
Abstract
Introduction
O
Oxygen is generally administered to patients with significant respiratory disease to enhance the delivery of oxygen to peripheral tissues. However, excessive oxygen inevitably causes acute lung injury (ALI). In hyperoxic conditions, overloading quantities of reactive oxygen species (ROS) generated by NADPH oxidase (NOX) family enzymes are responsible for ALI. Before the present study, the function of dual oxidase2 (DUOX2) in hyperoxia-mediated ALI had not been described despite the fact that it is a NOX homologue and a major source of ROS in lung epithelium. Our novel findings regarding the regulation of DUOX2-generated ROS may prove helpful in designing therapies to alleviate hyperoxia-induced ALI during treatment for patients who require supplementary oxygen therapy.
Hyperoxia-induced ROS can be generated by a range of mitochondrial chain transporter and NADPH oxidase (NOX) enzymes (31, 34). NOX-generated ROS were originally studied in the context of host defense in phagocytic cells (3, 30). However, after identification of several homologues of NOX2/gp91phox [NOX1, NOX3-5, dual oxidase 1 (DUOX1), and DUOX2], the known role of NOX-generated ROS has expanded into diverse cellular events, including cell proliferation, differentiation, apoptosis, and inflammation (4, 7, 28). Several reports have shown that different NOX isoforms are expressed in various lung cell types, and ROS generated from NOXs play a critical role in hyperoxia-induced ALI (11, 22, 45). In particular, NOX1 is required for hyperoxia-induced lung injury, inducing cell death in lung epithelial and endothelial cells (10). NOX2 and NOX4 participate in hyperoxia-induced damage by inducing cell migration and cell death in lung endothelial cells (13, 35, 36). However, until now, no function of DUOXs has been determined in hyperoxia-mediated ALI despite the fact that they are major NOX homologues and a significant source of ROS in lung epithelium. DUOXs were originally identified from the epithelium of the thyroid gland and are essential in thyroid hormone biosynthesis (8). In addition, DUOXs are expressed in epithelial cells of various tissues, including the airways, alveoli, salivary glands, intestinal tract, and prostate (16 –19, 21, 47). Recent studies have focused on the role of DUOX-generated ROS in the regulation of innate immune responses in airway epithelial cells (6, 20, 26, 27, 37, 42). Using DUOX2 mutant mice or mice in which DUOX2 gene expression has been suppressed in lung epithelium, we determined its specific role in the type II AECs that regulate hyperoxia-induced ALI. Our study demonstrates that ROS generated by DUOX2 during hyperoxia causes caspase-mediated cell death and ensuing lung injury by inducing ERK and JNK phosphorylation in type II AECs.
Results
DUOX2 plays a bigger role in hyperoxia-induced ALI than NOX1
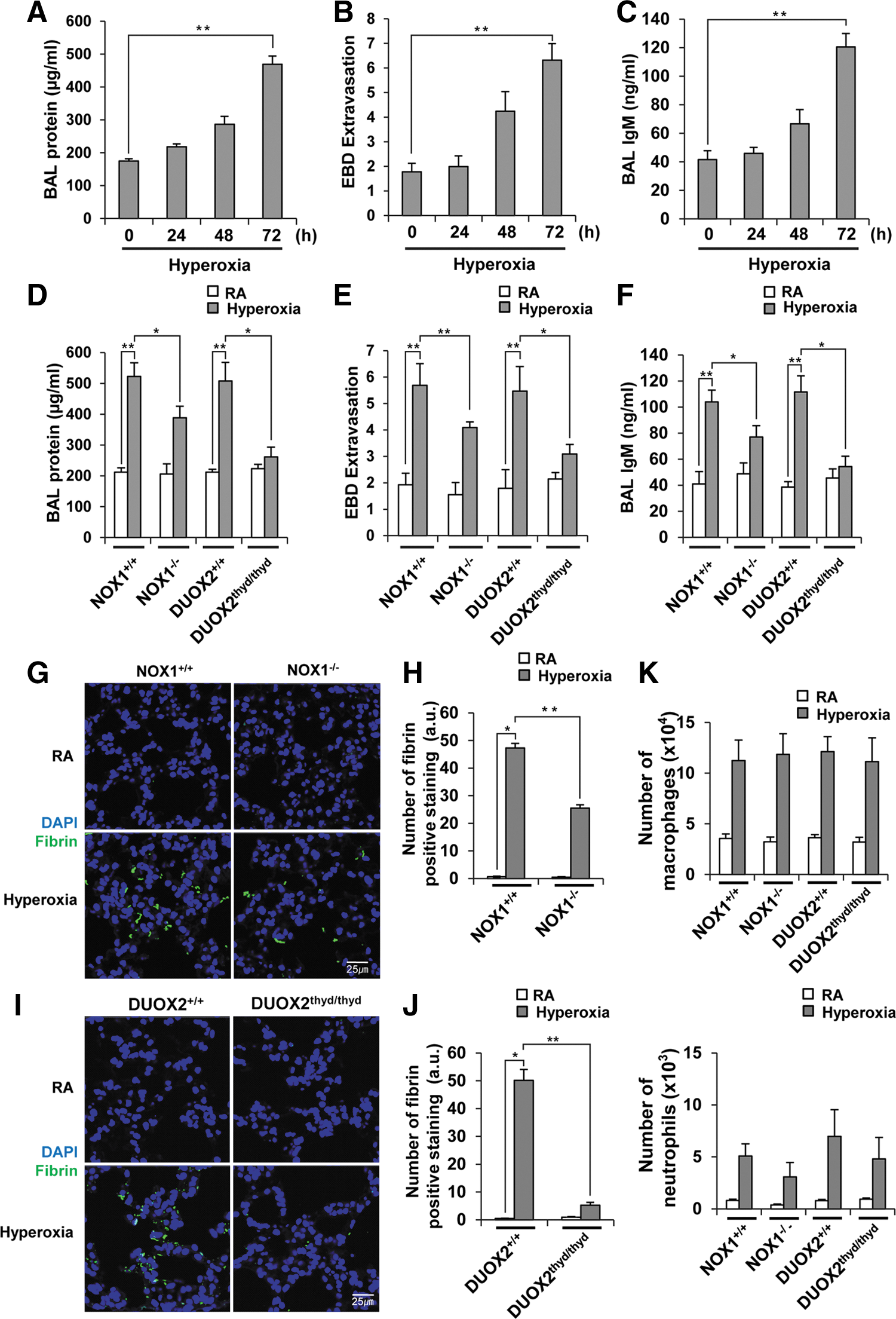
To establish a mouse model of hyperoxia-induced ALI, we exposed wild-type (WT) mice to room air or 100% oxygen at different time points, and measured the extent of lung damage. Lung injury was evaluated by quantifying vascular leakage using an extravasation assay and analyzing alveolar permeability by measuring the protein concentration and IgM levels in bronchoalveolar lavage (BAL) fluid. In agreement with previous studies, the degree of lung injury in WT mice increased in a time-dependent manner, reaching its peak at 72 h after oxygen exposure (Fig. 1A–C). To determine the role of DUOX2 in hyperoxia-induced lung injury, we compared the extent of lung injury in DUOX2thyd/thyd mutant mice with that of WT mice. Simultaneously, we compared the extent of hyperoxia-induced ALI in DUOX2thyd/thyd mice with that of NOX1-deficient mice (NOX1−/−) to determine which NOX enzyme plays a more prominent role in hyperoxia-induced ALI. NOX1 was selected for a comparison because it, rather than NOX2, has been reported to be responsible for hyperoxia-induced lung injury in mice (10). Though the extent of lung injury in NOX1−/− mice was less than the lung injury in NOX1+/+ mice (BAL protein: 25.6%, evans blue dye (EBD) extravasation: 28.0%, BAL IgM: 25.9%), DUOX2thyd/thyd mice (BAL protein: 48.5%, EBD extravasation: 43.5%, BAL IgM: 51.3%) had even less lung injury than NOX1−/− mice (Fig. 1D–F). In addition, fibrin deposition in the alveolar spaces of DUOX2thyd/thyd mice was much lower than that in NOX1−/− mice (Fig. 1G–J). All of the experiments with DUOX2thyd/thyd and NOX1−/− mice were carried out simultaneously under identical conditions with the relevant WT control mice (DUOX2+/+ and NOX1+/+). We then evaluated the lung inflammation induced by hyperoxia in NOX1−/− and DUOX2thyd/thyd mice. Hyperoxia caused an increase in macrophages (approximately threefold) and neutrophils (∼10-fold) in the BAL fluid of WT mice (Fig. 1K). However, the numbers of hyperoxia-induced macrophages and neutrophils in NOX1−/− and DUOX2thyd/thyd mice were similar to those of NOX1+/+ and DUOX2+/+ mice, respectively (Fig. 1K). We also examined inflammation on lung sections from DUOX2thyd/thyd and NOX1−/− mice via hematoxylin and eosin staining, and showed that hyperoxia-induced inflammation is not affected in DUOX2thyd/thyd or NOX1−/− mice (Supplementary Fig. S1A, B; Supplementary Data are available online at
DUOX2 is responsible for hyperoxia-induced ROS production in type II AECs
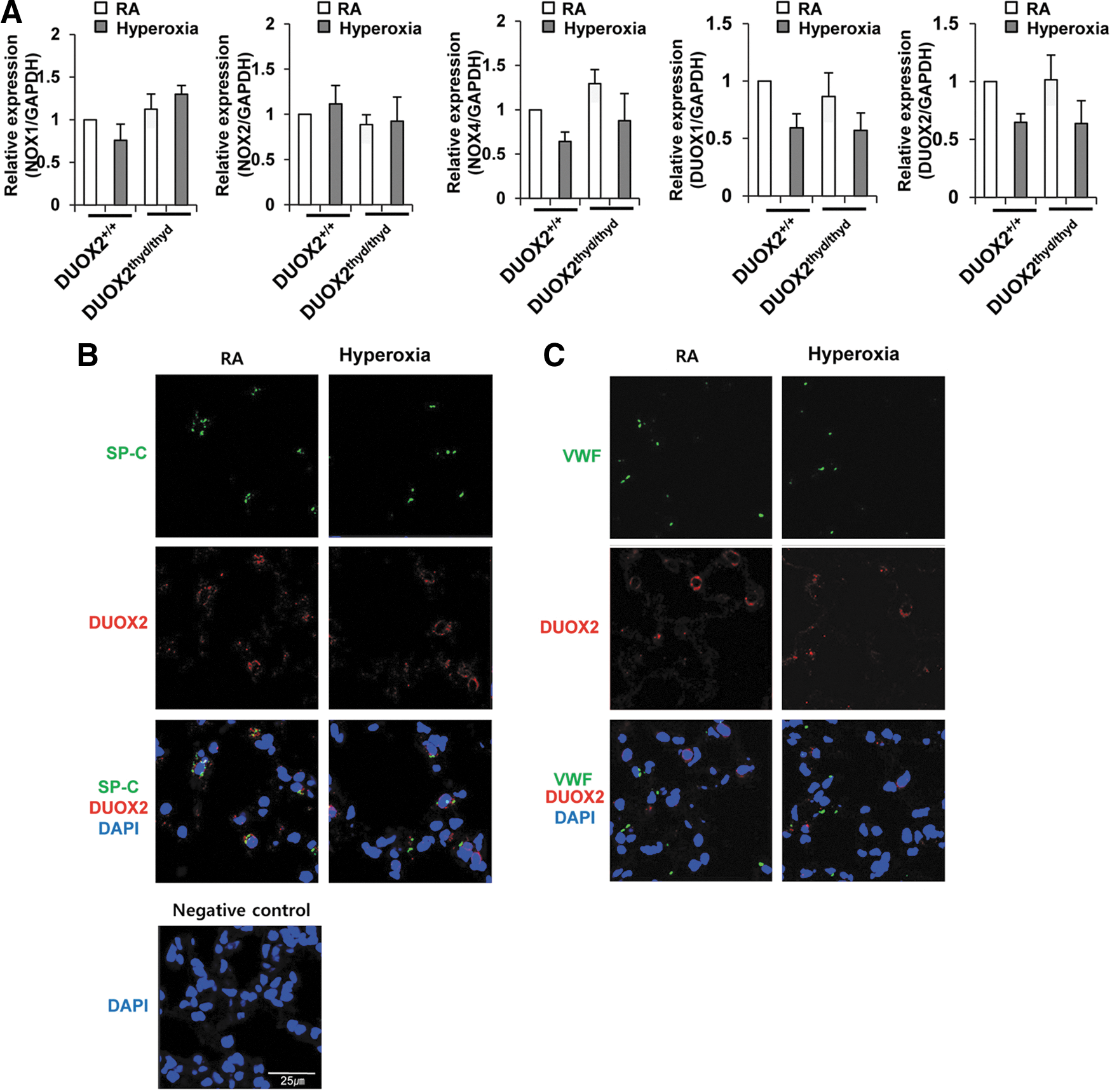
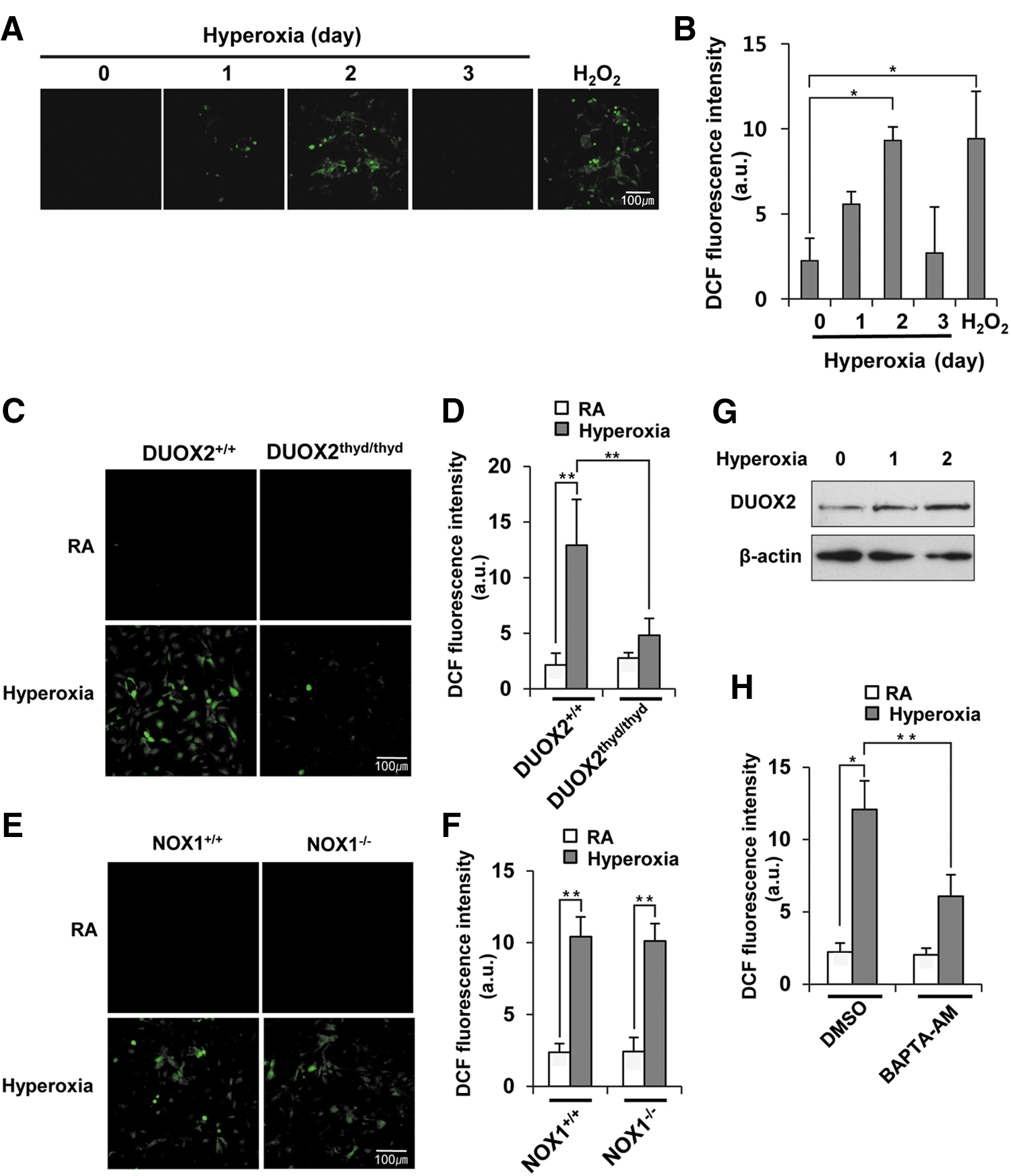
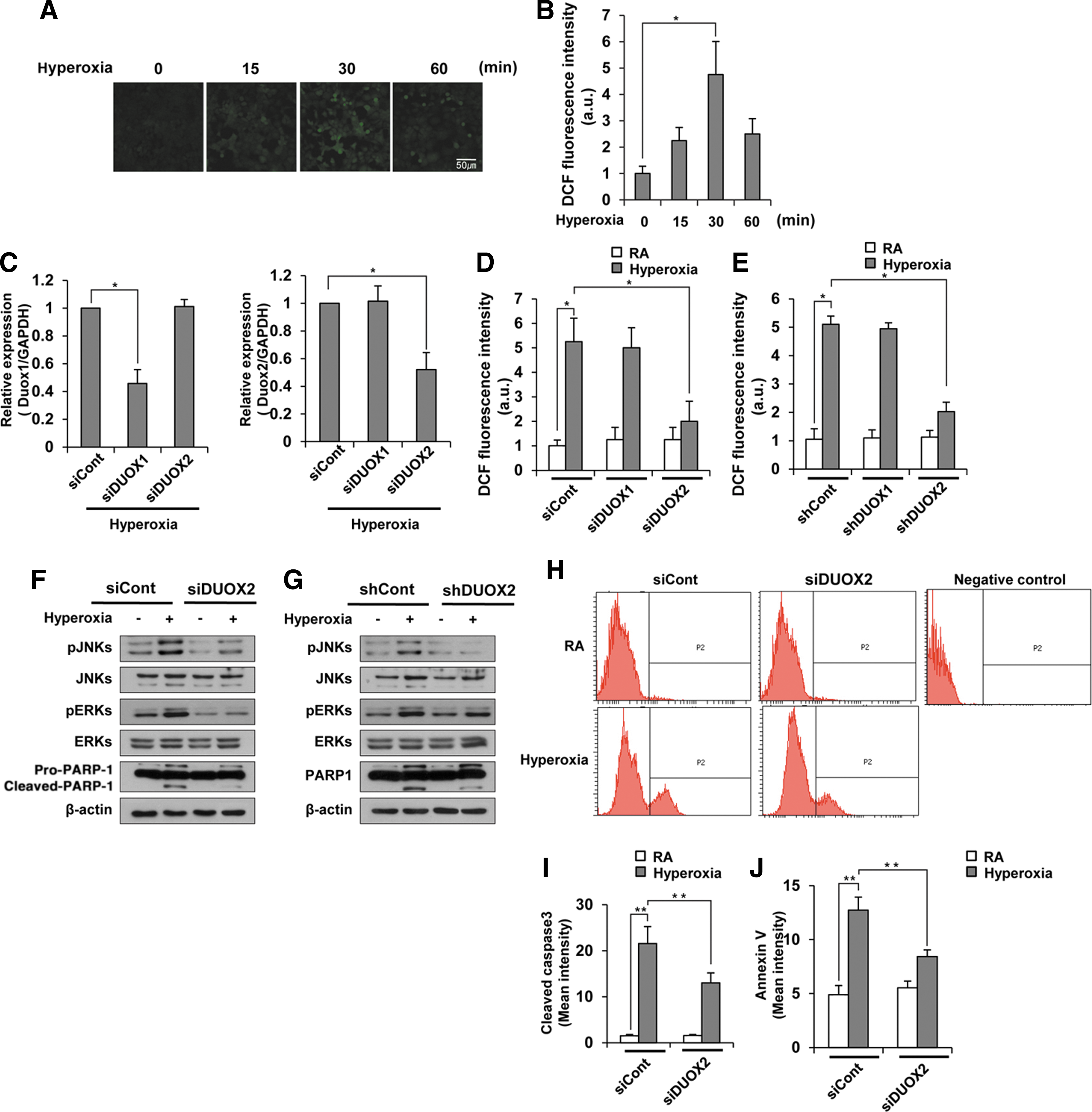
We checked the expression levels of NOX1, NOX2, NOX4, DUOX1, and DUOX2 in DUOX2thyd/thyd mice to determine whether the decrease in hyperoxia-induced lung injury in DUOX2thyd/thyd mice is attributable to downregulation of other NOX enzymes. There were no critical changes in the expression of NOX1, NOX2, NOX4, DUOX1, or DUOX2 genes in DUOX2thyd/thyd mice exposed to hyperoxia (Fig. 2A). In addition, there were no essential changes in the expression levels of NOX2, NOX4, DUOX1, or DUOX2 in NOX1−/− mice exposed to hyperoxia (Supplementary Fig. S2). The results suggested that expression levels of other NOXs were not affected in DUOX2thyd/thyd or NOX1−/− mice under normal or hyperoxia conditions. To examine the localization of DUOX2 expression, lung sections from WT mice were double stained with either anti-DUOX2 antibody and anti-surfactant protein-c (SP-C) (a type II AEC-specific marker) antibody, or anti-DUOX2 antibody and anti-von Willebrand factor (VWF) (an endothelial cell-specific marker) antibody. As shown in Figure 2B and C, DUOX2 was detected in SP-C-expressing cells, but not in VWF-expressing cells, indicating that DUOX2 is mainly expressed in type II AECs, rather than in endothelial cells. To determine the contribution of DUOX2 to hyperoxia-induced ROS production in primary type II AECs, we first examined the population of primary Type I AECs and Type II AECs from WT mice at different time points after hyperoxia exposure via FACS analysis. Two days after hyperoxia, 92.2% of the total cells were Type II AECs, and 5.3% of the total cells were Type I AECs (Supplementary Fig. S3). In this condition, we measured ROS production in primary type II AECs from WT mice at different time points after hyperoxia exposure and then compared the values in primary type II AECs from WT or DUOX2thyd/thyd mice utilizing Dichlorodihydrofluorescein (DCF) dye, which is known to be used to mainly detect hydrogen peroxide (H2O2). As a positive control, we showed that H2O2 treatment into type II AECs from WT mice increased ROS generation (Fig. 3A, B). H2O2 production started at 1 day after hyperoxia exposure and was at a maximum 2 days after hyperoxia exposure (Fig. 3A, B). Hyperoxia-induced H2O2 production was dramatically decreased in type II AECs from DUOX2thyd/thyd mice (Fig. 3C, D), while it was not affected in type II AECs from NOX1−/− mice (Fig. 3E, F). In contrast, when utilizing Dihydroethidium (DHE) dye, which is known to be used to mainly detect superoxide (O2 −), hyperoxia-induced O2 − production was not affected in type II AECs from DUOX2thyd/thyd mice (Supplementary Fig. S4A, B); while it was decreased in type II AECs from NOX1−/− mice (Supplementary Fig. S4C, D). These results suggested that hyperoxia-induced H2O2 production in type II AECs is mainly mediated by DUOX2, while O2 − production is primarily mediated by NOX1. To investigate that DUOX2 activation in type II AECs by hyperoxia exposure is caused by an increase in the DUOX2 expression or Ca2+ signaling or both, we first examined the mRNA expression and protein expression of DUOX2 in response to hyperoxia. The protein expression of DUOX2 was increased by hyperoxia for 2 days (Fig. 3G), whereas the mRNA expression level was not increased under the same conditions (Supplementary Fig. S5), suggesting that upregulation of protein levels of DUOX2 by hyperoxia might be controlled by post-transcriptional modifications, rather than regulation of transcriptional levels. We next measured the hyperoxia-induced ROS generation in type II AECs pretreated with Ca2+ signaling inhibitor (BAPTA-AM). Hyperoxia-induced ROS generation was decreased by Ca2+ signaling inhibitor in type II AECs (Fig. 3H). These results indicated that both the increase of DUOX2 expression and the activation of Ca2+ signaling are required for hyperoxia-induced ROS generation in type II AECs.
DUOX2 is required for hyperoxia-induced cell death in lung type II AECs
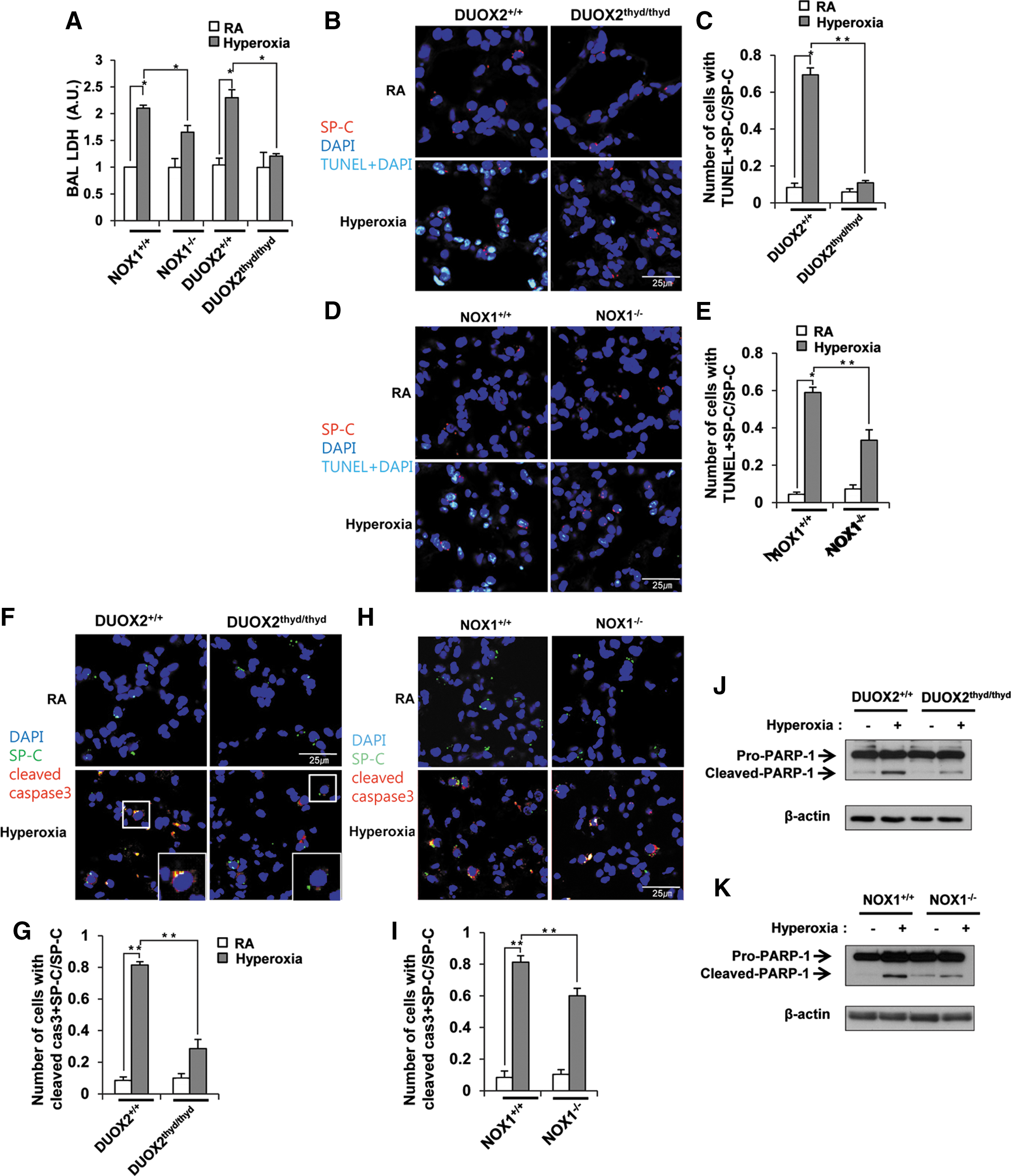
To investigate the role of DUOX2 in hyperoxia-induced cell death in lung epithelia, we examined BAL lactate dehydrogenase (LDH) release, an indicator of cell death, in DUOX2thyd/thyd mice during hyperoxia. BAL LDH release induced by hyperoxia was dramatically decreased in DUOX2thyd/thyd mice (Fig. 4A). Though the extent of LDH release in NOX1−/− mice is lower than in NOX1+/+ mice (21.4%), the decrease seen in DUOX2thyd/thyd mice (47.6%) was larger than that in NOX1−/− mice (Fig. 4A). These observations are in agreement with previous data (Fig. 1D–J). We hypothesized that the decrease in the cell death in DUOX2thyd/thyd mice lung by hyperoxia exposure was caused by cell death in type II AECs, because DUOX2 is responsible for hyperoxia-induced ROS production in type II AECs and ROS-dependent caspase3 activation in lung epithelium participates in hyperoxia-induced cell death (10, 51). To verify this, we double stained lung sections from WT or DUOX2thyd/thyd mice in situ with anti-SP-C antibody and TUNEL, or anti-SP-C antibody and anti-cleaved caspase3 antibody. Double-stained cells with anti-SP-C antibody and TUNEL (Fig. 4B, C), as well as with anti-SP-C antibody and anti-cleaved caspase3 antibody (Fig. 4F, G), were dramatically decreased in DUOX2thyd/thyd mice by hyperoxia exposure. Double-stained cells with anti-SP-C antibody and TUNEL (Fig. 4D, E), as well as with anti-SP-C antibody and anti-cleaved caspase3 antibody (Fig. 4H, I), were also decreased in NOX1−/− mice, though the extent of decline was less than that of DUOX2thyd/thyd mice. We also showed that cleaved PARP-1 induced by hyperoxia was decreased in lung lysates in DUOX2thyd/thyd and NOX1−/− mice, respectively (Fig. 4J, K). These results indicated that DUOX2 is mainly responsible for hyperoxia-induced cell death in lung type II AECs.
DUOX2 is responsible for hyperoxia-induced ERK and JNK phosphorylation in lung epithelium
Since MAPK phosphorylation has been reported to be responsible for hyperoxia-induced cell death (10, 51), we investigated the role of DUOX2 in the hyperoxia-induced activation of MAPK. Of the MAPK-related proteins tested, JNK and ERK were strongly phosphorylated by hyperoxia exposure (Fig. 5A), while p38 was not (data not shown). Hyperoxia-induced JNK and ERK phosphorylation was lower in DUOX2thyd/thyd mice than in WT mice (Fig. 5A); while JNK phosphorylation, but not ERK phosphorylation, was lower in NOX1−/− mice by hyperoxia exposure (Fig. 5B). These results indicated that hyperoxia-induced JNK activation is dependent on both DUOX2 and NOX1, but hyperoxia-induced ERK activation is dependent on DUOX2, but not on NOX1. To verify the effect of JNK and ERK activation on hyperoxia-induced cell death in type II AECs, we examined the hyperoxia-induced PARP-1 cleavage in murine transformed lung epithelial 12 (MLE12) cells pretreated with SP600125, a JNK inhibitor, and PD98059, an ERK inhibitor. Hyperoxia-induced PARP-1 cleavage was decreased by SP600125 treatment (Fig. 5C), whereas it was not decreased by PD98059 (Fig. 5D). Consistent with the data in MLE12 cells, hyperoxia-induced lung injuries were decreased in WT mice pretreated with SP600125 (Fig. 5E–G); while they were not decreased in those pretreated with PD98059 (Fig. 5H–J), indicating that JNK activation, rather than ERK activation, induces hyperoxia-induced cell death and lung injury. Together, these results demonstrated that DUOX2-induced cell death and lung injury by hyperoxia is mediated by JNK activation, though DUOX2 mediates activation of both JNK and ERK in response to hyperoxia exposure.
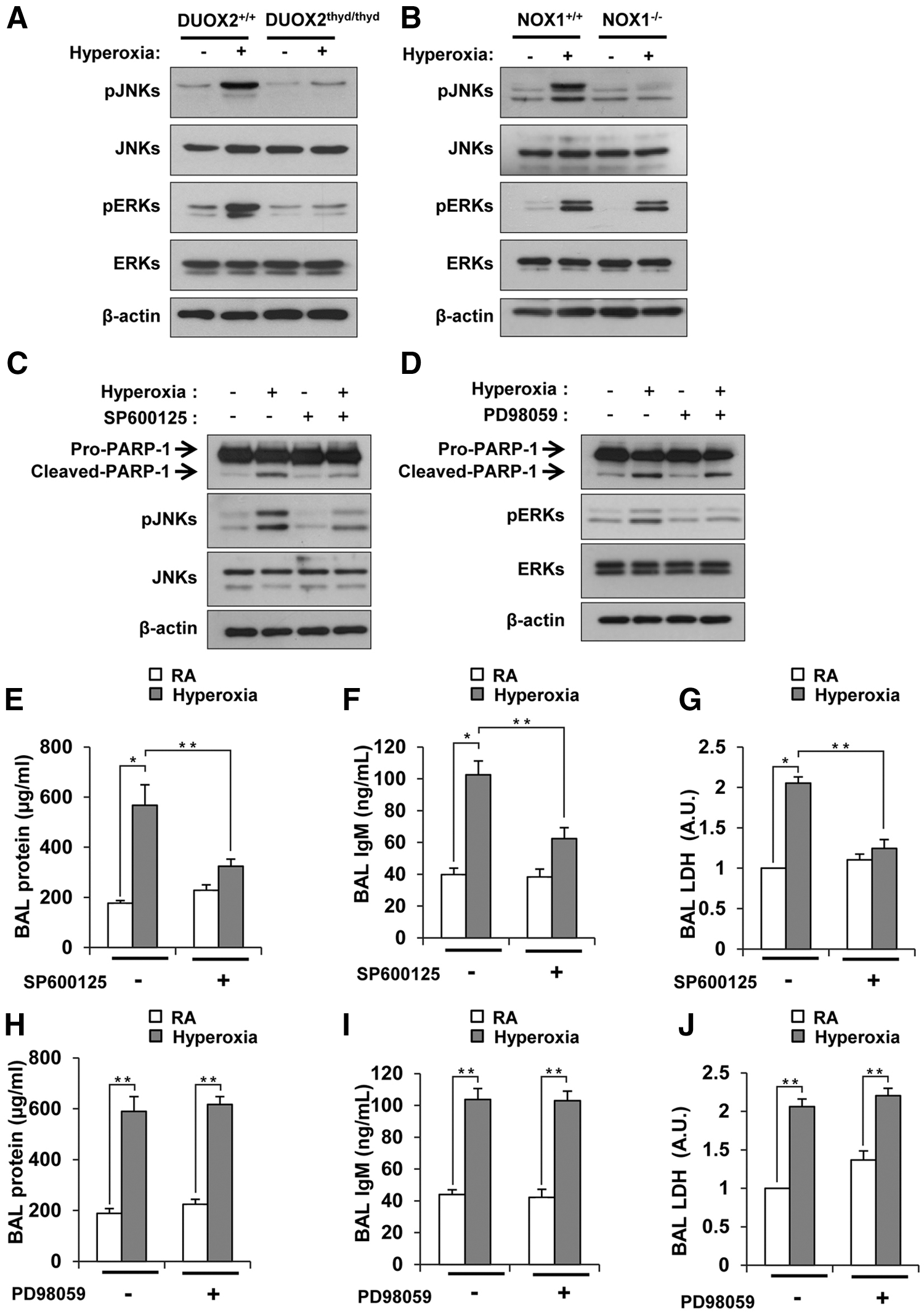
Hyperoxia-induced ROS generation in lung epithelium is required for ALI
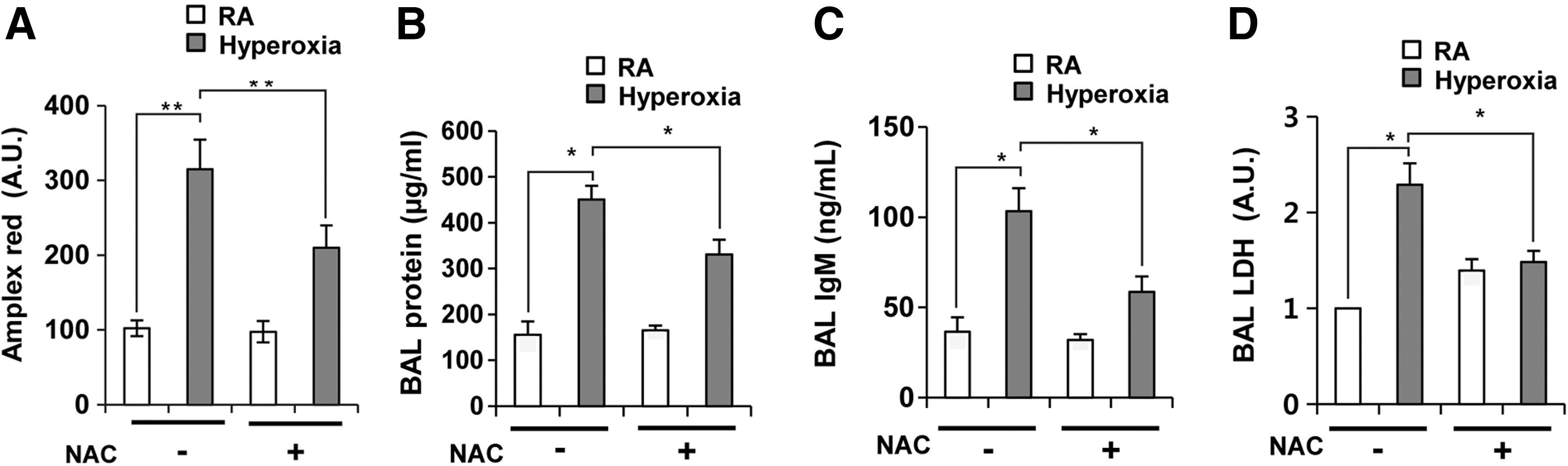
Since hyperoxia-induced H2O2 production and cell death were decreased in primary type II AECs from DUOX2thyd/thyd mice (Figs. 3C, D and 4B, C, F, G), we tested whether DUOX2-mediated H2O2 in mouse lung epithelium contributes significantly to hyperoxia-induced lung injury. We pretreated lung epithelium from WT mice with N-acetylcysteine (NAC), an ROS-scavenging chemical, and measured ROS production and hyperoxia-induced lung injuries. NAC treatment decreased ROS production (Fig. 6A), BAL protein (Fig. 6B), BAL IgM (Fig. 6C), and BAL LDH (Fig. 6D) in response to hyperoxia. As a positive control, we demonstrated that H2O2 treatment into mouse lung increased ROS generation in lung epithelium (Supplementary Fig. S6). These results suggest that hyperoxia-generated ROS in lung epithelium play an important role in hyperoxia-induced ALI
DUOX2, but not DUOX1, is responsible for hyperoxia-induced cell death in type II AECs
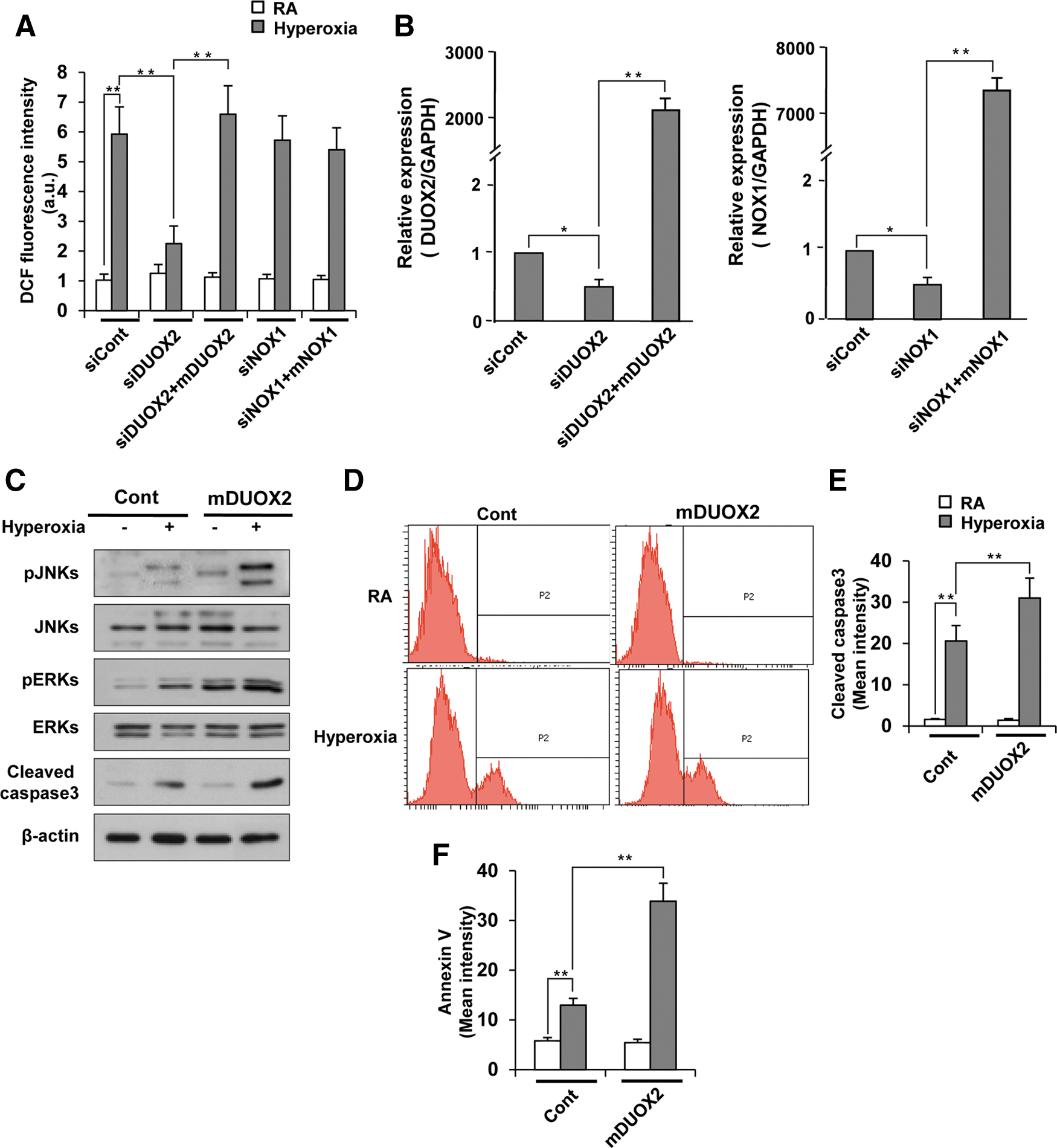
To determine which DUOX enzyme is more important in hyperoxia-induced H2O2 generation, we first measured H2O2 production in MLE12 cells at different time points after hyperoxia exposure. In contrast to primary mouse type II AECs, H2O2 generation in MLE12 cells started at 15 min after hyperoxia exposure and peaked at 30 min (Fig. 7A, B). In addition, hyperoxia-induced H2O2 generation was decreased by Ca2+ signaling inhibitor in MLE12 cells, indicating that hyperoxia-induced Ca2+ signaling is required for H2O2 generation in MLE12 cells (Supplementary Fig. S7). To specifically suppress the gene expression of DUOX1 and DUOX2 in MLE12 cells, we selected the most specific and effective suppressors of DUOX1 (siDUOX1-5) and DUOX2 (siDUOX2-4) from five candidate duplex siRNAs for each enzyme (Supplementary Fig. S8A, B). During hyperoxia, siDUOX1-5 treatment decreased the expression of DUOX1, but not DUOX2; whereas siDUOX2-4 treatment decreased the expression of DUOX2, but not DUOX1 (Fig. 7C). Suppression of DUOX2 expression dramatically decreased H2O2 generation induced by hyperoxia exposure, whereas suppression of DUOX1 had no effect (Fig. 7D). These results indicate that DUOX2, but not DUOX1, is required for hyperoxia-induced H2O2 production in type II AECs. Consistent with data obtained from the lung tissue of DUOX2thyd/thyd mice (Figs. 4J and 5A), suppression of DUOX2 expression in MLE12 cells decreased phosphorylation of JNK and ERK, PARP1 cleavage (Fig. 7F), cleaved caspase3 (Fig. 7H, I), and Annexin V staining (Fig. 7J). To confirm the effect of siDUOX2 on hyperoxia-induced cell death, we used a shRNA lentivirus gene suppression system. From the five candidates for each of shDUOX1 and shDUOX, we selected one duplex shDUOX1 (shDUOX1-3) and one shDUOX2 (shDUOX2-3) that most greatly decreased the expression of DUOX1 and DUOX2, respectively (Supplementary Fig. S9A, B). We also showed that the protein level of DUOX2 was decreased in DUOX2-knocked-down cells, but not in DUOX1-knocked-down cells, compared with shcont cells (Supplementary Fig. S10). Consistent with the siDUOX2 data, suppression of DUOX2 expression by treatment with DUOX2 shRNA lentivirus particles decreased H2O2 generation (Fig. 7E), ERK and JNK phosphorylation, and PARP-1 cleavage (Fig. 7G) in response to hyperoxia. To confirm the effect of DUOX2 on hyperoxia-induced H2O2 production in MLE12 cells, we examined hyperoxia-induced H2O2 production in DUOX2-knocked-down cells and DUOX2 overexpressed cells in DUOX2-knocked-down MLE12 cells. Hyperoxia-induced H2O2 production was decreased in DUOX2-knocked-down cells, and this decrease was rescued to the level of the control cells by hyperoxia in DUOX2 overexpressed cells in DUOX2-knocked-down cells (Fig. 8A, B). However, hyperoxia-induced H2O2 production was not decreased in NOX1-knocked-down cells (Fig. 8A, B). These results were previously confirmed by our results which showed that hyperoxia-induced H2O2 production is not decreased in type II AECs from NOX1−/− mice (Fig. 3E, F). To investigate the effect of overexpressed DUOX2 on hyperoxia-induced MAPK activation and cell death, we examined JNK and ERK activation, cleaved caspase3, and cell death by FACS analysis in DUOX2-trasnfected MLE12 cells. Activation of JNK, ERK, and cleaved caspse3 (Fig. 8C), as well as staining of cleaved caspase3 (Fig. 8D, E) and Annexin V (Fig. 8F) was further augmented in DUOX2 overexpressed cells by hyperoxia exposure. These results demonstrated that DUOX2-generated H2O2 in MLE12 cells activate hyperoxia-stimulated cell death.
DUOX2 in lung epithelia is required for hyperoxia-induced lung injury
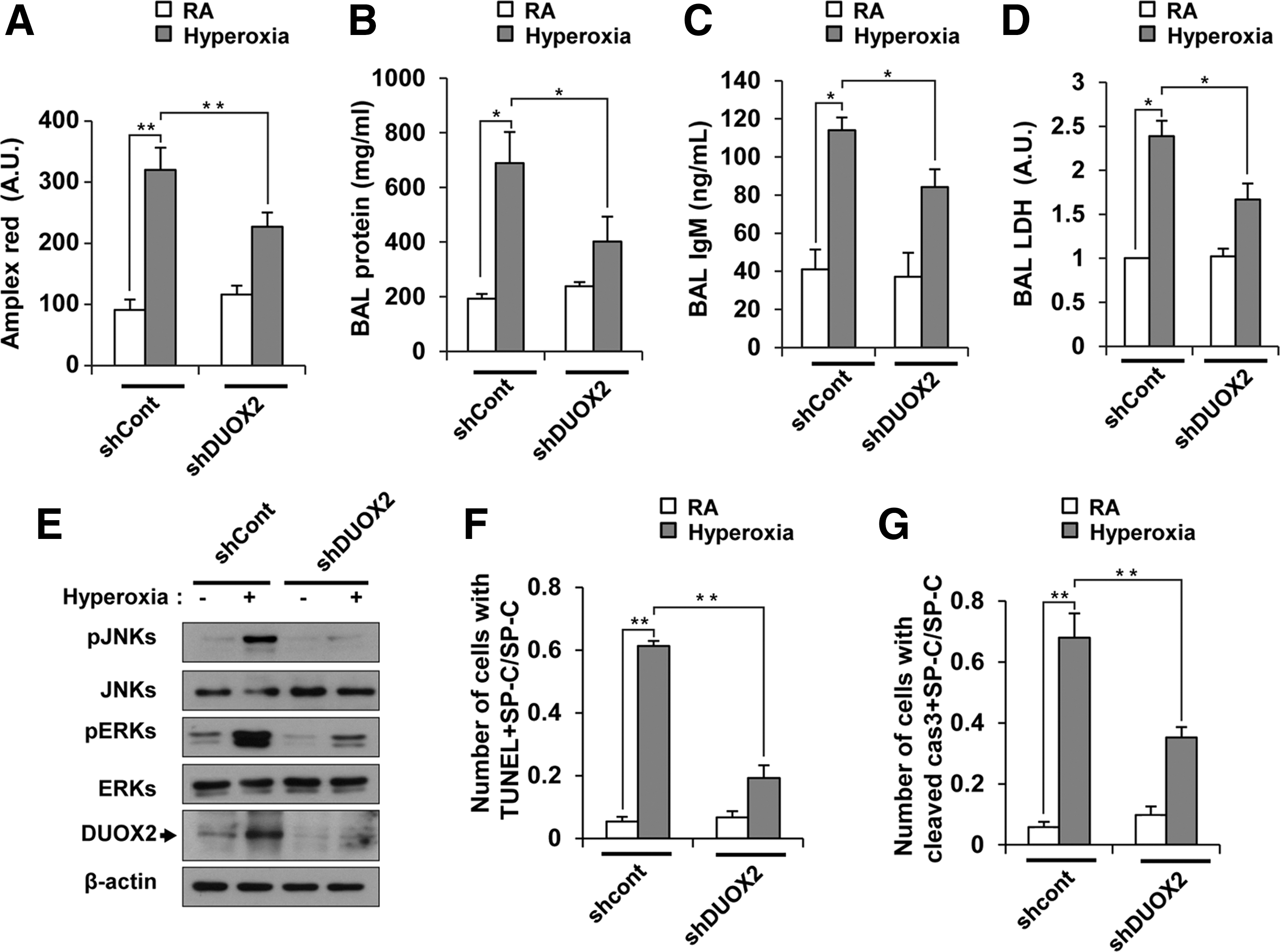
We also tested whether a transient decrease in DUOX2 expression in WT mouse lung epithelia has the same effect as the genetic lack of functional DUOX2 protein in DUOX2thyd/thyd mice. We pretreated the lung epithelia with DUOX2 shRNA lentivirus particles by intranasal challenge, and measured hyperoxia-induced lung injury. Suppression of DUOX2 expression in lung epithelia decreased H2O2 production (Fig. 9A), BAL protein (Fig. 9B), BAL IgM (Fig. 9C), and BAL LDH (Fig. 9D). In addition, hyperoxia-induced phosphorylation of JNK and ERK significantly decreased in lung epithelium pretreated with DUOX2 shRNA particles (Fig. 9E). We also investigated hyperoxia-induced lung injury and MAPK activation in NOX1-knocked-down mice that were pretreated with lentivirus particles with shNOX1 (shNOX1 mice). Expression level of NOX1 was decreased in shNOX1 mice (Supplementary Fig. S11A). H2O2 production, lung injuries, including BAL protein, BAL IgM, and BAL LDH (Supplementary Fig. S11B–E), as well as activation of JNK and ERK (Supplementary Fig. S11F) were not decreased in shNOX1 mice, which was different from the results in NOX1−/− mice (Figs. 1D–F and 5B). These results suggested that a decrease in NOX1 expression in shNOX1 mice might not be enough to decrease lung injuries and MAPK activation. In contrast, lung injuries and MAPK activation were decreased in shDUOX2 mice (Fig. 9B–E). To verify the effect of DUOX2 on cell death in situ, we double stained lung sections from shcont and shDUOX2 mice with anti-SP-C antibody and TUNEL or anti-SP-C antibody and anti-cleaved caspase3 antibody. Double-stained cells with anti-SP-C antibody and TUNEL (Fig. 9F), as well as with anti-SP-C antibody and anti-cleaved caspase3 antibody (Fig. 9G) were decreased in shDUOX2 mice. Specific suppression of DUOX2 expression in lung epithelium had a similar effect as the lack of a functional DUOX2 protein seen in DUOX2thyd/thyd mice, indicating that DUOX2 is a critical enzyme that regulates the lung epithelial response to hyperoxia and participates in ALI.
Discussion
In this study, we clearly demonstrated that DUOX2 is critical for hyperoxia-induced ALI, and that it increases ROS generation and subsequent cell death in AECs. This was done by showing that increased cell death and ALI caused by hyperoxia were decreased in both mutant mice that are devoid of DUOX2 activity (DUOX2thyd/thyd) and mice in which DUOX2 gene expression was knocked down in the lungs. In hyperoxia-induced cell damage, different NOX enzymes, including NOX1, NOX2, and NOX4, play an essential role by generating ROS in alveolar cells (10, 13, 34 –36, 44, 46, 51). With regard to NOX2, debatable studies have been reported, with regard to its role in lung injury by hyperoxia. It has been shown that hyperoxia-induced lung injuries, including pulmonary edema and inflammatory responses, are attenuated in NOX2−/− mice (35); whereas another report showed that lung injuries such as alveolar cell death and vascular leakage are not affected in NOX2−/− mice (10). Reflecting on these contrasting reports, it was suggested that NOX2 does not mediate hyperoxia-induced alveolar-capillary disruption, though it is involved in hyperoxia-induced lung inflammation, including infiltration of macrophages and neutrophils. Though both lung epithelial cell and endothelial cells are critical for maintaining alveolar-capillary barrier homeostasis in response to hyperoxia, many studies about the role of NOX in hyperoxia-induced cell damage have been focused on endothelial cells. However, it has recently been shown that NOX1 plays a pivotal role in hyperoxia-induced ALI by increasing ROS generation and cell death in epithelial and endothelial cells (10). Since DUOX2 is mainly expressed in lung epithelial cells and DUOX2-mediated ROS are essential for regulation of cell signaling in response to various stimuli, we compared the level of hyperoxia-induced ALI in DUOX2thyd/thyd mice with that in NOX1−/− mice. Indeed, hyperoxia-induced cell death and ALI were partially attenuated in NOX1−/− mice compared with NOX1+/+ mice, and the decrease thereof in DUOX2thyd/thyd mice was much greater than that in NOX1−/− mice. ROS generation and phosphorylation of ERK and JNK in response to hyperoxia in NOX1-suppressed MLE12 cells were not significantly decreased compared with those in DUOX2-suppressed MLE12 cells (data not shown). Given that NOX-generated ROS induce hyperoxia-stimulated phosphorylation of ERK and cell death in MLE12 cells (51), our experiments suggest that DUOX2-generated ROS, rather than those generated by NOX1, may have a primary role in modulating hyperoxia-induced cell death in lung epithelial cells. In addition, ROS generation was not affected in DUOX1-suppressed MLE12 cells, suggesting that DUOX2 is the main DUOX enzyme responsible for hyperoxia-induced cell signaling in lung epithelial cells.
The accumulation of inflammatory cells caused by hyperoxia was not affected in DUOX2thyd/thyd mice. In agreement with our findings, another study showed that NOX1 and NOX2 are not involved in the lung inflammation caused by hyperoxia (10). It should be noted, however, that these results about the role of NOX in hyperoxia-induced inflammation do not exclude a role for hyperoxia-induced oxidative stress in lung inflammation. NRF2, a transcription factor that induces several antioxidant enzymes and superoxide dismutase, protects the lung against inflammation and hyperoxia-induced ALI (33, 38, 39). Thus, an ROS source other than a NOX enzyme may be required for hyperoxia-induced inflammation. Meanwhile, previous work has demonstrated that DUOX2-generated ROS play an essential role in the innate immune response to bacterial or allergenic challenge in airway epithelial cells, leading to mucosal or allergic inflammation, respectively (6, 12, 17, 20, 23, 26, 27, 37, 42). This suggests that different NOX enzymes may specifically activate distinct signaling pathways by modulating their expression level (10, 35), subcellular localization (7), and interaction with membrane proteins (26, 42) in a signal-dependent manner.
Several lines of evidence showed that purinergic stimulation of lung epithelium increases DUOX-mediated H2O2 production, presumably via increasing intracellular [Ca2+] and consequent DUOX activation through regulation of its EF-hand domains (2, 19). Although we were unable to detect an increase in intracellular [Ca2+] subsequent to hyperoxia in type II AECs or MLE12 cells, we showed the effect of Ca2+ signaling on DUOX2 activation utilizing Ca2+ signaling inhibitor (BAPTA AM). We demonstrated that hyperoxia-increased DUOX2 expression and Ca2+ signaling are required for ROS generation in type II AECs, while Ca2+ signaling, rather than DUOX2 increase, is responsible for ROS generation in MLE12 cells. The difference between type II AECs and MLE12 cells may be caused by the existence of an unknown regulatory system that is capable of rapidly and efficiently activating DUOX2 without an increase in DUOX2 expression in MLR12 cells. The role of DUOX2 in hyperoxia-induced ALI and cell death was demonstrated both in mice genetically lacking functional DUOX2 and in a WT background using a transient DUOX2 suppression system in epithelial cells and mouse lung epithelium. This is imperative from a scientific point of view, as it proves that an acute decrease of DUOX2 expression is sufficient to inhibit ALI and cell death, and the resistance to hyperoxia-induced ALI was not due to chronic adaptation in mutant mice devoid of DUOX2 activity. Inhibition of NOX enzymes was expected to be efficient in hyperoxia-induced ALI, because it inhibits cell death in epithelial and endothelial cells by decreasing oxidative stress from hyperoxia. Moreover, inhibition of NOX4 had been reported to be a potential therapeutic target for ALI through suppression of endothelial cell death (9, 24). However, studies on repressors of hyperoxia-induced ALI via inhibition of epithelial cell death are largely unknown.
Our novel findings regarding the regulation of DUOX2-generated ROS in hyperoxia-induced ALI could have a significant effect on therapeutic developments, potentially favoring DUOX2-specific inhibitors mediated oversuppression of epithelial cell death signaling pathways in the treatment of ALI.
Materials and Methods
Mice
DUOX2 mutant mice were purchased from The Jackson Laboratory (25). The recessive thyd mutation developed spontaneously in a B6 (129)-Duox2 thyd/J mouse (Jackson Laboratory; Stock no. 005543). DUOX2+/+ littermates were used as wild-type controls. Nox1−/− mice were kindly provided by Dr. Yun Soo Bae (Department of Life Science, Ewha Woman's University, Seoul, Korea). NOX1+/+ littermates were used as wild-type controls. All mice used in this study were 8–10 weeks old and maintained in our animal facilities under specific, pathogen-free conditions. All experiments were approved by the Institutional Review Board of Yonsei University College of Medicine.
Hyperoxia exposure
Mice were exposed to hyperoxia in a large AtmosBag (Sigma-Aldrich) that was saturated with almost 100% O2 at a sufficient flow rate. Control mice were exposed to room air. All mice were allowed free access to water and food. Hyperoxic conditions for cultured cells were achieved by placing type II AECs and MLE 12 cells in a sealed plastic chamber filled with 95% O2 and 5% CO2 at 37°C. Controls were placed directly into the cell culture incubator, which was filled with room air supplemented with 5% CO2, resulting in a final ambient oxygen concentration of 20%.
Antibodies
We purchased antibodies for Western blotting and immunohistochemistry. We used antibodies to ERK (Cell Signaling; #9102), phospho-ERK (Cell Signaling; #9101), JNK (Cell Signaling; #9252), phospho-JNK (Invitrogen; 44-682G), PARP (Cell Signaling; #9542), caspase3 (cell signaling; #9662), and β-actin (Santa Cruz; sc-47778) for Western blotting. We used antibodies to SP-C (Santa Cruz; sc-7706), VWF (Santa Cruz; sc-365712), AQP5 (Santa Cruz; sc-9890), cleaved caspase3 (Cell Signaling; #9661), and fibrin (Dako; A0080) for immunofluorescence. Anti-DUOX2 antibody was kindly provided by Dr. Yun Soo Bae (Department of Life Science, Ewha Woman's University, Seoul, Korea).
Western blot analysis
For Western blots, mouse lung tissues were homogenized with ice-cold RIPA buffer (150 mM NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0.), protease inhibitor cocktail, and 1 mM PMSF. 30 μg of lysate was separated by 8% or 12% SDS-PAGE and transferred to PVDF (Bio-Rad). Membranes were blocked with 5% skim milk in TTBS (Tween-Tris Buffered Saline: 0.5% Tween-20 in 20 Mm Tris-HCl [pH7.5], 150 Mm NaCl), and then incubated overnight with primary antibodies and 5% skim milk in TTBS. After washing with TTBS, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies and 5% skim milk in TTBS for 1 h at room temperature. After washing with TTBS, the blot was visualized using the ECL system.
Immunofluorescence localization
Formalin-fixed mouse lung left lobes were dehydrated gradually in ethanol, embedded in paraffin, and cut into 5 μm sections and heat-induced epitope retrieval was performed before staining. The tissues were permeabilized with 0.2% Triton X-100 for 10 min and blocked with phosphate-buffered saline (PBS) containing 1% BSA and 10% normal goat serum. We then double stained with anti-SP-C and anti-DUOX2 antibodies or anti-VWF and anti-DUOX2 antibodies. After extensive washing with PBS, tissues were treated with Alexa568-conjugated anti-rabbit IgG and Alexa488-conjugated anti-goat IgG or Alexa568-conjugated anti-rabbit IgG and Alexa488-conjugated anti-mouse IgG. Co-localization of SP-C and DUOX2 or VWF and DUOX2 was analyzed by confocal microscopy (Carl Zeiss; LSM 700). For sections of fixed lung, tissue was treated with anti-fibrin antibody or anti-cleaved caspase3 antibody and examined using confocal microscopy.
BAL fluid collection
BAL fluid (BALF) was obtained from mouse lungs using 0.5 ml PBS after cannulation of the trachea. The collected BALF was centrifuged at 1000 g for 10 min. The cell pellet was deposited onto glass slides using a Cytospin centrifuge (600 g for 5 min) and used for differential cell counts by staining with a Diff-Quik Stain Set. The supernatant was collected and stored at −20°C or −80°C for BAL protein assays and LDH activity, respectively.
Assessment of lung injury
BAL protein was measured using a protein quantification assay (Pierce BCA Protein Assay) on BALF supernatant according to the manufacturer's instructions. Evans Blue Dye assay was performed as previously described (39). In brief, Evans Blue (Sigma-Aldrich; 30 mg/kg) was injected intraperitoneally and allowed to circulate for 2 h before mice were sacrificed. At the time of sacrifice, blood was sampled by heart puncture and centrifuged at 1600 g for 5 min to collect the plasma. Subsequently, lungs were perfused with PBS through the right ventricle to remove intravascular dye from the lung. Lungs were placed in 1 ml of formamide followed by incubation for 16 h at 60°C, and the absorption of Evans Blue was measured spectrophotometrically at a wavelength of 620 nm. Vascular leakage was expressed as OD620/gram of lung weight. IgM levels in BAL fluid samples were measured using a murine-specific IgM ELISA kit (Bethyl Laboratories).
Measurement of cell death
LDH activity was measured in BAL supernatant as previously described (14). BALF supernatant was measured at 490 nm using an LDH determination kit according to the manufacturer's instructions (Promega). For sections of fixed lung, DNA fragmentation and cell death were evaluated by terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) assay. TUNEL was performed with an in situ Cell Death Detection Kit (Fluorescein, Roche) according to the manufacturer's protocol.
Isolation and culture of mouse type II AECs
Type II AECs were isolated from adult mice (9–14 weeks old) lungs as described with slight modifications (15). In brief, mice were anesthetized using a mixture of Zoletil and Rompun, and the lungs were perfused with PBS. The trachea was cannulated, and 2 ml of dispase (BD Biosciences) was instilled followed immediately by 1 ml of 1% low-melt agarose. The lung was placed in ice-cold PBS for 2 min. Lungs were removed, immersed in 2 ml of dispase, incubated for 45 min at room temperature, and then minced in growth medium containing 0.01% DNase. The cell suspension was then filtered consecutively through 100-μm and then 40-μm cell strainers (BD Biosciences). Single-cell suspensions were centrifuged at 1200 rpm for 5 min, resuspended in DMEM+10% FBS, and incubated on anti-CD45- and anti-CD16/32- (clones 30-F11, 93, eBioscience)-coated plates for 2 h. Nonadhered cells were cultured directly on matrigel-coated cell culture plates in the presence of Keratinocyte Growth Factor (KGF, 10 ng/ml). Cells were cultured for approximately 5 days, and the media were exchanged every 2 days.
Murine transformed lung epithelial cell culture
Murine transformed lung epithelial cells (MLE12; American Type Culture Collection) were maintained in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were cultured at 37°C in a humidified atmosphere containing 5% CO2. All experiments were conducted in confluent and quiescent cells in a monolayer to avoid cell density variability.
DUOX1 and DUOX2, NOX1 silencing using siRNA and lentiviral shRNA in MLE12 cells and mouse lung epithelia
For DUOX1 and DUOX2 silencing in MLE12 cells, cells were transfected with control siRNA, DUOX1 siRNA (5′-GAGACAAAGUGAGAAUUAAUU-3′), and DUOX2 siRNA (5′-CAAAGAGAGUCUGAAGAAAUU-3′) purchased from Genolution Pharmaceuticals. NOX1 siRNA (number: 1392760) were purchased from Bioneer. For DUOX2 and NOX1 silencing in mouse lung epithelia using shRNA lentivirus particles (Thermo Scientific), mice were anesthetized with 50 mg/kg Zoletil (Virbac) and 10 mg/kg Rompun (Bayer AG) and given either mouse Duox2 shRNA (cloneID: V3LMM-425530), NOX1 shRNA (cloneID: V2LMM_33862) or scrambled shRNA lentiviral particles (3×107 TU/ml) intranasally in a total volume of 30ul. After 6 days, the mice were used for the experiments.
DUOX2 overexpression using full length of cDNA clones in MLE12 cells
For DUOX2 overexpression in MLE12 cells, cells were transfected with mouse DUOX2 cDNA clone purchased from Origene.
Real time-PCR
Total RNA was isolated from MLE12 cells using TRIzol (Invitrogen). cDNA was synthesized from 1 μg RNA with random hexamer primers using Moloney murine leukemia virus reverse transcriptase (Applied Biosystems). To quantify gene expression, fluorescence real-time PCR was executed using the SYBR Green qPCR kit (Kapa Biosystems). Primer pairs for real-time PCR were designed and manufactured by Bioneer. The unique primer sequences of the target genes were as follows: β-actin (sense: 5′-TCA CCC ACA CTG TGC CCA TCT ACG A-3′, antisense: 5′-GGA TGC CAC AGG ATT CCA TAC CCA-3′), Nox1 (sense: 5′-AGG TCG TGA TTA CCA AGG TTG TC -3′, antisense: 5′-AAG CCT CGC TTC CTC ATC TG -3′), Nox2 (sense: 5′-AGC TAT GAG GTG GTG ATG TTA GTG G -3′, antisense: 5′-CAC AAT ATT TGT ACC AGA CAG ACT TGA G -3′), Nox4 (sense: 5′-CCC AAG TTC CAA GCT CAT TTC C -3′, antisense: 5′-TGG TGA CAG GTT TGT TGC TCC T -3′), Duox1 (sense: 5′-TAC ATC AGC CAG GAG AAG ATC TGC -3′, antisense: 5′-TGC GTT GAA ACT TCT CTC GGT GTG -3′), and Duox2 (Sense: 5′-TCC ATT AGT GAG TCT GAT TGT C-3′, antisense: 5′-GTT TGT CAA GGA CCT GCA GAC T-3′). Real-time PCR was performed using the PE Biosystems ABI PRISM® 7300 sequence detection system. The thermocycling parameters were 50°C for 2 min and 95°C for 20 s, followed by 40 cycles of 95°C for 3 s and 60°C for 30 s. All reactions were performed in triplicate. The relative quantity of mRNA was determined using the comparative threshold method, and the results were normalized against β-actin as an endogenous control.
Intracellular ROS assay by DCF-DA or DHA
After exposing the confluent cells to hyperoxia, the cells were washed with Hanks' balanced salt solution (HBSS) and incubated for 10 min at 37°C in an incubator in the dark in HBSS containing 20 μM 2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA) (Molecular Probes) to detect intracellular hydrogen peroxide and 10 μM DHE (Sigma) to detect intracellular superoxide, respectively. Inside the cell, DCF-DA is converted into 2′7′- DCF by intracellular esterases and then oxidized to highly fluorescent 2′7′-DCF by reacting with hydrogen peroxide and DHE is converted to 2-hydroxyethidium (2-OH-E+) or a precursor by reacting with superoxide. The cells were washed with 1 ml of HBSS at one time to remove extracellular ROS. Culture dishes filled with 1 ml of fresh HBSS were examined with a Zeiss LSM 700 laser confocal microscope that was equipped with 10× and 20× objectives. DCF fluorescence was measured at an excitation wavelength of 488 nm and an emission wavelength of 515–540 nm.
DHE fluorescence was measured at an excitation wavelength of 488 nm and an emission wavelength of 585 nm. The fluorescences were visualized with an image size of 512×512 pixels, getting an average of 8 image frames with fast scanning speed. The mean fluorescence intensity of DCF and DHE was assessed by selecting randomly at least six fields of each dish using ZEN 2012 (Carl zeiss) software.
MLE12 cells were treated with BAPTA AM (10 μM) for 30 min and exposed to hyperoxia, and BAPTA-AM (30 μM) was treated to ATII cells for 30 min, in addition, just before measuring ROS.
Flow cytometry
Cells were trypsinized, washed with HBSS containing 1% FBS, and stained with anti-SP-C and anti-AQP5 antibodies for 30 min at room temperature. After washing with HBSS containing 1% FBS, cells were incubated with Alexa647-conjugated anti-rabbit IgG and Alexa488-conjugated anti-goat IgG for 30 min at room temperature. After washing twice with HBSS containing 1% FBS, flow cytometry was performed. To detect apoptosis, cells were stained with Annexin V-FITC and active caspase3-FITC using Annexin V FITC apoptosis kit and caspase3 active form apoptosis kit, respectively. Data were acquired and analyzed with a BD LSRII flow cytometer (BD biosciences).
Statistical analyses
Results are presented as the mean of three determinations±SEM. Wilcoxon rank-sum test was performed to determine differences between groups using R version 3.0.1 program (The R foundation for statistical computing, Vienna, Austria). p-values less than 0.05 were considered statistically significant.
Footnotes
Acknowledgments
The authors thank Yonsi-Carl Zeiss Advanced Imaging Center, Yonsei University College of Medicine, for technical assistance. They thank D.S. Jang for his excellent support with medical illustration. This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2013R1A1A200), by the Yonsei University College of Medicine (6-2012-0137 to J.-H.R.), by an NRF grant funded by the Korean government (MSIP) (No. 2007-0056092), and by an NRF grant funded by the Ministry of Science, ICT & Future Planning (2012M3A9C5048709), (NRF-2013M3A9D5072551).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.