
Abstract
Introduction
R
The present studies demonstrate the intrinsic repopulating defect of Parp1−/− hematopoietic stem cells (HSCs) under oxidative stress and identifies the tryptophan-glycine–arginine-rich (WGR) domain of poly(ADP-ribose) polymerase-1 (PARP1) required for Salidroside binding and PARP1 activation thereby preventing oxidative stress-induced premature HSC exhaustion. These findings not only provide a molecular explanation for Salidroside-stimulated PARP1 activation in HSC maintenance under oxidative stress, but also suggest new targets for therapeutically exploring the pathogenic role of oxidative stress in hematologic diseases.
Poly(ADP-ribosyl)ation (PARylation) is a post-translational protein modification by creating poly(ADP-ribose) (PAR) covalently attached onto target proteins that mediate gene transcription, DNA damage repair, and cell death signaling (4, 17, 23). Poly(ADP-ribose) polymerase-1 (PARP1) is the founding member of the PARP family with a highly conserved structure and six domains: three Zinc finger DNA-binding domain (DBD: Zn1, Zn2, and Zn3), the automodification domain (AD), the tryptophan-glycine–arginine-rich (WGR) domain, and the catalytic domain (CAT) that is composed of two subdomains—the helical subdomain (HD) and the ART subdomain (17, 23). It has emerged as a promising drug target for cancer therapy due to its role in maintaining genome stability (25). Although increasing evidence indicates that PARP1 is involved in oxidative DNA damage response (3, 7, 26, 45), how PARP1 functions in HSC maintenance under oxidative stress remains to be elucidated.
Salidroside, a phenylpropanoid glycoside, is the major active substance of Rhodiola rosea, which grows in dry and sandy ground and has been used traditionally and pharmacologically for a long period of time in many European countries in the 19th century (8, 38). The main use was as a brain tonic, as a roborant, and to alleviate headache (8, 38). Recently, it has been documented in experimental animals for protective effects of oxygen, cold, radiation, and heavy physical exercise (8), suggesting that Salidroside has various pharmacological properties, including antiaging, anticancer, anti-inflammation, hepatoprotective, and antoxidative effects (13, 16, 20).
We previously showed that Salidroside stimulated the PARP1 activity and protected HSCs from oxidative stress (26). To further understand the action of mechanism for Salidroside in PARP1 stimulation in the context of ODDR in HSCs, we constructed several truncated proteins lacking the defined functional domains of PARP1 and assessed the effect of these mutants in HSCs under oxidative stress and their binding to Salidroside. The results showed that Salidroside activated PARP1 through its binding to the WGR domain, while truncated proteins lacking the WGR domain or carrying R591K mutation in the WGR domain failed to bind and respond to Salidroside under oxidative stress. Functionally, Salidroside stimulated PARP1 activation in Parp1−/− HSCs expressing the wild-type (WT) PARP1 and prevented HSCs from H2O2-induced cycling and repopulating exhaustion. Taken together, we identified a specific binding domain of PARP1 required for Salidroside-stimulated PARP1 activation and a crucial role for PARP1 in maintaining HSC function under oxidative stress.
Results
Genetic dissection of Salidroside action on HSC function under oxidative stress
We previously showed that activation of PARP1 by Salidroside, a phenylpropanoid glycoside isolated from the medicinal plant R. rosea prevents the loss of HSCs in native mice and rescues HSCs repopulating in transplanted recipients under oxidative stress (26). To provide genetic evidence that Salidroside protects HSCs from oxidative stress through stimulating the PARP1 activity, we employed Parp1-deficient mice (Parp1−/−
). We first determined the optimal dose of hydrogen peroxide (H2O2); consistently with the previous finding (26), we found that 0.25 μmol/g body weight was the optimal dose that effectively induced oxidative stress, as evidenced by a dose-dependent increase in ROS (Supplementary Fig. S1A; Supplementary Data are available online at
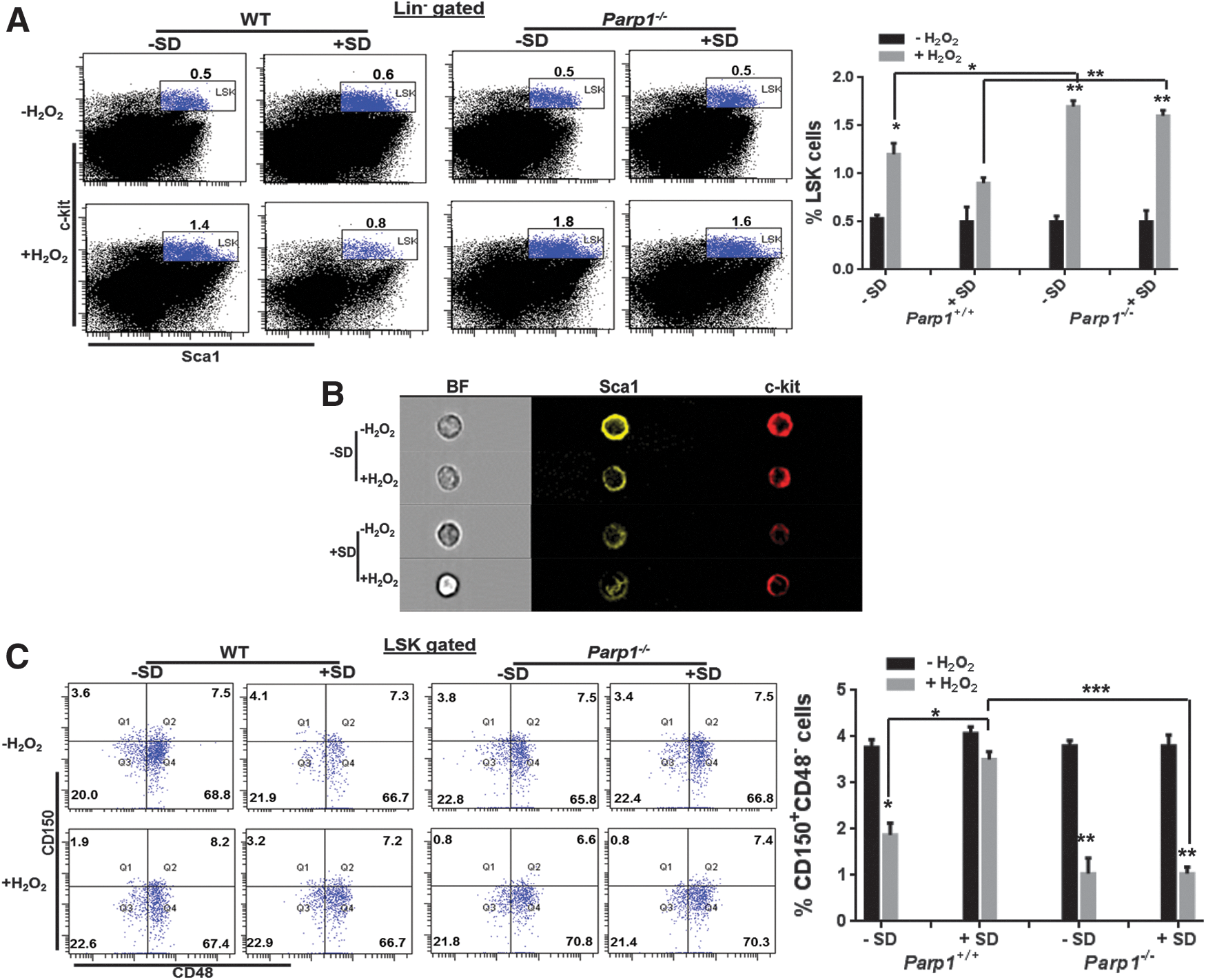
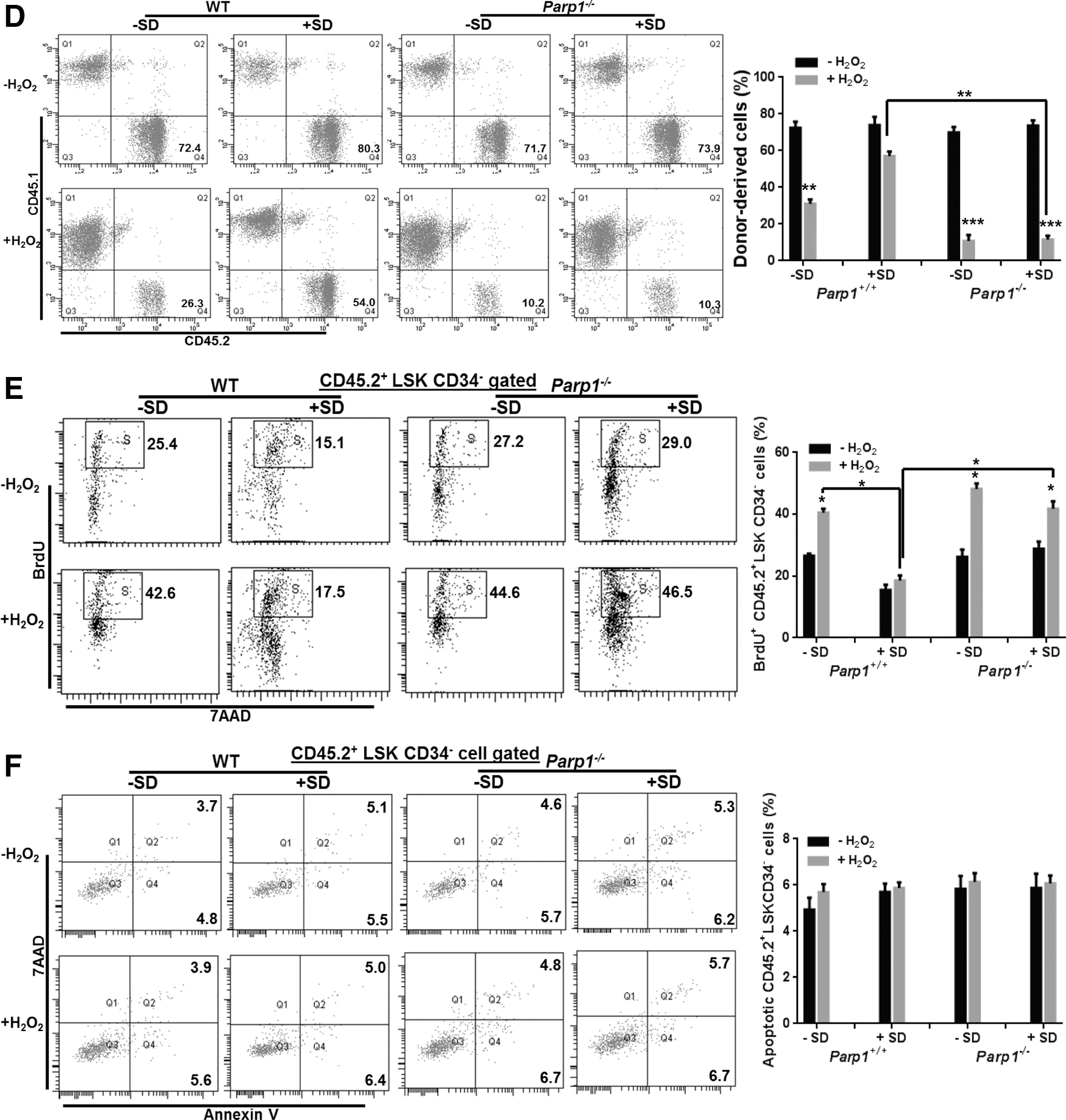
We then determined whether Salidroside could improve the repopulating ability of oxidative stressed Parp1−/− HSCs by transplanting LSK cells from H2O2-treated WT C57BL/6 mice or Parp1−/− mice (CD45.2+) pretreated with or without Salidroside into lethally irradiated congenic recipients (CD45.1+). We observed a significant decrease of donor-derived chimera (CD45.2+) in the recipients injected with LSK cells from H2O2-treated WT mice, which was partially reversed by Salidroside at 4 months after transplantation (Fig. 1D). Although HSCs from vehicle-treated Parp1−/− mice gave rise to comparable donor-derived chimera (72.37%±5.95% in the WT group and 69.93%±5.09% in the Parp1−/− group) in the recipients, oxidative stress compromised the repopulating capacity of Parp1−/− HSCs (10.83%±5.58%) more when compared with WT HSCs (31%±4.11%). Salidroside failed to restore the ability of stressed Parp1−/− HSCs in reconstituting mouse hematopoiesis in the recipient mice (11.5%±3.84%). Mechanistically, Salidroside appeared to prevent HSCs from oxidative stress-induced cycling (Fig. 1E) rather than apoptosis (Fig. 1F), as detected by bromodeoxyuridine (BrdU)/7-aminoactinomycin D (7AAD) and Annexin V/7AAD staining, respectively. These results collectively provide genetic evidence that PARP1 is required for the action of Salidroside in influencing HSC function under oxidative stress.
Salidroside binds to the WGR domain of PARP1
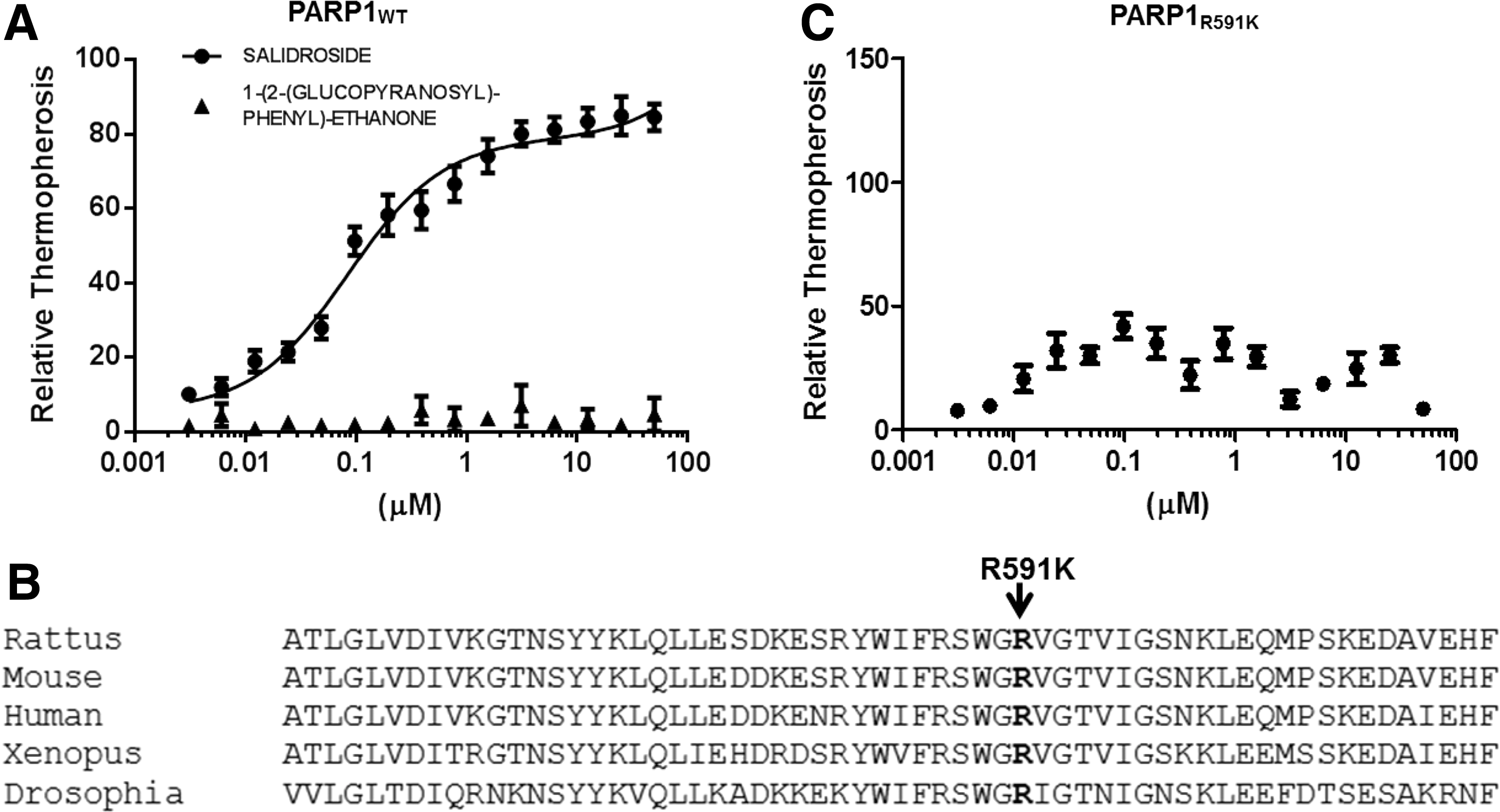
PARP1 has a highly conserved structural and functional organization, including an N-terminal zinc finger DBD, a nuclear localization signal, a central AD, and a C-terminal catalytic domain (4, 17, 23). We first determined whether Salidroside bound directly to PARP1. To this end, we constructed several truncated proteins lacking the defined functional domains of PARP1 (Fig. 2), and assessed their ability to bind to Salidroside. We employed microscale thermophoresis to determine the binding between Salidroside and the PARP1 derivatives. This technology probes for fluorescent changes in the hydration shell of molecules to measure protein–protein or protein–small molecule interactions with high sensitivity in near-native conditions (1). Thermophoresis of the protein in the presence of varying concentrations of chemicals was analyzed for 30 s. Measurements were performed at room temperature and standard deviation was calculated from three independent experiments. The results show that full-length PARP1 specifically bound Salidroside, but not 1-(2-(glucopyranosyl)-phenyl)-ethanone, a chemical with a similar structure to Salidroside (Supplementary Fig. S2), in titration assays and yielded a Kd value of ∼84 nM (Fig. 3A), while constructs without the WGR domain (PARP1 mutants deleted for C-terminus), including constructs 1 (PARP11–215, Zn1, and Zn2), 2 (PARP11ndash;375, Zn1, Zn2, and Zn3), 3 (PARP11–479), and 4 (PARP11–525, Zn1, Zn2, Zn3, and BRCT) failed to bind Salidroside (Supplementary Fig. S3 and Table 1). We also found that PARP1 mutants deleted for N-terminus, including constructs 5 (PARP1216–1014, N-terminal deletion of Zn1), 6 (PARP1376–1014, N-terminal deletion of Zn1 and Zn2), 7 (PARP1526–1014, WGR-CAT domain), and 8 (PARP1480–1014, DsDB-WGR-CAT domain, 37) had relatively comparable binding affinity to full-length PARP1 (Supplementary Fig. S3 and Table 1), suggesting that the C-terminal of PARP1 is required for Salidroside binding. To further reveal the binding between PARP1 and Salidroside, we mutated residue Arg591 in the WGR domain (Fig. 3B), which has been shown critical for the Zn1 DNA damage interface and the CAT (24). The result shows that mutagenesis of the WGR domain (R591K) disrupted the binding of PARP1 to Salidroside (Fig. 3C). Taken together, these results suggested that the WGR domain is required for Salidroside-PARP1 binding.
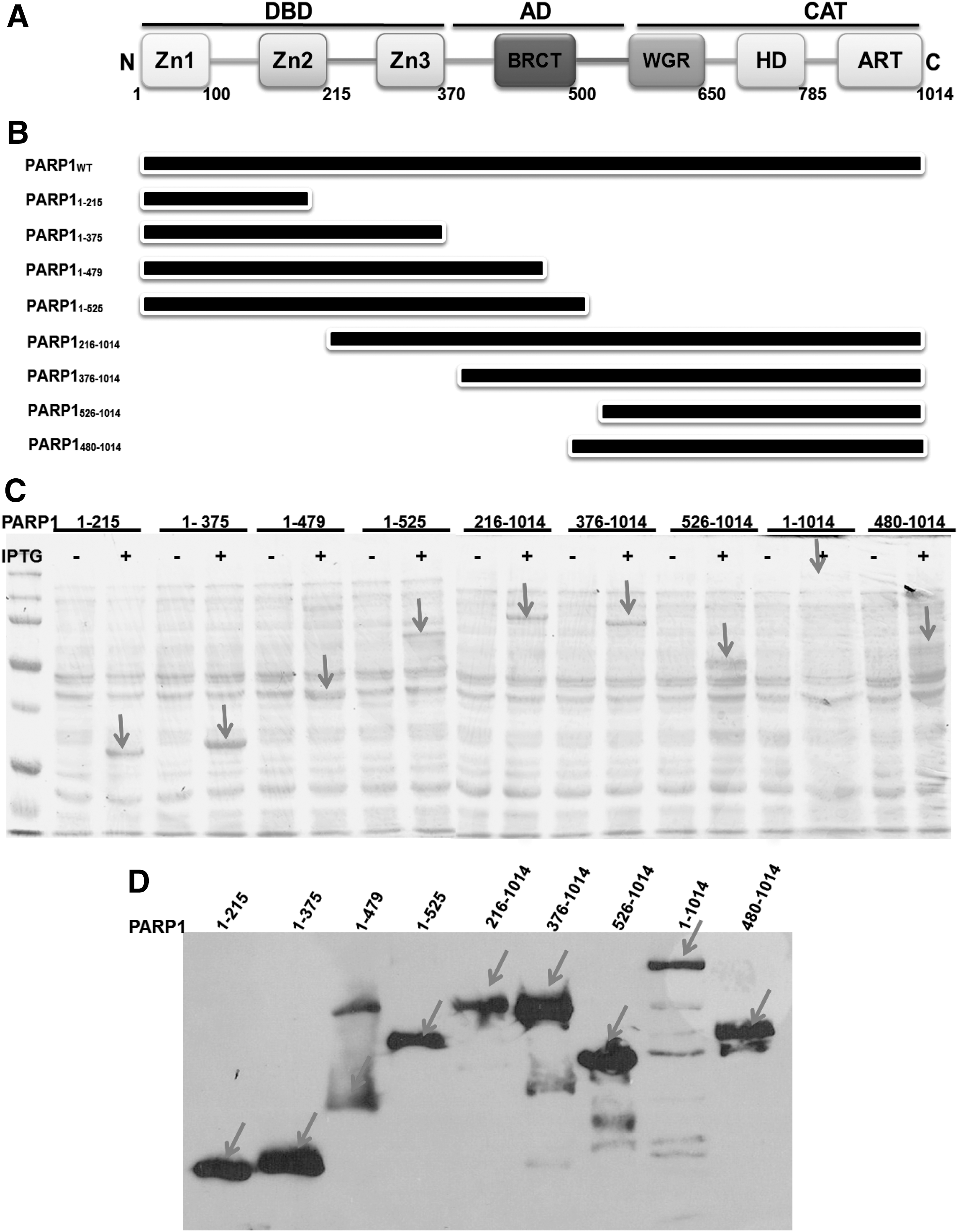
PARP1, poly(ADP-ribose) polymerase-1.
Salidroside activates PARP1 through binding to the WGR domain in vitro
To elucidate the action of mechanism for Salidroside in PARP1 stimulation, we isolated LSK cells from Parp1−/− mice and transduced them with the lentiviral vector expressing the Venus-only, Venus-PARP1WT, or Venus-PARP1R591K mutant. Transduction efficiency was comparable between groups (around 90%; Supplementary Fig. S4). We found that complementation of Parp1−/− cells with PARP1WT restored its activation under oxidative stress in response to Salidroside, as determined by in vitro PARP1 enzyme activity assay (Fig. 4A), which was correlated with accelerated DNA damage clearance as detected by immune blotting for γ-H2AX, a robust indicator of DNA strand breaks (Fig. 4B). Specifically, Salidroside stimulated PARP1 activity in LSK cells from Parp1−/− mice complemented with PARP1WT. Furthermore, the level of γ-H2AX decreased as early as 4 h after Salidroside treatment in Parp1−/− cells complemented with PARP1WT. In contrast, Parp1−/− cells expressing the WGR mutant (PARP1R591K) failed to respond to Salidroside and showed sustained accumulation of DNA damage up to 12 h post H2O2 treatment (Fig. 4B). Correspondingly, PARP1WT but not the PARP1R591K mutant restored in vitro the self-renewal capacity of Parp1−/− hematopoietic stem progenitor cells as determined by the colony-forming unit (CFU) assay (Fig. 4C). Together, these results indicate that Salidroside stimulates PARP1 activation through its binding to the WGR domain of PARP1.
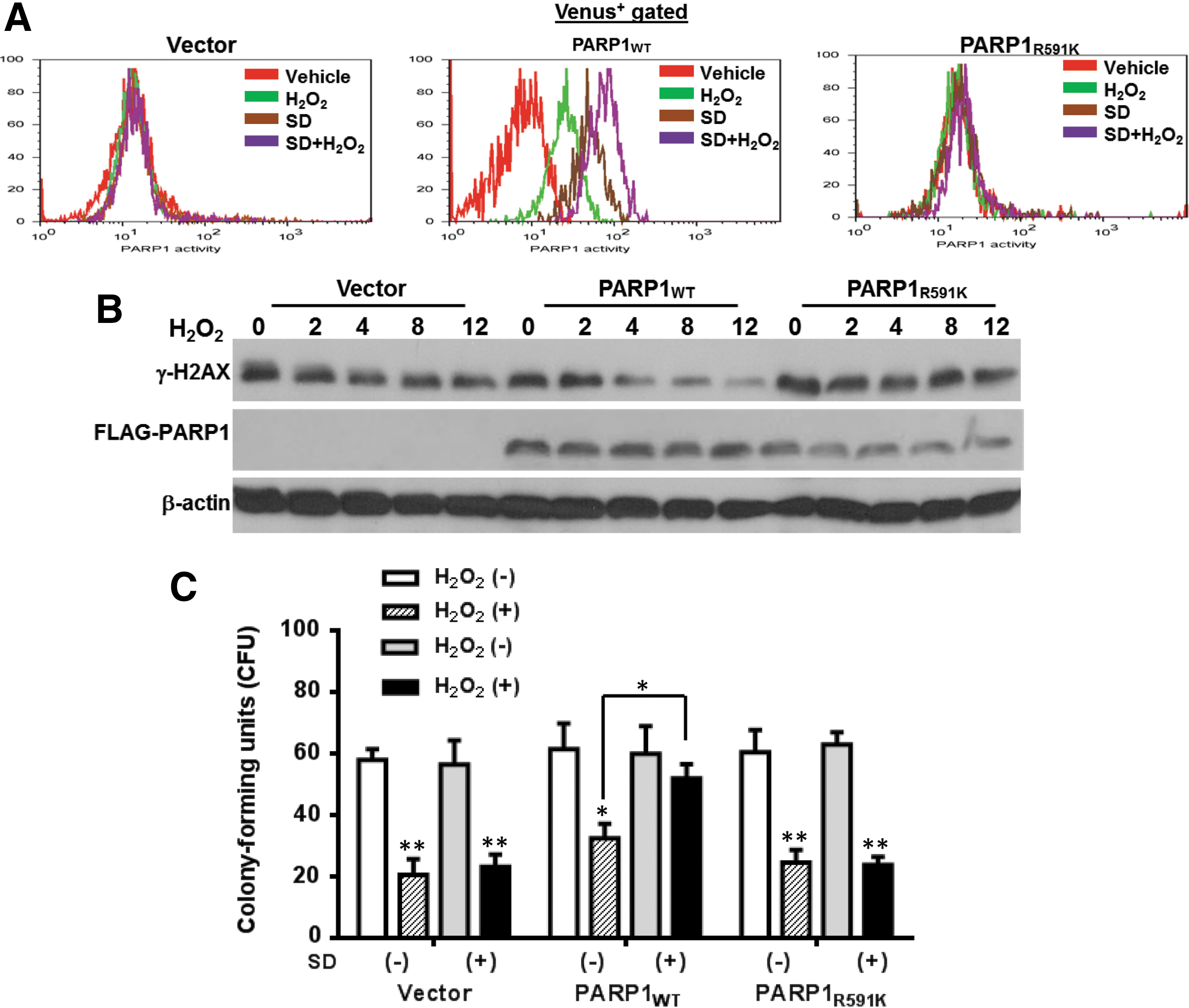
The WGR domain is critical for PARP1 activation by Salidroside in vivo
To substantiate the in vitro studies, we next sought to test whether the WGR domain of PARP1 was required for Salidroside-stimulated PARP1 activation in vivo. To this end, we transduced LSK cells isolated from Parp1−/− mice with lentiviral vectors expressing Venus-only, Venus-PARP1WT, or Venus-PARP1R591K mutant, followed by H2O2 treatment in the presence or absence of Salidroside. One thousand treated Venus+ LSK cells were then transplanted to lethally irradiated BoyJ recipients. Four months after bone marrow transplantation (BMT), donor-derived chimera was detected by flow cytometry. The results showed that cells expressing PARP1WT, but not the empty vector or the PARP1R591K mutant responded to Salidroside stimulation and succeeded in reconstituting recipient hematopoiesis, as determined by flow cytometry for CD45.2+ cell frequency in the BM of the recipient mice (Fig. 5A). We also examined the morphology of the donor-derived LSK cells after hematopoietic reconstitution by ImageStream analysis, and found no significant change in the morphological appearance of these LSK cells after reconstitution in the irradiated recipient mice (Fig. 5B).
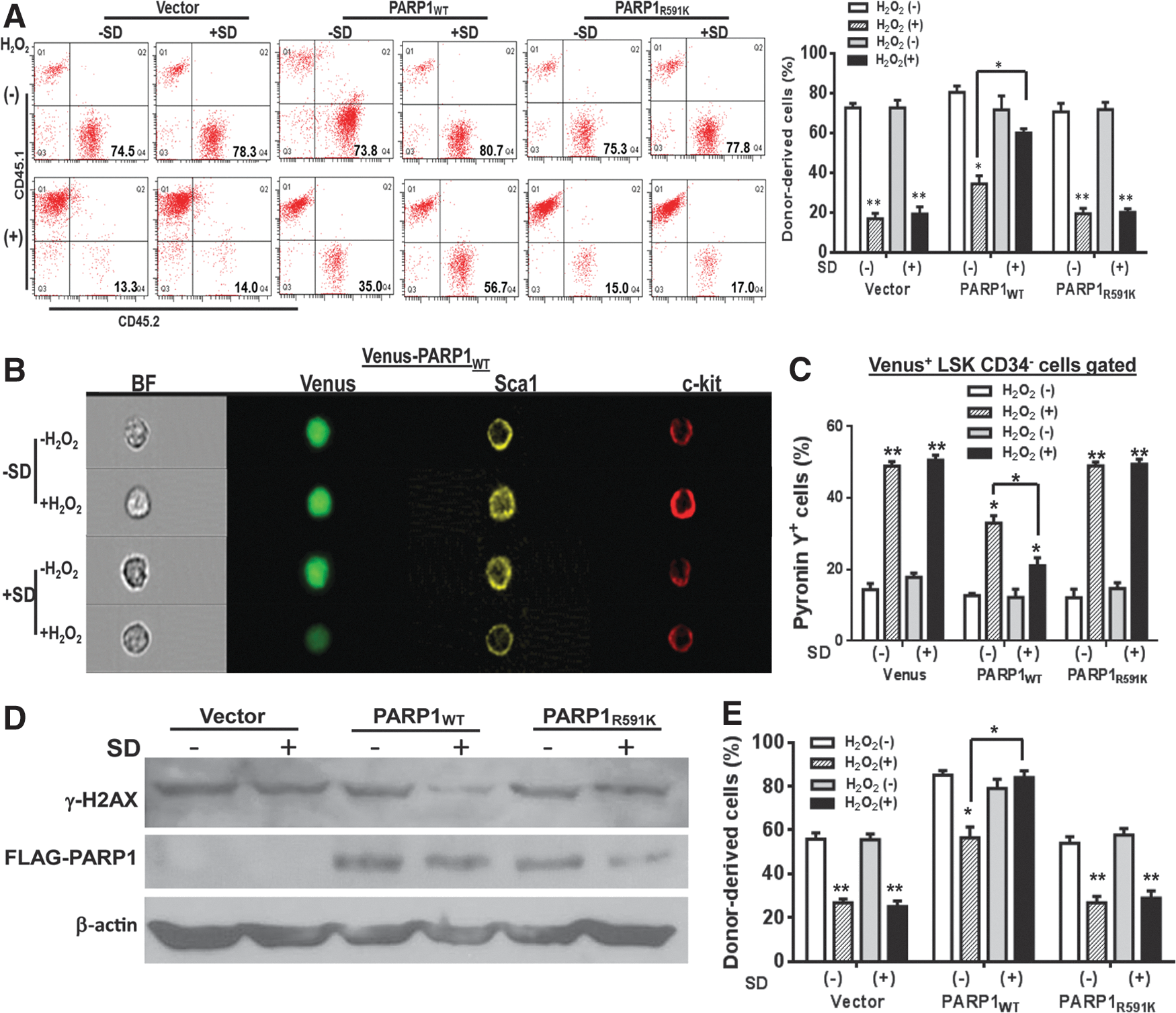
To provide functional evidence that binding to the WGR domain of PARP1 contributes to the antioxidant effects of Salidroside in HSC maintenance, we next determined the effect of WGR-Salidroside interaction on HSC quiescence in recipients transplanted with Venus-only-, Venus-PARP1WT-, or Venus-PARP1R591K-transduced LSK cells. We performed pyronin Y staining, an established dye to stain RNA and correlated to actively cycling cells (22). Upon H2O2 injection, we observed a significantly increased cell cycle progression (pyronin+) in empty vector- or the WGR mutant (PARP1R591K)-transduced Parp1−/− HSCs, which was not attenuated by Salidroside (Fig. 5C). Consistently, the level of DNA strand break marker, γ-H2AX, remained high in the presence of Salidroside in Venus-only or Venus-PARP1R591K mutant-transduced Parp1−/− HSCs (Fig. 5D). In contrast, PARP1WT-complemented Parp1−/− HSCs were prevented from H2O2-induced cycling by Salidroside (Fig. 5C) and showed reduced DNA damage (Fig. 5D). We also performed secondary BMT to confirm the functional relevance of binding to the WGR domain of PARP1 by Salidroside in LT-HSC maintenance. To this end, we transplanted 10 million whole bone marrow (WBM) cells from the Salidroside- and H2O2-treated primary recipients of Parp1−/− cells expressing Venus-only, PARP1WT, or the WGR mutant (PARP1R591K), into lethally irradiated secondary WT recipients. Four months post-transplantation, we analyzed hematopoietic reconstitution derived from donor (CD45.2+) cells. The results showed that Salidroside failed to enhance donor-derived hematopoiesis in recipients transplanted with Parp1−/− HSCs expressing the WGR mutant protein (Fig. 5E). Thus, the effect of Salidroside on HSC maintenance under oxidative stress requires the WGR domain of PARP1.
To explore the clinical significance of Salidroside in HSC maintenance in a disease model, we employed the Fanconi anemia (FA) mouse model, in which oxidative stress has been identified as a physiologic mediator of HSC loss (10, 29, 48). To this end, we treated Fanca−/− or Fancc−/− mice and their littermates (CD45.2+) with or without Salidroside followed by H2O2 injection. One thousand sorted LSK cells from the treated mice plus 1 million c-kit-depleted competitors (CD45.1+) were then transplanted into lethally irradiated recipients (CD45.1+). Consistent with previous reports that FA murine HSCs have defective reconstitution after replicative stress-generating BMT (48, 49), we found that Fanca−/− and Fancc−/− HSCs showed poor engraftment in the recipient mice and oxidative stress further compromised their reconstituting ability (Supplementary Fig. S5). Significantly, Salidroside enhanced the ability of FA HSCs in repopulating recipient hematopoiesis (Supplementary Fig. S5). These results suggest that Salidroside may have a therapeutic value for hematologic diseases like FA, where oxidative stress plays a pathogenic role.
Discussion
The present study investigated the role of PARP1 in oxidative DNA damage responses and identified the WGR domain as the structural element for Salidroside binding to PARP1. We propose that WGR-Salidroside binding is critical for PARP1 activation and protects quiescent HSCs from oxidative stress-induced cycling and subsequent exhaustion. We have provided several lines of evidence to support the following notions: (i) Salidroside binds to the WGR domain of PARP1 and stimulates the PARP1 activity in vitro; (ii) R591K mutation in the WGR domain of PARP1 disrupts the binding of Salidroside to PARP1; (iii) complementation of Parp1−/− cells with WT PARP1, but not the WGR mutant restores the Salidroside-stimulated PARP1 activation and ODDR; (iv) Parp1−/− HSCs expressing WT PARP1, but not the WGR mutant collaborate with Salidroside in long-term hematopoietic repopulation under oxidative stress.
We found that HSCs deficient in the Parp1 gene are more sensitive to H2O2 treatment (Fig. 1), which leads to significant long-term HSC loss and HSC exhaustion. Recently, premature stem cell exhaustion due to accumulation of ROS or DNA damage in mice deficient in several oxidative stress responses, such as Atm, Fancc, Fancd2, FoxO, or DNA damage repair, such as Lig4, Dna-pk, Ku80, Xpd, mTR genes, has been reported (28, 43, 46). We further show that the phenylpropanoid glycoside from medicinal plants, Salidroside, prevents LSK expansion and excessive stem cell cycling in WT mice, but fails to protect Parp1−/− HSCs from oxidative stress. This finding was confirmed by BMT using Salidroside-treated Parp1−/− LSK cells. These results provide genetic evidence that the antioxidant effect of Salidroside on the function of HSCs requires PARP1.
Poly(ADP-ribose) polymerase-1 (PARP1) is an enzyme that catalyzes post-translational modifications with ADP-ribose polymers, which play critical roles in multiple cellular processes, including gene transcription, DNA damage detection/repair, and cell death signaling (35). The molecular architecture of PARP1 has been a subject of intensive investigation (17, 23, 27). Among the structural domains, the function of WRG domain is less known. Recent study indicates that the WGR domain is a central component of the complex, interacting with Zn1, Zn3, CAT, and DNA, forming a network of interdomain contacts that links the DNA damage interface to the CAT domain (23). Structurally, a key Zn1-WGR contact is a salt bridge formed between Asp45 of Zn1 and Arg591 of WGR domain. Arg591 also interacts with the HD domain of PARP1 (23). In this study, we determined the binding of Salidroside to a set of truncated PARP1 peptides by in vitro microscale thermophoretic analysis and found that Salidroside specifically bound full-length PARP1 or the truncated proteins containing the WGR domain. Furthermore, mutagenesis of R591K in the WGR domain disrupted the binding (Fig. 3 and Supplemenaty Fig. S3), suggesting that the WGR domain is required for Salidroside binding. Functionally, Salidroside stimulated PARP1 activation and ODDR in Parp1−/− HSCs expressing only the PARP1 protein containing a functional WGR domain under oxidative stress (Fig. 4). This stimulation also enhanced the repopulating capacity of the stressed HSCs in irradiated recipients (Fig. 5). Taken together, our results indicate that the WGR domain of PARP1 is the structural determinant for Salidroside-PARP1 binding and PARP1 activation in the context of stressed hematopoiesis.
Another interesting finding of our study is that Parp1−/− HSCs display increased cycling under oxidative stress, probably due to accumulation of unrepaired DNA damage. Genomic DNA integrity is a limiting factor in the maintenance of a functional HSC (28). Recent studies using mouse models deficient for homologous recombination, nonhomologous end-joining, nucleotide excision repair, and telomere maintenance pathways have shown the importance of DNA repair pathways in maintaining the HSC function (9, 11, 37, 39). Although studies have begun to connect the molecular functions of PARP1 to specific physiological and pathological outcomes (27, 35), the role of PARP1 in HSC maintenance remains to be elucidated. Our present results suggest that oxidative stress compromises the self-renewal capacity of Parp1−/− HSCs (Fig. 1D), which is associated with sustained DNA damage accumulation (Fig. 4B), increased HSC cycling (Fig. 1E, C). In addition, by employing the FA mouse model, a blood disease associated with oxidative stress, our present study accentuated the therapeutic potential of Salidroside in HSC maintenance. Our findings are also consistent with recent studies that an intact DNA repair network is required for oxidative stress-induced DNA damage repair and HSC maintenance (28, 30, 34, 46). Although further studies are required to reveal the molecular mechanisms of PARP1 activation in protecting HSCs from oxidative stress-induced DNA damage, it is intriguing that Salidroside-stimulated PARP1 activity contributed to the accelerated repair of oxidative DNA damage in the stressed HSCs and the enhanced repopulating capacity in the recipients. In this context, our data lend support to the notion that DNA repair is essential for the maintenance of HSC self-renewal capacity (9, 11, 37, 39).
Materials and Methods
Mice
129SParp1tm1zqw /J (The Jackson Laboratory, Bar Harbor, ME) was backcrossing with WT C57BL/6 mice for eight generations before interbreeding heterozygous Parp1+/− mice to generate Parp1−/− mice. Fanca−/− and Fancc−/− mice were generated by interbreeding the heterozygous Fanca+/− mice (C57BL/6) (provided by Dr. Madeleine Carreau at Laval University) (44) or Fancc+/− mice (provided by Dr. Manuel Buchwald at Hospital for Sick Children, University of Toronto) (6), respectively. All the animals, including BoyJ mice, were maintained in the animal barrier facility at the Cincinnati Children's Hospital Medical Center.
Chemicals
For in vivo experiments, mice were injected with Salidroside (75 μg/g body weight; PhytoLab, Vestenbergsgreuth, Germany), or saline vehicle, intraperitoneally (i.p.) followed by H2O2 (0.25 μmol/g body weight; Sigma-Aldrich, St Louis, MO) (26). All experimental procedures conducted in this study were approved by the Institutional Animal Care and Use Committee of Cincinnati Children's Hospital Medical Center.
For thermophoresis analysis, Salidroside or 1-(2-(glucopyranosyl)-phenyl)-ethanone (Sigma-Aldrich), which shares a similar chemical construct with Salidroside, was titrated (3–50,000 nM) to a constant amount of labeled truncated PARP1 proteins (100 nM).
Molecular cloning and materials
Sequences corresponding to different PARP1 domains were cloned into the pET32a vector (a gift from Dr. Masahiko Satoh at Laval University Medical Center) (12). Proteins were expressed in HMS174 (DE3) pLysS (Stratagene, Santa Clara, CA) and purified using nickel-nitrilotriacetic acid agarose gel chromatography (Qiagen, Valencia, CA). In addition to His tag, FLAG tag was also attached at the N-terminus of each construct.
For lentiviral vector construction, FLAG-tagged full-length PARP1 cDNA was cloned into the pRRL-SIN-cPPT-MNDU3-MCS-IVW (TMND-IRES-Venus) vector (a gift from Dr. Punam Malik at the Cincinnati Children's Hospital Medical Center), which is a HIV-based self-inactivating (SIN) lentiviral vector containing the central polypurine and termination tract (cPPT). The expression of PARP1 is controlled by a modified MNDU3 promoter. The vector also carries the internal ribosome entry site (IRES) followed by Venus and woodchuck hepatitis virus post-transcriptional regulatory element (WPRE).
Mutagenesis was carried out using the QuickChange Lightening Site-Directed Mutagenesis Kit (Stratagene).
Microscale thermophoretic analysis
Purified different truncated PARP1 proteins were first labeled with Alexa-647 fluorescence dye (NanoTemper, München, Germany). Salidroside or 1-(2-(glucopyranosyl)-phenyl)-ethanone were titrated (3–50,000 nM) to a constant amount of labeled truncated PARP1 proteins (100 nM). The reaction was performed in 50 mM HEPES, 50 mM NaCl, 0.01% Tween20, and 2 mM MgCl2. Then, the samples were incubated in room temperature for 1 h before analyzing by microscale thermophoresis. A NanoTemper Monolith Instrument (NT.015; NanoTemper) was used for measuring thermophoresis. In this instrument, an infrared laser (IR-laser) beam couples into the path of light (i.e., fluorescence excitation and emission) with a dichroic mirror and is focused into the sample fluid through the same optical element used for fluorescence imaging. The IR-laser is absorbed by the aqueous solution in the capillary and locally heats the sample with a 1/e2 diameter of 25 μm. Up to 24 mW of laser power was used to heat the sample without damaging the biomolecules (42).
To analyze the thermophoresis of a sample, 4 μl was transferred in a hydrophilic-treated glass capillary (NanoTemper). Thermophoresis of the protein in the presence of varying concentrations of compound was analyzed for 30 s. Measurements were performed at room temperature and standard deviation was calculated from three independent experiments. Binding data were analyzed using Graphpad Prism to estimate Kd values. Data were normalized to either ΔFnorm [‰] (10*(Fnorm(bound) – Fnorm (unbound))) or Fraction bound (ΔFnorm [‰]/amplitude).
PARP1 enzyme activity
The PARP1 enzyme activity was detected following the established protocol (PARP1 Enzyme Activity Assay; Millipore, Billerica, MA) and the previous publications (21, 26) for the measurement of cellular poly(ADP-ribosyl)ation capacity. Briefly, cells (2×105 per data point) were centrifuged at 326 g for 5 min and resuspended in 100% ethanol and left at −20°C for at least 20 min. Cells were then permeabilized in 10 ml of buffer A (10 mM Tris-HCl pH 7.8, 1 mM ethylenediaminetetraacetic acid [EDTA], 4 mM MgCl2, and 30 mM 2-mercaptoethanol). Then, cells were centrifuged again at 815 g for 10 min and resuspended in buffer A again and transferred to a V-shaped 96-well plate on ice for at least 5 min. Then, 20 μl of 3× reaction buffer (with or without NAD+) plus 13 μl of 15 mM NaCl incorporating were added to the reaction mix followed by a 37°C incubation for 10 min. Controls (cells with a reaction buffer in the absence of NAD+) were always included in all the experiments to define the background. Then, second fixation was done by adding 60 μl of 4% formaldehyde/phosphate-buffered saline (PBS) for 20 min at room temperature. Sixty microliters of 100 mM glycine in PBS was then added to quench the reaction. Cells were then centrifuged at 815 g for 10 min and resuspended in 100 μl of primary antibody (anti-PARP1 antibody; Acris, San Diego, CA) diluted in a fluorescence-activated cell sorter (FACS) buffer (BD Biosciences, San Jose, CA) and incubated at 37°C for 1 h or overnight at 4°C. Then, the cells were washed and resuspended in 100 μl of diluted secondary antibody (PE-conjugated goat anti-mouse; Invitrogen, Grand Island, NY) followed by a 37°C incubation for 30 min in the dark. Cells were washed and resuspended for flow cytometry analysis. Similar levels of background were detected between samples.
Flow cytometry and cell cycle analysis
The lineage marker (Lin) mixture (BD Biosciences) for BM cells from treated or untreated mice included the following biotinylated antibodies: CD3ɛ (145-2C11), CD11b (M1/70), CD45R/B220 (RA3-6B2), mouse erythroid cells Ly-76 (Ter119), and Ly6G and Ly-6C (RB6-8C5). Other conjugated antibodies (BD Biosciences, San Jose, CA) used for surface staining included CD45.1 (A20), CD45.2 (A104), Sca1 (D7), c-kit (2B8), CD48 (HM48-1), and CD150 (9D1). Biotinylated primary antibodies were detected by incubation of antibody-coated cells with streptavidin-PerCP or fluorescein isothiocyanate (FITC) (BD Biosciences) in a two-step staining procedure. The percentage of apoptotic cells was determined by Annexin V and 7AAD staining (BD Biosciences) according to the manufacturer's instructions. For some of the experiments, pacific blue-conjugated CD45.2 (A104; BioLegend, San Diego CA) was used to determine donor-derived cells. For cell cycle analysis, cells were surface stained, fixed, and permeabilized in the BD Cytofix/Cytoperm Buffer (BD Biosciences), then stained with 5 μg/ml Hoechst 33342 and 150 ng/ml Pyronin Y (Sigma-Aldrich). Cells were then subjected to Flow Cytometric analysis (BD Biosciences).
BrdU incorporation assay
Subject mice were injected with BrdU (150 μl of 10 mg/ml) (40) i.p. followed by BM cells isolation 14 h later. BrdU-incorporated cells (S phase) were analyzed with the FITC BrdU Flow Kit (BD Biosciences), following the manufacturer's instructions. Briefly, cells were surface stained, then fixed, and permeabilized by the BD Cytofix/Cytoperm Buffer. After 1 h of incubation with DNase at 37°C, cells were stained with the APC-conjugated anti-BrdU monoclonal antibody. 7-AAD was added to each sample right before the flow cytometry analysis (BD Biosciences).
CFU assay
LSK cells isolated from Parp1−/− mice were transduced with lentivirus-expressing indicated construct. One hundred of transduced cells were seeded in duplicate and cultured in a cytokine-supplemented methylcellulose medium (MethoCult 3434; Stem Cell Technologies, Vancouver, Canada) in 35-mm gridded dishes. Colonies were counted on day 7.
Bone marrow transplantation
One thousand sorted LSK cells from H2O2-treated mice (CD45.2+) pretreated with or without Salidroside, along with 1 million BM cells (depleted of c-Kit+ cells to provide short-term hematopoiesis after irradiation using CD117 MicroBeads (Miltenyi Biotec, Auburn, CA) obtained from congenic WT mice (CD45.1+) were injected into lethally irradiated (11 Gy) congenic recipients (CD45.1+) by tail vein injection. Hematopoietic reconstitution in recipient mice by donor (CD45.2+) cells at 8 and 16 weeks post-transplantation was determined by the CD45.1-PE and CD45.2-FITC marker staining followed by flow cytometry FACS Canto I (BD Biosciences) analysis. For second transplantation, 10×106 bone marrow cells (BMCs) from the primary transplants (16 weeks after transplantation) were injected into lethally irradiated congenic (CD45.1+) recipients.
Preparation of cell extracts, immunoblotting
To prepare whole cell extracts, cells were washed with ice-cold PBS and resuspended in the ice-cold lysis buffer containing 50 mM Tris-HCL (pH 7.4), 0.1% NP40, and 1 M NaCl supplemented with protease and phosphatase inhibitors (10 μg/ml aprotinin, 25 μg/ml leupeptin, 10 μg/ml pepstatin A, 2 mM PMSF, 0.1 M NaP2O4, 25 mM NaF, and 2 mM sodium orthovandate) for 30 min on ice. Cell lysates were resolved on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), and immunoblots were analyzed with antibodies for the active form of γ-H2AX (Millipore, Billerica, MA), FLAG-M2, or β-actin (Sigma-Aldrich). Each lane contains protein from 30,000 cells. Signals were visualized by incubation with anti-mouse or anti-rabbit secondary antibodies followed by ECL chemiluminescence (Amersham Biosciences, Piscataway, NJ).
Measurement of oxidative stress
Surface marker-stained low-density bone marrow cells (LDBMCs) were incubated with CM-H2DCFDA (Molecular Probes, Eugene, OR; Invitrogen) in the dark for 15 min at 37°C. After washing, the cells were analyzed by flow cytometry using a FACSCanto (BD Biosciences). Data were analyzed using the FlowJo program (Tree Star, Ashland, OR).
Lipid peroxides were directly quantified using an ELISA assay that measures conversion of ferrous ions to ferric ions with the Lipid hydroperoxidase Assay Kit (Cayman, Ann Arbor, MI). Briefly, WBMCs isolated from H2O2-treated mice were resuspended with Extract R (Lipid hydroperoxidase Assay Kit) saturated methanol. Then, 1 ml of cold chloroform was added to the test tube followed by centrifugation at 1500 g for 5 min at 0°. Then, the bottom chloroform layer was collected by a syringe needle along the side of the test tube and transferred to another tube. The chromogen was prepared by mixing equal volumes of FTS Reagent 1 and FTS Reagent 2 in a test tube and vortex. Then, 50 μl of the freshly prepared chromogen was added to each assay tube and mixed well by vortexing followed by room temperature incubation for 5 min. Then, 300 μl of samples from each tube were transferred into the 96-well plate for the absorbance reading at 500 nm using SpectraMax Absorbance Microplate Reader (Molecular Devices, Sunnyvale, CA).
ImageStream analysis
LDBMCs isolated from Parp1−/− mice or donor-derived Venus+ cells from recipient mice, pretreated with or without H2O2 in the presence or absence of Salidroside, were stained for surface markers using antibodies against lineage markers (the lineage marker [Lin] mixture; BD Biosciences), Sca1, and c-kit (BD Biosciences) followed by ImageStream analysis (Amnis Corporation, Seattle, WA). The data were analyzed by IDEADS software (Amnis Corporation).
Statistical analysis
Paired or unpaired Student's t-test was used for two-group comparison and one-way ANOVA for more than two-group comparison. Values of p less than 0.05 were considered statistically significant. Results are presented as mean±SD. * Indicates p<0.05; ** indicates p<0.01; *** indicates p<0.001.
Footnotes
Acknowledgments
We thank Dr. Madeleine Carreau (Laval University) for the Fanca+/ − mice, Dr. Manuel Buchwald (Hospital for Sick Children, University of Toronto) for the Fancc−/− mice, Dr. Punam Malik (Cincinnati Children's Hospital Medical Center) for the lentiviral vector, Dr. Satoh Masahiko (Laval University Medical Center) for the pET32a-PARP1 plasmid, the Vector Core of the Cincinnati Children's Research Foundation (Cincinnati Children's Hospital Medical Center) for the preparation of lentivirus, and the Comprehensive Mouse and Cancer Core of the Cincinnati Children's Research Foundation (Cincinnati Children's Hospital Medical Center) for bone marrow transplantation service. This work was supported by the National Natural Science Foundation of China (81370608), the Guangdong Natural Science Foundation (S2013010013350), the Visiting Scholarships from South China Normal University, and the Shanghai Children's Medical center. WD is supported by a NIH T32 grant.
Authors' Contributions
XL, designed research, performed research, analyzed data, and wrote the article; OE, LL, QY, AW, performed research; WD, designed research, analyzed data, and wrote the article.
Author Disclosure Statement
The authors declare no conflicts of interest.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.