
Abstract
Tetrahydrobiopterin (BH4) functions as a cofactor for several important enzyme systems, and considerable evidence implicates BH4 as a key regulator of endothelial nitric oxide synthase (eNOS) in the setting of cardiovascular health and disease. BH4 bioavailability is determined by a balance of enzymatic de novo synthesis and recycling, versus degradation in the setting of oxidative stress. Augmenting vascular BH4 levels by pharmacological supplementation has been shown in experimental studies to enhance NO bioavailability. However, it has become more apparent that the role of BH4 in other enzymatic pathways, including other NOS isoforms and the aromatic amino acid hydroxylases, may have a bearing on important aspects of vascular homeostasis, inflammation, and cardiac function. This article reviews the role of BH4 in cardiovascular development and homeostasis, as well as in pathophysiological processes such as endothelial and vascular dysfunction, atherosclerosis, inflammation, and cardiac hypertrophy. We discuss the therapeutic potential of BH4 in cardiovascular disease states and attempt to address how this modulator of intracellular NO-redox balance may ultimately provide a powerful new treatment for many cardiovascular diseases. Antioxid. Redox Signal. 20, 3040–3077.
I. Introduction
(6R) 5,6,7,8-
The biological synthesis of pteridines was first discovered in 1889, when Sir Fredrick Gowland Hopkins isolated the yellow pigments from the wings of English butterflies (114). The identity of these compounds was not recognized until the 1940s, when three compounds were isolated and shown to share a novel pyrimidine ring system. The name pterin was given to these compounds after the Greek name Ptera, meaning wing, from which these molecules were first isolated.
Kaufman and coworkers were the first to demonstrate a cofactor function for BH4 in mammalian biology. In the liver, BH4 was shown to be an essential cofactor for the metabolism of phenylalanine to tyrosine by phenylalanine hydroxylase (187) and was later found to play a similar cofactor role for the two other mammalian AAAH, tyrosine hydroxylase and tryptophan hydroxylase (24). These findings implicated BH4 in the biosynthesis of neurotransmitters, including epinephrine, norepinephrine, dopamine, and 5-HT (Fig. 2). The observation that BH4 synthesis is dramatically induced in mammalian cells by cytokines puzzled researchers for many years, as there was no known enzyme that was both inducible and dependent on BH4. This led to the discovery that BH4 is an essential cofactor for inducible NOS (iNOS) activity (146, 270). With the groundbreaking discovery of the endothelial-derived relaxing factor (EDRF), and subsequently the critical role for BH4 in the synthesis of EDRF, the investigation of BH4 and its implication in cardiovascular homeostasis soared—indeed, to date, more than 650 publications are highlighted using the search term “cardiovascular tetrahydrobiopterin” in PubMed.
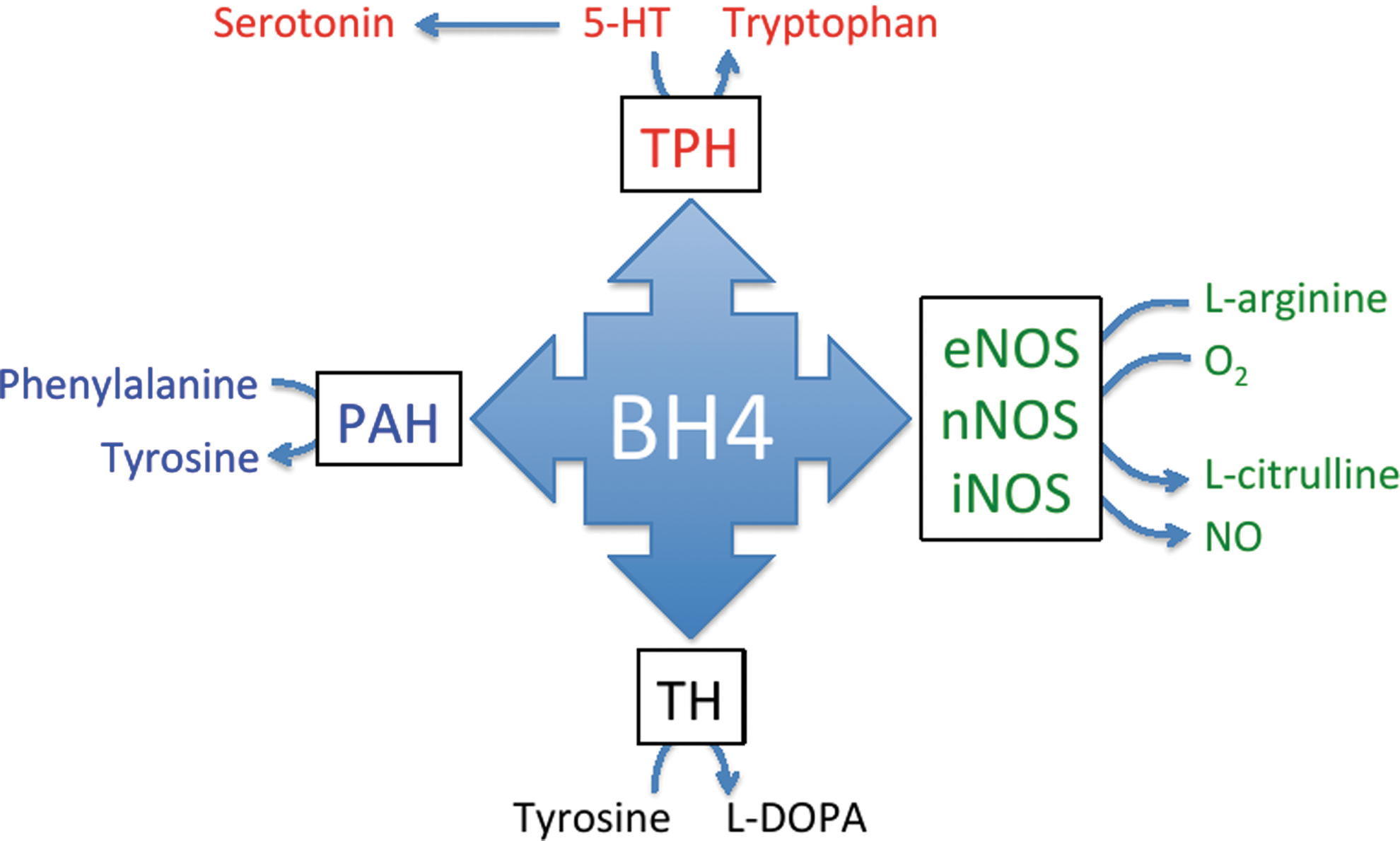
Cardiovascular disease is the largest cause of mortality and morbidity in Western societies, and it is also emerging as a major health burden in developing countries. This comprehensive review will discuss the physiological and pathophysiological roles of BH4 in cardiovascular health and disease. We will describe the mechanistic importance of BH4 to NOS catalysis that underlies NOS uncoupling, the role of BH4 in the maintenance of cardiac and vascular homeostasis, and describe in detail the deleterious effects of diminished BH4 levels on cardiovascular pathophysiology and inflammation. We will then consider how alterations in BH4 bioavailability may modulate the development of cardiovascular diseases such as atherosclerosis, heart failure, and hypertension (Fig. 3). This review brings together the biochemical and mechanistic roles of BH4 in the cardiovascular health, inflammation, and disease states, and discusses current BH4-based therapeutic strategies and methods that are used to alleviate vascular and cardiac dysfunction. More focused reviews of NOS biochemistry and regulation can be found elsewhere and will, therefore, not be discussed in further detail here.
II. Biochemistry
A. Tetrahydrobiopterin
The pterins were first described by Hopkins in 1895 (114) and characterized by Watt in the 1960s (304). Initially identified as a cofactor for the AAAH (phenylalaninehydroxylase (EC 1.14.16.1), tyrosine 3-hydroxylase (EC 1.14.16.2), and tryptophan 5-hydroxylase (EC 1.14.16.4)), BH4 was revealed as a required cofactor for the NOS enzymes by Stuehr and co-workers (146) and Tayeh and Marletta (270) in 1989. BH4 is also a cofactor for AGMO, which is the only enzyme known to cleave the ether bond of alkylglycerols and lyso-alkylglycerol phospholipids, including lyso-platelet activating factor (PAF), and current studies are only now unraveling the physiological role of this enzyme (302, 303). Although currently not implicated in cardiovascular homeostasis or disease, future investigations and the development of AGMO knockout mice may reveal a cardiovascular role for this protein.
1. De novo synthesis of BH4 by guanosine triphosphate cyclohydrolase I
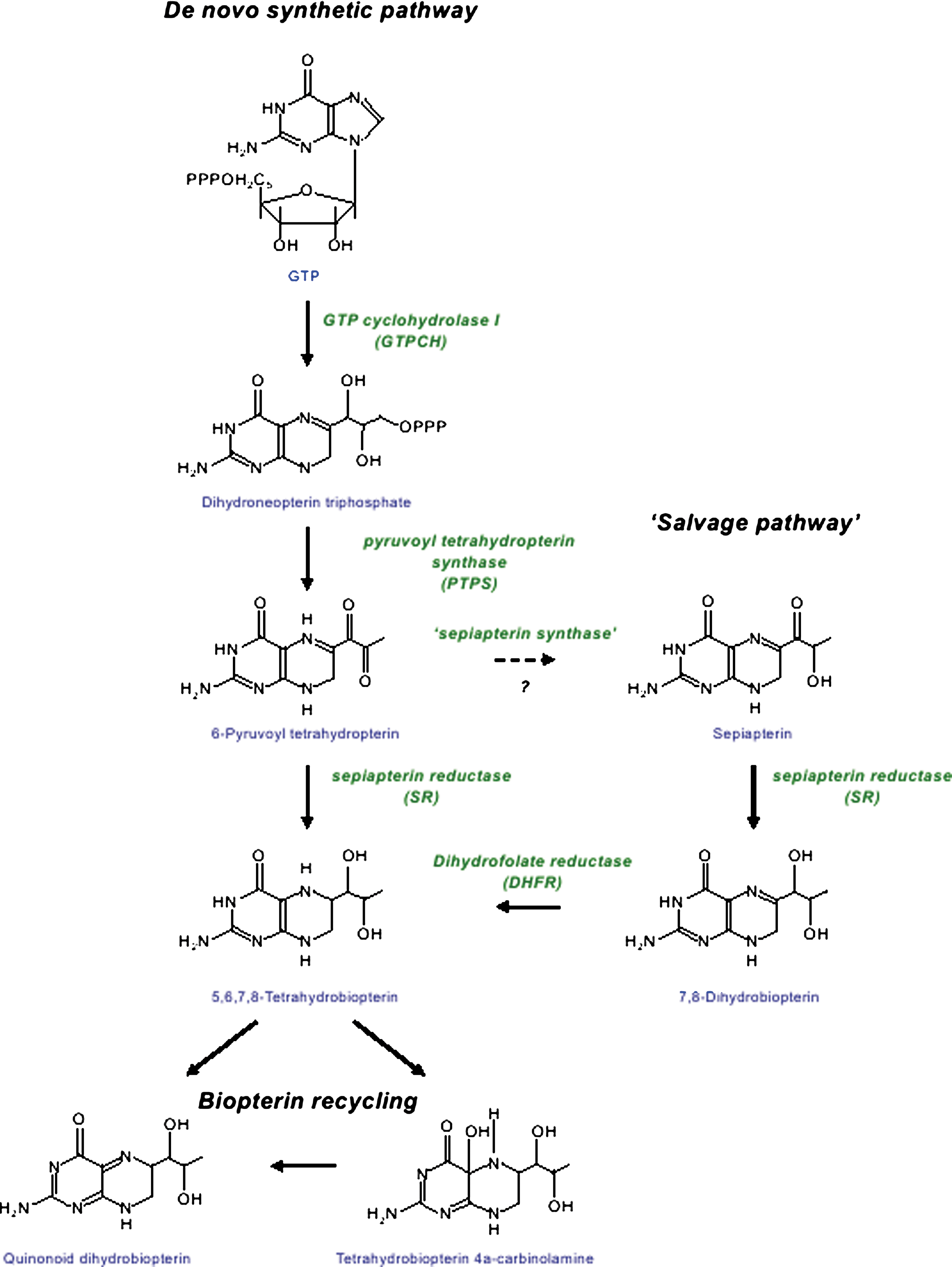
BH4 is synthesized de novo from guanosine triphosphate (GTP) by the sequential action of three enzymes: GTP cyclohydrolase I (GTPCH, EC 3.5.4.16), 6-pyruvoyl tetrahydropterin synthase (PTPS, EC 4.6.1.10), and sepiapterin reductase (SR, EC 1.1.1.153). Outlined in Figure 1, the first and rate-limiting reaction is catalyzed by GTPCH, producing 7,8-dihydroneopterin triphosphate (DNTP) from GTP. This pivotal first step in BH4 biosynthesis is complex and highly regulated at the transcriptional, translational, and post-translational levels (93). GTPCH-catalyzed formation of DNTP is a common initial step in the biosynthesis of unconjugated pterins, folates, and riboflavin, but not molybdopterin (a cofactor of sulfite oxidase, xanthine dehydrogenase, and aldehyde oxidase in man).
Continuing along the de novo BH4 synthesis pathway, H2NTP is next converted to 6-pyruvoyl tetrahydropterin by the zinc-dependent enzyme, PTPS. Although GTPCH is rate limiting to BH4 synthesis in most cells, PTPS has been suggested to be rate limiting in some, most notably human hepatocytes. PTPS may become rate limiting in other tissues and cells, after stimulation with cytokines and other immunological stimuli that induce BH4 synthesis by up-regulation of GTPCH expression (274).
The final reaction in the pathway is catalyzed by SR and involves two sequential NADPH-dependent reductions; a side-chain carboxyl of 6-pyruvoyl tetrahydropterin is initially reduced and rearranged to form the intermediate 6- lactoyl tetrahydropterin, and then reduced on a second side-chain carboxyl to BH4 (274).
2. Role of BH4 in NOS catalysis
The role of BH4 in the catalysis of the AAAH enzymes for the synthesis of tyrosine, L-3,4-dihydroxyphenylalanine (L-DOPA), and serotonin is well established; is excellently described in several key reviews (274, 315); and, therefore, will not be further discussed in the review.
The three isoforms of NOS are encoded by distinct mammalian genes (328). Two of the three isoforms are constitutively expressed in cells and synthesize NO primarily in response to transient elevations in intracellular Ca2+ levels, a process that is mediated by the binding of calcium-calmodulin (CaM) (74). These constitutive enzymes are designated neuronal NOS (nNOS or NOS I) and endothelial NOS (eNOS or NOS III) after the cell type and order in which they were discovered. The third isoform, or inducible NOS (iNOS or NOS II), is typically synthesized in response to inflammatory stimuli, but has been shown to be constitutively expressed in some tissues, such as lung epithelium (63). The main difference between iNOS and the constitutive NOS isoforms is the high-affinity binding of CaM to iNOS such that it remains bound even at subnanomolar levels of Ca2+, rendering iNOS constitutively active (35).
The first NOS to be isolated, nNOS, was purified from rat brain (22). nNOS was found to be dimeric, with each monomer consisting of an N-terminal oxygenase domain and a C-terminal reductase domain, bridged by a short (∼30 amino acid) calmodulin-binding peptide. The oxygenase domain contains binding sites for heme, the required cofactor tetrahydrobiopterin (BH4) and L-arginine, forming the active site where NO synthesis takes place. The reductase domain shares ∼40% sequence homology with NADPH cytochrome P450 reductase (22) and contains binding sites for FMN, FAD, and NADPH (23, 260). During NO synthesis, the flavins in the reductase domain of NOS serve to transfer electrons from NADPH to heme iron in the oxygenase domain (triggered by CaM binding in nNOS and eNOS), enabling the activation of heme-bound molecular oxygen for NO synthesis (169, 170). Subsequent purification and cloning of the two other NOS isoforms showed that they share the same basic bidomain structure where each is an obligate dimer, with the reductase domain of one monomer supplying electrons to the oxygenase domain of the other (Fig. 4).
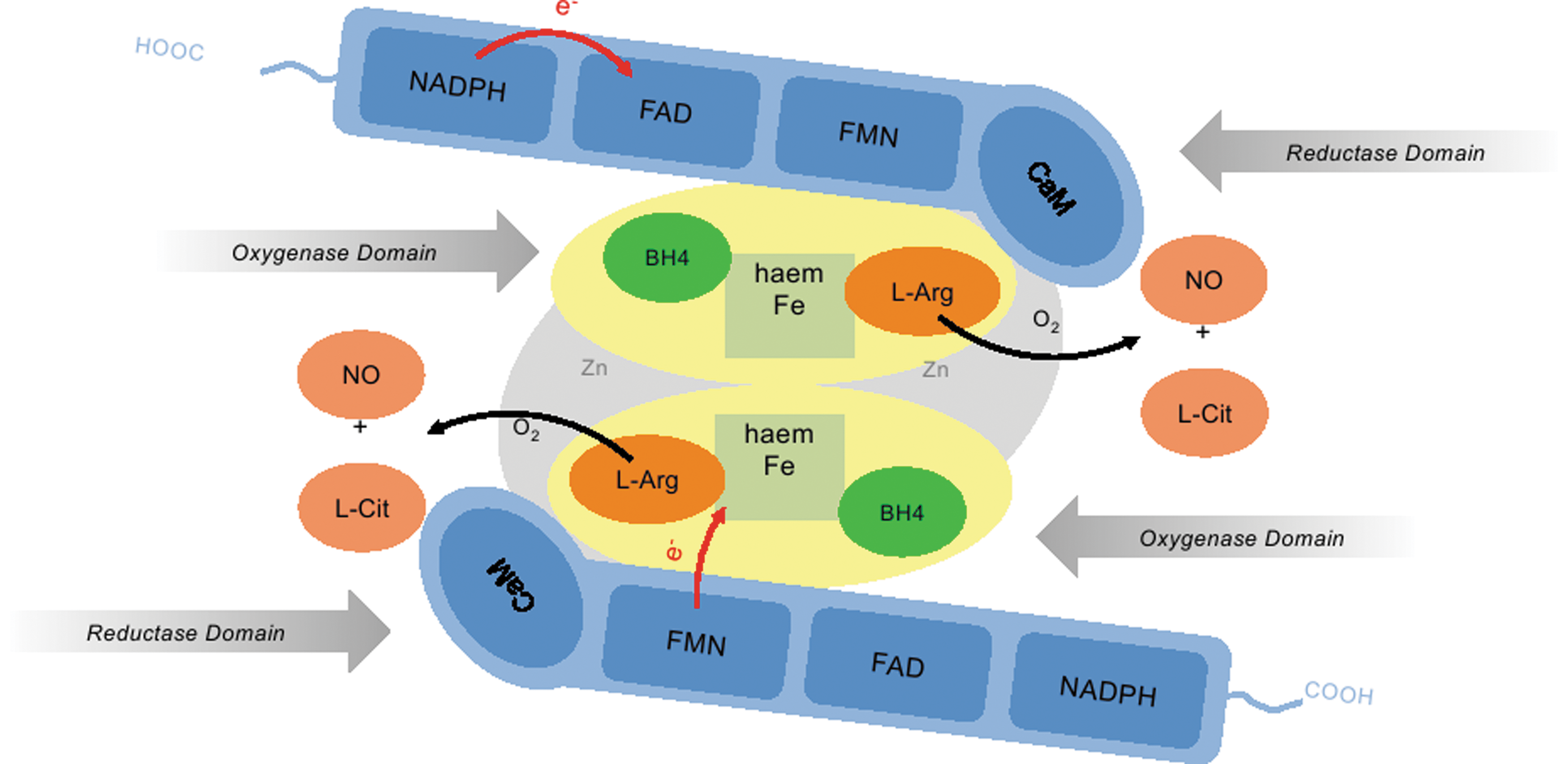
After the discovery of BH4 as an essential cofactor in NOS catalysis, it became clear that BH4 adopted a different role in NOS catalysis when it was discovered that NOS contained a cytochrome P450-type heme, a moiety which was able to support the activation of oxygen without the need for a pterin cofactor. The main evidence against a direct redox role for BH4 came from the observation that citrulline formation is not stoichiometric with BH4 consumption and the fact that BH4 had little effect on the initial rate of NOS catalysis (88). A comparison of eNOS (72, 213), iNOS (46), and nNOS (287) oxygenase domains with crystal structures of the AAAHs has revealed fundamental structural differences between the BH4 binding sites. In NOSs, BH4 functions, in part, as an allosteric modulator of arginine binding. The binding of BH4 to NOS elicits a conformational change that increases the affinity for the binding of arginine-based ligands (139, 158). Support for an allosteric role of BH4 was confirmed by spectrophotometric and electron paramagnetic resonance (EPR) studies, which show that BH4 binding converts the heme iron from a low-spin to a high-spin state (90). Another possible role of BH4 was considered in dimer assembly (86). Although proved not to be essential for dimer assembly (221), BH4 binding plays a role in dimer stabilization (140).
The allosteric effects mentioned earlier do not fully explain the essential role of BH4 in NO synthesis, and electron donation from BH4 to the heme iron remains almost certain. Indeed, only fully reduced pterins such as tetrahydrobiopterin have ever been shown to support catalysis (212, 219, 288). Moreover, tetrahydrobiopterins with modifications that would cause them to be redox silent are unable to catalyze NO synthesis (107, 310). Spectral and EPR studies have revealed the presence of a trihydrobiopterin (•BH3) radical intermediate, which strongly supports a redox role for BH4 in NOS catalysis and may explain the ability of eNOS-bound BH4 to limit the release of oxygen as O2 − (17).
3. BH4 bioavailability and eNOS uncoupling
Many studies have focused on the potential role of BH4 oxidation to BH2 and other oxidized biopterin species in reducing BH4 bioavailability for eNOS. Although superoxide (O2
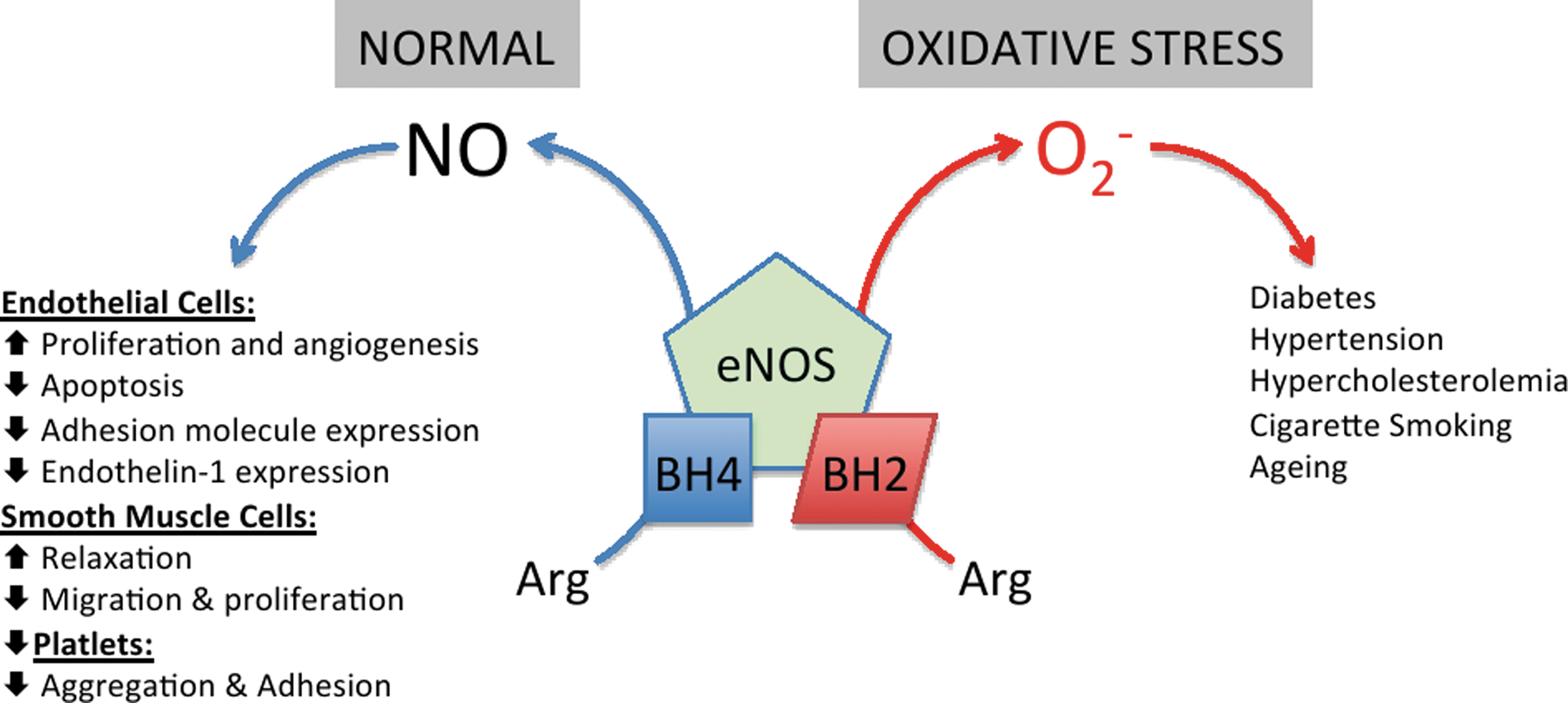
Depletion of BH4 in oxidatively stressed endothelial cells can result in eNOS “uncoupling,” where electron transfer from NOS flavins becomes “uncoupled” from L-arginine oxidation, the ferrous-dioxygen complex dissociates, and O2 − is produced from the oxygenase domain (258, 287, 293, 294). Indeed, it is now believed that the intracellular BH4:BH2 ratio, rather than absolute concentrations of BH4, is the key determinant of eNOS uncoupling (42, 43, 287, 292).
O2 − generation by eNOS has been implicated in a variety of experimental and clinical vascular disease states of the heart and vascular system, including heart failure, ischaemia reperfusion injury, atrial fibrillation (AF), diabetes (39, 95), cigarette smoking (104), hypertension (148), and atherosclerosis (163), shown in Figure 5 and described in greater detail throughout this review.
4. Allosteric regulation of GTPCH by GTPCH feedback regulatory protein
It was appreciated for many years that GTPCH is subject to feedback inhibition by the pathway end-product, BH4 (237). However, it was the demonstration that the activity of pure recombinant GTPCH protein was not inhibited in vitro by BH4 (97) that proved to be the most interesting. This led to the discovery of a novel protein which co-purifies from rat liver with GTPCH that, when added to purified recombinant GTPCH in the presence of BH4, is able to reconstitute feedback inhibition (97)—this protein was named GFRP, for GTPCH Feedback Regulatory Protein (177). In the presence of recombinant rat GFRP, BH4 elicits a concentration-dependent inhibition of GTPCH activity, confirming that GFRP is both necessary and sufficient for BH4-mediated feedback inhibition of GTPCH (334). Stimulation of GTPCH activity is apparently specific to Phe and can occur in the presence or absence of bound BH4.
Immunostimulant-induced NO synthesis was found to be markedly suppressed in many cell types when de novo synthesis of BH4 was inhibited at the level of GTPCH. It seemed unlikely that the GFRP feedback inhibition mechanism was functioning to influence NOS activity under these conditions, given that the Km of BH4 for NOS is very low (30–100 nM) compared with threshold BH4 levels reported to be necessary for the engagement of feedback inhibition (1–10 μM) (97, 177). Nonetheless, GFRP mRNA was detectable by RT-PCR at basal levels in RASMCs, apparently inducible by immunostimulants, and Phe was observed to increase BH4 synthesis, suggestive of basal GFRP-dependent inhibition of GTPCH (327). In contrast, levels of GFRP mRNA in human myelomonocytoma cells, measured by northern blotting, were reported to be significantly down-regulated after lipopolysaccharide (LPS) treatment (314). qRT-PCR analysis of GFRP mRNA in LPS-treated human umbilical vein endothelial cells (HUVEC), and in various tissues extracted from rats treated with LPS, also indicated down-regulation of GFRP mRNA (314). Recently, it was shown that NO can suppress GFRP protein expression in hepatocytes, resulting in the activation of GTPCH and increased immunostimulant-induced NO production due to increased iNOS subunit dimerization and elevated levels of intracellular BH4 (201).
5. BH4 recycling
Depending on its cofactor function for the AAAH versus the NOS enzymes, BH4 turnover products and regeneration reactions differ. In the aromatic amino acid hydroxylase reactions, BH4 is oxidized to tetrahydrobiopterin-4a-carbinolamine; BH4 is subsequently regenerated in a two-step process. Actions of pterin-4a-carbinolamine dehydratase (EC 4.2.1.96; PCD) lead to the production of the quinonoid dihydrobiopterin intermediate, which is subsequently reduced by dihydropteridine reductase (EC 1.6.99.7; DHPR). Both enzymes are expressed in mammalian liver (100), kidney, and brain (54), and mutations in the PCD and DHPR genes are associated with clinical systemic BH4 deficiency and hyperphenylalaninemia.
As a cofactor for NOS, BH4 is not oxidized to tetrahydrobiopterin-4a-carbinolamine but, during the transfer of electrons to the ferrous-dioxygen complex in the NOS active site, it forms the protonated trihydrobiopterin cation radical (BH3.H+), which is subsequently reduced in the next catalytic cycle by electron transfer from eNOS flavins (120, 290). Since the BH3.H+ radical returns to the BH4 state after each electron transfer step, continuous BH4 regeneration by PCD and DHPR is not a requirement for short-term eNOS activity in endothelial cells. However, ultimately, this regenerative cycle is not sufficient to maintain cellular BH4 levels, and on activation, eNOS is reliant on the intracellular BH4 pool and, therefore, the presence of these recycling enzymes becomes critical.
B. Dihydrofolate reductase
In addition to key roles in folate metabolism, dihydrofolate reductase (DHFR; E.C. 1.5.1.3) can reduce BH2, regenerating BH4 (173, 194). Thus, it is likely that net BH4 cellular bioavailability reflects the balance between de novo BH4 synthesis, loss of BH4 by oxidation to BH2, and the regeneration of BH4 by DHFR, shown in Figure 1. In human liver extracts, DHFR has been shown to reduce BH2 to BH4 as a part of the salvage pathway for biopterin synthesis (51). Several studies implicate the biopterin recycling pathway in the regulation of steady-state BH4 levels—In a Chinese hamster ovary cell mutant lacking dihydrofolate reductase (DUKX-BII), endogenous formation of BH4 proceeds normally, but unlike the parent cells that express DHFR, extracts do not convert sepiapterin or BH2 to BH4 (190). In vitro and in vivo studies reveal that this BH2 reductase activity of DHFR is crucial in determining cellular BH4 homeostasis, NO bioavailability, and, ultimately, eNOS coupling in cultured endothelial cells (44). Exposure to angiotensin II down-regulated DHFR expression, decreased BH4 levels, and increased eNOS uncoupling, which was restored by overexpression of DHFR (32). Pharmacological inhibition of DHFR activity by methotrexate (MTX), or genetic knockdown of DHFR by RNA interference, reduced intracellular BH4 and increased BH2 levels, resulting in enzymatic uncoupling of eNOS in endothelial cells. In cells expressing eNOS with low biopterin levels, DHFR inhibition or knockdown further diminished the BH4:BH2 ratio and exacerbated eNOS uncoupling (44, 262).
The importance of DHFR activity in vivo was has been investigated in mice after the inhibition of DHFR with MTX. In these studies, using models of BH4-deficiency (hph-1 mice, developed by ENU mutagenesis, have diminished GCH1 mRNA and total GTPCH protein, leading to approximately 90% decrease in GTPCH activity) and GTPCH-1 overexpression specifically within the endothelium (GCH-Tg mice), DHFR activity was required to maintain eNOS coupling in vivo and this effect was more prominent in conditions of overall BH4 deficiency. BH4 augmentation protected against these deleterious effects of DHFR inhibition by MTX. Moreover, it has also been observed that MTX treatment in patients with inflammatory disease leads to a striking accumulation of BH2 in plasma, further illustrating the requirement for DHFR activity in the maintenance of BH4 homeostasis in humans in vivo (45).
Interestingly, some previous studies have shown that DHFR levels or activity are diminished in experimental models of cardiovascular disease states, suggesting that insufficient recycling of BH2 to BH4 by DHFR is at least, in part, responsible for the reduced BH4 levels and the accumulation of BH2, leading to eNOS uncoupling. For example, DHFR protein levels are significantly decreased in streptozotocin (STZ)-induced diabetic mice, and diabetes-induced impairment of cardiac myocyte function is exacerbated after treatment of mice with the DHFR inhibitor, MTX (216). Furthermore, reduced DHFR activity in adult cardiac myocytes underlies their limited capacity to synthesize BH4 after cytokine stimulation after treatment of rat cardiac allograft recipients with sepiapterin (126). Insufficient DHFR activity might also explain impaired vasorelaxation in atherosclerotic vessels from hypercholesterolemic rabbits, as exposure to sepiapterin that increases BH4 levels through the DHFR-dependent conversion of BH2 failed to reverse the impairment (291).
C. Dihydropteridine reductase
DHPR, along with PCD, is critical for the regeneration of BH4 that is required for activity of the AAAHs. DHPR concentrations compared with those of the AAAHs are relatively high. The enzyme is almost ubiquitously expressed, and its presence in tissues lacking enzymes of the AAAHs suggests that DHPR may be involved in other metabolic processes. DHPR may preserve tetrahydrofolate levels in the brain, where the concentration of DHFR is comparatively low (135). The presence of DHPR in the lung and liver may be protective in BH4 homeostasis, catalyzing the reduction of q-BH2 to BH4 to maintain NO production by NOS. Deficiency in DHPR is an autosomal recessive condition, and it has been shown to cause hyperphenylalaninemia due to BH4 deficiency.
D. Sepiapterin reductase
In contrast to other BH4 metabolic enzymes, SR is involved in both the synthetic and salvage pathways of BH4 synthesis (Fig. 1). SR was first discovered in chicken and rat liver by Matsubara and Akino, and purification of SR from rat erythrocytes showed it to consist of two 28-kDa sub units (171). Exogenous sepiapterin can be reduced in all cells by SR to 7,8-dihydrobiopterin (BH2) and further by DHFR to form BH4. Known as the salvage pathway, this has been exploited by many investigators as an approach to increase BH4 levels by pharmacological supplementation of sepiapterin (190).
Little is known about the expression and regulation of mammalian SR. The only evidence for a regulatory role of SR in NO synthesis comes from a study by Gao and co-workers, where knockdown of SR in endothelial cells was associated with dramatic decreases in both BH4 content and NO levels (82). Evidence for the sepiapterin synthesis pathway in humans comes from a recent study of rare patients with SR deficiency, in which sepiapterin levels were elevated in cerebrospinal fluid (CSF), which suggests endogenous production of sepiapterin (344).
III. Homocysteine, Folate, and ADMA
Homocysteine is dependent on S-adenosyl-methionine, which is responsible for many intracellular methylation reactions. The by-product of these reactions, S-adenosyl-homocysteine, is then hydrolyzed to form homocysteine. When there is an excess of methionine, homocysteine is metabolized via trans-sulfurylation, producing cystathionine and, eventually, cysteine. Cystathionine b-synthase converts homocysteine to cystathione and requires vitamin B6 as an essential co-factor. Under conditions of methionine deficiency, homocysteine is remethylated into methionine in the liver via betaine-homocysteine-methyltransferase; however, in most tissues, homocysteine is remethylated into methionine by methionine synthase, which uses vitamin B12 as a co-factor and 5-methyltetrahydrofolate (5-MTHF) as a substrate. 5-MTHF synthesis is catalyzed by methyltetrahydrofolate reductase, which uses tetrahydrofolate as a substrate (Fig. 6).
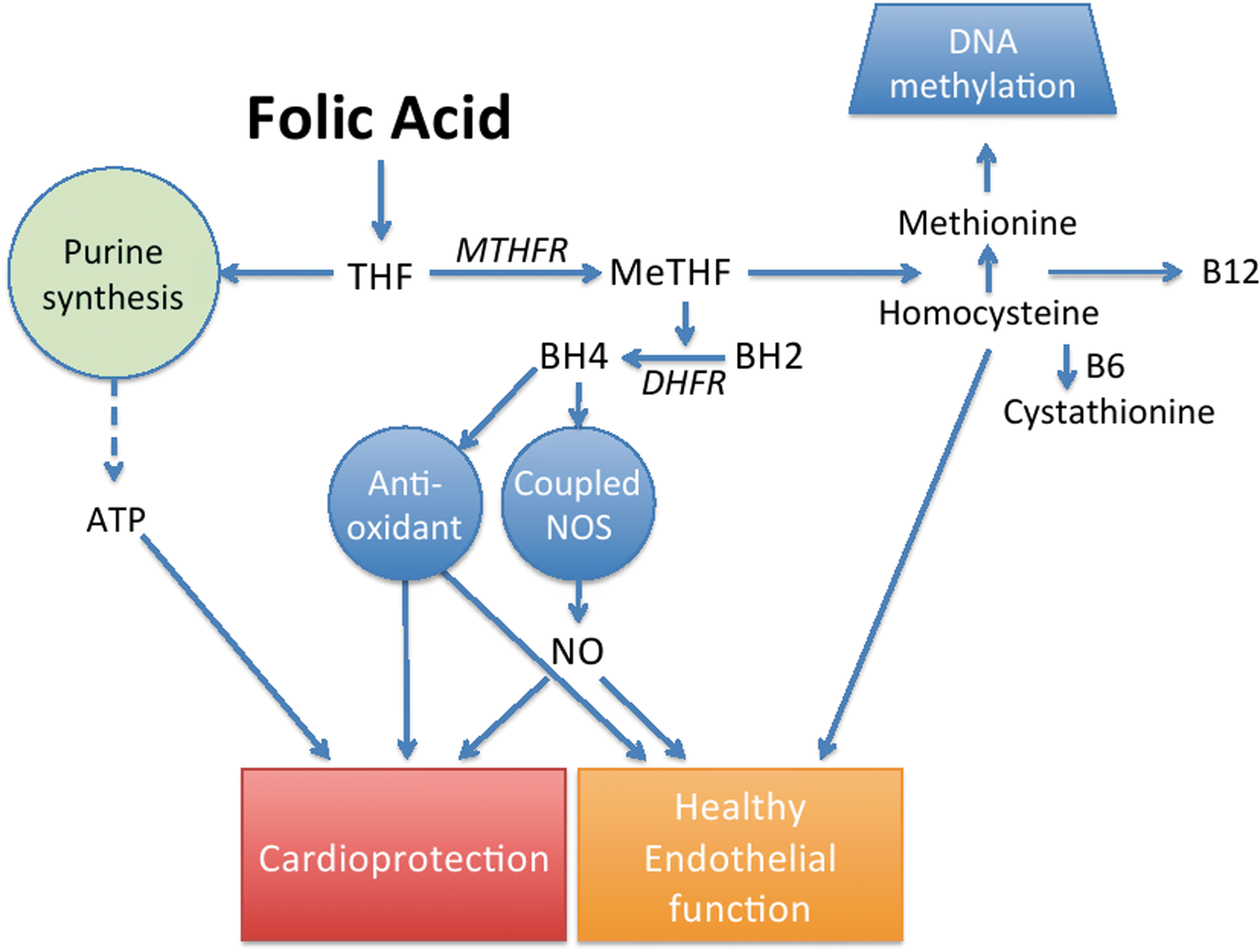
Plasma levels of homocysteine and folate are related to cardiovascular risk and endothelial dysfunction, which may, in part, be mediated by their influence on BH4. Homocysteine decreases BH4 levels as well as its synthesis by GTPCH, and clinical trials have demonstrated that decreasing homocysteine levels with folate supplementation may retard developing atherosclerosis. However, three larger-scale clinical trials in patients with stable coronary artery disease, myocardial infarction, or in patients with stroke have found that folate treatment failed to improve clinical outcome, thus suggesting the possibility that the increased risk may only be a marker for a related pathophysiological process. Some studies have shown that mechanisms alternative to homocysteine lowering may be important factors in the relationship between folate and cardiovascular disease. The close association between homocysteine and endothelial dysfunction is largely dependent on its impact on eNOS activity. An observed decrease in arginine availability that has been demonstrated to accompany elevated homocysteine levels has been shown to induce eNOS uncoupling in vascular endothelial cells in culture. Furthermore, the conversion of methionine to homocysteine is associated with an activation of arginine-protein-methyltransferases, the group of enzymes responsible for the methylation of L-arginine and the subsequent formation of asymmetric dimethyl arginine (ADMA), which is a known endogenous inhibitor of eNOS. ADMA represents a novel independent predictor for all-cause cardiovascular mortality. The activities of both protein arginine N-methyltransferase (PRMT, type I) and the ADMA-degrading enzyme dimethylarginine dimethylaminohydrolase (DDAH) are redox sensitive. In cultured endothelial cells, rat models, and humans, oxidative stress has been shown to increase the activity of PRMT(s) and decrease that of DDAH, leading to increased concentrations of ADMA. Thus, an increased production of reactive oxygen species (ROS) could be the reason for increased ADMA levels. Elevated ADMA may inhibit NO synthesis by eNOS or even result in eNOS uncoupling, which would further enhance oxidative stress (75). Chronic ADMA treatment inhibits NOS enzymes with the expected effects of a nonselective NOS inhibitor; elevating blood pressure, causing vasoconstriction, impairing endothelium-dependent relaxation, and increasing endothelial cell adhesiveness. Animal models of prolonged treatment with ADMA have the potential to have similar effects to other NOS inhibitors, enhancing atherogenesis and sustained hypertensive organ damage. In the heart, ADMA reduces heart rate and cardiac output (283).
IV. Role of BH4 in Cardiovascular Development
BH4 is an essential co-factor for multiple enzymes that play a key role in embryonic development. Indeed, BH4 levels are higher at birth and during development than in adults (18, 335). Loss-of-function mutations in the GCH1 gene lead to rare congenital neurological diseases such as dystonia, DOPA-responsive dystonia, and hyperphenylalaninemia. The majority of mutations are heterozygous autosomal dominant with a smaller number of autosomal recessive mutations (275). Both disorders are characterized by low CSF levels of BH4 (80). Due to the critical role for BH4 in the function of multiple enzymes, it is unsurprising that nonfunctional mutations resulting in complete deficiency of BH4 have not been described. This interesting observation perhaps indicates that absolute loss of BH4 is embryonically lethal.
Mouse models of BH4 deficiency, and those models where the enzymes responsible for the biosynthesis of BH4 have been knocked out, indicate a key role for BH4 in embryonic development (Table 3). Although a knockout mouse model of GCH deficiency is not currently available, mice deficient in other synthetic enzymes, including PTPS, have been created. PTPS knockout mice are born with the expected medelian ratio but die within 48 h of birth. As expected, knockout mice have low levels of biopterin and catecholamines compared with wild type (WT) litter mates, but perhaps surprisingly show no gross organ defects. Postpartum lethality in PTPS pups can be rescued with BH4 and neurotransmitter replacement therapy (64, 263). This supports studies of mutations in the human GCH1 gene in patients in whom symptoms of GCH deficiency can be significantly improved with BH4 therapy (9).
eNOS, endothelial nitric oxide synthase; KO, knockout; PTPS, 6-pyruvoyl tetrahydropterin synthase; SR, sepiapterin reductase.
Despite no obvious gross organ defects at birth, PTPS knockout mice are significantly bradycardic when compared with their WT littermates (263). It is thought that the bradycardia observed in PTPS knockout mice is due to a deficiency in catecholamine levels, as tyrosine hydroxylase (TH) knockout mice (L-DOPA, dopamine, noradrenaline, and adrenaline) show significant bradycardia at E12.5 (342). As in the PTPS knockout mice, TH knockout embryos are deficient in catecholamine synthesis and show no gross organ abnormalities, hinting that that cause of lethality is physiological rather than anatomical (263, 273, 342). Other studies of catecholamine-deficient mice have indicated that during development, catecholamines are released by the embryo in response to in utero stress such as hypoxia which acts to increase heart rate via the β-adrenergic receptor (211). TH knockout mice and dopamine β-hydroxylase knockout mice show a more severe phenotype than PTPS knockout mice with significant embryo lethality noted and the majority of knockout embryos dead by E16.5-E18.5 (141, 273, 342). Catecholamine-deficient embryos also show signs of an altered development of the vasculature and the myocardium, along with blood congestion and thinning of the myocardium. There is also a greater heterogeneity in cardiac myocyte size and cellular orientation in catecholamine-deficient embryos (273, 342).
Since BH4 is critical for the function of TH, it may be expected that the PTPS knockout mouse would also be embryonically lethal. However, this is not the case and may be explained by observations from several studies that BH4 can cross the placenta (295), is also found in breast milk (306) and that maternal transfer of both BH4 and catecholamines can occur (273).
Dysfunctional development of the cardiovascular system is not restricted to mouse models of cofactor production and, indeed, is also observed in the eNOS knockout mouse. Unlike the PTPS and TH knockout mice, eNOS knockout mice are viable, although significantly more embryos were found to be reabsorbed between days E8.5 and 13.5 (198). eNOS knockout mice display cardiovascular abnormities in utero; at E12.5, eNOS knockout mice have coronary artery hypoplasia, thus indicating poor cardiac perfusion (159). Moreover, elevated cardiomyocyte apoptosis and myocardial caspase-3 activity are also observed in the myocardium of eNOS knockout mice (70). At birth, cardiovascular defects persist; with reports of spontaneous myocardial infarction in eNOS knockout mice postpartum most likely due to a combination of reduced coronary artery diameter, vessel density, and volume, as well as increases in ventricular wall thickness and myocardial volume (159). In neonates, congenital atrial and ventricular septal defects have been observed, along with heart shortening (70). Vascular defects in eNOS knockout mice are also observed within the pulmonary circulation—severe pulmonary congestion has been described along with a lack of distal arterial branches and regions of capillary hypoperfusion. This occurs in conjunction with a misalignment of the pulmonary veins (96, 159). From E17.5 and after birth, eNOS knockout mice are significantly smaller than WT mice, indicating in utero growth restriction in knockout embryos (102, 284). Restricted growth in utero may be due to mal-adaptation of the uterine artery to pregnancy, as uterine arteries from eNOS knockout dams are significantly smaller and show signs of impaired smooth muscle cell dedifferentiation and proliferation (284).
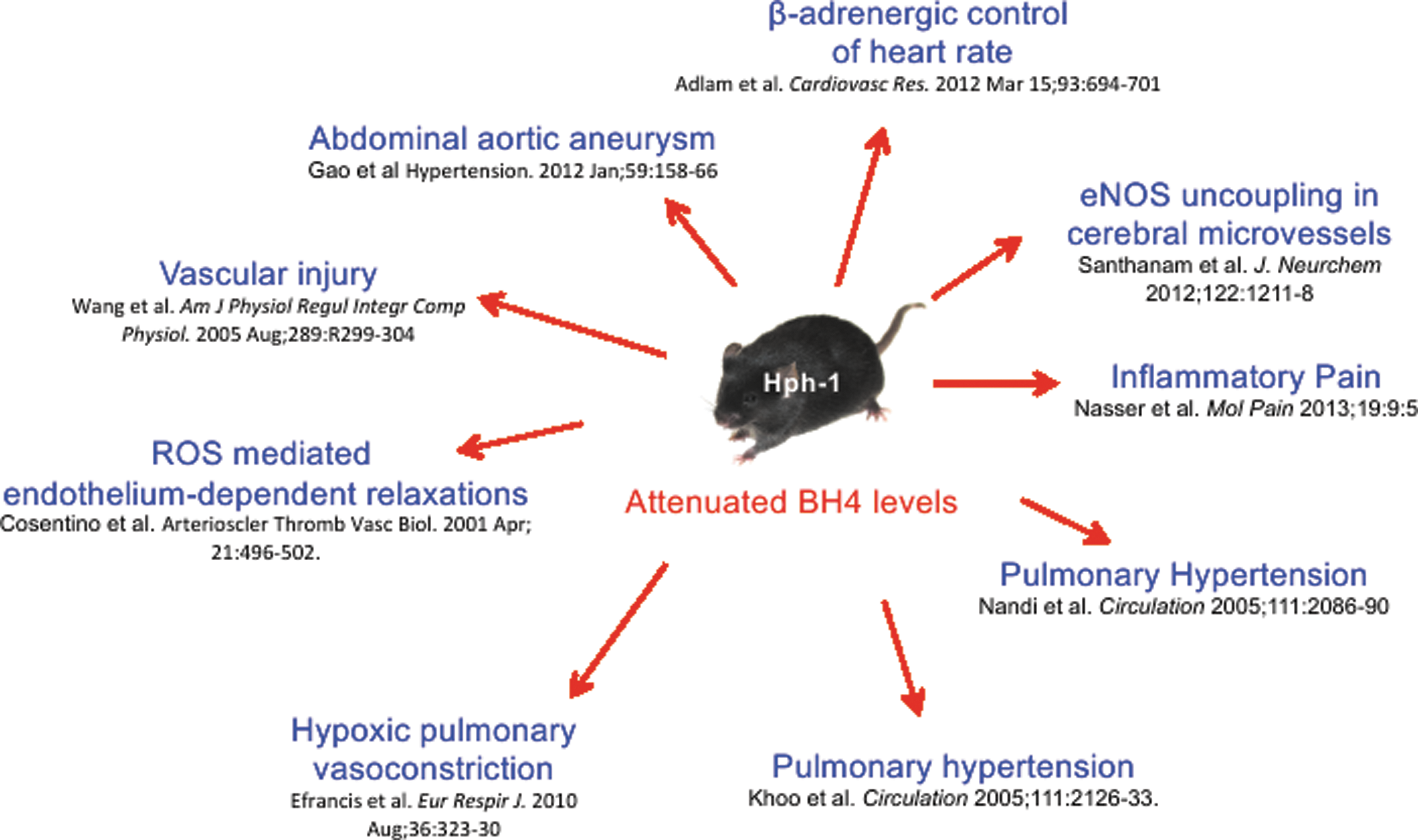
Models of BH4 deficiency and studies of genetic mutation in human subjects have indicated that even a partial diminution in the levels of BH4 during development can lead to the onset of cardiovascular abnormalities. The hph-1 mouse, developed by ENU mutagenesis, has diminished GCH1 mRNA and total GTPCH protein, leading to a 90% decrease in GTPCH activity in the heart of adult mice (2). hph-1 mice have higher pulmonary vascular resistance, increased media thickness, and pulmonary hypertension at birth (18).
In summary, BH4 has been demonstrated to play a critical role in cardiovascular development while having both NOS-dependent and -independent roles with the production of catecholamines shown to be critical to maintain heart rate in the developing heart. Generation of a tissue-specific GCH1 knockout mouse model would elucidate any cell autonomous role of BH4 in developmental biology and define whether there is an absolute requirement for BH4 in development.
V. BH4 in the Vascular System
A. BH4 and the regulation of blood pressure and vascular tone
Regulation of blood pressure and vascular tone is critical for the maintenance of proper organ perfusion, and one of the key regulators of blood pressure and organ perfusion is vascular resistance. A decrease in the diameter of the blood vessel due to increased sympathetic drive and/or decreased production of vasodilators results in an increased vascular resistance, causing an increased blood pressure upstream, and reduced blood flow downstream of the constriction.
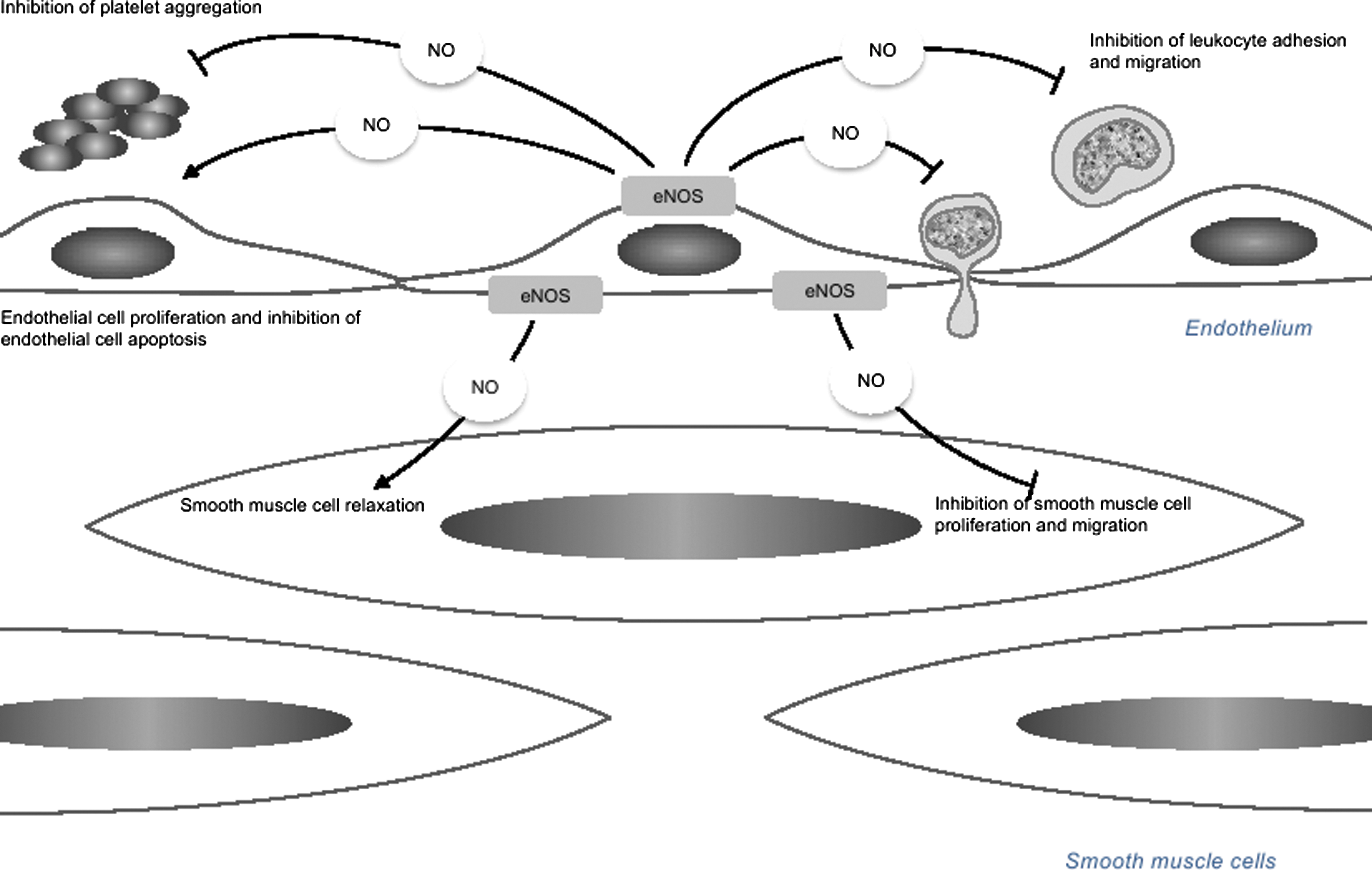
Physiological stimuli, such as laminar shear stress, and receptor-mediated agonists such as bradykinin, estrogen, and acetylcholine, act on endothelial cells to increase the production of NO. The locally produced NO diffuses into the vascular smooth muscle cells of the vessel wall, where it activates multiple second messenger pathways through the activation of guanylate cyclase and direct S-nitrosylation of cysteine residues (76).
Endothelial dysfunction is a hallmark of multiple disease states such as diabetes, atherosclerosis, and hypertension, and it is characterized by impaired endothelial dependent vasodilatation (69). Given the key role of BH4 in the production of vasoactive substances such as NO and noradrenaline, it is not surprising that alteration in the bioavailability of BH4 alters blood pressure and endothelial function. A common GCH1 variant, C+243T in the 3′ untranslated region, has been reported to be associated with decreased NO production, increased blood pressure and heart rate, and a dysfunction of baroreflex coupling (305). Studies of a haplotype defined by three single nucleotide polymorphisms (SNP) have shown that the X haplotype is associated with lower vascular levels of GCH1 mRNA and decreased levels of BH4 in plasma and the vasculature. This attenuation of BH4 is associated with increased vascular O2 − production and reduced endothelial dependent vasodilatation in arterial and venous segments from coronary artery disease patients (7).
As previously discussed, very few genetic models of GTPCH deficiency exist—in animal models, the treatment of 2,4-diamino-6-hydroxypyrimidine (DAHP), a selective inhibitor of BH4, leads to a significant increase in blood pressure and, ultimately, endothelial cell dysfunction. Endothelial dysfunction, characterized by reduced endothelial cell-dependent aortic relaxation, could be reversed by incubation with the BH4 precursor sepiapterin (179). siRNA knockdown of GCH1 in mice has been shown to result in endothelial dysfunction, increase vascular O2 − production, increase the expression of adhesion molecules, and significantly increase blood pressure (299). Studies in the hph-1 mouse, with globally diminished BH4 levels, have shown that these mice have increased blood pressure compared with WT, nonlittermate controls (40). However, another study has not been able to replicate this finding and shows no difference in blood pressure between hph-1 mice and littermate controls (326); however, hph-1 mice develop pulmonary hypertension (137). A summary of the key reports using the hph-1, BH4-deficient mouse is shown in Figure 7. These studies have highlighted the importance of BH4 in the maintenance of normal vascular function. The generation of a tissue-specific GCH knockout model would help elucidate the role of GTPCH in vascular function.
BH4 availability is regulated at both the gene and protein levels, and GTPCH protein expression has been shown to be altered by both physiological and pathophysiological stimuli. ROS such as H2O2 and ONOO− increase GCH expression in a concentration-dependent manner, leading to an increase in cellular BH4 levels after a transient decrease (243). H2O2-mediated induction of GCH has been shown to be mediated, in part, by the JAK2-Stat1 pathway (244), and it is implicated in endothelial cell growth, proliferation, and survival (282). Numerous studies have shown that GCH expression is increased by inflammatory cytokines such as TNF-α (222) and pathogen-associated stimuli such as LPS (297), discussed in greater detail later in this review. Laminar shear stress is an important regulator of basal NO production within the vessel, and it acts as a stimulus to increase vasorelaxant production in response to increased blood flow. Laminar but not oscillatory shear stress has been shown to increase GTPCH protein expression, cellular BH4 and prevent eNOS uncoupling (154, 317). As previously discussed, GFRP has been shown to be a negative regulator of GTPCH activity. GFRP has an important regulatory role in endothelial cells—in vitro studies in HUVEC have shown that GFRP modulates phosphorylation of GTPCH and BH4 levels. In these cells, laminar shear stress promotes the dissociation of GFRP from GTPCH, elevated phosphorylation resulting in increased enzymatic activity, and, subsequently, BH4 production (154). In vivo models have confirmed a critical role for GTPCH in the response to shear stress—Oscillatory shear stress decreases phosphorylation of GTPCH and decreases BH4 levels (154). In contrast, increased vascular flow in vivo has been shown to increase GCH1 expression, BH4 levels, and eNOS expression (147). Increased GCH1 expression in response to flow is critical to maintain normal vascular function by optimizing the generation of NO from eNOS, preventing eNOS uncoupling, the production of O2 −, and further diminished NO bioavailability.
The importance of BH4 availability in disease conditions has been highlighted within conditions such as diabetes. In the coronary artery endothelial cells from diabetic rats, eNOS levels remain unchanged; however, NO production is decreased compared with endothelial cells from nondiabetic rats. This diminished production of NO can be restored by the supplementation of BH4 (175). Reduced vascular BH4 levels have also been shown in other experimental animal models of vascular disease such as the spontaneously hypertensive mouse (113), DOCA salt-induced hypertension (148), spontaneously hypertensive rats (150), STZ-induced diabetes (29), insulin-resistant fructose-fed rats (245), and in arteries from models of aging (248, 332). However, it is not just the level of BH4 that is critical—studies also suggest that the ratio of BH4 to BH2 can also have a significant impact on eNOS function. Increasing cellular BH2 levels in the presence of a constant eNOS:BH4 ratio have been shown to be sufficient to induce eNOS uncoupling (20). It has been suggested that since the Km values of BH4 and BH2 are similar for eNOS, BH2 may compete with BH4 for the binding site (43). Thus, significant eNOS uncoupling in disease may still be observed in parallel with no change in absolute BH4 levels if BH2 accumulation becomes excessive.
The importance of eNOS in the vasculature is highlighted by the finding that both pharmacological (214, 253) and genetic (118, 142, 286) inhibition of eNOS activity results in elevated blood pressure. Evidence for eNOS uncoupling has been observed in animal models of impaired vascular function; for example, in mesenteric arteries from young versus old mice (332), hyperlipidemic mice (149), stroke-prone SHR rats (136), angiotensin-II-induced hypertension (183), DOCA-salt hypertension (148), and streptozotcin-induced diabetes (110).
eNOS uncoupling has also been observed in clinical conditions that are commonly associated with impaired endothelial cell function—saphenous veins and internal mammary arteries from patients with diabetes mellitus have increased endothelial-dependent O2 − production compared with nondiabetic controls. This enhanced O2 − production was, in part, inhibited by the NOS inhibitor N(G)-nitro-L- arginine methyl ester (L-NAME), indicating NOS uncoupling. Critically, the NOS-dependent O2 − production could be inhibited by incubation with sepiapterin, indicating a role for BH4 in regulating eNOS coupling (95). In vascular tissue obtained during coronary artery bypass graft surgery, decreased vascular BH4 content is associated with increased NOS-dependent O2 − production and attenuated endothelial-dependent relaxation (6).
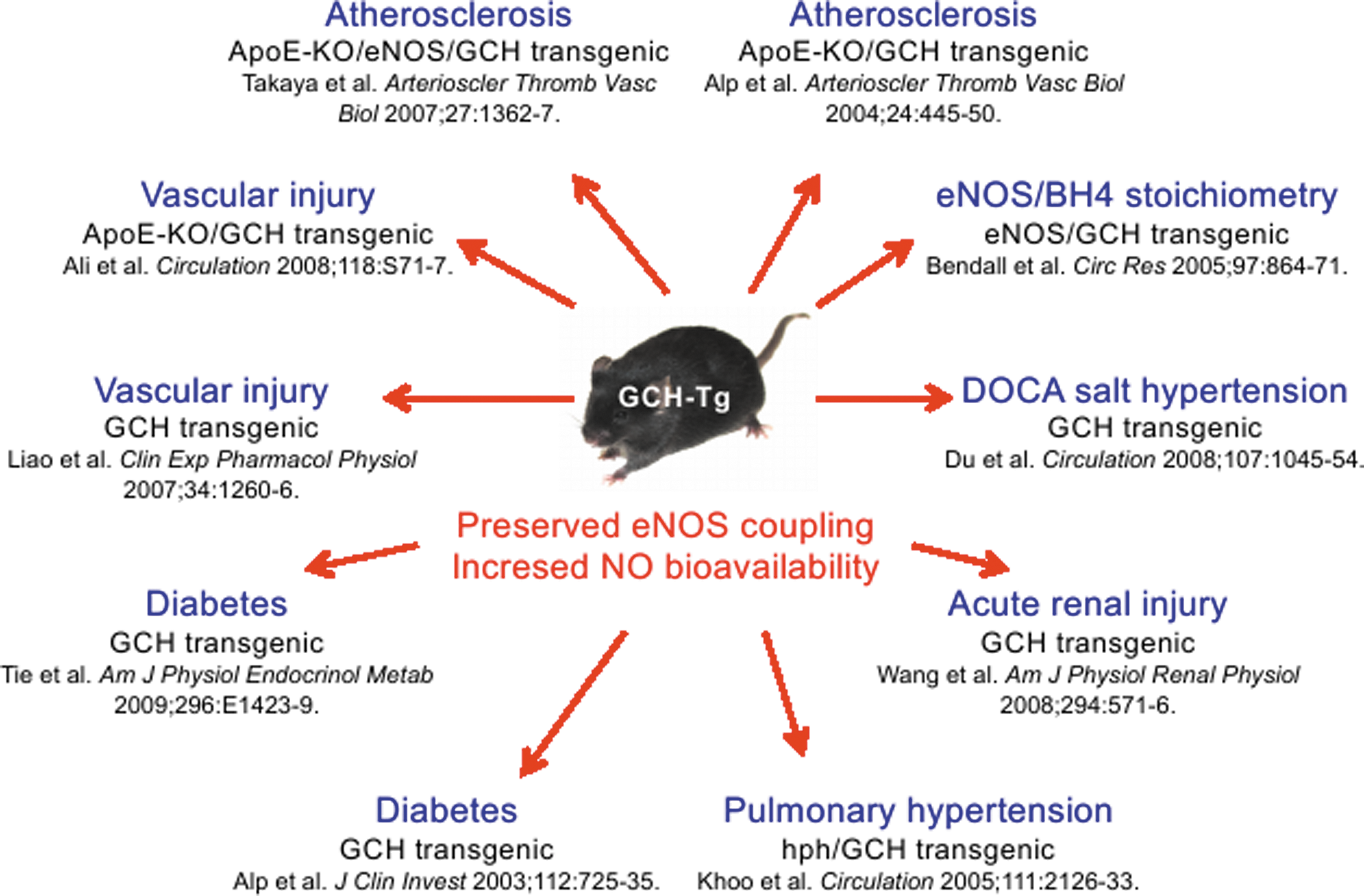
Given the information indicating the pathological consequences of reduced BH4 levels, animal studies have aimed at determining whether vascular disease states can be improved by increasing vascular GCH expression. The treatment of Zucker fatty rats with adenovirally delivered GCH plasmid has been shown to improve aortic GCH expression and BH4 levels, and this was associated with improved endothelial-dependent relaxation, decreased aortic O2 − levels, and augmented eNOS activity (246). Mice that overexpress GCH1 specifically within the endothelial cell layer (GCH-Tg mice) have been used to investigate the effects of increased endothelial BH4 on vascular function. GCH-Tg mice have significantly elevated aortic BH4 and eNOS activity, as measured by arginine to citruline conversion (60). In healthy animals, no difference in O2 − production is detected between GCH-Tg mice and their WT littermates; however, in disease states such as diabetes, atherosclerosis, and DOCA salt-induced hypertension (326), GCH-Tg mice show significantly decreased ROS production versus WT mice. In diabetic mice and in the apolipoprotein E knockout mouse model of atherosclerosis (ApoE−/− mice) (5), overexpression of GCH-Tg is associated with reversal of diabetes and atherosclerosis-induced endothelial dysfunction (14). Improved endothelial function is observed in GCH-Tg mice, which also have a significant abrogation of hypertension in response to DOCA salt compared with the control group (326). Endothelium-specific overexpression of GCH1 has also been shown to reverse the pulmonary hypertension in hph-1 mice (137). Endothelial-specific overexpression of GCH has been shown to not alter blood pressure compared with WT littermates, thus indicating that in healthy vessels with normal vascular function, increasing vascular BH4 levels is not a potential hazard and is unlikely to cause aberrant hypotension (1). A summary of studies showing the importance of the GCH-Tg mouse is depicted in Figure 8.
B. Role of NO and BH4 in endothelial cell proliferation and survival
eNOS, or NO-mediated signaling pathways have been demonstrated to regulate endothelial progenitor cell (EPC) mobilization and function, participating in vasculogenesis by differentiating into endothelial cells and enhancing angiogenesis under ischemia. Several recent studies indicate that the number of EPCs may serve as a surrogate marker for cardiovascular risk, affecting the progression of cardiovascular diseases, including hypertension. Re-endothelialization has been shown to be a key event in vascular repair after vascular injury (89, 164, 255), reducing the risk of thrombosis and the development of neointimal hyperplasia; whereas accelerated endothelial cell apoptosis and endothelial denudation are linked to the formation of thrombotic atherosclerotic plaques (162, 265).
eNOS and NO have been shown to mediate VEGF-induced endothelial cell proliferation (153, 200) and angiogenesis (185, 200) and to play a critical role in the prevention of endothelial cell senescence (196, 279) and apoptosis (58, 131, 224, 322). These critical roles of eNOS in endothelial cell growth and survival also implicate BH4 in angiogenesis. There is increasing evidence that BH4, as an essential eNOS cofactor, improves endothelial cell proliferation (81, 87, 165, 241) and tube formation (81, 87). In vitro supplementation with the BH4 precursor sepiapterin has been shown to enhance proliferation in coronary endothelial cell from diabetic rats (165), bovine aortic endothelial cells (241), and HUVEC (87). These effects may be mediated by either a direct antioxidant effect of BH4 or/and improved eNOS function with increased NO availability and reduced production of ROS. Indeed, eNOS uncoupling impairs EPC number and function in diabetes and in the GCH transgenic mouse, endothelial BH4 augmentation reduces neointima formation that is induced by vascular injury with endothelial denudation (148). Moreover, EPC number is decreased and angiogenesis is impaired in both the hph-1 and DOCA-salt mouse models, which can be rescued by endothelial overexpression of GTPCH in GCH-Tg mice (326). In genetic models of colon cancer, GTPCH inhibition either by DAHP or by GCH-targeted siRNAs markedly attenuated tumor cell proliferation and survival, as well as inhibited tube formation of HUVEC in response to hypoxia (204).
These studies provide important evidence that BH4 is not only critical for the effects of NO on vascular reactivity, but also BH4 plays a key role in other functions of NO and NO-mediated pathways such as the formation of new blood vessels and the proliferation of endothelial cells within the vasculature; thus, the benefits of BH4 supplementation on the vasculature may be, at least in part, due to its pro-vasculaogenic or pro-angiogenic effects.
C. Sepsis
In contrast to the benefit of increasing BH4 to offset the pathological effects of restricted endothelial bioavailability in the context of vascular disease, under conditions of septic shock, restricting the availability of BH4 may be beneficial. Within intensive care medicine, septic shock is a leading cause of mortality (202). Sepsis is a systemic inflammatory reaction to bacterial products and endotoxins such as LPS. The presence of LPS and the cardinal cytokines induced by this potent pro-inflammatory mediator (IL-6, TNF-α, and IFNγ) cause a widespread induction of iNOS, including within the vessel wall (138). This induction of iNOS expression within the vascular system, particularly within endothelial cells and smooth muscle cells, is associated with a profound hypotension that can be largely refractory to aggressive fluid resuscitation and treatment with cardiac inotropes and vasopressors (55).
The role of BH4 is mediating septic pathology by iNOS has been highlighted by a number of studies. The addition of LPS to ex-vivo rat aorta was shown to cause a strong induction of both iNOS and GCH1 mRNA and biopterins, and to cause the generation of an L-Arg inducible relaxation in vessel rings, which could be prevented using DAHP (240). In a model of Staphylococcus aureus postburn sepsis, rat GCH1 mRNA was induced in multiple tissues and significantly correlated with multiple organ dysfunction (240). Septic patients have been shown to have significant elevations in plasma biopterins as well as nitrites and nitrates (115). A similar pattern is seen in LPS-treated rats, and is reduced when following plasma endotoxin adsorption onto polymyxin B fibers (115).
Strategies to reduce BH4 availability in animal models have been assessed as potential interventions to limit the profound hypotension seen in septic patients. Treatment of rats with the GTPCH inhibitor 2,4-Diamino-6-hydroxyoyrimidine (DAHP) reduces plasma nitrate and nitrite levels and nitric oxide production in response to LPS treatment (27), coincident with reduced biopterin levels. This intervention was sufficient to reduce the degree of hypotension seen at 6 h after LPS administration.
The use of a 4-amino analogue of BH4 (4-ABH4), which acts as a competitive inhibitor of BH4 binding to NOS, improves survival in a 6 day model of endotoxaemia, being more potent than the NOS inhibitor NMMA (13). Follow-up studies using 4-ABH4 observed improvements in cardiac index and stroke volume, but did not reproduce the positive effects on blood pressure (73). However, 4-ABH4 reduced LPS-induced damage to liver, lung edema, and intestinal necrosis.
The disconnect in these studies between effects on survival and on hypotension may be due to opposing local effects in the microcirculation. Perturbation of organ microcirculation or local deficiency of NO within the microcirculation causes hypoperfusion, exacerbates organ failure, and can manifest as a worsening of outcome following strategies to inhibit NO production in sepsis (160). This phenomenon would, in contrast to the original rationale for targeting nitric oxide production in sepsis, support local supplementation of nitric oxide function. To address whether BH4 has any potential to cause a positive modification of sepsis through effects on the microcirculation, tetrahydrobiopterin supplementation in an ovine model of peritoneal sepsis has been evaluated. Sheep received two bolus treatments with BH4 during the induction of sepsis through an injection of fecal material into the peritoneum (101). BH4 treatment, in addition to attenuating the decreases in cardiac index and blood pressure, maintained small vessel perfusion and microvascular flow index. Increased survival in the BH4-treated animals was accompanied by preserved gas exchange and kidney function (101).
These data underline that nitric oxide biology in sepsis is complex, but that achieving the right balance between reducing hypotension, while maintaining organ microcirculation may have significant therapeutic potential in the treatment of septic patients.
D. BH4 therapy
Given the evidence for a critical role of BH4 in vascular function, it is unsurprising that studies supplementing BH4, by either genetic or pharmacological means, have shown improvements in vascular function (Table 1).
Treatment with BH4 has been shown to reverse the disease-related redox disequilibrium observed with BH4 deficiency. For example, high glucose treatment of human aortic endothelial cells results in the uncoupling of eNOS and profound BH4 oxidation, both of which were reversed after transfection with GCH1 DNA. (29). Similarly, in isolated vessels, incubation with BH4 improves endothelial function—isolated coronary arterioles from pigs fed a high-fat diet, versus pigs fed standard chow show a significant improvement in endothelial-dependent relaxation on incubation with sepiapterin (277). Treatment of arteries from both streptozotcin-induced diabetic rats (205) and spontaneously hypertensive rats (94, 113, 191) with BH4 ex vivo improves endothelial dependent relaxation but does not affect relaxation of control vessels. Incubation with BH4 has also been shown to increase NO production by aortas from DOCA-salt hypertensive mice (336). Studies utilizing diseased arteries and veins from patients have also demonstrated beneficial effects of acute ex vivo incubation with BH4-. Coronary arterioles from patients with coronary artery disease have significantly reduced endothelial-dependent relaxation compared with controls (aortic value replacement patients), which could be increased in arteries from atherosclerotic patients by treatment with sepiapterin (277). This finding was confirmed by a second study which showed that mammary arteries and saphenous veins from patients with coronary artery disease demonstrated a significant improvement in endothelial-dependent relaxation after exposure to BH4 (296).
1. Animal models of chronic BH4 supplementation
Chronic BH4 treatment has been shown to reduce angiotensin-II-induced hypertension and endothelial dysfunction (130, 133), and to decrease blood pressure while improving endothelial-dependent relaxation in SHR rats (113). Renal failure-induced hypertension, due to partial nephrectomy, has also been shown to be reduced with chronic BH4 treatment (166, 329), which promotes an increase of resistance artery eNOS protein expression levels (166). Chronic BH4 treatment also improves metabolic endothelial dysfunction—improving endothelial function in insulin-resistant fructose fed rats (245, 300) and the ob/ob mouse model of obesity (148). Improved endothelial-dependent relaxation was also observed in hyperlipademic ApoE−/− mice, along with an attenuation of aortic O2
Not all studies have shown that chronic BH4 therapy improves blood pressure and endothelial function. Indeed, chronic oral treatment with BH4 neither altered blood pressure in ApoE−/− mice (226) nor reduced the hypertension observed in fructose-fed rats (300), although improved endothelial function was observed. This could, in part, be due to the use of eNOS-derived H2O2 from uncoupled NOS as an endothelial-dependent vasodilator. A shortage in vascular BH4 may shift the balance from the use of eNOS-derived NO to eNOS-derived H2O2 as vasodilators. In the hph-1 mouse, endothelial-dependent relaxation was found to be abolished by inhibition with L-NAME; however, this relaxation was also inhibited by the H2O2 scavenger catalase and was enhanced by superoxide dismutase, presumably due to the conversion of O2 − to H2O2 (40). H2O2-mediated relaxation was found to be inhibitable by L-NAME in rabbit aortic rings (338) and in rat aorta (333). In canine arteries, incubation with DAHP (to inhibit GTPCH) significantly reduced endothelial-dependent vasodilatation, indicating a dependence on H2O2 as a vasodilator (38). In DOCA salt-induced hypertension, endothelial-dependent relaxation also appears to be mediated, at least in part, by H2O2 derived from uncoupled NOS (148). Care should be taken in the interpretation of these studies—due to the near diffusion reaction rate between NO and superoxide, it is unlikely that eNOS produces solely H2O2, and catalase has recently been shown to decompose peroxynitrite (85), which provides a constant “peroxide tone” (12) through its action on cyclooxygenase.
The relative contribution of NO or H2O2 to vasorelaxation may be, in part, determined by eNOS protein levels—in the early stages of hypertension, there is a compensatory increase in eNOS levels due to decreased NO production resulting from increased ROS production (61), leading to an elevation in NO synthesis in the presence of BH4, or H2O2 in the absence of sufficient BH4. In more advanced disease where BH4 is limiting and hence a greater proportion of eNOS is uncoupled, this O2 − production could become pathological and, thus, lead to the down-regulation of eNOS expression as a compensatory mechanism to decrease the pathological production of ROS.
2. Clinical studies of acute BH4 supplementation
Clinical studies have used acute pharmacological BH4 supplementation to assess whether treatment with BH4 has the capacity to reverse endothelial dysfunction. Treatment of BH4, either orally or by infusion, usually through the brachial artery has marked positive effects on endothelium-dependent vasodilatation and vessel relaxation in several models (66,105, 108, 109, 124, 206, 256, 280). For example, in chronic smokers, acute BH4 administration improves endothelial-dependent relaxation (280) and in patients with type II diabetes, an augmented acetylcholine-mediated dilation after infusion of the brachial artery with BH4 (500 μg/min) is observed. Furthermore, BH4 improved the postprandial endothelial-dependent vasodilatation in healthy subjects after an oral glucose challenge (124). In the heart, coronary artery infusion of BH4 in patients with coronary artery disease (79, 163, 233, 320), chronic heart failure (234), and vasospastic angina (78) has been shown to improve acetylcholine response in the coronary artery.
There are several unifying features of these studies that are of concern and should be considered. These studies of acute supplementation of BH4 are done with super-physiological concentrations of BH4, which are much higher than those found in healthy subjects and patients with atherosclerosis, which may mean that BH4 has nonspecific effects other than those purely of an NOS cofactor (6). The beneficial effects of BH4 are also not sustained and often disappear a few minutes after withdrawal of exogenous BH4. This is surprising, as it would be expected that if vasodilatation is determined by eNOS coupling, and the beneficial effects observed are through the restoration of eNOS-derived NO production, then the effects of BH4 on vasodilatation would be prolonged. BH4 is a potent antioxidant, and it may also be possible that these improvements could be due to the anti-oxidant capacity of BH4 rather than its role as an NOS cofactor. This is supported by a study in healthy volunteers which shows that acute beneficial effects of BH4 may be due to scavenging of ROS—this study found no effect of BH4 on acetycholine-mediated dilation at baseline; however, ischemia reperfusion (which is associated with increased ROS)-induced decreases in dilation responses to acetylcholine can be rescued by the infusion of BH4 (500 μg/min). This dilatory response could be restored by both 6R- and 6S-BH4, as well as by amino-BH4 (NH4) that has the same ROS scavenging capacity as BH4 while being unable to catalyze the production of NO, thus indicating that the improvement observed in this study was likely due to the scavenging of ROS (172).
Most attention for the investigation of BH4 in the cardiovascular system focuses on how alterations in BH4 levels or oxidation state alter eNOS function. However, more recent evidence has also shown that nNOS may also have a role in maintaining basal vascular NO production (230). It is, therefore, relevant to consider that BH4 may both enhance agonist-induced vasodilatation via eNOS and improve basal nitric oxide production via its action on nNOS.
3. Clinical studies of chronic BH4 supplementation
The breadth of preclinical and acute clinical data implicating BH4 as a key regulator in endothelial function suggests that oral BH4 therapy may be able to prevent or treat cardiovascular disease. Although acute studies have provided valuable data on the role of BH4 in vascular health and disease states, these studies do not inform us of the efficacy of BH4 treatments after chronic administration. Long-term studies of BH4 have been hampered by its chemical instability; however, a recent preparation of oral BH4 has been developed by BioMarin, which has significantly improved stability at room temperature compared with BH4 available from other sources (152). This new preparation could potentially make future longer-term studies of BH4 more viable.
Chronic treatment with BH4 has been shown to improve hypertension. Hypertensive subjects receiving 5 or 10 mg/Kg/day for 8 weeks, or 400 but not 200 mg/Kg/day for 4 weeks showed a significant decrease in blood pressure and improved endothelial function. However, care should be taken when interpreting these results, as this was a relatively small study (8–16 patients per group) and did not have a placebo control (210). In hypercholesterolaemic patients (low-density lipoprotein, LDL >4.5 mM), 4-week oral treatment with BH4 (400 mg twice daily) or placebo (11 BH4 vs. 10 placebo) significantly improved endothelial-dependent vasodilatation (41). The oxidative stress marker 8-F2 isoprostane was decreased in the plasma of BH4-treated patients. BH4 therapy (dose escalation 2.5–20 mg/kg) has also been found to improve 6 min walking distance in patients with pulmonary hypertension at doses more than 5 mg/kg, although care should be taken, as this study was primarily designed to address safety, not efficacy and no placebo was used (220).
A recent randomized placebo-controlled trial of oral BH4 in patients with coronary artery disease has shown no benefit of BH4 therapy (50). In this study, treatment with low (400 mg/day) or high (700 mg/day) BH4 for 2–6 weeks did not alter arterial stiffness, endothelial-dependent relaxation, or O2 − production. Treatment with BH4 significantly increased plasma and saphenous vein BH4 levels; however, it also significantly increased levels of the oxidized pterin product BH2 that lacks eNOS cofactor activity. Thus systemic oxidative stress may play a critical role in determining the degree of oxidation of BH4 to BH2 and, hence, the ratio of BH4:BH2 and the efficacy of the treatment (50).
A discordance has been shown between plasma BH4 levels and endothelial function, with increased plasma BH4 levels being associated with decreased endothelial-dependent relaxations (6). This indicates a differential regulation of plasma and vascular BH4 levels. Total biopterins, including all oxidized species, have been shown to be correlated with the inflammatory biomarker C-reactive protein (CRP) (6). This is interesting, as multiple studies have shown that the stimulation of primary endothelial cells with inflammatory cytokines such as TNF-α, INF-γ, and/or pathogen-associated stimuli such as LPS results in increased GCH expression and, therefore, GTPCH protein activity and increased BH4 levels (222, 316). However, as previously discussed, many vascular disease states are associated with reduced vascular BH4 levels. It could be that systemic inflammation leads to a cytokine-induced increase in plasma BH4 levels, whereas inflammation within the vessel wall may result in loss of BH4, possibly due to oxidation by ROS.
E. Alternative strategies to increase BH4
Studies discussed in the previous section have used chronic supplementation with BH4 to re-couple NOS and improve vascular function. However, there are alternative pharmacological approaches that may reverse eNOS uncoupling by preventing BH4 oxidation, increasing BH4 biosynthesis, or augmenting the regeneration of BH4 from BH2.
In HUVECs, treatment with fluvastatin and cerivastatin increases GTPCH mRNA and intracellular BH4 levels (98). In STZ-induced diabetic rats, atorvastatin results in an increased expression of GCH and normalized vascular dysfunction (307). In rats, treatment with simvastatin has been shown to increase GTPCH activity, BH4 production, and nitric oxide levels in the kidney-clip model of hypertension (340). In patients with cardiovascular disease, atrovastatin has been shown to improve flow-mediated dilation in association with an increase in plasma BH4/BH2 ratio (269). Patients with coronary artery disease treated with atrovastatin have increased vascular BH4 levels, improved vascular nitric oxide bioavailability, and reduced vascular O2 −; while ex vivo atrovastatin was found to increase GTPCH expression and activity, independently of its action on LDL (8). In ApoE−/− mice, treatment with resveratrol (a natural phenol) was associated with increased expression of GCH, increased cardiac expression of GCH, and a reversal of eNOS uncoupling (325).
Inhibition of Angiotensin I receptor has also been shown to increase GCH expression and to improve eNOS coupling. In STZ-induced diabetic rats, treatment with the AT-1 receptor antagonist telmisartan resulted in increased aortic expression of GCH, decreased aortic O2 − production, and improved endothelial-dependent vasodilatation (308). Treatment with the AT-1 receptor antagonist losartan has also been shown to increase serum BH4 levels and to increase GCH expression in the glomerulus of STZ-induced diabetic rats (225). Finally, exercise may also improve eNOS coupling by increasing BH4 levels—exercise was found to increase flow-stimulated nitric oxide production and to increase BH4 content in arteries from old rats versus old sedentary rats (248).
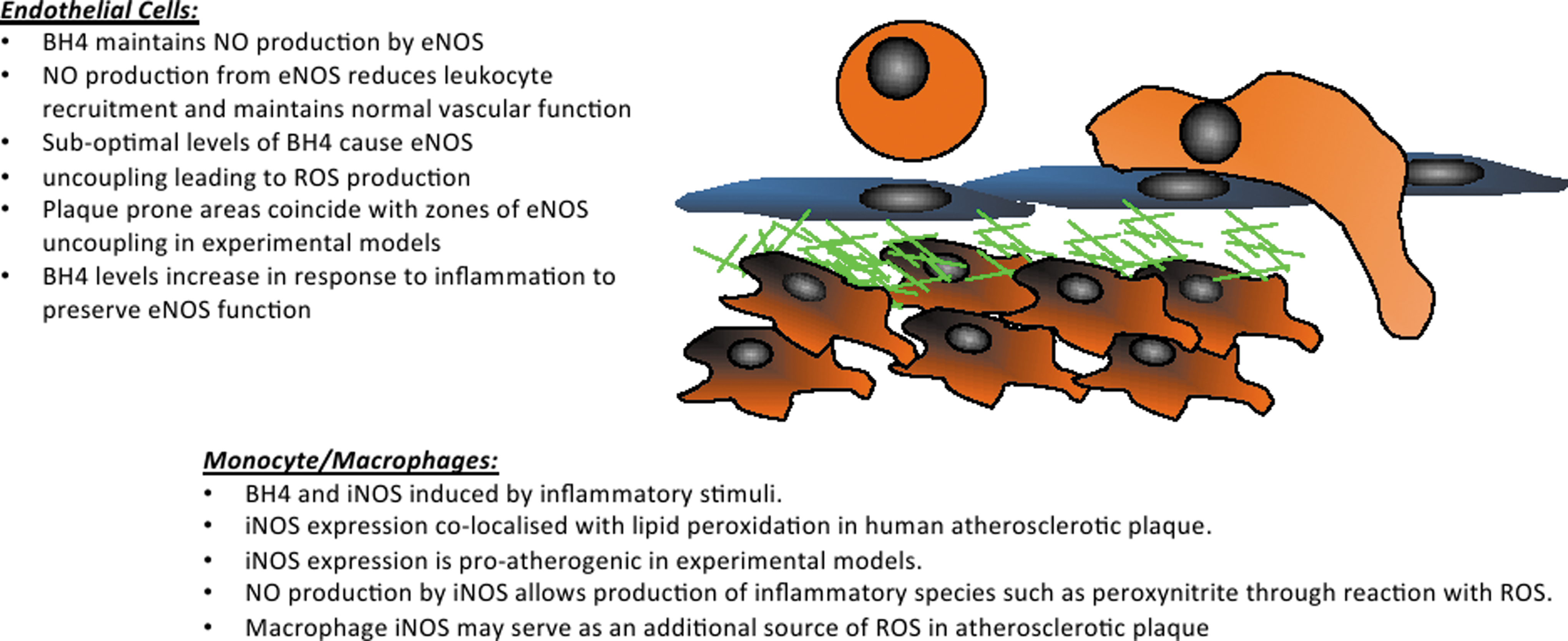
VI. BH4 in Vascular Inflammation and Atherosclerosis
Atherosclerotic vascular disease is typified by the accumulation of lipid-laden macrophage foam cells in the arterial wall. The vascular endothelium at atherosclerosis prone sites shows increased expression of cell adhesion molecules. This expression supports the recruitment of mononuclear phagocytic cells to the vessel wall, where they accumulate oxidized lipids (68). The uptake of lipid causes cellular activation and further local inflammation within the vessel wall. This inflammation progresses through the production of cytokine and chemokine signals that cause the recruitment of further inflammatory cells, both monocytes and other leukocytes, including adaptive immune cells such as T lymphocytes (174). While atherosclerotic disease causes a defined local inflammation driven by the oxidized lipid, cytokine production, and cell death within the core of the plaque, atherosclerotic disease is also accompanied by systemic inflammation. Systemic inflammation has been linked to atherosclerosis not only in general terms, with the correlation of inflammatory biomarkers such as CRP with disease burden and risk (132, 218), but also as a specific driver of disease, as is demonstrated by the strong relationship of chronic inflammatory conditions such as arthritis and systemic lupus erythematosus with high disease risk and severe disease pathogenesis (223, 238). The complexities of how local and systemic aspects of inflammation are involved in the disease process are paralleled by the biology of BH4 in inflammatory vascular disease. While atherosclerotic sites are assumed to lack sufficient BH4, causing NOS uncoupling, many inflammatory signals up-regulate BH4 levels and systemic markers of the BH4 biosynthetic pathway, namely neopterin levels, are robustly elevated in CAD patients (228).
A. Regulation of BH4 availability by inflammatory stimuli
GTPCH gene and protein expression are induced by multiple inflammatory stimuli in many of the key cells types involved in cardiovascular inflammation, including endothelial cells, leukocytes, and smooth muscle cells (92, 312). BH4 biosynthesis is induced by inflammatory stimuli such as TNF-α, LPS, and hydrogen peroxide (134, 242). Cytokine induction of GCH1 gene and GTPCH protein expression is minimal when individual cytokines such as TNF-α, IL-1, or interferon-γ are applied, but the application of cytokine cocktails causes robust induction of BH4 biosynthesis through elevations in GTPCH protein levels (117). This reliance on multiple activation pathways was shown to be due to a requirement for both NF-κB and STAT1 transcription factors (117). The use of knockout cells and specific inhibitors has demonstrated that activation of the cardinal pro-inflammatory transcription factor NF-κB alone does not cause GTPCH protein production, while signaling via the Jak/STAT pathway can cause some GTPCH production. The co-ordinated activation of both pathways was shown to cause robust elevations in biopterins.
While the association of BH4 production and iNOS induction, a hallmark of inflammatory M1 macrophages, has been studied in detail, the expression of GTPCH in other leukocyte subsets has been less fully explored. The recent observation is that a macrophage cell line could express AGMO, and that this enzyme may be controlling production of the important inflammatory mediator PAF (278). This study indicated that the down-regulation of AGMO by LPS resulted in a decreased metabolism of Lyso-PAF, meaning that more Lyso-PAF was available for conversion into PAF. Given that this regulation requires BH4 in macrophages before inflammatory activation (and resulting iNOS expression) indicates that the exploration of GTPCH expression and BH4 levels may reveal other NOS-independent roles for BH4 in macrophage biology. A recent study from the cancer field has shown that treatment of tumor-bearing mice with DAHP, thus inhibiting GCH1, resulted in an increase in CXCL10 and CCL5 in the plasma and a decrease in IL-10, hallmarks of a switch away from an M2 phenotype (204). This change in systemic markers was accompanied by a reduction in tumor macrophages of an M2-like phenotype. This study implicates that GTPCH may, indeed, have important roles in other cell types than in controlling iNOS activity in inflammatory M1 macrophages. Given this finding, more investigation of monocytes, the cells from which macrophages arise, may reveal altered expression and regulation in the different monocyte subpopulations Ly-6CHI and Ly-6CLO, both of which have been implicated in experimental atherosclerosis models (116).
In addition to activation by these cardinal cytokines, GCH1 gene expression is also regulated within the adaptive immune system with induction by IL-2 or Phytohaemagglutin, both of which are potent inducers of lymphocyte activation and division, in T-cell lines (OVA) or human T cells (227, 343). Recent studies have shown that BH4 synthesis is also induced by T-cell activation via CD3 and the co-stimulatory signal (33). In addition to the regulation of BH4 levels through the control of GTPCH, pro-inflammatory signals can also modulate BH4 availability through the control of other elements of the biosynthetic pathway, such as PTPS, which is induced in endothelial cells by IL-1 beta or via GFRP, which is down-regulated in monocytic cells by LPS (77, 313).
B. Regulation of BH4 levels in atherosclerosis
Atherosclerotic disease occurs at sites of disturbed blood flow, typically in the curvature of vessels or at branching points (125, 188, 189). In other sites, smooth laminar blood flow induces nitric oxide production, which has multiple anti-atherosclerotic functions such as suppressing adhesion molecule expression by endothelial cells, inhibiting platelet aggregation, and inhibiting inflammatory cytokine production (123). This protection is reduced or absent at atherosclerosis-prone sites. Laminar sheer stress has been shown to enhance tetrahydrobiopterin levels through phosphorylation of GTPCH protein (317). Oscillatory sheer stress, however, reduces this phosphorylation event, meaning that endothelial cells at atherosclerosis-prone sites have lower intracellular BH4 content (154). In vivo studies using partial ligation of the left common carotid artery to model low and oscillatory sheer stress have demonstrated that NOS uncoupling occurs at sites of disturbed flow, as shown by reduced eNOS dimer formation and enhanced L-NAME inhibitable O2 − production (155). This uncoupling was attributed to insufficient BH4 levels by the restoration of normal NOS function by tetrahydrobiopterin supplementation (155).
Genetic models of atherosclerotic disease such as the ApoE−/− mouse are typified by a chronic inflammation in the vessel wall that is associated with high levels of local oxidative stress (28). The induction of NADPH oxidases in multiple plaque cell types, including endothelial cells and macrophages, by plaque components such as oxidized LDL (11, 103) causes a high local production of ROS. The net effect of atherogenesis on cellular and tissue BH4 levels is the integration of competing stimuli that have well-defined positive and negative effects of BH4 availability. While proinflammatory stimuli can increase GCH1 gene expression, oscillatory sheer stress decreases GCH1 gene expression, GTPCH enzymatic activity, and the increased oxidative stress within the vascular wall can cause an oxidative loss of BH4. Accordingly, contrasting findings are found in experimental tissues, possibly relating the stage and severity of disease. Since multiple plaque-derived cells are potential sources of BH4, the relative contribution of each may vary according to local plaque biology.
When BH4 levels and those of the oxidized form BH2 are studied in young versus older animals, disease burden increases total biopterin levels in many tissues such as the lung and heart, as would be expected given the induction tetrahydrobiopterin by inflammatory stimuli (281). BH4 levels are unchanged, whereas there is a dramatic increase in BH2 levels (281). This demonstrates increased oxidation of BH4 and thus a loss of the NOS-catalyzing form of the molecule. The up-regulation of tetrahydrobiopterin in atherosclerotic disease has also been addressed in the diseased organ itself (52). When BH4 levels, GTPCH protein, activity, and gene expression are measured in the aorta, all are increased as the animal ages and the disease progresses. This time-dependent regulation of aortic BH4 is not seen in normal C57bl6/J animals till 14 months of age. In this study, no increased level of oxidized biopterins was seen in the older diseased animals. Removal of the endothelium demonstrates that medial BH4 production is increased eight-fold in ApoE−/− animals, whereas 80% of the aortic BH4 production is localized to the endothelial cell layer in normal mice (52). Contrastingly, Ozaki et al. showed a 50% loss of aortic BH4 in fat-fed ApoE−/− compared with WT mice (197). These data on high-fat-fed ApoE−/− mice show a far less dramatic regulation of BH4 levels by atherosclerosis than the findings in rabbits fed a high-fat diet for 10 weeks (291). In that study, aortic segments from lipid-fed animals showed a 27-fold decrease in BH4 levels and an increase in the BH2 content from 30% of total biopterins to 63% and were associated with marked alterations in ex vivo vascular function.
To assess the degree to which BH4 levels are negatively affected by oxidative stress in atherosclerotic disease, studies of long-term supplementation with Vitamin C have been undertaken in wild-type and ApoE−/− mice. These studies demonstrate that vitamin C could increase aortic BH4 availability in C57bl/6J mice. Decreased accumulation of oxidized biopterins in ApoE−/− mice was also observed, although without a corresponding increase in aortic BH4 (268). However, the net effect was to improve the BH4 ratio. This improvement was accompanied by increased eNOS activity without an alteration in eNOS protein levels and a significant improvement in NO-mediated endothelium-dependent relaxations in the ApoE−/− animals.
Most of the existing atherosclerosis studies have been limited to experimental models of atherogenesis and disease progression. The Reversa mouse is a genetic model of atherosclerosis regression in which Apob100-driven hyperlipidemia can be reversed by inducible knockout of Mttp, thus switching off hepatic lipoprotein secretion (157). This model enables the effect of disease reversal to be elucidated. Normalizing hypercholesterolemia reduced impairment of endothelial function in ex vivo studies after prolonged periods of high-fat feeding (176). High-fat feeding in this model caused increased expression of GTPCH and reduced DHFR expression, and these changes were not seen when mice underwent a 6 month period of reversal compared with expression levels before reversal or in age-matched high-fat-fed controls (176). These data show that the changes in BH4 biosynthesis in atherosclerosis are not irreversible.
C. The role of BH4 and NOSs in atherosclerosis
BH4 availability can impact atherogenesis through the modulation of all three members of NOS enzymes. While eNOS is the predominant form of NOS detected in normal blood vessels, all forms of NOS are expressed in atherosclerotic vessels (319). A lack of eNOS in atherogenic models causes an exacerbation of disease and an increase in aortic lesion area, abdominal aneurysm, and cardiac fibrosis when mice are fed a high-fat diet (143). Despite the proatherogenic potential of uncoupled eNOS, eNOS−/−/ApoE−/− mice demonstrate that eNOS has a net protective effect with these mice showing increased leukocyte-endothelial cell interactions, vascular cell adhesion molecule 1 (VCAM-1) expression, and macrophage infiltration into the vessel wall (209). Deficiency in nNOSα causes an exacerbation of disease, with increased lesion area seen in ApoE−/−/nNOSα−/− animals (144), although nNOSγ expression remains, indicating a possible further function for nNOS in atherogenesis that cannot be studied using this partial knockout. In contrast to the protective roles of eNOS and nNOS, as seen by the exacerbation of disease in their absence, iNOS expression is deleterious in atherosclerotic disease. iNOS RNA and protein is reported in human atherosclerotic lesions (319), where its presence has been shown to co-localize with areas of lipid oxidation and protein nitration (47, 161). In experimental ApoE−/− models of atherosclerosis, iNOS expression has been found to exacerbate disease, with iNOS−/− animals showing a decreased disease burden after high-fat feeding (34, 56, 142, 181). iNOS is expressed by multiple cell types within atherosclerotic plaque, including both macrophages and smooth muscle cells (161), with data from bone marrow chimera showing that iNOS expression by both leukocytes and parachenymal cells is proatherogenic (208). The pro-inflammatory role of iNOS has been ascribed to the high enzymatic output of nitric oxide, which can, in turn, react with ROS to produce damaging reactive nitrogen species (RNS). While a lack of iNOS expression has been shown to cause a decrease in 3-nitrotyrosine levels and lipid peroxidation after high-fat feeding (281) (142), the source of O2 − that is required to produce RNS is assumed to be traditional ROS-producing enzymes. More recent studies have shown that O2 − production itself is reduced in the atherosclerotic vasculature when iNOS protein is acutely inhibited or absent (208). This provides some in vivo evidence that iNOS uncoupling may play a pathological role in vascular disease as an additional source of ROS. Given the opposing roles of NOS enzymes in disease pathogenesis and how the coupled and uncoupled forms of NOS alter output species, the regulation of BH4 availability is likely the key in controlling disease (Fig. 9).
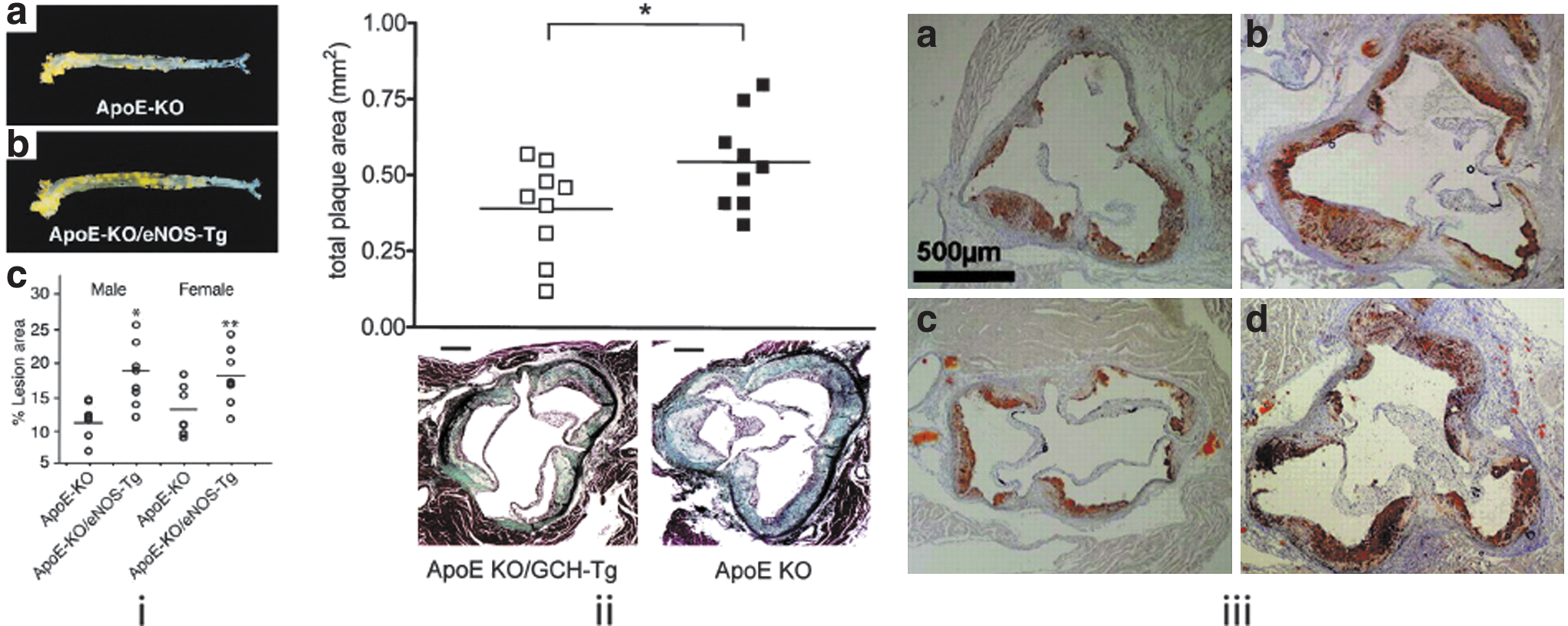
Transgenic overexpression of eNOS in the endothelium has dramatically shown the importance of BH4 availability in vascular disease (197). In ApoE−/− mice harboring the transgene, rather than seeing a beneficial effect of increasing expression of the “atheroprotective” eNOS molecule, a histological examination of the aortic root showed a two-fold or greater increase in plaque burden in both male and female animals. This was despite an evident decrease in blood pressure and an increase in NOS enzymatic activity. The involvement of uncoupling in this phenomenon is demonstrated by the absence of any increase in NO-producing activity in the aorta of eNOS transgenic mice and instead a 3.3-fold increase in aortic O2 − production (197). That this pathological phenotype was driven by a disparity in NOS enzyme expression and available BH4 co-factor is underlined by the effect of crossing this mouse with one overexpressing GCH in the endothelium. The endothelial GCH transgenic mouse (GCH-Tg) has endothelial specific overexpression of the human GCH1 gene (14). When crossed with an ApoE−/− mouse, the animals carrying the GCH1 transgene show both an increased aortic BH4 content and an improved ratio of BH4:total biopterins. These animals show the hallmarks of improved NOS function, namely reduced vascular O2 − production and improved vascular function. Atherosclerotic disease in these animals has been shown to be reduced by 28% in the aortic root. However, it is the cross of this animal with the eNOS transgenic that shows the power of BH4 to limit inflammation due to eNOS uncoupling (268). In these animals, the pathological effect of eNOS overexpression is completely reversed when GCH1 is co-expressed in the endothelium. Key figures from these very important reports are compared in Figure 10.
D. BH4 supplementation in models of atherosclerosis
The beneficial effects of BH4 mediated through eNOS have led to multiple studies attempting to “improve” eNOS function by enhancing BH4 availability using supplementation approaches. Short-term and acute protocols have been extensively used in vascular function studies, as discussed in the “BH4 in the vascular system” section, but some studies have addressed the ability of BH4 to modify vascular disease (Table 2).
The ability of exogenous BH4 to have positive effects on plaque biology has been shown by the increase in vascular NO production and reduced O2 − levels when ApoE−/− eNOS overexpressing mice were supplemented with 10 mg/kg/day BH4 (197). This study also demonstrated that BH4 supplementation could reduce plaque formation in ApoE−/−/eNOS transgenic mice. While this study also showed some positive effect of BH4 supplementation on atherosclerosis in female ApoE−/− mice, it has been later studies that have probed the ability of BH4 to reduce atherosclerosis in the absence of NOS manipulation. Hattori et al. showed that supplementation with 10 mg/kg/day BH4 in drinking water caused both rescue of impaired acetylcholine-induced vasodilatation to wild-type levels and a reduction in lesion area in the aortic root by 25% (99). A similar study by Schmidt et al. showed that a pharmaceutical preparation of 10 mg/kg/day BH4 dosed daily by gavage also caused the expected attenuation of atherosclerotic disease seen in the earlier study (226). This study profiled the effect of BH4 supplementation on the inflammatory biology of atherosclerosis in greater detail showing decreased plaque macrophage and T-cell content as well as a supporting decrease in aortic VCAM-1 expression (226). Hattori et al. also described a positive modification of inflammatory gene expression, with reduced expression of adhesion molecules, MCP-1 and NADPH oxidase subunits. This demonstrates that increasing BH4 levels in the diseased tissue enables a correction of upstream disease events such as the endothelial cell activation which supports leukocyte recruitment. This finding is strengthened by the observation that leukocyte content in atherosclerotic plaque is reduced when BH4-deficient hph-1/ApoE−/− animals are crossed with mice overexpressing GCH1 in the endothelium (226).
E. Vascular injury models
The formation of neointima and restenosis after vascular injury and stenting are another model of vascular inflammation in which BH4 and NOS biology play a key role. One of the simplest experimental models of this type is femoral wire injury. In the wire injury model, endothelial cells lining the vessel are denuded, and neointima formation develops over 14 weeks after injury. The expression of both eNOS and iNOS is seen after vascular injury, with eNOS expression predominating (298). The response to vascular injury encompasses macrophage recruitment to the media, smooth muscle cell hyperplasia, and re-endothelialization. A deficiency of BH4, in this case the hph-1 mouse model, is associated with enhanced intimal and advential macrophage accumulation and increased cell proliferation, resulting in a seven-fold increase in neointimal area (298). Treatment with 10 mg/kg/day BH4 ip caused a significant blunting of the enhanced injury response in the hph-1 mice; however, whether BH4 supplementation was sufficient to reduce vascular injury in this model under wild-type conditions was not studied. Similar studies have been performed using the carotid artery wire injury model. In this model, re-endothelialization occurs between 3 and 7 days post injury, as measured by Evans blue staining in vivo (156). In this model, overexpression of GCH in the endothelial cell layer resulted in a smaller medial and intimal area post injury (156). Whether these effects were manifested through reduced inflammation or by positive effects on re-endothelialization was not directly assessed.
More extensive studies of the effect of endothelial GTPCH overexpression have been performed in vein-graft accelerated arteriosclerosis. Wild-type and GCH transgenic mice underwent carotid interposition bypass grafting using the vena cava from ApoE−/− or ApoE−/−/GCH-Tg mice (4). Vessel wall area was reduced by 69% 28 days postinjury and by 53% 56 days post injury in mice overexpressing GCH. Vein graft tissue showed decreased macrophage infiltration and decreased tissue MCP-1 levels, indicating that elevated endothelial BH4 content had an anti-inflammatory effect in this model (4). Apart from protective effects being mediated through the effects on inflammatory gene expression and leukocyte recruitment, the overexpression of GCH has also been shown to reduce vascular injury by promoting endothelial cells repopulation in a model of stent deployment (60). Ex vivo aortic segments undergo balloon angioplasty or the placement of stainless steel stents before carotid-interposition grafting in the murine arterial stent-graft model. Endothelial cell repopulation was significantly increased in GCH-Tg ApoE−/− mice compared with ApoE−/− littermates, in addition to significantly diminished formation of neointima (60). This study shows that elevated endothelial BH4, in addition to reducing inflammatory pathways, can mediate enhanced endothelial cell repopulation after injury.
An acute model of atherogenesis is partial carotid ligation; in this model, approximately three of the four caudal branches of the common carotid artery are ligated, causing altered blood flow and sheer stress and resulting in occlusive atherosclerotic plaque formation within 3 weeks of surgery. As discussed earlier, this is associated with NOS uncoupling through limiting BH4 availability. In this model, treatment with 10 mg/kg day BH4 in drinking water prevented plaque formation, as compared with vehicle (vitamin C) controls (155). To further demonstrate that BH4 had an effect through NOS-dependent activity, the authors used NH4 (a chemically similar compound with similar anti-oxidant but no NOS-binding activity) and this had no ability to prevent plaque formation. Conversely, BH4 treatment was shown to reduce the infiltration of both lymphocytes and myeloid cells (155).
F. Tetrahydrobiopterin in human cardiovascular inflammation
Addressing the regulation of tetrahydrobiopterin in human vascular inflammation is limited by the availability of relevant tissue samples. Many studies draw parallels between plasma biopterin levels and CAD pathologies; however, the systemic and local vascular regulation of BH4 levels may be different. The relationship between plasma and vascular biopterins was directly assessed using patients undergoing elective coronary artery bypass grafting (CABG). Samples of saphenous vein or internal mammary artery were obtained during the CABG procedure along with fasted preoperative plasma samples (6). These samples revealed an inverse relationship between plasma and vessel BH4 levels, despite a positive correlation of plasma BH4 levels with CRP. In these patients, low vascular BH4 was associated not only with decreased vascular function and increased vascular O2 −, but also with elevated plasma BH4 and CRP levels. These data would indicate that plasma BH4 is associated with systemic inflammation, in a similar manner to the use of neopterin as a biomarker in cardiovascular diseases (261).
That plasma biopterins are driven by systemic inflammation was established using innoculation with Salmonella typhi vaccine as an acute stimulus of systemic inflammation (9). Vaccination caused an acute peak in both markers of inflammation, IL-6 and CRP, and in plasma biopterins. This peak in inflammation was associated with a decrease in vascular function, with flow-mediated dilatation (FMD) in the brachial artery being reduced by 50% 8 h post inoculation.
Genetic variation in the GCH1 gene is associated with alterations in the regulation of BH4 availability by inflammation. Individuals harboring the XX haplotype first described in Tegeder et al. (271, 272) are unable to increase biopterins in response to inflammation (9) when compared with the OO haplotype. This is not caused by a generalized lack of response to inflammatory stimuli, as XX patients had a similar induction of IL-6 to the OO individuals. These individuals showed a more dramatic drop in FMD after vaccination, indicating that the ability to up-regulate BH4 levels acts to preserve vascular function in the context of inflammation (9). These data would indicate that biopterin are induced both locally and systemically by inflammatory stimuli, with the local induction acting to maintain vascular function in the face of inflammation.
VII. Cardiac BH4
A. NO in the heart
It is well known that NO generated constitutively by eNOS in endothelial cells influences vascular homeostasis and function. Ever since, it has been established that agonist-stimulated release of NO in the coronary endothelium exerts paracrine effects on cardiomyocytes, affecting both cardiac morphometric and functional properties. A significant recent advance has been the finding that NO is also generated in the heart not just by the coronary endothelium but by myocytes themselves, making us rethink previous interpretations of studies undertaken in the intact heart (15). Furthermore, unlike in vascular and inflammatory cells, nNOS is expressed in cardiomyocytes. eNOS is found in coronary and endocardial endothelial cells and cardiomyocytes (71), and nNOS has been localized to cardiac autonomic nerves, ganglia, and cardiomyocytes (53, 71). iNOS is not expressed in the myocardium under normal circumstances, although it may be induced under certain stress conditions, such as ischemia (48). The effects of changes in NOS function on myocardial contractile function may, therefore, reflect changes in coronary flow, myocyte function, and/or autonomic transmission. By virtue of their distinct subcellular locales within the cardiomyocyte, eNOS and nNOS are likely to couple to distinct effector molecules and to elicit quite different responses after enzyme activation (53, 71, 254). This is because the diffusion distance of NO within cardiac myocytes is limited to a local environment by both a high cytoplasmic concentration of myoglobin (which has a high affinity for NO) and, particularly in disease states, an abundance of O2 −, which reacts with NO to limit its bioavailability (203, 229, 236).
It is well established that NO influences myocardial function and morphometry through its activation of soluble guanylate cyclase (sGC), leading to an increase in cyclic guanosine monophosphate (cGMP) production and subsequent PKG activation. NO binds to the heme moiety of sGC to provoke a conformational change, thereby increasing its affinity for MgGTP, which gets converted to cGMP (252). Intracellular accumulation of cGMP in cardiomyocytes leads to negative inotropy, possibly due to reduced myofilament Ca2+ responsiveness (289) and enhanced relaxation (309). Evidence suggests that the NO-elicited effects on cardiac inotropy are biphasic. Low levels of NO stimulate adenylyl cyclase and increase cyclic adenosine monophosphate (cAMP) levels, therefore increasing Ca2+ levels (311). In contrast, high levels of NO cause sGC activation, thus reducing myofilament Ca2+ sensitivity (289).
Recently, NO has been shown to elicit effects on cardiac contractility independent of sGC activation and cGMP production through s-nitrosylation (167) [the nonenzymatic attachment of NO to a reduced thiol, causing post-translational modification of protein conformation and function, akin to those induced by phosphorylation (249)]. Although sGC activation produces profound biological effects, it is now considered that the majority of the biological actions of NO in the heart are via SNO modification due to the relatively short half life of NO (167, 266). The most prominent targets of s-nitrosylation in the heart are the ion channels, particularly the ryanodine receptor Ca2+ release channel (RyR2) (122, 266), SR Ca2+ ATPase (SERCA) (26), and the L-type Ca2+ channel (217, 323).
A change in NOS activity can lead to a dysregulation of s-nitrosylation in the heart. Knockout of s-nitrosoglutathione reductase has been shown to increase cardiac levels of SNO proteins and to significantly attenuate heart failure in mice (10).
B. BH4 in the heart
Comparatively few studies have focused on the role of BH4 in the heart; investigations to date have largely focused on its role in inflammation, vascular function, and vascular pathologies. However, BH4 is likely to play a very specific role in the cardiomyocyte by virtue of the fact that (i) this cell type has a unique function which is regulated by NOS-derived NO as outlined earlier; (ii) its basal levels are relatively low compared with in other cell types (126), indicating that it delicately regulates NOS coupling/uncoupling (rather than being in excess); (iii) both eNOS and nNOS are expressed, unlike as in vascular and inflammatory cells (19); and (iv) eNOS and nNOS have distinct subcellular localizations where their derived NO or ROS may modify key target proteins nearby, such as calcium handling proteins, to influence cellular function (121).
To investigate the impact of BH4 on the myocardial NO-redox balance, contractile function, and calcium fluxes, Carnicer et al. (30) generated a cardiomyocyte-specific GCH1-overexpressing mouse (mGCH-Tg). mGCH-Tg mice had an increase not only in BH4 but also in the levels of oxidized products such that the ratio of BH4:BH2+B was actually reduced compared with wild types. However, myocardial nNOS activity was still elevated and O2 − production was attenuated in the transgenic mice, with eNOS activity largely unchanged. Furthermore, NOS inhibition had no effect on O2 − levels, indicating that NOS remained coupled under these conditions. Functionally, both the speed of relaxation and the rate of decay of the [Ca2+]i transient were faster in mGCH-Tg mice that were associated with an nNOS-dependent increase in PKA-mediated PLB phosphorylation at serine16. These findings suggest that GCH1 and BH4 may be therapeutic targets for the treatment of diastolic dysfunction.
C. BH4 in cardiac pathologies
An increase in ROS production and diminished NO bioavailability have been associated with many cardiac diseases. Since NOS uncoupling leads to both of these phenomena, recent studies have focused on the role of BH4 in many heart diseases, including ischemia/reperfusion (49, 239), cardiac hypertrophy (182), diabetes (14, 25), and hypertension (91, 128, 151, 341) with the hypothesis that reversing myocardial NOS uncoupling could be a therapeutic tool. However, the cardiomyocyte-specific roles of BH4 in cardiac pathologies remain uncertain, as targeted gene deletion models are lacking and, as this review highlights, the requirements for BH4 in cardiovascular diseases are cell specific with divergent mechanisms and effects in different cells, such as endothelial and inflammatory cells. Furthermore, NOS knockout models are unable to provide us critical information on the role of BH4, as neither the consequences of NOS coupling nor NOS uncoupling, a potential redox switch, can be assessed.
D. Ischemia/reperfusion injury and infarction
Myocardial ischemia-reperfusion (I/R) injury occurs commonly in the clinical setting after pharmacological or mechanical reperfusion, such as during angioplasty, thrombolysis, and cardiac bypass. I/R injury is characterized by myocardial apoptosis, necrosis, and endothelial dysfunction that are associated with an increased production of ROS. Many cardioprotective strategies to counter perioperative I/R have focused on the NO pathway, and, recently, the effects of I/R on BH4 oxidation and the therapeutic potential of BH4 supplementation have been examined. Dumitrescu et al. (62) demonstrated that cardiac ischemia followed by reperfusion resulted in significant BH4 oxidation, NOS uncoupling, endothelial dysfunction in coronary arterioles, and impaired coronary blood flow in isolated rat hearts. Exogenous BH4 supplementation partially inhibited O2
Diabetic hearts are known to be particularly susceptible to I/R injury due to elevated oxidative stress that is associated with this condition (31). The enhanced oxidative stress in diabetes promotes iNOS up-regulation, BH4 oxidation, and, consequently, iNOS uncoupling, thus further exacerbating the oxidative/nitrosative stress (186, 251). However, Okazaki et al. showed that BH4 treatment was sufficient to restore iNOS coupling in isolated diabetic rat hearts and the iNOS-derived NO conferred cardioprotection against I/R injury in terms of both infarct size and left ventricular function (21, 83, 112, 195). The cardioprotective effect of BH4 was abrogated by treatment with a thiol-reducing agent (dithiothreitol), but not an NO-sensitive guanylyl cyclase inhibitor (ODQ), suggesting that iNOS-derived NO-mediated cardioprotection occurs through protein S-nitrosylation and not cGMP-dependent signaling in the diabetic heart (112).
A number of studies have focused on the role of BH4 in the phenomenon of late-phase ischemic preconditioning (IPC). IPC is an innate phenomenon in which brief exposure to sublethal ischaemia induces a profound protection from a subsequent ischemic insult in various organs, including the heart (180). IPC is associated with two forms of protection: the “early phase” lasting ∼2 h after the preconditioning ischemia followed by a “late phase” of protection occurring a day later and lasting ∼3 days. Studies have implicated NO as an important mediator in IPC, with NO derived from constitutive NOS being implicated in the early phase and NO derived from iNOS being involved in the late phase of IPC (127). The inhibition of BH4 synthesis in rats (using DAHP to inhibit GTPCH) has also been shown to abolish the protective effect of IPC in rat myoardium (259). This study revealed that IPC increases cardiac BH4 through the up-regulation of GCH1 via the H2O2-Jak2 signaling pathway. Similarly, in rabbits, Tang et al. (269c) showed that left coronary artery IPC resulted in increased myocardial BH4 content in the ischemic area after I/R injury, but not in the nonischemic zone. They also demonstrated that both pharmacological inhibition of BH4 synthesis and hypercholesterolaemia abolished both the rise in BH4 and the infarct-limiting effect of late IPC. The increase in ROS associated with hypercholesterolaemia is likely to be responsible for the diminished BH4 bioavailability in this setting. Recently, Ge et al. (84) used a mouse model of cardiomyocyte-specific GCH1 overexpression to demonstrate that increased BH4 bioavailability preserves the ability of IPC to elicit myocardial protection in the hyperglycemic state.
E. Cardiac hypertophy and function
Cardiac hypertrophy is an adaptive response of the heart to pressure overload and is, thus, a common pathological feature of many cardiovascular diseases, including hypertension and myocardial infarction. Although cardiac hypertrophy may initially be beneficial, providing compensation to the stress, sustained hypertrophic stimulation becomes maladaptive, leading to increased morbidity and mortality due to congestive heart failure and sudden death (232). Growing evidence suggests that oxidative and nitrosative stresses modulate the transition from adaptive to maladaptive hypertrophy [reviewed by Takimoto et al. (269a)]. The production of ROS is linked to inducers of hypertrophy such as Gαq/G11-coupled agonists (e.g., phenylephrine, angiotensin) (76, 183), signalling kinases and phosphatases (267), and mechanotransduction (207). ROS have also been shown to induce fetal gene re-expression (a marker of hypertrophy) (301) and to contribute to chamber remodeling by activating MMPs. As such, the role of BH4 and NOS (un)coupling in the pathogenesis of cardiac hypertrophy has been brought into question.
Takimoto and Kass (269b) demonstrated that pressure overload induced by transverse aortic constriction (TAC) in mice reduces cardiac BH4 levels and triggers eNOS uncoupling, which, in turn, leads to myocyte hypertrophy, cardiac dilation, interstitial fibrosis, and ventricular dysfunction. Perhaps controversially, eNOS-knockout animals exhibited neither dilation nor impaired systolic function and only showed mild hypertrophy; however, this is likely, because eNOS-derived ROS is abolished. Treatment with oral BH4 prevented the NOS uncoupling and reduced the TAC-induced hypertrophy in control mice. Furthermore, a separate study by Moens et al. (182) revealed that exogenous BH4 recoupled eNOS, thus reversing pre-existing advanced cardiac hypertrophy and fibrosis caused by TAC-induced pressure overload (Fig. 12).
Most recently, Silberman et al. (247) studied the role of NOS uncoupling in a mouse model of diastolic dysfunction. Diastolic dysfunction can cause heart failure with a preserved ejection fraction, and there is a strong epidemiological association between hypertension and diastolic dysfunction (16). Mild hypertension with diastolic dysfunction in the absence of hypertrophy and systolic dysfunction was generated by unilateral nephrectomy with DOCA-saline treatment (247). The resulting mice had increased cardiac oxidative stress, predominantly from uncoupled nNOS, and an attenuated BH4:oxidized biopterins ratio. Feeding with BH4 improved total cardiac BH4 levels and redox status. Furthermore, treatment with BH4 (but not NH4) both prevented and reversed diastolic dysfunction. In addition, BH4 treatment in DOCA animals augmented the phosphorylation status of phospholamban, a key regulator of sarcoplasmic reticulum Ca2+ pump activity and cytosolic Ca2+ levels, suggesting a possible underlying mechanism (247). Similarly, in a separate study, BH4 was able to restore diastolic dysfunction and LV remodeling in ovariectomized mRen2.Lewis rats (which have depleted myocardial BH4) that were associated with a decrease in nNOS-derived O2 − production (129).
Importantly, a recent study by Moens et al. (181a) highlighted that the effective dose range of BH4 for ameliorating pressure overload-induced hypertrophy and heart failure may be more limited than expected. On oral administration, BH4 is largely oxidized to BH2 and biopterin, and the BH2 can then be re-reduced once in the cell. Since BH2 can compete with BH4 to impair NOS coupling, higher doses of BH4 might tip the balance toward more reduced BH2 such that any physiological benefit of a higher initial dose of BH4 is offset. Moens et al. (181a) revealed a bimodal dose response of BH4 for the treatment of hypertrophic heart disease; a mid-range dose of 36 mg/kg/day BH4 was found to be beneficial, whereas higher doses (approximately 400 mg/kg/day BH4) became ineffective. The beneficial doses correlated with an enhanced BH4:BH2 ratio.
Together, these studies highlight the importance of BH4 to maintain NOS coupling in cardiac hypertrophy, failure, and dysfunction and to support BH4 as a potential new therapeutic approach.
F. Cardiac autonomic function
Many cardiovascular disease states, including heart failure and myocardial infarction, are associated with changes in cardiac autonomic function. Furthermore, abnormalities in the autonomic control of the heart, such as reduced baroreflex sensitivity and heart rate variability, are predictors of adverse events in conditions such as ventricular arrhythmias (37). Evidence suggests that endogenous NO regulates both sympathetic and parasympathetic cardiac control (36). Under physiological conditions, NO appears to inhibit sympathetic activity and to increase vagal tone. In pathophysiological conditions such as diabetes and heart failure, a decreased NO bioavailability is concomitant with autonomic dysfunction.
BH4 is likely to be a key mediator of cardiac autonomic function, as it is a critical cofactor for both TH and NOS enzymes, thus controlling both L-DOPA-derived and NO neurotransmitter activity (57, 111). Zhang et al. (339) investigated whether a common genetic variation of the GCH1 gene alters autonomic function in humans. An SNP, C+243T, was found to predict reduced urinary catecholamine excretion and NO production and was associated with decreased baroreflex sensitivity and heart rate variability. In addition, this SNP had a significant association with higher blood pressure in women. Although BH4 levels were not measured in this study, this SNP is in tight linkage disequilibrium with the “X” GCH1 haplotype described by Tegeder et al. (271) and Antoniades et al. (7), indicating that this observed autonomic phenotype may be BH4 mediated (59).
Adlam et al. (2) studied the effect of BH4 deficiency on the autonomic regulation of heart rate in the hph-1 mouse model. Reduced GCH1 expression and subsequent BH4 deficiency in hph-1 mice was associated with a resting tachycardia before and after exercise training despite normal blood pressure and normal heart rates during exercise. This resting tachycardia was linked to enhanced β-adrenergic sensitivity with no effect on vagal function (57). The resting tachycardia and increased cardiac sympathetic sensitivity of these BH4-deficient hph-1 mice is similar to that observed in humans with BH4 deficiency, including patients with dopa-responsive dystonia and those with the C243T polymorphism in GCH1(305, 337).
AF, the most common sustained cardiac arrhythmia, has also been linked to a decrease in myocardial BH4 and NOS uncoupling. In both a goat model of pacing-induced long-standing AF and in patients with persistent AF, Reilly et al. (215) demonstrated increased uncoupled NOS activity and a diminution in both BH4 bioavailability and the BH4:BH2+B ratio.
These insights into the influence of BH4 on cardiac autonomic function are likely to have important clinical implications, as the autonomic regulation of heart rate plays a fundamental role in cardiovascular health.
VIII. Concluding Remarks
In summary, BH4 is a critical regulator of cardiovascular homeostasis; abnormalities in the absolute levels and oxidation state of BH4 are a hallmark of vascular and cardiac dysfunction; and current research indicates that maintaining adequate BH4 levels in the endothelium is likely to be critical in regulating the balance of NO and O2 − synthesis by eNOS. Strategies to maintain vascular BH4 availability may include measures to increase BH4 biosynthesis or to reduce oxidative degradation, and ongoing studies attempting to restore intracellular BH4 levels via gene therapy and direct supplementation are showing promising results. However, the clinical efficacy of improving BH4 bioavailability also may depend on other biochemical determinants of NOS uncoupling, including cellular concentrations of L-arginine, methylarginines, and oxidized glutathione. Future studies are, therefore, needed to understand how BH4 bioavailability and these factors interact with one another to determine NOS activity at biochemical, cellular, and systemic levels. To move forward and develop BH4 as a successful therapeutic for cardiovascular disease, we need to better understand the interaction of BH4 and the NOSs with downstream NO-ROS signaling in both endothelial cells and inflammatory cells. Resulting advances could lead to significant improvements in the treatment of the wide range of cardiovascular disease states in which NOS uncoupling is a central pathogenic mechanism.
Footnotes
Acknowledgments
The authors wish to thank Ruth Rinze for help with several figures, and they acknowledge support from the British Heart Foundation (BHF) Center of Research Excellence, Oxford. This research was supported by a BHF Center of Research Excellence Intermediate Fellowship award to Mark J. Crabtree (RE/08/004) and a BHF Program Grant (RG/12/5/29756).