
Abstract
Introduction
H
The results of this study point to the complexity and interdependence of the mechanisms employed by radiation-resistant cells to withstand and repair the damage induced by radiation at the center of which stands the regulation of reactive oxygen species by the antioxidant system. The matched model of radiation resistance for head and neck cancer discussed here provides a valuable opportunity to investigate the molecular mechanisms of response to combined radiation and Erlotinib in a preclinical setting.
Most reported studies investigating the resistance to radiation involve a comparative analysis of cancer cell lines established from patients with distinct genetic backgrounds and complex medical and treatment histories (32). A better understanding of resistance to radiation can be achieved by investigating the molecular and cellular features that characterize a clonal population which is resistant to radiation in matched cell lines. In this study, we generated a radiation-resistant head and neck cancer cell line (rSCC-61) from the radiation-sensitive SCC-61 cell line by fractionated radiation. We characterized the two cell lines in terms of their proteomic composition, survival in response to radiation and Erlotinib treatment, metabolic features, and a number of other parameters to unveil the mechanisms of resistance to radiation and response to Erlotinib in HNSCC. The most unexpected findings of our studies were the increased sensitivity to Erlotinib and the emergence of epithelial phenotype in rSCC-61 cells relative to the parental SCC-61 cells, which were Erlotinib resistant and had mesenchymal properties. This is significant in the context of recent studies showing the presence of both mesenchymal and epithelial cells in tumors of advanced-stage HNSCC (5, 39).
Thus, the findings presented here have profound clinical implications for identifying molecular markers of radiation resistance that are associated with epithelial cell types in HNSCC and may offer selective avenues for drug targeting of epithelial radiation-resistant HNSCC cells in tumors.
Results
Generation of a matched model of radiation resistance for HNSCC and its primary characterization
The radiation-sensitive SCC-61 cells previously derived from an HNSCC tumor located at the base of the tongue (52) were treated in vitro with fractionated radiation (2 Gy) for a cumulative total of 16 Gy. The resulting cell population was plated at a low density on soft agar, and eight single-cell-derived colonies were picked and expanded in culture. The clone R8E, hereafter called rSCC-61, was randomly selected for further investigation.
Cell morphology
The first noted difference between the SCC-61 and the rSCC-61 cells was their morphology. The SCC-61 cells were round and larger in size, while the rSCC-61 were spindle shaped and smaller. To quantify the change in morphology, we calculated the nucleus-to-cytoplasm ratio (NCR) using imaging analysis (Fig. 1A). The results show an approximately twofold higher NCR in rSCC-61 (1.1±0.05) compared with SCC-61 (0.5±0.01) (Fig. 1B). A potential explanation for the increased NCR in rSCC-61 is provided by the quantitative proteomics analysis below, indicating the downregulation of proteins involved in cytoskeletal organization in rSCC-61.
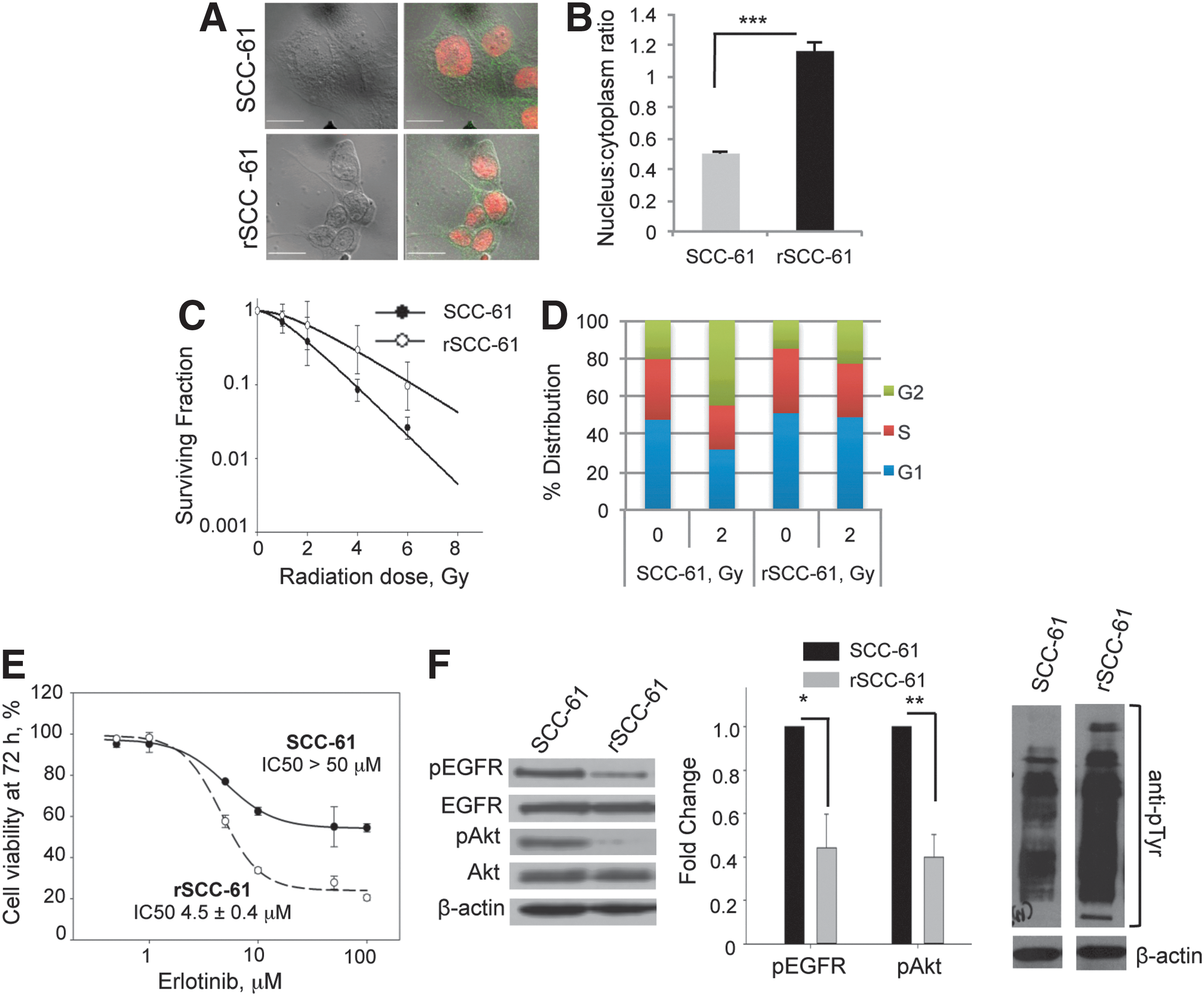
Response to radiation treatment
Clonogenic assays were performed to determine the radiation response parameters for the SCC-61 and rSCC-61 cells (Fig. 1C and Supplementary Table S1; Supplementary Data are available online at
Cell cycle analysis
Cell cycle analysis was performed at 24 h after treatment with ionizing radiation. In the absence of radiation, the SCC-61 and rSCC-61 cells showed a similar distribution of cells in the G1, S, and G2/M phases of the cell cycle (e.g., rSCC-61 51.0%±5.2%, 34.1%±0.1%, and 14.9%±5.3%, respectively) (Fig. 1D). Treatment with 2 Gy radiation induced increased G2/M arrest in SCC-61 cells (45%±15.4%) compared with rSCC-61 (22.9%±8.9%).
Response to Erlotinib treatment
The resistance to Erlotinib treatment of SCC-61 cells has been previously reported (30, 45). To determine whether the acquired resistance to radiation in rSCC-61 has affected the response to Erlotinib, cell viability assays were performed at increasing concentrations of Erlotinib (0.5 to 100 μM). The IC50 for rSCC-61 was 4.5±0.4 μM compared with >50 μM for SCC-61, suggesting a more than 10-fold increase in sensitivity to Erlotinib in rSCC-61 (Fig. 1E). Tumors become resistant to targeted inhibitors of EGFR through diverse mechanisms, which include acquisition of oncogenic mutations (e.g., mutations that inhibit the binding of inhibitors to EGFR but do not decrease its kinase activity), activation of bypass pathways, epithelial-to-mesenchymal transformation (EMT), and others [reviewed in Ref. (13)]. Here, we first explored the status of EGFR and Akt expression and activation, as reports have correlated increased activation of EGFR/Akt with resistance to EGFR inhibitors (11, 19). The phosphorylation status of EGFR and its downstream signaling molecule Akt in SCC-61 and rSCC-61 cells was monitored using Western blot analysis. The results show a decrease in EGFR signaling in rSCC-61 cells by ∼50% despite an overall increase in tyrosine phosphorylation in rSCC-61 (Fig. 1F). The difference in EGFR phosphorylation is consistent with the response to Erlotinib and literature cited earlier. In addition to EGFR phosphorylation, the response to Erlotinib has also been linked to the regulation of integrin expression (27), lipid rafts content (25), and EMT (16). The results described next for the SCC-61/rSCC-61 system are consistent with all these independent studies. The finding of acquired sensitivity to Erlotinib further increases the value of the SCC-61/rSCC-61 cell model that now enables the investigation of the mechanisms of response to Erlotinib in the background of a matched radiation-resistant and radiation-sensitive cell model. It also points to the importance of pursuing clinical trials to investigate the treatment of head and neck tumors with Erlotinib, as tumors that are resistant to radiation may respond to Erlotinib treatment.
Quantitative proteomic analysis of SCC-61 and rSCC-61
To identify the underlying molecular mechanisms of the acquired resistance to radiation in rSCC-61, a quantitative proteomics analysis of SCC-61 and rSCC-61 was performed using stable isotope labeling with amino acids in cell culture (SILAC) and mass spectrometry. The experimental design is summarized in Figure 2A. The SCC-61 and rSCC-61 cells were cultured in medium containing light (SCC-61) and heavy (rSCC-61) isotopes of lysine and arginine. The SCC-61 and rSCC-61 lysates were then normalized with regard to their protein concentration, combined in a 1:1 ratio, and analyzed using a Thermo LTQ Orbitrap mass spectrometer. The data were processed using the Proteome Discoverer 1.2 (Thermo Fisher Scientific) and searched against the UniProtKB human database. The results were filtered using a false discovery rate of 1%, which yielded quantitative data for 965 proteins. Data in Figure 2B show the distribution of protein ratios in rSCC-61 versus SCC-61 (heavy/light) for 920 proteins whose ratios range from 0.017 to 50.5. The remaining 45 proteins listed in Supplementary Table S2 had a ratio of less than 0.01, with the lower limit set in our quantitation algorithm. These are proteins that are more than 100-fold downregulated in rSCC-61 and were considered separately in an effort to avoid bias in the downstream computational analysis. Ingenuity Pathway Analysis (IPA) was used to analyze the 920 and 45 proteins datasets (i) to determine the distribution of subcellular locations and protein functions, and (ii) to identify biological networks and molecular functions that are enriched in proteins significantly which are up- or downregulated in rSCC-61.
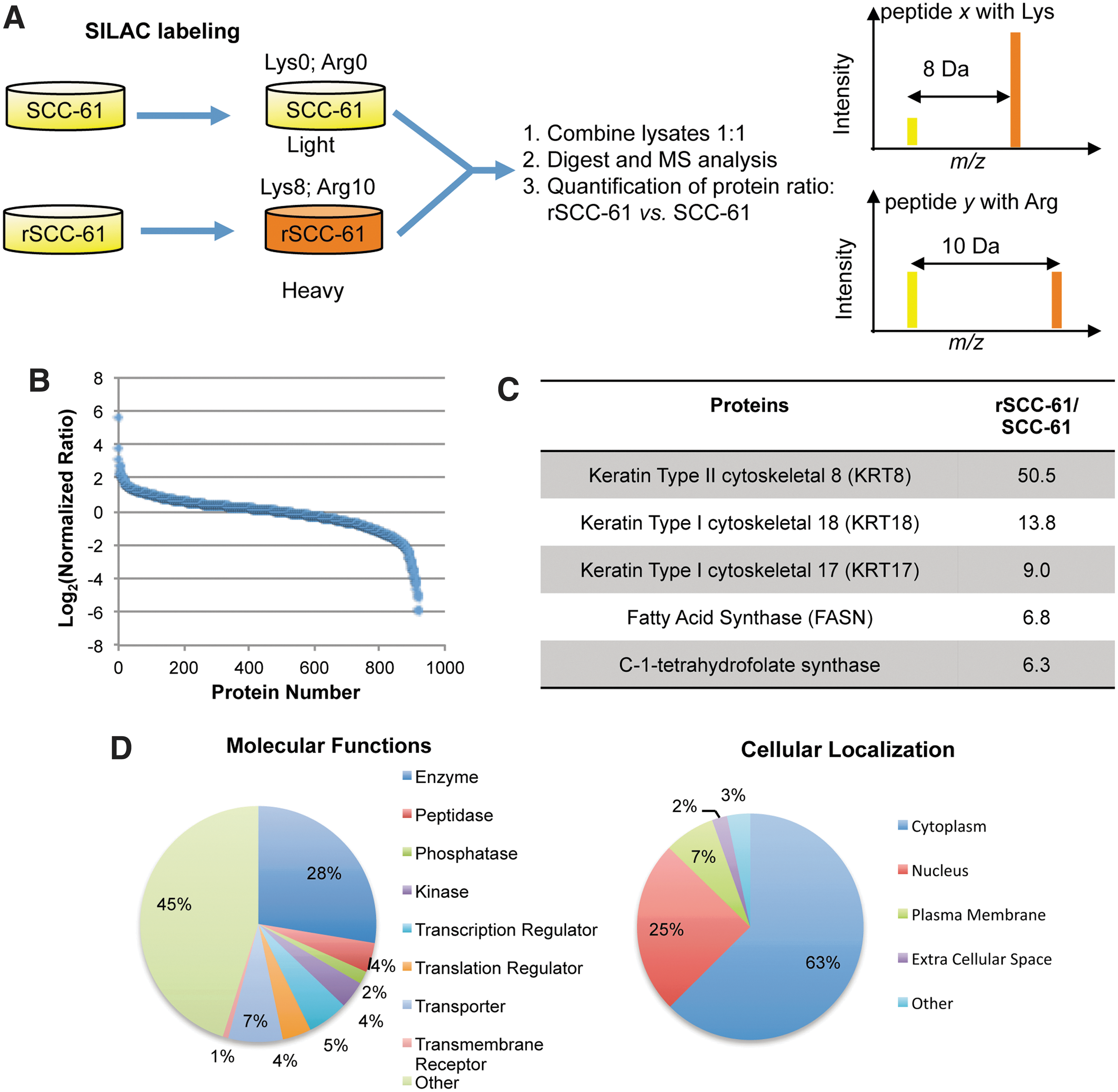
Overall, the most upregulated proteins in rSCC-61 were keratins (>8-fold upregulation) followed by a 6.8-fold increase in the fatty acid synthase (FASN) (Fig. 2C) and a number of other proteins such as the deoxyuridine 5′-triphosphate nucleotidohydrolase (DUT), peroxiredoxins (PRX), and GSK3β with a potential function in mediating the resistance to radiation in rSCC-61 (an extended list is included in Supplementary Table S3) [e.g., GSK3β (33); FASN (29); PRX (41)].
The distribution of subcellular locations and functions of the 920 proteins is shown in Figure 2D. The IPA core analysis of this dataset identified that among proteins which were down-regulated in rSCC-61 cells, there was significant enrichment of proteins involved in cell death (p<0.001; z score: −1.174) and apoptosis (p<0.001; z score: −2.016) (Supplementary Table S4). Similarly, the IPA analysis of the 45 protein dataset indicated a decrease in cell death (p<0.001), a decrease in cell migration (p<0.001; z score: −2.354), and downregulation of EGF signaling in rSCC-61 (p<0.001; z score: −2.574). These results are consistent with the increased survival of rSCC-61 in response to radiation and the anti-proliferative effect of Erlotinib in rSCC-61. Interestingly, 36% (16 proteins) of the 45 downregulated proteins are localized in the extracellular space (Supplementary Fig. S1). Among these, integrin alpha 6, thrombospondin 1, and vitronectin are required for the regulation of focal adhesion and extracellular matrix (ECM)-receptor interactions.
Mapping of proteomics data to KEGG and HPRD human interaction networks and functional analysis
Cytoscape version 2.8.2 (47) was used to mine and visualize interactions present in the 920-protein dataset as described in the “Materials and Methods” section. Two interaction databases—the Kyoto Encyclopedia of Genes and Genomes (KEGG) (28) and the Human Protein Reference Database (HPRD) (43)—were used in the analysis. The KEGG database contains known canonical pathway interactions, while HPRD comprises known protein–protein interactions derived from experimental data, thus providing overlapping as well as distinct information. In order to identify which parts of these networks contained significantly altered proteins in rSCC-61 cells, all proteins with an absolute fold change of 2.0 or greater were extracted, along with first neighbors and all associated interactions, to create the KEGG interaction subnetwork containing 130 nodes and 264 interactions (Supplementary Fig. S2) and the HPRD interaction subnetwork containing 184 nodes and 141 interactions (Fig. 3). Annotation analysis was performed on the lists of UniProt accessions representing the proteins present in the KEGG (130 proteins) and HPRD (184 proteins) networks using the tools provided by the Database for Annotation, Visualization, and Integrated Discovery (DAVID) (23, 24). A number of significantly regulated canonical pathways (p<0.05) in rSCC-61 cells were identified by these analyses: DNA replication and base excision repair, ECM-receptor interaction, cell cycle, focal adhesion, and regulation of actin cytoskeleton (Fig. 3 and Supplementary Table S5A–E). Complementary data to support these results and the significance of the findings to the radiation-resistant phenotype of rSCC-61 are presented in the next few sections.
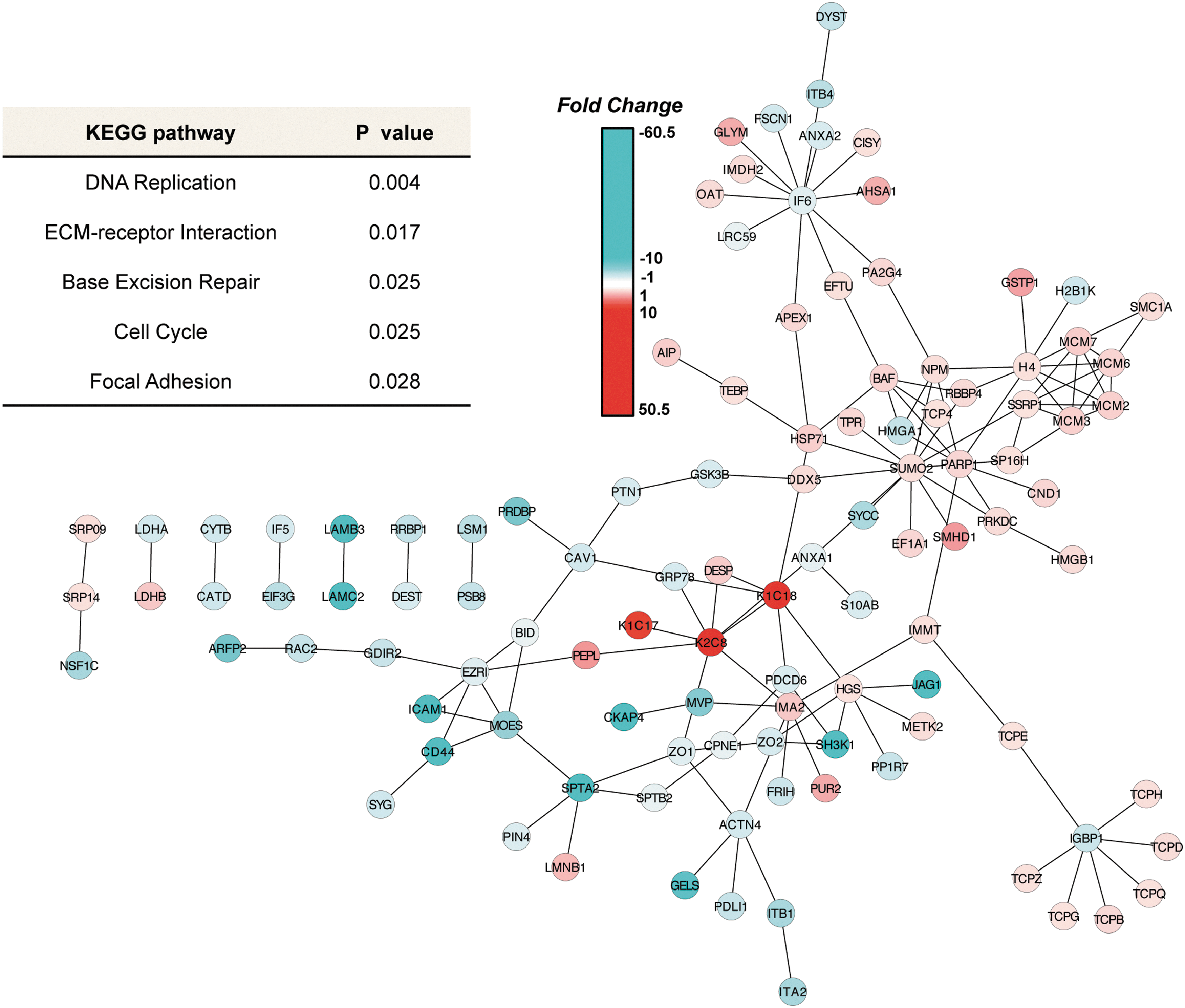
Mesenchymal-to-epithelial transition in rSCC-61
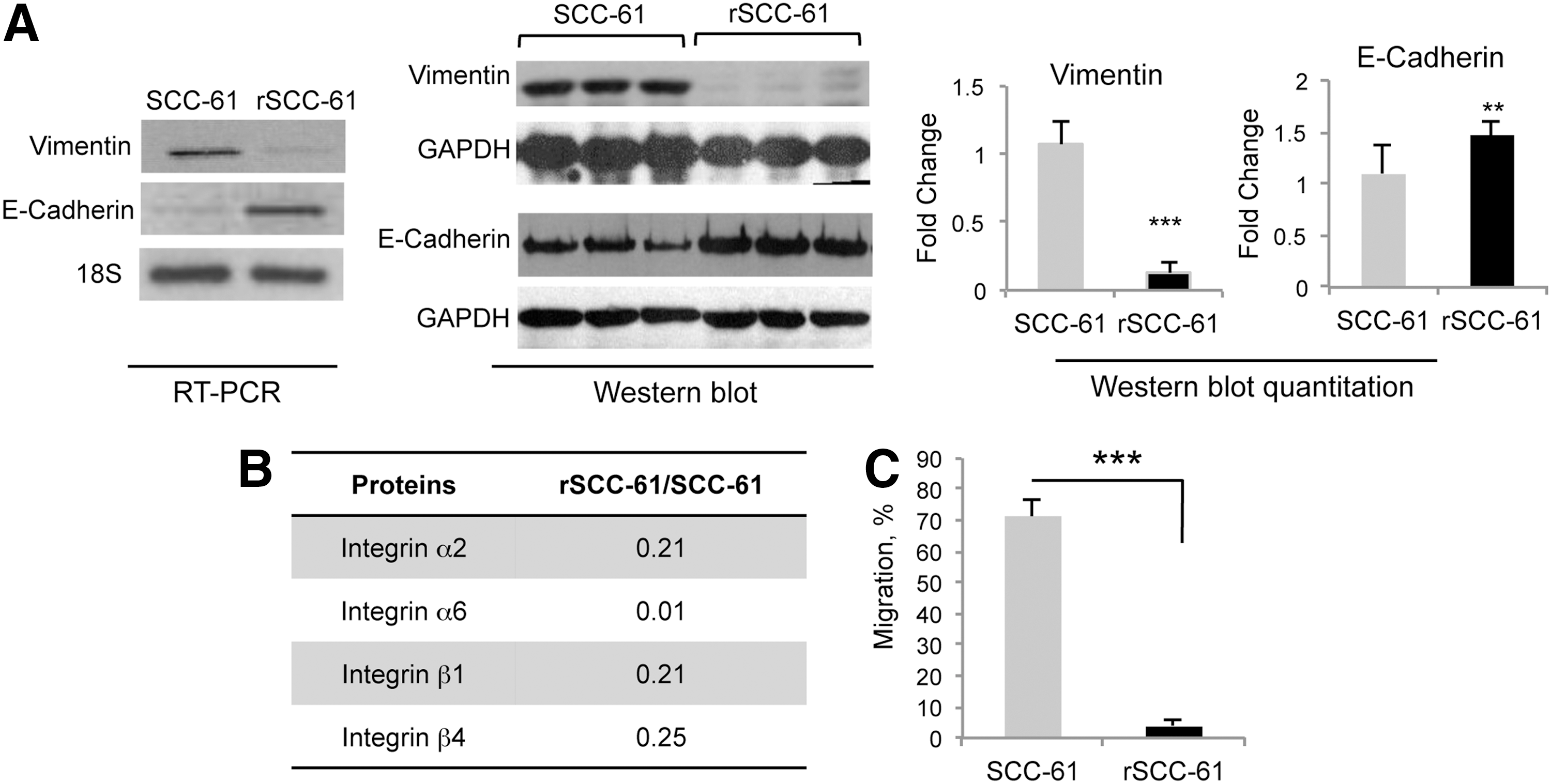
Functional annotation of the KEGG and HPRD networks identified proteins involved in the regulation of actin cytoskeleton, focal adhesion, and ECM-receptor interaction (p<0.05) in rSCC-61 to be significantly downregulated. Combined with the noted morphological differences between SCC-61 and rSCC-61 cells, and the observation that proteins such as keratins (KRT8, KRT18) and periplakin are upregulated in rSCC-61, these events point to a mesenchymal-to-epithelial transition in rSCC-61. To further confirm the epithelial phenotype in rSCC-61, we performed semi-quantitative PCR and Western blot analysis to evaluate the expression of traditional markers of mesenchymal-to-epithelial transition: E-cadherin and vimentin. In comparison to SCC-61, the rSCC-61 cells displayed a higher expression of E-cadherin and a lower expression of vimentin, which was further confirmed by Western blot analysis (Fig. 4A). Another hallmark that differentiates between mesenchymal and epithelial cells is their migration properties. From the proteomics analysis, it was evident that rSCC-61 exhibits reduced ECM-receptor interaction and focal adhesion, which are known mediators of migration (relative change of integrins and other proteins mapping to the ECM-receptor interaction and focal adhesion networks is shown in Fig. 4B and Supplementary Tables S5D and S5E) (48, 49). A trans-well migration assay was performed and confirmed the significantly lower migration of rSCC-61 relative to the SCC-61 cells (p<0.001; Fig. 4C). Cumulatively, these results point to a transition from mesenchymal phenotype in SCC-61 toward an epithelial phenotype in rSCC-61.
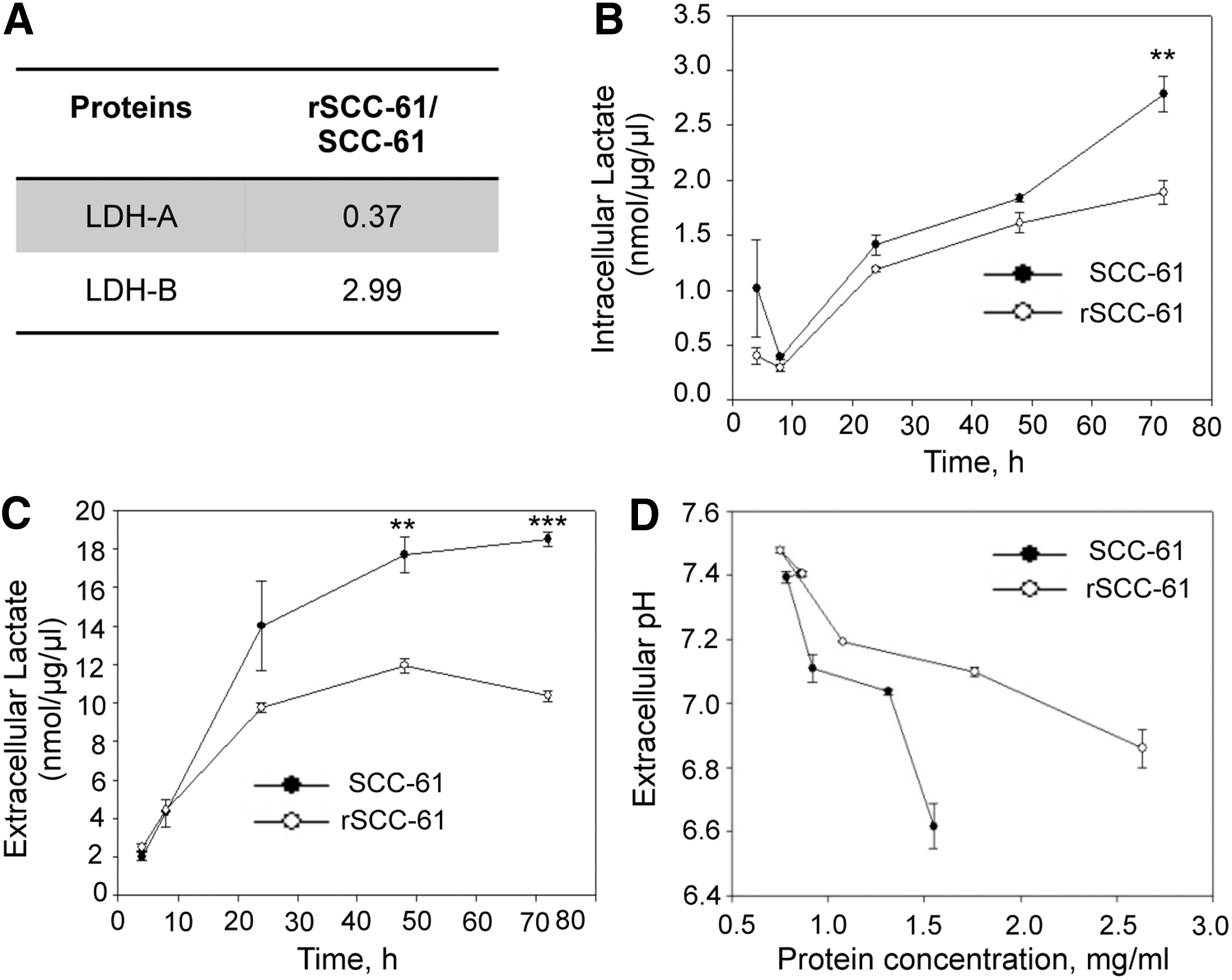
Measurement of intracellular and extracellular lactic acid
Lactic acid has been reported as one of the metabolic factors affecting migration (21). The proteomics data showed down-regulation of lactate dehydrogenase A (LDH-A, threefold) and upregulation of lactate dehydrogenase-B (LDH-B, threefold) in rSCC-61, suggesting a shift from glucose routing into lactic acid synthesis in SCC-61 to pyruvate in rSCC-61 (Fig. 5A). To determine the intracellular and extracellular lactic acid in rSCC-61 and SCC-61 cells during cell growth, the lactic acid assay was performed as described in the “Supplementary Materials and Methods” section. Significantly higher levels of both intracellular and extracellular lactate were observed in SCC-61 cells at 48 and 72 h of cell growth (Fig. 5B, C). Extracellular pH of the two cell lines was also monitored at 4, 8, 24, 48, and 72 h after cell seeding, and the results show increased acidification of the extracellular environment in SCC-61 culture compared with rSCC-61 (Fig. 5D). Both the efflux of lactate and H+-ions from inside the cell to the extracellular medium are likely contributing to the pH change (42). The mechanism of this dynamic process is not completely understood, and we will be investigating other potential sources that could lead to the observed differences in media acidification between the SCC-61 and rSCC-61 cells.
Reactive oxygen species, the antioxidant system, and DNA damage in SCC-61 and rSCC-61 cells
Under normal conditions, physiological levels of reactive oxygen species (ROS) are under the tight control of the cellular antioxidant system, which is considered responsible for maintaining ROS below the threshold at which DNA damage can occur while enabling temporal and localized accumulation of these species when needed during cell growth. Cancer cells, however, are known to accumulate intracellular ROS, and numerous studies have linked ROS in cancer cells to processes such as DNA damage, response to therapies, cellular plasticity, and others (18). The proteomics analysis described here revealed the upregulation of a number of antioxidant proteins in rSCC-61 (Supplementary Table S6). The upregulation of proteins such as glutathione S-transferase pi (GSTpi) and PRX was also previously reported in a comparison study of the radiation-resistant AMC-HN9 and the radiation-sensitive AMC-HN3 head and neck cancer cell lines (32). Thus, one of our first goals was to assess the intracellular ROS in the SCC-61 and rSCC-61 cells and to validate the upregulation of antioxidant proteins. The dichlorofluorescein (DCF) staining and imaging analysis showed lower ROS in rSCC-61 compared with SCC-61 (Fig. 6A). Western blot analysis showed increased levels of antioxidant proteins such as Prx1, Prx 2, and GSTpi, thus further corroborating the proteomic findings (Fig. 6B). In addition, the higher ratio of superoxide dismutase (SOD) to catalase in SCC-61 offered reasoning for the higher ROS accumulation in SCC-61. We have further monitored the hyperoxidation of Prx proteins (Prx-SO2/3, markers of oxidative stress) and PTEN expression and oxidation. PTEN is a known negative regulator of Akt and to be consistent with the pAkt results in Figure 1F, we posed that PTEN oxidation would be lower in rSCC-61 as a result of lower ROS and/or there would be increased expression of PTEN in rSCC-61. The analysis in Figure 6B shows a higher expression of PTEN in rSCC-61, thus confirming a part of our hypothesis. Due to the difference in protein expression, we could not reliably conclude whether there is a shift in protein migration (oxidized PTEN migrates slightly lower than the reduced protein). However, given the lower ROS in rSCC-61, it is unreasonable to hypothesize higher PTEN oxidation in this cell line. Taking this together with increased expression of PTEN in rSCC-61, the results clearly point to combined EGFR and PTEN-mediated downregulation of Akt activity. The contribution of ROS to EGFR/Akt signaling will be further investigated in future using complementary approaches.
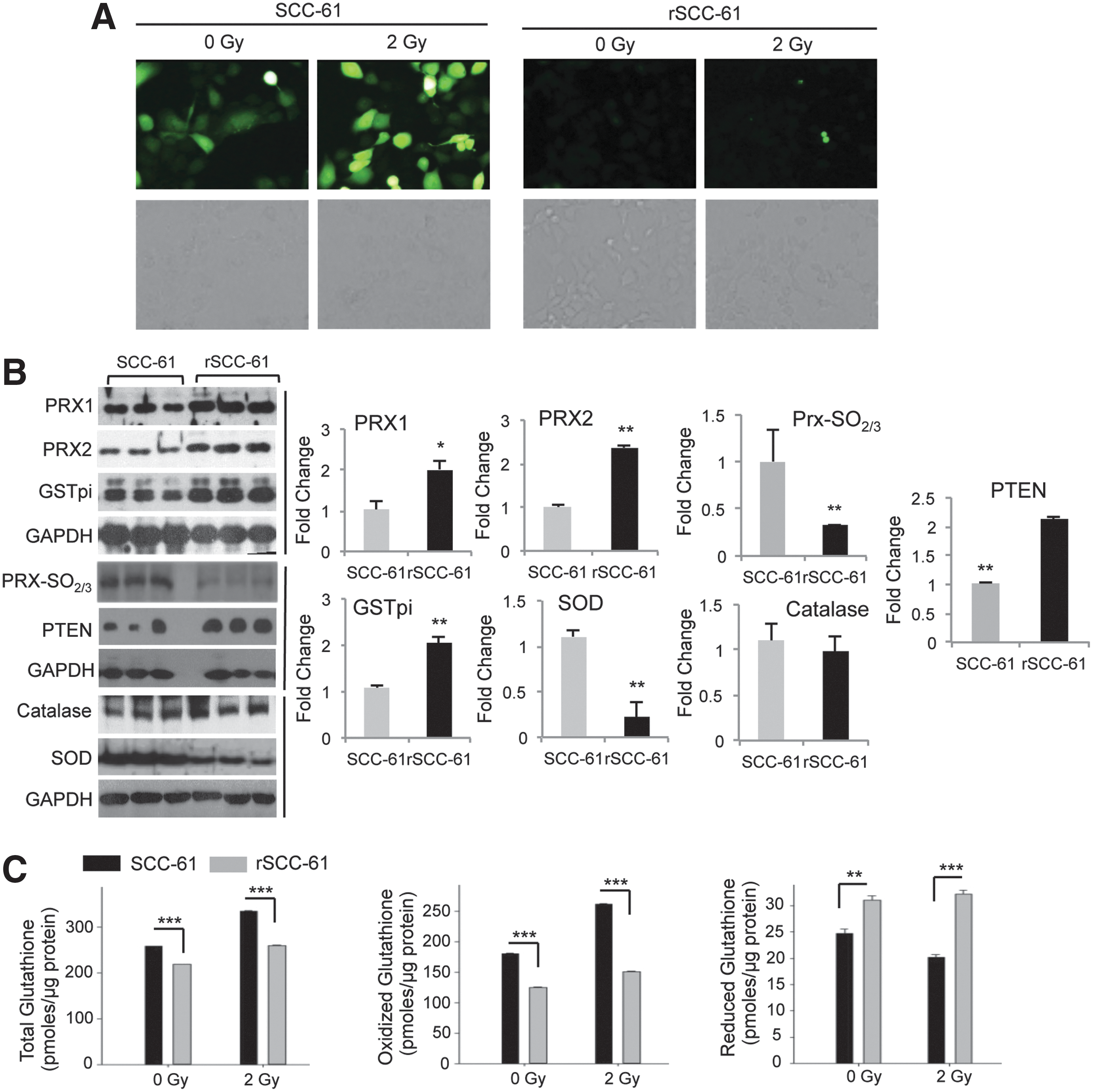
Glutathione is another component of the antioxidant system that is implicated in the response to radiation (10, 37). We quantified total, oxidized, and reduced glutathione using the glutathione assay kit as described in the “Supplementary Materials and Methods” section. While the total glutathione was significantly less in rSCC-61 than in SCC-61, the endogenous reduced glutathione was significantly higher in rSCC-61 (Fig. 6C). Nevertheless, the majority of glutathione resided in oxidized state in both cell lines, suggesting a lesser redox buffering capacity by this system in response to an ROS challenge.
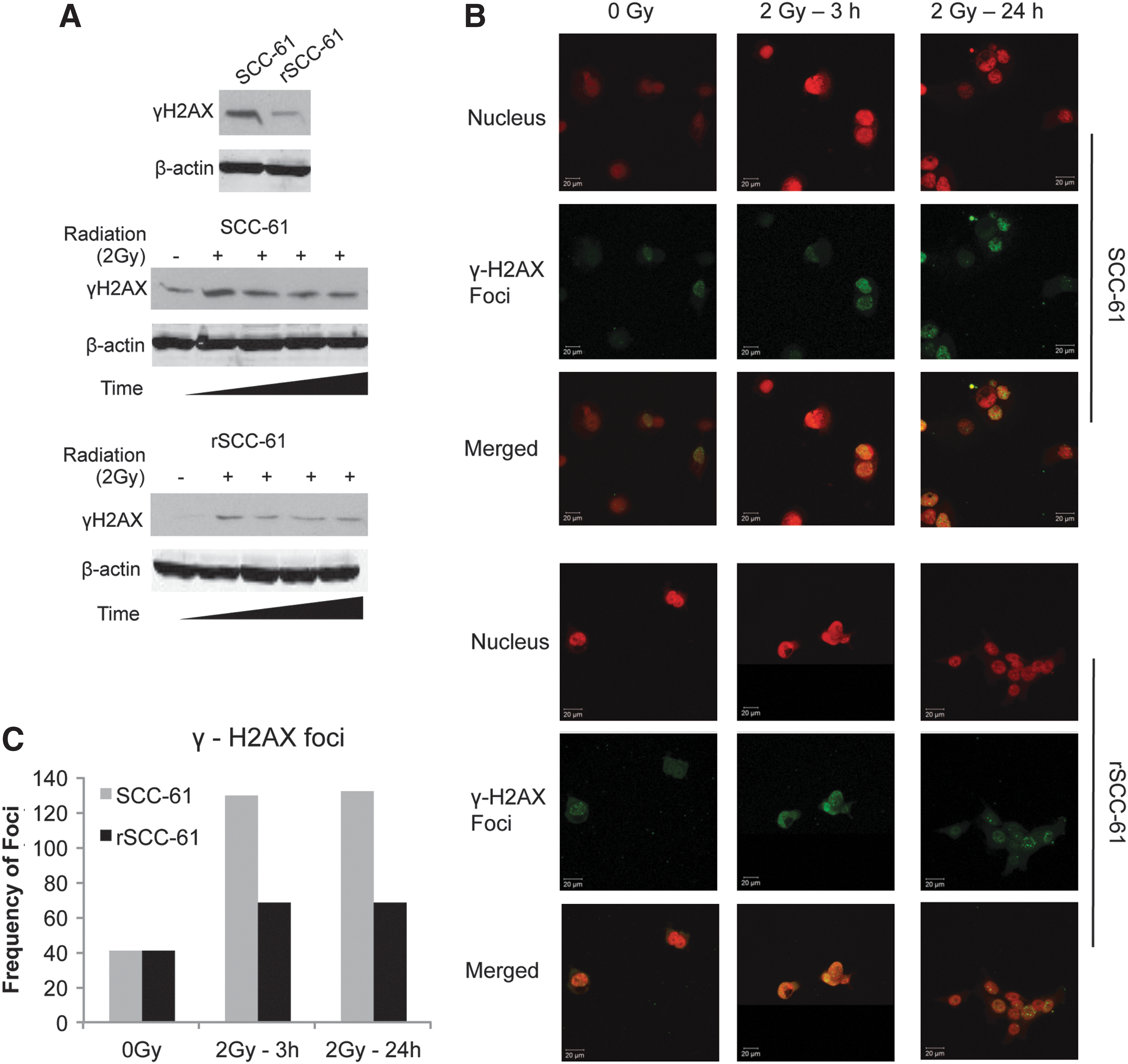
To determine whether the combined effects of increased ROS scavenging capacity in rSCC-61, and the upregulation of proteins involved in DNA replication and base-excision repair result in lesser radiation-induced DNA damage, we monitored the γH2AX protein, a known marker of DNA damage. As shown in Figure 7, there was a lower number of γH2AX foci formation in rSCC-61 compared with SCC-61 in response to radiation treatment, indicating decreased DNA damage in rSCC-61 compared with SCC-61 [Western blot analysis (Fig. 7A); Immunofluorescence analysis of γH2AX foci (Fig. 7B, C)]. This was further supported by the IPA analysis of the proteomic data using the function prediction tool, in which the fold change of a number of proteins listed in Supplementary Table S7 pointed toward reduced DNA damage in rSCC-61.
Relationship between ROS, lipid rafts, and the response to radiation and Erlotinib treatment
Another significant consequence of radiation-induced ROS is triggering the coalescence of lipid microdomains in the plasma membrane to form enlarged lipid rafts. Both resistance to radiation and increased sensitivity to EGFR inhibitors have been independently correlated to decreased lipid rafts and decreased localization of EGFR within these structures. The decreased basal and radiation-induced ROS in rSCC-61 coupled with the sevenfold upregulation of FASN in this cell line (Supplementary Table S3) led us to explore the potential differences in the lipid raft arrangements in SCC-61 and rSCC-61 cells. The cells were stained for lipid rafts and EGFR. Unlike rSCC-61, the SCC-61 cells showed increased lipid raft formation and EGFR co-localization within these structures in both control and radiated cells (Fig. 8A).
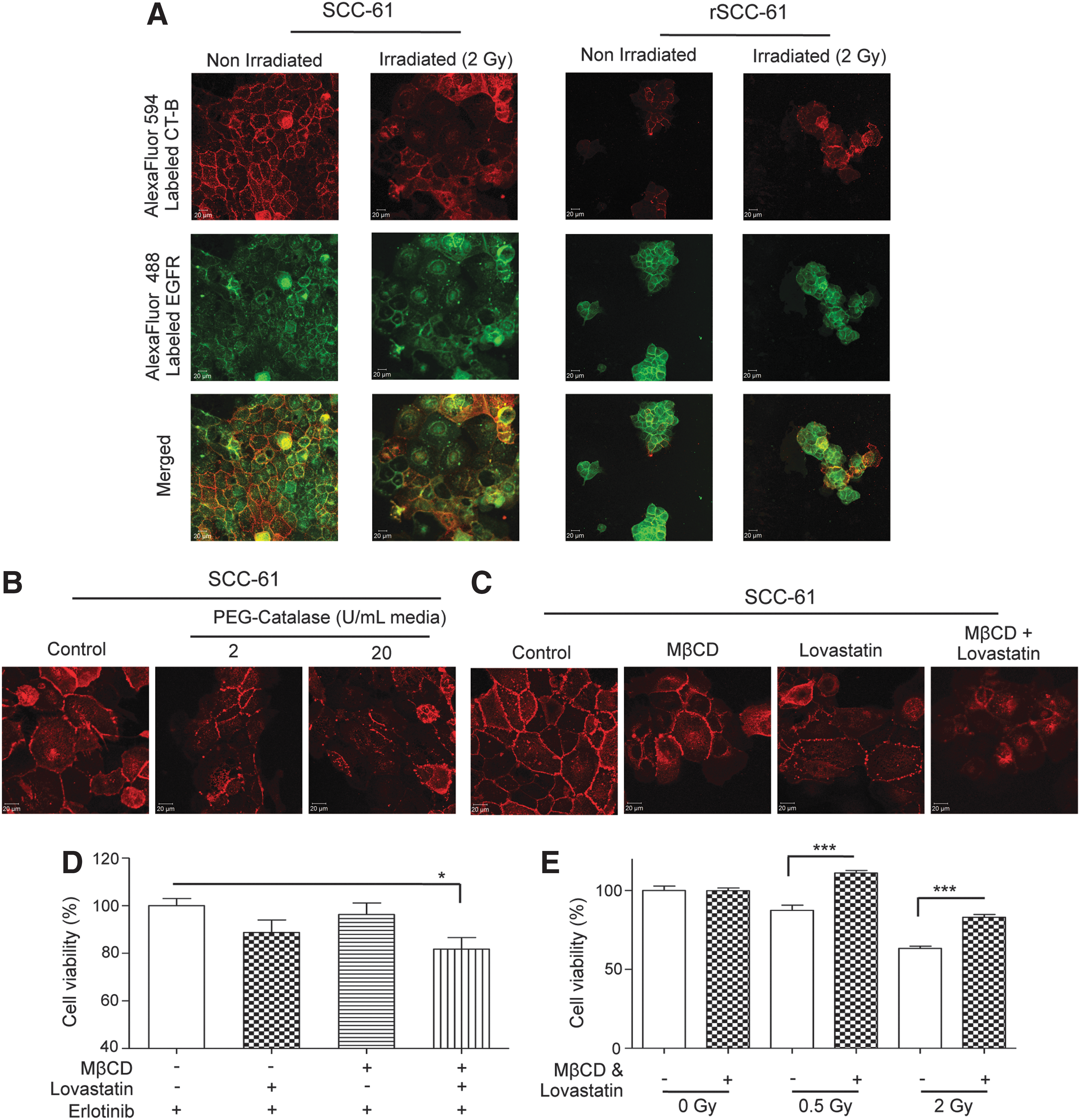
We then asked whether a decrease in the intracellular ROS in SCC-61 would influence the formation of lipid rafts and whether the disruption of lipid rafts would affect the response to Erlotinib or radiation. The SCC-61 cells were treated with polyethylene glycol-catalase, and the lipid rafts were stained with cholera toxin subunit B (Fig. 8B). A gradual disruption of lipid raft structures was observed on increasing catalase activity (2 and 20 U, respectively). The results were comparable with those obtained by the treatment of cells with a cholesterol extracting cyclodextrin MβCD and/or Lovastatin, an inhibitor of cholesterol biosynthesis (Fig. 8C) (14). Further experiments were performed to investigate the impact of lipid rafts in SCC-61 on the response to Erlotinib and radiation. As shown in Figure 8D and E, the disruption of lipid rafts increased the sensitivity of SCC-61 to Erlotinib and rendered the cells more resistant to radiation, a phenotype characteristic of rSCC-61.
Evaluation of redox balance in HNSCC clinical samples
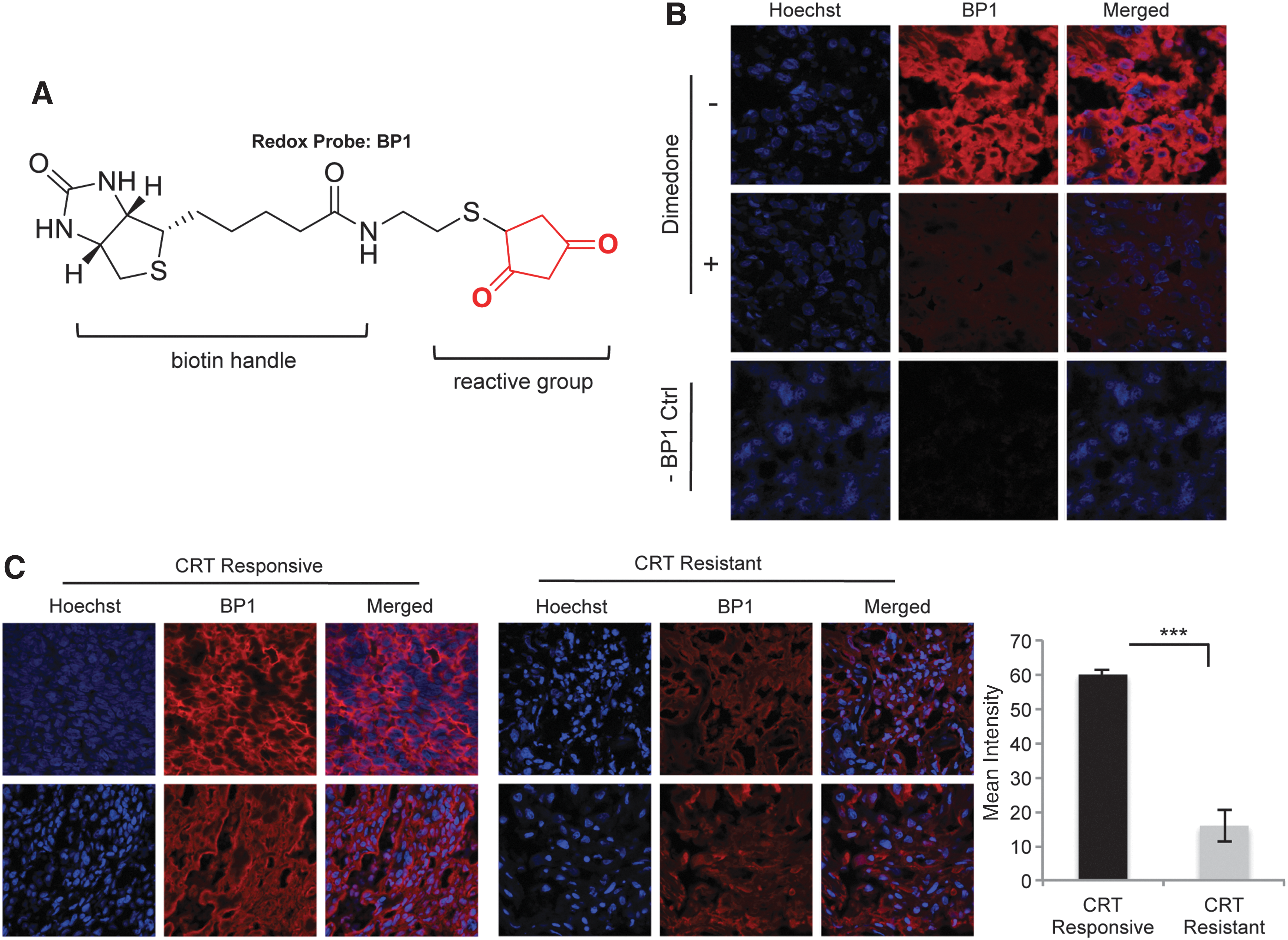
Given the in vitro data showing a mechanistic connection between the control of ROS and resistance to radiation, we next investigated whether this has relevance in vivo. We have used the redox biotin-tagged probe BP1 [Fig. 9A, (44)] to investigate the protein oxidation as a marker of ROS in a set of HNSCC tumor samples. The criteria for tumor selection and grouping into the treatment “responsive” and “resistant” groups are presented in the “Materials and Methods” section. The control experiments showing the selectivity of BP1 for oxidized proteins and the lack of signal in the absence of BP1 are presented in Figure 9B. The quantification of BP1 staining in the radiation- and chemoradiation-resistant and -responsive HNSCC groups shows statistically significant differences in protein oxidation, providing initial validation of the in vitro findings (Fig. 9C).
Discussion
Surgery, radiation, and chemotherapy are the major modes of treatment for HNSCC, and resistance to radiation or chemotherapy poses significant problems in disease management. While the newer EGFR therapies show significant promise, only ∼10% of HNSCC tumors respond to Cetuximab; for example, despite the fact that more than 80% of these tumors have increased EGFR (9, 20). It is, therefore, highly important to identify the critical molecular features involved in response to radiation, chemotherapy, and targeted therapies. These would facilitate the discovery and validation of clinical biomarkers to predict the response to a particular treatment in HNSCC patients; to date, such biomarkers are not available in clinics.
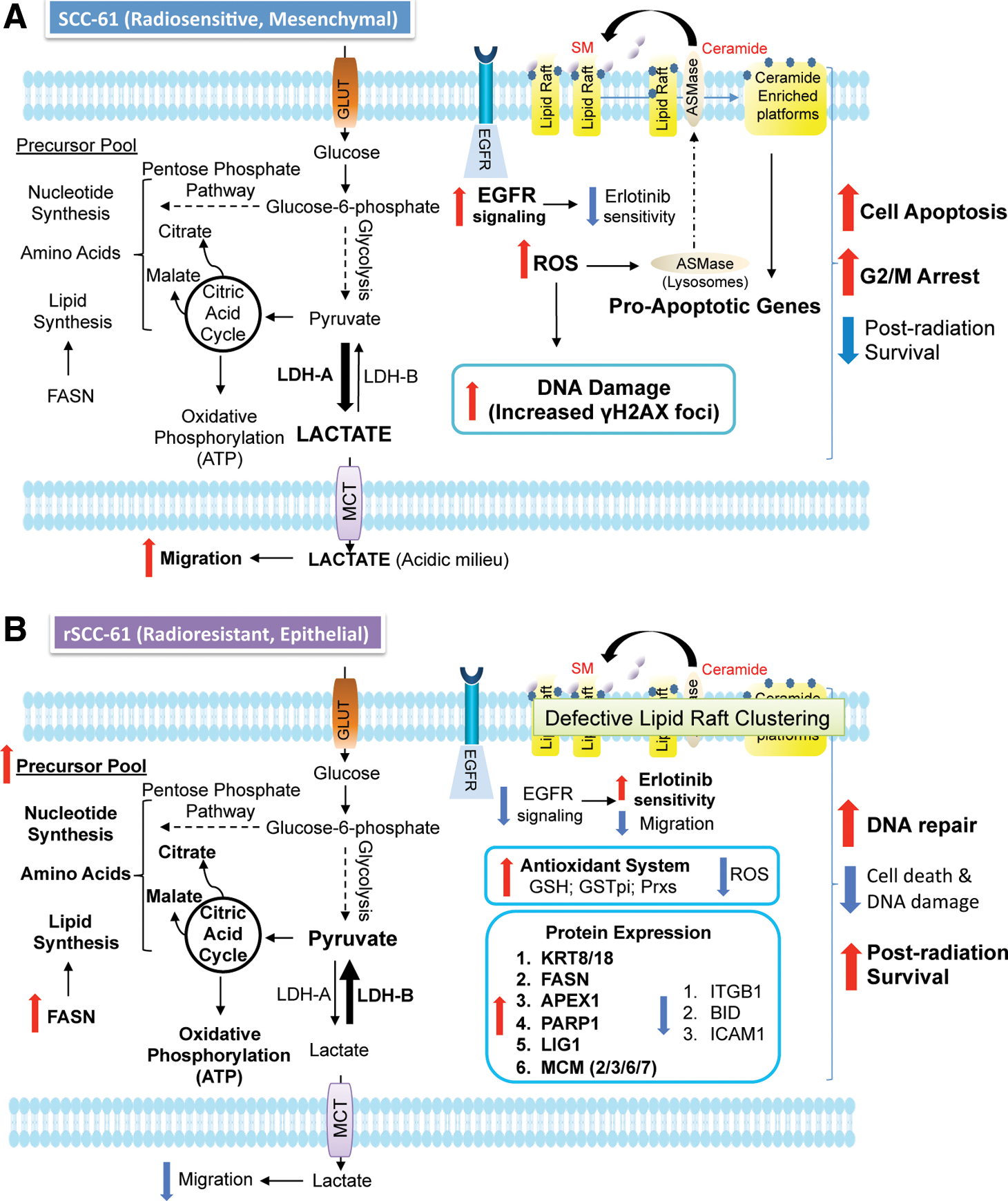
Our studies show a complex mechanistic connection between cellular phenotype (epithelial/mesenchymal), extracellular environment (pH, ROS), and the response to radiation or Erlotinib in HNSCC cells. A summary of the results is presented in Figure 10A and B, and the mechanistic connections are further discussed next.
Reversible epithelial-to-mesenchymal transition and response to radiation and Erlotinib
The epithelial-to-mesenchymal transition (EMT) has gained attention because of its role in the acquisition of cancer stem cell properties. In this process, the cells undergo cytoskeletal remodeling, loss of E-cadherin, and switch from keratin to vimentin-type intermediate filaments. The HNSCC tumors have been traditionally classified as mesenchymal characterized by loss of keratin and a substantial increase in vimentin (31). More recent studies, however, demonstrate the presence of both mesenchymal and epithelial cells in advanced-stage HNSCC tumors (5). There is extensive evidence that connects the epithelial or mesenchymal phenotype to the response to radiation, chemotherapy, or EGFR inhibitors. Examples include (i) the EMT transition was proposed to be responsible for the modest response of HNSCC tumors to small-molecule EGFR inhibitors such as Gefitinib (Iressa) and Erlotinib (Tarceva) (16); (ii) the increased keratin—a hallmark of epithelial phenotype—was correlated to resistance to DNA-damaging drugs (2, 12); (iii) in a study involving patients with advanced HNSCC, the nonkeratinizing HNSCC responded better to combined radiation and chemotherapy than the tumors that expressed keratin (15, 36). These observations are consistent with the properties of the rSCC-61 cells described here. The upregulation of keratins, periplakin, and E-cadherin in rSCC-61, the decrease in vimentin, and the decreased migration in rSCC-61 are the main results that support the transition from a mesenchymal phenotype in SCC-61 to an epithelial phenotype in rSCC-61. This conclusion is also supported mechanistically by the quantitative proteomics analysis that showed down-regulation of functional pathways involved in migration and cell morphology in rSCC-61 such as the ECM-receptor interaction, actin cytoskeleton regulation, and focal adhesion signaling. While the response of SCC-61 and rSCC-61 to radiation and Erlotinib is in accordance with the studies described earlier, the mechanisms involved are complex. We discuss next a potential network that may connect the cellular phenotype to the observed response to radiation and Erlotinib.
EGFR phosphorylation, ROS, lipid rafts, and response to radiation and Erlotinib
Observations from Figure 1F show that despite increased total phosphoprotein content in rSCC-61, there is significant downregulation of EGFR signaling in rSCC-61. The decreased phosphorylation of EGFR in rSCC-61 cells has two major consequences: (i) It results in reduced phosphorylation of downstream signaling proteins such as Akt, and the inhibition of Akt was previously shown to induce mesenchymal-to-epithelial transition in HNSCC cells (22); downregulation of Akt in rSCC-61 is further enforced by upregulation of PTEN, a negative regulator of Akt, in this cell line (Fig. 6B). (ii) Decreased phosphorylation of EGFR, along with the upregulation of antioxidant proteins, contributes to lower intracellular ROS through mechanisms that may involve decreased activation of NADPH oxidase and decreased glycolysis/mitochondrial electron transport chain, two major sources of ROS.
The regulation of redox microenvironment in rSCC-61 is important to explain both the resistance to radiation and the sensitivity to Erlotinib in rSCC-61. The Western blot analysis of γH2AX indicates reduced DNA damage in rSCC-61, and the cell cycle analysis shows reduced G2/M arrest with 2 Gy radiation in rSCC-61. Another contributing factor to reduced DNA damage with radiation results from upregulation of proteins involved in base-excision repair and DNA replication pathways shown by the proteomics data. In addition, both resistance to radiation and increased sensitivity to EGFR-inhibitors have been independently correlated to impaired structural rearrangement and formation of lipid rafts. Lipid rafts are a hub for the amplification of receptor signaling, including death receptor signaling, and are regulated by ROS—increased ROS have been linked to increased lipid raft formation (35, 38, 50). Our data show decreased ROS in rSCC-61 and increased FASN in these cells. FASN is the main enzyme for synthesis of palmitate and lipogenesis, and its upregulation has been linked to resistance to chemotherapy and radiation (34, 53). FASN inhibition was also proposed to contribute to increased levels of ceramide, a major component of lipid rafts (4). The studies described here show lower lipid staining at the cell membrane in rSCC-61, which may contribute to the increased cell survival and sensitivity to EGFR inhibitors. Indeed, the disruption of lipid rafts in SCC-61 resulted in gain of resistance to radiation and improved sensitivity to Erlotinib. The increased sensitivity of rSCC-61 to Erlotinib is contrary to previous studies which show that cells resistant to EGFR-targeted inhibitors are cross-resistant to ionizing radiation (6, 17). Our results show that the connection between the resistance to radiation and EGFR inhibitors is not bidirectional and emphasizes the significance of adding targeted therapies to radiation or chemotherapy for improving clinical response to treatment. The analysis of protein redox status in clinical samples shows a clear difference between tumors that responded well to radiation and chemoradiation treatment and tumors which did not respond to treatment. This provides the necessary justification for further exploring protein oxidation as a potential biomarker of response to radiation and chemoradiation treatment.
In conclusion, we generated a matched model of radiation resistance in HNSCC that shows increased sensitivity to EGFR inhibition by Erlotinib. Cumulatively, quantitative proteomics, computational, and mechanistic investigations of this system show a convergence of signaling and metabolism networks to elicit protection against ionizing radiation. In some cases, such as the system described here, the acquired resistance to radiation can be accompanied by phenotypic changes or altered sensitivity to other treatments (e.g., targeted inhibitors). The preliminary analysis of clinical samples using newly developed chemical probes for protein oxidation is consistent with the in vitro findings. Prospective clinical studies are ongoing and will address the predictive value of the potential biomarkers of response identified by the studies described here. These include global protein redox status, FASN level, EGFR phosphorylation, and tumor epithelial–mesenchymal composition.
Materials and Methods
Reagents and details of standard methods used in the study are included in the “Supplementary Materials and Methods” section
Cell culture and radiation treatment
All cells used in this study were cultured in the DMEM/F12 medium that was supplemented with 10% fetal bovine serum (FBS; Invitrogen) at 37°C and 5% CO2. The head and neck cancer cell line SCC-61 was kindly provided by Ezra Cohen, Department of Medicine, University of Chicago. Fresh medium was added to cultured cells every 2 days. Subconfluent cells (∼60%–80% confluency) were subjected to radiation with different doses as indicated for each experiment. Radiation was performed using a 444 TBq 12,000 Ci self-shielded 137Cesium (Cs) irradiator. Culture dishes were placed on a styrofoam insert within the chamber of the irradiator, such that the distance from the Cs source results in a homogenous dose distribution over the desired field with a dose rate of 392 rad/min. From the dose rate, the exposure time required to deliver the desired dose was calculated and input into the irradiator.
Establishment of the radiation-resistant rSCC-61 clone
The radiation-sensitive SCC-61 cells were irradiated using a 2 Gy radiation dose. After the radiation treatment, the cells were cultured, split 1:2, allowed to achieve 60% confluence, and then exposed to another cycle of 2 Gy radiation. This process was repeated for a cumulative total of 16 Gy. The resulting cell population was plated at a low density on soft agar, and single cell colonies were picked and expanded in culture. The studies here were focused on the clone R8E, called rSCC-61.
Stable isotope labeling of amino acids in cell culture
The SCC-61 and rSCC-61 cells were cultured in DMEM/F12 media containing the light and heavy isotopes of Lys and Arg, respectively, and supplemented with 10% dialyzed FBS and 200 mg/L proline to prevent the conversion of isotope-coded arginine to proline in cells (7, 40). The cells were passed in their respective media to achieve a minimum of 97% incorporation of the isotope labeled Lys and Arg. The cells were then lysed in modified RIPA buffer (50 mM Tris-HCl, pH 7.4; 1% NP40; 0.25% Sodium deoxycholate; 15 mM NaCl; 1 mM EDTA; 1 mM NaF; supplemented with Roche protease and phosphatase inhibitor tablets). Protein concentration was determined using the bicinchoninic acid (BCA) assay (Thermo Scientific). The SCC-61 and rSCC-61 lysates were then mixed in a 1:1 ratio, precipitated using chloroform/methanol to concentrate the sample, and resuspended in 0.1% SDS. The mixed lysates were resolved on 12% SDS-PAGE and stained with Coomassie Brilliant Blue (R-250). The entire lane was divided into 15 gel bands, which were then digested with trypsin following standard in-gel digestion protocols (1). The resulting tryptic peptides were analyzed on a nanoLC system that was coupled with a LTQ Orbitrap mass spectrometer. Peptide separation was performed on a Thermo Scientific Acclaim PepMap RSLC column (15 cm, 2 μm particle sizes, 100 Å pore sizes) with a flow rate of 300 nL/min and using a 65 min gradient of solutions A (0.05% formic acid in water) and B (80% acetonitrile, 20% water, 0.05% formic acid). The mass spectrometer was operated in the data-dependent mode. The first MS scan was acquired in the Orbitrap at 60,000 resolution (m/z 300–2000). The following MS/MS scans were collected in the ion trap for the top five most intense ions using collision-induced dissociation. The raw MS files were analyzed by Proteome Discoverer 1.2 software (Thermo Fisher Scientific) using MASCOT search engine and the UniProtKB human database. The results were filtered using a false discovery rate of 1%.
Pathway and network analysis
IPA software (
Analysis of clinical samples
Tissue samples
Previously collected and deidentified tumor samples (n=10, flash frozen) were obtained from the Tumor Tissue Core Laboratory at Wake Forest School of Medicine (IRB # 00022263). All patients signed consent forms to permit the use of tumor specimens for scientific, developmental technology, research, and education purposes. Each of these 10 samples were squamous cell carcinomas of the base of tongue. Each patient had radiation as a component of upfront definitive treatment, whether it was radiation, chemoradiation, or surgery followed by radiation or chemoradiation. Treatment-responsive samples (n=5) were classified as those patients who had no evidence of any local or distant failure, or residual or persistent disease (such as noted in a neck dissection) within 1 year after completion of radiation. Of these five radiation sensitive samples, one patient underwent definitive chemoradiation, two patients underwent surgery with immediate adjuvant chemoradiation, and two patients underwent surgery with immediate adjuvant radiation alone. Treatment-resistant samples (n=5) were those who were found to have biopsy proven residual or persistent disease within 1 year of completion of radiation. Of these five radiation-resistant samples, one patient underwent definitive radiation and the remaining four patients underwent definitive chemoradiation. The biotin-tagged redox probe BP1 was synthesized in our laboratory following the procedure previously published (44).
Cryosectioning of frozen tissue samples
The flash frozen tissue blocks were stored at −80°C until the time of sectioning. Before sectioning the frozen tissue, blocks were transferred to a cryotome cryostat (e.g., −20°C) and allowed to equilibrate to the temperature of the instrument. The tissue sections (8 μm thickness) were placed on poly-L-lysine-coated glass slides.
BP1 staining for protein oxidation
The tissue sections were fixed by immersing the slides of 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS) for 20 min at room temperature followed by washing 5×5 min with 0.1 M PBS. After fixation, the samples were permeabilized and stained with BP1 (500 μM) in 0.01 M, pH 7.4 PBS containing 0.2% Triton X-100 (PBT) for a total of 80 min. After this incubation, the slides were washed 5×5 min with PBS and then incubated with Streptavidin-AlexaFluor594 (Red) (2 mg/ml; 1:100 dilution in PBT). The slides were washed with PBS followed by nuclear staining with Hoechst (1:10,000 in 0.01 M, pH 7.4 PBS). Two control experiments were performed: 1. Dimedone, a nontagged reagent for protein oxidation (1 mM, 1 h) was added as blocking reagent before the addition of BP1; and, 2: the same staining procedure was followed as described earlier but in the absence of BP1.
Image collection, processing, and data analysis
A Zeiss 510 or 710 confocal microscope was used for the collection of images as indicated for each study. For each tissue sample, the 40×images were taken at a laser intensity setting of 1% for all samples. All fluorescent excitation and emitted light collection settings were carefully held constant between samples to facilitate equivalence for intensity comparisons. LSM image browser was used for processing the confocal images. The mean fluorescence intensity in sections stained for BP1 was quantified using ImageJ.
Statistical analysis
Statistical analysis (t-test, one-way analysis of variance) was based on a minimum of three biological replicates using SigmaPlot v. 12.0. Asterisks indicate statistically significant changes compared with untreated controls (α=0.05, p-values of 0.01–0.05 [*], 0.001–0.01 [**], or <0.001 [***]).
Footnotes
Acknowledgments
Research reported in this article was supported by the National Cancer Institute of the National Institutes of Health under award number R01 CA136810 to C.M.F. The authors also acknowledge financial support from the Wake Forest School of Medicine (development funds to C.M.F. and TRADONC fellowship to B.C.). An NSF Major Research Instrumentation award supported purchase of the LSCM used to generate images for the clinical samples included in this article (MRI-0722926) within the WFU Microscopic Imaging Core Facility. The clinical samples were obtained from the Tumor Tissue Core Laboratory of the Wake Forest University Comprehensive Cancer Center (grant number P30 CA12197).
Author Disclosure Statement
The redox probe BP1 is manufactured in the laboratory of C.M.F. and distributed by KeraFast. No competing financial interests exist for any of the other authors.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.