
Abstract
Introduction
D
This study is the first to elucidate the mechanism by which oxidative stress and inflammation modulate the circadian system through the key circadian protein Rev-erbα. We demonstrate an opposing effect of oxidative stress, via NrF2, and inflammatory stress, via nuclear factor kappa B (NFκB), on circadian regulation. Since the circadian system synchronizes cell metabolism and proliferation, our results provide important insights into the dynamics between oxidative or inflammatory stress and cellular homeostasis.
One of the clock proteins, Rev-erbα (encoded by the Nr1D1 gene) is a nuclear receptor that links circadian behavior and metabolic output (14). Not only it is it important in generating circadian rhythmicity (10) but it also regulates genes encoding enzymes that are important to metabolism and heme synthesis (75). Multiple transcription factor binding sites are present on the promoter region of Rev-erbα, including an E-box and retinoid-related orphan receptor response elements (ROREs), which are essential for its circadian regulation (65) and nuclear receptor function (31). Other signaling pathways can also act on the RORE site to up-regulate Rev-erbα transcription as demonstrated in adipocyte differentiation (13), or to repress Rev-erbα transcription as in myocyte differentiation (19) and with exposure to glucocorticoids (64). In addition, Rev-erbα gene expression can be inhibited by its own gene product (1). Given that AP-1 and NFκB are involved in CCG regulation with phase specificity in a variety of tissues, they may also influence the regulation of key circadian genes such as Rev-erbα in the context of oxidative stress or inflammation.
Hyperoxia is known to cause type II cell injury, inhibit cell proliferation, and mediate inflammation in the lung, leading to decreased alveolar septation, increased terminal air space size, and increased fibrosis akin to what is observed in bronchopulmonary dysplasia (6, 67). In the neonatal period, this condition has long-lasting consequences, as alveolarization continues for many weeks postnatally (48). Nonetheless, neonatal rodents are more tolerant to hyperoxia than adults (25). Some of this may be related to NFκB signaling, because the neonatal lung readily activates NFκB in hyperoxia, which targets BCL2 gene transcription and protein expression and prevents apoptosis in lung cells (71). Whether other NFκB downstream signaling is also involved in neonatal hyperoxic tolerance is still not known.
Although Rev-erbα is highly expressed in the adult lung (8), little is known about its regulation during lung development and its function in the context of oxidative stress or inflammation. A recent report demonstrated that mouse lung Rev-erbα gene expression is down-regulated by cigarette smoke (66), suggesting that it can be modulated by oxidative stress. In many other clinical conditions, there are diurnal variations in the biological response to pathological conditions that may be due to oxidative stress or inflammation. For example, the onset of pain in patients admitted with myocardial infarction varied in frequency with the time of day (47). Asthma exacerbations also vary with the circadian rhythm [reviewed by Martin (44)]. More recently, investigators have described a loss of circadian gating of the endotoxin response in Rev-erbα null mutant mice and in cultured macrophages (29). Diurnal variation of hepatic antioxidant gene expression in response to oxidative stress also varies with the time of day (70).
It has been recently demonstrated that there is a link between the oxidation–reduction cycle of peroxiredoxin and cellular time keeping, which has co-evolved from an ancient time in all domains of life (21). This redox homeostasis integrates with the circadian rhythm that does not require nuclear involvement as documented in the erythrocytes (52, 53). How oxidative stress and/or inflammation influence circadian gene regulation at the transcriptional level is not yet clear. Given the importance of Rev-erbα in linking metabolic function, gating to the inflammatory response, and circadian regulation, we investigated whether oxidative stress and inflammation can alter Rev-erbα expression. Using hyperoxia as an inducer of oxidative stress, we identified an AP-1 site on the distal region of the Rev-erbα promoter, which functions as an NrF2-binding site regulating oxidative stress-induced Rev-erbα promoter activity and transcription. In addition, this site, along with two adjacent putative NFκB binding sites, was responsible for TNFα-mediated down-regulation of Rev-erbα. This mechanism was further tested and verified in the lungs of neonatal mice exposed to hyperoxia.
Results
Oxidative stress activates Rev-erbα promoter activity via the N2 NFκB/NrF2 binding site
To test whether oxidative stress regulates Rev-erbα promoter activity, the pGL4Nd0.9luc plasmid (0.9 kb) (see “Materials and Methods” section) was transiently transfected into the Mlg cells and exposed to hyperoxia. Reporter activity peaked at 4 h and remained elevated for the duration of the 48 h exposure as compared with the air-exposed controls (Fig. 1A and Supplementary Fig. S1; Supplementary Data are available online at
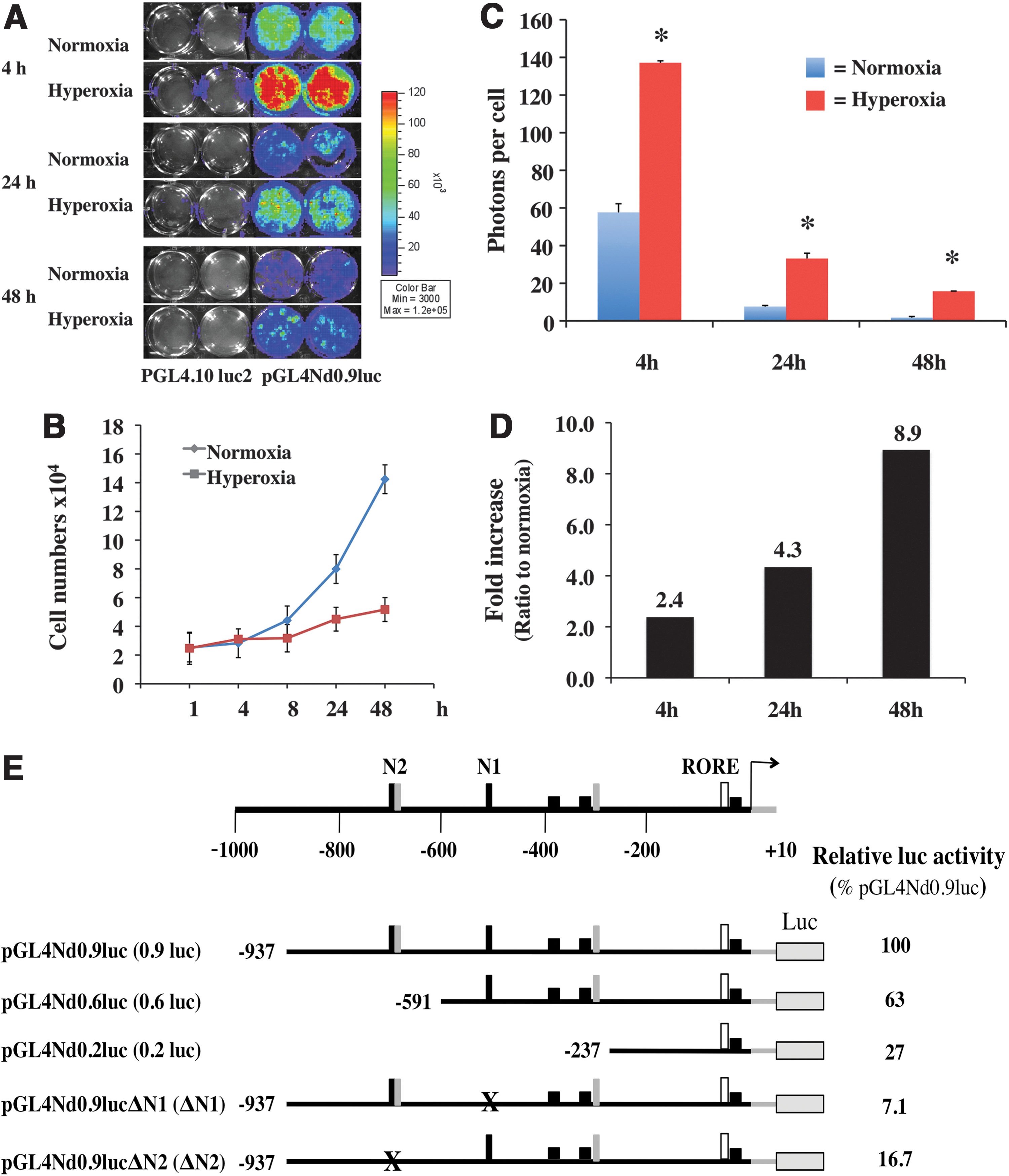
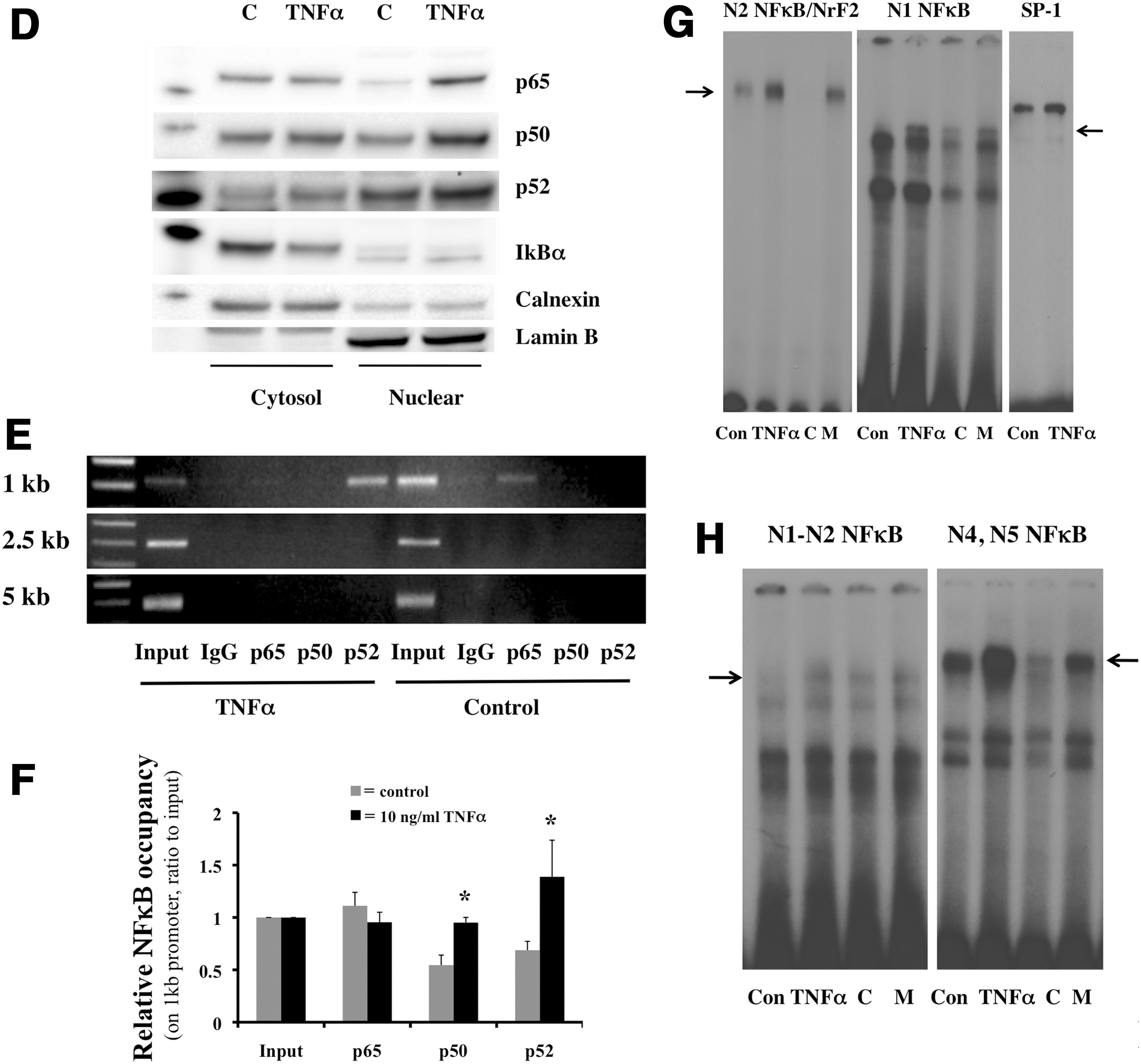
We next investigated whether the increased 0.9 kb luciferase reporter activity in hyperoxia was mediated via the conserved NFκB- or NrF2-binding sites seen on the Rev-erbα promoter. Two NFκB (N1 and N2) binding sequences and two NrF2-binding sites were identified on the 0.9 kb promoter. The N2 site overlaps with a conserved NrF2 site (Fig. 1E). To test whether these sequences were essential to Rev-erbα promoter activity, linear deletion of the 0.9 kb promoter was performed. The 0.6 kb luc construct excluded the N2 site, and the 0.2 kb luc excluded both N1 and N2 (Fig. 1E). Targeted deletion mutants that disrupted 10 bp of the N1 (ΔN1) and N2 (ΔN2) sites were also generated. At basal levels, the 0.6 and 0.2 kb constructs retained only 63% and 26% of the 0.9 kb luc activity, respectively. The ΔN1 and ΔN2 resulted in a reduction of the 0.9 kb luc activity by 93% and 83% respectively (Fig. 1E). When the 0.6 or 0.2 kb construct was transfected into the Mlg cells, the hyperoxia-induced promoter activity was significantly reduced in the 0.6 kb and almost eliminated in the 0.2 kb compared with the 0.9 kb promoter (Supplementary Fig. S3A). These results suggest that the N2 and, to a lesser extent the N1 site, is essential for hyperoxia-induced promoter activation.
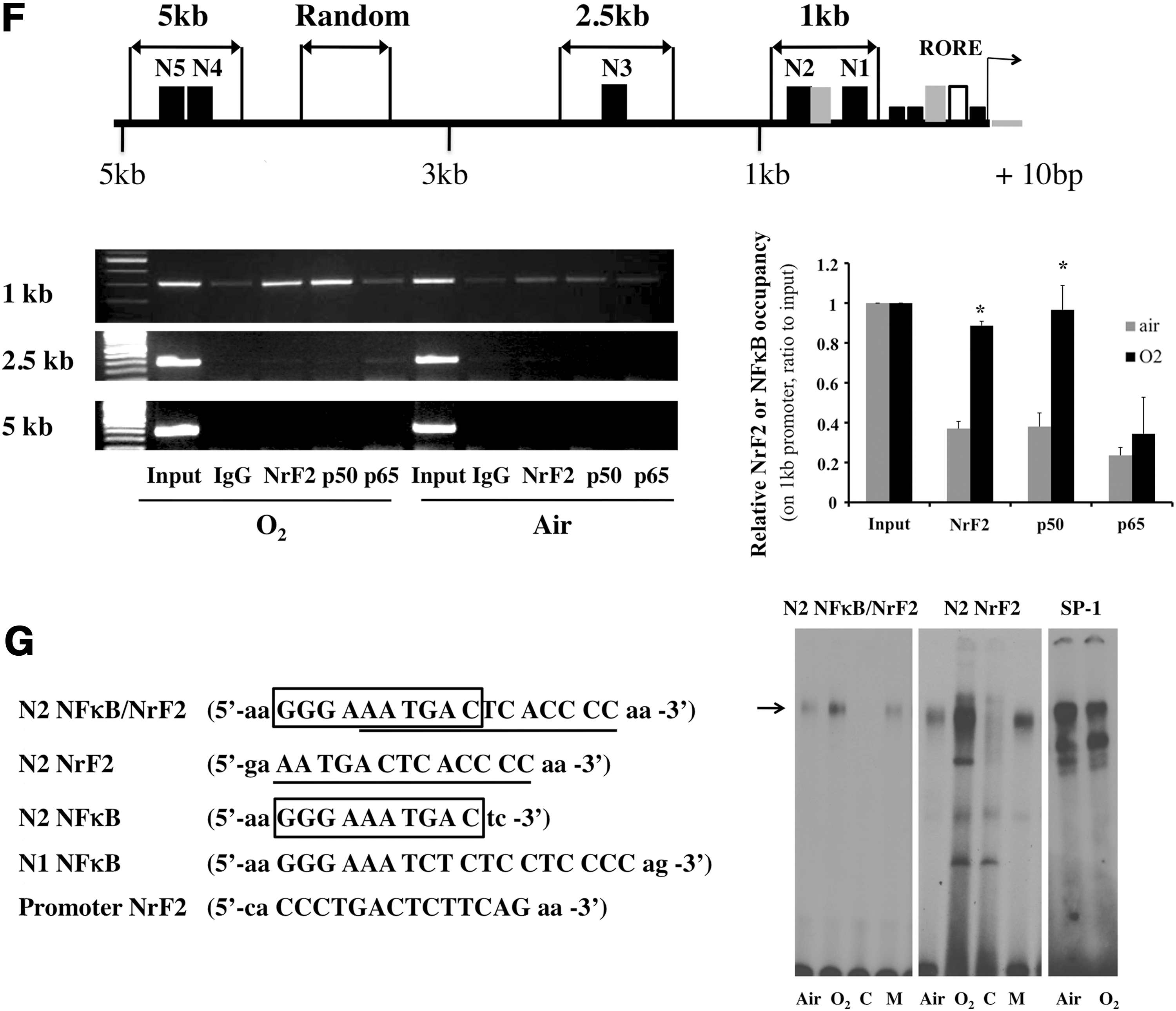
Primers designed to amplify the regions, including the N1 and N2 sites (Table 2), were used to test whether NrF2 and NFκB were recruited to this region (Fig. 1F, upper panel). As demonstrated with the chromatin immunoprecipitation (ChIP) assay, occupancy of NrF2 and only the p50 NFκB subunit were increased after 24 h of hyperoxic exposure (Fig. 1F, lower panel). Since distal enhancer regions could also influence transcription, we queried the distal portion of the promoter for approximately 2.5 and 5 kb. There is one conserved NFκB site (N3) and two conserved NFκB sites (N4 and N5) on the 2.5 and 5 kb regions respectively. There was no increase in NrF2, p65, or p50 occupancies on the N3 region as well as in the N4 and N5 regions after hyperoxia (Fig. 1F). When the entire 2.5 kb promoter was cloned into the luc reporter, there was no incremental luc activity observed in hyperoxia (Supplementary Fig. S3B). These results suggest that the N1 and N2 sites are the predominant responsive elements of the Rev-erbα promoter in hyperoxia.
We next synthesized DNA oligomers that were specific to the N1 and N2 sequences (Table 3) and performed electrophoretic mobility gel shift assay (EMSA) from nuclear extracts obtained from cells exposed to hyperoxia or normoxia for 24 h. The overlapping NFκB (box) and NrF2 (underline) regions on the N2 site were synthesized and tested separately (Fig. 1G). Increased DNA binding was observed on the N2 NFκB/NrF2 site, and this was further enhanced when using N2 NrF2 as a probe (Fig. 1G). No binding was observed when the N1 or N2 NFκB probe (Supplementary Fig. S4B, C), or the promoter NrF2 probe was investigated (Supplementary Fig. S4A). This further corroborated that the N2 NrF2 site was the major site regulating hyperoxia-induced Rev-erbα promoter activity.
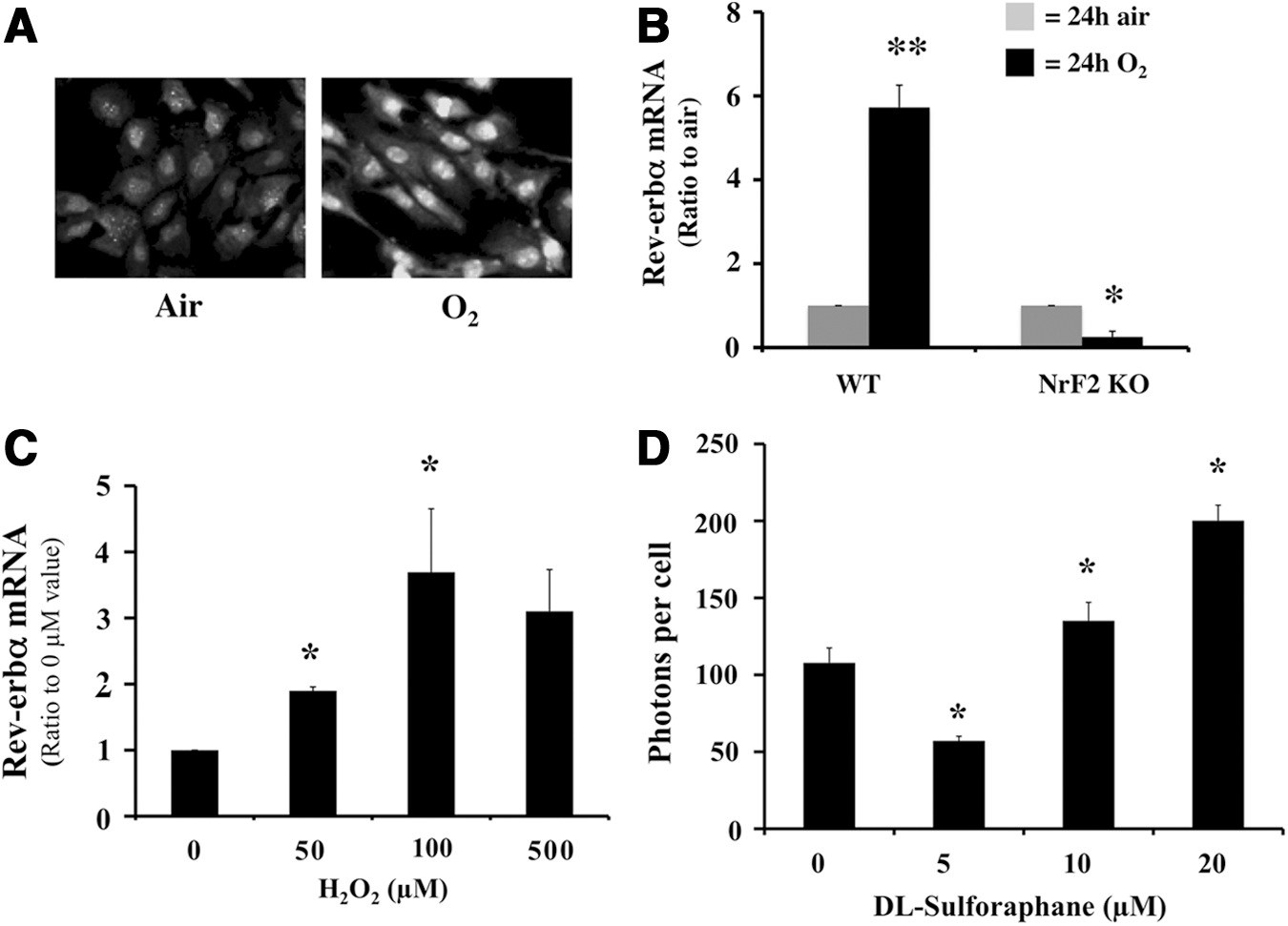
Since the putative N2 NrF2 site lacks a cytosine and a guanine nucleotide, it cannot traditionally be classified as an NrF2 site (23). Nonetheless, a 4-h hyperoxic exposure activated NrF2 in the Mlg cells, as demonstrated by enhanced NrF2 immunostaining in the nucleus (Fig. 2A). When mouse embryonic fibroblast (MEF) cells with disrupted NrF2 were exposed to hyperoxia, Rev-erbα mRNA levels were attenuated in contrast to the WT (Fig. 2B). In addition, H2O2, a known oxidative inducer of NrF2 (24), increased Rev-erbα mRNA in a dose-dependent manner at 0–100 μM (Fig. 2C). At 500 μM H2O2, Rev-erbα mRNA did not further increase, suggesting that a high concentration of H2O2 might have been too toxic to the cells. This was confirmed in the comet assay, which showed that 500 μM H2O2 resulted in a 100% DNA damage in the Mlg cells (Supplementary Fig. S5). Using a thiol-modifying NrF2 activator sulforaphane, Rev-erbα promoter activity was also increased in a dose-dependent manner from 5 to 20 μM (Fig. 2D) despite a decrease between 0 and 5 μM concentrations. This might have resulted from the normalization to the cell numbers, as increased cell proliferation was observed at 5 μM sulforaphane in the Mlg cells as well as in other cell lines (41). These data provide strong evidence that NrF2 directly regulates Rev-erbα promoter activity and expression.
Oxidative stress alters the rhythmicity of Rev-erbα and other core circadian genes
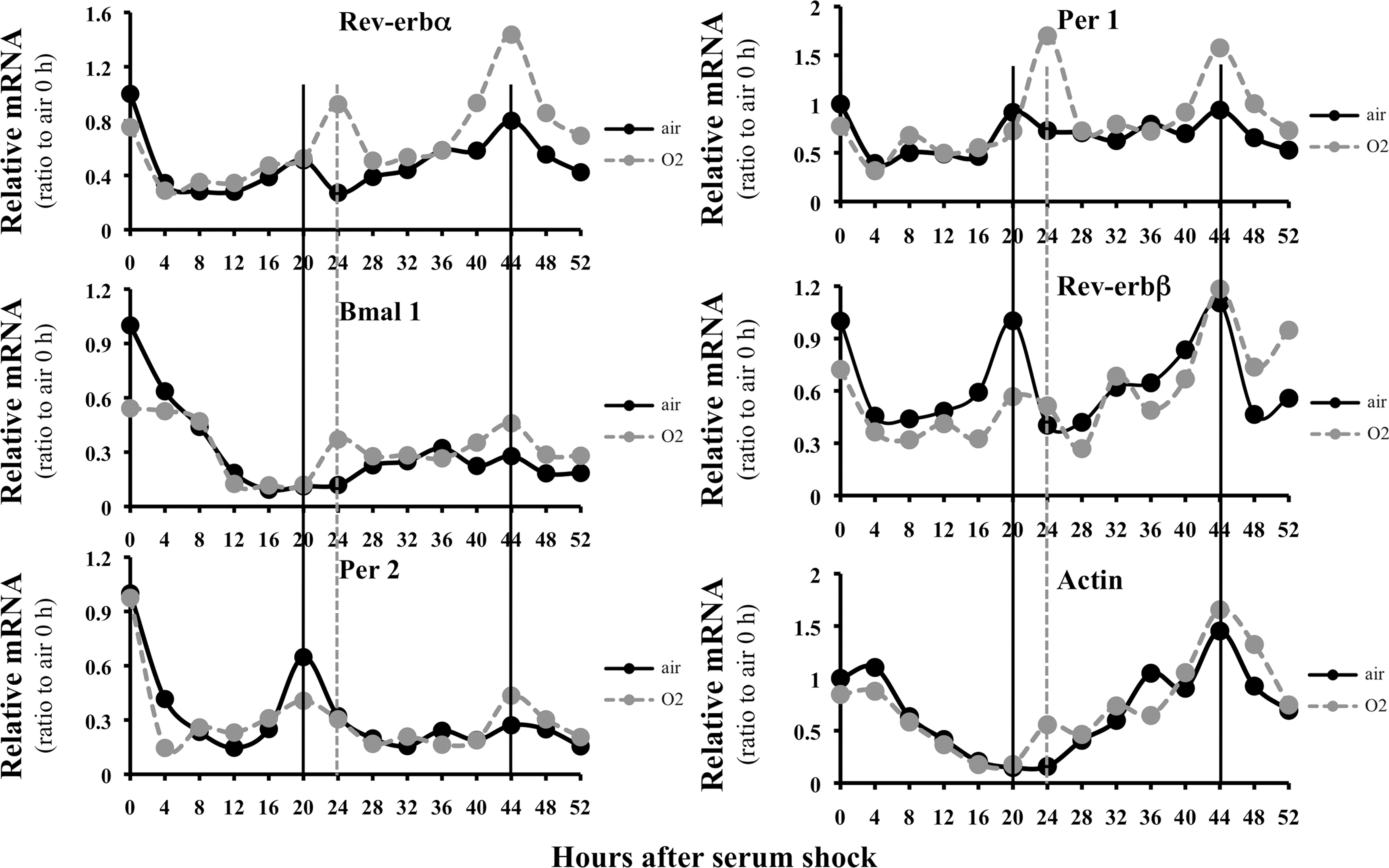
The Rev-erbα promoter is regulated by two major mechanisms: transcriptional activation via E-boxes with circadian rhythm regulation (65) and transcriptional repression via RORE sites in its role as a nuclear receptor (31). It is thought that close proximity of the E-boxes and RORE sites does not enable simultaneous modulation of the two regulatory systems. Since the putative NrF2 site is located several hundred base pairs away from the E-boxes and RORE sites, we predicted that transactivation via the NrF2 site could synergistically enhance rhythmic transcription of Rev-erbα. To test this hypothesis, we used serum shock as a model to generate rhythmicity (4). Pre-exposure to hyperoxia for 12 h not only phase-shifted Rev-erbα expression in the first circadian cycle but also increased the amplitude of its oscillations in both the first and second circadian cycles (Fig. 3, upper left panel). Downstream Bmal 1 gene expression reached the lowest point in both the first and second circadian cycles in air-exposed cells (middle left panel) in agreement with the literature (17, 30). However, it did not increase beyond these points, suggesting that this gene expression was not rhythmic in our model. Nevertheless, hyperoxia either phase-shifted or changed the amplitude of the circadian genes Per 1 and Rev-erbβ (upper and middle right panels). Per 2 amplitudes were moderately altered (lower left panel). Although an overall increase in actin gene expression was observed 24 h after serum shock, it did not shown a rhythmic pattern in either air- or hyperoxic-exposed cells (lower right panel). These data suggest that hyperoxic exposure alters rhythmicity of core circadian genes.
Inflammatory stimuli attenuate Rev-erbα promoter activity and expression
Since all conserved sites (N1–N5) contain NFκB sequences, we hypothesized that the overall impact of these cis elements in inflammation would be to activate Rev-erbα expression. Surprisingly, when TNFα was incubated with the Mlg cells, both the 0.9 kb promoter activity and mRNA of Rev-erbα were attenuated (Fig. 4A, B). In addition, lipopolysaccharides (LPS), another inflammatory stimulus-activating NFκB, also reduced the 0.9 kb Rev-erbα promoter activity in a dose-dependent manner (Supplementary Fig. S6). We next elucidated which NFκB site was responsible for the down-regulation of Rev-erbα expression. First, we tested the N1 and N2 sites by transfecting the 0.9, 0.6, or 0.2 kb luc constructs into the cells and incubating them with TNFα for 12 h. The decreased 0.9 kb luc activity was minimized in the 0.6 and diminished in the 0.2 kb luc-transfected cells (Fig. 4C), suggesting that the N2 and N1 sites are required for the TNFα inhibitory response. Next, nuclear extracts from the TNFα incubated cells were tested for protein levels of NFκB subunits. TNFα clearly decreased the inhibitor I-κBα in the cytosol and increased nuclear protein levels of p65, p50, and p52 (Fig. 4D). The recruitment of these subunits was also tested in the regions of N1 to N5 as described in Figure 1F. Enhanced p52 and moderately increased p50 occupancy were observed in the N1 and N2 regions (Fig. 4E, F). In addition, p65 occupancy was relatively higher than that of p50 and p52 in untreated cells (Fig. 4E), and remained the same under stimulated conditions (Fig. 4F). The sequence corresponding to the N1 NFκB site induced DNA binding in the nuclear extract with TNFα (Fig. 4G). Although the N2 NFκB sequence did not show binding (Supplementary Fig. S4D), the N2 NFκB/NrF2 demonstrated enhanced DNA binding with TNFα (Fig. 4G). These results indicated that the NFκB binding sites on N1 or N2 were able to bind NFκB complexes. However, this enhanced NFκB sequence-specific binding resulted in Rev-erbα down-regulation. Since there was enhanced recruitment of p52 and p50, the only two subunits of NFκB without transactivations domains, we reasoned that these transcriptional repressors would require co-repressors to down-regulate Rev-erbα. We, therefore, evaluated occupancy of HDAC1, a known corepressor of the p50/p50 homodimer (55); BCL3, a known p50/p50 or p52/p52 binding partner (51) and a corepressor (11); as well as I-κBα, the cytosolic NFκB inhibitor known to translocate to the nucleus and repress the PPARγ promoter (27). None of these proteins showed enhanced occupancy on the N1 to N5 (Supplementary Fig. S7A), despite increased abundance of BCL3 in the nucleus (Supplementary Fig. S7B).
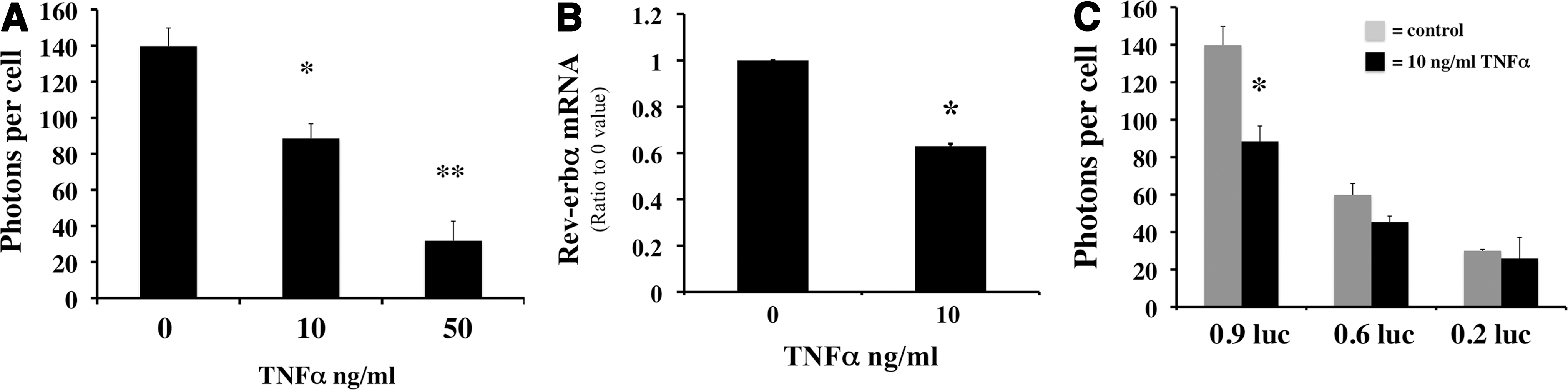
p50 specific binding sites are required for TNFα-induced Rev-erbα down-regulation
The p50 NFκB subunit was initially described as sequence specific on promoter regions of downstream genes (45). To understand how TNFα-mediated NFκB transcriptional activity down-regulated the Rev-erbα promoter without recruiting co-repressors, we evaluated whether the abundance of p50 homodimer-binding sites on the promoter could correlate with the repressive ability of this NFκB subunit protein. We performed an analysis for conserved transcription factor binding sites focused only on the 1 kb region of the Rev-erbα promoter. Indeed, a p50 homodimer-specific binding site was identified within the N1 and N2 (Table 3). This sequence was able to bind to the TNFα-induced NFκB complex as shown by EMSA (Fig. 4H). However, its binding affinity was much lower than that of the N4 or N5 p50-specific sequence (Fig. 4H). When p50 was silenced (Fig. 5A), TNFα-mediated attenuation of the Rev-erbα promoter was abolished (Fig. 5B).
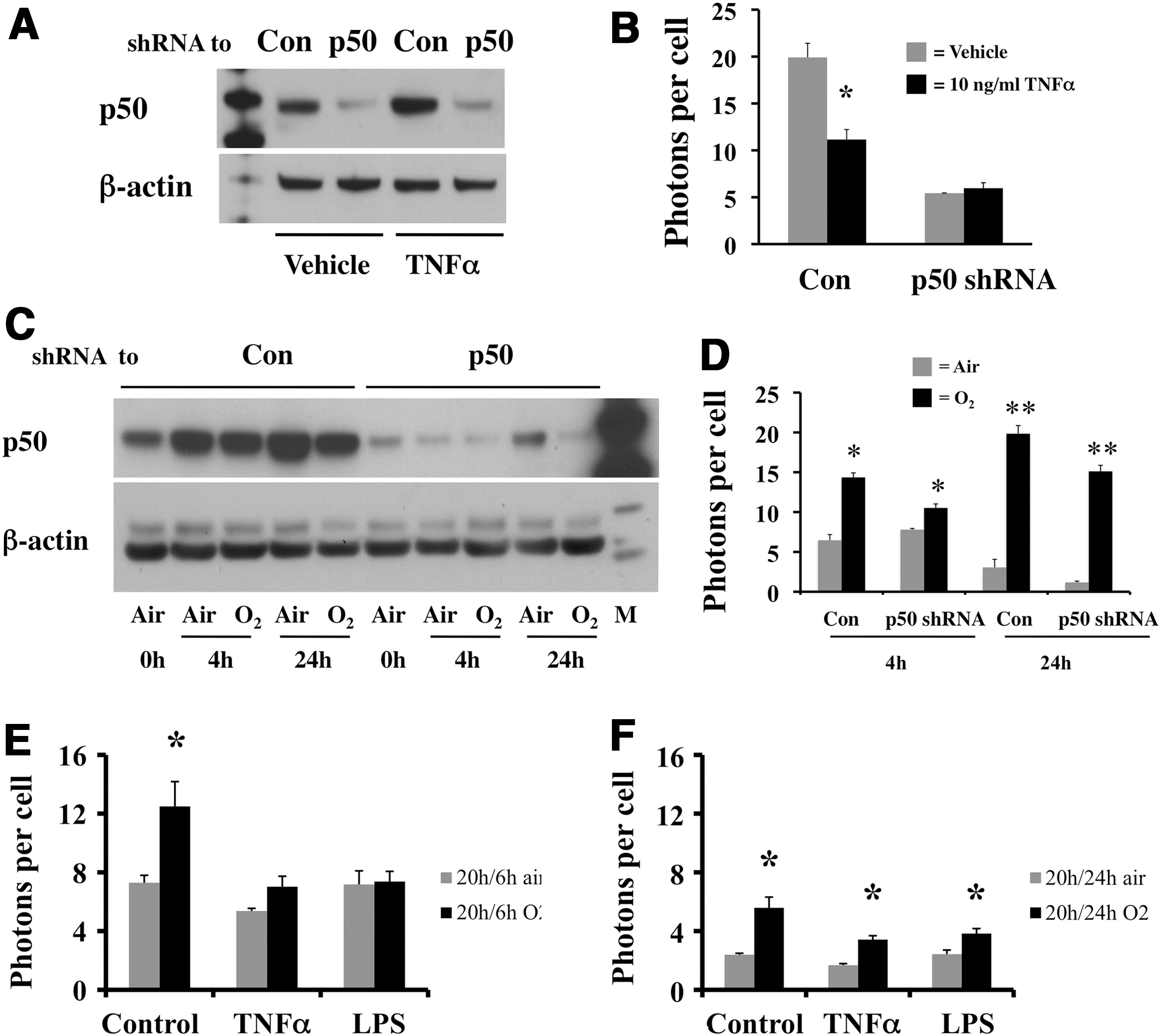
Occupancy of p50 was also enhanced in hyperoxia (Fig. 1F), suggesting that this site mediates both the oxidative and inflammatory response. However when the p50 shRNA-infected cells were exposed to hyperoxia (Fig. 5C), Rev-erbα promoter activity was increased to a similar extent as in the control shRNA-infected cells (Fig. 5D), further demonstrating that NrF2 transcriptional activation overrides all other stimuli in hyperoxia.
Hyperoxia overcomes the inhibitory effect of inflammatory stimuli on the Rev-erbα promoter
To further understand how the Rev-erbα promoter is regulated when both oxidative and inflammatory stress was present, cells were pre-incubated with either TNFα or LPS, at the dose and duration seen to inhibit Rev-erbα promoter activity when used alone (Fig. 4A, B and Supplementary Fig. S6), and subsequently exposed to hyperoxia. Hyperoxia-induced promoter activity was diminished in the TNFα or LPS pre-treated cells after 6 h of exposure (Fig. 5E) but induction was observed with longer (total of 24 h) exposures in the pre-incubated cells (Fig. 5F), suggesting that the inhibitory effect of inflammatory stimuli was temporary and was superceded by the activating effect of oxidative stress.
Disruption of p50 alters the rhythmicity of Rev-erbα and other core circadian genes
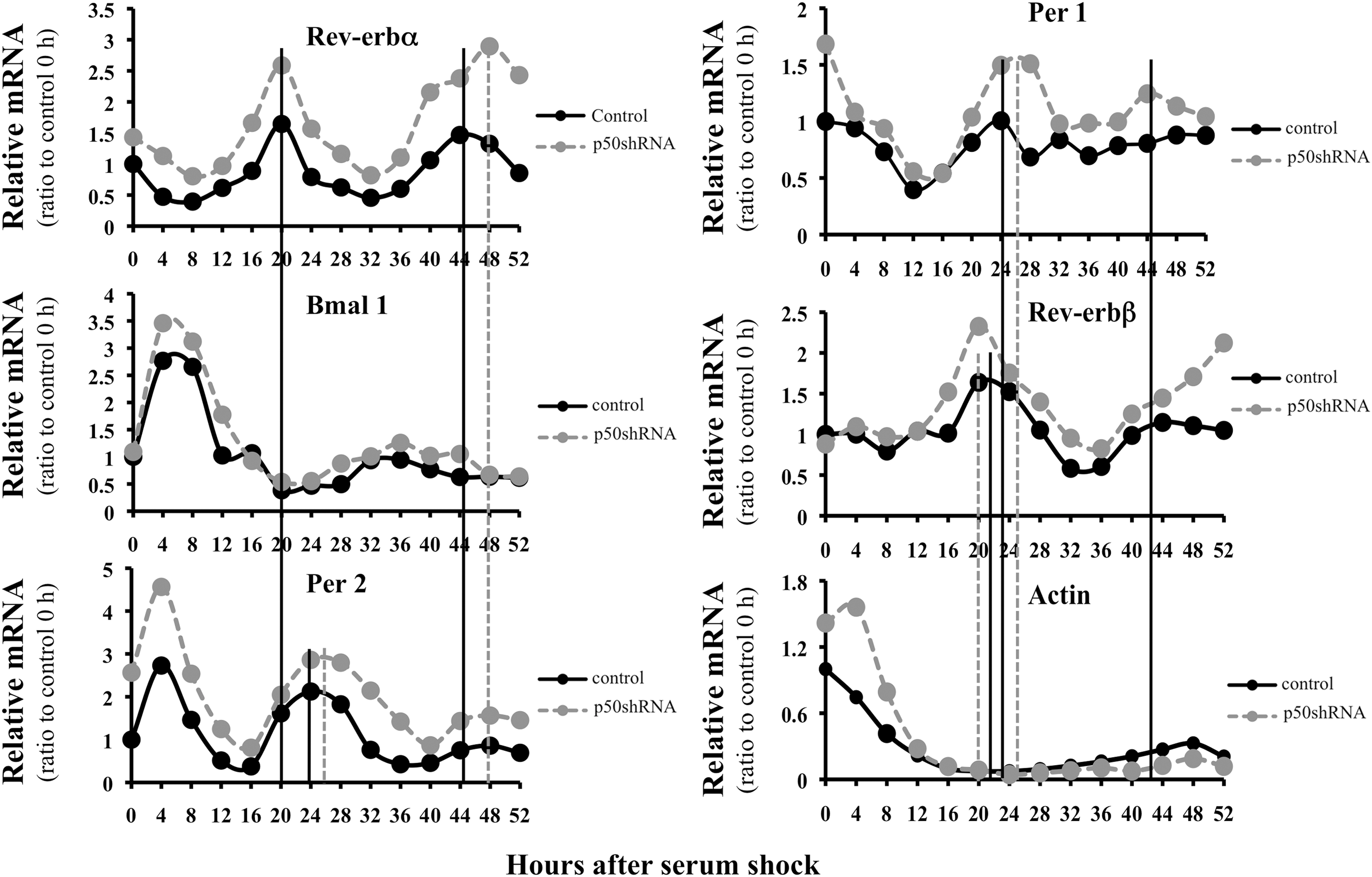
To understand whether the repressive effects of inflammation alter circadian rhythmicity of Rev-erbα, p50 shRNA-infected cells were subjected to serum shock to induce circadian rhythmicity. With disruption of p50, the amplitude of the rhythmic oscillations was increased in both circadian cycles and phase shifted during the second cycle (Fig. 6, upper left panel). Mild oscillations of Bmal 1 were observed and these were anti-phasic to Rev-erbα in air-exposed cells, suggesting that Rev-erbα represses Bmal 1 expression (middle left panel). However, Bmal 1 gene expression did not further decrease with p50 inhibition, suggesting that other factors regulate Bmal 1 suppression. In addition to altered Rev-erbα mRNA, the rhythmicity of Per 2 (lower left panel), Per 1, and Rev-erbβ gene expression (upper and middle right panels) showed either a phase-shift or an altered amplitude during the two circadian cycles. In contrast, the non-circadian gene actin showed no oscillations in both control and p50 shRNA cells (lower right panel). In addition, cells infected with p50 shRNA, where p50 mRNA was reduced by 50% showed no oscillations (Supplementary Fig. S8).
Verification of the inhibitory effect of NFκB on Rev-erbα transcription in vivo
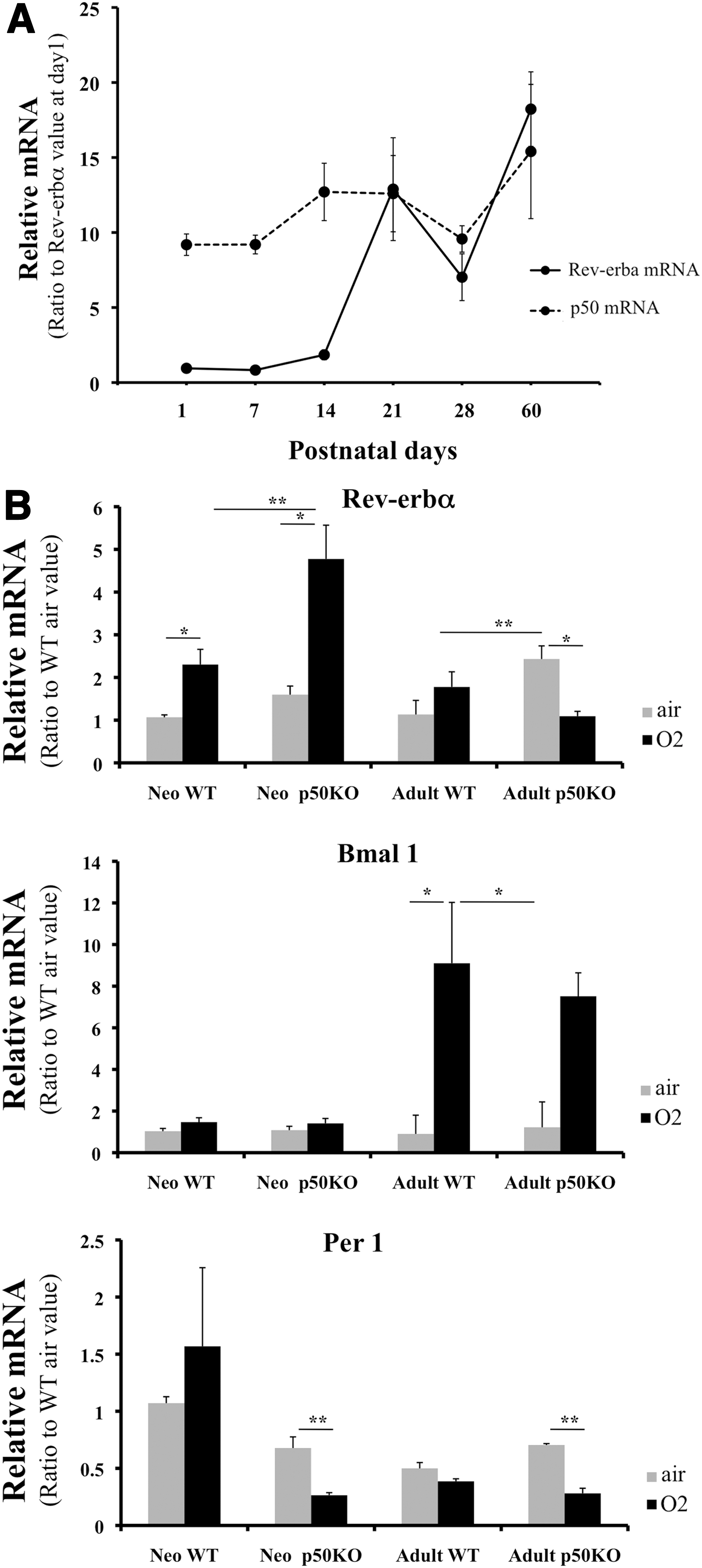
Inflammatory and oxidative stimuli often coexist in the clinical setting. To test whether the bimodal nature of Rev-erbα regulation was observable in vivo, WT and p50KO adult and neonatal mice were exposed to hyperoxia for 72 h. Disruption of p50 further enhanced hyperoxia-induced Rev-erbα mRNA in the neonates (Fig. 7B). This disinhibition of Rev-erbα mRNA correlated with a decrease in Per 1 mRNA, a downstream circadian gene. In contrast, no change in the mRNA of the circadian gene Bmal 1 was observed. In the adults, p50 disruption did not lead to enhanced hyperoxia-mediated Rev-erbα activation (Fig. 7B), suggesting that the hyperoxia response is developmentally regulated. Supporting this hypothesis, lung Rev-erbα mRNA was up-regulated and paralleled lung p50 mRNA levels until postnatal day 14 but not beyond (Fig. 7A), suggesting that the inhibitory effect of p50 on Rev-erbα is most prominent in early postnatal life.
Discussion
The influence of the circadian rhythm on the response to inflammation and oxidative stress has been shown in many biological and clinically relevant situations. As an example, TNFα expression in peripheral blood cells undergoes circadian rhythmicity after exposure to endotoxin (60). In addition, asthma is influenced by changes in pulmonary function that occur during sleep (44). The signaling mechanisms by which this occurs are not clearly understood. Conversely, it is not known whether oxidative stress and inflammation modulate rhythmicity of core circadian proteins, including Rev-erbα. In this article, we demonstrated direct effects of both oxidative stress and inflammation on the expression of the key circadian gene Rev-erbα. These effects were mediated via conserved NrF2- and NFκB-binding sites on a specific region of the Rev-erbα promoter.
Oxidative stress regulates gene expression predominantly via the transcription factor NrF2. In non-stimulated cells, NrF2 is associated with Keap-1 in the cytosol and is ubiquitinated by E3 ligase, which targets it for degradation (37). Redox-sensitive sulfhydryl (-SH) groups on Keap-1 can be oxidatively modified (63), leading to the release of NrF2 and enabling it to migrate to the nucleus where it dimerizes to small Maf proteins with subsequent activation of downstream gene expression [reviewed by Itoh et al. (34)]. Inflammation is predominantly regulated by NFκB signaling through the NFκB family proteins, which serve as dominant regulators of inducible gene expression in virtually all cell types. Five subunits of the NFκB family, Rel A (p65), p50, p52, RelB, and cRel, from which all transactivation components are formed as either hetero- or homo-dimers, regulate downstream gene transcription. In quiescent cells, NFκB remains sequestered in the cytoplasm bound to members of the I-κB family of inhibitory proteins. On stimulation, I-κB is degraded to release NFκB, which enables its translocation to the nucleus, binding to NFκB recognizing sequences, and transcription of downstream genes (32). How NrF2 and NFκB influence the regulation of the Rev-erbα promoter was explored here.
A binding site comprising the core AP-1 consensus sequence TGACTCA was identified on the Rev-erbα promoter. This site lacks the cytosine and guanine bases on either side of the extension that is required to classify this site as an NrF2 motif (23). However, this site was necessary for hyperoxia-induced Rev-erbα promoter activity, and when NrF2 was disrupted, hyperoxia-induced Rev-erbα transcription was abolished. In agreement with the fact that NrF2 activation requires modification of the -SH group on the Keap-1 protein, DL-sulforaphane, a known -SH modifier of Keap-1 (18), induced the Rev-erbα promoter activity in a dose-dependent manner. In addition, H2O2, another oxidative stress stimulus known to activate NrF2 (24), induced Rev-erbα promoter activity and mRNA levels in a dose-dependent manner. All of these lines of evidence support that NrF2 directly regulates Rev-erbα in oxidative stress.
As to inflammation, given the multiple NFκB-binding sequences on the Rev-erbα promoter, we expected that these sites would enhance Rev-erbα transcription when NFκB was activated. Surprisingly, when either TNFα or LPS was incubated with Mlg cells, Rev-erbα promoter activity or transcription was decreased. When these sequences were individually tested, all of them enhanced NFκB binding with TNFα. This would suggest that Rev-erbα is a direct target of NFκB. Indeed, an in silico analysis designed to study pancreatic cells under stress revealed that Rev-erbα was among hundreds of newly identified NFκB targets (49). How NFκB directs its downstream target gene expression may depend on several factors [reviewed Hayden and Ghosh (33)]. One such mechanism could be sequence-specific responses to p50/p50 (46) or p52/p52 (16) homodimers as a means of suppressing Rev-erbα transcriptional activation as well as that of its downstream genes. Indeed, the two distal NFκB binding sites on the Rev-erbα promoter, N4 and N5, have an identical sequence (GGGGCATCCCC) specific for p50/p50 homodimers. However, these sites do not increase occupancy of any tested NFκB subunit proteins when evaluated with a relatively preserved chromatin structure as with the ChIP assay. A sequence (GGGAGTCCC) able to preferentially bind p50 (28) was found between the N1 and N2 (N1-N2 NFκB). Although this sequence showed a moderate binding affinity to NFκB with TNFα stimulation (Fig. 4H), the site containing this sequence preferentially recruited p50 or p52 subunits and maintained basal p65 occupancy, suggesting that Rev-erbα transcription was regulated at a higher structural level rather than just through sequence recognition. Our data could not preclude the possibility of recruitment of p105 or p100, the precursor proteins of p50 or p52, which also have a repressor function for NFκB (59). Perhaps the involvement of these lager proteins is important in specific cell conditions as previously demonstrated (74). This needs to be further verified.
Another factor contributing to the decreased gene expression of NFκB targets is the recruitment of transcriptional corepressors such as the HDAC1 (22), BCL3, or I-κBα, as demonstrated in the regulation of TNFα (55), cyclin D1 (69), and PPARγ promoter (27), respectively. However, the lack of occupancy of HDAC1, BCL3, or I-κBα in the present model (Supplementary Fig. S7) excludes the possibility that these factors govern NFκB regulation of the Rev-erbα promoter.
The unique sequence of the N2 NFκB/NrF2 enables this site to respond to either oxidative stress or inflammatory signaling as depicted in Figure 8. It seems counterintuitive that the region containing this site has bimodal effects on Rev-erbα expression (i.e., up-regulation in oxidative stress and down-regulation in inflammation). Perhaps a higher-order regulation at the chromatin level is involved with NFκB regulation (50). Indeed, p50/p50 homodimers were found to bind to the nucleosome, a basic unit of chromatin that can inhibit transcription (2). To support this hypothesis, we demonstrated that the TNFα-mediated decrease of Rev-erbα promoter activity was abolished when p50 was disrupted (Fig. 5B). In addition, using a DNA-sequence based nucleosome prediction web software (35) (
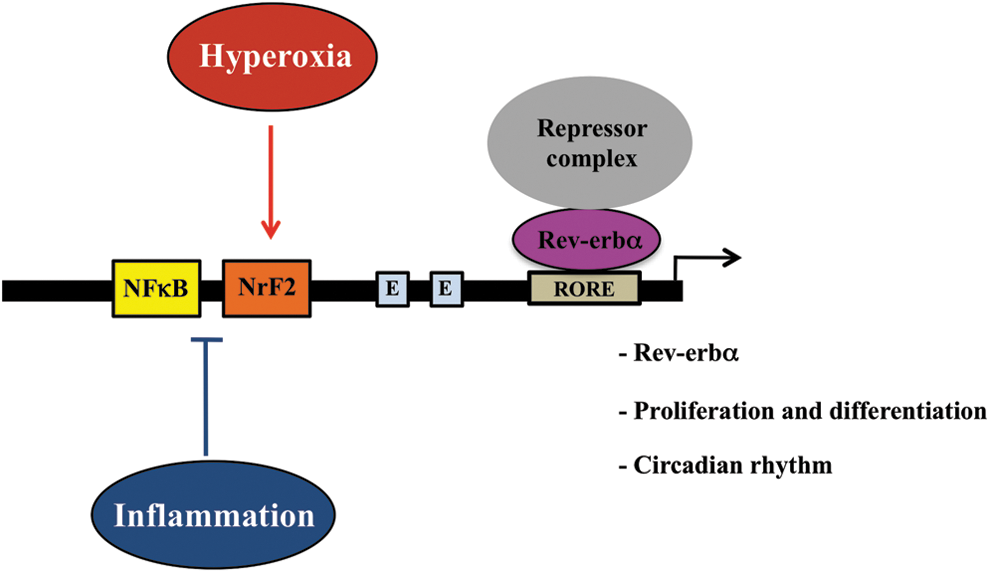
Diurnal gating of circadian output and correlation with oxidative defense have been reported in Drosophila (39). Other redox mechanisms such as peroxiredoxin integrate with the circadian system (61) and this is conserved in human, mice, Caenorhabditis elegans, and marine algae (21, 56). Our findings that oxidative stress and inflammatory stimuli also affect rhythmicity of Rev-erbα and other core circadian genes in mammalian cells substantiates that oxidative stress and inflammation can alter circadian regulation at the transcriptional level.
Hyperoxia enhances the production of reactive oxygen species (3, 12), leading to DNA strand breaks, chromosomal aberrations (5, 40), and inhibition of cell cycle progression in the lung (7, 54). In the neonatal period, this injury could have long-lasting consequences, because alveolarization continues for several weeks postnatally (48). Transcription factors such as AP-1 and NFκB are important in the neonatal lung's response to hyperoxia (71, 73). The reversal of the correlation between p50 and Rev-erbα mRNA expression between postnatal day 1 to 14 (Fig. 7A) suggests that there is a developmental window for NFκB-mediated Rev-erbα repression in the lung. Although we do not know what regulates this developmental expression pattern, we speculate that modulation of Rev-erbα in the neonatal lung by both oxidative stress and inflammation impacts postnatal lung repair after injury. This is partly supported by the fact that neonatal lung Rev-erbα mRNA is readily responsive to hyperoxia and is further up-regulated when NFκB is disrupted in contrast to the adult lung and that this is associated with altered Bmal 1 transcription. The latter is important in regulating oxidative stress and DNA damage responses, leading to cellular senescence in vivo (38). Overall, these data indicate that oxidative and inflammatory regulation of Rev-erbα and other circadian genes is physiologically relevant in vivo. This could result in the modulation of metabolic and homeostatic responses pertinent to hyperoxic lung injury. Thus, vulnerability to circadian dysregulation by external stimuli in the early postnatal period may be of clinical importance. Further in vivo delineation of these mechanisms is needed.
Materials and Methods
Animals and hyperoxia exposures
Null mutant mice for p50 were purchased from the Jackson Laboratory and cross-bred with c57Bl/6 mice for five generations. Littermates of WT and p50 KO neonatal (<12 h old) or adult mice (2-month-old) of mixed genders were exposed to either 21% oxygen or room air or 95% oxygen (O2) for 72 h as previously described (72). All procedures were reviewed and approved by the Animal Fair and Care Community of the Children's Hospital of Philadelphia.
Evaluation of circadian gene mRNA levels
Lung mRNA levels of Rev-erbα, Bmal 1, or Per 1 were determined using quantitative real-time PCR (qRT-PCR) as previously described (72).
Cell lines
Mlg cells
Neonatal mouse lung fibroblasts were obtained from American Type Culture Collection (Manassas, VA). Cells were maintained in DMEM medium with 10% FBS and 1% antibiotic–antimycotic (Gibco, Life Technologies, Grand Island, NY). Cells were discarded after 10 passages.
p50 and control shRNA cell lines
Five lentiviral-based p50 shRNA clones were purchased from Thermo Scientific (Waltham, MA). Based on the results of a selection process, one clone resulted in a 90% reduction of the p50 protein, and was chosen for generating viral particles that were used as infecting stocks to obtain a stable p50 knockdown cell line. Mlg cells were infected with the p50 shRNA stock or the empty viral vector and selected with puromycin to obtain stable cell lines.
WT and NrF2 KO MEF cells
These were a gift from Dr. Melpo Christofidou-Solomidou (University of Pennsylvania Medical Center, Philadelphia, PA).
Identification of putative NrF2- and NFκB-binding sites on promoter regions of mouse Rev-erbα
Putative NrF2 and NFκB binding sites 5000 bp upstream of the Rev-erbα promoter that are conserved between mouse and human were initially screened using a web-based DNA sequence analysis built on the Mouse July 2007 (mm9) assembly in alignment with the Human February 2009 (hg19) assembly (Whole genome RVista,
Generation of mouse Rev-erbα promoter luciferase reporter constructs
Mouse genomic DNA was extracted and used as a template to amplify the 0.9 and 2.5 kb regions upstream of the transcription initiation site. The resultant PCR products were ligated into a promoterless luciferase vector pGL4.10[luc2] (Promega, Madison, WI) to obtain the pGL4Nd0.9luc (0.9 kb luc) and pGL4Nd2.5luc (2.5 kb luc) plasmids. Subsequent linear deletions to exclude the N1 (pGL4Nd0.6luc or 0.6 kb luc) and both the N1 and N2 (pGL4Nd0.2luc or 0.2 kb luc) sites were generated using the 0.9 kb luc as a template. Targeted deletion mutants of N1 (pGL4Nd0.9lucΔN1) or N2 (pGL4Nd0.9lucΔN2) were achieved by a two-step cloning process using designed primers as described in Table 1. All constructs were verified by DNA sequencing.
Transfection and detection of luciferase activity
All constructs were transiently transfected into the Mlg cells using lipofectamine 2000 (Invitrogen, Grand Island, NY) and were grown on 6- or 12-well plates overnight. To control for the transfection efficiency of each construct, a Renilla vector (pRL-TK; Promega) was co-transfected into the Mlg cells. Luciferase activity was measured using either a live-cell imaging system as described earlier (43) or a luminometer using a dual luciferase system (Promega). The photons were normalized to cell numbers.
Models to generate oxidative stress
Hyperoxic exposure
Cells were exposed to hyperoxia (O2, 95% O2/5% CO2) or normoxia (air, 95% air/5% CO2) for 0–48 h using a C-Chamber (Biospherix, Redfield, NY).
Incubation with H2O2
Cells were incubated with 0–500 μM hydrogen peroxide (H2O2) for 1 or 4 h.
Incubation with DL-sulforaphane
Cells were incubated with 0–20 μM DL-sulforaphane for 12 h h.
Models to activate NFκB
Incubation with TNFα
Cells were incubated with 0–50 ng/ml TNFα for 0–24 h.
Incubation with LPS
Cells were incubated with 0–1 μg/ml LPS for 4 h.
Determination of cell viability and DNA damage
Trypan blue exclusion
Cells were washed, trypsinized, and centrifuged. The pellets were re-suspended in PBS and counted in a solution containing 0.2% trypan blue using a hemacytometer (Hausser Scientific, Horsham, PA). The percentage of cell death was calculated as the ratio of blue cells over totals.
Comet assay
This was performed according to the published method (5). Briefly, an aliquot of the cells was fixed with 1% agarose, incubated with a lysing solution, electrophoresed in a alkaline solution, and stained with Sybr gold (Trevigen, Gaithersburg, MD). Individual cell DNA was visualized under a Nikon E800 fluorescence microscope. Images were captured with a SPOT-RT digital camera. Metamorph analysis software was used to measure tail moment {(TM)=tail length×[DNA in tail/(total DNA in head+tail)]} in 50 cells. The mean value of the % DNA measurements in tail was then calculated.
Elucidation of binding regions and sequences of putative NFκB and NrF2 on the promoter
Chromatin immunoprecipitation
Primer sets selection for amplifying regions containing NrF2- and/or NFκB-binding sites on the Rev-erbα promoter was guided by basic principles for primer designs and synthesized by Integrated DNA Technologies, Inc. (Coralville, IA) (Table 2 and Fig. 1F). Live cells were cross-linked with 10% formaldehyde and shredded by sonication as described per manufacturer's instructions (ChIP assay kit, #17-295; Millipore, Temecula, CA). The antibodies used for IP or for western analysis were purchased as follows: NrF2 (C20, #sc722), I-κBα (C21, #sc-371), and Lamin B (C20, #sc6216) from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); ChIP grade p65 (#7970-1) and p50 (#7971) from Abcam (Cambridge, MA); HDAC1 (#06-720), normal rabbit IgG (#12-370), normal mouse IgG (#12-371), and BCL3 (MAB2350) from Millipore; p52 (#4882), HDAC2 (#5113), and HDAC3 (#3949) from Cell Signaling Technology (Boston, MA); and calnexin (pAb-ADI-SPA-865) from Enzo Life Sciences (Farmingdale, NY).
Preparation of nuclear extracts
This was performed according to published methods (42).
Electrophoretic mobility gel shift assay
The corresponding DNA oligomer sequences for NFκB and NrF2 binding sites (Table 3) were synthesized as duplexes by IDT. The quality of the duplex was verified by 2% agrose gel and labeled with gamma p32 ATP. EMSA assay was performed according to previous publications (42).
Obtained from Promega.
Underlines indicate mutated bases.
All oligonucleotides other than SP-1 were synthesized as duplexes by IDT technologies.
NFκB, nuclear factor kappa B; EMSA, electrophoretic mobility gel shift assay.
Verification of transcription of Rev-erbα and other core circadian genes
RT-PCR
Steady-state mRNA levels of Rev-erbα were assayed by qRT-PCR using the ABI Taqman gene expression system as describe earlier (72).
Generating rthymicity of Rev-erbα and other core circadian genes
Serum shock
Cells were seeded in 6 cm plates at 2.5×104 density for 3 days. The media were composed of 5% FBS in DMEM and 1% antibiotic/antimycotic. At the end of 3 days, the cells were exposed to either hyperoxia or normoxia for 12 h. Media were then replaced with 50% horse serum for 2 h. Subsequently, the media was replaced with 0.5% FBS and cells were collected at 1 h after the replacement and thereafter every 4 h for the 48 h period.
Statistical analysis
For a comparison between treatment groups, the null hypothesis is that there is no difference between the treatment means. This was tested by a single factor analysis of variance (ANOVA) for multiple groups or an unpaired t-test for two groups after testing for normal distribution (InStat 3; GraphPad Software, Inc., La Jolla, CA). Statistical significance was accepted to be p<0.05. A comparison between and within groups was determined by Tukey's method of multiple comparisons.
Footnotes
Acknowledgments
This work was funded by grants from National Institutes of Health (HL-58752 and HL-111907, P.A.D.). The authors thank Hayato Go, Rarinthip Supapannachart, Mekdes Yohannes, and Monica Williams for their assistance to collect the samples for the serum shock experiments. They also thank Fumihiko Namba for his technical expertise and assistance with the Comet assay. They thank Melpo Christofidou-Solomidou from the Department of Medicine at the University of Pennsylvania Medical Center for providing the NrF2 null mutant cells.
Author Disclosure Statement
All authors state that no competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.