
Abstract
The present work provides the first clinical evidence about the neuroprotective potential of a therapeutically safe chelation treatment on early-stage Parkinson's disease (PD) patients who were already stabilized for dopamine and responded significantly to treatment in both brain iron deposits and indicators of disease progression. The novel treatment relied on oral administration of deferiprone (DFP) that by chelation of labile iron it conferred upon oxidation-stressed animals increased striatal dopamine levels and improved motor functions while essentially sparing systemic iron. The paradigmatic modality of chelation with DFP in PD should prompt multicenter studies in various disorders of regional iron accumulation or misdistribution.
Introduction
I
The regional siderosis patterns revealed by MRI have also been used as diagnostic markers of neurodegenerative disorders (14, 16, 26, 52, 69)—despite the lack of proof of a causal relationship between brain siderosis and disease initiation/progression (73). In Parkinson's disease (PD), excess iron is detected primarily in the substantia nigra pars compacta (SNc) (9, 13, 14, 15, 26, 52, 69), where dopaminergic neurons are exposed to high levels of ROS produced by the metabolism of dopamine (6, 27, 43, 47). By catalyzing ROS formation from the by-products of oxygen consumption, excess labile iron can further affect neuronal functions either by directly damaging cell components or by chemically affecting signal mediators that (like dopamine) are prone to oxidation (27, 43, 47, 74). In various animal models of neurodegeneration and in PD in particular (54, 74), regional brain siderosis has served as marker of disease and as target of metal chelators for attaining neuroprotection or neurorescue (12, 29, 34, 39, 46, 70, 73, 75).
We reasoned that chelation strategies targeted to any type of locoregional siderosis, should, as prerequisite, spare any systemic metal loss (4, 33), particularly when dealing with the elderly populations affected by anemia, as in some cases of PD (58). It is because of the following properties that we selected deferiprone (DFP) as a paradigm of a membrane-permeant bidentate chelator suitable for conferring neuroprotection in experimental models of PD and in the clinical setting: (i) its ability to rescue iron-overloaded cells by scavenging labile iron (24, 64), especially from mitochondria, the organelles most affected by cell iron accumulation and ensuing labile iron-mediated oxidative damage (3, 24, 32, 40, 50, 59, 64, 71); its neuroprotective action (ii) against MPP+ in the human neuroblastoma catecholaminergic SHSY-5Y cell line (44) and (iii) against 6-OHDA effects in the rat PD model in which systemic chelator administration significantly attenuated dopaminergic neuron loss and striatal dopamine content (12); (iv) its capacity to deliver chelated metal to extracellular apotransferrin (64), so that systemic iron losses are largely avoided and hematological indices are affected as little as possible; (v) its blood–brain barrier crossing ability (30) that facilitates accessing to foci of iron accumulation, as demonstrated in Friedreich's ataxia (2, 67) and neurodegeneration with brain iron accumulation patients (1).
Moreover, DFP's good clinical record worldwide in the treatment of hemosiderosis (20, 8) (and particularly in the rescue of patients with severe cardiac iron overload) (51, 72) facilitated the regulatory and ethical approval of a pilot study (the FAIRPARK study) in PD patients. We also demonstrate here for the first time that a relatively safe pharmacological approach to attenuating ROS production (via the reduction of labile iron pools) with DFP confers in vivo protection against oxidative insults on dopaminergic neurons and dopamine. The approach was initially assessed in a murine model of PD and then translated into a clinical trial setting, to evaluate DFP's potential for attenuating disease progression while not affecting hematological or other systemic parameters.
Results
Pharmacological effects of DFP in a translational model of PD
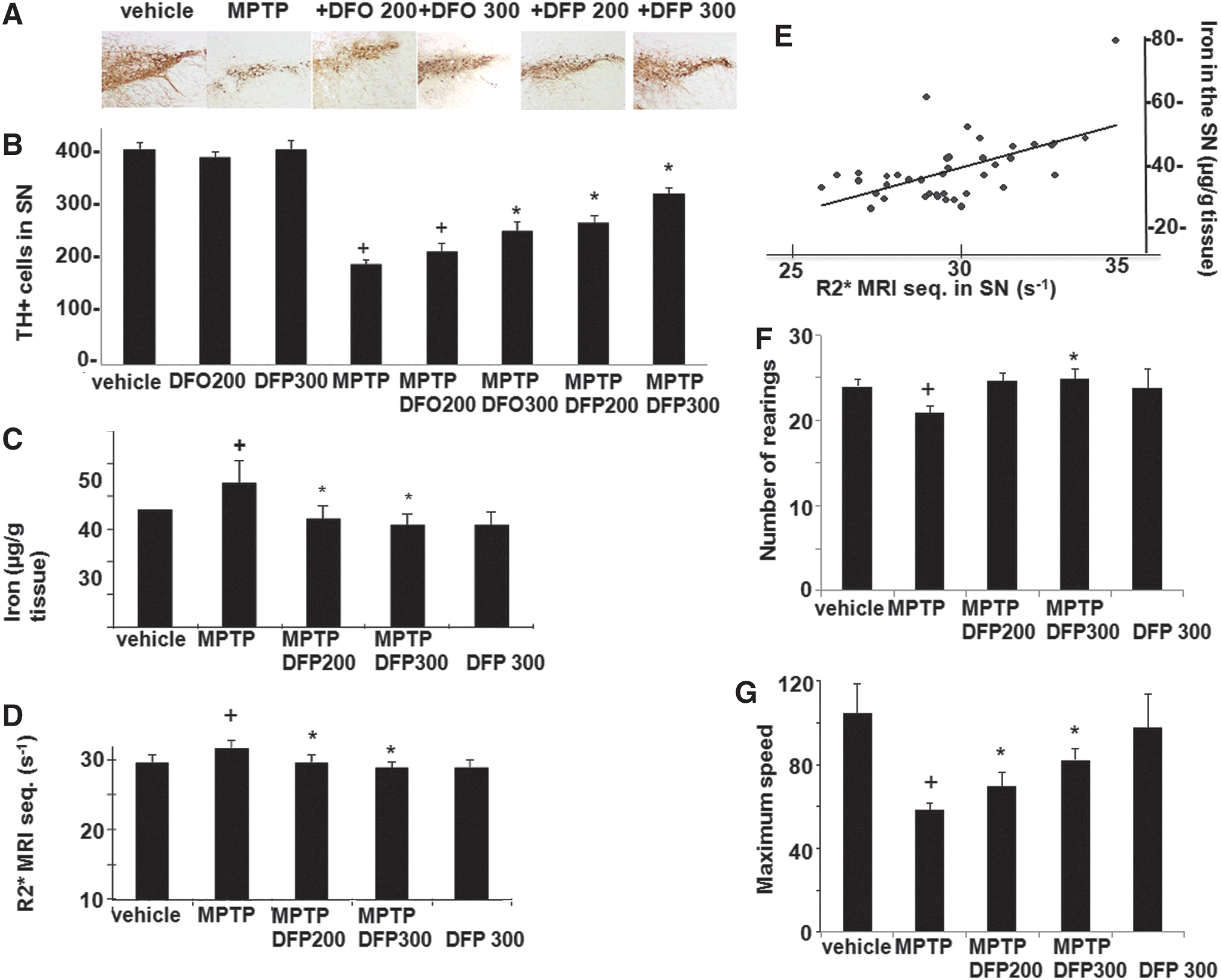
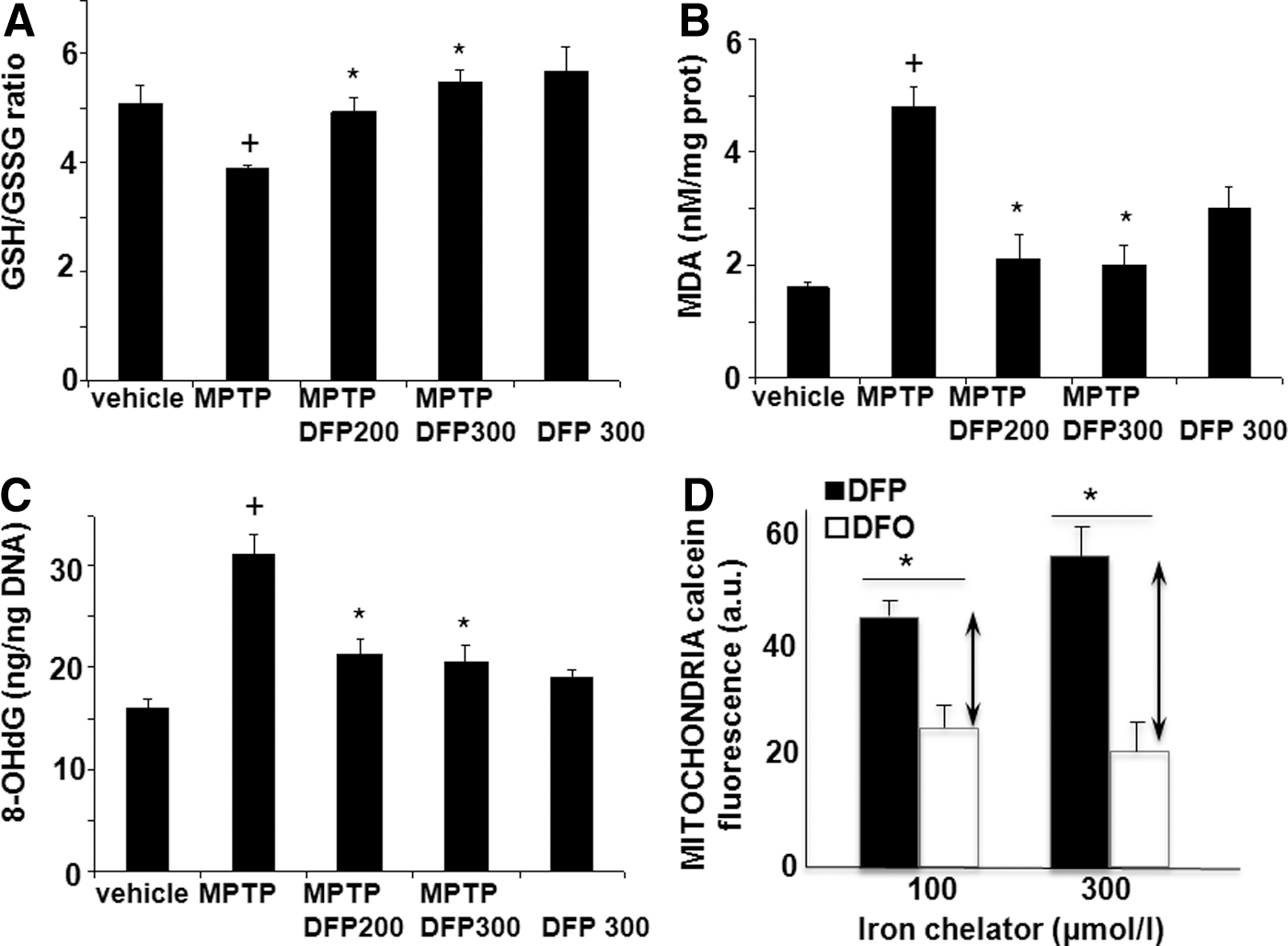
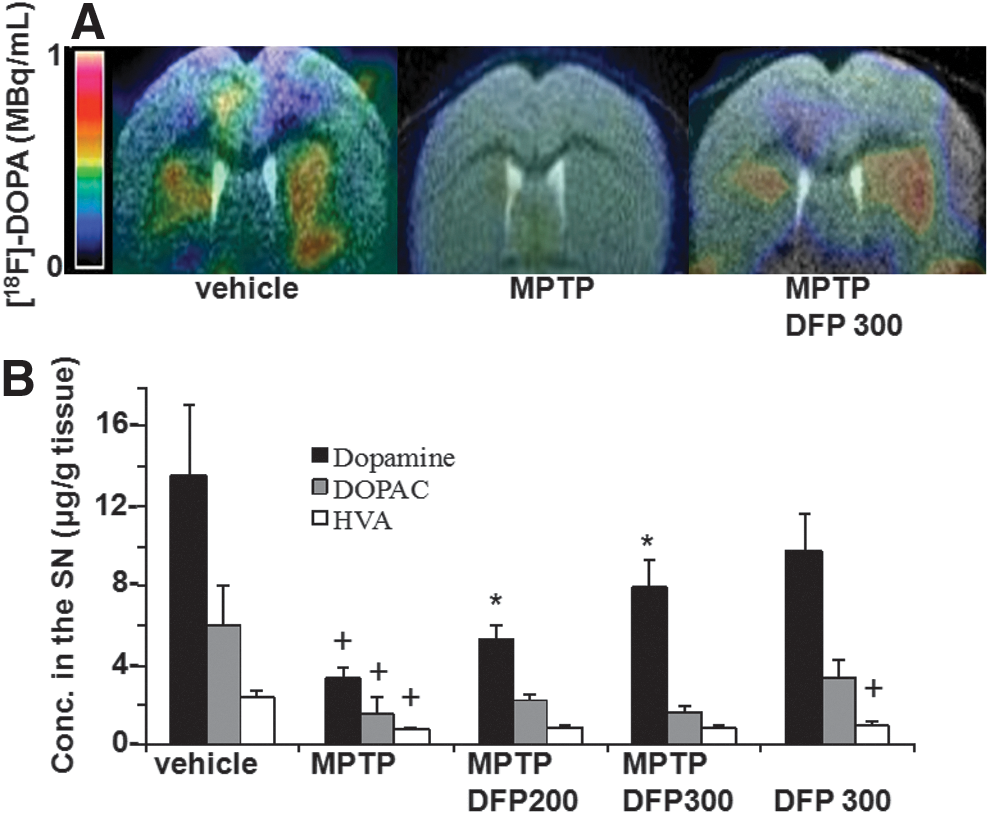
The effect of DFP treatment on brain parameters with relevance to PD was initially studied in the acute 1-methyl-4-phenyl-1,2,3,6-tetrapyridine (MPTP) mouse (Figs. 1 –3). This model recapitulates several features of the human disease (34, 54). Oral administration of the membrane-permeant, bidentate chelator DFP partially relieved the oxidative damage generated within the SN by MPTP treatment, as reflected by the increase in the number of dopaminergic [i.e., tyrosine hydroxylase (TH)-positive] cells (Fig. 1A, B). Per os treatment with DFP afforded twice as much protection (i.e., ≈60%) as the intraperitoneally administered hexadentate deferoxamine (DFO) (which binds Fe(III) with a 1:1 stoichiometry) did (i.e., ≈30%). DFP's ability to reach the SN was deduced from the observed reduction in iron accumulation in MPTP-intoxicated mice, as measured in situ by MRI (Fig. 1D, E) or in isolated tissue by atomic absorption spectrometry (Fig. 1C). Importantly, the pharmacological effects of oral DFP administration were reflected as an improvement in the animals' motor function, the number of rearing, and the maximum speed (Fig. 1F–G). Similar to what has been previously observed in the MPTP mice model with clioquinol (34), we found that the DFP pretreatment did not significantly affect the MPTP to MPP+ conversion, as reflected in the respective MPP+ striatal levels (ng/mg protein) of control (saline) versus DFP-pretreated mice: 0.4±0.2 and 0.3±0.1 (n=12, p>0.05). DFP also led to increased levels of reduced glutathione (GSH) relative to the oxidized glutathione (GSSG) (Fig. 2A) and to reduced oxidation products of lipid (i.e., malondialdehyde-MDA formation) (Fig. 2B) and of DNA (i.e., 8-oxo-deoxyguanosine formation) (Fig. 2C). As with other cases of toxicity resulting from iron accumulation in cell organelles (24, 33, 63), we found that DFP demonstrably neutralized mitochondria labile iron pools (measured in organelles isolated from mice brains and calcein labeled with the aid of calcein-AM) (Fig. 2D). We also looked at whether DFP's ability to chelate labile cell iron (and thereby reduce ROS formation and ensuing oxidative stress) was associated with a reduction in dopamine depletion in dopaminergic neurons affected by the MPTP treatment. Indeed, as shown in Figure 3A and B, MPTP treatment caused a major decrease in striatal [18F]-DOPA distribution and dopamine level. DFP partly rescued this dopamine depletion and significantly modified dopamine's metabolic conversions (as reflected by the levels of DOPA metabolites).
Pharmacological effects of DFP in cell-based models
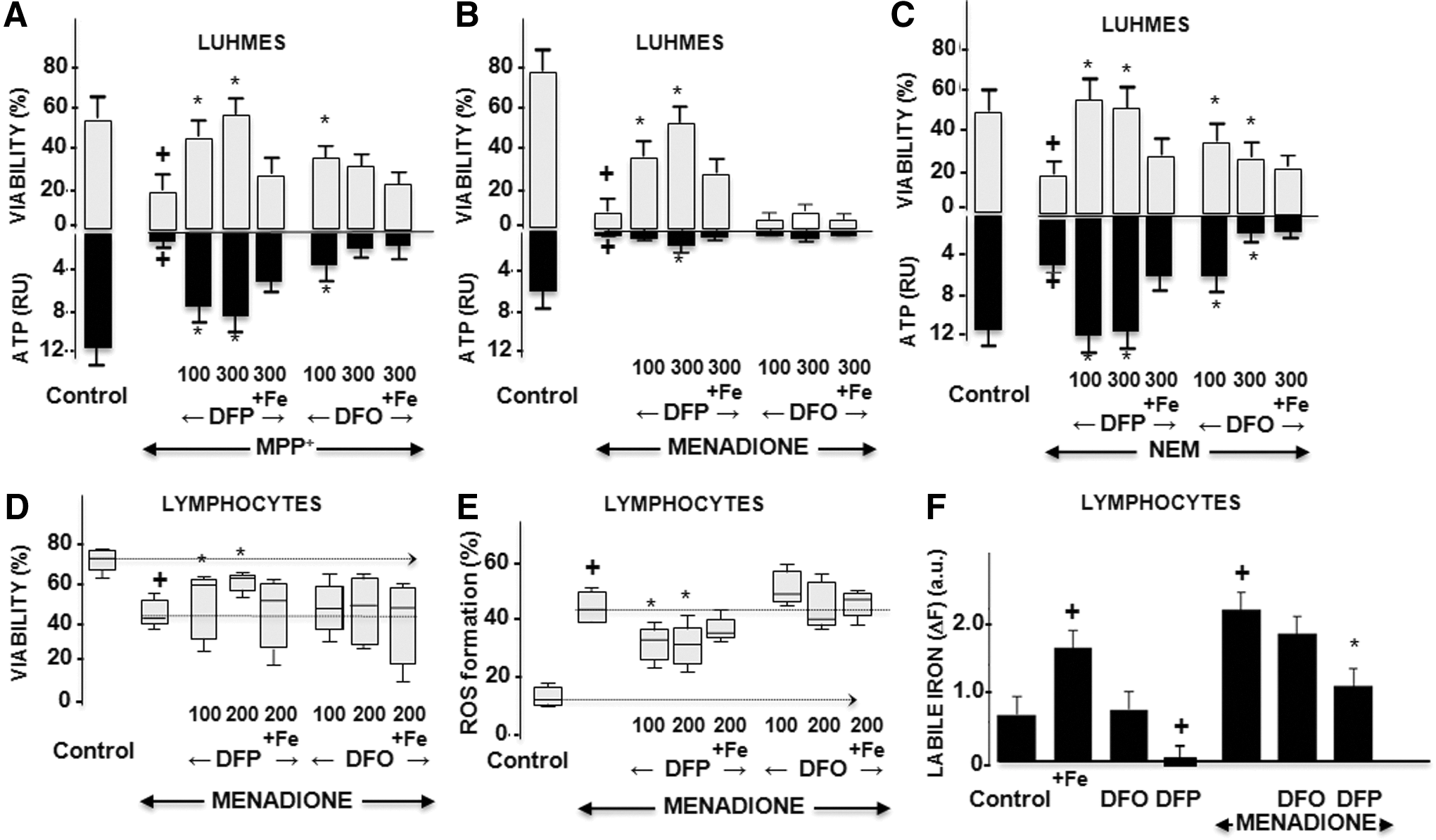
DFP's chelating ability in intact human dopaminergic neurons, a Lund human mesencephalic (LUHMES) cell line (62), was studied under oxidative stress conditions that simulated various aspects of the PD brain (Fig. 4A–C). Treatment of cells with 1-methyl-4-phenyl-pyridinium (MPP+, which affects mitochondrial complex I activity), menadione (which induces aberrant mitochondrial redox cycling), or N-ethylmaleimide (NEM, which depresses the cell's antioxidant capacity by blocking GSH) resulted in adenosine triphosphate (ATP) depletion and an ensuing drop in cell viability. Treatment with DFP conferred cytoprotection from various oxidative insults, which, in part, might be associated (directly or indirectly) with chelation of labile iron, as demonstrated in a variety of model systems (5, 24, 33). In the present study, the DFP protective features observed in cells subjected to different oxidative insults were largely abrogated by precomplexation with exogenous Fe(III). DFP's ability to increase the survival of pro-oxidant-challenged cells was also demonstrated in human lymphocytes (Fig. 4D–F), which are reportedly modified in PD patients receiving dopaminergic/L-dopa treatment (7). As shown in Figure 4, the application of DFP to human lymphocytes challenged with pro-oxidants resulted in higher survival (Fig. 4D) and lower ROS formation (Fig. 4E). We attribute this partial protective effect of DFP to its ability to reduce the levels of labile cell iron (Fig. 4F).
Clinical effects of DFP in PD patients (FAIRPARK trial)
Study design and procedures
Proof of concept in the translational PD model (whereby systemic administration of DFP demonstrably reduced the levels of accumulated iron in the SN but avoided systemic iron loss) served as the rationale for exploring the drug's potential in early-stage PD patients (61) (i.e., within 3 years of disease onset) with an established, stable dopaminergic treatment regimen. We used the accumulation of iron in the patients' SN as a marker for DFP's ability to act at sites associated with disease progression. To that end, we used multiple-echo/spin-echo sequences in a 3-Tesla MRI system to estimate R2* proton relaxation rates (=1/T2*) and thus levels of agglomerated iron. The latter is generally associated with ferritin (9, 25, 51, 66, 72) and/or hemosiderin derived from ferritin degradation (36). Hence, changes in the MRI T2* value at 6-month intervals served as the primary indicator of DFP's ability to access affected brain areas, whereas the Unified Parkinson's Disease Rating Scale (UPDRS) motor score was used as a measure of changes in motor handicap (i.e., disease progression). With a view to distinguish between (i) possible DFP-induced neuromodulating effects and (ii) symptomatic effects due to DFP's inhibition of catechol O-methyltransferase (COMT) (68), we applied a delayed-start (DS) paradigm (49, 55, 60). In the initial phase, 40 early-stage PD patients were randomly assigned to receive either oral liquid DFP (15 mg/kg in the morning and evening) for 12 months (the early-start [ES] group) or placebo for 6 months followed by DFP for the next 6 months (the DS group) (Supplementary Fig. S1 and Supplementary Table S1; Supplementary Data are available online at
Changes in iron accumulation in the nigrostriatal system
On study entrance, the R2* values in the SN and putamen (Pu) were significantly higher in PD patients than in age- and gender-matched control individuals (Supplementary Table S2), as previously reported for the SN (14, 15, 52, 69). After 6 months of treatment with DFP, there was a significantly greater decrease in SN R2* values in the ES group than in the DS group. A further decrease in SN and Pu R2* values was observed after pursuit of the treatment for 6 additional months (i.e., completing 12 months of DFP treatment for the ES group and 6 months for the DS group) (Fig. 5 and Supplementary Fig. S2 and Table 1, upper part).
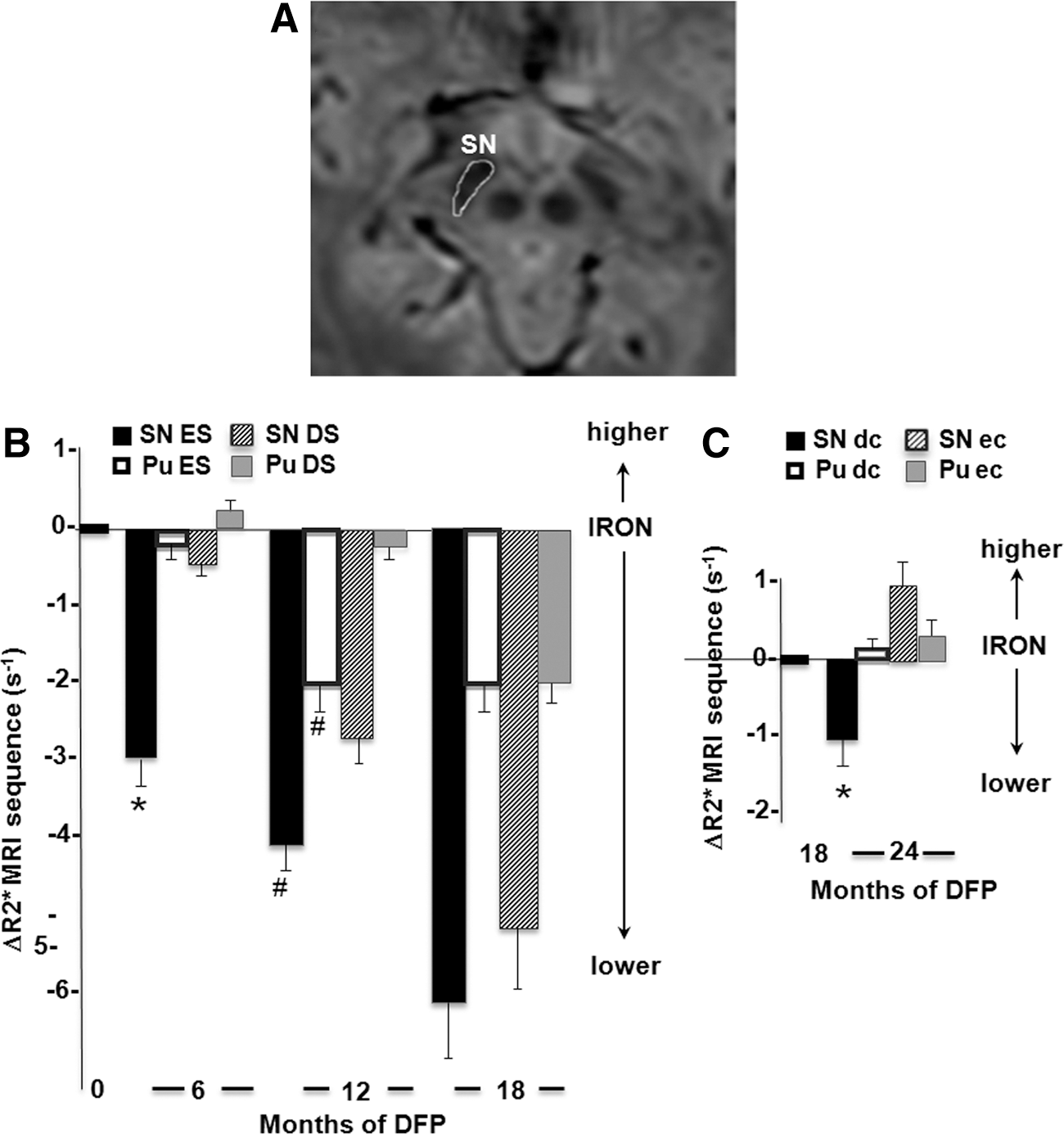
Mean±SD or median values [1st quartile–3rd quartile] are indicated. Covariance analyses at 6 and 12 months were adjusted for baseline values and weighted according to the sample size (p-size effect: small effect size ≤0.20 and large ≥0.80). Nonserious adverse effects: transient diarrhea (n=3 DFP; n=1 placebo), gastritis (n=1 DFP; n=1 placebo), transient headache (n=1 DFP), and slight proctorrhagia (n=1 DFP; n=1 placebo). Serious adverse effects under DFP: agranulocytosis (n=1 at 3 month) and neutropenia (n=2 at 3 and 7 months). CSF, cerebrospinal fluid; DFP, deferiprone; GPX, glutathione peroxidase; SOD, superoxide dismutase; 8-oxdG, 8-oxo-7,8-dihydro-2′-deoxyguanosine; carbonyl groups are given per mg protein. chemical=Trolox mM equivalents as described in Materials and Methods section.
Although a few minor adverse events occurred during the first 12 months of treatment, those related to DFP treatment were not frequent (Table 1, legends). Serious adverse events that resolved within 2 weeks of DFP withdrawal only occurred within the first months of treatment and comprised a single case of agranulocytosis in the ES group and two cases of neutropenia. These cases were detected through the weekly blood count monitoring routinely performed on patients on DFP (8, 20). However, no serious adverse events occurred during the 12-month treatment extension phase (i.e., months 13–24).
Effects on biochemical, pharmacological, and hematological parameters
We observed that DFP can act both systemically and within the brain (directly or indirectly) by studying changes in levels of circulating ferritin in the cerebrospinal fluid (CSF) and blood plasma (Fig. 6A, +inset, F, respectively) and the iron levels in urine (Table 1). Although sideruria was significantly higher in the ES group than in the DS group, the daily iron loss induced by chelation was relatively low and represented no more than 5% of the 1 mg of daily iron gain/loss (56). In pharmacological terms, it is important to note that the chelation regimen applied here did not interfere with iron, copper, or zinc levels in plasma or CSF or with iron-related metabolic processes within the brain [i.e., there were no significant changes in the scales presented in Supplementary Table S1 (data not shown)] or the whole body (Supplementary Table S3). All the enrolled PD patients showed normal hematological indices and stable iron levels throughout the study. The decline in serum ferritin following DFP treatment might be construed as an indication of iron store depletion. However, it is more likely to represent increased hepatic iron turnover elicited by moderate chelation, which affected neither plasma iron levels (Supplementary Table S3) nor the erythron (Fig. 6E). These findings support our choice of DFP as a paradigm for a safe conservative chelation therapy. In all PD patients, liver functions were normal at baseline and remained normal during the 24 months of study (Supplementary Table S3, legends).
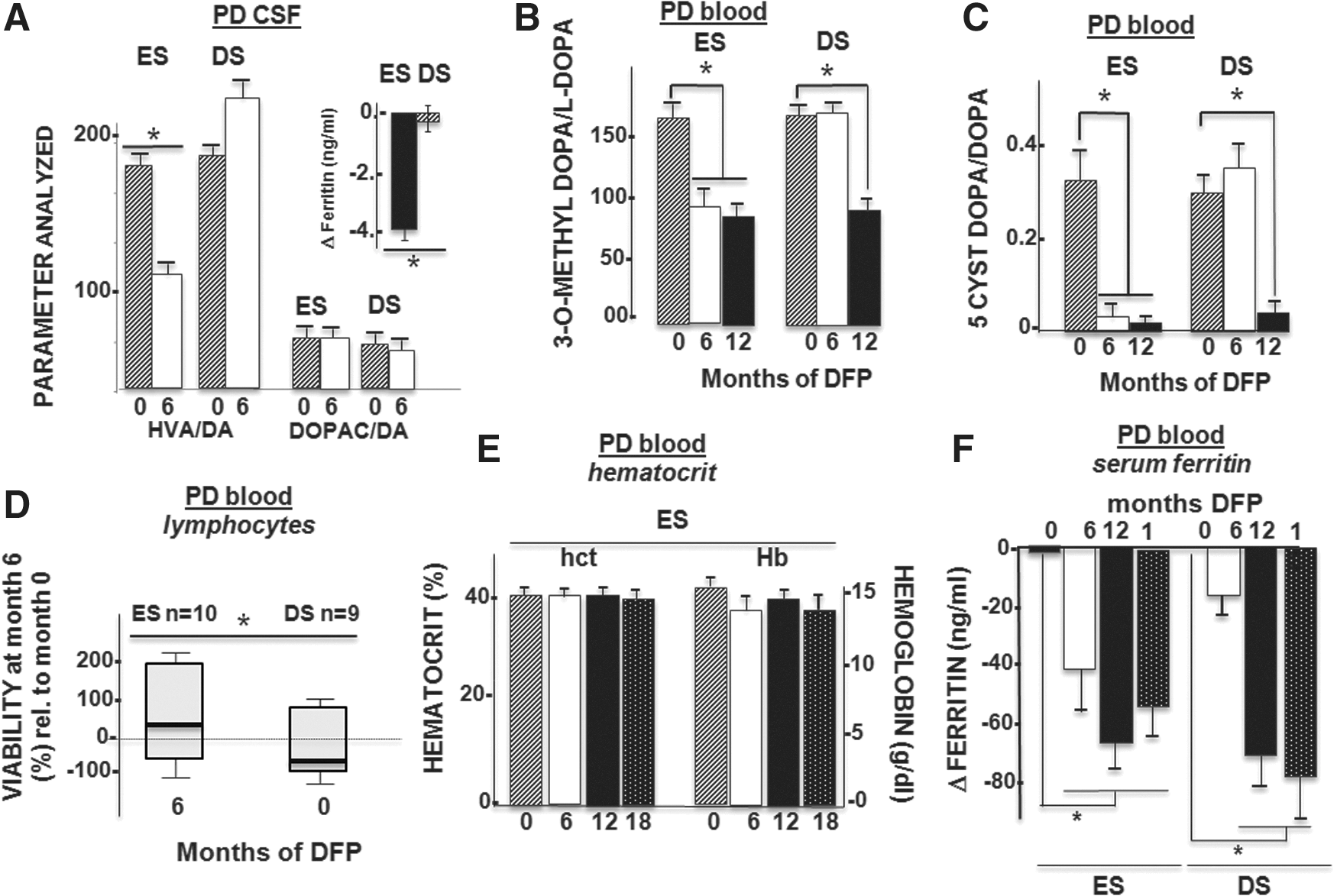
With respect to other brain areas involved in the pathophysiology of PD (the caudate nucleus and pallidum) and those not directly involved [the frontal cortex and bulbar (data not shown)], the patients' baseline R2* values did not differ significantly from those in healthy controls and were not significantly different between the ES group and DS group (Table 1 and Supplementary Table S2). However, DFP likely affected the COMT activity [as suggested by the lower CSF homovanillic acid/dopamine ratio in the CSF (Fig. 6A)] but not monoamine oxidase B activity [based on the insignificant changes in the 3,4-dihydroxyphenylacetic acid (DopaC)/dopamine ratio (Fig. 6A)]. Peripheral COMT inhibition by DFP was also observed in PD patients on L-dopa treatment, as shown by the lower of L-dopa to 3 O-methyl-dopa ratio in plasma (Fig. 6B). This was apparently associated with a lower peripheral 5-cysteinyl-dopamine/dopamine ratio (Fig. 6C), indicating a concomitant decrease in nonenzymatic dopamine auto-oxidation. This type of protection is probably the result of a DFP-evoked reduction in labile iron. The latter form of iron usually promotes dopamine auto-oxidation and the formation of harmful dopamine quinones that react with the thiol groups of cysteine and/or glutathione to form cysteinyl-dopamine derivatives (42). The fact that CSF glutathione peroxidase and superoxide dismutase activities increased in DFP-treated patients (Table 1) provides an additional indication of the antioxidant response underlying the chelator's mode of action. Other changes elicited by DFP in PD patients undergoing dopaminergic treatment were manifested in circulating lymphocytes, whose ex vivo ability to withstand oxidant stressors was significantly improved in patients treated with DFP (Fig. 6D).
DFP treatment of PD patients: clinical outcomes and potential disease-modifying properties
Patients who started DFP treatment immediately (the ES group) showed a significantly better motor performance at either 6 or 12 months than patients in the DS group. Moreover, the patients in the DS group showed a significant worsening in motor performance during the first 6 months on placebo; only during the following 6 months of DFP treatment did they show a relative improvement (Fig. 7 left part; Table 1, upper part; Supplementary Table S4). To gain further insights into DFP's mode of action in PD patients, we used the DS paradigm applied in the recent ADAGIO trial of rasagiline (49, 55) and the PROUD trial of pramipexole (60). Supplementary Table S4 expands on the data shown in Table 1 and summarizes the DFP treatment outcomes as changes over 24-week periods and indicates the statistical significance on the basis of a covariance analysis (adjusted for baseline differences) (Fig. 7 and Table 1 and Supplementary Table S4). Within the constraints of the present pilot study, DFP met the three hierarchical criteria for invoking a disease-modifying effect (49, 55), as follows:
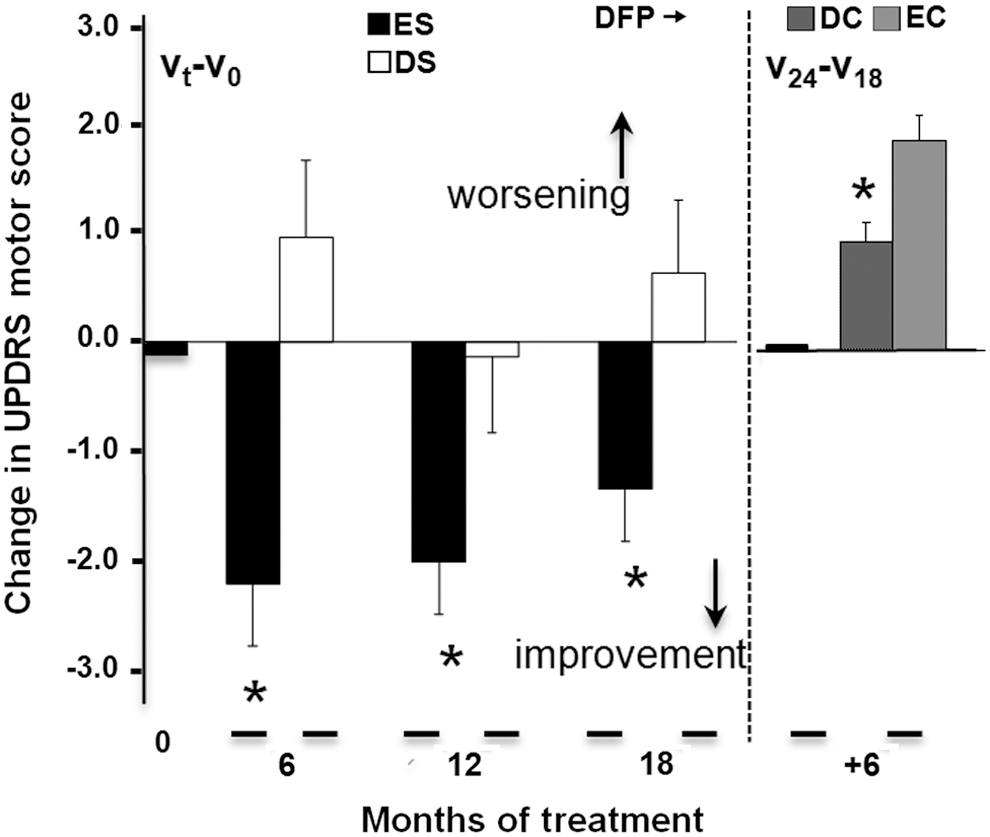
First endpoint: Significant benefit, with a smaller change in the UPDRS score during the first 24 weeks (i.e., about 6 months) in the ES group (−2.3±0.6) than in the DS group (+1.0±0.73) (i.e., placebo). Similar benefit was observed for the change in the DS group after the first 6 months of DFP treatment, that is, months 6–12 (−1.9±0.3) relative to placebo (+1.0±0.73).
Second endpoint: Significant benefit, with a smaller change in the UPDRS score from baseline to the end of the study (month 18) in the ES group (−1.4±0.5) than in the DS group (+1.0±0.7).
Third endpoint: Persistence of benefit throughout the study was based on a comparison of the change in the UPDRS score in the ES and DS groups during the last 6 months of treatment (+0.6±0.2 and +0.67±0.20, respectively).
Based on the significant clinical benefits achieved in the initial study, we extended the DFP treatment to assess its longer-term efficacy and safety and study the consequences of treatment cessation. Hence, in the extension phase (months 13–24), all patients continued the DFP treatment until month 18. At this point, the patients were re-randomized to either early cessation at month 18 or delayed cessation at month 24 (study end). In the second phase of the extension (between months 19 and 24), the improvement in motor function in patients on continuous DFP waned (relative to that attained in the initial study, that is, months 1 to 6 or months 6 to 12) but remained significantly greater than that seen in patients for whom DFP treatment was withdrawn at month 18) (Fig. 7, right and Supplementary Table S5).
Discussion
Here, we present the first clinical demonstration of a novel strategy for treating PD. DFP targeted regional iron accumulation but did not significantly interfere with iron-dependent mechanisms essential for normal physiological functions. The selection of DFP as the pharmacological tool for differentially reducing foci of excessive iron accumulation was not solely based on its ability to scavenge labile cell iron and thereby confer oxidative-stress protection since this type of protection might also have been achieved with other permeant chelators that can remove iron (and most likely other transition metals) from organs (12, 30, 39, 41, 46, 70, 75). A unique feature of DFP-like chelators is their ability to donate chelated iron to unsaturated transferrin (64, 65), thus avoiding the body iron losses generated by other chelators in clinical use. Although we have no direct evidence of iatrogenic brain iron loss or redistribution, we can assume that DFP treatment did not cause CNS iron deprivation because none of the DFP-treated PD patients developed new neurological signs, particularly restless legs syndrome (10). Moreover, the 12-month course of DFP significantly reduced foci of accumulated iron in the SN of PD patients and did not cause any abnormal decrease in R2* brain areas not directly involved in the pathophysiology of PD or detectable changes in systemic levels of iron or other transition metals (Table 1 and Supplementary Tables S2 and S3).
We noted that after several months of daily chelation, the levels of iron deposition determined by MRI stabilized at a lower level than at the onset of treatment without a commensurate functional improvement, but also that following cessation of chelation iron deposition reappeared. These features that are indicative of the ongoing iron dynamics of the pathological state (Fig. 5) led us also to consider future readjustment of the chelation regimen after the first 12 months treatment as a possible means to reduce future risks while preserving the attained benefits.
Our clinical justification for a pilot clinical study with DFP on PD patients resided largely on: (i) the experience gained in previous safety/tolerability assessments of DFP in the treatment of nonsystemic iron-overloaded disorders (2, 67) and (ii) the continuation of an already established optimal dopaminergic regimen (i.e., either a dopamine agonist or small doses of L-dopa) for each PD patient (61). This setting was chosen for several reasons: (i) first and foremost, to avoid enrolling atypical dopa-unresponsive patients in the study, (ii) to maintain patient comfort and promote good compliance throughout the entire length of the study, and (iii) to allow the assessment of possible interactions between DFP and dopaminergic drugs. Accordingly, we expected to see slower disease progression in DFP-treated patients than in patients treated only with dopaminergic agents and, conversely, greater disease progression upon withdrawal of chelation (despite the continuation of the dopaminergic treatment). We deliberately restricted the pilot study to a relatively small group of patients (n=∼40) who could be comprehensively monitored in a single study center. This enabled us to perform all the clinical examinations (motor handicap assessment and MRI-based measurements of SN iron levels), specimen withdrawals (including repeated lumbar punctures), and clinical laboratory assessments by the same personnel and according to the same protocols. We consider that these factors contributed to the achievement of the statistically significant changes associated with DFP treatment in 100% of the ES patients (with 12 months of DFP treatment) and in 66% of the DS patients (with 6 months of DFP treatment). Moreover, the symptomatic benefits of DFP were obtained by switching the DS group from placebo to DFP and, conversely, were reduced upon cessation of DFP treatment. Thus, when patients were re-randomized from DFP treatment to either placebo or DFP (early or late cessation groups), the benefits of DFP were still evident.
In terms of the drug's mode of action, our cell and translational studies indicate that DFP alone has a protective effects on neuronal viability on at least two levels: (i) via a direct reduction in cellular labile iron pools and the ensuing detoxification and/or indirectly via induction of cell protective measures [as proposed recently for other chelators (38, 70)] and (ii) via a reduction in enzymatic dopamine catabolism and/or through nonenzymatic oxidation of the naturally produced dopamine or supplemented dopamine substitutes. We base the latter assumption on the fact that DFP decreased the 3-O-methyl-DOPA/dopamine ratio in a dose-related manner in control and MPTP-intoxicated animals, indicating the possible inhibition of COMT activity via metal chelation and/or by interaction with the enzyme, due to the similarity between DFP hydoxypyridine structure and the enzyme's native substrates (68). Moreover, the fact that neither in DFP-treated PD patients nor in the DFP-pretreated mice subjected to MPTP there was any significant change in DOPAC/dopamine levels is interpreted as an indication for the unlikeness of MAO-B inhibition by DFP as contributor to its effect on PD patients or to MPTP to MPP+ conversion in the animal model of disease. Some additional insights could be gained from future studies of oxidized metabolites in plasma and CSF, as shown in similar studies in cystic fibrosis (45).
At this stage, we are not in a position to differentiate between clinical benefits associated with iron chelation alone and those associated with a permissive effect of chelation on dopaminergic treatments nor with chelation-independent features of the hydroxypyridin-on DFP per se. DFP can be shown to confer upon cells protection from various oxidative insults, as reflected in the partial reduction of both cell labile iron, cell ROS production per se, as well as in cell parameters related to or affected by ROS (Figs. 2 and 4). This would indicate that only some of the cell protected properties might be associated (directly or indirectly) with DFP chelation of labile iron per se. Among these properties, we differentiate between labile-DFP-chelatable iron that catalytically enhances ROS production in cell compartments to that which confers chemical activity to key enzymes (e.g., proline-hydroxylases) and whose chelation triggers cell cytoprotective measures, as proposed earlier for the action of DFP in Friedreich ataxia (2). Such indirect effects of chelators have been implicated in experimental models of PD, whereby cell protective mechanisms are activated by hypoxia-inducible factors (38, 70) or by a reduction in an α-synuclein-induced pathology (18).
The present pilot clinical study was originally designed to establish a proof of concept from cell to human study. Although it was not powered for investigating DFP's mechanism of action in the clinical setting of PD, its reliance on the delayed-onset drug therapy paradigm allowed us to conduct the analysis used in trials of other drugs for differentiating between symptomatic effects and disease-modifying effects (49, 55, 60). To the extent that the fulfillment of the three key endpoint criteria by rasagiline 1 mg/day monotherapy in the ADAGIO study (49, 55) is suggestive of a disease-modifying mechanism, we can tentatively draw a similar conclusion regarding DFP 30 mg/kg/day effects on dopamine-treated PD patients (Supplementary Table S4). These results are interpreted as sufficiently significant to prompt multicenter clinical studies of chelation that (either alone or in combination with dopaminergic treatment) might provide clinical benefits without compromising systemic iron properties. Such studies would also provide more definitive information on DFP's putative mechanisms of action.
Materials and Methods
The acute MPTP mouse model
Saline- and MPTP-treated mice (5-month-old male C57Bl/6J mice, weighting 28–30 g, from Janvier Le Genest St Isle, France) (MPTP 4×20 mg/kg i.p. over 24 h) received 100 or 150 mg/kg i.p. DFO or p.o. DFP (ApoPharma) twice a day for 10 days (starting 3 days before saline or MPTP intoxication).
Spontaneous motor activity in the mice was recorded over a 10-min period in an actimeter equipped with Actitrack analytical software (Panlab, Barcelona, Spain). The transparent Plexiglas open field was equipped with two frames of infrared beams for measuring horizontal motor activity (distance travelled, speed, and type of movement) and vertical motor activity (rearing). Body weight was measured on treatment days and actimetry measurement days.
Brain section analyses were performed on 6-μm-thick coronal sections prepared from the SN/ventral tegmental area using a microtome (Leica, Nussloch, Germany). Three sections were taken (−2.92, −3.16, and −3.4 mm relative to the bregma), as previously described (37), and the number of TH neurons per sample was counted “treatment blinded” under a light microscope (400×magnification) by a highly trained scientist (C.L.). The results of cell body protection of DFP against MPTP neurotoxicity were systematically validated by stereological examination on randomly selected samples. The 1-methyl-4-phenyl pyridinium (MPP+) was measured in the striatum tissue homogenates 4 days after MPTP intoxication as previously described (31).
MRI and positron emission tomography image analysis in the mouse model
The MRI T2* value was analyzed through an exponential curve described by 12 echoes on a 7-T MRI machine (Biospec from Bruker, Ettlingen, Germany). [18F]-DOPA positron emission tomography (PET) imaging was performed with mouse-adjusted doses.
Human cell model
A LUHMES cell line was generated by immortalizing human embryonic mesencephalic cells with a v-myc oncogene driven by tetracycline-activatable promoter and maintained as described elsewhere (62). Peripheral blood mononuclear cells from healthy donors were prepared from buffy coats diluted in RPMI 1640 (Life Technologies, Paisley, Scotland) that were layered over a Ficoll gradient (Pharmacia, Upsala, Sweden), centrifuged (400 g×30 min), harvested, washed, and resuspended in 100 ml in 0.9 mg/ml NaCl. The lymphocytes isolated by elutriation (Avanti J-20 XP, Beckman Coulter, Inc., Brea, CA) were cultured in 75-cm2 flasks for 2 h at 37°C at a density of 25–50×106 cells per milliliter RPMI supplemented with 10% fetal calf serum and antibiotics.
Flow cytometry
Cell viability, ROS generation, and the intracellular labile cell iron pool were determined with the supravital dye propidium iodide (0.5 μM), hydroxyethidium (2.5 μM), DIOC6(3) (40 nM), and acetomethoxy derivate of calcein (calcein-AM) (1 μM), respectively. Analyses were performed on samples with >10,000 cells using a CANTO II flow cytometer with a 488 nM argon laser (BD Immunocytometry Systems, San Jose, CA) and running DIVA software (BD Immunocytometry Systems).
Mitochondrial labile iron pool
Mitochondria (40 μg protein per assay) purified from mouse brain homogenates by differential centrifugation were stained with calcein-AM (1 μM, 10 min, 37°C), washed three times, and resuspended in 10 mM HEPES, pH 7.5, containing 0.05 M sucrose, 1 mM ATP, 0.08 mM ADP, 0.05 mM sodium succinate, 2 mM K2HPO4, and 1 mM DTT. Calcein fluorescence was assessed spectrofluorimetrically (excitation: 485 nm; emission: 535 nm) (66). It should be noted that although calcein labeling of cells with its acetomethoxy precursor calcein-AM occurs primarily in the cytosol (17, 24), the removal/repression/prevention of cytosolic fluorescence by quenching of the probe with added Co(II) (53), cold loading (48), or cell permeabilization (63) exposes iron-sensitive calcein-fluorescence within mitochondria.
Clinical trial (registered as ClinicalTrials.govNCT00943748)
We performed a randomized clinical trial with DFP versus placebo on early-stage PD patients [<3 years after diagnosis, with stable early stage (i.e., no dyskinesia/motor fluctuations)] receiving an optimized dopaminergic regimen (i.e., either dopamine agonist and/or low doses of L-dopa, with no change in the course of the study) and with no overt anemia, iron deficiency, or other hematological disorders. An initial DFP dose escalation of 300 mg twice a day was applied every third day until the final dose of 30 mg/kg/day was reached. Balanced, computer-based randomization lists with allocation sequences based on block sizes of six were generated by the Lille University Medical Center's Statistics Department and sent to the hospital pharmacy for dispensing identical liquid formulations of DFP and placebo (provided by ApoPharma, Inc., Toronto, Canada). All medical investigative and administrative personnel were masked to treatment assignment. The compliance (>90%) was assessed by questioning and the inspection of dispensed medication packages.
Study design
We performed a randomized, double-blind, placebo-controlled, parallel-group single-center trial. We adopted a DS design: patients received either DFP (30 mg/kg/day in two doses, morning and evening) for 12 months or placebo for 6 months and then DFP for 6 months. All patients continued DFP treatment until month 18 and then entered a 6-month randomized drug cessation phase (whereby some patients switched to placebo and the others continued the DFP treatment).
Parameters assessed
R2* proton relaxation rates (=1/T2*) were obtained with a 3-Tesla MRI system (Achieva from Philips, Best, Netherlands) using a 2D fast-field echo multishot sequence of 15 echoes, starting at a Echo Time1 of 3.6 ms, with an echo spacing of 3.3 ms and a repetition time of 1803 ms. Two stacks were acquired subsequently in the axial plane (orientated along the anterior and posterior white commissure); 17 slices for each (slice thickness: 2 mm; isotropic, no gap; field of view: 230×190 mm, matrix: 116×95, number of signal averages: 2) to cover a volume between the floor of the fourth ventricle floor and the corpus callosum convexity. Images were processed using a T2* tool on an IDL virtual machine (V2,
Safety assessment
Complete blood counts were performed weekly to monitor for the iatrogenic risk of agranulocytosis. The patient's general health status (as reflected by the body weight, systolic/diastolic arterial blood pressures, heart rate, electrocardiogram, and standard blood biochemistry profile) was assessed every 2 months, and adverse event reports were reviewed (anonymously) by a safety monitoring board.
Sample size selection or R2* measurements in the SN
For a 6-month DFP treatment to elicit a change in SN R2* relative to placebo that has 5% alpha risk and 80% power, we estimated the involvement of 24 subjects per group. If the correlation coefficient between consecutive measurements at 6 months intervals is set to 0.4 and the data are adjusted for baseline levels, then the group size could be reduced to 20.
Oxidative stress parameters
Protein carbonyls were assayed following centrifugal filtration-concentration with a kit (Immunodiagnostik AG, Bensheim, Germany). Both MDA concentrations and glutathione status (i.e., glutathione disulfide, GSSG/GSH) were determined in tissue homogenates by using high-performance liquid chromatography with fluorescence detection, as published elsewhere (21). DNA adduct 8-OHdG concentrations were studied in tissue homogenates using commercially available enzyme immunoassays (Highly Sensitive 8-OHdG Check, Gentaur France SARL, Paris, France) as previously described (22). Enzymatic activities of SOD and GPx in whole blood and chemical antioxidant capacity of plasma using Trolox as standard (Sigma Chemical Co, Saint Quentin Fallavier, France) were carried out as published elsewhere (23).
L-dopa and dopamine
Dopamine and metabolites were determined by HPLC using a 6100 column (Chromsystems, Gräfelfing, Germany) and 5001 mobile phase (Chromsystems) with colorimetric detection (Coulochem III, ThermoFisher, Sunnyvale, CA).
Statistical analyses
We used a covariance analysis (adjusted for baseline differences) to estimate clinical outcomes after 6 months of treatment. For nonnormally distributed data, the robustness of the results was checked after log transformation. The threshold for statistical significance was set to p<0.05. The analysis of variance assumption was validated by residual analysis, and the size effect was adjusted for baseline differences. In cell-based and animal studies, we performed nonparametric tests (with Kruskal–Wallis and Mann–Whitney tests for comparisons of three or more nonrelated and two nonrelated groups, respectively). For comparisons of two related samples (e.g., repeated measurements on a single sample), we used the Wilcoxon signed-rank test. All statistical analyses were performed with SAS software (version 9.3, SAS Institute, Inc., Cary, NC).
Study approval
All clinical investigations were performed in accordance with the tenets of the Declaration of Helsinki. All patients provided their written informed consent to participation. A local institutional review board approved the aims and procedures of the main study (national reference number: 2008-006842-25;
Footnotes
Acknowledgments
The authors thank the French Ministry of Health for funding PHRC grants 2008-006842-25; Apopharma for providing DFP and placebo formulations; the French Parkinson's Disease Association and Fédération de la Recherche Clinique du CHU de Lille for promoting the study; the Centre d'Investigation Clinique of Lille, Marie Delliaux, and Salomé Devos for data acquisition; Francine Niset, Carine Piatek, and Valerie Vasseur for their assistance and Dr. David Fraser (Biotech Communication, Damery, France) for editing the article; AFIRNE (Paris) and the DellaPergola Chair in Life Sciences of HUJI for support of Z.I.C., and Olivier Rascol for critical reading of the article.
Authors Contributions and Accountabilities
David Devos, Caroline Moreau: The research project—conception, organization, execution; the article—writing of the first draft, review and critical comment, final draft.
Jean Christophe Devedjian, Jérome Kluza, Charlotte Laloux: The research project—organization, execution; the article—writing of the first draft, review and critical comment.
Aurélie Jonneaux, Maud Petrault, Gilles Ryckewaert: The research project—organization, execution; the article—writing of the first draft.
Guillaume Garçon, Nathalie Rouaix, Patrice Jissendi, Florent Auger, Laura Ravasi, Lucie Hopes, Guillaume Grolez, Bernard Sablonnière, Isabelle Strubi-Vuillaume, Wance Firdaus, Jean-Christophe Corvol, Dominique Pöltl: The research project—execution; the article—review and critical comment.
Kathy Dujardin: The research project—conception, organization, execution; the article—review and critical comment.
Alain Duhamel: The article—review and critical comment and biostatistical analysis.
Noel Zahr: The research project—execution.
Alain Destée: The article—review and critical comment.
Jean-Christophe Corvol: The research project—execution; the article—review and critical comment.
Z. Ioav Cabantchik: The article—writing of the first draft, review and critical comment, final draft.
Christian Rose: The research project—organization; the article—review and critical comment.
L. Defebvre: The research project—execution; the article—review and critical comment.
Philippe Marchetti: The research project—organization, execution; the article—review and critical comment.
Régis Bordet: The research project—conception; the article—writing of the first draft, review and critical comment.
The indicated authors take responsibility for data collection and analysis and the principal investigator (D.D.), who had full access to all the study data, takes full responsibility for submitting the final work for publication.
Author Disclosure Statement
The authors have no financial disclosures to make or potential conflicts of interest to report in relation to this investigator-driven study. The study was funded by the French Ministry of Health (PHRC (Projet Hospitalier Recherche Clinique) grants: Protocol ID: 2008-006842-25). Apopharma provided DFP and advice on the molecule.
David Devos served on the Scientific Advisory Board for Novartis and Aguettant and has received PHRC grants from the French Ministry of Health and research funding from the ARSLA charity and honoraria from pharmaceutical companies for consultancy and lectures.
Caroline Moreau served on the Scientific Advisory Board for Aguettant.
Kathy Dujardin served on the Scientific Advisory Board for Novartis and received a grant from the MJ Fox Foundation for Parkinson's research.
Alain Destée worked with Laboratories Novartis, Boehringer-Ingelheim, Schering-Plough, Merck-Serono, GSK, Merck-Scherring, Teva, Sanofi-Aventis, Lundbeck, Janssen-Cilag, GE Healthcare, Schawrtz, Bioprojet, Neurosearch, Synosia, Holmes, and Impax.
Jean-Christophe Corvol received research grants from INSERM and APHP and speaker's honoraria and travel reimbursements from Lundbeck, Biogen, and Schering-Plough.
Z. Ioav Cabantchik was supported by the A&M Della Pergola Chair in Life Sciences, served on the Scientific Advisory Board of Novartis ESB, received in the past research grants from NIH, EEC (5 and 6 framework), Israel Science Foundation, Novartis, and Apopharma (also lecture honoraria from the last two) and is presently a consultant for Aferrix, Ltd and Hinoman, Ltd.
Christian Rose served on the Scientific Advisory Board for Novartis, received various honoraria from Novartis and Genzyme for lectures.
Luc Defebvre served on the Scientific Advisory Board for Novartis and Aguettant and received honoraria from pharmaceutical companies for consultancy and lectures.
Régis Bordet is funded by the French Ministry of Research. He has received honoraria from pharmaceutical companies for consultancy and lectures at symposia.
The other authors have no disclosures to report.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.