
Abstract
Introduction
H
Under normoxia, in each cell of the body, including neurons and astrocytes, HIF-1α is constitutively transcribed and translated into protein. A set of HIF prolyl hydroxylases (HIF-PHDs) destabilizes HIF-1α in normoxia by hydroxylating it at proline 402 and proline 564. HIF prolyl hydroxylation targets HIF-1α for recruitment and polyubiquitination by the Von Hippel-Lindau (VHL) E3 Ubiquitin ligase complex, leading to degradation by the proteasome (8, 21). Under conditions of hypoxia, the HIF-PHDs cease to function, and HIF-1α is, thus, not hydroxylated and degraded. The stabilized HIF-1α dimerizes with its partner, HIF-1β, translocates to the nucleus, and induces expression of a host of genes, including erythropoietin (EPO) and vascular endothelial growth factor (VEGF), both of which are neuroprotective (14, 18, 38, 41). Since the identification of this adaptive response, a number of groups, including our own, have established that activators of the HIF pathway are neuroprotective, especially those targeting HIF-PHDs (27, 35, 37, 40, 50).
By screening a hypoxia-inducible factor (HIF) reporter expressed in a hippocampal neuroblast line against a library of clinically approved drugs, we have defined antihelminthic benzimidazoles as novel inducers of the transcriptional adaptive response to hypoxia in neurons. In contrast to cancer cells where tubulin-binding drugs inhibit HIF activation, we have shown that the on-target effect of these drugs in activating hypoxic adaptation and protecting from oxidative neuronal death is free β-tubulin. Altogether, these results define a novel target for manipulating adaptive transcriptional responses in neurons and preventing oxidative death. Since antihelminthic benzimidazoles are utilized around the world for treatment of parasitic infections, our findings suggest that they could be employed readily in humans as hypoxia mimics and to treat brain injury associated with oxidative stress.
To identify novel activators of the HIF pathway in neurons with known safety in humans, we stably transfected a hypoxia response element (HRE) upstream of a luciferase reporter (HRE-Luc) in immortalized hippocampal neuroblasts. We subsequently screened these cells against a library of 1040 FDA-approved compounds, including drugs, nutriceuticals, and reference chemicals from MicroSource Spectrum Collection (Microsource Discovery, Inc.). Here, we describe the results of our screen and the identification of antihelminthic benzimidazoles as unexpected activators of the host adaptive response to hypoxia in neurons in vitro and in the brain in vivo. Since these compounds are extensively used in clinical practice, our findings suggest they could be interesting pharmacological candidates for the treatment of neurological conditions involving oxidative or hypoxic stress.
Results
Identification of FDA-approved activators of hypoxic adaptive response
To screen for activators of HIF-mediated transcription, we used a luciferase promoter-reporter construct driven by a 68 bp region of enolase promoter containing a wild type HRE (HRE-Luc) stably expressed in immortalized hippocampal neuroblasts (HT22-HRE) (Fig. 1A). We have previously shown that enolase is upregulated by hypoxia or deferoxamine (DFO) in primary cortical neurons at both mRNA and protein levels, and validated the reporter by showing that DFO (10 μM) induces a nearly twofold increase in luciferase activity in HT22 cells transiently transfected with HRE-Luc. Importantly, mutations in the HRE sequence abrogated the response to DFO, while mutations outside the HRE did not affect DFO-induced reporter activity (1, 61).
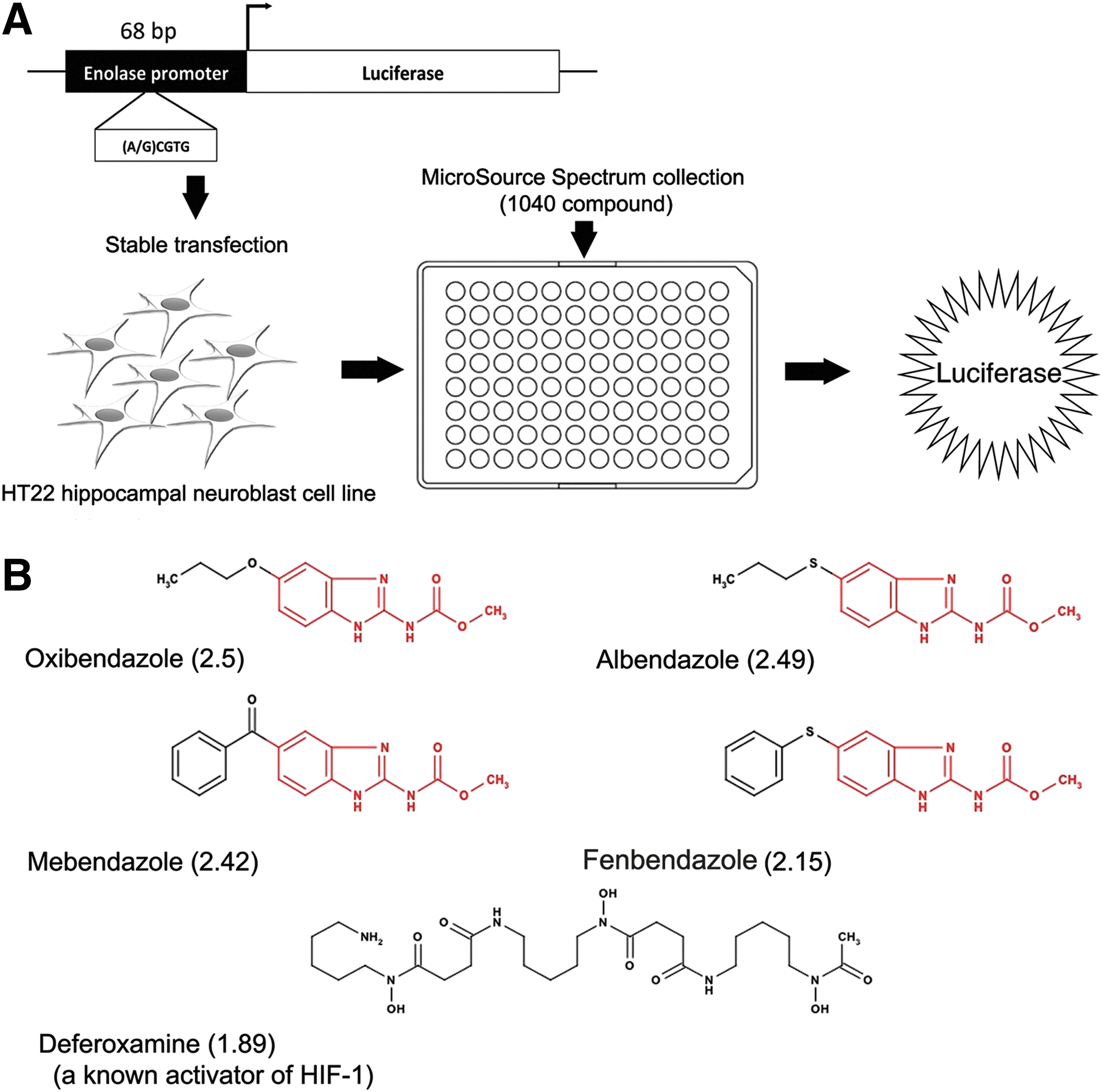
To identify inducers of HRE-dependent transcription with known safety in humans, we screened the 1040 compound Microsource Library Collection using the HRE-Luc-expressing HT22 cells. Each compound was added to cells in 96-well plates at a final concentration of 10 μM, including DFO as a positive control. Luciferase activity was normalized to cell viability in each well as measured by MTT assay. Using this strategy, we identified a number of known activators of the HIF pathway, including the anti-viral agent tilorone (45), the antifungal ciclopirox (28, 53), and the antihypertensive hydralazine (23), which validates our screening tools (Table 1). Of note, the antihelminthic benzimidazoles, a group of structurally similar compounds, show multiple hits: oxibendazole (2.5-fold), mebendazole (2.42-fold), albendazole (2.49-fold), and fenbendazole (2.15-fold). Interestingly, each drug induced a greater degree of HRE activity than DFO (1.89-fold at 10 μM) (Fig. 1B).
HIF, hypoxia-inducible factor.
Benzimidazoles stabilize HIF-1α and induce HIF-regulated gene transcription
HIF-1α stability is primarily governed by HIF-PHDs, which in the presence of oxygen hydroxylate HIF-1α within its oxygen-dependent degradation domain (ODD). To establish whether albendazole and fenbendazole interfere with HIF-PHD-regulated pathways, we tested these agents with a reporter containing the HIF-1α ODD fused to luciferase (ODD-Luc). The ODD-luc construct encoded in pcDNA3.1 plasmid vector was constructed as previously described (47). Both albendazole and fenbendazole increased ODD-Luc activity in a concentration-dependent manner in HT22 cells (Fig. 2A), while having no effect on a constitutive cytomegalovirus (CMV) luciferase construct (not shown). HRE activation customarily results from stabilization of HIF-1α followed by binding of a HIF-1α/HIF-1β heterodimer to the HRE. To establish whether increased activity of HRE-Luc reporter by benzimidazoles reflects the stabilization of HIF-1α protein in HT22 cells, we performed immunoblots using an antibody specific for HIF-1α. As a positive control, we used a 100 μM DFO, which was 10-fold higher than our screening dose of benzimidazoles. For validation purposes, we used albendazole and fenbendazole, two agents commonly applied in clinical practice in humans and animals. As shown in Figure 2B, they induce HIF-1α protein at the screening dose (10 μM) in HT22 cells.
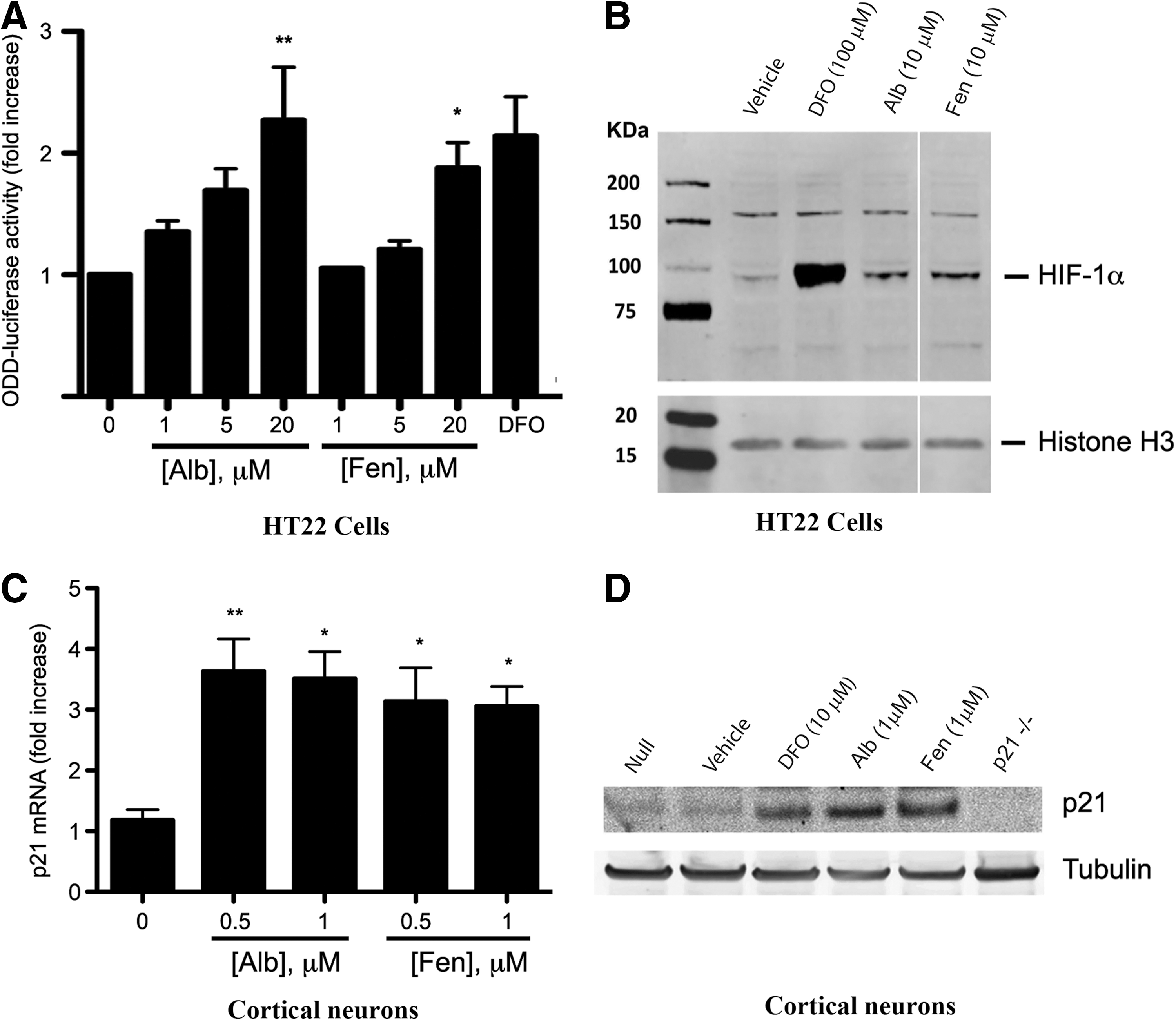
These findings suggest that benzimidazoles are interdicting an aspect of canonical oxygen-dependent signaling. To verify these results in post-mitotic neurons, we treated primary mouse cortical neurons with albendazole and fenbendazole, and found that each induced mRNA levels of p21cip1/waf1 (p21), an established HIF-1-regulated gene (Fig. 2C). As expected, albendazole and fenbendazole also increased p21 protein levels (Fig. 2D). Together, these data suggest that benzimidazoles induce HIF stability via a canonical HIF-PHD-regulated mechanism, resulting in increased HIF-dependent gene expression.
Benzimidazoles do not regulate HIF activity through inhibition of mitochondrial function
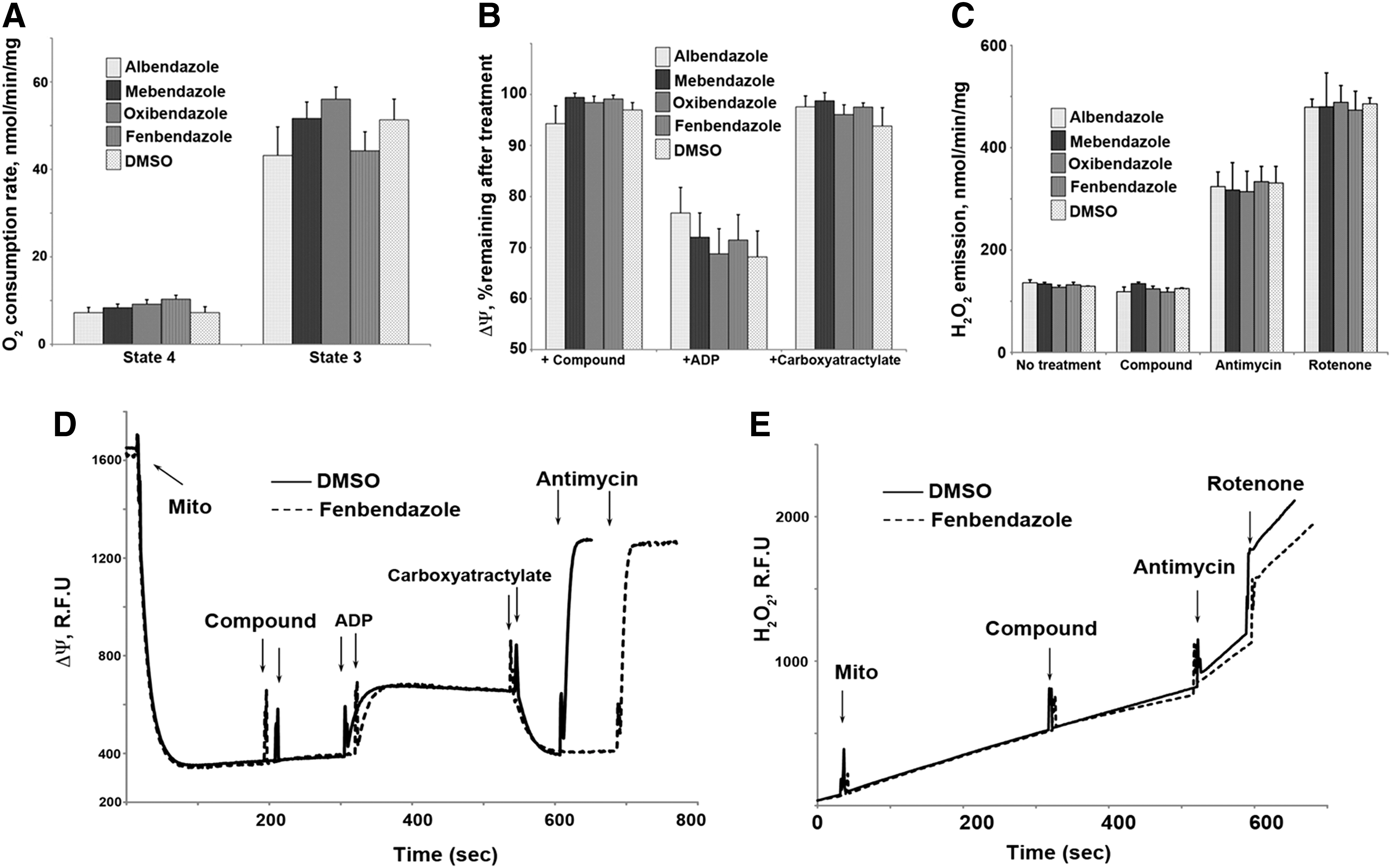
To understand how benzimidazoles might stabilize HIF, we focused on their known activity in killing parasites. One possibility is having global effects on mitochondrial function. To investigate the global effects on mitochondrial function, we measured mitochondrial membrane potential, oxygen consumption rate, and reactive oxygen species (ROS) production in isolated mitochondria and found that these parameters were unchanged by exposure to benzimidazoles (Fig. 3). A second mechanism is the inhibition of worm mitochondrial enzymes, such as fumarase, that results in energy depletion (33). In mitochondria, fumarase participates in the tricarboxylic acid (TCA) cycle and is essential for oxidative ATP synthesis. Inhibition of fumarase in worms can lead to mitochondrial dysfunction, immobilization, and death of the parasite (42). In humans, activity-inhibiting mutations of fumarase lead to an accumulation of fumarate and succinate in the cytoplasm and mitochondria. These metabolites are proposed to promote HIF-1α stability by inhibiting HIF-PHDs through competition with 2-oxoglutarate, a necessary cofactor for prolyl hydroxylase activity (17, 48). To determine whether benzimidazoles stabilize HIF-1α by inhibiting a TCA cycle enzyme, we measured the effect of benzimidazoles on fumarase activity. We found that of the four benzimidazoles tested, only mebendazole inhibited fumarase activity (Supplementary Fig. S1A; Supplementary Data are available online at
Benzimidazoles affect HIF stabilization/activity through tubulin-binding activity
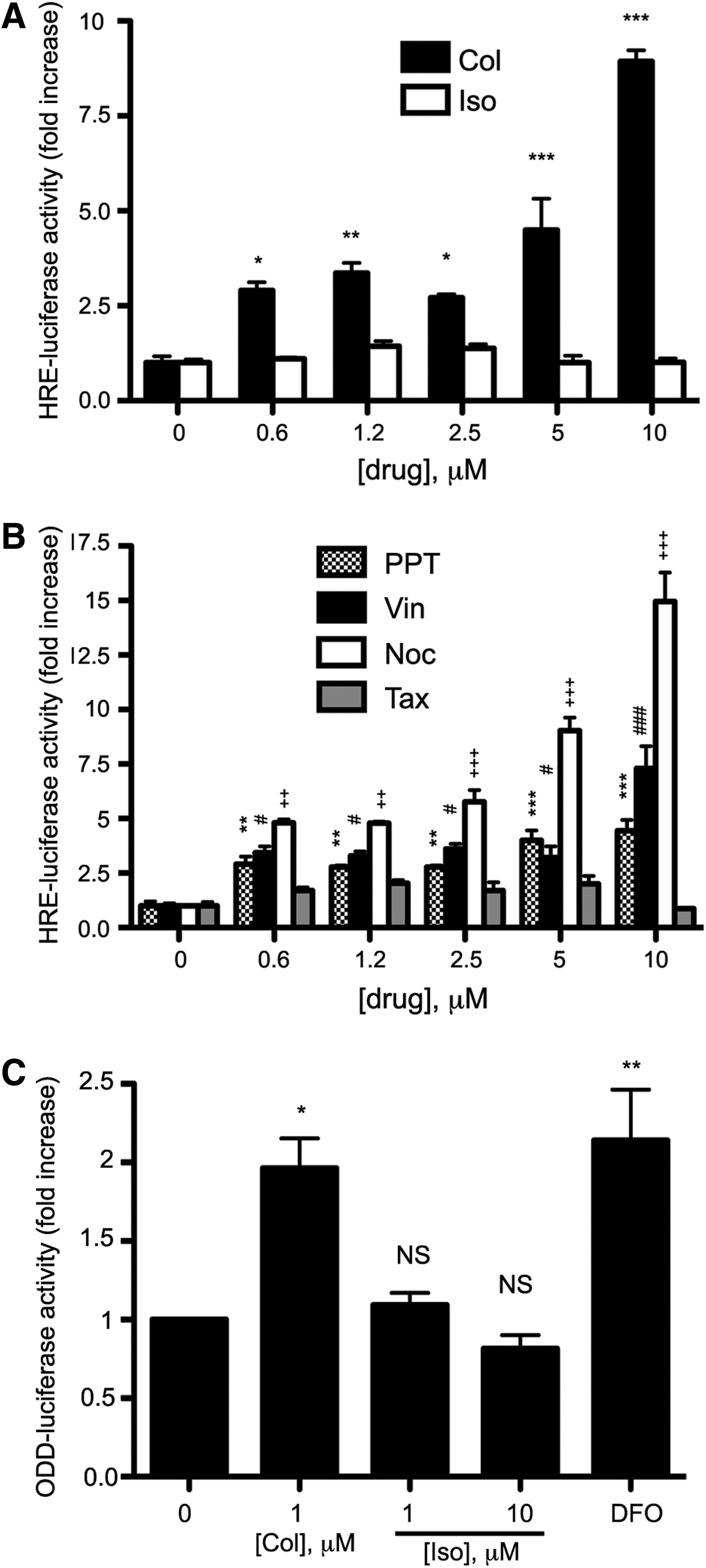
Another well-established and important target of antihelminthic benzimidazoles is tubulin (12, 25, 29). Antihelminthic benzimidazoles bind to tubulin and prevent its polymerization and subsequent formation of microtubules, although less robustly than classical microtubule depolymerizing agents (22) (Supplementary Fig. S2). Since microtubules are important cytoskeletal components, they are essential for neuron structure, growth, and axonal trafficking. Drugs and mutations that disrupt the dynamic equilibrium between free and polymerized tubulin may alter neuronal physiology, including cell-signaling pathways (7). In re-examining our hits (Table 1), we noted that colchicine, a prototypical tubulin-binding drug, also induced HRE-Luc activity to levels similar to benzimidazoles. To assess whether tubulin binding could be the on-target effect of benzimidazoles and colchicine in increasing HRE-Luc activity, we examined isocolchicine, a semi-synthetic structural isomer of colchicine with transposed C-ring carbonyl and methoxy groups. This structural modification decreases isocolchicine affinity for tubulin by 500-fold (16). Thus, if tubulin binding is an on-target effect, isocolchicine should not affect HRE-Luc activity at concentrations equimolar to colchicine. Indeed, while colchicine activated HRE-Luc in HT22 cells starting at 0.6 μM, isocolchicine had no effect (Fig. 4A). Based on this model, we predicted that other compounds which target the colchicine-binding site of tubulin would also induce reporter activity. Two such compounds, podophyllotoxin and nocodazole, were found to significantly increase HRE-Luc activity. Of note, we found that vinblastine, which depolymerizes microtubules by binding to a distinct site on β-tubulin, also activated the reporter in a dose-dependent manner. In contrast, Taxol, which stabilizes microtubules by binding to polymerized tubulin, had no effect (Fig. 4B). Consistent with these observations, colchicine, but not isocolchicine, increases ODD-Luc activity (Fig. 4C). This suggests that HIF-activation after treatment with tubulin-binding agents is through HIF-1α stabilization, likely via the inhibition of prolyl hydroxylation. Taken together, these observations suggest that benzimidazoles, similar to other agents which bind monomeric tubulin, induce HIF-dependent gene expression under normoxia by stabilizing HIF-1α through an ODD-dependent mechanism.
Benzimidazoles and other agents that bind to tubulin protect embryonic cortical neurons against oxidative stress
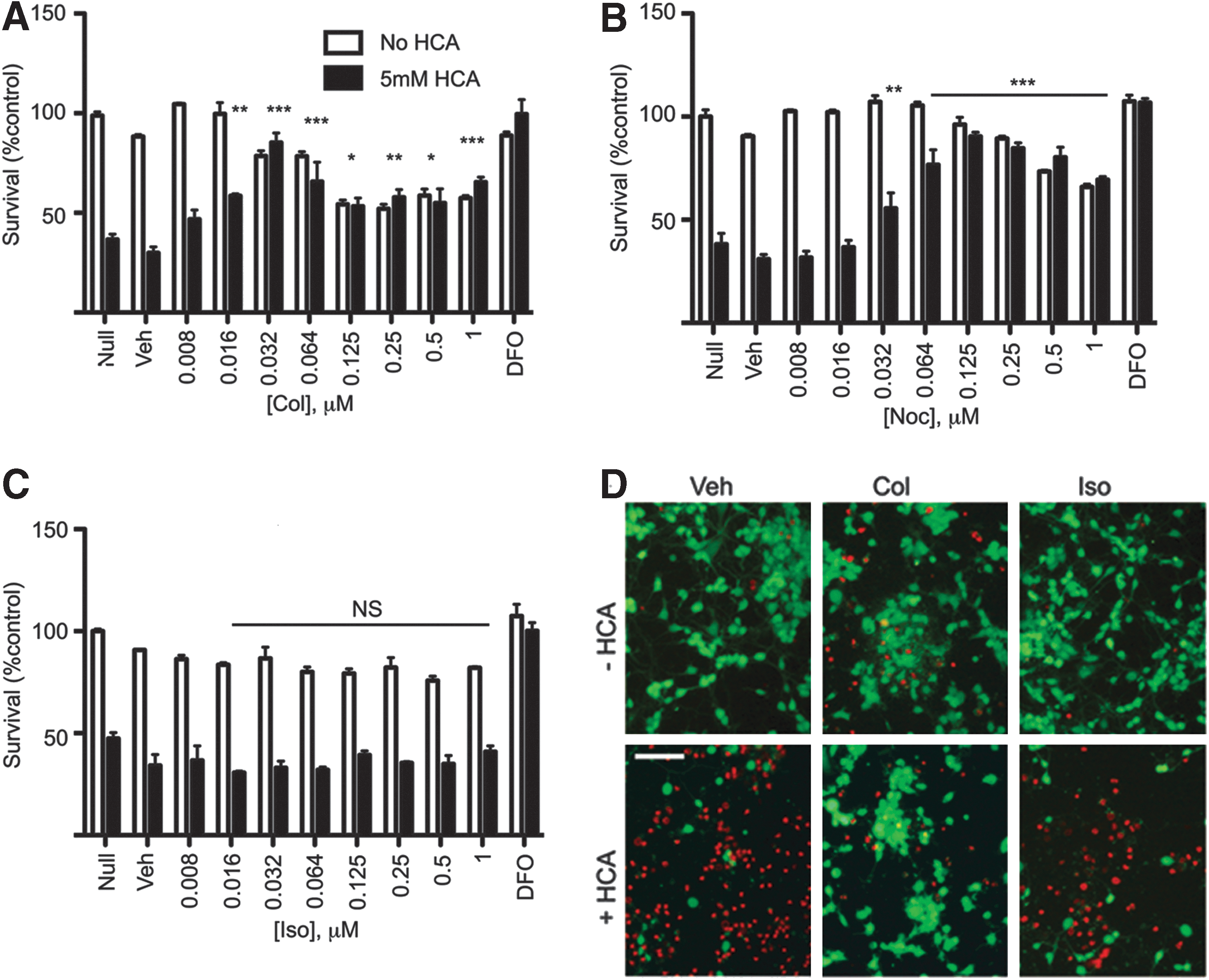
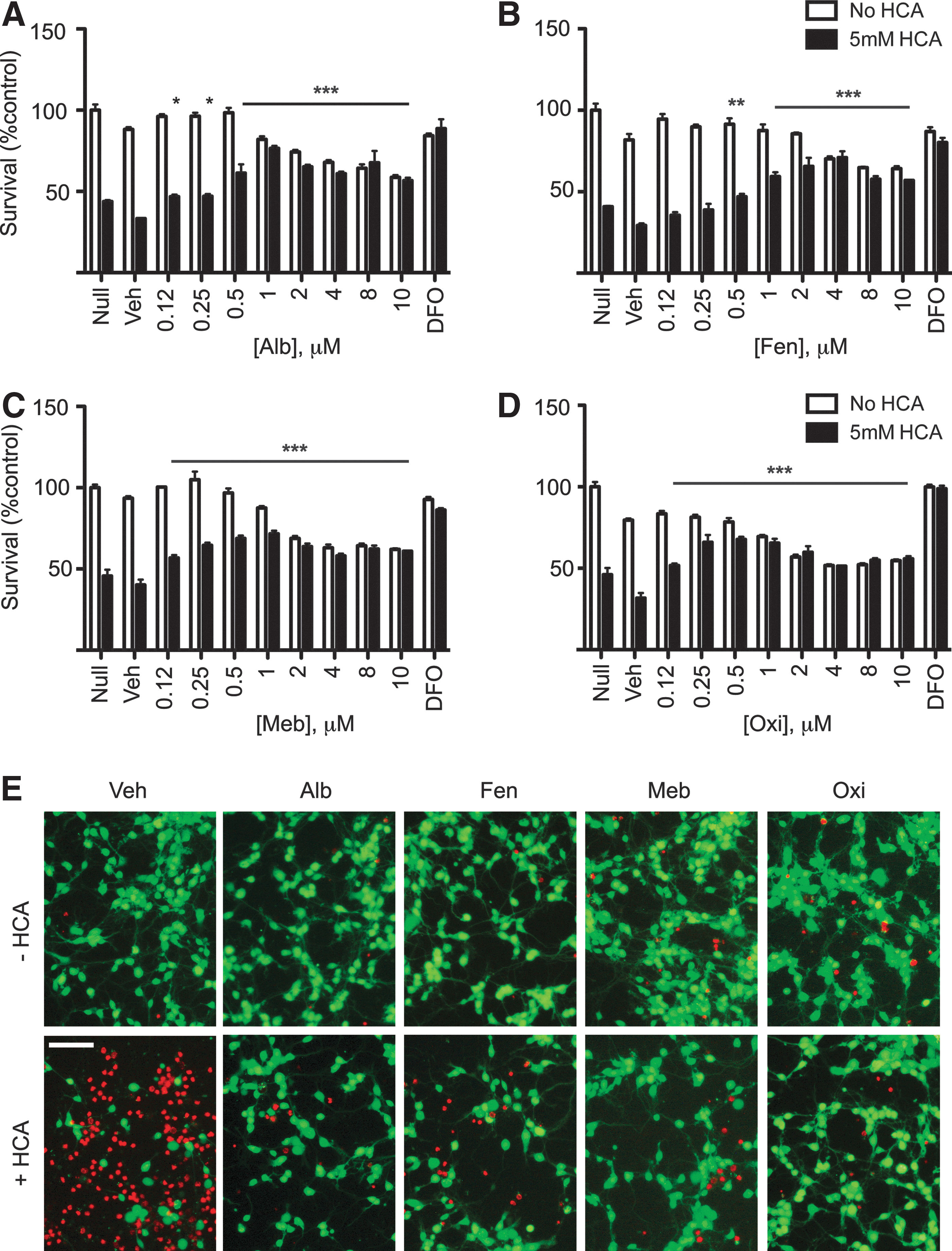
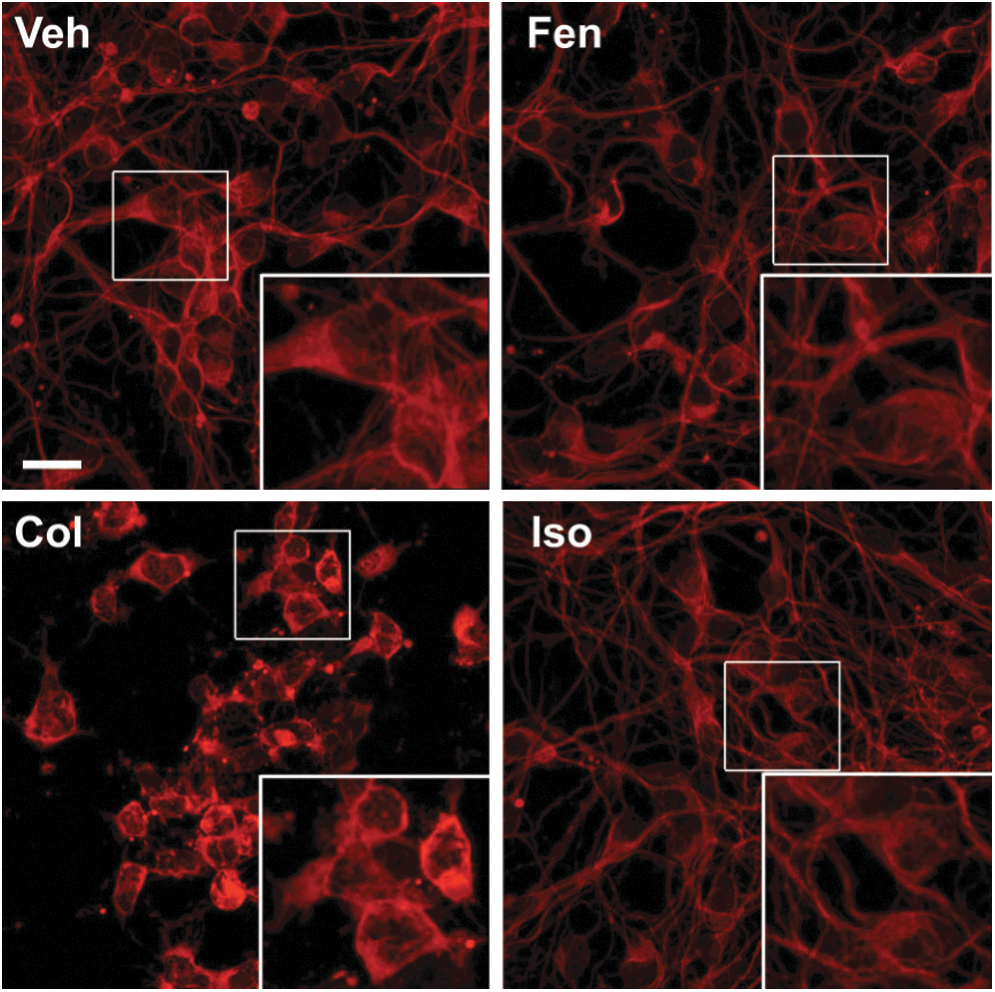
To examine whether activation of the hypoxic adaptive response by benzimidazoles is associated with neuroprotection, we tested them in a glutathione depletion model of oxidative stress in immature primary neurons using the glutamate analog, homocysteic acid (HCA). Extracellular HCA inhibits cystine uptake by the system Xc− antiporter, and depletes intracellular glutathione by reducing its precursor, cysteine. This leads to the disruption of the oxidant-antioxidant balance in favor of oxidants and, ultimately, leads to cell death. Our lab and others have used this model to identify a number of novel pathways and drugs, including inhibitors of the HIF-PHDs, that have shown neuroprotection in preclinical models of acute and chronic neurodegeneration (3, 26, 30, 44, 52). We found that the tubulin-binding drugs colchicine and nocodozole protected neurons from HCA-induced oxidative stress (Fig. 5A, B, and D), while isocolchicine had no effect at the same concentrations (Fig 5C, D). Each benzimidazole identified in our screen protected neurons against oxidative stress in a dose-dependent manner (Fig. 6A–E). However, higher concentrations of benzimidazoles (≥1 μM for oxibendazole and ≥2 μM for others) show a gradual negative impact on the basal viability. We also observed that while colchicine protects the soma during oxidative stress, it disrupts the neurites (Fig. 5D). To examine the effects of benzimidazoles on microtubule-dependent neuronal structures such as axons and dendrites, we stained embryonic cortical neurons after treatment with benzimidazoles with an antibody that recognizes a neuron-specific tubulin, βIII tubulin. Treatment with fenbendazole within the neuroprotective dose did not change the morphology of neurons (Fig. 7). Together, our data support a model in which tubulin-binding drugs, including benzimidazoles, activate hypoxic adaptation and show neuroprotective activity against oxidative stress without gross changes in neuronal morphology.
Pretreatment with Taxol inhibits neuroprotective activity of tubulin-binding agents
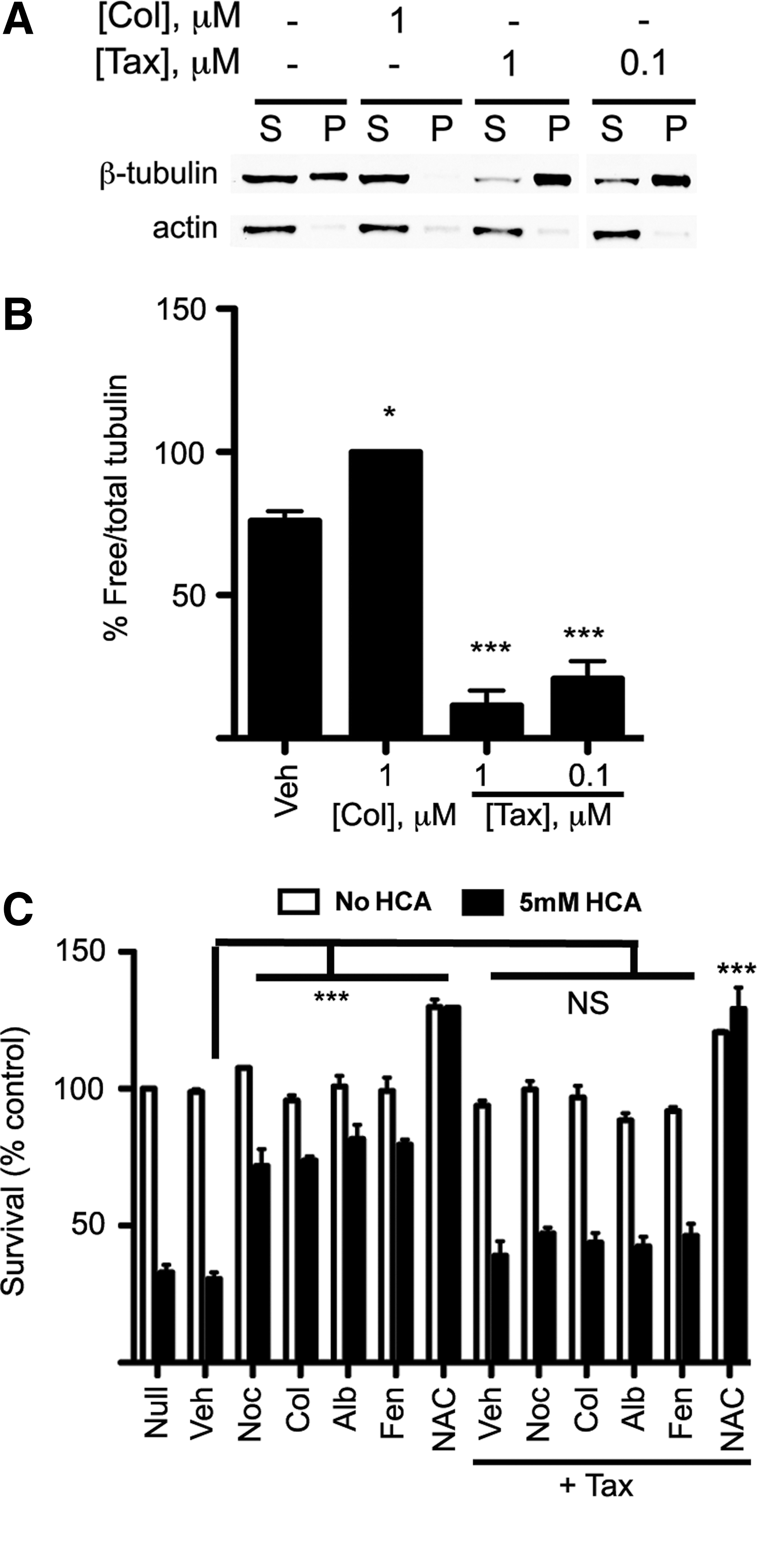
If binding to free tubulin by benzimidazoles is necessary for the initiation of the signaling cascade that activates hypoxic adaptation and neuroprotection from oxidative stress, then reduction of free tubulin should abrogate the protective actions of these drugs. To test the hypothesis, we first verified that Taxol reduces free tubulin levels in cortical neurons (Fig. 8A, B). We then pretreated primary neurons with 0.1 μM Taxol, 30 min before treatment with the tubulin-binding agents. This dose of Taxol does not cause significant neurotoxicity in cortical neurons (Fig. 8C). We used doses of tubulin-binding agents previously shown to provide maximum neuroprotection (Figs. 5 and 6). Taxol promotes microtubule stability, driving tubulin monomer into polymer (Fig. 8A); pretreatment with Taxol completely abrogated the neuroprotection conferred by tubulin-binding compounds, including benzimidazoles (Fig. 8C). To verify that reversal of protection was due to a specific effect of Taxol, we examined the effect of Taxol pretreatment on neuroprotection from HCA by an agent not known to bind tubulin, N-acetylcysteine (NAC). We have previously shown that NAC prevents glutathione depletion-induced death by enhancing intracellular glutathione levels (43). As expected, Taxol did not change the neuroprotective effect of NAC, suggesting that Taxol's ability to reverse protection is specific to that induced by drugs which bind to tubulin monomer. Moreover, the ability of Taxol to nullify both protective effects of classical tubulin binders and benzimidazoles is consistent with these agents working via a similar mechanism.
Mebendazole inhibits degradation of ODD-Luc, a surrogate marker of HIF activation, in vivo
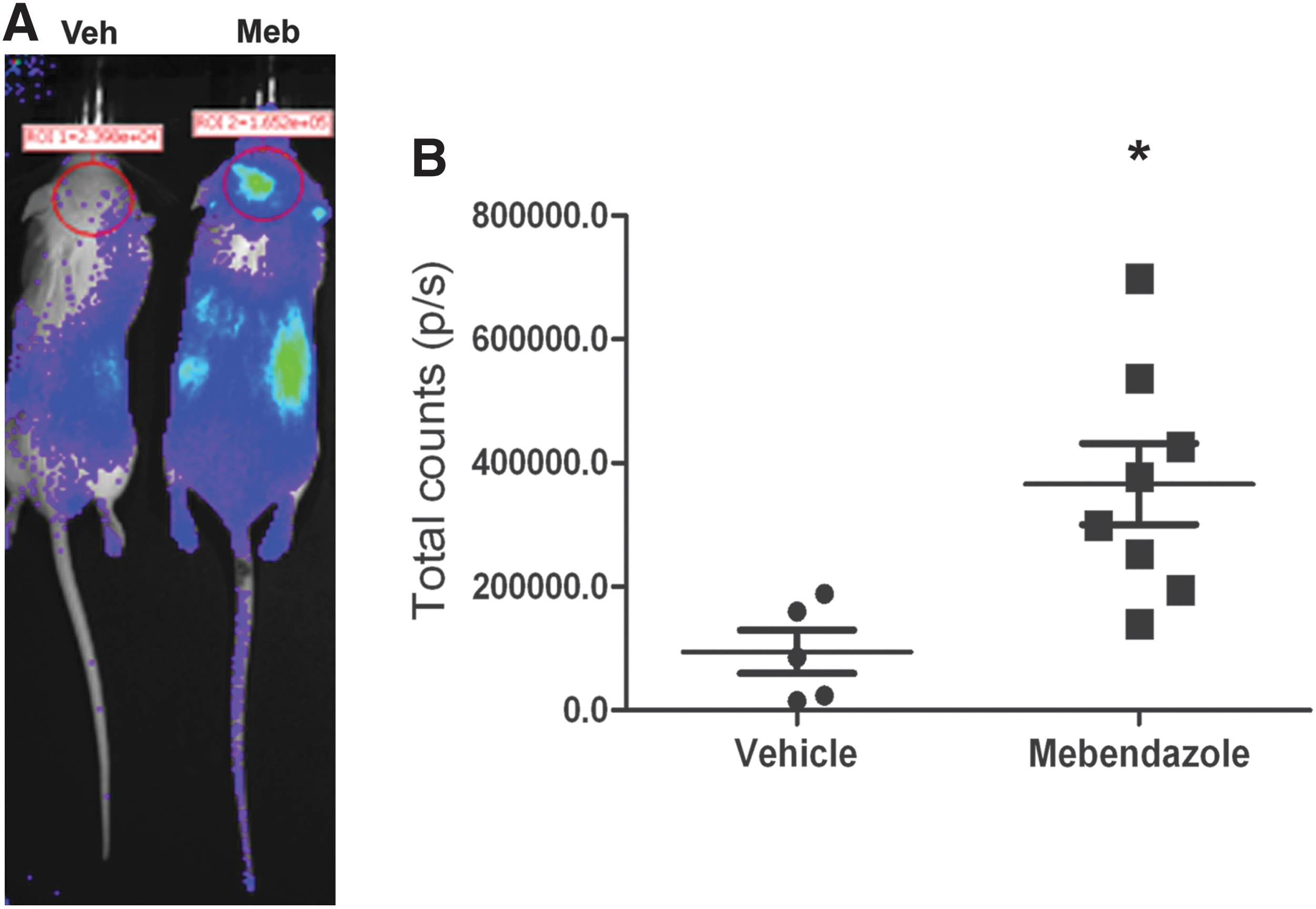
Our findings suggested that benzimidazoles activate hypoxic adaptation and reduce oxidative death in vitro. However, one of the many challenges in translating the results of cell-based and invertebrate assays to more clinically relevant models in mammals is the unpredictable bioavailability of these compounds. Indeed, clear demonstration that the concentration of drug delivered safely to an animal affects a biomarker of drug activity studied in in vitro systems is essential. For example, benzimidazoles such as mebendazole are taken orally in clinical settings. The question remained as to whether oral mebendazole reaches sufficient concentrations to stabilize HIF-1α there. To evaluate HIF stabilization in the brain after treatment with these drugs, we used a transgenic mouse strain that ubiquitously expresses the HIF reporter ODD-Luc (47). We administered albendazole and mebendazole via oral gavage, daily for 5 days. After 6 h of the last dose, mice were anesthetized and then underwent live bioluminescence imaging using an IVIS-100 system. We observed that mebendazole, at a dose of 50 mg/kg, increased ODD-Luc activity fourfold in the brain compared with the control group (Fig. 9). By contrast to mebendazole (5), albendazole showed only a weak signal increase in the liver with no detectable effect in the brain at a dose of 150 mg/kg (data not shown).
Discussion
Antihelminthic benzimidazoles are used throughout the world to treat parasitic infestations in a host of species and in diverse organs, including the central nervous system (CNS). An on-target effect of these agents is β-tubulin; specific mutations in this gene can confer resistance to antihelminthics (24). The study of the interaction of antihelminthic benzimidazoles with mammalian tubulin and its biological effect on the host, previously underappreciated, has developed into an area of growing interest. Indeed, two groups described the ability of benzimidazoles to modulate expression of PGC-1α, a co-activator involved in mitochondrial biogenesis, and to decrease ROS formation in non-neural cells (4, 58). Here, we expand our understanding of the host effects of benzimidazoles in neurons and show that these agents stabilize HIF-1α and augment HIF-dependent transcription. We correlate these changes with the ability of benzimidazoles to confer resistance to oxidative death in primary cortical neurons. Three lines of evidence suggest that free β-tubulin is targeted to achieve these biological effects. First, the effects of benzimidazoles on HIF-dependent gene expression and neuroprotection can be mimicked by structurally diverse agents that bind tubulin monomer, including those which interact with colchicine- or vinca alkaloid-binding sites. Second, the effects of colchicine on hypoxic adaptation and neuroprotection are abrogated by chemically modifying it to isocolchicine, which possesses a 500-fold lower affinity for tubulin than the parent compound. Third, reducing free tubulin levels with Taxol counteracts the protective effects of benzimidazoles.
Several models could explain the ability of drugs that bind β-tubulin to mimic hypoxia and prevent death in neurons. It is possible that these agents act to shift microtubule-tubulin equilibrium in favor of depolymerization, although we do not observe a gross change in neuronal architecture assessed by βIII tubulin staining in benzimidazole-treated neurons (Fig. 7). Small changes in microtubule dynamics are known to modulate signaling events that can lead to de novo transcription. For example, the cytoplasmic inhibitor of the transcription factor NF-κB, IκB can be sequestered by microtubules in the axonal initial segment. On microtubule depolymerization, IκB is degraded, unmasking the nuclear localization sequence of NF-κB and allowing it to translocate to the nucleus where it can up-regulate a number of genes, including HIF-1α (19). Increases in HIF-1α transcription could overwhelm the ability of neurons to degrade HIF-1α and lead to its stabilization. In this scheme, HIF-1α induction may be a biomarker of other genes that are known to abrogate oxidative stress, such as MnSOD (9), rather than being responsible for protection itself (49). Of course, it is also possible that benzimidazoles act post-transcriptionally rather than transcriptionally to enhance the interaction of HIF-1α (and other proteins) with Septins, a group of microtubule polymerizing guanine nucleotide-binding proteins which lead to HIF-1α stabilization in tumors (2). Since some of the earlier reports indicate that uptake and intracellular trafficking of iron and iron transporting proteins such as transferrin receptor and metal transporter protein 1 (MTP1) is microtubule dependent (34, 60), it is possible that benzimidazoles, through their tubulin-binding activity, might interrupt the intracellular trafficking of iron, an important cofactor for the activity of prolyl hydroxylases. Cytosolic iron starvation could lead to loss of HIF-PHD activity, stabilization of HIF, activation of HIF target genes, and HIF-independent neuroprotection (49, 50, 61).
The antihelminthic benzimidazoles are the first line treatment for parasitic infestations and are believed to be vermicidal by disrupting worm microtubules, leading to parasite immobility and death (57) without similarly affecting host cells. An unexplored possibility consistent with the results presented here is that host cells, unlike parasites, augment genetic adaptation in response to benzimidazole exposure that prevents extensive microtubular disruption. Indeed, a recent study shows that cytoskeletal insult through depolymerization of synaptic microtubules can lead to reduction of FOXO via activation of Akt to help limit microtubule disruption (39). Irrespective of the mechanism for selective toxicity in parasitic helminths by benzimidazoles, our findings show that they protect neurons in the nanomolar range. Finally, using a well-validated reporter, we demonstrate that mebendazole stabilized HIF-1α in the brain, a biomarker of hypoxic adaption. In clinics, the drug of choice to treat neurocysticercosis is albendazole. The concentration of its main metabolite, albendazole sulfoxide achieves levels in the brain that are sufficient to eradicate parasites (20). However, our finding that (i) albendazole sulfoxide is a weak neuroprotective agent compared with the parent compound (Supplementary Fig. S3), and (ii) albendazole failed to activate the HIF pathway in the brain, makes albendazole a less favorable therapeutic candidate than mebendazole in evaluating the effects of these agents on neurological conditions.
In addition to the novel potential applications for antihelminthic benzimidazoles, our studies also introduce numerous compounds with current clinical approval and short- and long-term safety data in humans that have the unexpected ability to activate hypoxic adaptation. A closer look at the list of our hits reveals at least two other groups of pharmacophores, isoflavones and phenylpropionic acids (ibuprofen and indoprofen). Of note, zomepirac, a non-steroidal anti-inflammatory drug, also a hit in our screen, binds to tubulin and mildly inhibits microtubular assembly (6). While the results of our primary screen have yet to be fully vetted, drugs such as hydralazine, atenolol, aminopyridine, and hydroxyzine with known applications in stroke prevention (11), functional recovery (10), or as CNS antihistamines (13) are of particular interest as novel activators of the adaptive program to hypoxia.
Materials and Methods
Chemicals
Albendazole, mebendazole, fenbendazole, oxibendazole, colchicine, nocodazole, citrate synthase and malic dehydrogenase, fumarase, oxaloacetic acid, acetyl-CoA, β-NADH, β-NAD,
Cell culture
Cortical neurons were harvested from E.15 CD-1 mouse embryos as previously described for rat cortical cultures (36). Dissociated neurons were plated in minimum essential media supplemented with glutamax, 10% fetal bovine serum (FBS), and 5% horse serum and seeded in 96-well (9×104 cells/well) or 6-well plates (2×106 cells/well), or 10 cm dishes (1×107 cells/dish) that were precoated with poly
ODD-luciferase activity
The ODD-luc construct encoded in pcDNA3.1 plasmid vector was constructed as previously described (47). Hydroxylation of prolines P402 and P564 within the HIF-1α ODD by HIF-PHDs permits binding to the VHL protein that targets it for proteasomal degradation. Targeted P402A and P564A mutation within the ODD prevents hydroxylation and confers stability to HIF-1α or the ODD-Luc construct under normoxia. ODD-Luc activity can, therefore, be used as a surrogate for HIF-1α stability regulated by the HIF-PHDs (47, 53). To make stably expressing ODD-Luc hippocampal mouse neuroblast HT22 cells, we co-transfected the plasmid construct along with a puromycin resistance plasmid (p-BABEpuro). Transfected cells were grown in the presence of 4 μg/ml of puromycin. Luciferase activity was measured by luciferase assay kit (Promega) using an LMaxII™ microplate luminometer (Molecular Devices). ODD-Luciferase activity was normalized to the protein content of each well measured by bicinchoninic acid (BCA) protein assay (Pierce).
Neuronal viability assay
After 1 day in culture in 96-well plates, neurons were co-treated with an indicated concentration of each drug and homocysteic acid (HCA, 5 mM) to induce neuronal death via glutathione depletion (43). The compounds were solubilized in dimethyl sulfoxide (DMSO). To keep benzimidazoles soluble during serial dilution, we used 50% ethanol. The final concentration of ethanol in the culture media was 0.1%. For microtubule prestabilization experiments, the neurons were exposed to 100 nM of Taxol for 30 min before co-treatment with the compounds and HCA. Viability was assessed at 16 h after treatment by the MTT assay, or calcein AM/ethidium homodimer-1 staining (live-dead assay) (Invitrogen) under fluorescence microscopy according to the manufacturer's protocol. The viability has been calculated as the ratio of MTT absorbance in three treated wells divided by the average of three untreated wells in each plate and presented as percent to control.
Western blot analysis
Cells were plated in 10 cm plates, treated with the indicated compounds overnight, and then lysed with NP-40 buffer supplemented with protease inhibitors. For HIF-1α blots, nuclear proteins were enriched using a nuclear/cytoplasmic subcellular fractionation kit (Pierce). Protein content was measured by BCA protein assay (Pierce). Thirty microgram of total protein for p21cip1/waf1 (p21) and 100 μg of nuclear protein for HIF-1α were loaded on 10% or 4%–12% gradient Bis-Tris polyacrylamide gels respectively (Invitrogen), resolved, and transferred to nitrocellulose membranes. Membranes were probed by antibodies against p21 (1:200; Santa Cruz), β-tubulin III (1:10,000; Epitomics), HIF-1α (1:500; Novus Biologicals), or histone H3 (1:1000; Cell Signaling Technology) and developed by IR-dye-conjugated antibodies (1:15,000) using the LI-COR system. Blots were imaged with the Odyssey IR quantitative Western blot detection system (LI-COR Biosciences).
Immunocytochemistry
Cortical neurons were treated with indicated compounds, fixed with 4% paraformaldehyde, and incubated with anti-β-tubulin III antibody (1:200; Epitomics), followed by secondary antibody as previously described (15).
Isolation of mitochondria
Non-synaptic mouse brain mitochondria were isolated by a modified isopycnic centrifugation procedure employing Percoll density gradient (32, 51). Briefly, cortex brain tissue was homogenized in the MSEGTA buffer composed of 225 mM mannitol, 75 mM sucrose, 20 mM HEPES (pH 7.4), 1 mM EGTA, and 0.2% (w/v) fatty acid-free bovine serum albumin (BSA) and centrifuged at 2000 g×4 min. The supernatant was collected and centrifuged at 12,000 g×10 min. The pellet was re-suspended in MSEGTA, layered over 25% (v/v) Percoll prepared in MSEGTA mixture, and centrifuged at 30,000 g×10 min. Purified mitochondria fraction was collected at the bottom of the tube, re-suspended in MSEGTA without added BSA, and washed twice by centrifugation at 12,000 g×10 min. Final mitochondrial pellet was re-suspended in MS buffer comprising 225 mM mannitol, 75 mM sucrose, and 20 mM HEPES (pH 7.4) and stored on ice. Protein content was estimated by a commercial BCA assay (Pierce Biotechnology/Thermo Scientific).
Mouse liver mitochondria were isolated by differential centrifugation. Liver was homogenized in MSEGTA buffer and centrifuged at 900 g×10 min; the supernatant was collected and centrifuged at 10,000 g×10 min; and the pellet was re-suspended in MSEGTA buffer with BSA omitted and centrifuged at 10,000 g×10 min. The final pellet was re-suspended in MSEGTA buffer without BSA to 15–20 mg mitochondria protein per milliliter and stored on ice during the experiment.
Functional assays
The base KEGTA incubation buffer employed in all assays was composed of 125 mM KCl, 20 mM HEPES (pH 7.2), 0.2 mg/ml BSA (fatty acid free), 4 mM KH2PO4, 0.2 mM EGTA, 5 mM glutamate, and 2 mM malate. For the measurements of the mitochondrial membrane potential, KEGTA was supplemented 2×10−6 M Safranine O. Mitochondria at 0.1–0.12 mg/ml were added to 1 ml of incubation buffer in a stirred thermostated (t=37°C) cuvette, and the changes in the membrane potential were recorded by following the fluorescence of Safranine O with excitation and emission wavelengths of 495 and 586 nm, respectively, using a Hitachi 4500 (“Hitachi”) spectro-fluorimeter as described earlier (54, 55). For the measurements of mitochondrial ROS emission, KEGTA was supplemented with 4 U/ml of horseradish peroxidase, 40 U/ml of Cu, Zn superoxide dismutase, and 1×10−5 M Amplex RedUltra (Invitrogen). A change in the concentration of H2O2 in the medium was detected by fluorescence of the oxidized Amplex RedUltra product using excitation and emission wavelengths of 555 and 581 nm, respectively (54, 55). The response of Amplex RedUltra to H2O2 was calibrated by sequential additions of known amounts of H2O2. The concentration of commercial 30% H2O2 solution was calculated from light absorbance at 240 nM employing E 240=43.6 M −1×cm−1; the stock solution was diluted to 0.1 mM with water and used for calibration immediately. The rates of resting and phosphorylating respiration were recorded with Oxytherm oximeter (Hansatech), at 37°C and 0.5–0.7 mg/ml of mouse liver mitochondria. All compounds were dissolved in DMSO at 25–50 mM.
Fumarase assay
Fumarate production from malate was measured with continuous monitoring of optical density at 240 nm, corresponding to the maximum absorption of fumarate (E=2440 M −1×cm−1) in 0.1 M K-phosphate buffer, pH 7.6, in the presence of varied concentrations of benzimidazole and malate (0.05–0.4 mM). The data were plotted in a double-reciprocal format to compare inhibitory effects of various benzimidazoles, and on Dixon coordinates to determine the inhibition constant (Ki ).
RNA extraction and real-time polymerase chain reaction
Cortical neurons were cultured in six-well plates and treated with indicated drugs. After 8 h, neurons were harvested with Trizol (Invitrogen). The levels of p21cip1/waf1 (cdkn1a) mRNA were analyzed using One-step Taqman PCR assay mix and primers (Mm00432448_m1) on a 7500 Fast Real-Time PCR system (Applied Biosystems). β-Actin was used as an endogenous control (Mm00607939_s1).
Tubulin polymerization assay
The levels of free and polymerized tubulin were assessed at 3 h after treatment with indicated microtubule-binding agents using a microtubule/tubulin in vivo assay kit (Cytoskeleton).
Mebendazole treatment and in vivo bioluminescence imaging
Adult FVB.129S6-Gt(ROSA)26Sortm2(HIF1A/luc)Kael/J ODD Luc transgenic mice purchased from Jackson Laboratories were used to evaluate the inhibition of HIF-PHDs in vivo. For drug treatments, mebendazole tablets were powdered, mixed in a vehicle that was composed of phosphate-buffered saline and sesame oil 1:1 (Sigma), and delivered via oral gavage (100 μl) to adult ODD Luc mice. Animals were treated with mebendazole (50 mg/kg) or vehicle once a day for 5 days. Six hours after the last treatment, animals received an intraperitoneal injection of firefly D-luciferin potassium salt (75 mg/kg; i.p.) and were anesthetized with a mixture of 2.5% isoflurane and oxygen. The mice were placed in the chamber, where anesthesia was maintained with isoflurane 2%, administered through a nosecone via a gas anesthesia system. Luciferase luminescence was evaluated by a 100 Series IVIS Imaging System (Caliper Life Sciences, PerkinElmer). Mice were imaged on the dorsal side at 10 min post D-luciferin injection. The bioluminescent signal was integrated every 10 min for 40 min, with the peak signal observed at 20 min in most of the animals. Integral luminescence from a defined region of interest around each animal's brain was measured using the Living Image Software and expressed as photons per second of light.
Statistical analysis
Data were analyzed using Prism (GraphPad software). Quantitative data are presented as mean±standard error of mean of at least three experiments. A comparison between the groups and control was performed by one-way analysis of variance (ANOVA), followed by post hoc Dunnett multiple-comparison test or Student's t-test for two-group analysis.
Footnotes
Acknowledgments
This work was supported by National Institutes of Health (P01 NIA AG014930, Project 1 to R.R.R.), the Dr. Miriam and Sheldon G. Adelson Program in Neurorehabilitation and Neural Repair, the Thomas Hartman Foundation for Parkinson's Research, the New York State Center of Research Excellence on Spinal Cord Injury, and the Burke Foundation. The authors wish to thank Dr. Natalia Smirnova for construction and evaluation of ODD-Luc reporter, Jimmy Payappilly and Dr. Hengchang Guo for providing technical assistance, and Dr. Gregg Gunderson for helpful scientific discussions.
Author Disclosure Statement
The authors declare no competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.