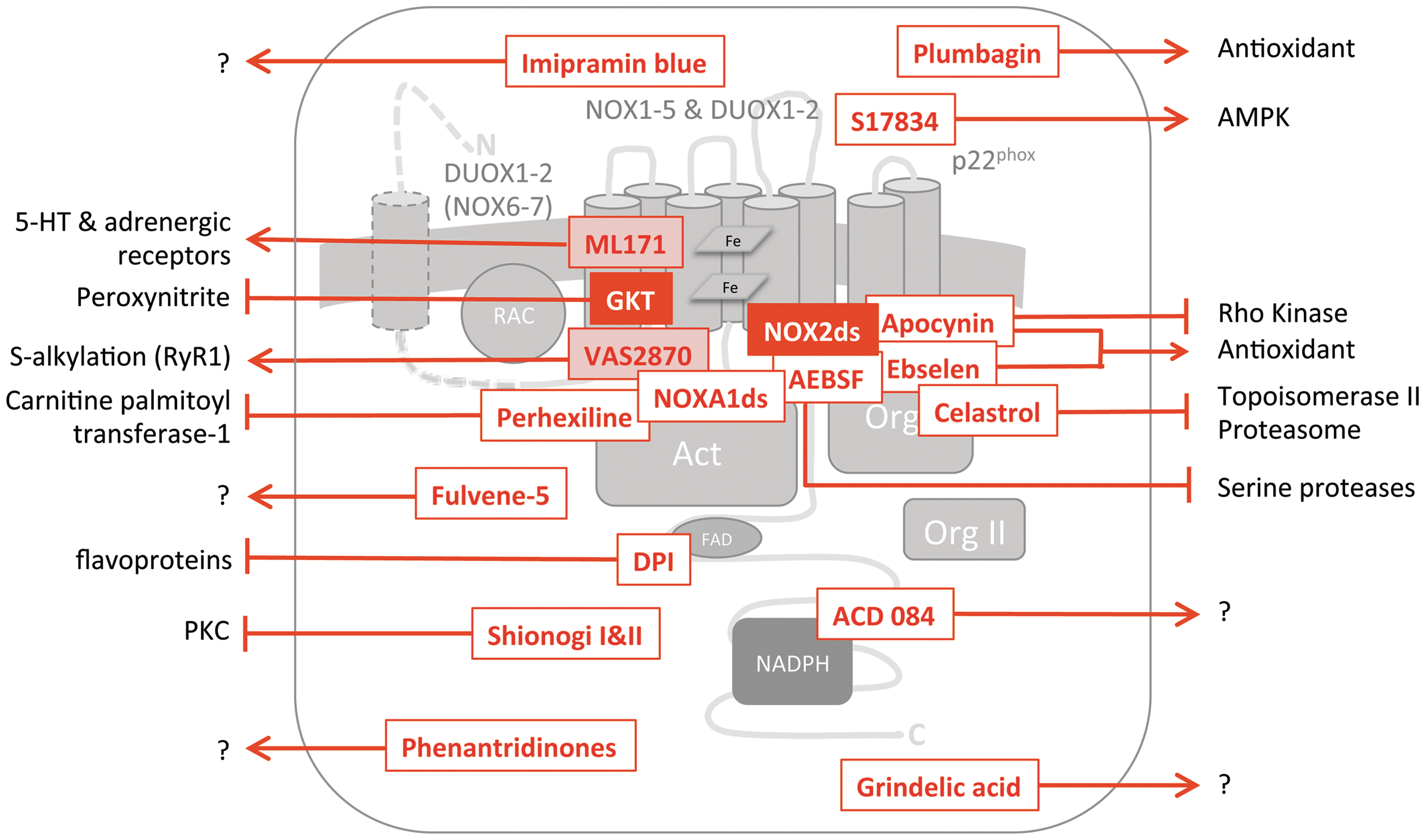
Several small molecules have been and are still being used as direct NADPH oxidase inhibitors. Although many of these suggested inhibitors inhibit NADPH oxidase activity, the majority of these historical inhibitors are unspecific, due to several off-target effects or the inhibition of features of NADPH oxidases that are not unique for NOX enzymes but also occur in other (ROS generating) enzymes. These unspecific inhibitors include the most frequently used NOX inhibitors, diphenylene iodonium (DPI) and apocynin, which have been proved to be unspecific as reviewed earlier in detail (2, 77, 157, 182). Briefly, DPI acts as a general flavoprotein inhibitor and, therefore, also inhibits eNOS, xanthine oxidase, and proteins of the mitochondrial electron transport chain (2, 124, 125). Apocynin shows intrinsic antioxidant activity, that is, ROS-scavenging properties (70), and it inhibits rho kinases (151). Other proposed NOX inhibitors such as 4-(2-aminoethyl)-benzenesulphonyl fluoride (AEBSF) or plumbagin (44) are used less frequently. However, they do not only have low potencies for NADPH oxidase but also unspecific effects. For example, AEBSF inhibits serine proteases (42), and plumbagin acts as an antioxidant and exerts several other unspecific effects such as NF-kappa-B inhibition and bactericidal actions [reviewed in ref. (127)]. In addition, none of the inhibitors mentioned here exhibits significant selectivity for any of the NOX isoforms. Furthermore, some inhibitors interfere with ROS detection dyes (181). Therefore, their use likely results in overestimations of the actual NADPH oxidase-linked effects. In conclusion, historical NADPH oxidase inhibitors have several weaknesses, and their use does not emable drawing any conclusions on the involvement of NOX in a given system. Obviously, more specific compounds are needed.
Novel small-molecule NADPH oxidase inhibitors
Characteristics of the ideal NADPH oxidase inhibitor
The ideal NADPH oxidase inhibitor should neither scavenge ROS, that is, not have antioxidant actions, nor inhibit other flavoproteins or NADPH-dependent proteins. NADPH oxidase inhibitors should further not influence the expression levels of NOX or their respective binding partners. They also should not interfere with upstream signaling pathways of NOX activation but rather inhibit NADPH oxidase activity directly. In addition to NADPH oxidase specificity, isoform selectivity for one of the NOX isoforms is desirable.
Recently, several new small-molecule NADPH oxidase inhibitors have been identified by drug screening approaches and characterized with regard to NOX isoform selectivity and potential unspecific effects (52, 58, 92). Growing knowledge on the biochemical properties of NADPH oxidases and their mechanisms of activation also enabled rational inhibitor design (145) followed by the identification of further inhibitors (76, 164). In the next few chapters, we will discuss these novel small-molecule NADPH oxidase inhibitors. We summarize their specificity for NOX enzymes (Table 2), NOX isoform selectivity (Table 1), likely mechanisms of action (Fig. 2), reported off-target effects, and their potential feasibility for in vivo proof-of-concept studies.
IC50 values of NOX inhibitors for different NOX isoforms and XO or AO activity are presented as determined in different cellular or cell-free assays as described in the respective publication and indicated with the superscription. The IC50 values that suggest relative NOX isoform selectivity are shown in bold. Only some inhibitors were tested for XO inhibition or ROS-scavenging effects, and inhibition was not significant (n.s.) for some of these compounds.
IC50 values were determined in acell-free or lysate assay; b K i were published; coverexpressing cells; dnative cells; eno significant inhibition for concentrations of approximately 30 μM or f100 μM, g20 μM, h10 μM, i5 μM.
AO, antioxidant; DUOX, dual oxidase; IC50, concentration with 50% inhibition of activity; NADPH, nicotinamide adenine dinucleotide phosphate; NOX, catalytic subunit of NADPH oxidases; NOX2ds, NOX2 docking sequence; XO, xanthine oxidase.
Proof for a direct interference with the NADPH oxidase complex is suggested when the compound is active in cell-free assays (+). Cell-free NOX1 and NOX2 assays use isolated membranes of NOX1- or NOX2-expressing cells and the respective recombinant cytosolic binding partners, or they use cytosolic fractions containing these binding partners instead of recombinant proteins. Assembly of the active NADPH oxidase complexes was induced with LiDS, SDS, or arachidonic acid. Compounds marked with (A) were added before assembly of the active NADPH oxidase-1 or -2 complexes as indicated. They may not inhibit NOX activity when added after complex assembly. Inhibition of NOX4 and NOX5 that do not require external binding partners has been determined using isolated membranes of transfected cells or whole cell homogenates. Some compounds have been applied in vivo in animals or humans intravenously (i.v.), intraperitoneally (i.p.), per os (p.o.), or intrathecally (i.t.). For Celastrol and ACD084, oral application is likely, as whole plant extracts are used orally in case of Celastrol and ACD084 is found in edible plants. ROS-scavenging effects of the different compounds were determined by measuring the decay of applied radicals itself or by measuring the interference with ROS derived from xanthine oxidase as a measure of superoxide [O2 •− (via XO)] scavenging. Ebselen did not interfere with XO but scavenged H2O2 at micromolar concentrationsa (IC50 ∼80 μM). Off-target effects were either examined systematically using commercially available librariesb or were found by accident. Successful completion of phase I clinical studies is indicated.
CNS, central nervous system; eNOS, endothelial nitric oxide synthase; GPCR, G-protein coupled receptor; LiDS, lithium dodecylsulfate; ROS, reactive oxygen species; SDS, sodium dodecyl sulfate.
GKT136901 and GKT137831
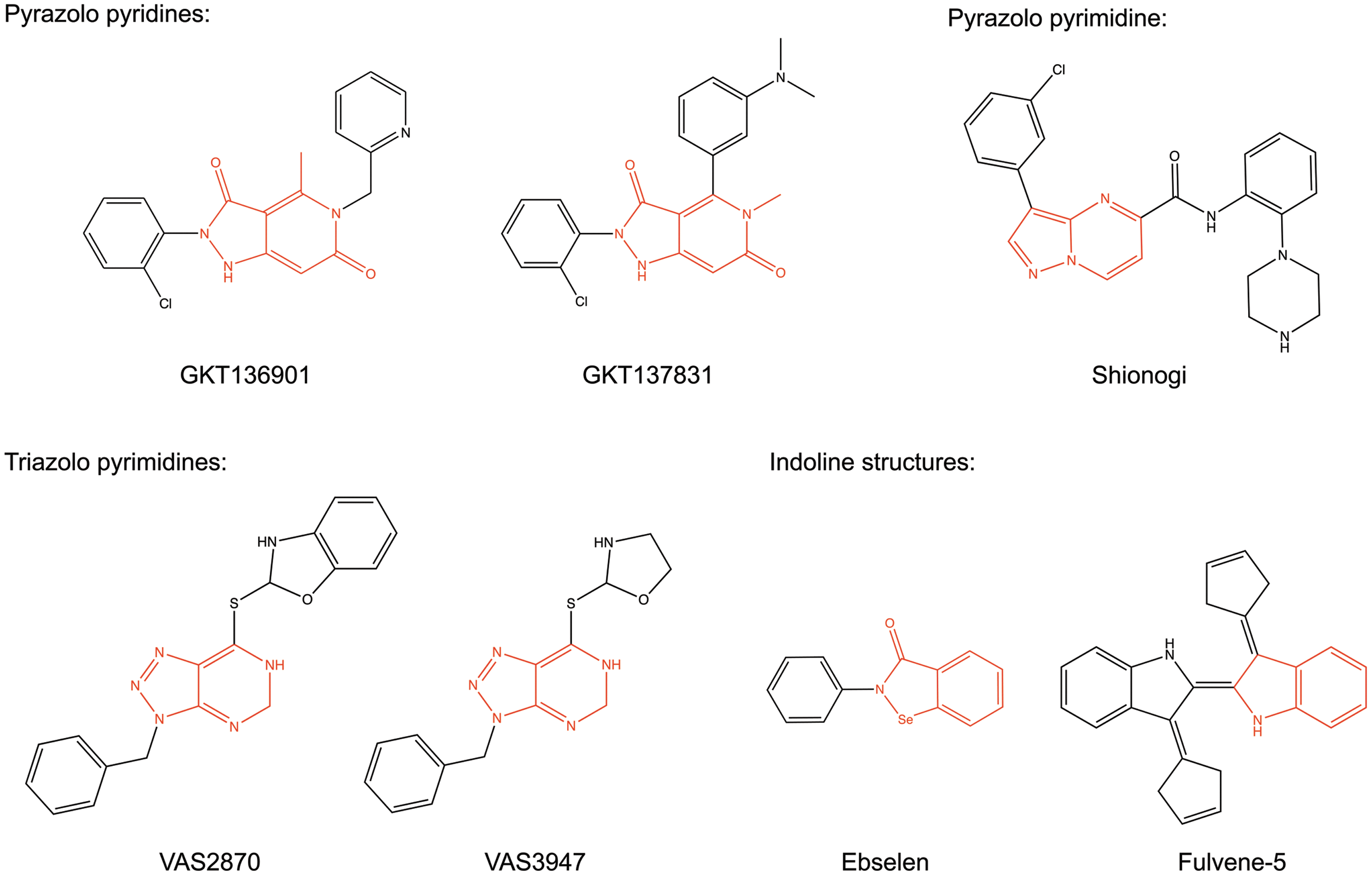
GenKyoTex developed the two inhibitors, GKT136901 and GKT137831, exploring structure–activity relationships (SAR) around pyrazolopyridine dione derivatives, which were identified in a high-throughput screen for NOX4 inhibitors (92) (Fig. 3). The GKT compounds potently inhibit NOX1, NOX4, and NOX5 with concentrations with 50% inhibition of activity (IC50) values in the three digit nanomolar range and show ∼10–15-fold higher IC50 values for NOX2 inhibition (Table 1) (5, 53, 92, 154). Inhibition profiles of DUOX1 and DUOX2 are not published. Both compounds only inhibit xanthine oxidase-derived ROS formation with high IC50 values of ∼100 μM (5, 154) [These IC50 values were estimated from the published concentration response curves (5, 154)]. However, GKT136901 potently scavenges peroxynitrite, a reactive nitrogen species created from the reaction of superoxide with nitric oxide (150) and dose dependently decreases Amplex Red fluorescence (own, Unpublished observation). While this might be beneficial for the therapeutic efficiency of the compound, it complicates the interpretation of obtained results regarding the participation of NOX enzymes. GKT137831 remains to be tested for this type of radical scavenging properties. With regard to NADPH oxidase inhibition, only K i (inhibition constant) values of GKT compounds, generated using the Cheng-Prusoff equation, are published (92). The Cheng–Prusoff equation describes the inhibition constant of a competitive inhibitor with an agonist or substrate (33). To our knowledge, the competitive character of GKT compounds with NADPH has not been analyzed. Thus, the interpretation of these inhibition constants is complex. Nevertheless, GKT136901 and GKT137831 are the best characterized NOX inhibitors currently available, for example, with regard to off-target effects and pharmacokinetics. GKT compounds did not influence tested off-target proteins, including redox-sensitive enzymes, G-protein-coupled receptors (GPCRs), kinases, ion channels, and other enzymes. They are orally bio-available and have favorable ADME profiles (5, 92). If GKT137831 does not scavenge free radicals, it is the currently best compound for in vivo proof-of-concept studies on the role of NOX enzymes in disease. Furthermore, GKT137831 is already in clinical development (79). Thus, it seems that its properties are more suitable for in vivo use compared with GKT136901.
ML171
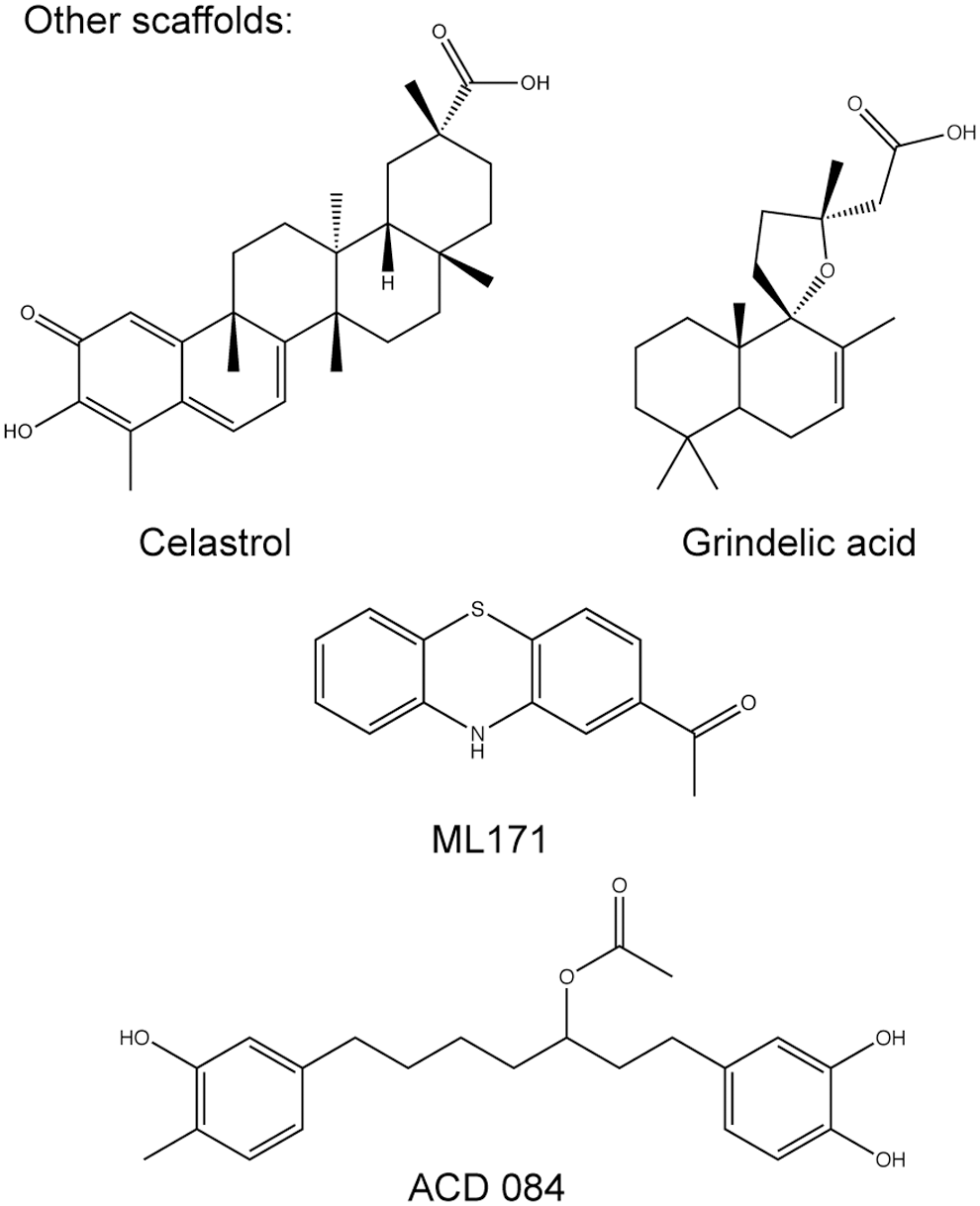
The Scripps Research Institute screened 16,000 compounds for NOX1 inhibition and performed limited SAR analysis with commercially available phenothiazines around the most promising hit, 2-(trifluoromethyl)-phenothiazine. They identified 2-acetylphenothiazine (ML171) (Fig. 4) as a potent NOX1 inhibitor (58). ML171 showed IC50 values of 130–250 nM for NOX1, and of 3–5 μM for NOX2–4 (Table 1) as well as for xanthine oxidase (58). It is known that several phenothiazines (e.g., chlorpromazine) act as antagonists on dopamine and serotonin receptors and are clinically used as anti-psychotic drugs. Therefore, ML171 was tested for its ability to inhibit a set of GPCRs, ion channels, and transporter proteins. While several compounds of the promazine class of drugs did not inhibit NOX enzymes, ML171 showed some inhibition of serotonin and adrenergic receptors. Although high K i values for this off-target inhibition of ML171 were reported, the effects on these receptors need to be excluded before using ML171 for in vivo proof-of-concept studies. However, due to the high degree of similarity to currently used drugs, the pharmacokinetics and safety data of ML171 likely enable the use of this compound in vivo.
VAS2870 and VAS3947
The NOX inhibitor VAS2870 is a triazolo pyrimidine (Fig. 3) that was developed by the company Vasopharm GmbH in a screening approach for NOX2 inhibitors (52, 165). The close derivative VAS3947 is slightly improved for solubility but shows a similar NOX inhibition profile (Fig. 3) (3, 181). For VAS2870, only IC50 values for NOX2 inhibition were published (52, 55), but it was also shown that NOX4 and NOX5 activity was inhibited by the compound (3, 88). VAS3947 showed IC50 values around 10 μM for NOX1, NOX2, and NOX4 in cell-free assays and no inhibition of xanthine oxidase or eNOS (181). The mechanism of NOX2 inhibition seems to be inhibition of the active complex assembly as shown in an assay using a semi-recombinant system (52). In contrast to other agents, VAS2870 only inhibited NOX2 activity in this assay when added before the induction of NOX2 active complex assembly (3) but not when added after complex assembly (3, 55). Notably, VAS2870 did not interfere with the translocation of p47phox (55). While this concept can be adapted easily for NOX1, the inhibition of NOX4 and NOX5 may be explained by the inhibition of an intra-molecular interaction of different domains of the enzymes (3) (illustrated in Fig. 2). Unfortunately, the poor solubility and lack of pharmacokinetic and specificity data for these substances limit their in vivo use. Recently, a potential off-target effect was reported: VAS2870 thioalkylated cysteine residues in the ryanodine receptor Ca2+ channel (RyR1) and GSH in vitro (167). Further analyses are required to find out whether this mechanism can be translated to the in vivo situation or be observed in the presence of physiological concentrations of GSH.
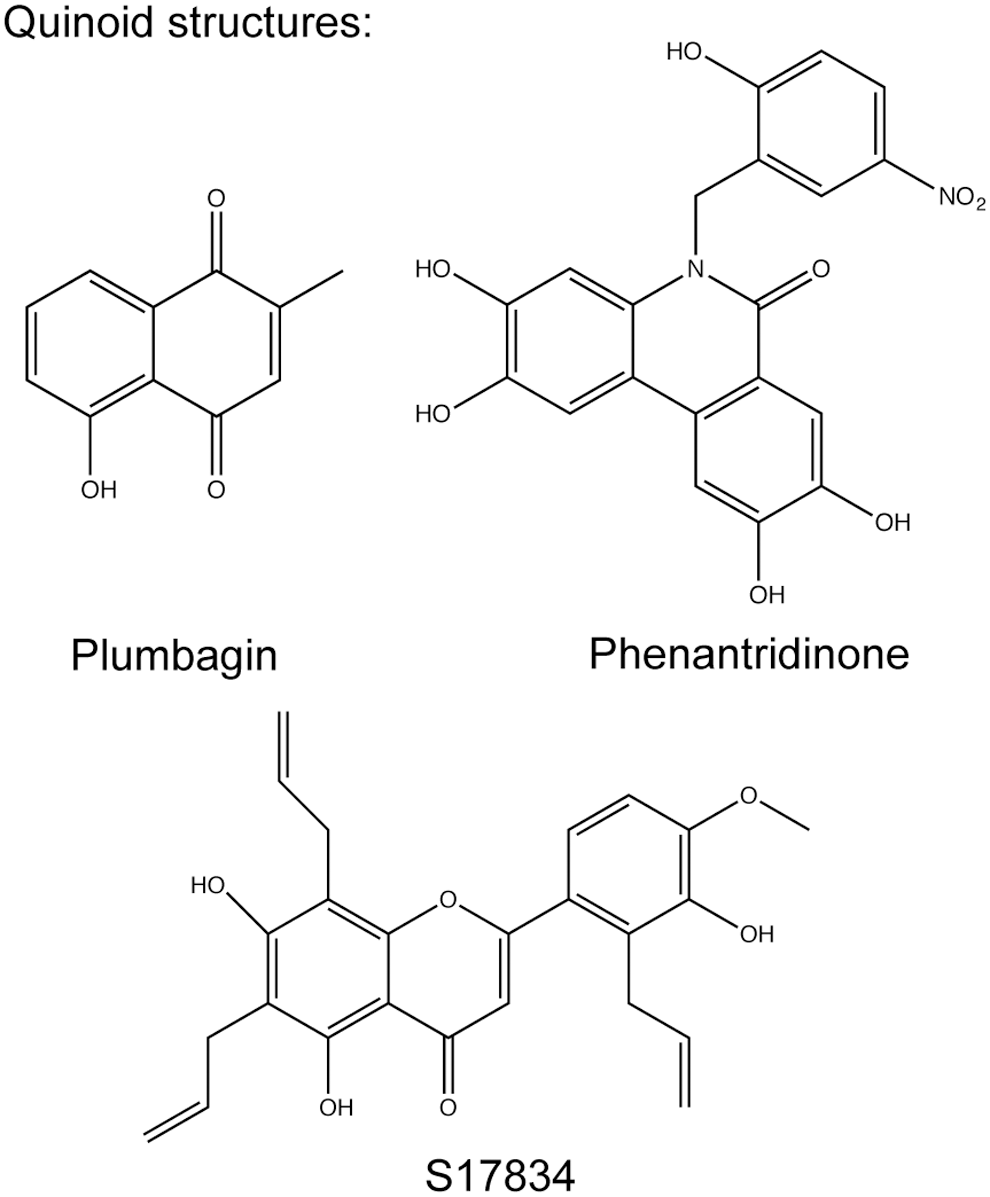
S17834
The polyphenol S17834 (Fig. 5) was developed by Servier and proposed to inhibit NOX enzymes based on the inhibition of superoxide formation in human umbilical vein endothelial cells (HUVECs) membranes. As reported, it neither scavenges superoxide nor inhibits xanthine oxidase or eNOS (27). Unfortunately, S17834 has not been further characterized with regard to its NOX isoform selectivity. However, it can be assumed that NOX2 and/or NOX4 are inhibited, as these are the main NOX isoforms expressed in HUVECs (157). Although no pharmacokinetic or safety data have been published, mouse studies suggest satisfying oral bioavailability and safety profiles (27, 96, 138, 187). These studies also revealed that S17834 activates adenosine monophosphate-activated protein kinase (AMPK) more potently than it inhibits NOX (187). Although a contribution of the NOX inhibition capabilities cannot be ruled out, the beneficial effects of S17834 in animal models of diabetes and atherosclerosis are mainly attributed to the activation of AMPK (96, 138, 183, 187).
Fulvene-5
Fulvene-5 (Fig. 3) inhibits NOX2 and NOX4-mediated ROS production at 5 μM concentration by 40% in a cellular assay (15). Neither the selectivity of this compound for other NOX isoforms, potential direct ROS-scavenging effects, nor inhibition of other flavoproteins was analyzed. Fulvenes are highly water soluble, but no pharmacokinetic data and safety profiles have been reported.
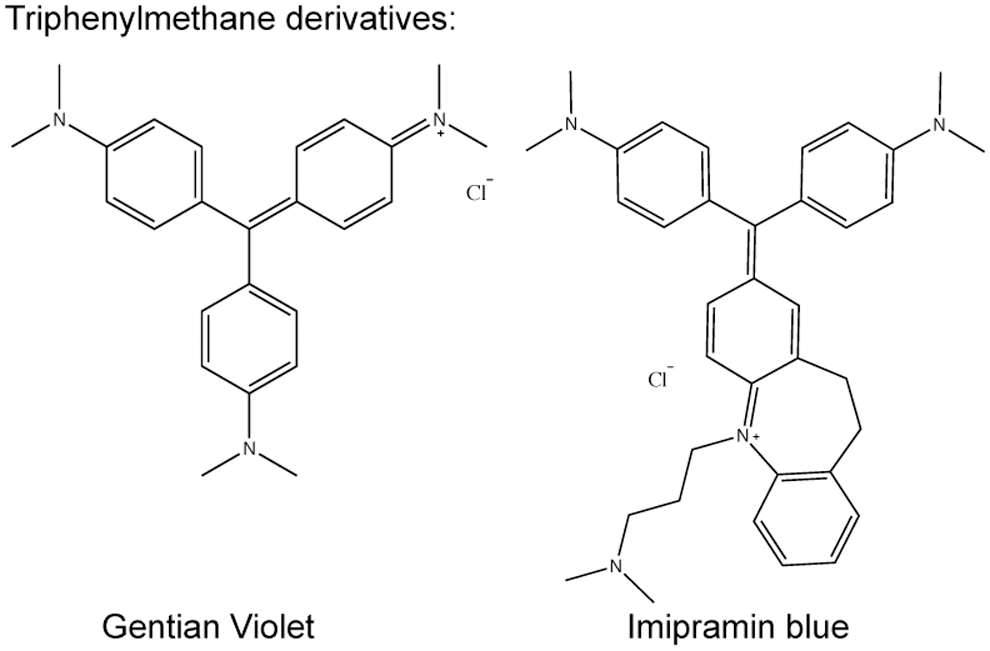
Triphenylmethane derivatives
The triphenylmethane derivatives Brilliant green, Gentian violet, and Imipramin blue (Fig. 6) were examined for NOX inhibition because of structural similarity to diphenyl iodonium (117, 134), a weak flavoprotein inhibitor compared with its derivative DPI (own observation). All three compounds were shown to inhibit NOX4 in cellular assays in different concentrations. Brilliant green and Gentian violet were additionally tested for NOX2 inhibition. Brilliant green showed a high potency and some selectivity for NOX2 compared with NOX4, while Gentian violet inhibited both NOX isoforms with a comparably low potency (134). Imipramin blue inhibited NOX4-derived ROS generation by ∼50% at 5 μM (117). However, no concentration response curves and IC50 values were reported, and unspecific effects such as direct ROS-scavenging properties or inhibition of other flavoproteins were not assessed. Thus, such actions cannot be excluded. In particular, the latter would be essential due to the compounds' structural similarity to diphenyl iodonium. Since the cationic character of these compounds might hinder membrane penetration, they may interfere with extracellular domains of NOX (Fig. 2). Nevertheless, Gentian violet is FDA approved for topical applications as an antiseptic for humans, and some toxicity data were determined for Imipramin blue (117). While this might enable in vivo proof-of-concept studies for the respective indications, the unclear specificity and NOX isoform selectivity does not enable linking the effects of these compounds to NOX enzymes.
Diarylheptanoids
Kofler et al. screened a selection of active compounds from edible plants to search for NOX4 inhibitors (89). They identified diarylheptanoid structures with four-substituted phenolic structures as the main structural feature for NOX inhibition. One of these compounds ACD 084 (Fig. 4) inhibited NOX4 with IC50 values of 3 μM, but it did not inhibit NOX2 or NOX5. Three similar diarylheptanoids not only showed higher potencies for NOX4 but also inhibited NOX2. While none of the mentioned compounds directly scavenged ROS, the effect on flavoproteins such as xanthine oxidase or eNOS was not analyzed. In addition, assessing a potential NOX1 inhibition by these compounds would have been important, particularly as another described potent NOX4 inhibitor class, GKT compounds, typically inhibits NOX1 and NOX4 with the same potencies (5, 92). This may indicate that it is difficult to obtain inhibitors with selectivity for NOX1 or NOX4. No data on the compounds' mechanism of actions have been published. However, ACD084 inhibited ROS production of the purified NOX4 dehydrogenase domain. This implies interference with FAD or NADPH binding as the mechanism of action, which is most likely not only specific for NOX4 but might also affect other NOX isoforms and other enzymes containing FAD or NADPH. The substances fulfil Lipinski's rule of five for drug-like molecules (97), and ACD084 seems to have a promising ADME profile (89).
Grindelic acid
The same screen resulting in diarylheptanoids also identified grindelic acid (Fig. 4) as an NOX4 inhibitor (IC50 2 μM) (89). Grindelic acid did not inhibit NOX2 and NOX5 and was not a direct ROS scavenger. However, similar to the heptanoids, the potential inhibition of NOX1 or other flavoprotein remains to be determined. Grindelic acid's mechanism of action is unknown, but it inhibited neither ROS in a cell-free membrane-based NOX4 assay nor from the purified NOX4 dehydrogenase domain. Similar to ACD 084, it seems to have promising ADME properties. Taken together, the diarylheptanoids and grindelic acid need further validation with regard to NOX specificity and selectivity. Nevertheless, these compounds may contribute to our understanding of the structural features of NOX inhibition.
Phenantridinones and flavonoids
Phenantridinones (Fig. 5) and flavonoids were identified as potent inhibitors of NOX4 in a cell-based screening of ∼1000 compounds (20). The most active compounds of the tested phenantridinone derivatives exhibited IC50 values around 200–600 nM. They neither reduced cell viability nor acted as a direct hydrogen peroxide scavenger. However, no further determination of the compounds' NOX isoform selectivity or flavoprotein inhibition potential is reported. The authors also described several flavonoids as potent NOX4 inhibitors, which are not scavenging hydrogen peroxide. However, one of the tested flavonoids was found to be an intrinsic antioxidant. Thus, these compounds should be used with caution. Flavonoids have a long history as antioxidants scavenging free radicals, lipid peroxides, and peroxynitrite (136, 147). Thus, although for example Quercetin [compound 7a in Borbely et al. (20)] does not seem to scavenge hydrogen peroxide according to this publication, one needs to consider the large body of literature stating antioxidant activity for this compound [reviewed in ref. (69)]. This underlines that special care should be taken to validate potential NOX inhibitors, especially natural compounds, which often show pleiotropic and direct antioxidant effects. In this regard, also off-target screens for identified compounds would be required before they could be used as reliable tools for in vivo proof-of-concept studies on NOX enzymes. Nevertheless, the authors used the structural properties of the identified NOX4 inhibitors to construct a pharmacophore model, which might help design and synthesize new NOX inhibitors, although most likely with limited NOX isoform selectivity.
Shionogi I and II
The pharmaceutical company Shionogi and Co Ltd. patented pyrazolo pyrimidine derivatives (Fig. 3) as NOX inhibitors. They claimed that this class inhibits NADPH oxidase activity in bovine aortic membrane fractions as well as in vivo in neutrophils and blood vessels (77, 162). From the inhibition of ROS release from neutrophils, the inhibition of NOX2 could be concluded, which was, indeed, recently confirmed (55). However, the authors also reported that these compounds are not direct NOX inhibitors but rather inhibit protein kinase C, which induces NOX2 complex assembly via phosphorylation and membrane translocation of p47phox. Thus, these compounds do not fulfil the requirements of direct NADPH oxidase inhibitors.
Ebselen
Smith et al. identified ebselen (Fig. 3) and derivatives as potent NOX2 inhibitors using an innovative approach: a recombinant protein encompassing the tandem SH3 domain of p47phox linked to a proline-rich p22phox peptide. This enabled them to assay direct binding of the two NADPH oxidase binding partners, p47phox and p22phox (Fig. 2) (164). It was the first published fully recombinant assay used for NOX inhibitor screening purposes (12). Hits were validated in cell-free NOX2 assays and cellular assays for NOX1, NOX2, NOX4, and NOX5. Several derivatives were used to gain insights into SAR of NOX inhibition. None of the tested compounds exhibited significant activity on NOX4. The majority of the active compounds showed selectivity for NOX1 and NOX2 compared with NOX5. One compound (JM-77b) exhibited promising NOX2 selectivity. Ebselen itself was among the most potent derivatives inhibiting NOX1, NOX2 and NOX5 with a similar potency (IC50 values 150–700 nM). It neither influenced xanthine oxidase activity nor scavenged hydrogen peroxide (see Table 1). Interestingly, the concentration of ebselen that is required for the inhibition of NOX1 and NOX2 in cellular assays is magnitudes lower than the concentrations required for the long-known action of ebselen as a glutathione peroxidase mimetic at concentrations around 10 μM (116). However, ebselen also is a potent peroxynitrite scavenger in vitro (concentration with 50% of the maximal effect [EC50] of 150 nM) (22) and a moderate eNOS inhibitor in endothelial homogenates (IC50 8.5 μM) (188). This might complicate the interpretation of results from in vivo studies obtained with ebselen if used as an NOX inhibitor. An advantage is that ebselen is orally available and that its safety profile enabled clinical development up to phase III for the treatment of cerebral ischemia injury (109, 184). Nevertheless, the drug was never brought to the market.
Celastrol
Plant extracts from Thunder God Vine (Tripterygium wilfordii Hook F.) are used in Traditional Chinese Medicine to treat states of chronic inflammation. Celastrol (Fig. 4), one of the active compounds isolated from this plant, was identified to inhibit NOX2 in neutrophils. A thorough characterization of its NOX enzyme inhibition profile showed a slight selectivity for NOX1 (IC50 0.41 μM) and NOX2 (IC50 0.59 μM) compared with NOX4 (IC50 2.79 μM) and NOX5 (IC50 3.13 μM) in live cell assays. It also inhibited NOX2 (IC50 1.24 μM) and NOX5 (IC50 8.4 μM) in cell-free assays (76). The authors experimentally linked this selectivity to a mechanism of NOX inhibition with Celastrol binding to p47phox and disrupting the binding of p47phox to p22phox. Interestingly, NOX4 and NOX5, which act independent of the classical organizer and activator binding proteins, are also inhibited. This suggests a more complicated mechanism of action. However, Celastrol shows a broad variety of effects and targets, such as the inhibition of topoisomerase II (119) or proteasomes (185). It is also known to covalently bind to cysteine residues [reviewed in ref. (148)]. These effects might complicate the interpretation of in vivo results obtained with Celastrol when used as an NADPH oxidase inhibitor.
Perhexiline
Perhexiline is a prophylactic anti-anginal agent that is mainly prescribed in New Zealand and Australia. Mechanistically, it was suggested to inhibit carnitine palmitoyl transferase-1 (86), resulting in a shift in myocardial energy usage from fatty acid to glucose metabolism. However, a part of its beneficial effects was also attributed to the inhibition of NOX2 in neutrophils (IC50 1.5–3.6 μM) as well as in different cardiovascular tissues and cells (85). A later study confirmed the inhibition of NOX2 in neutrophils and in an assay measuring purified semi-recombinant NOX2 activity (IC50 13.2 μM). Xanthine oxidase was not inhibited, and superoxide was not directly scavenged (55). This indicates direct NOX2 inhibition. However, effects on other NOX isoforms have not yet been published. Since perhexiline is an approved drug, its use in animal studies should be straightforward. However, it will be difficult to attribute results obtained in in vivo experiments to certain actions of perhexiline.
Biologicals As NADPH Oxidase Inhibitors
Biologicals as drugs include activity regulating peptides and therapeutic antibodies. The situation regarding specific antibodies against the different NOX isoforms is unsatisfying (3). So far, only a few antibodies with a suggested capacity to inhibit NOX2 (24) or NOX4 (189) were published, but they have neither been used as biologicals in experiments, nor have they been fully validated against other NOX isoforms. Peptides as drugs or inhibitors face the major limitation not only of potentially causing antigenic responses and exhibiting poor in vivo stability, but also of not being able to penetrate through cell membranes. The screening of larger peptide libraries or peptide walking along the sequence of the respective proteins in the search for modifiers of enzymes, therefore, relies on simple reproducible cell-free assays. In case of the NAPDH oxidase enzyme family, such an assay only exists for NOX2 [reviewed in ref. (40)]. Several NOX2-derived peptides have been created and used to analyze the mechanisms of NOX2 activation (49). Some of these inhibited NOX2 activity. Especially peptides targeting the FAD and NADPH binding domains of NOX2 showed inhibitory effects. However, they are most likely not selective for NOX2, as all NOX isoforms show homologous FAD and NADPH binding sites (39). This might explain why only three NOX peptides have been linked to tat peptides, an HIV trans-activator protein that enables cell membrane penetration. One of these peptides is a part of the rac binding domain of NOX2 and inhibits its activity in intact neutrophils (83). However, this rac binding site is also present in NOX1 and NOX3, and, therefore, this peptide might inhibit not only NOX2 but also the other NOX isoforms. In addition, rac GTPases interact with a plethora of proteins. Thus, other unspecific effects are not unlikely. Another peptide, resembling a phosphorylation site required for the activation of p47phox, was found to inhibit NOX2 activity in neutrophils by interference with the assembly to an active complex (41). In a similar approach, very recently, a peptide termed NOXA1ds was designed and shown to inhibit NOX1 activity (143). It was also shown that NOX4 activity could not be inhibited with specific peptides against its C-terminal tail, its B-loop, or the N-terminal tail of p22phox (37, 100). To our knowledge, no attempts to find peptides directed against NOX3, NOX5, or DUOX1 and DUOX2 have been published. The only NOX-derived peptide, which was examined and extensively used, is NOX2 docking sequence (NOX2ds)-tat.
NOX2ds-tat
The first rationally designed biological NOX inhibitor is the 18-amino-acid peptide NOX2ds-tat (originally named gp91ds-tat). The peptide contains a nine-amino-acid sequence of the NOX2 intracellular B-loop that binds the NOX organizer protein p47phox, thereby preventing assembly of the active NOX2 complex (74, 99, 133, 145, 194, 195) (Fig. 2). This B-loop sequence of NOX2 shows high homology with the B-loop sequences of NOX1 and NOX4. Despite this, it was recently shown that NOX2ds-tat, indeed, is selective for NOX2 compared with NOX1 and NOX4 (36). However, this proof is based on a reconstituted cell-free assay. Therefore, the inhibition of NOX1 or NOX4 in vivo in the presence of all activators and potential co-factors, which might not even have yet been discovered, cannot be excluded. Cell permeability is conferred by linking a nine-amino-acid tat peptide from the human immunodeficiency virus to the peptide. Although the tat peptide was shown to localize to the cytoplasm of neutrophils (35), NOX2ds-tat inhibited superoxide formation released by intact neutrophils only by 35%; while it inhibited 80% of superoxide generated in cell-free assays (145) with an IC50 of 0.74 μM (36). Still, NOX2ds-tat was shown to be effective in several in vivo animal models, either applied intravenously or expressed via adenoviral infection, indicating that it reaches its destination (74, 99, 133, 194, 195). To rule out effects caused by the tat peptide itself (25), scrambled nine-amino-acid peptide sequences linked to the tat peptide were used as controls in most of the studies (74, 133, 145, 194). In summary, NOX2ds-tat is a good tool that could be used to investigate the role of NOX in diseases, at least in acute situations.
NOXA1ds
The first peptide inhibitor of NOX1 activity was developed in a similar approach as the one that led to the identification of NOX2ds-tat (143). This peptide contains the 11-amino-acid sequence of the docking sequence of NOXA1 to NOX1. It was shown to bind to NOX1 and to prevent assembly of the active NOX1 complex (143). The inventors chose an amino-acid-sequence that markedly differs from similar sequences in NOX2 to provide NOX isoform selectivity. Conclusively, they showed that the peptide does not inhibit NOX2, NOX4, or NOX5 in assays using lysates of transfected cells (143). Further, no inhibition of xanthine oxidase or direct superoxide scavenging by NOXA1ds was observed. However, as mentioned earlier for NOX2ds-tat, the inhibition of different NOX isoforms in living cells with intact signaling pathways and potential activating factors was not determined. Although the peptide was shown to penetrate through cellular membranes, its in vivo efficacy remains to be determined.
NOX Isoforms—Are They Really Therapeutic Targets?
ROS and NADPH oxidases as one of their main sources were suggested to be involved in many diseases. However, for a long time, a validated physiological function could only be found for NOX2 and DUOX2 based on mutations in humans or mice leaving a dysfunctional protein and therefore leading to chronic granulomatous disease (CGD) or severe hypothyroidism, respectively (47, 80, 113). The role of the other NADPH oxidase isoforms in health and disease were for a long time—and for NOX5 still are—based on reported up-regulations of NOX expression levels in certain disease conditions or based on results from cultured cells. The solid validation of NADPH oxidases as potential drug targets (Fig. 7) only started with the introduction of the first NOX knockout (KO) mice. The first NOX KO mice, NOX2 KO, was published in 1995 (137). It took another 10 years until a NOX1 KO mouse was published (56, 103). Only in 2010, four reports of NOX4 KO mice came out, with all four using different targeting strategies (26, 88, 91, 190). In the same year, a DUOX1 KO mouse was published (45). Since NOX5 is not expressed in mice and rats, no KO animals exist. In case of the other NOX isoforms, the availability of KO mice enabled a more detailed validation of the roles of different NOX isoforms in health and disease. However, most KO models are constitutive. Thus, the deleted gene is already absent before the induction of a disease in a mouse model, which—with the exception of human monogenic diseases such as CGD—does not mimic the human disease pathology. In addition, the long-term absence of the gene might trigger compensatory changes, masking the actual effect of the lacking gene product. Thus, for the validation of NOX enzymes as drug targets, not only the effect of a deletion of a NOX isoform but also the inhibition of its activity by small molecules or peptides in a disease model is required.
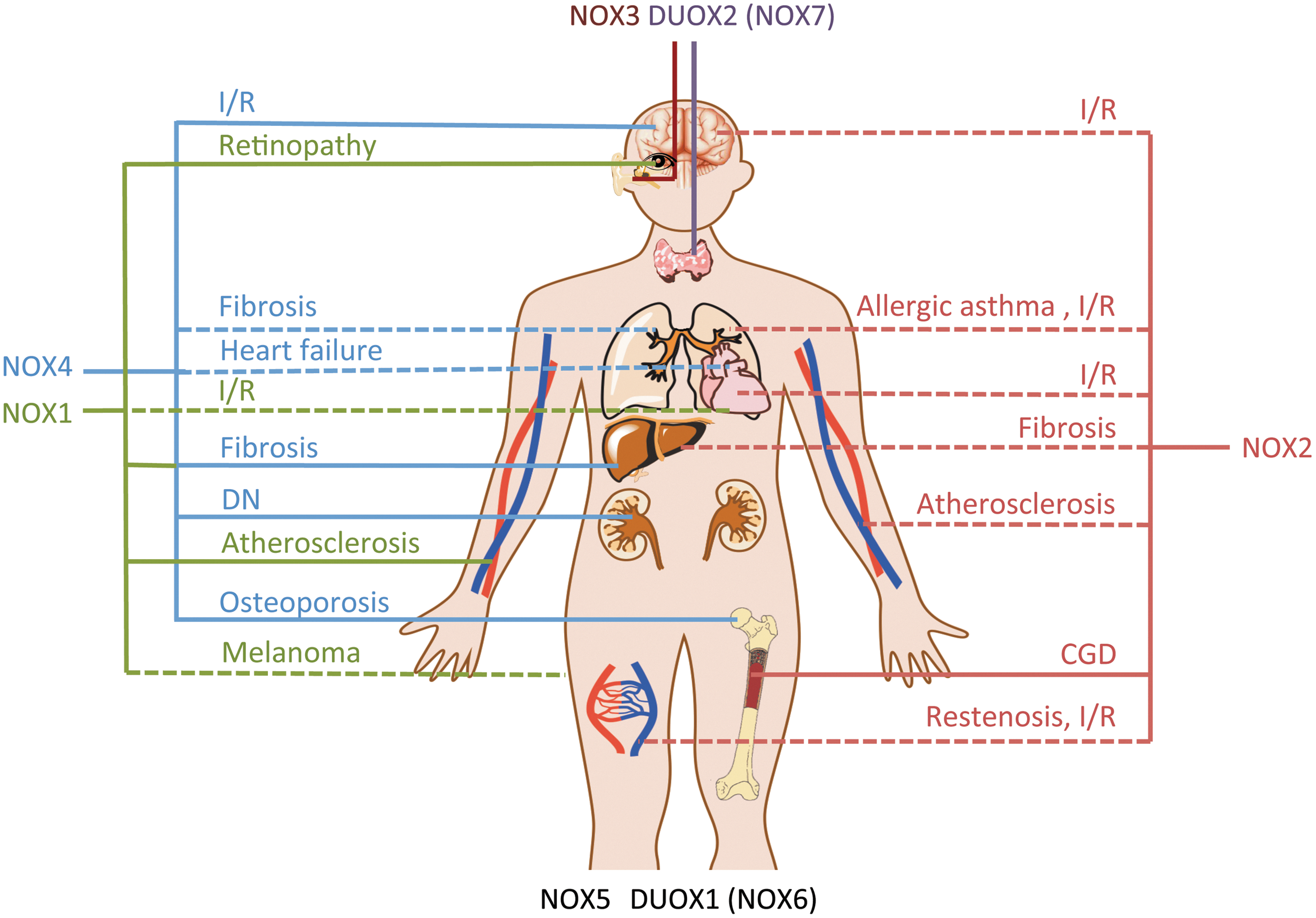
The history of the in vivo use of novel NOX inhibitors is short
The number of validated and largely specific NOX inhibitors that have been applied for proof-of-concept studies is limited. Although DPI and—even more—apocynin have been extensively used to inhibit NOX enzymes in numerous disease models [reviewed in refs. (142, 157)], they are inadequate to determine the role of NAPDH oxidases due to their unspecific effects mentioned earlier. The NOX2 inhibitory peptide NOX2ds-tat and GKT136901/GKT137831 have been used as relatively specific NOX inhibitors in vivo and, to a lesser extent, also VAS2870, Fulvene-5, and Imipramine blue.
NOX1 as a therapeutic target for diabetic atherosclerosis, ischemic retinopathy, and liver fibrosis
NOX1 has been suggested to play a role in vascular disorders such as hypertension, atherosclerosis, and ischemic injuries, but final validation is warranted. More recently, NOX1 has been validated as a pathologic player in diabetic atherosclerosis (64). Both treatment of streptozotocin (STZ)-treated apolipoprotein E (ApoE) KO mice with the NOX1/4 inhibitor GKT137831 and deletion of NOX1 in STZ-treated ApoE KO mice resulted in a profound anti-atherosclerotic effect. In contrast, the deletion of NOX4 in STZ-treated ApoE KO mice was not protective. A role of NOX1 in atherosclerosis is further supported by two other studies. The first showed decreased lesion area and macrophage infiltration, two hallmarks of atherosclerosis, in NOX1/ApoE double KO mice after high fat diet feeding compared with ApoE KO mice (160); the second showed decreased atherosclerotic lesions and cellular surface adhesion factor CD44 in ApoE KO mice treated with GKT136901 (176). Very recently, NOX1 was also validated as a therapeutic target in ischemic retinopathy (179). Using a rodent model of retinopathy of prematurity, which is a major cause of blindness caused by damage to the retinal microvasculature, retinas of NOX1 KO mice showed reduced signs of retinopathy, such as neovascularization and avascular retina. In contrast, retinas of NOX2 and NOX4 KO mice were not protected. In addition, a subcutaneous injection of the NOX1/4 inhibitor GKT137831 protected rat retinas from retinopathy when applied during the hyperoxic or during the next normoxic phase (179). This effect in post-hyperoxic rats confirms the therapeutic potential of NOX inhibition in ischemic retinopathy. With regard to ischemia-reperfusion injury in the heart, another study reported that NOX1 and NOX2 KO mice, as well as combined NOX1/NOX2 KO, but not NOX4 KO mice were protected (21). Notably, no protection was seen using a permanent ischemia model. However, to validate the role of NOX1 and NOX2 in heart ischemia-reperfusion, data on the pharmalogical inhibition of these NOX isoforms is warranted. Despite a mild hypotensive phenotype of NOX1 KO mice (56), a role of NOX1 in blood pressure regulation and hypertension has not yet been fully validated. Most studies exploring a role of NOX1 in blood pressure used angiotensin II (AngII)-mediated hypertension models (56, 103). However, AngII infusion does not necessarily reflect the human situation of hypertension, as unphysiologically high concentrations of AngII are required to increase the blood pressure and induce end organ damage. Therefore, the proposed role of NOX1 in hypertension might not be translatable to the human situation. In addition, NOX1 deletion did not change blood pressure in a hypertension model using a transgenic mouse expressing human renin (186). No differences in blood pressure have been detected in mice treated with the NOX1/NOX4 inhibitor GKT136901 (156).
Next to cardiovascular diseases, a role of NOX1 in liver fibrosis evolved. Treatment with the NOX1/NOX4 inhibitor GKT137831 attenuated hepatic fibrosis in bile duct ligation and hepatotoxin models (5). The authors showed that NOX1 up-regulation leads to increased expression of NOX4 and suggested the inhibition of both NOX isoforms as a promising therapeutic strategy in hepatic fibrosis. The same group validated this role of NOX1 by showing attenuation in both NOX1 and NOX2 KO mice (129). The role of NOX4 in liver fibrosis was confirmed by another study showing that GKT137831 treatment and NOX4 deletion attenuated fibrosis in mice (79). Based on these results, a causal interaction between NOX1 and NOX4 seems likely in disease progression. Therefore, both isoforms represent potential therapeutic targets for these liver pathologies.
NOX1 was further suggested to mediate tumor angiogenesis after an injection of two different tumor cells into mice (54). Surprisingly, tumor angiogenesis by melanoma cells was attenuated in NOX1-deficient mice, while GKT136901 treatment decreased tumor angiogenesis and weight in the other tumor model. A more detailed analysis on the role of NOX1 in tumor angiogenesis is, therefore, required before declaring NOX1 as a validated therapeutic target for cancer. Based on observations in NOX1-deficient mice, NOX1 may also be relevant in models of neointima formation after wire-induced injury of the femoral artery (95), of neuroinflammation (34), and of hyperalgesia (73). Nevertheless, since a therapeutic branch was not explored for the latter disease models, NOX1 cannot yet be considered a validated therapeutic target for the treatment of these diseases.
NOX2 inhibition—one to cure them all?
NOX2 KO mice display an impaired immune defence against pathogens and represent a validated model of CGD (137). Therefore, complete abrogation of NOX2 activity might not be acceptable in humans. This should be kept in mind when considering the inhibition of NOX2 for therapeutic purposes. Up-regulated or overly active NOX2 has been implied in a plethora of diseases, especially diseases of the cardiovascular and cerebrovascular system [reviewed in ref. (47)]. The role of NOX2 in the innate and adaptive immune response might help explain this involvement of NOX2 in the majority of tested disease models. Overall, NOX2 has been linked to almost every animal disease model, with many of them involving a significant inflammatory component. The protective effects often observed by deleting NOX2 in mice may, thus, be mediated by an attenuated inflammatory response. On the inhibitory side, NOX2 is the rare exception among the NOX enzymes, as with NOX2ds-tat a selective inhibitor is available. Unfortunately, the low bioavailability of peptides limits its use for in vivo applications and potential therapeutic options in humans. Due to the large number of studies using NOX2 KO mice or the NOX2ds-tat peptide, we will focus on indications that were assessed by several groups or which were validated using both NOX2 inhibitors and KO mice.
NOX2 inhibition was shown to be effective in different cerebrovascular regulations and disturbances in the preventive branch by NOX2 KO and in the therapeutic branch by NOX2ds-tat. These indications include age-dependent neurovascular de-regulation (131), AngII-induced de-regulations in cerebral blood flow, and neurovascular coupling (59, 84), as well as Alzheimer's disease models, namely in amyloid precursor protein- (133) and amyloid beta peptide-induced de-regulations (132). However, in these models, NOX2ds-tat was applied via a cranial window, which almost excludes these studies as valuable proof of concepts for NOX inhibition in human therapy of these diseases. Several studies suggested a protection by deleting NOX2 in brain ischemia reperfusion injury, some by applying unspecific inhibitors such as apocynin, DPI (120), or ebselen (90) [reviewed in ref. (140)]. Next to the unspecific character of these inhibitors, the time point of application, namely pre-stroke, presents the major drawback of these studies, as this does not reflect a realistic treatment option. The role of NOX2 in stroke was partly confirmed by protection with NOX2ds-tat in rats when applied before (144) or even after ischemia reperfusion (191). In contrast, another study found no protective effect of deleting NOX2 KO in stroke (88). However, the protective effect of NOX2 inhibition or NOX2 deletion in these studies was rather moderate, and the studies were largely underpowered (139). In conclusion, NOX2 may play a role in stroke, but the effect is still under discussion. Therefore, at present, NOX2 cannot be considered a validated stroke target. This also holds true for many other diseases. Remarkably, the use of NOX2 KO animals and studies using NOX2ds-tat (98, 99, 194) suggest that NOX2 is not involved in blood pressure regulation or hypertension in mice. In contrast, a role of NOX2 in hypertension in spontaneously hypertensive rats (SHRs) based on NOX2 inhibition by NOX2ds-tat was suggested (158, 195). Thus, NOX2-derived ROS might be involved in baroreceptor reflex regulation in these rats (158). However, a further analysis is required to clarify the precise role of NOX2 in SHR and in blood pressure regulation in different species. A role of NOX2 in vascular disorders such as restenosis after arterial injury (30, 46, 74, 177), endothelial dysfunction, and atherosclerosis (11, 81, 98, 99, 145, 175, 194) was suggested based on experiments with NOX2 KO models and by the inhibition of NOX2 with NOX2ds-tat. However, the respective various studies used slightly different disease models. Accordingly, a direct comparison is not straightforward. Therefore, NOX2 should not be considered a fully validated target for these diseases. Furthermore, chronic treatment with NOX2 inhibitors would always carry the risk of impairment of the immune system. Diabetic NOX2 KO mice (STZ model), for example, showed increased susceptibility to Gram-negative infections and died at 20 weeks after diabetes induction unless treated with antibiotics (64). Importantly, even a protective role of NOX2-derived ROS in the severity of autoimmune diseases such as rheumatoid arthritis has been suggested (149).
NOX3—a target candidate for cisplatin-induced hearing loss
Based on a mutant mouse study, a functional role of NOX3 in otoconia formation in the inner ear was described (128). NOX3 was also suggested to be the main source of ROS in cisplatin-induced hearing loss (7, 115). This effect was prevented by in vivo application of NOX3 small interfering RNA (siRNA) into the rat ear (114) and of a small molecule that inhibited NOX3 mRNA expression after cisplatin treatment (161). With the exception of ML171 (IC50 of 7.5 μM), none of the novel NOX inhibitors has been tested for its ability to inhibit NOX3. It would be interesting to know whether the other inhibitors attenuate NOX3 activity. If so, their use may provide a final proof of concept for a critical role of NOX3 in cisplatin-induced hearing loss.
NOX4 as a therapeutic target for ischemic stroke, diabetic nephropathy, and osteoporosis
With four different KO mice and several proposed inhibitors, NOX4 seems to be the best validated drug target among the NOX isoforms. NOX4 KO or an intrathecal injection of the NOX inhibitor VAS2870 significantly reduced mortality and infarct size and improved neurological outcome in a mouse brain ischemia-reperfusion model (88). A role of NOX4 in diabetic nephropathy has already been suggested in 2005 (61), and the GKT inhibitors are developed by GenKyoTex for this indication since several years. Therefore, the validation of NOX4 as a target in diabetic nephropathy came rather late. Diabetic nephropathy was significantly reduced in terms of albuminuria, collagen IV accumulation in glomeruli, fibronectin accumulation, and vascular endothelial growth factor expression in NOX4 KO and wild-type mice treated with GKT137831 in an STZ model of insulin-independent diabetes (78). Along with an earlier study conducted in db/db mice that already suggested a role for diabetic nephropathy based on results obtained with GKT136901 (155), NOX4 can be considered a validated target for diabetic nephropathy, at least in mice. Recently, NOX4 was validated as another therapeutic target. Both inducible genetic KO of NOX4 and NOX1/4 inhibition with an NOX inhibitor by GenKyoTex attenuated bone loss in an ovariectomy model of murine osteoporosis (60). The effect of pharmacological NOX inhibition on bone loss in this mouse model was less pronounced than the effect of the gold standard therapeutic bisphosphonate (60). Nevertheless, NOX4 KO mice showed significantly increased bone density, and higher NOX4 protein levels have even been detected in bones of human osteoporosis patients. Furthermore, NOX4 was suggested to be the main ROS source in fibrotic diseases of different organs, including the lung (68), liver (10), and heart (38). As already discussed in the section on NOX1, the role of NOX4 in hepatic fibrosis was reported in NOX4 KO mice and confirmed by GKT137831 treatment (5, 79). Surprisingly, NOX4 is not fully validated as a therapeutic target in pulmonary fibrosis, although GKT137831 was granted orphan drug status for the treatment of idiopathic pulmonary fibrosis. While genetic deletion in a NOX4 KO mouse (26) or genetic silencing using siRNA versus NOX4 in vivo (68) improved bleomycin-induced pulmonary fibrosis, the in vivo application of a pharmacological NOX4 inhibitor for the treatment has not been reported. However, the authors described a reduction of the collagen content in lungs of bleomycin-treated mice after intratracheal application of a close analogue of GKT137831. However, histological data clearly showing fibrosis in these lungs are not presented in this publication (53). In general, the bleomycin model only enables limited statements on drug efficacies against lung fibrosis. Only compounds that prevent or stop progression of fibrosis have the potential to be translated into the clinic (110). Importantly, fibrosis does not ordinarily progress in this model (153). Another role for NOX4 in pulmonary vessels was suggested, as GKT137831 treatment prevented hypoxia-induced right ventricle hypertrophy, thereby attenuating pulmonary hypertension (65). However, an involvement of NOX4 in pulmonary hypertension still awaits confirmation in NOX4 KO animals. The role of NOX4 in heart failure is discussed controversially. A cardiac-specific NOX4 KO mouse was protected from heart failure that developed after thoracic aortic banding (91), a rather severe model of heart failure progression. In contrast, a constitutive NOX4 KO mouse showed more pronounced heart failure after abdominal aortic banding (190), a comparably mild model of heart failure. This suggests that NOX4 is beneficial in chronic heart failure by mediating angiogenesis, an effect that has also been shown in hindlimb ischemia (152). In contrast, acute activation of NOX4 in a severe heart failure model may result in toxic ROS concentrations. Other proposed pathological roles of NOX4 include the progression of tumor invasion of glioblastoma cells into healthy tissue and the growth of hemangiomas. While the first could be inhibited with imipramine blue (117), the second could be inhibited with Fulvene-5 (15). However, the unclear isoform specificity of these compounds does not enable any attribution of these functions to NOX4.
In conclusion, we recommend the use of NOX KO animals in parallel to inhibitors in order to validate NOX4 as a therapeutic target for any diseases. Finally, NOX4 inhibition was suggested as therapy for the treatment of neuropathic pain (82). The effectiveness of preventions was shown using NOX4 KO animals. However, again, inhibitor studies are required to validate NOX4 as a therapeutic target for neuropathic pain.
Is NOX5 a neglected therapeutic target?
Since NOX5 is not expressed in mice and rats, its role is less clear than the role of any of the other NOX isoforms. Since KO strategies in larger animals, such as rabbits, have only recently become available and are expensive, the use of selective NOX5 inhibitors might be a more promising approach while analyzing its role in larger animals. So far, only GKT compounds were tested or shown to exhibit potent inhibition of NOX5 in a cellular system (5). Since NOX5 likely is the only NOX isoform in human spermatozoa, the inhibition of ROS formation by GKT137831 in spermatozoa was attributed to NOX5. As a first hint on a physiological role of NOX5, it was shown that NOX5 inhibition decreased spermatozoa motility (118). However, the knowledge on NOX5 is still too limited to consider it as a therapeutic target for any disease.
DUOX1/NOX6 and DUOX2/NOX7—only relevant for hypothyroidism?
The physiological and pathophysiological roles of DUOX1 are widely unknown. A recently published DUOX1 KO mouse showed no obvious phenotype (45). However, a role of DUOX1-derived hydrogen peroxide for pressure responses in the urinary bladder was suggested (45). Dysfunctional DUOX2 leads to severe hypothyroidism in humans and mice. To our knowledge, nothing is known about the dysregulation of DUOX2 in hyperthyroidism, which would represent the only imaginable therapeutic use for a DUOX2 inhibitor at this time. In addition, no selective inhibitors for DUOX1 or DUOX2 have been identified, and none of the developed NOX inhibitors have been tested on their abilities to inhibit DUOX1 or DUOX2. Finally, one publication reported the inhibition of DUOX-derived hydrogen peroxide by VAS2870 in a zebra fish wound-healing model (122). However, whether there is a role for DUOX1 (NOX6) or DUOX2 (NOX7) in higher animals remains to be determined.
Future NOX Inhibitors
Need for future of small-molecule NOX inhibitors
Until now, the role of NOX isoforms in disease has only been validated in animals for NOX1 in diabetic atherosclerosis, liver fibrosis, and ischemic retinopathy; for NOX2 in CGD and probably inflammatory diseases; for NOX4 in stroke, diabetic nephropathy, osteoporosis, and liver fibrosis; and for DUOX2 in hypothyroidism. However, for all other suggested diseases, the final proof for a role of NOX isoforms is lacking. Apart from clear physiological roles of NOX2 and DUOX2, we still do not know exactly what physiological functions the other NOX isoforms fulfil, neither in mice let alone in humans. A role of NOX4 in hypoxia-induced angiogenesis seems likely (152), NOX5-derived superoxide might be responsible for sperm motility (118), and DUOX1 might be involved in pathogen defence in the lung (173) as well as play a role in bladder contractions (45). The currently available NOX inhibitors lack the clear NOX isoform selectivity that would be required to exclusively rely on data obtained with inhibitors. Therefore, for complete validation of a physiological or pathophysiological role of a specific NOX isoform and its validation as a drug target, both need to be studied: NOX KO animals and NOX inhibitors in parallel and in combination.
Although clearly isoform selective inhibitors might not be required for acute treatment of diseases such as stroke, most other indications of NOX inhibition, currently liver fibrosis, osteoporosis, diabetic atherosclerosis, and diabetic nephropathy, would require chronic treatment with NOX-inhibiting drugs. Since the physiological functions of most NOX isoforms are still unknown, unpredictable side effects of non-selective NOX inhibition in long-term treatment cannot be ruled out. In conclusion, for both, validation of the role of NOX isoforms in health and disease and for development into drugs against these diseases, future NOX inhibitors with clear isoform selectivities are required. In addition, oral bioavailability would be desired.
Potential for novel biologicals—therapeutic antibodies
The field of therapeutic antibodies for use in humans is flourishing and provides new exciting therapeutic options. Currently, therapies using these novel biologicals are mainly available for the treatment of cancer, autoimmunity, and inflammatory diseases (94). One of their main strengths is their high potential for target specificity combined with a low toxicity. Therefore, therapeutic antibodies also have a high prospective for treating diseases that involve certain NOX isoforms. An antibody that inhibits NOX isoform activity would have to be directed against extracellular regions of NOX, and it would have to be generated in large amounts in a reproducible and high quality. However, so far, hardly any specific antibodies against the different NOX isoforms exist (3), with NOX2 being the exception. Here, four promising monoclonal antibodies targeting the extracellular loops of NOX2 have been created and shown to inhibit NOX2 activity in living neutrophils and a reconstituted cell-free assay (24). As soon as the NOX antibody dilemma for other NOX isoforms is solved, the road for therapeutic antibodies may be paved. Recently, a first step in this direction was the development of a monoclonal antibody against the extracellular E-loop of NOX4 (189). Although this antibody was not able to inhibit NOX4 activity in a cell-free assay (189), it moderately inhibited NOX4-derived hydrogen peroxide production in transfected cells that expressed NOX4 in the plasma membrane (169). However, none of the monoclonal antibodies against NOX2 or NOX4 are freely available yet, and they would still need to be validated in NOX KO models and to be tested against other NOX isoforms. In conclusion, this progress may present a first step in the direction of therapeutic antibodies against NADPH oxidases.
Conclusions
Much information is already available suggesting pathologically relevant implications of NOX isoforms in several diseases. However, a complete validation of the role of individual NOX isoforms in diseases will require further studies. These should show not only beneficial effects in KO animals of the respective NOX isoform but also in parallel proof of the therapeutic potential by attenuation of the disease using a specific inhibitor in a given disease model. Ideally, the role of other NOX isoforms in the respective model should be excluded by using other NOX KO animals.
Based on specificity for NOX proteins, isoform selectivity, and toxicity of currently available NOX inhibitors, we recommend the GKT compounds and NOX2ds-tat for NOX1/4 and NOX2 inhibition, respectively. For additional validation, ML171 and the VAS compounds are a good choice. However, in future, fully isoform selective NOX inhibitors with favorable toxicity and bioavailability are still required. Biologicals, such as peptides and therapeutic antibodies, have provided the potential for specific, selective, and effective therapeutic options for the treatment of diseases that are linked to the over-activation of NOX enzymes.
Footnotes
Acknowledgments
The authors would like to acknowledge J.J. Rob Hermans and Tessa S. van Kempen for carefully reading this article. H.H.H.W.S. is supported by a Marie Curie International Reintegration Grant, an ERC Advanced Investigator Grant, and the Dutch Kidney Foundation. H.H.H.W.S. chairs and K.W. is a management committee member of the COST Action BM1203.
Author Disclosure Statement
H.H.H.W.S. declares that he holds shares in vasopharm GmbH (Würzburg, Germany), which pharmaceutically develops NADPH oxidase inhibitors. H.H.H.W.S. and K.W. are inventors of a patent (US8236809) on VAS2870 and VAS3947, which is owned by vasopharm GmbH (Würzburg, Germany). No competitive financial interests exist for S.A., K.A.R., and P.W.M.K.