
Abstract
The paracellular cleft within epithelia/endothelia is sealed by tight junction (TJ) proteins. Their extracellular loops (ECLs) are assumed to control paracellular permeability and are targets of pathogenes. We demonstrated that claudin-1 is crucial for paracellular tightening. Its ECL1 is essential for the sealing and contains two cysteines conserved throughout all claudins.
Introduction
T
Cld1 is important for paracellular tightening (64), which is achieved by homo- and heterophilic trans-interactions of their ECLs (39). The ECL1 of Clds may directly be involved in barrier function (58). In rats, Cld1 protein expression and immunoreactivity were specifically decreased via metalloproteinase-9 and low-density lipoprotein-receptor-related protein-1 after perineurial application of hypertonic saline, which is known to open the perineurial barrier for anesthetics (18, 21). This correlates with opening of the paracellular cleft. The first ECL of Cld1 consists of about 50 amino acids (aa) and contains two cysteines (cysteine motif, position 54 and 64, human sequence), which are strongly conserved in all Clds and reported to be essential for the sealing function (58). A similar cysteine motif exists in the ECL2 of all TAMPs. It is thought that these cysteines which are oxidizable form an intramolecular disulfide bridge in both Clds (26) and TAMPs (6). In addition, we hypothesize that the entire Cld1-ECL1 constitutes a functional, redox-sensitive secondary structure and is able to bind to other ECL1s of this and other Clds. Earlier studies indicated that a peptide comprising parts of Cld1-ECL1 (human aa 53–80), when applied to a T84 cell monolayer, reduced transcellular electrical resistance (TER) (36). The structure of the peptide (and of the entire Cld1-ECL1)—as well as the cellular target of the peptide—remained unclear.
A first experimentally based molecular model of claudin-1 (Cld1)-extracellular loop 1 (ECL1) is suggested as a template for the cysteine motif conserved in all Cld-ECL1 and tight junction-associated marvel protein (TAMP)-ECL2. Cld1-ECL1 constitutes a functional β-sheet binding surface, stabilized by an α-helix and a shielded redox-sensitive disulfide bridge. Cld1-ECL1 binds to the ECL1 of claudin-1 and other Clds in a highly affine manner and redox dependently.
A novel peptidomimetic of Cld1-ECL1 (C1C2-4aaC) has been developed, which forms a similar structure and interacts with ECL1 of Cld1 and other Clds in a highly affine manner and redox dependently. It opens Cld1-controlled tissue barriers and improves drug delivery in the rat.
Recent data indicate that specific, selective, and transient modulation of TJ integrity is a promising strategy to improve drug delivery across paracellular barriers, in order to better overcome tissue barriers expressing Cld1 (43a, 64). Accordingly, the aim of this study was a functional and structural characterization of the ECL1 of Cld1, to understand the molecular binding mechanisms under reducing conditions, and to improve the drug delivery effect of Cld1-ECL1 peptidomimetics. Our investigations utilized a recombinant ECL1 construct of Cld1 (human Cld131–81) and a peptide based on the aa sequence of the second part of the ECL1 of Cld1 (C1C2, murine aa 53–81). We investigated their binding behavior to the widely expressed Clds1–5 and to stably Cld1-transfected human embryonic kidney 293 cell line (HEK-293) cells. Truncated C1C2 peptides were generated, and their effect on cells both in vitro (human epithelial colorectal adenocarcinoma, Caco-2) and in vivo (rat perineurium) was studied. Data were acquired on the secondary structure of these peptide variations and on the recombinant Cld131–81 protein, which provided an experimental basis for the generation of a structural and functional model of the ECL1.
Results
Binding studies with peptides from Cld1-ECL1
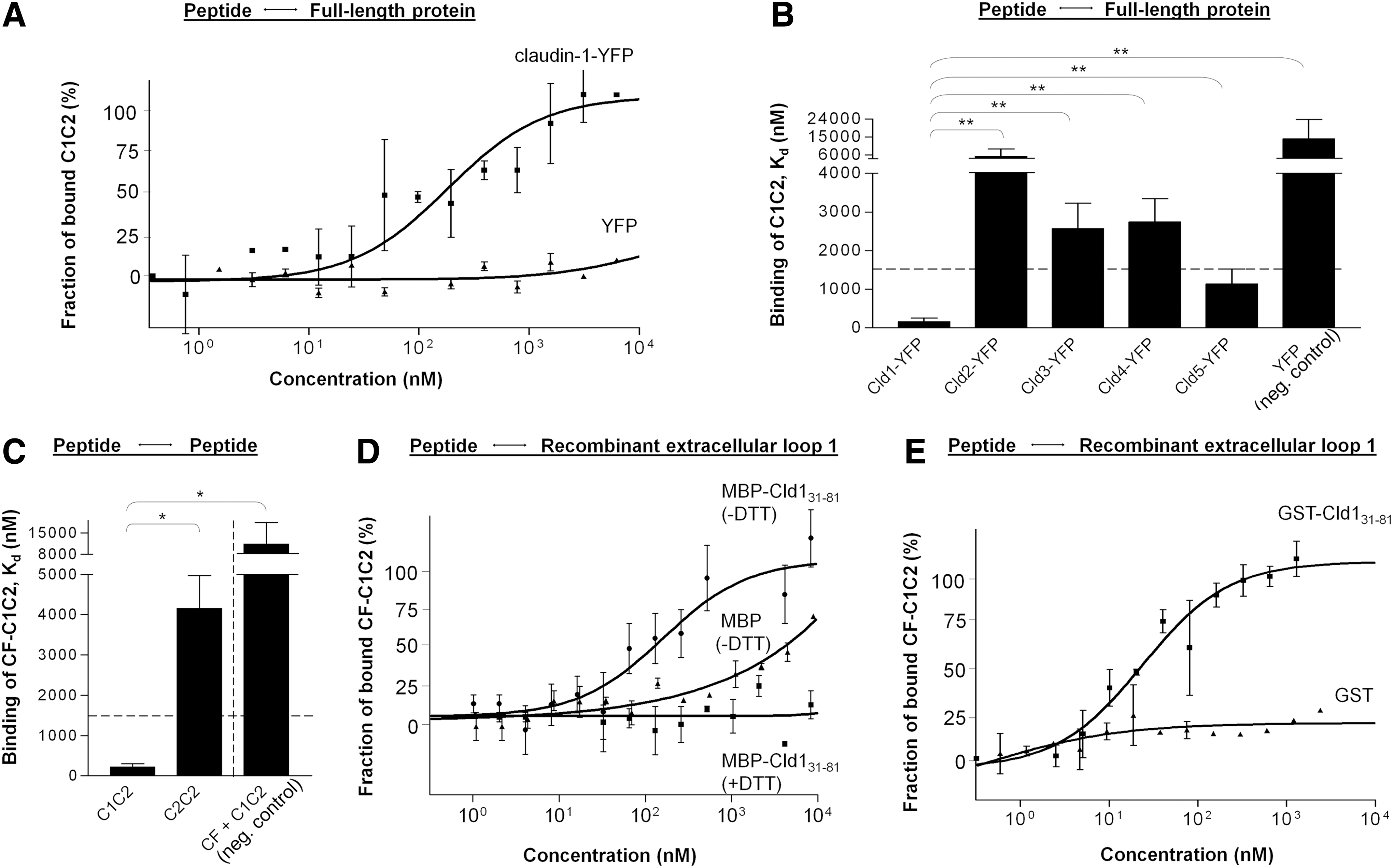
The dissociation constants (Kd) of the peptide C1C2 were determined by microscale thermophoresis (MST). The results show that C1C2 binds selectively to full-length Cld1-yellow fluorescent protein (YFP; Kd=157±98 nM) (Fig. 1A, B) and to Cld5-YFP (Kd=1133±391 nM) but nonselectively to Cld3 (Kd=2574±654 nM) and Cld4 (Kd=2745±603 nM). Untagged YFP was used as a negative control (14,040±9772 nM) (Fig. 1B). Carboxyfluorescein-labeled C1C2 (CF-C1C2) bound to itself, that is, C1C2 without CF (Kd=213±84 nM), but not to a peptide with a high homology (C2C2), taken from Cld2-ECL1 (Kd=4155±816 nM). In addition, C1C2 exhibited nonselective Kd of 11,332±7068 nM to CF alone (Fig. 1C). Binding studies with recombinant maltose binding protein (MBP)-Cld131–81 and glutathione-S-transferase (GST)-Cld131–81 confirmed the results. Here, CF-C1C2 bound selectively (Kd=133±14/39±3 nM) to MBP-/GST-Cld131–81, but not to MBP/GST (Fig. 1D, E). Dithiothreitol (DTT) prevented the binding (Fig. 1D).
Effect of truncation of C1C2 on its structure, claudin-1 binding, and barrier modulation
Truncated peptides were designed to determine which part of the C1C2 sequence and which structural element is crucial for its function. As suggested (64), C1C2 had the potential to form β-sheet and α-helical segments (Fig. 2A). We found that truncation at the C-terminal end by 4, 6, or 8 aa (-4aaC, -6aaC, -8aaC) resulted in a significant decrease in the helical content, whereas the β-sheet content slightly increased. However, N-terminal truncation did not greatly affect the structure. The helix content did not decrease in either of the N-terminally truncated peptides (Fig. 2A).

MST showed that the -4aaC peptide had a similar affinity to full-length Cld1 as untruncated C1C2, at 1 h after incubation (87±54 and 157±98 nM, respectively). C-terminal truncation by 5 (-5aaC: 348±130 nM) or 6 aa (-6aaC: 1116±969 nM) reduced the affinity to Cld1. In contrast, -4aaN exhibited a high Kd=5637±4248 nM, suggesting that the lost aa are important for binding. C2C2 showed no selectivity for Cld1-YFP, Kd=12,497±4819 nM (Fig. 2B).
Functional studies exhibited that C1C2 significantly reduced the TER and Cld1 membrane location of a Caco-2 monolayer at 24 h after application (64), whereas stepwise C-terminal truncation of C1C2 resulted in a successive loss of the TER reducing effect (Fig. 2D). If, however, the cell monolayers were incubated with N-terminally truncated peptides (-4aaN, -8aaN), no effect on TER was observed (Fig. 2C). Again, this emphasizes functional importance of these four N-terminal aa.
C1C2-4aaC but not C1C2-4aaN facilitated opioid-induced regional analgesia and decreased perineurial expression of claudin-1
Two truncated peptides were examined in rats. Increased paw pressure threshold (PPT), as seen with C1C2 (64), was not observed at 48 h after perineural administration of the N-terminally truncated peptide C1C2-4aaN and of the μ-opioid receptor agonist [D-Ala2, N-MePhe4, Gly5-ol]-enkephalin (DAMGO) (Fig. 3A). In contrast, a perisciatic injection of C1C2-4aaC for 48 and 24 h and then DAMGO elicited a significant analgesic effect (Fig. 3B, C) similar to that observed for C1C2 (64). A parallel reduction in the Cld1 level after C1C2-4aaC but not after C1C2-4aaN treatment was detected by a reverse transcriptase–polymerase chain reaction (RT-PCR) and Western blotting in perineurium-containing nerve preparations (Fig. 3D, E). This reduction in mRNA and protein expression was confirmed by immunohistochemistry. Cld1 immunoreactivity was greatly reduced in the perineurium after administration of C1C2-4aaC (Fig. 3F). The same treatment enhanced permeability of the perineurium to Evans blue-albumin (EBA) whose fluorescence became detectable within the nerve (endoneurium), while EBA was confined to the perineurium in untreated samples (Fig. 3G).

Secondary structure model of C1C2
Different modeling procedures were performed with C1C2 and its truncations, in order to develop a basic structural model of the peptide. Then, the model was optimized according to circular dichroism (CD) spectroscopical, binding, and functional results (Figs. 1
–3). The resulting model consists of two β-strands forming a β-sheet at the N-terminal section and an α-helical tail at the C-terminus (Fig. 4 and Supplementary Fig. S2; Supplementary Data are available online at

Redox-sensitive oligomerization and structural properties of Cld1-ECL1
Oligomerization of the recombinant MBP-ECL1 (MBP-Cld131–81) was examined using different separation conditions in sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). We observed that this construct formed dimers under oxidative conditions (no DTT), which disappeared after addition of the reductant. MBP-Cld131–81 was identified in a monomer and dimer band by liquid chromatography–mass spectrometry (LC-MS)/MS in (15% SDS-PAGE, Fig. 5A). Separation in 10% SDS-PAGE achieved better separation of larger oligomers. An MBP-Cld131–81 dimer dominated, but tetrameric and hexameric bands were also detected and their identity was confirmed by LC-MS/MS (Fig. 5A, B, left) and Western blot (Fig. 5B, right). Addition of DTT strongly reduced the oligomer amounts (Fig. 5B). Trimers or pentamers were not found, indicating a preference for even oligomers. Site-directed mutagenesis of Gln61Ala, located on the proposed redox-sensitive β-sheet binding face of Cld1-ECL1, diminished oligomerization of this ECL (Supplementary Fig. S1). This shows disturbed homophilic affinity due to a changed surface of the β-sheet stabilized by the disulfide bridge between the two β-strands, supporting the assumption of a redox-dependent structural and functional binding interface of Cld1-ECL1.

Secondary structure investigations of the tag-free Cld131–81 protein using CD spectroscopy exhibited that about half of the ECL forms α-helical and β-sheet segments. SDS, a β-sheet supporting agent, doubled the β-sheet content; whereas 2,2,2-trifluoroethanol (TFE), which supports helix formation, had no effect on the helicity but slightly increased β-sheet formation (Fig. 5C). This indicates that the ECL1 structure is flexible and has a great β-sheet potential. On the basis of the aa sequence of Cld131–81, a molecular model was developed and optimized according to the oligomerization, redox (Figs. 5A, B and 6A, B), and CD (Fig. 5C) data obtained. The secondary structure prediction fitting best to the results mentioned earlier exhibited α-helix and β-strand portions in the C-terminal half of the ECL1; the N-terminal half appeared nonstructured. The unstructured N-terminus and the helix-carrying C-terminus were directed toward the membrane surface, as they border on transmembrane domains 1 and 2, respectively. Arrangement of the two β-strands to a β-sheet was completed via the two cysteines, each on either strand, which were also oriented toward the cell surface. The cysteines were shielded inside the ECL as aggregates of the construct were not found, enabling a redox-sensitive intramolecular disulfide bridge (Fig. 5D and Supplementary Fig. S2, side views). In addition, the β-sheet was flanked by the C-terminal helix, which further shielded the cysteine residues. The remaining free side of the β-sheet, suggested to be important for the binding and tightening function (Fig. 2), was oriented away from the membrane (Fig. 5D, E). Thus, trans-interactions should become possible with other Cld-ECLs (Figs. 5A, B and 6A–C, G) or ECL peptidomimetics (Fig. 1D, E and Supplementary Fig. S2) to this β-sheet.

Binding of the recombinant Cld1-ECL1 to the full-length protein and to the surface of claudin-expressing cells
It was found that the Kd for the recombinant MBP-Cld131–81 and Cld1-YFP was strongly affected by increasing DTT concentrations and rose from 150±97 to >6000 nM in the presence of 100 mM DTT. The Kd of tag- and DTT-free Cld131–81 was 47.0±0.7 nM (Fig. 6A). Co-immunoprecipitation also demonstrated an association between MBP-Cld131–81 and full-length Cld1-YFP transfected in HEK-293 cells (Fig. 6B, top), as well as binding to endogenous Cld1 derived from Caco-2 cells (Fig. 6B, bottom). Both associations were abolished by DTT (Fig. 6B, lower lines lane 4). Co-localization and a strong interaction was detected for GST-Cld131–81 at the cell membrane of HEK cells transfected with Cld1-YFP (Fig. 6C, top), or to a lesser degree with Cld5-YFP (Fig. 6C, bottom). GST alone showed almost no binding to either Cld (Fig. 6D). The used anti-GST antibody (Fig. 6E), as well as GST-Cld131–81 (Fig. 6F), did not attach to HEK-293 wild-type cells. Quantification of these associations revealed a highly significant attraction of GST-Cld131–81 to either full-length Cld, compared with the respective GST controls (Fig. 6G).
Discussion
Claudins form the backbone of the TJs and are directly involved in their barrier function; Cld1, in particular, is expressed in various tissues, for example, the kidney, liver, and brain (54). It is responsible for the barrier tightness in epithelial cells, which is achieved by homo- and heterophilic trans-interactions of their ECLs (26, 39). The ECL1 of Clds directly contributes to paracellular sealing (58), conserves two cysteines, and is assumed to form a redox-sensitive intramolecular disulfide bridge (26).
Studies with peptides derived from TJ proteins (6, 61) or specific Cld ligands (52, 60) report interactions with the ECLs. The Cld1-ECL1 peptide C1C2 has recently been shown to cause a temporary increase in the paracellular permeability for ions and larger compounds in a cellular barrier model and in an epithelial barrier in vivo (43a, 64). Consequently, transient modulation of Cld-Cld interactions is an appropriate strategy for opening the paracellular cleft. The objective of this study is the exploration of the molecular basis of redox-sensitive interactions between the ECL1 of Clds and the optimization of a Cld peptidomimetics, which binds to Cld1, temporarily opens the TJ barrier, and facilitates drug delivery.
We have found that both a peptide and a recombinant protein of the ECL1 of Cld1 selectively associate with C1C2, the ECL, and the entire Cld1 in cell lysates and on the cell surface. The common binding core, in both peptide and ECL, is a β-sheet stabilized by a helix and, in the native ECL, by a disulfide bond. After internalization (65) of the peptide, the expression of Cld1 is decreased at the cell–cell contact, resulting in the opening of cellular barriers for small and large molecules. These data lead to the model of a functional β-sheet in the ECL1 of Clds.
Our preferred method for analyzing Cld peptide/protein associations, MST, is applied to cell contact proteins for the first time. MST offers the advantage that the protein examined does not need immobilization (59), and interactions can be measured under close-to-native conditions even in cell lysates. In addition, MST requires a very small protein amount compared with other methods, such as nuclear magnetic resonance and surface plasmon resonance spectroscopy. Determination of dissociation constants by MST provides an innovative possibility to quantify binding properties and to evaluate structural predictions. However, for large dissociation constants, higher peptide concentrations are necessary, sometimes resulting in precipitation. High protein concentrations in cell lysates may contribute to nonspecific interactions with other proteins, which can lead to outliers in the sigmoidal binding curve and incorrect dissociation constants. We, therefore, included proper controls and confirmed the data with independent methods and systems. Nevertheless, MST can successfully be applied to Clds, as demonstrated with anti-Cld1 antibodies exhibiting a typical Kd of several nM for such antibody-antigen interactions (42). Thus, Kd values found for peptides and recombinant ECL ∼100 nM are also considered as exhibiting a high affinity. C1C2 derived from Cld1-ECL1 binds strongly to Cld1 but does not to other, less homologous Clds. Since the Kd of the peptide to other Clds is at least one magnitude higher, the homophilic peptide-Cld interaction is highly selective. Kd≥1500 nM is considered nonselective binding, as proved by the negative control C2C2 being functionally inactive (64), both in cell systems and in rats. The association of the ECL to the full-length Cld is also highly selective, as Cld1-ECL1 bound to Cld1 with similar Kd. The ECL binding properties have been verified by co-immunoprecipitation of Cld1, in both transfected HEK and endogenously expressing Caco-2 cells, as well as by detection of binding at the surface of living cells.
The study shows that C1C2-4aaC has full structural, binding, and functional potential compared with C1C2 and can be used as an optimized version because it is easier to synthesize. The structure of the truncated peptides was, similar to C1C2, very flexible, as shown by addition of SDS or TFE. This supports the assumption that an interaction partner is essential for a functional secondary structure. Nevertheless, the C-terminal section of the peptide that is potentially α-helical has an important function for the overall structure and cannot be removed completely. The helix stabilizes the functionally essential β-sheet at the N-terminal part, which constitutes the binding surface to the ECL1, and, hence, is indirectly responsible for the barrier opening activity (Fig. 4). However, peptide binding does not affect intercellular ECL-ECL association immediately, as peptide and Cld, bound together, are internalized (64) via the clathrin pathway (65). Obviously, this complex is quite stable and inhibits proper transcription and localization of the Cld. This mode of action explains the redistribution of Cld1 away from cell–cell contacts (64), as well as the reduced Cld1 expression in rat perineurium and the paracellular opening effect both in vitro and in vivo at only 24 h after the administration. Due to endocytosis after the internalization, only a part of the peptide amount is able to inhibit the transcription, which would explain the micromolar concentration/dosage needed and the late onset of effect.
The perineurium acts as a barrier between the normal tissue and nerve bundles, and prevents paracellular permeation, for example, of hydrophilic agents. There are a few approaches to open that barrier, but only by relatively crude methods such as hypertonic solutions (57) or cholic acid (53), which are nonspecific and used at high concentrations. As far as known, Cld1 is the major sealing Cld expressed in the perineurium surrounding peripheral nerves (41). This obviously plays an important role in paracellular tightening, as decreased expression is reported in rats under pathological conditions (21), which correlates with opening of the perineurial barrier. Selective and transient opening of the perineurium in rats is observed after administration of C1C2 (64). Our in vivo studies indicate that the C-terminally truncated C1C2 peptide (-4aaC) decreased the Cld1 level and increased the permeability of the perineurium for medium-sized molecules such as albumin. As a consequence, the -4aaC peptide (but not -4aaN) facilitates regional opioid-mediated analgesia. This supports the functional importance of these N-terminal residues. Perineural application of hydrophilic opioids has been shown to cause analgesia without impairing the motor function of the central nervous system or causing side effects such as sedation, nausea, and respiratory depression. C1C2-4aaC, therefore, provides an interesting vehicle for drug delivery in nociception-specific analgesia.
Previous secondary structure considerations (26) and initial experimental data (64) have revealed that the ECL1 of Clds consists of a mixture of β-sheet and α-helical segments. The active C-terminal peptide C1C2 also shows this structure, whereas the inactive C2C2 lacks β-elements (64). Thus, our findings confirm such a model for Cld1-ECL1. However, the CD-data indicate that only 50%–60% of the isolated loop is structured. The modeled structures adapted to the experimental results suggest an unstructured N-terminal half of the ECL and a structured and functionally active C-terminal half (Fig. 5D, E). Interestingly, the peptidomimetics C1C2 taken from the latter region of the Cld1-ECL1 exhibits structural and functional similarities: Both peptide and ECL have a β-sheet binding surface flanked and stabilized by an α-helix, and both associate homo- and heterophilically with each other and bind to full-length Cld1. Peptides from the N-terminal half of the ECL lack functional efficiency and proper structural folding (36, 64).
Studies on other Clds support the structural and functional importance of the C-terminal half of the ECL1, in which we describe a β-sheet from about Met52 to Val66 in Cld1. Thus, the cation pore between first ECLs of two Cld2 molecules is caused by Asp65, just at the C-terminal surface of the proposed β-sheet (2). The homologous Lys65 in Cld1-ECL1 is also outwardly directed in our model, suggesting that this structure is a part of the binding interface (Supplementary Fig. S2). Ile66 in Cld2 is a part of the pore surface (2) and, in the Cld1 model, the respective Val66 is related to the β-sheet surface. The charged aa Lys65 in Cld4 (9), Arg65 in Cld10a (27), Lys65 in Cld10b (55), and Lys65 in Cld17 (28) are comparable with the Asp65 in Cld2 and also crucial for ion selectivity. The similarities let us assume that all these Clds share an analogous basic structure.
The Cld1-ECL1 contains two cysteines at position 54 and 64 hypothesized to form an intramolecular disulfide bridge (26) that stabilizes the β-sheet and determines the binding behavior of the entire ECL1. Generally, cysteines are able to form disulfide bonds within ECLs (37) if localized in a suitable position, as normal extracellular space has an oxidizing environment. In SDS-PAGE, the Cld1-ECL1 displays dimers, tetramers, and hexamers similar to those found for different Clds (25), which disappear in the presence of reductants. Aggregates are not observed. This favors the idea of a disulfide structure within the ECL1, which is shielded and, hence, cannot be formed intermolecularly. ECLs of other membrane proteins may also constitute intraloop disulfide bridges without establishing intermolecular disulfide bonds (50). Previous studies have shown that addition of DTT results in reduction of intramolecular disulfide bonds within ECLs and prevents their dimerization (20). This effect may explain the disintegration of the ECL1 oligomers as well as the rise in paracellular permeability (Supplementary Fig. S3) at constant cell levels of Cld1 (Supplementary Fig. S4) after DTT addition. Thermophoresis measurements support the idea that a reducing environment prohibits formation of the intraloop disulfide structure of the recombinant ECL1 and, hence, of the proper binding surface as well as oligomer formation. The disulfide structure could be relevant for the Cld turnover. The reducing intracellular state would cause free cysteine residues until cell membrane insertion and after beginning of endocytosis, and would prevent oligomerization. The generation of reducing equivalents by, for example, mitochondrial oxidative phosphorylation is intensified under conditions such as hypoxia. Then, the extracellular space also becomes reduced, resulting in loss of disulfide structures in ECLs.
In Cld2, an intramolecular disulfide bond located near the interface of two molecules involved in this Cld's homoassociation, stabilizing the folding of the ECL1, is also reported (30). Cld19 known to self-associate forms hydrogen bonds via Gln57 and Gln61 (24). In our Cld1-ECL1 and C1C2 models, these residues are outwardly directed on the β-sheet. This agrees with reduced oligomerization after Gln61Ala replacement, supporting functional and structural relevance of this ECL1 area for binding to the peptide or another ECL. The cysteines and the proximate aa sequence forming the binding surface are conserved throughout classic Clds, such as Cld1–9, 14, 17, and 19 (26). We, therefore, conclude that the basic structure and function of the segment around the disulfide bond can be generalized, at least, to this Cld subgroup. Since the two cysteines are conserved in one ECL of all four-transmembrane TJ proteins—including also all other claudins (26), occludin, tricellulin, and marvelD3 (6, 26)—which may interact heterophilically (10), the Cld1-ECL1 might have a model character for such TJ proteins as well.
For the claudin family, a two cysteine-containing signature sequence within the ECL1 has been recently identified (17). This is homologous with other cell contact proteins: (i) peripheral myelin protein 22 (PMP22) expressed in TJ-forming Schwann cells; (ii) the pfam00822 family in Drosophila involved in septate junction organization and epithelial barrier function (3); (iii) PMP22/Cld-domain proteins supporting cell adhesion and polarization in Caenorhabditis elegans (46); or (iv) Cld-like proteins in yeast related to membrane organization (11), membrane fusion (7), and Ca2+ flux (12). The signature motif is highly conserved within Clds, further suggesting that the ECL1 has a well-defined structure and serves an important conserved function of Clds. Clds also interact with the TAMP members occludin, tricellulin, and marvelD3 (10). In addition, ECL1 of Clds and ECL2 of TAMPs share a receptor function. Thus, it is demonstrated that binding of the hepatitis C virus to Cld1-ECL1 (13) and occludin-ECL2 (40) is essential for the infection. Moreover, the functional structure of occludin's ECL2 (4) is very similar to that of Cld1-ECL1. These similarities are of general significance for the structure and function of cell contacts from yeast to man, and for pathological conditions from oxidative stress to infection, including pharmacological aspects.
In conclusion, the C-terminal half of the ECL1 of Cld1 is composed of an N-terminally localized binding surface with a β-sheet character in which two β-strands are kept together by a shielded intraloop disulfide bond, and by a C-terminal α-helix. This conformation is in agreement with a crystal structure of Cld15 (51). The functional ECL structure, its oligomerization and peptidomimetic binding properties, respectively, are redox sensitive due to the possibility of formation of a disulfide bridge. A peptide derived from the second half of Cld1-ECL1 exhibits a very similar structure, namely β-sheet segment stabilized by a C-terminal α-helix, as well as comparable binding properties. The aa sequence responsible for structure and binding was specified, and homo- and heterophilic interactions to other Clds were disclosed. We introduced the novel well-defined Cld1-ECL1 peptidomimetic C1C2-4aaC and demonstrated its efficacy to increase paracellular permeability both in vitro and in vivo. This is a new approach and it has the potential to be applied for different therapeutic areas as a drug enhancer through tissue barriers. Targeting the paracellular barrier may provide a possibility for improving pharmacological intervention under normal conditions in tissues that have been inaccessible for water-soluble drugs.
Materials and Methods
Recombinant proteins
Human Cld131–81, with N-terminal GST- or MBP-tag (Table 1), was expressed in Escherichia coli (5), harvested, and resuspended in binding buffer, for MBP-constructs, 20 mM Tris/HCl, pH 7.4, 200 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA; Merck, Darmstadt, Germany) (5), and for GST-constructs, 20 mM Tris/HCl, pH 7.9, 125 mM NaCl, 5 mM MgCl2, 10% glycerol, 0.1% TX-100, 0.1 mM phenylmethanesulfonyl fluoride (Sigma-Aldrich, Taufkirchen, Germany), and 0.5 mM DTT (Bio-Rad, Munich, Germany). After sonication (300 W) and centrifugation (39,000 g), the supernatant was incubated (1 h, 4°C) with amylose resin (MBP-proteins; NEB, Ipswich, MA) and glutathione sepharose 4B (GST-proteins; GE Healthcare, Buckinghamshire, United Kingdom) to immobilize and purify the constructs. MBP-proteins were eluted with the binding buffer containing 10 mM maltose and GST-constructs in 50 mM Tris/HCl, pH 8.0 with 10 mM reduced glutathione.
Further truncations were synthesized and named analogously.
CF, 6-carboxyfluorescein; Cld1, claudin-1; GST, glutathione-S-transferase; MBP, maltose binding protein; MW, molecular weight.
To separate the ECL from the purification tag, the GST-Cld131–81 construct, bound to glutathione sepharose 4B beads (56), was incubated (20 h, 22°C) with a factor Xa mixture (Serva, Heidelberg, Germany). After centrifugation, the collected supernatant contained the tag-free construct.
Peptide synthesis
Peptides (Table 1) were generated automatically (ACTIVOP11; Activotec.com, Cambridge, United Kingdom) by solid-phase synthesis using Fmoc/tBu chemistry. Synthesis followed in a batch-wise mode at a distinct temperature with coupling times of 10 min at 70°C and Fmoc-removal for 7 min at 70°C on PEG-resins (SRAM, 0.2 mmol/g, Rapp-polymere; Sigma-Aldrich). Cleavage from the resin employed trifluoroacetic acid (TFA)/H2O (9/1) for 3 h at room temperature (RT). Peptides were precipitated with cold diethyl ether and purified by preparative reversed-phase high-performance liquid chromatography (HPLC, C-18 column; Dionex, Sunnyvale, CA) with acetonitrile gradients in aqueous 0.1% TFA. Purified peptides were quantified and characterized by LC-ESI-MS (LC: ACQUITY UPLC, C18 column; MS: LCT Premier; Waters, Eschborn, Germany), showing sufficient purity (220 nm, HPLC) and expected masses. C1C2 represented the second half of the ECL1 of mouse Cld153–81 and C2C2, the homologous sequence of mouse Cld253–81. Peptides were synthesized with serine substitution of the cysteines in position 54 and 64 (64).
Eukaryote cells, paracellular tightness, and cell lysates
HEK-293 cells were cultured (10) in Dulbecco's modified Eagle's medium (DMEM, 1 g/L glucose, 4 mM
For TER measurements (61), Caco-2 cells were seeded (135,000 cells/cm2) on 24-well Transwell filters coated with rat tail collagen (Millicell-CM; Millipore, Eschborn, Germany). Monolayers were treated bilaterally with 300 μM peptide or peptide-free medium (control). Since medium half time of the peptide was ∼20 h in presence of the cells and as the effect was reversible after 72 h (65), preferred test time was 24 or 48 h.
For cell lysates, nearly confluent cells were washed (2×phosphate-buffered saline [PBS]), scraped off, and centrifuged (10 min, 250 g). Pellets were resuspended in lysis buffer: 20 mM Tris/HCl, pH 7.5, 100 mM NaCl, 1 mM MgCl2, 5% glycerol, 5 units benzonase, containing protease inhibitors (Roche Diagnostics, Mannheim, Germany), pressed 5× through a 25-G needle, and centrifuged (10 min, 10,000 g, 4°C).
Co-immunoprecipitation, SDS-PAGE, Western blotting, and RT-PCR
MBP-Cld131–81 bound to amylose resin was washed and incubated (1 h, 4°C) with eukaryote cell lysate containing the potential interaction partner. Beads were pulled down (500 g), washed (3×PBS), resuspended in 50 μl SDS sample buffer (0.125 M Tris/HCl, pH 6.8, 4% SDS, 20% glycerol, 0.2 M DTT, and 0.02% bromophenol blue), and heated (5 min, 95°C). For input controls of bait- and prey protein, one-thousandth part of the MBP-Cld131–81 lysate and one-hundredth part of the cell lysates, respectively, were used. Eluted recombinant proteins were quantified by Pierce 660 nm protein assay (Thermo Fisher Scientific, Waltham, MA) and loaded on a 6% or 15% PAGE. The gel was stained in Coomassie solution [0.04% Coomassie G-250 (Serva), 3.4% perchloric acid, and 3.5% ethanol] overnight. Co-immunoprecipitation samples were separated in 15% PAGE and transferred (30 min, 20 V) onto a nitrocellulose membrane (Hybond ECL; GE Healthcare). Proteins were detected using anti-Cld1 and horseradish peroxidase (HRP)-conjugated antibody (Life Technologies). The HRP detection was performed by lumi-imager F1 (Boehringer Ingelheim, Germany).
Sciatic nerves with perineurium before and at 48 h after a perisciatic injection of a peptide were harvested and homogenized in lysis buffer for TX-100 soluble proteins (25 mM Tris/HCl pH 7.6, 120 mM NaCl, 2 mM EDTA, 25 mM NaF, 1% TX-100, containing protease inhibitors; Roche Diagnostics). Cytosol fractions were obtained by homogenization with minipistil and sonification (3×5/3 s breaks), centrifugation (200 g), and subsequent centrifugation of the supernatant (20,000 g, 60 min). TX-100-insoluble pellet was resuspended in an equal volume of extraction buffer (25 mM HEPES, pH 7.6, 2 mM EDTA, 25 mM NaF, and 1% SDS). Extracted protein was quantified as described earlier. Aliquots were fractionated on 12% SDS-PAGE and blotted onto polyvinylidene fluoride membranes (PerkinElmer, Boston, MA). Proteins were detected by mouse monoclonal anti-Cld1 antibody (Life Technologies), β-actin (Sigma-Aldrich) loading control, and visualized as described (64). For RT-PCR, sciatic nerve tissue was dissected, RNA was extracted using Trizol (Invitrogen, Karlsruhe, Germany), and cDNA synthesis was performed using Maxima First-Strand Synthesis Kit (Thermo Fisher Scientific). Claudin-1 mRNA was semiquantified by Step One PCR-System (Applied Biosystems, Foster City, CA). Actin RNA was used as a reference gene for quantification.
Live cell imaging and peptide application
Living, stably transfected HEK-293-Cld1-YFP and HEK-293-Cld5-YFP cells were cultured on poly-
Microscale thermophoresis
Association between peptides and/or proteins (ECL eluates or cell lysates) was measured with MST, a sample-efficient method based on the Soret effect, capable of analyzing interactions of proteins or small molecules in biological liquids (23). An infrared laser creates a temperature gradient, in which the directed motion of the molecules is very sensitive to changes in their size, charge, and solvation shell. One binding partner carries a suitable fluorophore, and the other one is added in defined amounts as titration series (peptides in PBS, 15 dilution steps). After incubation (1 h, 22°C), samples were loaded into standard treated capillaries, and measured with Monolith™ NT.115 (light-emitting diode power 50%, infrared laser power 80%, laser-on time 35 s). Evaluation was performed with NT Analysis software 1.2.229 (NanoTemper, Munich, Germany). Binding was characterized by a dissociation constant (Kd). Determination was achieved using saturation-binding curves obtained at equilibrium. [L0] is the amount of added ligand at each data point, [B0] total concentration of binding sites, [BL] concentration of formed complexes between the binding sites [B], and the ligand, [L]. The fitting function
Secondary structure prediction
Prediction of secondary structure of the recombinant protein Cld131–81 was performed by I-TASSER (
CD spectroscopy
Peptides (50 μM) and recombinant Cld131–81 (25 μM) were dissolved in phosphate buffer (PB; 10 mM NaH2PO4, 10 mM Na2HPO4 pH 7.4), PB with 60% TFE (Sigma-Aldrich) or PB with 0.5 mM SDS. CD-spectra were measured at RT in 0.1 cm silica cuvettes, using a J-720 spectrometer (Jasco, Tokyo, Japan). Wave length regions, 195–265 nm, were used to accumulate six spectra. The solvent baseline was subtracted from the peptide/protein spectra (64).
Protein identification by MS
Proteins were separated using SDS-PAGE. After Coomassie staining, bands of interests were excised. Digestion of proteins and nano-LC-MS/MS was performed as described (29). Briefly, proteins were analyzed by a reversed-phase capillary LC system (Eksigent NanoLC Ultra; Axel Semrau, Sprockhövel, Germany) connected to an LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific). Spectra were acquired in a data-dependent mode with one MS survey scan (resolution 60,000) in Orbitrap and MS/MS scans of the five most intense precursor ions in the linear trap quadrupole. The processed MS/MS data and MASCOT server (version 2.0; Matrix Science Ltd, London, United Kingdom) were used to search in house against the SwissProt database [Version 2010_10, 2010_10 (521,016 sequences; 183,900,292 residues)].
In vivo pain behavior experiments in rats
Experiments were approved by the animal care committee (Lower Franconia Regional Council, Germany) according to International Association Pain Studies (63). In rats, nociceptive inputs from the hind paw mainly travel through the sciatic nerve. Under brief isoflurane anesthesia, the right sciatic nerve in male Wistar rats, 180–220 g, was located using a 22-G needle connected to a nerve stimulator (Stimuplex Dig RC; Braun, Melsungen, Germany) (18). Three hundred microliters test compound (400 μM) in 0.9% NaCl was injected perineurally (i.e., 605 nmol/kg). Measurements were undertaken at 0 and 48 h after the injection of C1C2-4aaC or C1C2-4aaN, as the effect was expected between 24 and 96 h (64). Mechanical thresholds were determined using the paw pressure algesiometer (modified Randall–Selitto test; Ugo Basile, Comerio, Italy) (48), increasing pressure through a piston onto the dorsal surface of the paw until the rat withdrew its paw (PPT; cutoff at 250 g). Averages were calculated from three measurements each. To evaluate the effect of opioid receptor agonists, PPT was measured at 10 min after an additional injection of 100 μl containing 30 μg (514 g/mol) of DAMGO (Sigma-Aldrich). Controls were solvent injections.
Immunohistochemistry and EBA permeation in peripheral nerves
Immunostaining and EBA permeation in sciatic nerves were performed at 48 h after a perisciatic injection of C1C2 (64). For Cld1 immunostaining, frozen tissue sections were blocked (60 min, PBS containing 2% goat serum, 1% bovine serum albumin [BSA]) and incubated with polyclonal rabbit anti-claudin-1 antibody (recognizing cytosolic C-terminal sequence; Invitrogen), followed by Alexa Fluor 594 goat anti-mouse and Alexa Fluor 488 goat anti-rabbit (Invitrogen) antibody. ProTags Mount-Fluor was employed for mounting the sections (Biocyc, Luckenwalde, Germany), and confocal microscopy was performed (Zeiss LSM 510 META, Zeiss LSM Image Browser Software). For EBA permeation, treated sciatic nerves were immersed ex vivo in 2 ml EBA (5% BSA labeled with 1% EB; Sigma-Aldrich) for 1 h. Samples were fixed with 10% formaldehyde, and serial 10 μm-frozen sections were prepared for confocal microscopy.
Statistical analysis
Results are mean±SD, obtained by Mann–Whitney test (one- or two tailed), which was chosen as normal distribution was not detectable. Differences were considered significant if p<0.05, n≥4. For repeated rat measurements, Student–Neuman–Keuls test (one-way-RM-ANOVA) was applied and differences were considered significant if p<0.05, n=6.
Footnotes
Acknowledgments
This work was supported by the EU project JUSTBRAIN #241861 and Else Kröner-Fresenius Stiftung FKZ2010_A52. The authors wish to thank J. Cording, Berlin for valuable comments and discussions; N. Milicevic, Berlin for experimental support; and M. Schümann, Berlin for expert help with MS.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.