
Abstract
Introduction
R
While the role of vascular NADPH oxidases has been well described in pathology, their physiological functions remain less clear. We have recently gained substantial insight into the contribution of individual NADPH oxidase homologues in the maintenance of normal vascular function. In particular, the role of Nox4 in the regulation of endothelial function was clearly defined (166). This review focuses on the role of vascular NADPH oxidases in physiological and pathological processes in the vasculature, with particular emphasis on recently elucidated mechanisms such as the role of NADPH oxidases in vascular protection, vascular inflammation, pulmonary hypertension, and tumor angiogenesis. Finally, we briefly discuss the possibilities of pharmacological regulation of vascular NADPH oxidases and inhibitors being developed, in both the laboratory and clinical wards.
Localization, Structure, and Basic Functions of Major Nox Isoforms in Vasculature
Vascular Nox isoforms have six transmembrane domains, including alpha helices with cytosolic N- and C-termini, which participate in electron transfer, leading to the reduction of molecular oxygen to superoxide anion. Electron flow and thus ROS production is tightly controlled by the interactions of Nox subunits with other proteins, subunit phosphorylation, or elevation of intracellular calcium (15). There are seven isoforms of NADPH oxidases expressed in mammals: Nox1, Nox2, Nox3, Nox4, Nox5, Duox1, and Duox2. Four (Nox1, Nox2, Nox4, and Nox5) are most commonly expressed in vascular cells, while other homologues have not been found or are expressed at very low levels; thus, their role has not been established so far.
Nox2
Initially termed gp91phox, it has been cloned and identified as a phagocytic respiratory burst oxidase, critical for the initial nonspecific host defense. In addition to phagocytes, it is the most widely expressed vascular NADPH oxidase isoform. It is expressed in vascular smooth muscle cells (VSMCs), adventitial fibroblasts, endothelial cells, and perivascular adipocytes (92, 149, 188). This NADPH oxidase homologue has been characterized in detail and consists of the following subunits: gp91phox (glycoprotein-91 kDa phagocytic-oxidase, newly termed Nox2), p22phox, p47phox, p67phox, p40phox, and the GTPase Rac1. The gp91phox and p22phox subunits are membrane bound and together form cytochrome b558, located in cytoplasmic vesicles and the plasma membrane (20). The structure of this oxidase in vascular cells is similar to that found in phagocytes, although it may have additional or different regulatory subunits in selected conditions. In particular, Nox organizer protein 1 (NoxO1) and Nox activator protein 1 (NoxA1), initially discovered as Nox1 regulators (in place of p47phox and p67phox, respectively), may also have modest activating properties toward Nox2 (100). In endothelial cells, Nox2-derived ROS are important for p38 MAP-kinase-mediated proliferation and vascular endothelial growth factor (VEGF)-induced migration (187). Nox2 that is expressed in endothelial cells is involved in the regulation of numerous functions of the endothelial cell. For example, Nox2 activation affects NO bioavailability, and modulates expression of adhesion molecules during inflammation and angiogenesis. These will be further discussed next.
Nox1
Nox1 (Mox1, NOH1) has been identified as the first homologue of Nox2 (7). It shares a 60% amino-acid sequence identity with Nox2. Similar to its phagocytic homolog, Nox1 contains six transmembrane domains and conserved motifs corresponding to binding sites of heme, flavin, and NADPH. Nox1 is expressed in endothelial, smooth muscle, and adventitial cells of the vasculature. Most of the studies using recombinant Nox1 protein show localization in cell membranes, particularly in the plasma membrane (85). Using immunofluorescence microscopy, endogenous Nox1 protein displayed surface distribution along the cellular margins where it co-localized with caveolin-1 (33, 87). Other studies showed endogenous or overexpressed Nox1 protein localized to the nucleus, cytosol (28), endoplasmic reticulum (ER) (2), and mitochondria (40). Nox1 activity requires p22phox, NoxO1 (or possibly p47phox in some cases), and NoxA1, and the small GTPase Rac. Nox1-dependent ROS generation has been shown to play a pivotal role in cell signaling, cell growth, angiogenesis, and cell motility (6). Interestingly, ROS-generated via Nox1 have been reported to contribute to a growing number of diseases involving vasculature, including atherosclerosis, hypertension, neurological disorders, inflammation, and cancer (35, 142), which will be further discussed next. However, while highly expressed in animal models of vascular disease, Nox1 appears to be only expressed at low levels in human peripheral or coronary vessels (79, 80).
Nox4
This Nox isoform is a 67 kDa protein sharing a 39% amino-acid sequence identity with Nox2. It was initially detected in the kidney; therefore, it was termed Renox or kidney oxidase (KOX) (62). However, it was soon identified in vascular walls, particularly VSMCs, fibroblasts (15), and endothelial cells (64, 65). Studies of human coronary arteries have shown Nox4 to be expressed predominantly in the media (170). It has been suggested to be involved in stress signal transduction in the kidney (67) and SMCs (151). Nox4 mediates transforming growth factor β (TGF-β)-induced differentiation (173), insulin signaling (130), oxygen sensing (115), cardiac differentiation (116), and transcriptional regulation (107). Nox4 is predominantly localized at the ER (142, 151) and in the nucleus (40, 166). In addition, co-localization of Nox4 with the cytoskeleton and focal adhesions has been demonstrated (36, 87, 128). Enzymatic activity of Nox4 mainly depends on the expression level of Nox4 and p22phox and likely does not need any further activation (131). Currently, it is unclear which type of ROS is predominantly generated by Nox4. Unlike Nox1 or Nox2 that primarily produce O2 −•, Nox4 has been shown to produce hydrogen peroxide (H2O2) (43, 85). Production of H2O2, rather than O2 −•, by Nox4 is possible due to a highly conserved histidine residue in the E-loop of Nox4 that promotes rapid dismutation of O2 −• before it leaves the enzyme (177). This aspect of Nox4 biology needs to be further clarified. Physiological roles of Nox4 differ depending on cell type and stimulus. Nox4 might have an antagonistic function to Nox1 and Nox2 (166). Nox4 is implicated in both pro- and anti-apoptotic pathways. It is needed for 7-ketocholesterol- induced apoptosis in SMCs (151), while silencing of the protein induces apoptosis in pancreatic cancer cells (136). Nox4 is important for the differentiation of cardiac cells from mouse embryonic stem cells (116) and myofibroblasts from cardiac fibroblasts (37). Differentiated VSMC require Nox4 to maintain expression of differentiation markers, smooth muscle major histocompatibility complex (MHC), alpha -actin, and calponin (36). Recent studies raise important questions regarding the major physiologic and pathologic roles of Nox4. While Nox4 contributes to oxidative stress, compelling evidence from Nox4−/− mice indicates that endogenous Nox4 protects the vasculature during ischemic or inflammatory stress (166). Interestingly, we have recently found that Nox4 expression is decreased in human abdominal aortic aneurysm (AAA) in spite of largely increased oxidative stress in the walls of AAAs (74). Moreover, Nox4 immunoreactivity has been demonstrated in the nucleus and nucleolus of VSMCs by confocal microscopy (87). A recent study has identified a novel nuclear-localized Nox4 splice variant with a size of 28-kDa, Nox4D, which lacks putative transmembrane domains (3). The possible functional role of nuclear Nox4 has been previously described (87). Interestingly, Nox4D overexpression results in increased NADPH-dependent ROS production and causes increased phosphorylation of extracellular signal-regulated kinase1/2 and the nuclear transcription factor Elk-1 (3). Thus, Nox4D may have important pathophysiologic effects through the modulation of nuclear signaling and DNA damage; however, its role in vascular biology should be further elucidated.
Nox5
Nox5 is expressed in primates but does not naturally occur in rodents. It has been found in leukocytes, in the testis (during development), and in lymphatic tissue. Expression of lesser extent has been reported in ovaries, pancreas, and placenta (8, 32). Recent studies have also identified the presence of Nox5 in vasculature, namely in endothelial cells and VSMCs, where it is localized in the ER and the plasma membrane (16). Nox5 has been reported to produce both superoxide and H2O2; Nox5 is activated by Ca2+ and does not appear to require other subunits, although it may associate with p22phox. The basic transmembrane structure of Nox5 is similar to that described for other Nox isoforms, but what distinguishes Nox5 is a unique N-terminus which encodes four calcium-binding EF hands (Fig. 1) (8). We have identified a calcium-dependent Nox5 protein and mRNA in human coronary vasculature, in both endothelium and vascular media, which was functionally linked to calcium-dependent NADPH-oxidase activity (75).
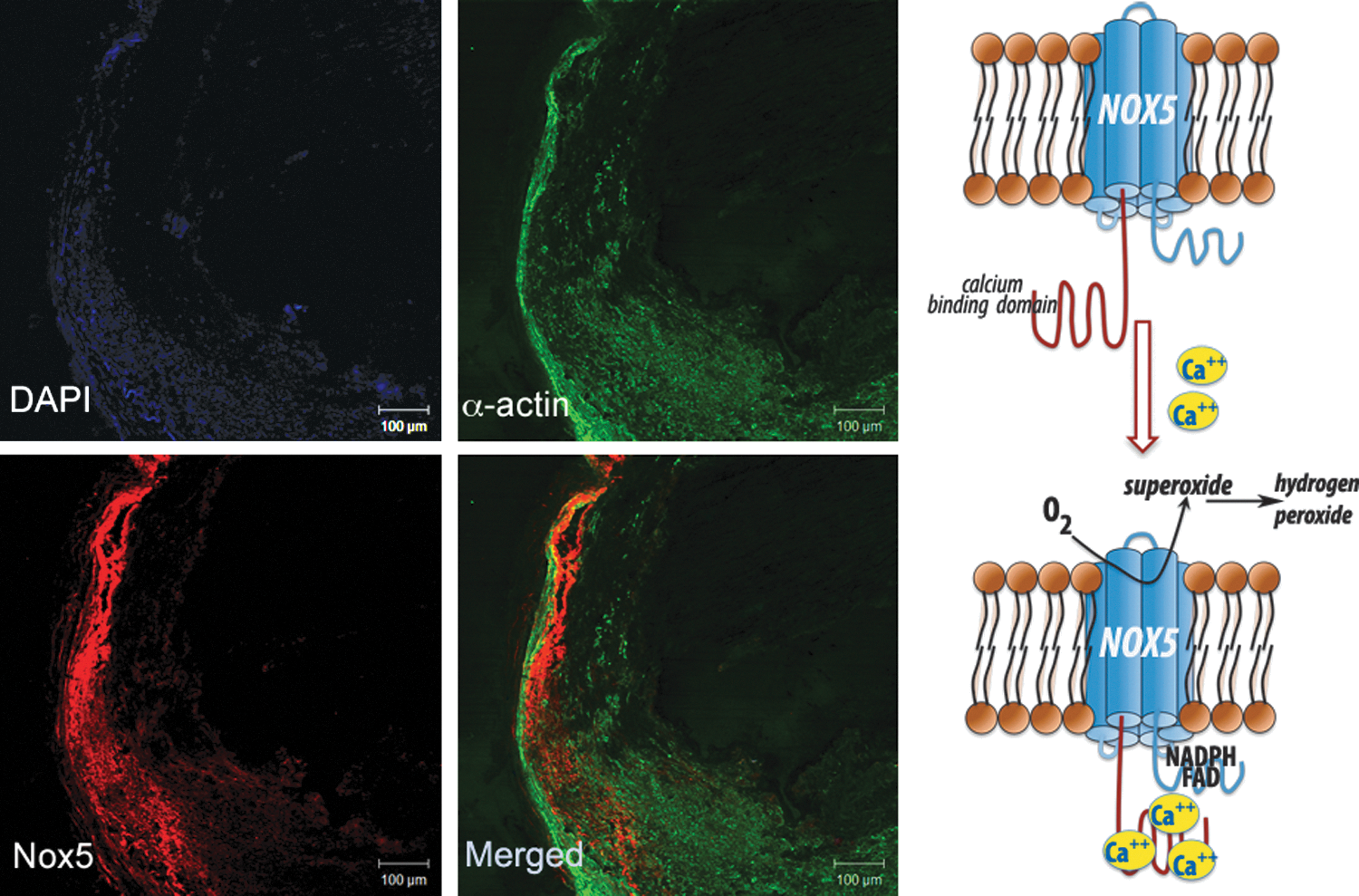
Diverse Mechanisms of Activation and Regulation of Vascular NADPH Oxidases
Despite similarities in core structures, vascular Nox homologues have different mechanisms of activation. Activation as well as its consequences may be different in the endothelium (Fig. 2) or VSMCs (Fig. 3) or other cells such as perivascular adventitial fibroblasts or adipocytes Nox1 and 2 require association with cytosolic components (p47phox, p67phox or NoxO1 and NoxA1), Nox4 is constitutively active, and Nox5 is activated by an elevation in intracellular Ca2+. The activation mechanism of Nox2 is most clearly defined and similar to that described in phagocytes. On activation, p47phox is phosphorylated and translocates to the membrane, through the formation of a complex with p67phox and p40phox. Phosphorylation of p47phox induces a conformational change in a tandem SRC Homology 3 (SH3) domain that enables binding to a proline-rich region in the cytosolic C-terminus of the transmembrane subunit p22phox. Independently, the GTP-binding protein Rac also moves to the membrane and activation occurs (15). In contrast to Nox2, the Nox4-based oxidase appears to be constitutively active and does not require p47phox, p67phox, or Rac.
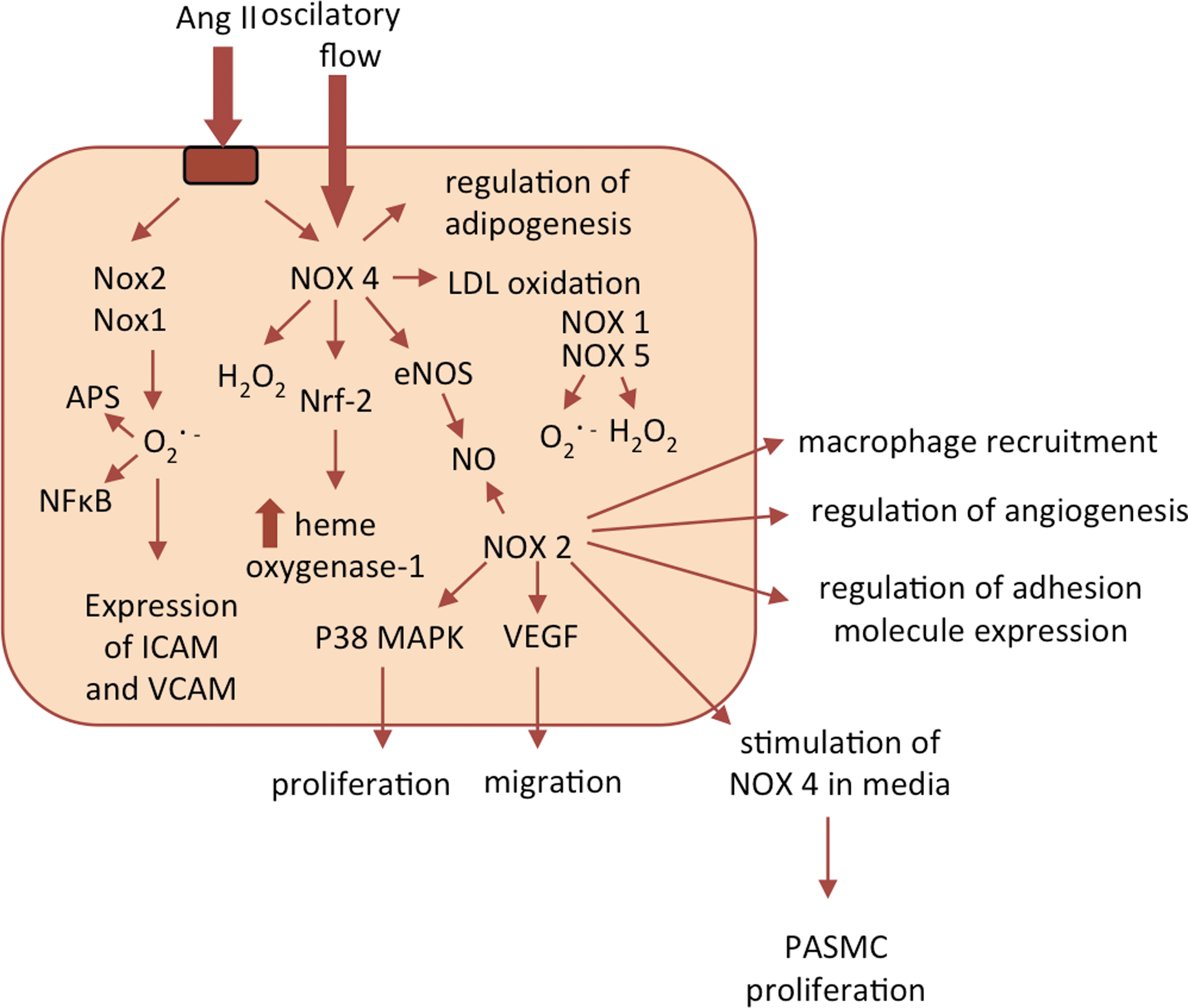
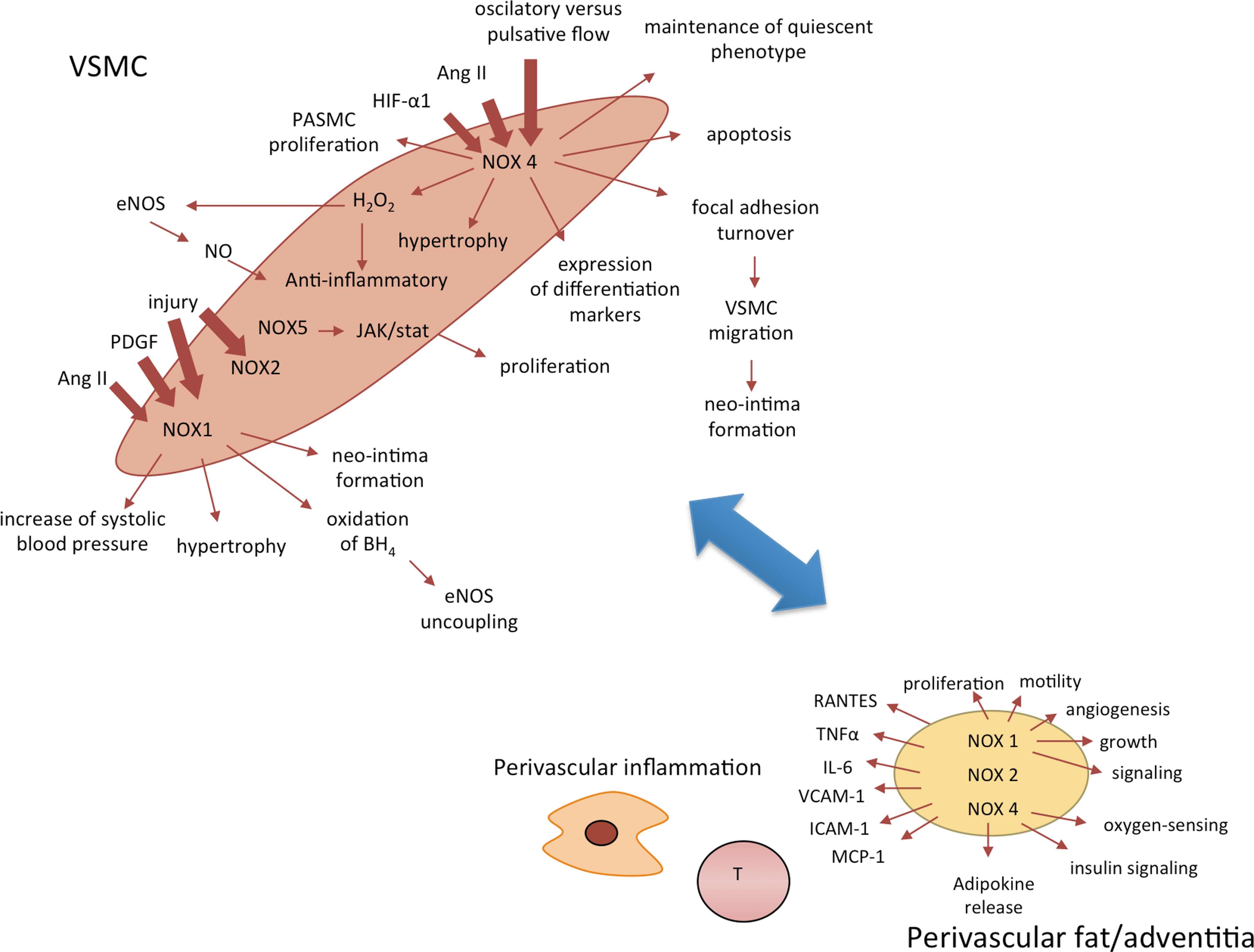
Sustained activation of vascular NADPH oxidases occurs in response to numerous agonists. Vascular NADPH oxidases are responsive to several growth (platelet-derived growth factor [PDGF], epidermal growth factor [EGF], and TGF-β), cytokines (e.g., tumor necrosis factor, interleukin-1 [IL-1], and platelet aggregation factor), mechanical forces (cyclic stretch, laminar, and oscillatory shear stress), metabolic factors (hyperglycemia, hyperinsulinemia, free fatty acids, and advanced glycation end products), and G protein–coupled receptor agonists (serotonin, thrombin, bradykinin, endothelin, and angiotensin II [Ang II]) (71, 152, 154). In addition, c-Src, p21Ras, protein kinase C (PKC), phospholipase D, and phospholipase A2 (PLA2) have been demonstrated to play key roles in signaling involved in vascular NADPH oxidase activation (1, 12, 39).
Ang II is one of the key agonists stimulating Nox1 and Nox2 oxidase subunit expression as well as oxidase activation in vascular cells and neutrophils (148, 156, 187). However, its effects on Nox4 and Nox5 are much less pronounced and perhaps indirect, which will be further discussed next (37, 111, 139). Ang II-induced production of ROS by VSMCs is initially PKC-dependent (early phase), whereas the prolonged phase (>30 min) is dependent on Rac, Src, and phosphatidylinositol 3-kinase (167). Ang II increases mRNA levels not only of Nox proteins but also of p22phox and p67phox (55, 148). Ang II-dependent or H2O2-induced activation of Nox4 in mesangial cells or cardiac fibroblasts has been reported to be dependent on PLA2 and release of arachidonic acid (37, 86).
Cytokines have also been shown to regulate vascular NADPH oxidases, which links inflammation with oxidative stress. In particular, tumor necrosis factor-α (TNF-α) stimulates NADPH oxidase Nox1, Nox2, and Nox4 expression and activation in a variety of vascular cells (4, 11, 138). Other stimuli that induce vascular NADPH oxidases include ER stress (151), shear stress (91), or an elevation of intracellular Ca2+, which could act as an upstream signal to activate Nox during cellular stress. The Nox5 isoform contains multiple EF-hand Ca2+-binding domains in the N-terminal region, allowing its activation through calcium sensing (8). Calcium binding induces a conformational change in the N terminus of Nox5, resulting in the exposure of a hydrophobic motif, which leads to a direct interaction between the regulatory N-terminus and the catalytic C-terminus. This intra-molecular interaction is likely to be responsible for Ca2+-induced Nox5 activation (9), as it enables electron flow to the heme moieties and, consequently, drives superoxide production.
Redox regulation of vascular NADPH oxidase activation provides both negative feedback and feed-forward regulation mechanisms. For example, Rac1 protein turnover is strongly dependent on the redox status of the cell, creating a negative feedback mechanism (106). At the same time, a feed-forward mechanism exists (118), by which exogenous exposure of SMC or fibroblasts to H2O2-activated NADPH oxidase stimulates them to produce endogenous O2 −•, thereby amplifying the vascular injury process. The self-limiting mechanism could predominate during physiologic conditions, and be involved in maintaining low output of the nonphagocyte NADPH oxidase, whereas the feed-forward mechanism may have a role in NADPH oxidase-dependent oxidative stress in a variety of diseases, including atherosclerosis and inflammation.
NADPH Oxidases in Vascular Pathobiology
The complexity of vascular NADPH oxidase systems makes it difficult to unequivocally define the role of individual Nox oxidases in vascular physiology and pathology. Several genetically modified mouse strains with either removed or overexpressed Nox proteins have been used to investigate the roles of NADPH oxidases in the vasculature (Table 1). Such studies have clearly linked Nox1, Nox2, and Nox4 to the pathogenesis of vascular diseases, while other Nox isoforms showed little to no vascular expression or involvement. Obviously, this approach is not optimal for studies involving Nox5, which would require a more complex humanized model approach. Numerous studies in animal models and in humans have also characterized the role of Nox proteins in the most common vascular disease states such as atherosclerosis, hypertension, and diabetes, which will be discussed next.
ApoE, apolipoprotein E; NADPH, nicotinamide adenine dinucleotide phosphate; VSMC, vascular smooth muscle cell.
Atherosclerosis
A number of studies have investigated the role of individual NADPH oxidases in atherosclerosis in animal models. Nox2 levels were increased in the aortas of apolipoprotein E (ApoE)−/− atherosclerotic mice, while Nox4 levels were not significantly different between ApoE−/− and wild type (97). However, studies using deletion of the Nox2 gene in ApoE−/− mice have not been fully consistent. Nox2/ApoE double knockout mice, which had been fed a high fat diet, were found to have no obvious protection against atherosclerosis when examining aortic sinus sections, in spite of decreased vascular superoxide formation (102). Similar surprising results were reported in global p47phox knockout mice crossed with ApoE−/− mice (90). Endothelial-targeted Nox2 overexpression in ApoE−/− mice was sufficient to increase vascular superoxide production, endothelial cell activation, and subsequently increased macrophage recruitment. This initial increase in macrophage recruitment did not alter the progression of atherosclerosis (50). In contrast to these studies, Barry-Lane et al. found that total aortic lesion area (from arch to bifurcation) was reduced in p47phox knockout mice crossed with ApoE−/− mice, suggesting that Nox1 and/or Nox2 may be involved in atherogenesis (10). Such discrepancies in the findings regarding the vascular consequences of p47phox−/− could be due to several reasons. An example is the inclusion of assessments of different areas from aortic lesions (sinus vs. whole aorta). Another reason for the differences may also be related to the removal of p47phox, which causes immunodeficiency. These mice are prone to sub-clinical infections that are characterized by splenomegaly and inflammation which could counteract the anti-atherosclerotic effect of the loss of NADPH oxidase. While studies in animal models are conflicting, a genetic hereditary deficiency of NADPH oxidases, particularly Nox2 (gp91phox), occurs in humans with chronic granulomatous disease (CGD). In humans, the genetic deficiency of Nox2 is associated with enhanced endothelium-dependent flow-mediated vasorelaxation, and decreased markers of vascular aging and oxidative stress (192). Importantly, diminished atherosclerosis burden has been described in these patients, which emphasizes the possible role of Nox2 oxidase in atherosclerosis (191). Although promising, these observations should be interpreted carefully, as CGD is a serious disease leading to important changes in the immune system, which could affect vasculature in a number of additional ways.
Nox4 may also participate in atherogenesis, although much less is known in this regard. It has been shown, however, that Nox4 regulates VSMC migration and differentiation, which is critical for neo-intima formation (128). In human atherosclerosis, Nox4 expression is increased in intimal lesions of coronary arteries (170). Similarly, Nox1 overexpression in the media results in increased neo-intima formation on vascular injury (114).
Numerous studies have looked at vascular NADPH oxidases in human atherosclerosis. Our group as well as others have characterized NADPH oxidases in human coronary arteries with evident atherosclerotic plaques (75, 79, 170), as well as in peripheral arteries and veins without plaques but characterized by systemic endothelial dysfunction (73, 74, 80 –82). The NADPH oxidase is predominantly Nox2-based in veins, whereas an Nox4-based oxidase appears proportionately more important in human mammary or radial arteries (80). In veins, the predominance of Nox2 expression may also suggest the major contribution of endothelium and adventitia to total vascular ROS production, as these contain Nox2-based oxidases (14), potentially contributing to endothelial dysfunction (160, 170, 175). However, the cellular expression of individual oxidases has not been clearly established. While initial studies in human vessels suggest that Nox2 is not usually present in medial VSMCs, it has been demonstrated that human microvascular SMCs can express Nox2 in response to Ang II (182). Meanwhile, Nox4 expression is increased by oscillatory versus pulsatile flow, which could be particularly relevant to the development of atherosclerotic plaques in the regions of turbulent flow (91). This clearly illustrates that Nox isoform expression in human vascular cells is regulated in a complex manner that depends on the cell type predominating in different vessels and on the nature of pathophysiologic stimuli.
Still, it is vital to emphasize that overall, enzymatic sources of O2 −• in coronary arteries are similar to those of peripheral arteries (18, 78). In summary, Nox2, Nox4, Nox5, p22phox, and, to a lesser extent, Nox1 are expressed in human peripheral and coronary vessels, which may contribute to atherosclerosis (80, 170). Nox1 expression directly alters cell proliferation (174), and treatment of VSMC with Ang II or PDGF up-regulates Nox1, while down-regulating Nox4 (111). Vascular injury increases expression of Nox1, Nox2, and p22phox, while Nox4 increases later (175), coinciding with a reduction in the rate of VSMC proliferation. Taken together, these findings suggest that while Nox1 and Nox2 are involved in acute response to injury or to Ang II stimulation, Nox4 is involved in maintaining the quiescent VSMC phenotype (175). Thus, a high Nox2/Nox4 ratio in veins could partially account for the susceptibility to intimal hyperplasia, remodeling, and accelerated atherosclerosis in vein grafts (197); whereas a lower Nox2/Nox4 ratio in mammary artery grafts could convey less susceptibility to atherosclerosis. Functional consequences of increased NADPH oxidase activity also include the effects on lipid oxidation, which is a critical step in the initiation of atherosclerotic plaque development. For instance, Nox4 expression elevates in relation to oscillatory flow and coincides with increased oxidative stress and low-density lipoprotein (LDL) oxidation (91), while NADPH oxidase activity is correlated with oxidized-LDL in carotid plaques. Despite the differences in the expression levels between individual Nox homologues, the expression of both p22phox and Nox4 mRNA is strikingly correlated in human arteries and veins (80). This indicates that risk factors for atherosclerosis may be critical for the systemic regulation of NADPH oxidase expression (80). Indeed, activity and expression of major NADPH oxidase components is correlated to the number of major risk factors for atherosclerosis in humans (82).
Finally, Nox5 is an important source of ROS in atherosclerosis (75). Nox5 mRNA and Nox5 protein are dramatically increased in human coronary arteries obtained from patients with coronary artery disease. These correlate with the Ca2+-dependent NADPH oxidase activity in arteries. Nox5 was expressed in the endothelium of early-stage lesions and in VSMCs in the intima of advanced coronary lesions (Fig. 1) (75).
In summary, differential generation of ROS by functionally distinct NADPH oxidase isoforms expressed in different vascular cell types in atherosclerosis may be used as a therapeutic advantage. For example, Nox2-based production of ROS predominates in the endothelium and adventitia, whereas Nox1 and/or Nox4 could be much more important in VSMC (158), as their expression and activity differs depending on the stage of the disease. Thus, one could postulate, that if we were able to specifically inhibit these individual oxidases, we would be able to affect distinct stages in vascular disease processes.
Hypertension
Vascular NADPH oxidases are probably characterized in most detail in relation to hypertension. This was one of the first pathologies in which NADPH oxidases were clearly implicated (156). Ang II plays an important role in the development of hypertension and is one of the most important inducers of increased NADPH oxidase-dependent superoxide production in VSMCs (48) and throughout the vascular wall (141). The effects of Ang II on NADPH oxidases are mediated primarily via the angiotensin receptor type 1 (AT1) receptor (79). NADPH oxidases also participate in Ang II-stimulated intracellular H2O2 production, which mediates vascular hypertrophy (205). Ang II increases the expression of several NADPH oxidase homologues (Nox1, Nox2, Nox4, and p22phox) (156), all of which may to some extent participate in the pathogenesis of hypertension and associated vascular dysfunction. Nox1 is increased in the aortas of aged spontaneously hypertensive rats (SHRs) in parallel with increased NADPH oxidase activity. While Nox2 was also up-regulated, Nox4 protein levels were unchanged or even decreased in the aortas of SHRs (198). Overexpression of Nox1 in mouse VSMCs caused a marked increase in systolic blood pressure and hypertrophy in response to Ang II (44). In contrast, deletion of Nox1 in mice results in a blunted hypertensive response to Ang II and protects from development of endothelial dysfunction, oxidative stress, vascular hypertrophy, and aortic dissection (60, 61, 132). Overexpression of p22phox in VSMCs leads not only to up-regulation of Nox1-based oxidase in the vessel wall, but also to increased hypertensive responses to Ang II (112, 195). At the same time, silencing of p22phox in rats prevents a slow pressor response to Ang II (137). Similarly, p47phox knockout mice, which have deficient Nox1 (and Nox2) activation, exhibit reduced hypertension and preserved endothelial function after chronic Ang II treatment (108, 109). VSMCs from p47phox-deficient mice do not produce superoxide (113). Moreover, local overexpression of Nox1 in VSMCs has profound effects on other cell types in the vessel wall. For example, this can lead to the oxidation of tetrahydrobiopterin (BH4) in endothelial cells and endothelial nitric oxide synthase (eNOS) uncoupling, resulting in a decrease of NO bioavailability and causing endothelial dysfunction (45). In contrast, Nox2−/− mice were reported to have only modestly altered (193) or even unaltered (183) blood pressure responses to Ang II, suggesting that this isoform may be less important in blood pressure regulation, but still showing effects on vascular hypertrophy (193) and endothelial dysfunction during experimental hypertension (98). In addition, Nox2 was critical for cardiac hypertrophy in Ang II-dependent hypertension (17), but not in a pressure-overload-dependent model, where hypertrophy might be related to other homologues such as Nox4 (25). While gene-specific deletion or overexpression approaches have proved useful in defining the role of Nox1and Nox2 in the vascular system, the functional importance of Nox4, while undeniable, has been raising the most controversy and discussion. Endothelium-targeted Nox4 overexpression causes enhanced vasodilation and reduced basal blood pressure, suggesting a protective role for Nox4 that has been linked to increased H2O2 production by Nox4 (157). In the absence of pathogenic stimuli, Nox4 knockout mice do not have an obvious phenotype and are normotensive (176). However, more recent data have convincingly shown that in vascular pathologies, Nox4 may serve as a protective oxidase (22, 166). In the conditions associated with stress induced by ischemia or by Ang II, loss of Nox4 resulted in reduction of eNOS expression, NO production, and heme oxygenase-1 (HO-1) expression. These changes were associated with apoptosis and inflammatory activation in Nox4−/− mice (166). These effects were partially related to Nrf-2 induction. Consequently, there is now clear evidence that endogenous Nox4, in contrast to Nox1 and Nox2, may protect the vasculature during ischemic or hypertensive stress (166). However, this may not be the case in the brain, where Nox4 has been shown to contribute to oxidative stress related to stroke and other pathologic conditions (103). Hence, further studies are needed to determine whether Nox4 is always protective or whether it can become dysfunctional.
Moreover, while the role of Nox1 in the pathogenesis of hypertension appears to be relatively well documented, chronic models of high renin transgenic overexpression do not show significant effects of Nox1 on blood pressure in this model of hypertension (204).
Diabetes
Diabetes is associated with a variety of metabolic abnormalities, such as insulin resistance and hyperglycemia. However, cardiovascular complications are the major cause of mortality in diabetic patients. ROS generated during hyperglycemia are implicated in the development of endothelial dysfunction and progression of diabetic vascular complications. Endothelial dysfunction characterized by increased NADPH oxidase-dependent ROS generation has been demonstrated in animal models of diabetes (88), and in patients with diabetes (82). In aortas from the streptozotocin (STZ)-induced diabetic ApoE−/− mice, levels of Nox2 and Nox4 are increased (47). Similarly, in db/db mice, Nox1 or Nox4 are up-regulated, which is associated with increased ROS production and inflammation, indicating a potential role of Nox1 and Nox4 in diabetic macrovascular disease (119). Another recent study has elegantly addressed the role of Nox1 in diabetic vasculopathy using the specific Nox inhibitor GKT137831, as well as Nox1−/− mice on an ApoE−/− background in which diabetes has been induced (69). Deletion of Nox1, but not Nox4, had a profound anti-atherosclerotic effect correlating with reduced ROS formation, diminished chemokine expression, reduced vascular adhesion of leukocytes, reduced macrophage infiltration, and reduced expression of proinflammatory and profibrotic markers (69). Similarly, treatment of diabetic ApoE-deficient mice with GKT137831 attenuated atherosclerosis development (69).
Hyperglycemia itself can induce vascular NADPH oxidases. Incubation of human endothelial cells with red blood cells isolated from patients with type 1 diabetes, but not from normal volunteers, activated endothelial NADPH oxidase and increased endothelial ROS generation, which was mediated by advanced glycation end products on the surface of red blood cells from diabetic patients (194). Hyperglycemia increases NADPH oxidase expression, levels of oxidative stress markers, and apoptosis (155) of human endothelial cells in relation to increased NADPH oxidase subunit expression (for example, p22phox and p47phox) (46). Similarly, in arteries and veins, superoxide anion production is strongly increased in diabetic patients, most prominently in the endothelium (78). Superoxide anion produced in large quantities by NADPH oxidases reduce NO bioactivity by direct scavenging.
While NADPH oxidases play a key role, uncoupled eNOS has also been shown to be a particularly important source of ROS in diabetic blood vessels (88). Importantly, superoxide derived from vascular NADPH oxidases leads to eNOS uncoupling through oxidation of BH4 (88). Ang II is also involved in the activation of vascular NADPH oxidases in diabetes. AT1 receptor-mediated NADPH oxidase activation appears to contribute to vascular insulin resistance, endothelial dysfunction, apoptosis, and inflammation (196). While vascular Nox4 does not appear to have a direct role in diabetic vasculopathy (69), it may have indirect effects through the regulation of adipogenesis (preadipocyte differentiation in response to insulin) in metabolic syndrome (165).
Consequently, vascular NADPH oxidases have emerged as potentially important targets contributing to the pathogenesis of long-term cardiovascular complications of diabetes. Of the numerous pathologies and vascular disease states, diabetic vasculopathy may prove be the most interesting area for the future application of NADPH oxidase inhibitors.
In summary, genetically manipulated mouse models as well as studies in human vasculature have been very useful in defining the functional importance of individual NADPH oxidases. Further studies characterizing individual knockout and overexpression of Nox isoforms, specifically in endothelial cells, VSMCs, and fibroblasts will undoubtedly provide a better understanding of the complex mechanisms involved in NADPH oxidase activation and regulation. Studies of Nox5 expression in various models are now warranted to further understand its role in the vasculature using these valuable molecular tools.
Vascular NADPH Oxidases in Pulmonary Hypertension
Recently, significant progress has been made in our understanding of the role of vascular NADPH oxidases in pulmonary hypertension. This is crucial, considering the clinical consequences of pulmonary hypertension and a lack of successful therapies. The major pathologic process in pulmonary hypertension is chronic hypoxia-induced vascular remodeling. It is characterized by malignant SMC hypertrophy and proliferation within the pulmonary vessel media (123). This leads to a decrease in vascular luminal area, increased vascular resistance, and thus development of pulmonary hypertension and increased right ventricular pressure. A role for Nox2-based NADPH oxidase in this process has been suggested, as all of these phenotypes are attenuated in Nox2−/− mice (123). This is consistent with the role of this oxidase in VSMC hypertrophy, particularly pulmonary artery smooth muscle cells (PASMCs) (93, 173).
However, the role of NADPH oxidases in pulmonary hypertension appears to be more complex than simply stimulating VSMC hypertrophy and proliferation. They may be involved at very early stages of disease pathogenesis. ROS derived from vascular Nox isoforms, in particular Nox2 and Nox4, are involved in long-term responses of pulmonary vasculature to hypoxia (52, 135). Thus, NADPH oxidases have been suggested to serve as oxygen sensors in the lung. Interestingly, Nox4 has been reported to be the predominant homolog in human airways and in PASMCs (49). PASMCs are particularly sensitive to oxygen availability and are responsible for acute hypoxic vasoconstriction and the development of pulmonary hypertension in response to chronic hypoxia (72). Nox4 expression level has been increased in mice exposed to chronic hypoxia and in isolated PASMCs exposed to hypoxia. Diebold et al. have described a putative hypoxia responsive element in the human Nox4 promoter and demonstrated that this sequence is indispensable for the binding of hypoxia-inducible factor-1α (HIF-1α) (42). In fact, RNA interference against HIF-1α suppressed the hypoxia-induced expression of Nox4 (42). In patients with idiopathic pulmonary arterial hypertension, the level of Nox4 expression has been up-regulated in the lung. Treatment of PASMCs with an anti-Nox4 siRNA significantly reduced their proliferation.
Interestingly, it appears that Nox2 and Nox4 play coordinated roles in the development of hypoxia-induced pulmonary hypertension. Theoretically, endothelial ROS generation by Nox2 may stimulate Nox4 up-regulation in the vessel media, which would be important for hypoxia-dependent PASMC proliferation (135). Hypoxia also increases the level of TGF-β (97), which has been shown to induce Nox4 expression. Inflammatory processes can also contribute to the induction of Nox4 in pulmonary hypertension. In particular, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) is involved in the hypoxia-induced expression of Nox4 in PASMC, in that hypoxia increases the binding of NF-κB p65 to the putative NF-κB binding site adjacent to the putative hypoxia-responsive element. Furthermore, activation of the nuclear hormone receptor peroxisome proliferator-activated receptor gamma (PPARγ) attenuated hypoxia-induced Nox4 expression, pulmonary hypertension, and pulmonary vascular remodeling (143). Rosiglitazone, a synthetic agonist of PPARγ, attenuated hypoxia-induced pulmonary hypertension and vascular remodeling in the mouse and reduced hypoxic Nox4 induction and ROS generation in the lung (127). Thus, while both Nox2 and Nox4 seem to be involved in pulmonary hypertension, it appears that Nox4 may be involved in responses to hypoxia as well as inflammation, and may be an important target for novel pulmonary hypertension therapies.
Vascular NADPH Oxidases in Tumor and Ischemic Angiogenesis
Another field in which vascular NADPH oxidases (mostly endothelial and endothelial progenitor cells) seem to be gaining importance is angiogenesis. It encompasses a wide range of conditions ranging from physiological situations such as embryogenesis and wound repair, to pathological conditions such as diabetes, arthritis, and cancer. In endothelial cells within the tumor, ROS have a critical role in neovascularization during tumor growth (140), while the thiol-antioxidant, N-acetylcysteine (NAC), attenuates endothelium cells invasion and angiogenesis in an in vivo tumor model (26). Vascular NADPH oxidases respond to various pro-angiogenic factors such as VEGF, making them prime targets in anti-angiogenic therapies. For example, Nox2-derived ROS are important in signaling for cytoskeletal organization in endothelial cell migration (186). Similarly, Nox1, Nox4, and Nox5 expression is increased in melanoma cells, and affects neovascularization and angiogenesis in the tumor (68). While being targets for pro-angiogenic factors, vascular NADPH oxidases are important modulators of the release of various angiogenesis-related factors, which are redox sensitive. These include VEGF-A, HIF-1α, p53, and matrix metalloproteinases (MMPs). Nox1-induced H2O2 increases VEGF and VEGF receptor expression, and MMP activity, markers of the angiogenic switch, thereby promoting vascularization and rapid expansion of the tumors (6). Increased VEGF expression and enhanced angiogenesis in experimental atheromas have been demonstrated in transgenic mice overexpressing Nox1 and Nox2 (101). Implantation of tumorigenic B16F0 melanoma cells subcutaneously in wild-type mice has resulted in vascularized tumors after 10 days. In Nox1−/− mice, the tumors that have developed have been smaller and less vascularized. This reduction in angiogenesis was associated with a reduction in the expression of several genes, including VEGF, MMP-2, MMP-9, and a reduction in NF-kB activity (58).
In a similar fashion, NADPH oxidases participate in ischemic revascularization. Nox2 knockout mice showed reduced VEGF-induced new vessel formation (186) and delayed the blood flow recovery in ischemic hind limbs (181). It is noteworthy that Nox2 deficiency-induced impairment of ischemic angiogenesis was associated with reduced endothelial progenitor mobilization from the bone marrow in response to ischemic insults (185). Impaired blood flow recovery in Nox2−/− mice has been rescued by transplantation of wild-type bone marrow cells into Nox2-deficient animals. This indicates the therapeutic potential of targeting Nox in endothelial progenitors in the treatment of ischemic cardiovascular diseases. The role of Nox5 in angiogenesis has also been recently described. In human aortic SMCs, silencing of Nox5 abrogated PDGF-induced proliferation via a JAK/STAT signaling pathway (96). Nox5 also mediates thrombin-induced formation of capillary-like structures in endothelial cells, suggesting an additional role in angiogenesis (16).
While mechanisms of angiogenesis are well defined in both ischemia and cancer, the role of vascular NADPH oxidases is becoming clearer. Use of novel Nox inhibitors may turn out to be a valuable strategy in future anti-angiogenic therapies, although possible adverse effects on ischemic angiogenesis should also be taken into consideration.
Seasonal Variation of Vascular NADPH Oxidase Activity
A recent study has reported an interesting phenomenon that could have important implications for studies of vascular NADPH oxidases (105). Activity and expression of vascular NADPH oxidases may vary seasonally in both rats and mice (and possibly in humans). Seasonal variation in endothelial dysfunction and oxidative stress has been noted in humans and rats, suggesting it to be a common phenomenon of potential clinical relevance (89). The mechanism is still not fully understood and has been related to changes in physical activity. Interestingly, Langendorff-perfused guinea-pig and rat heart preparations generated close to 100% more O2 −•, while human subjects excreted 65% more 8-isoprostane in the summer versus other seasons (105). Inhibitors of NADPH oxidase, xanthine oxidase, and NO synthase prevented seasonal O2 −• overproduction (105). In the summer, NADPH oxidase and xanthine oxidase activity, and protein expression were increased. Meanwhile, endothelial NO synthase and superoxide dismutases were down-regulated, which was accompanied by adhesion molecule up-regulation and the endothelial glycocalyx destruction associated with these changes (105). Moreover, during the summer, STZ-induced diabetes was associated with greater vascular disturbance, in spite of similar hyperglycemia (105). These findings are intriguing, as they may indicate rhythmic regulation of Nox expression and activity, which should be further investigated and verified.
NADPH Oxidases in the Regulation of Vascular Inflammation
During the past several years, it has become apparent that inflammation and oxidation are two basic processes underlying the pathogenesis of most disease states in humans. Furthermore, it is now evident that these two distinct mechanisms are in a constant interplay, with interactions particularly evident in the vessel wall (121, 134, 176). Vascular oxidative stress regulates the development of vascular and renal inflammation that has been recently implicated in the pathogenesis not only of atherosclerosis but also of hypertension and hypercholesterolemia (84).
The role of vascular NADPH oxidases in the regulation of vascular inflammation has been initially defined in the pathogenesis of atherosclerosis. The expression of endothelial adhesion molecules such as vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), and P- and E-selectins on large vessel arterial lumen endothelial cells is redox sensitive and dependent on Nox1, Nox2, and Nox5 NADPH oxidases. This is in part, because they contain an NFκB site within the promoter region that is known to be regulated in a redox-sensitive fashion (169). It is the earliest and most critical step in the inflammatory process, which leads to transmigration of leukocytes into the subendothelial space (120, 144). This occurs not only in large vessels during atherosclerotic lesion formation, but also in the endothelium of the vasa vasorum, leading to accumulation of leukocytes in the adventitia and outer media. It appears that perivascular inflammation characterized by macrophage, dendritic cell, and T- and B-cell accumulation, in the form of tertiary lymphoid structures, is a key feature of early atherosclerosis (56, 99, 104) or hypertensive vascular dysfunction (77, 190). Although the precise mechanism underlying this early process is not entirely clear, NADPH oxidase-derived ROS are increasingly being recognized as important players (117, 150). Studies of coronary arteries from diabetic pigs have shown that diabetes-induced increases in NADPH oxidase activity are accompanied by up-regulation of inflammatory cytokines (IL-6 and TNF-α), chemokines (monocyte chemoattractant protein-1 [MCP-1]), and vascular cell adhesive molecules (VCAM-1), providing further support for the role of NADPH oxidase in vascular inflammation (206). In addition, NADPH oxidases, via overproduction of O2 −•, mediate expression and release of chemokines such as regulated on activation, normal T cell expressed and secreted (RANTES), or chemokine (C-C motif) ligand 2 (CCL2) from endothelium or VSMCs, essential for leukocyte recruitment to the vascular wall (29). ROS derived from adventitial fibroblasts may also serve as a direct chemotactic signal for leukocytes (122) and increase another chemotactic molecule, MCP-1 (201). Scavenging of ROS by SOD and catalase suppresses the induction of MCP-1 mRNA by TNF-α (31). Ang II, endothelin 1 (ET-1), and inflammatory mediators can modulate basal NADPH oxidase-induced O2 −• production by affecting both activation of the enzyme and expression of NADPH oxidase subunits (55). Liu et al. have shown that Ang II infusion for 1 week increased NADPH oxidase-derived ROS production in rat aortas, and enhanced ICAM-1 expression and subsequent adventitial macrophage infiltration (122). Similarly, Ang II infusion in mice induced a convergence of macrophages as well as T cells to the adventitia (24). Finally, recent mechanistic studies have shown a particular importance of Nox5 in the regulation of vascular inflammation, which has been linked to Fraktalkine gene expression (161). These effects are particularly important in the light of recent observations, showing that inflammatory cell activation is critical for the development of hypertension (190) and vascular dysfunction (83). However, these are not limited to the vasculature, as NADPH oxidases and ROS are essential in regulating inflammation in other organs such as the lungs (66).
While vascular NADPH oxidases are important in the regulation of inflammatory responses, NADPH oxidases are also expressed in immune cells. Ang II-induced hypertension is associated with increased Nox2-mediated superoxide production and Nox2 expression in T cells and monocytes in peripheral blood. This has been linked to the activation of these cells and may be important in the pathogenesis of Ang II-mediated hypertension. Furthermore, Nox4 is critical in monocyte priming for chemotaxis and recruitment to the vessel wall (184).
Understanding the complex interactions between vascular oxidative stress and vascular inflammation has become possible, thanks to the development of novel strategies for quantitative assessment of vascular inflammation in relation to Nox activity and expression. Nevertheless, a further understanding of pro- and anti-inflammatory roles for vascular oxidases is now necessary (176).
NOX-Derived ROS and Central Regulation of Blood Pressure
Blood pressure regulation is a complex process involving renal, vascular, and central mechanisms. The brain is essential for processing and integrating neurohumoral signals from the periphery to maintain pressure and fluid homeostasis. NADPH oxidases have been identified as major producers of ROS in the brain and are expressed in numerous brain structures (19). Increased ROS production is seen in several regions of the brain in hypertension, including the circumventricular organs (CVOs) (210), hypothalamic centers, and the brainstem (146). Nox2 as well as Nox4 are the predominant homologues expressed in the fore-, mid-, and hindbrain of mice, while Nox1 is detectable at very low levels (92). However, Ang II induces expression of individual NADPH oxidase homologues, particularly Nox2 in the brain, which may be related to increased sympathetic nervous system activation (92). Taken together, it is possible that an increase in NADPH oxidase-derived ROS in CVOs, hypothalamic nuclei, and brainstem sites plays a central role in the neurocardiovascular dysfunction observed in hypertension (21). Vascular NADPH oxidases could also be important, as Nox2 appears to be a prominent mediator of the harmful effects of Ang II in the cerebral circulation during hypertension (34). In addition, knock down of Nox2 and Nox4 proteins in the paraventricular nucleus attenuated the development of Aldosterone/NaCl-induced hypertension (202). Silencing Nox2 or Nox4 alone in the subfornical organ (SFO) significantly attenuated the central Ang II-induced pressor response, while silencing both Nox2 and Nox4 together abolished this response. In contrast, silencing Nox2, but not Nox4, significantly attenuated the central Ang II-induced dipsogenic response, showing the different roles of Nox2 and Nox4 in the regulation of hypertension and the dipsogenic response (153). Nox2 and its cytosolic components (p40phox, p47phox, and p67phox) are expressed in the rostral ventrolateral medulla (RVLM) of rabbits, and are increased by intracerebroventricular Ang II administration (57). Similar findings were described in rat RVLM (30), additionally showing that Ang II increased the serine phosphorylation of p47phox, suggesting NADPH oxidase assembly and activation. Lob et al. showed, in turn, that deletion of p22phox from the SFO brain region diminished the hypertensive response and eliminated peripheral vascular inflammation caused by Ang II (126). Inhibition of NADPH oxidase in the SFO repressed the cardiovascular and dipsogenic effects to intracerebroventricular administration of Ang II (208). In addition, there is compelling evidence that superoxide anion is necessary to elicit the vasopressor, bradycardic, and dipsogenic responses produced by the intracerebroventricular administration of Ang II (209). Selective deletion of superoxide dismutase 3 (SOD3) in the SFO specifically increases Ang II-induced vascular T-cell and leukocyte infiltration in addition to increasing sympathetic modulation of heart rate, blood pressure, and vascular O2 − (125). These studies provide compelling evidence for the role of ROS in the central regulation, not only of hypertension but also of vascular inflammation. However, we now need to clearly identify whether oxidative stress leads to increased sympathetic nervous system activation or whether it is the latter that controls induction of oxidative stress in hypertension, atherosclerosis, or other cardiovascular disorders.
NADPH Oxidase Inhibitors—Vascular Effects
Several classes of drugs currently used in clinical practice, such as medications affecting the renin-angiotensin-aldosterone system (133), statins (5, 162), and calcium channel blockers (203), have been shown to decrease NADPH oxidase activity and/or expression (76, 164). Moreover, some naturally occurring polyphenols may effectively inhibit NADPH oxidases in the vasculature or platelets, apart from simple scavenging of ROS (163). Understanding the importance of vascular NADPH oxidases and their potential value as therapeutic targets has sparked a search for specific and efficient Nox enzyme inhibitors. This is reviewed in depth elsewhere in this Forum. We have summarized selected vascular effects of NADPH oxidase inhibitors in Table 2. However, it should be noted that the specificity and selectivity of many of these compounds is under constant revision, and this aspect should be very carefully considered when designing new studies and trials.
17DMAG, 17-dimethylaminoethylamino-17-demethoxygeldanamycin; AEBSF, 4-(2-aminoethyl)-benzenesulfonylfluoride; Ang II, angiotensin II; DGLA, dihomo-gamma-linolenic acid; DPI, diphenylene iodonium; eNOS, endothelial nitric oxide synthase; HUVECs, human umbilical vein endothelial cells; ICAM-1, intercellular adhesion molecule 1; LDL, low density lipoprotein; MPO, myeloperoxidase; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NoXO1, Nox organizer protein 1; PDGF, platelet-derived growth factor; PPARγ, peroxisome proliferator-activated receptor gamma; ROS, reactive oxygen species; S17834, 6,8-diallyl 5,7-dihydroxy 2-(2-allyl 3-hydroxy 4-methoxyphenyl)1-H benzo(b)pyran-4-one; SH3, SRC Homology 3; SHR, spontaneously hypertensive rat; TNF-α, tumor necrosis factor-α; VCAM, vascular cell adhesion molecule; XJP-1, 7,8-Dihydroxy-3-methyl-isochromanone-4.
Conclusion
NADPH oxidase-derived ROS can act as vital signaling molecules or can cause toxicity depending on the time and cellular localization of Nox enzyme expression. This is particularly evident in the cardiovascular system. Therefore, a systematic and detailed characterization of NADPH oxidases in relation to the complex setting of cardiovascular diseases is warranted. Within recent years, we have further understood the complexity of this problem. For example, it is very difficult to draw a clear line between the protective and damaging effects of Nox4-derived ROS. In addition, discrepancies between studies looking at the consequences of Nox4 inhibition or Nox4 knockout should be further explained. We have also understood that expression and activity of NADPH oxidases should be very closely regulated, as either too much or too little of NADPH oxidase-derived ROS is detrimental. Thus, we need to learn how to tightly control/carefully affect their activity. Particularly, we need development of Nox oxidase-specific inhibitors, which would enable us to differentiate between various Nox homologues. Fortunately, much progress has been made in this field, and we are getting closer to this important goal.
Footnotes
Acknowledgments
This work has been supported by the National Science Foundation of Poland Grant No 2997/B/P01/2009/36 and the Foundation for Polish Science Grant for T.J.G. and A.S. (Welcome02/09). T.J.G. is supported by The Wellcome Trust Senior Research Fellowship.