
Abstract
Introduction
I
Fe and Heme Homeostasis
Fe homeostasis is regulated by a series of integrated mechanisms acting at cellular and systemic levels (78). Disruption of Fe homeostasis is often associated with defective erythropoiesis and/or anemia as well as with tissue Fe overload, tissue damage, and disease. As such, mechanisms that control Fe homeostasis are critical to prevent these pathological outcomes (57, 66).
Cellular Fe homeostasis is controlled by several evolutionary conserved genes that act in a concerted manner to regulate intracellular Fe uptake, trafficking, and export (6, 78) (Fig. 1). Mammalian cells acquire Fe bound to transferrin (TF), a plasma homodimeric beta-globulin that binds two ferric Fe molecules with exceedingly high affinity (Kd=10−20 M) (3). Soluble TF-Fe complexes are recognized by transferrin receptor 1 (TFR1) and 2 (TFR2) (81), two transmembrane disulfide-linked glycoproteins encoded by distinct genes sharing 45% homology (90) (Fig. 1). The affinity of TFR2 for diferric TF is 30 times lower to that of TFR1 (90), suggesting that the contribution of TFR2 to intracellular Fe uptake is not as critical compared to TFR1. Cubilin is another TF-Fe receptor expressed in more restricted cellular subsets, including small intestine epithelial cells, renal proximal tubule cells, visceral yolk sac cells, and placenta cytotrophoblasts (Fig. 1) (38).
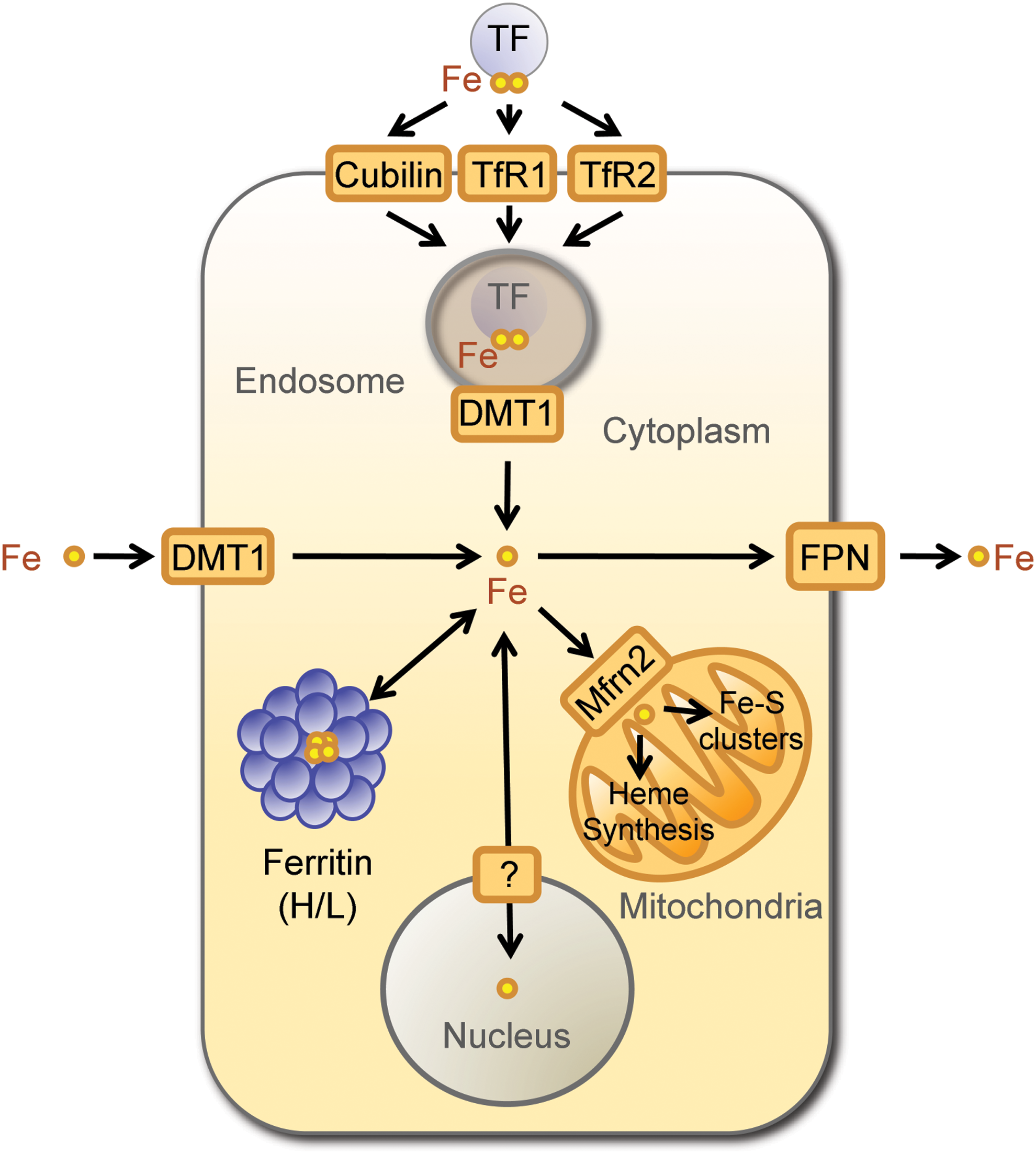
Fe is extracted from TFR1/TF-Fe or TFR2/TF-Fe complexes through acidification of the endolysosomal compartment, leading to conformational changes in the tertiary structure of TFR1/2. This decreases the affinity for TF-Fe and allows for Fe release from TF (81). Endosomal Fe3+ is reduced into Fe2+ by metalloreductases, including the six-transmembrane epithelial antigen of the prostate family member 3 (STEAP-3) (121) and transported into the cytoplasm by the Fe2+ regulated divalent metal transporter 1 (DMT1) (68). Cytoplasmic Fe2+ can be shuttled to the mitochondria, either bound to chaperone proteins or via interorganelle interactions (139), being internalized by the mitochondria via mitoferrins, of which mitoferrin 2 is ubiquitously expressed (124). In the mitochondria, Fe is used essentially for heme biosynthesis and Fe-sulfur cluster assembly (139) (Fig. 1). Alternatively, Fe can be stored in the mitochondria, by mitochondrial ferritin (105).
When intracellular labile Fe increases above a certain threshold level, its pro-oxidant activity must be controlled to avoid cytotoxicity. This is achieved, in large measure, via a mechanism regulated by iron regulatory proteins (IRPs) 1 and 2 (IRP1/2) (79). Both IRPs act as Fe sensors and bind to short conserved cis-regulatory stem-loop iron responsive elements (IRE) in the untranslated mRNA regions (UTR) of genes, where expression is regulated by IRPs (12, 76, 77). Binding of IRPs to IRE located in the 3′UTR increases mRNA stability and promotes translation (71), whereas translation is repressed upon binding of IRPs to IRE located in the 5′UTR (44, 72, 181).
Ferritin H (heart/heavy) chain (FTH) is a prototypical gene regulated by IRPs, encoding a 21 kDa protein that catalyzes the conversion of Fe2+ into ferric Fe3+ and allowing for intracellular storage of inert Fe3+ (72) (Fig. 1). The FTH gene contains one 5′UTR IRE, recognized under low cellular Fe content by IRP1/2, repressing FTH translation (71). Although TFR1 is also a gene regulated by IRPs, it contains several IRE in its 3′UTR that are recognized under low Fe by IRP1/2, promoting its translation (71). This is not the case for TFR2, where expression is not regulated by Fe (56), consistent with the lack of IRE in TFR2 mRNA (56). Another gene regulated by IRPs is ferroportin (FPN), which encodes a 62 kDa transmembrane Fe transporter that exports Fe2+ from cells (45) (Fig. 1). The FPN gene has a 5′UTR IRE regulated by IRPs that represses its translation under low cellular Fe (79). The regulation of FTH, TFR1, and FPN by Fe, sensed by IRPs, allows for rapid adaptation to changes in intracellular Fe content. Regulated expression of FTH appears to be of particular importance for cellular adaptation to Fe overload in the context of inflammation and immunity, as discussed in the next sections (66, 141, 167, 168, 182).
Ferritins are multimeric complexes (∼450 kDa) made of FTH and FTL (liver/light) chains (72). These form heteropolymeric nanocage-like structures comprising of 24 subunits, which assemble as a hollow shell providing a central 80-Å diameter storage cavity that can incorporate up to 4500 Fe atoms (72), in the form of inorganic ferrihydrite aggregates (94). The proportion of the two FTH and FTL subunits composing ferritin multimeric complexes is regulated in a tissue-specific manner with FTL-rich nanocages being produced mainly by hepatocytes and FTH-rich nanocages by the brain and muscle cells (72). Proportion of FTH and FTL subunits is also regulated in response to inflammation, with proinflammatory cytokines inducing FTH expression and hence enriching its content in ferritin nanocages (141, 168, 182). This creates several isoferritins with different Fe storage capacity as well as other physiological properties (72).
FTH has ferroxidase activity (28, 80), which catalyzes the reaction: 4Fe2++4 H++O2=4Fe3++2H2O, converting reactive Fe2+ into Fe3+ and forming inert ferrihydrite aggregates that do not partake in the production of free radicals via the Fenton chemistry (9, 50). The ferroxidase activity of FTH is provided by a core of evolutionary conserved amino acids, that is, Glu-27, Glu-62, and His-65 (80, 103). These are absent from FTL, which does not have ferroxidase activity (8). FTL, however, is essential to promote Fe nucleation by ferritin nanocages, a process leading to the formation of inorganic ferrihydrite aggregates (107). FTL also confers stability to multimeric ferritin, protecting the nanocage from elevated temperatures and denaturants (145). While FTL can form homopolymers (106), this is not the case for FTH, which promote aggregation and degradation of ferritin nanocage (65).
As mentioned above, FTH and FTL are prototypical Fe-dependent genes, which expression is regulated essentially at post-transcriptional level by the dissociation of IRP1/2 from mRNA 3′UTR IREs (71). In response to high cellular Fe content, IRP1/2 are degraded via the 26S proteasome pathway (71, 86) promoting mRNA stability and hence FTH and FTL translation. Physiological modulators of FTH translation, via the IRP/IRE system, include nitric oxide (NO) as well as reactive oxygen species and hypoxia (77). Expression of FTH is also regulated at the transcriptional level via the activation of the transcription factors nuclear factor kappa B (NF-κB) (100, 127) and NF-E2-related factor 2 (NRF2) (129). More recently, FTH expression was show to be post-transcriptionally regulated by miR-200b, which reduces its expression (151).
Presumably, when the levels of cellular Fe are reduced, redistribution of the Fe sequestered inside the ferritin nanocages occurs through a mechanism regulated at least partially by FTH and FTL proteolytic degradation (10). The mechanisms involved in Fe release from ferritins are not well defined, but are thought to involve cysteine and/or serine proteases acting in acidic or autophagic lysosomes (11, 99, 166). Additional mechanisms involved in ferritin disassembly and Fe release from ferritin include FTH ubiquitin-dependent 26S proteasomal degradation (117, 143), a notion in keeping with our own observation that pharmacological inhibition of the 26s proteasome increases FTH expression in cells exposed in vitro to heme plus tumor necrosis factor (TNF) (Raffaella Gozzelino, unpublished).
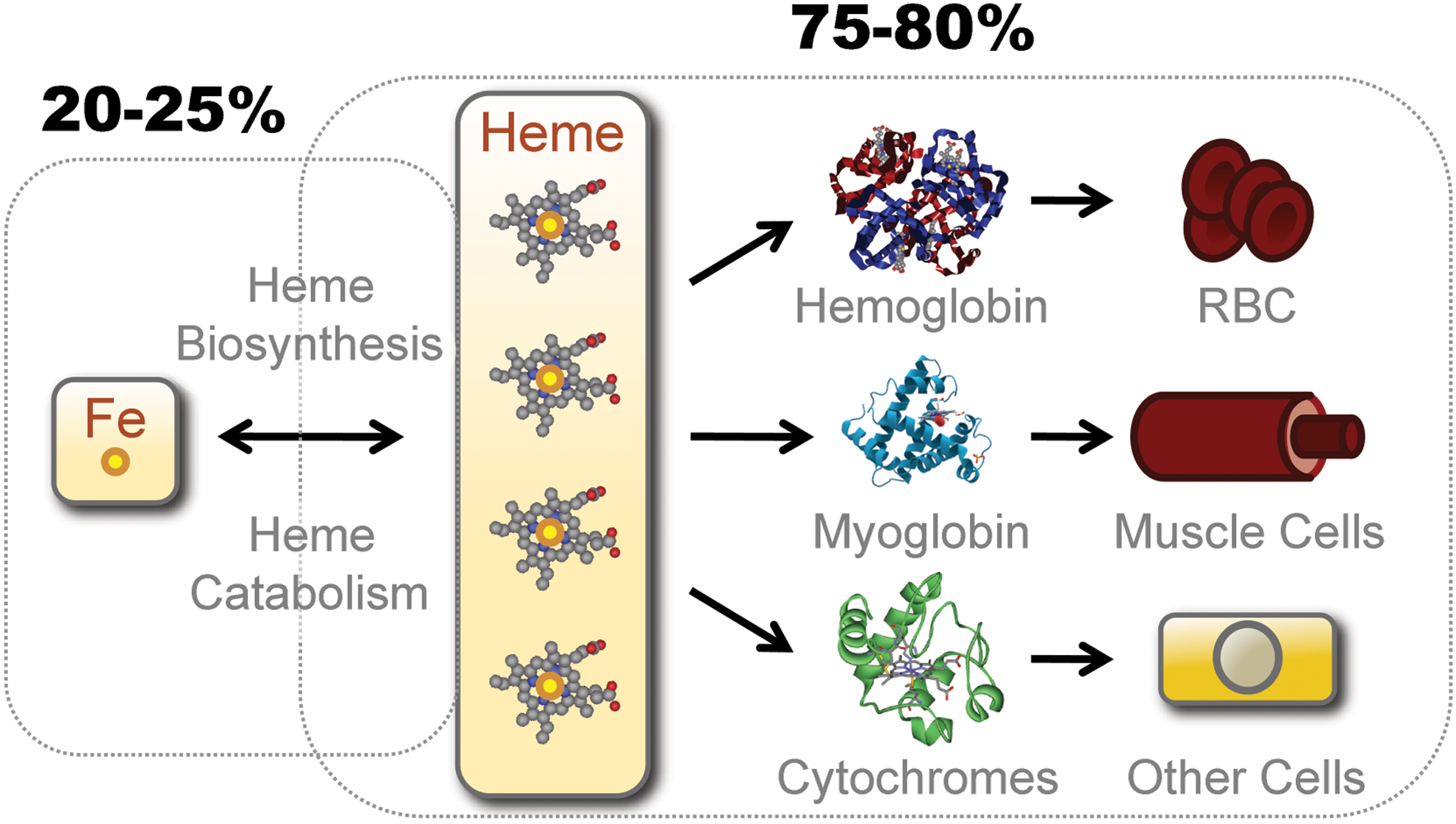
More than 80% of bioavailable Fe in mammals is contained within the hydrophobic methane-bridged tetrapyrrole ring of heme, where it is used within the prosthetic groups of hemoproteins (Fig. 2). The most abundant hemoproteins in mammals are hemoglobin (Hb) and myoglobin, expressed in red blood cells (RBCs) and muscle cells, respectively (67) (Fig. 2). Another significant pool of hemoproteins is formed by ubiquitously expressed cytochromes (67) (Fig. 2).
Under conditions of oxidative stress, noncovalently bound heme can be released from hemoproteins, as demonstrated for Hb (32, 123). The redox activity of nonhemoprotein-bound heme, referred hereby as “free heme,” is no longer controlled by the heme pockets of hemoproteins, and thus, free heme becomes pro-oxidant and cytotoxic (66, 67, 102, 148). This deleterious effect is restrained by two proteins expressed at high levels in human plasma, namely haptoglobin (36–195 mg/dL) and hemopexin (40–150 mg/dL). Binding of haptoglobin to cell-free Hb (Kd≈10−15) prevents heme release from Hb, with the resulting heterodimeric complex being recognized by the macrophage (Mø) transmembrane receptor CD163 (98). Hemopexin scavenges free heme (Kd<10−12 M in humans), forming a heterodimeric complex recognized by the macrophage transmembrane low-density lipoprotein (LDL) receptor-related protein 1 (LRP1/CD91) (82) (Fig. 3). Cellular Hb uptake can also occur irrespectively of haptoglobin or hemopexin via engulfment of senescent RBC by hemophagocytic Mø in the spleen (88).
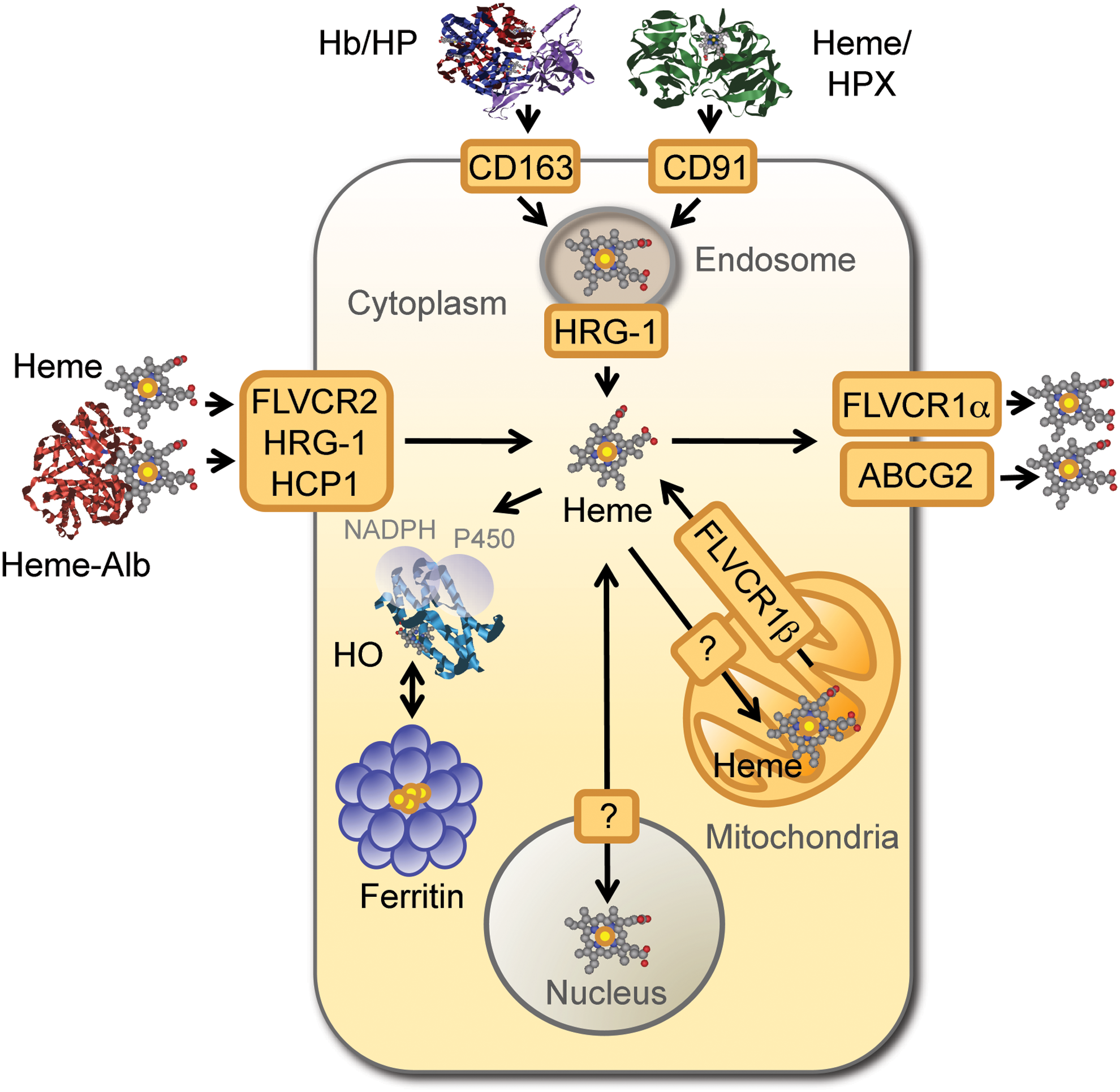
In a similar manner to TF-Fe, cellular uptake of haptoglobin/Hb and hemopexin/heme complexes converges at the endolysosomal compartment, where heme is extracted (82, 98) (Fig. 3). Subsequent transport of heme into the cytoplasm is mediated by the heme transporter heme responsive gene-1 (HRG-1) (137), as demonstrated in hemophagocytic Mø (184) (Fig. 3). Hrg-1 gene deletion impairs erythroid development, as demonstrated in zebrafish (137) and attenuates heme transport from the phagolysosomal compartment of mouse erythrophagocytic Mø (184), illustrating the nonredundant role of these heme transporter in heme/Fe homeostasis (184).
Presumably, most of the cytoplasmic heme transits to the mitochondria via a mechanism involving specific heme chaperones and mitochondrial transporters (189). Several other genes are known to control the fate of cytoplasmic heme, including HO, which catabolize heme degradation (165). There are two HO isoforms, namely HO-1 (≈32 kDa) and HO-2 (≈36 kDa), encoded by the HMOX1 and HMOX2 genes respectively (25, 113, 171). HO-2 is constitutively expressed in most cells (171), whereas excess intracellular heme induces the expression of the stress-responsive HO-1 isoform, which is constitutively expressed by hemophagocytic Mø as well as by natural regulatory T cells (29, 190).
Intracellular heme is exported from cells via the feline leukemia virus C receptor 1 (FLVCR1) (91) (Fig. 3). There are two FLVCR1 isoforms, namely FLCVR1α (≈60 kDa) and FLVCR1β (≈28 kDa), generated via alternative splicing of the same FLCVR gene (35, 91). FLCVR1α drives heme extracellular transport, whereas FLVCR1β mediates mitochondria heme export (35). At least two other ABC transporters can regulate intracellular heme trafficking, namely ABCG2 (≈72 kDa), which promotes heme extracellular transport (96) and ABCB6 (≈94 kDa), which is involved in intracellular heme import (92, 97) (Fig. 3). The heme carrier protein 1 (HCP1) is another putative heme transporter, probably involved in intracellular heme import (149).
A functional system ensuring heme transport and catabolism is essential to sustain systemic Fe homeostasis, as demonstrated unequivocally for HO-1 deficiency in mice (131, 132) and humans (188). In both cases, the lack of HO-1 expression is associated with impaired Fe recycling, anemia, vascular damage, and depletion of hemophagocytic Mø (95), presumably driven by heme cytotoxicity (59). Moreover, HO-1 deficiency is also associated with increased expression of FLVCR in the kidneys (160), suggesting that mechanisms regulating heme catabolism and transport cross-regulate each other to maintain Fe homeostasis. Flvcr gene deficiency is lethal in mice, due to defective erythropoiesis and severe macrocytic anemia (91). This is probably owed to the lack of mitochondrial FLVCR1 expression, given that specific deletion of the Flvcr1α gene variant is dispensable for erythropoiesis, although necessary to prevent edema and hemorrhage (35). FLVCR1 mutations in humans are associated with the development of posterior column ataxia and retinitis pigmentosa (136), whereas FLVCR2 mutations have been associated to proliferative brain vasculopathy, such as observed in the Fowler syndrome (47). Conversely, mutations in ABCG2 and ABCB6 do not impair erythroid differentiation, suggesting a redundant role for these transporters in erythropoiesis (75, 144, 189).
Systemic Fe homeostasis relies on continuous Fe intake from diet (Fig. 4), that is, average of 1–2 mg of Fe per day in humans (70). Fe is absorbed in the form of Fe3+, which is then reduced by cytochrome B reductase (Dcytb) expressed in gut epithelial cells, that is, enterocytes (114). This produces Fe2+, which is transported into the cytoplasm by DMT1 (68) and exported from enterocytes by FPN (45) (Fig. 1). Expression of FTH in enterocytes is required to sustain this process (176) (Fig. 1). Extracellular Fe2+ is delivered to TF and consequently to TFR1-expressing erythroblasts (81), where it is used in heme biosynthesis and loaded into nascent Hb in the process of erythropoiesis (Fig. 4). Dietary Fe can also be extracted from heme, which involves most probably the catabolism of heme by HO, allowing for Fe extraction and transport.
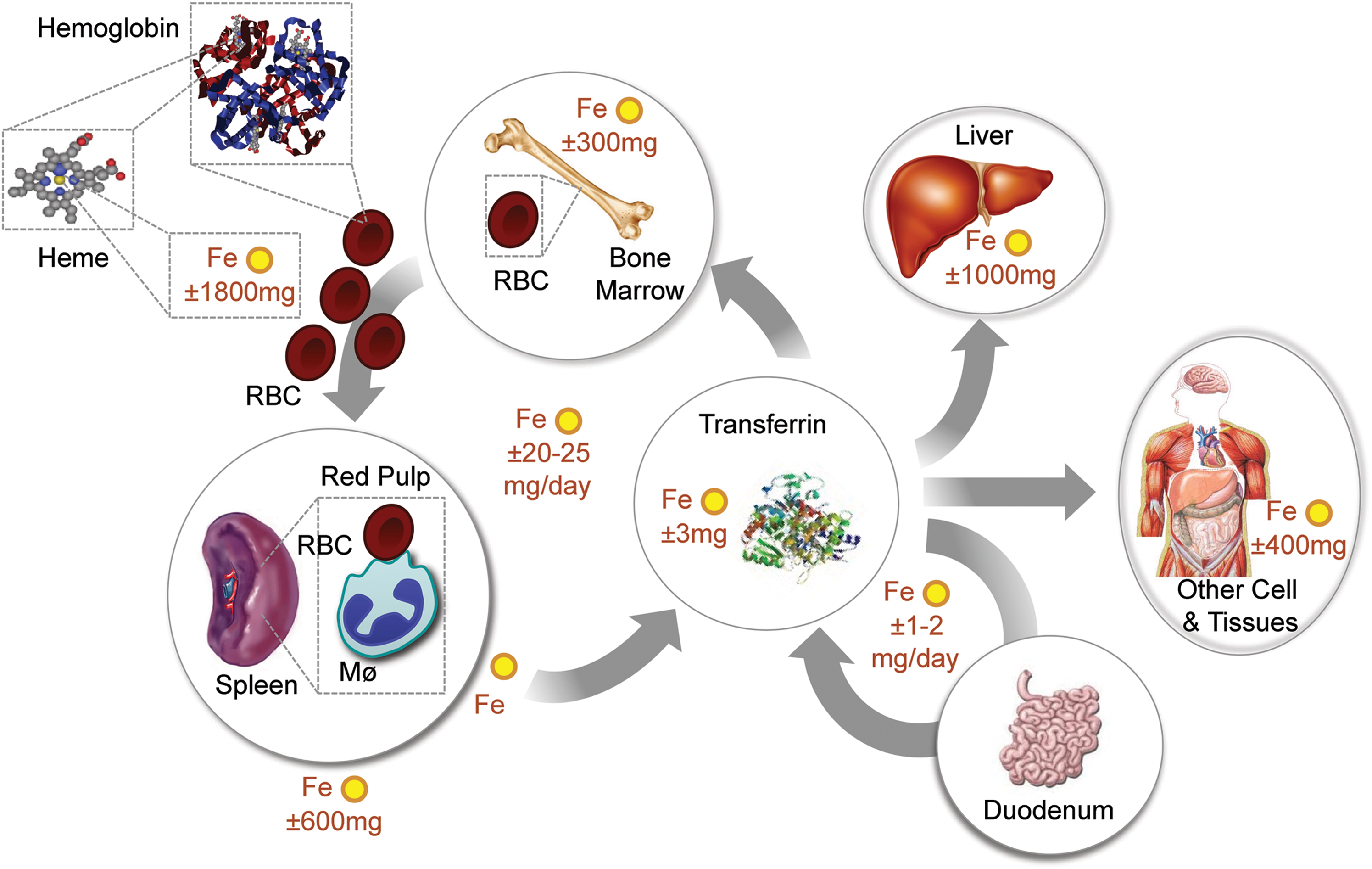
Recycling of the Fe extracted from the prosthetic heme groups of Hb in senescent RBCs is essential to maintain Fe homeostasis (Fig. 4). This is achieved by hemophagocytic Mø in the red pulp of the spleen, via a mechanism involving the heme transporters HRG-1 (137) and presumably FLVCR2 (189). These deliver heme from the phagocytic endosomes or plasma membranes, respectively, into the cytoplasm allowing its catabolism by HO-1, which extracts the Fe that is then exported by FPN (45). The Fe captured by TF in plasma is incorporated into TFR1-expressing erythroblasts, sustaining erythropoiesis (81) (Fig. 4). Expression of FTH by hemophagocytic Mø is most probably not required to this process, as suggested by the deletion of the mouse Fth allele specifically Mø (data not shown) (66).
Fe concentration in plasma is regulated by hepcidin (62), an acute-phase protein produced mainly by hepatocytes (46, 64). Hepcidin is also produced by innate immune cells, for example, neutrophils and Mø (126). This occurs in response to pathogen recognition by pattern recognition receptors (PRR), for example, toll-like receptor (TLR) 2 or 4, or in response to cytokines, for example interleukin-6 (IL-6), IL-22, and type I interferon or IL-1β (46, 64, 126). Hepcidin expression occurs via a mechanism regulated essentially at transcriptional level by the transcription factor signal transducer and activator of transcription 3 (STAT3) (46) as well as by SMAD proteins (64, 138), adjusting the requirement of Fe for erythropoiesis (122).
Hepcidin binds to and triggers FPN degradation (119), thus suppressing cellular Fe export and dietary Fe uptake, while retaining intracellular labile Fe systemically (119). When the levels of hepcidin in plasma increase above a certain threshold level, such as observed during inflammatory and immune reactions, cellular Fe export by FPN is no longer an available option to reduce cellular Fe overload (46, 61, 183). Presumably, this explains the central role played by FTH in preventing the deleterious effects of cellular Fe overload in the context of inflammation and immunity (66). The role of hepcidin in the control of Fe homeostasis will not be discussed in further detail hereby, and the reader is kindly directed to recent reviews on the subject (46, 55, 63, 64, 128).
Heme Cytotoxicity
Inflammation and immunity are associated with the production of free radicals, aimed at destroying evading pathogens and providing host resistance to infections (116). The evolutionary trade-off of this defense strategy is that free radicals can lead to oxidative stress in host tissues and eventually cause tissue damage and disease (116). Under oxidative stress, noncovalently bound heme can be released from hemoproteins, producing free heme (32, 66, 123) (Fig. 5). This phenomenon is well illustrated for Hb, which can donate its prosthetic heme groups to albumin or hemopexin (32) as well as to LDL (15, 87). Many factors associated with inflammation and immunity can act directly or indirectly to promote varying degrees of RBC lyses and as such the generation of cell-free Hb and the subsequent release of its prosthetic heme groups (Fig. 5). Moreover, other hemoproteins containing noncovalently bound prosthetic heme groups, such as myoglobin, can probably act in a similar manner, although this remains to be established.
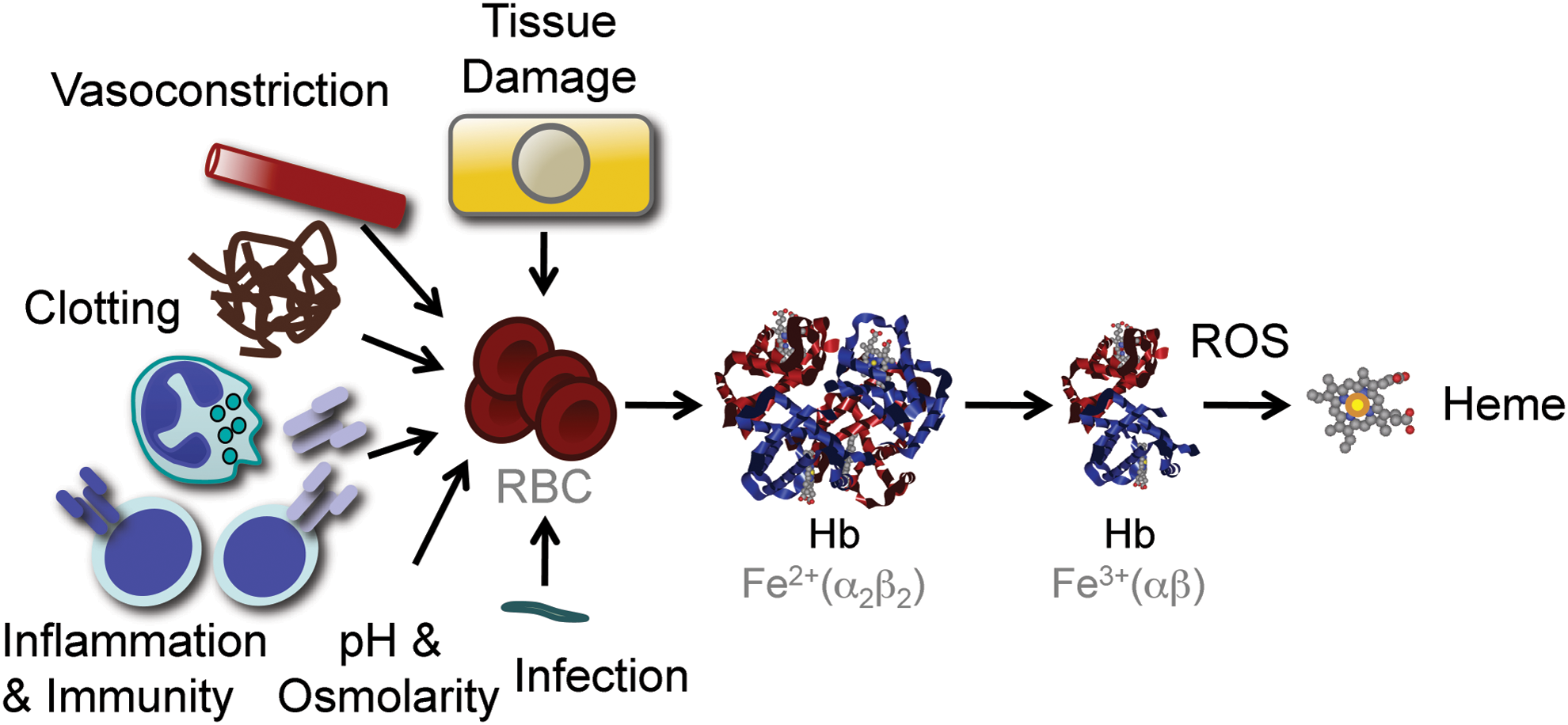
While not cytotoxic per se, free heme can sensitize nonhematopoietic cells to undergo programmed cell death in response to proinflammatory agonists, such as demonstrated for TNF (148), among others (67, 102) (Fig. 6). This deleterious effect is driven by Fe (66), although it is not clear if Fe must be released from heme to become cytotoxic or whether cytotoxicity is exerted by Fe within the context of the protoporphyrin ring of heme or both (67).
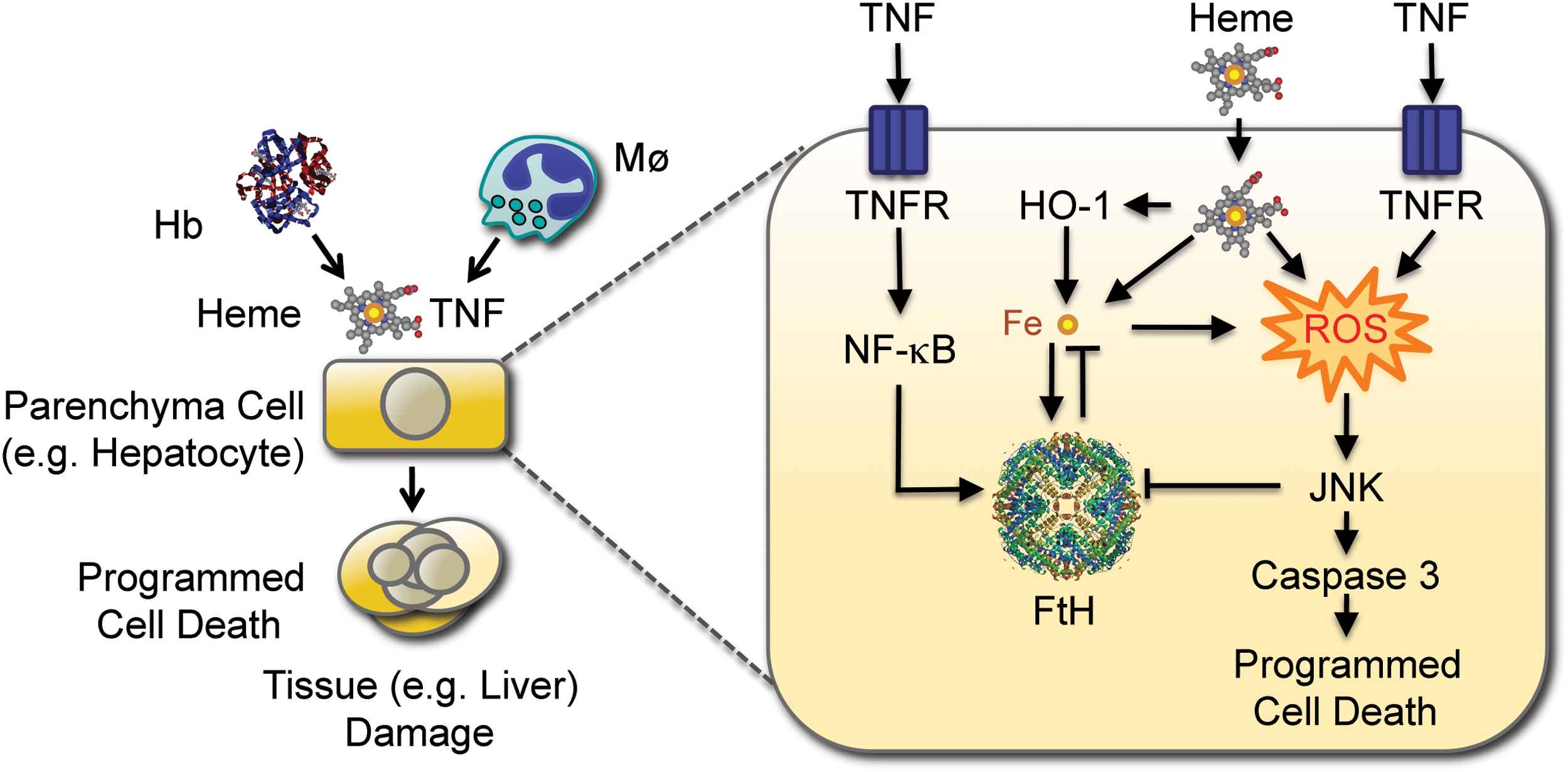
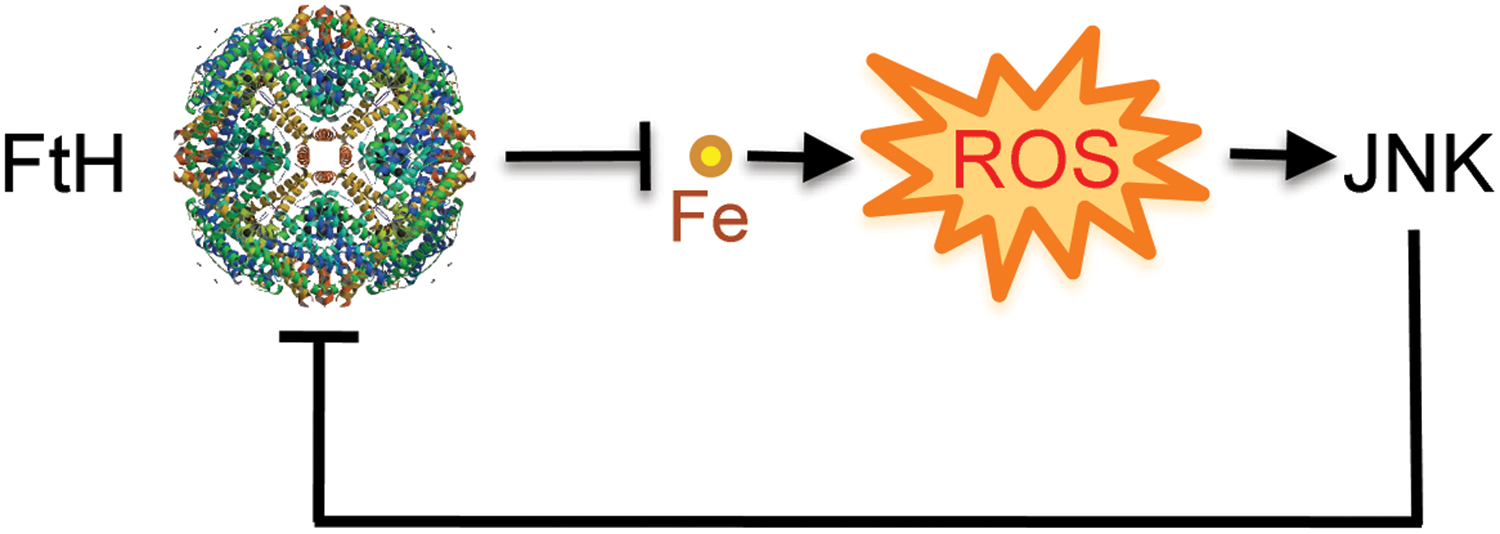
The mechanism underlying Fe-heme-mediated cytotoxicity involves the sustained activation of the c-Jun N-terminal kinase (JNK) signaling transduction pathway (66). This is in keeping with the notion that JNK activation plays a central role in the mechanism via which cytotoxic agonists associated with the generation of free radicals induce programmed cell death, as illustrated for TNF (111) and UV radiation (170), among others (110). However, under pathophysiological conditions, TNF cytotoxicity is prevented via a mechanism involving the activation of NF-κB, a transcription factor that uncouples inflammation from programmed cell death (20). The cytoprotective action of NF-κB is exerted via the expression of the so-called protective genes (13), which include manganese superoxide dismutase (186), A20 (40, 51, 104), GADD45 (43, 125), and FTH (127), all of which control cellular accumulation of free radicals in response to TNF (89). The antioxidant effect of these NF-κB-dependent genes restrains JNK activation and as such the induction of programmed cell death in response to TNF (30, 164). The mechanism underlying this cytoprotective effect involves the activation of redox-sensitive phosphatases inhibiting JNK activation, via the conversion of their catalytic cysteine residues to sulfenic acid upon exposure to free radicals (89, 127). This explains why restraining the participation of Fe in the Fenton chemistry prevents sustained activation of JNK and programmed cell death in response to TNF (21, 66, 127) (Fig. 6).
The cytoprotective effect exerted by NF-κB-dependent genes and in particular by FTH was so far only observed under nonphysiological conditions, such as when specific components of the NF-κB family of transcription factors and/or when NF-κB-responsive genes are deleted in vitro (127). Our recent findings that the pro-oxidant effect of intracellular heme can bypass this protective pathway, sustaining JNK activation and triggering programmed cell death (66), provides evidence for a pathophysiological relevance for this phenomenon. This cytotoxic effect of free heme is strictly dependent on the accumulation of free radicals (66), most likely produced by the mitochondria. These observations provide a physiological context in support of the notion that tissue Fe overload can drive the oxidative activation of JNK and promote programmed cell death (66), a deleterious effect that plays a central role in the pathogenesis of inflammatory diseases as illustrated for severe malaria (52, 102, 148) as well as for severe sepsis (102) (Fig. 6).
Heme sensitization to TNF-mediated programmed cell death is partially suppressed by pharmacological inhibition of caspase-3 (66, 101, 148), indicating that heme sensitizes cells to undergo a caspase-dependent form of programmed cell death, that is, apoptosis (49). However, heme sensitization to TNF-mediated programmed cell death is also suppressed by pharmacological inhibition of the receptor interacting serine/threonine kinases 1 (RIPK1) as well as by deletion of the Rip3 gene (Raffaella Gozzelino and Ana Ribeiro unpublished), suggesting that heme sensitizes cells to undergo programmed cell death by necrosis, that is, necroptosis (175). This is in keeping with the recent finding that heme can signal via TLR-4 to induce TNF-mediated necroptosis in Mø (59) as well as with the notion that sustained JNK activation shifts TNF-mediated programmed cell death from apoptosis to necrosis (175, 177). The cytotoxic effect of free heme and in particular its ability to trigger necroptosis may be particularly relevant in the context of intracellular pathogens, as illustrated for Mycobacterium tuberculosis (140, 153). We suggest that heme sensitization to TNF-mediated programmed cell death acts in a rather peculiar way, in that it can probably drive, simultaneously, the activation of two antagonistic cytotoxic signal transduction pathways, that is, apoptosis and necroptosis. The mechanisms via which this occurs remain to be elucidated and are currently subjects of our studies.
Cytoprotective Effect of Heme Catabolism by HO-1
Although heme oxidation can release Fe from its protoporphyrin ring (18), this process is strongly catalyzed by HO (165). Of particular relevance to this process is the stress-responsive HO-1 isoform whose expression is regulated essentially at the transcriptional level via several stress-responsive transcription factors (36), including NRF2 (5). Under steady-state conditions, NRF2 is constitutively targeted for proteolytic degradation by the 26s proteasomal pathway, driven by its binding to the Kelch-like ECH-associated protein (Keap1) (84). Keap1 has several redox-sensitive cysteine residues that form disulfide bounds when exposed to free radicals, altering its tertiary structure and disrupting its interaction with NRF2 (85, 93). While this allows for NRF2 nuclear translocation, it is not sufficient per se to promote the transcription of the HMOX1 gene encoding HO-1 (84). The reason for this is that HMOX1 transcription is constitutively repressed by the binding of BRCA-1 associated carboxy C-terminal helicase (BACH-1) to DNA Maf responsive elements (MARE) present in its promoter (163). BACH-1, however, is a heme sensor targeted for 26s proteosomal degradation upon cognate heme binding (5, 83). This releases the MARE in the HMOX1 promoter and allows nuclear NRF2 to drive HMOX1 transcription (5, 83). Although NRF2 appears to play a central role in the transcriptional regulation of the HMOX1 gene, several other stress-responsive transcription factors also contribute to the regulation of HO-1 expression, as reviewed elsewhere (36, 67). More recently, HO-1 expression was also shown to be under the control of several micro-RNAs, including miR-155 and miR122, which promote HO-1 expression by inhibiting BACH-1 (133, 134), whereas miR-217, miR-377, and miR-378 inhibit HMOX1 translation via direct interaction with the 3′UTR in HMOX1 mRNA (19, 154).
HO-1 confers cytoprotection to different forms of programmed cell death (157, 179), including apoptosis and/or necroptosis driven by heme and TNF (67, 102, 148). This cytoprotective effect is driven by heme degradation per se as well as by its end products, including the gasotransmitter CO (27) and the antioxidants biliverdin/bilirubin (147). CO can exert cytoprotective effects via the modulation of cellular signal transduction pathways, including the p38 mitogen activating protein kinase (MAPK) (26, 27, 152, 159). In addition, CO can bind Fe in the heme pockets of hemoproteins, inhibiting heme release and preventing the its cytotoxic effects, as illustrated for Hb, to which CO binds avidly to suppress its oxidation as well as heme release (73, 123). While the conversion of biliverdin into bilirubin, catalyzed by biliverdin reductase, may contribute to the antioxidant and cytoprotective effect of heme catabolism by HO-1 (147), this is probably not sufficient to per se to override the pro-oxidant effect of the labile Fe produced via heme catabolism. It is also possible that the cytoprotective effect of HO-1 is mediated to some extent, in the absence of its catalytic activity, via regulation of gene transcription consequent to C-terminal cleavage and nuclear translocation (39, 109).
We have argued that cytoprotective effects of HO-1 contribute critically to its salutary effects exerted in a variety of immune-mediated inflammatory diseases (156, 158), including the rejection of transplanted organs (155), autoimmune diseases (37), subclinical recurrent abortions (191), or infectious diseases, such as severe malaria (52, 123, 148) and sepsis (102). In the context of systemic infections, induction of HO-1 expression by the infected host provides protection against the cytotoxic effects of free heme released from Hb (52, 67, 123, 148). This cytoprotective effect is essential to support host survival and yet it does not appear to exert a negative impact on the pathogen, a phenomenon referred to as disease tolerance (116).
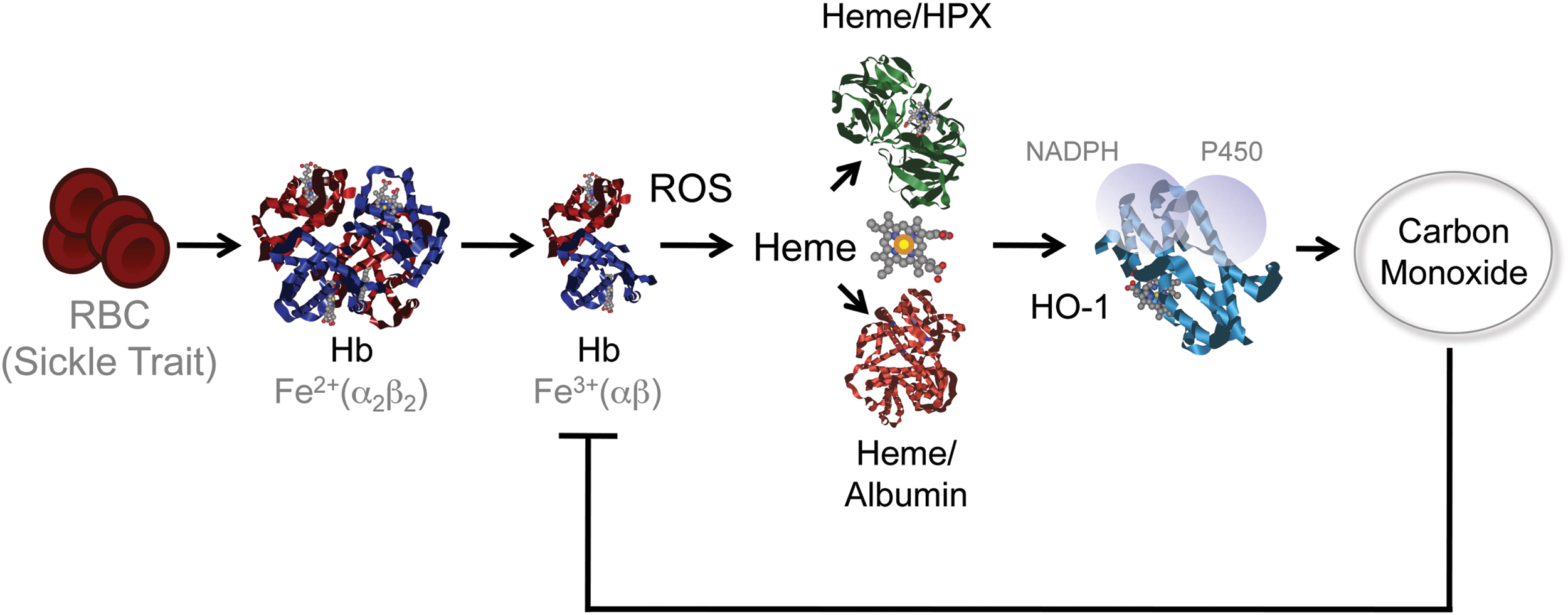
The relative importance of this interplay, between heme and HO-1, to the pathogenesis of severe forms of malaria is strongly supported by the finding that sickle Hb, a mutation in the beta chain Hb that confers protection against Plasmodium infection in human populations (31), does so via a mechanism that relies on the induction of HO-1 expression, through the activation of the transcription factor NRF2 (Fig. 7) (53, 142). Moreover, the production of the gasotransmitter CO, such as induced by sickle Hb, prevents heme release from Hb, conferring protection against severe forms of malaria (53, 123, 142). This protective effect acts irrespectively of pathogen load, and as such is said to confer disease tolerance to malaria (53, 116) (Fig. 7).
Fe, Heme, and Infection
Microorganisms, including invading pathogens, are sensed by host PRR, which trigger inflammatory responses aimed at restricting microbial growth and when necessary achieve their clearance, while limiting tissue damage (115). Through the perspective of a microbial organism, a successful infection depends strictly on its capacity to divert components of host metabolism into its own metabolic pathways (88). One of the strategies via which inflammatory responses limit the growth of invading pathogens is through the deployment of mechanisms limiting microbial access to key metabolic components such as Fe, which are essential for microbial growth (33, 183). These host defense mechanisms act in a concerted manner to provide systemic (i) neutralization of circulating Fe, (ii) inhibition of cellular Fe delivery, and (iii) intracellular Fe retention (7).
Systemic neutralization of circulating Fe in response to infection is mediated via a mechanism involving lactoferrin, a potent soluble Fe chelator secreted by activated neutrophils, which limits Fe availability to pathogens while exerting antimicrobial activity (24, 173, 174, 180). Pathogens evolved strategies to counter this host defense mechanism, via the expression of siderophores, that is, low-molecular-weight Fe-binding complexes that can extract Fe from lactoferrin as well as from TF (120). These virulence factors are themselves countered by siderocalin/lipocalin-2, a host acute-phase Fe-binding protein that sequesters siderophores and decreases susceptibility to infection (58). Pathogens also evolved mechanisms to circumvent this host defense strategy, including the production of stealth siderophores, precluding siderocalin binding (1, 135).
Expression of natural resistance-associated Mø protein 1 (Nramp-1) in late phagolysosomes is another host defense strategy that reduces Fe availability to intracellular bacteria (34, 88, 172, 178), conferring resistance to infection (60). Moreover, Nramp-1 modulates immune responses to microbial organisms, regulating cytokine production and recruitment of phagocytic cells to the site of the infection (60).
Pathogens can also acquire Fe from host heme, using HO-1 homologues, including ChuS (161), HemO (192), HugZ (69), and HmuO (185), for Fe extraction. While essential to the establishment of host–microbe interaction, the strategies employed by microbes to acquire Fe from the host and those used by the host to starve microbes from Fe will not be reviewed in further detail hereby as these have been recently reviewed elsewhere (33).
Systemic intracellular Fe retention by the infected host is achieved through the action of hepcidin, via a mechanism involving the phosphorylation, internalization, and subsequent lysosomal degradation of the Fe cellular exporter FPN (46, 63). The trade-off to this host defense strategy is deregulation of host Fe homeostasis with cellular Fe retention interrupting Fe recycling and reducing the levels of circulating Fe available not only to microbes but also to host erythropoiesis (88). In keeping with this notion, high levels of hepcidin in plasma and low levels of FPN are associated with impaired erythropoiesis, anemia, and hypoferrimia (46), as well as with cellular and tissue Fe overload (88). Moreover, systemic inhibition of FPN expression also causes intracellular Fe accumulation leading to oxidative stress and tissue damage (7). It should be noted that deregulated Fe metabolism, such as driven by the sustained production of hepcidin is not specific to infection, but rather to inflammation. As such it can be associated to a variety of immune-mediated inflammatory diseases as well as with cancer progression (162, 169), where it leads to poor prognosis, unfavorable outcome, and metastasis (130).
The deleterious effects of sustained hepcidin expression are exacerbated under inflammatory conditions associated with hemolysis and the generation of cell-free Hb. While cell-free Hb can provide some level of resistance against bacterial pathogens (23), the evolutionary trade-off of this defense mechanism is the release of its prosthetic heme groups (32). This leads to systemic heme loading into host cells, eventually causing oxidative tissue damage and disease (52, 67).
Heme induces the expression of HO-1, which inhibits hepcidin expression via a mechanism mediated by CO (150), presumably restoring FPN expression and hence Fe cellular export. NO can also induce FPN expression (118), contributing to the antimicrobial activity of this gasotransmitter (22, 118), while allowing Fe to recycle as to be used in erythropoiesis. Despite these salvage pathways, high levels of hepcidin still promote the systemic accumulation of intracellular Fe. Moreover, while cytoprotective per se, heme catabolism by HO-1 produces intracellular labile Fe (14, 16, 50, 67, 102, 123, 148), which cannot be readily exported due to inhibition of FPN expression enforced by hepcidin (46). Therefore, under inflammatory conditions, the intracellular pool of labile Fe must be neutralized by the induction of FtH expression (14, 21, 48, 167).
FTH and Infection
Ferritin is an acute-phase protein induced during systemic infections, via a mechanism regulated at both transcriptional and post-transcriptional levels (167) (see Fe and Heme Homeostasis section). FTH expression is regulated at a transcriptional level by NF-κB (100, 127), a transcription factor that plays a central role in the regulation of inflammation and immunity. This argues strongly for the integration of Fe metabolic adaptation as an intrinsic component of inflammation and immunity, presumably contributing to host protection against infection (46, 116). FTH transcription is also regulated by the transcription factor NRF2 (112, 129), suggesting yet another level of integration between cellular adaptation to oxidative stress and Fe metabolic adaptation during infection. The transcription factor hypoxia-inducible factor alpha and heat shock factor 1 also regulate FTH expression in Caenorhabditis elegans (2), arguing for yet a broader level of integration between Fe metabolic adaptation and cellular responses to different forms of stress associated with inflammation and immunity. Whether this level of integration is evolutionary conserved, as to be extrapolated from C. elegans to mammals, remains to be established.
A functional effect of ferritin in the outcome of systemic infections has only recently started to be elucidated. FTH acts via a mechanism involving its ferroxidase activity to provide metabolic adaptation to tissue Fe overload during systemic infections, as demonstrated for malaria (66) and severe sepsis (Sebastian Weiss and Rasmus Larsen, unpublished) in mice. Expression of FTH prevents intracellular Fe accumulation from sustaining JNK activation and hence from sensitizing nonhematopoietic cells to undergo programmed cell death (66). This finding is in line with previous demonstration of a cytoprotective effect of FTH in vitro (14, 41, 127, 187) and in vivo (21). The cytoprotective effect of FTH against heme is apparently at odds with the notion that FTH cannot target Fe inside heme (66). An alternative interpretation, however, is that the cytotoxic effect of heme acts via a mechanism driven by labile Fe released from, which can be targeted by FTH, and not by the Fe contained within its protoporphyrin ring, which cannot be targeted by FTH (67). This is in keeping with the notion that labile Fe fuels the activation of JNK in response to TNF (127), a cytotoxic effect repressed by the ferroxidase activity of FTH (66, 127).
Deletion of the Fth allele in mice promotes tissue Fe overload, oxidative stress and damage in response to systemic infections, despite normal levels of HO-1 expression (67). This suggests that FTH is required to support the salutary effects of HO-1 in the context of systemic infection (148), uncoupling heme catabolism from Fe-driven cytotoxicity (14, 66). Moreover, when HO-1 expression is deleted, induction of FTH expression can be impaired (66), which may explain the high lethality of these mice in response to systemic infections (66, 148). To what extent the protective effect of FTH is required to sustain the salutary effects of HO-1 against other immune-mediated inflammatory diseases remains to be established.
The combination of heme and TNF produced during systemic infections inhibits the expression of FTH via a mechanism that involves JNK activation, as demonstrated in vitro (66, 127) as well as in vivo (66) (Fig. 8). This reduction in FTH expression leads to cellular Fe overload and oxidative stress, ultimately involved in the mechanism via which JNK triggers programmed cell death during systemic infections (66, 127). This observation reveals the existence of a functional link between JNK activation and metabolic adaptation to cellular Fe overload during infection, in which TNF acts via JNK activation to promote tissue Fe overload (66) (Fig. 8). Moreover, inhibition of FTH expression also contributes to explain how JNK activation in response to TNF reinforces the accumulation of free radicals, an effect that can shift programmed cell death from apoptosis to necrosis (177). The molecular mechanism via which JNK activation inhibits FTH expression and deregulates Fe homeostasis remains, however, to be elucidated (Fig. 8).
Conclusion
Host defense strategies limiting Fe availability to pathogens should be considered as an integral component of inflammation and immunity providing host resistance to infection. Moreover, metabolic adaptation to tissue Fe overload, as conferred by the expression of FTH should be taken into account as an integral component of host protection against infection, promoting disease tolerance to infection (66, 116).
Footnotes
Acknowledgments
Authors are indebted to Birte Blankenhaus and Sebastian Weis for critical review of the article as well as all other members of the inflammation laboratory for intellectual input over the years. Authors work is supported by Fundação para a Ciência e Tecnologia (Portugal), SFRH/BPD/44256/2008 to R.G. and PTDC/BIA-BCM/101311/2008, PTDC/SAU-TOX/116627/2010, HMSP-ICT/0022/2010, RECI/IMI-IMU/0038/2012, European Research Council ERC-2011-Advanced Grant no. 294709 DAMAGECONTROL to M.P.S.