
Abstract
Introduction
T
Recent studies have highlighted a fundamental role for tumor suppressor p53 in the formation and development of atherosclerosis (36). As a transcription factor, p53 controls the expression of its target gene glutathione peroxidase-1 (GPX1) (53), the most abundant isoform of the GPX family that detoxifies H2O2. The GPX family belongs to a sophisticated antioxidant enzyme defense system in the cells. In this system, superoxide dismutases (SODs) convert O2 •− into H2O2, whereas GPX and catalase convert H2O2 into water (28). Recent attention has focused on the role of GPX1 in H2O2 detoxification in endothelial cells (ECs) because catalase is reportedly not expressed or expressed at very low levels in these cells (32, 59). At physiological levels, p53 is required to maintain a normal basal level of GPX1 transcription (45). Suppression of p53 results in a significant decrease in basal GPX1 expression, which increases cellular ROS levels and subsequently leads to oxidative damage. Restoring physiological levels of p53 upregulates the antioxidant enzymes and decreases the ROS production (33, 45).
Free fatty acids (FFA)-induced oxidative stress causes cardiovascular diseases, yet molecular mechanisms remain undefined. Here, we showed, for the first time, that nuclear phosphatase and tensin homolog deleted on chromosome 10 (PTEN) plays a role in the transmission of signals of FFA and induction of endothelial oxidative stress. Identification of S6K as an upstream kinase for PTEN suggests a possible avenue for targeted therapy for hyperlipidemia-related cardiovascular diseases. Furthermore, our finding that S380 phosphorylation affects PTEN monoubiquitination via PTEN-USP7 interaction represents the first example of coordinated regulation of PTEN nuclear localization via phosphorylation/ubiquitination. Understanding the role of individual modification sites in PTEN will allow design of specific therapies.
Previous studies in our laboratory show that nuclear phosphatase and tensin homolog deleted on chromosome 10 (PTEN) plays an important role in maintaining high level of p53 acetylation and activates the p53 transcription activity (26). PTEN is a tumor suppressor gene localized to human chromosome 10q23.31, a genomic region frequently lost in glioblastoma and prostate cancer (27). PTEN classically converts phosphatidylinositol-3,4,5-trisphosphate (PIP3) in the cytoplasm to phosphatidylinositol-4,5-bisphosphate (PIP2), thereby directly antagonizing the activity of phosphatidylinositol 3-kinase (PI3K) (44). Despite its well-defined role in signaling at the plasma membrane, PTEN is found preferentially localized to the nucleus in differentiated or resting cells and to the cytoplasm in advanced tumor cells (9, 30). Thus, the regulation of PTEN's subcellular localization seems to be critical to its tumor-suppressing functions. Posttranslational modifications are believed to control PTEN tumor suppressor functions and nuclear localization (51). PTEN appears to be phosphorylated upon a cluster of serine and threonine residues in a highly acidic stretch of the C-terminal tail (3). In addition to phosphorylation, PTEN also undergoes ubiquitination through the neural-precursor-cell-expressed developmentally downregulated 4-1 (NEDD4-1) ligase and WW domain-containing protein 2 (WWP2) ligase (35, 54). In general, monoubiquitination has been implicated in nuclear import (54) and polyubiquitination in proteasome-mediated degradation (48). Monoubiquitination of PTEN can be reversed by the deubiquitinating enzyme herpesvirus-associated ubiquitin-specific protease (HAUSP/USP7) (50), demonstrating dynamic nature of the regulation. Despite extensive investigation, little is known about the regulation of PTEN nuclear export.
As the most abundant fatty acids present in human diet and serum is the saturated palmitic acid (PA, 16:0), we examined the effects of PA on GPX1 expression and ROS accumulation in human umbilical vein endothelial cells (HUVEC) and human aortic endothelial cells (HAEC). In addition, C57BL/6J mice fed with high-fat atherogenic diet (HFAD) as a model for high-fat diet-induced atherosclerosis were employed to confirm the related mechanism in vivo. Our results suggest that PA inhibits the p53 activity and GPX1 expression through a novel mechanism that is involved in mammalian target of rapamycin (mTOR)/S6K activation, PTEN phosphorylation and nuclear export, and faulty p53 acetylation and GPX1 expression. Consequently, our study provides new insight into the regulation of PTEN nuclear function and suggests a possible avenue for targeted therapy for hyperlipidemia-related cardiovascular diseases.
Results
PA treatment increases ROS accumulation via PTEN, p53, and GPX1
To investigate the role of GPX1 in PA-induced damage in ECs, we treated HUVEC with 0.4 mM PA, a concentration that mimics hyperlipidemia condition, for various time and assessed GPX1 protein and mRNA levels. In addition, the protein levels of p53, a transcription activator of GPX1, were also detected. As shown in Figure 1A and B, prolonged incubation (8–16 h) of HUVEC with PA caused significant decreases in both p53 and GPX1 levels. Significantly, intracellular H2O2 levels are also increased concurrent with decreased GPX1 protein levels (Fig. 1A, right), suggesting that PA-mediated GPX1 inhibition may lead to ROS accumulation in HUVEC. To better mimic vascular bed affected by high FFA, we assessed the effect of PA in HAEC and obtained similar results (Supplementary Fig. S1A; Supplementary Data are available online at
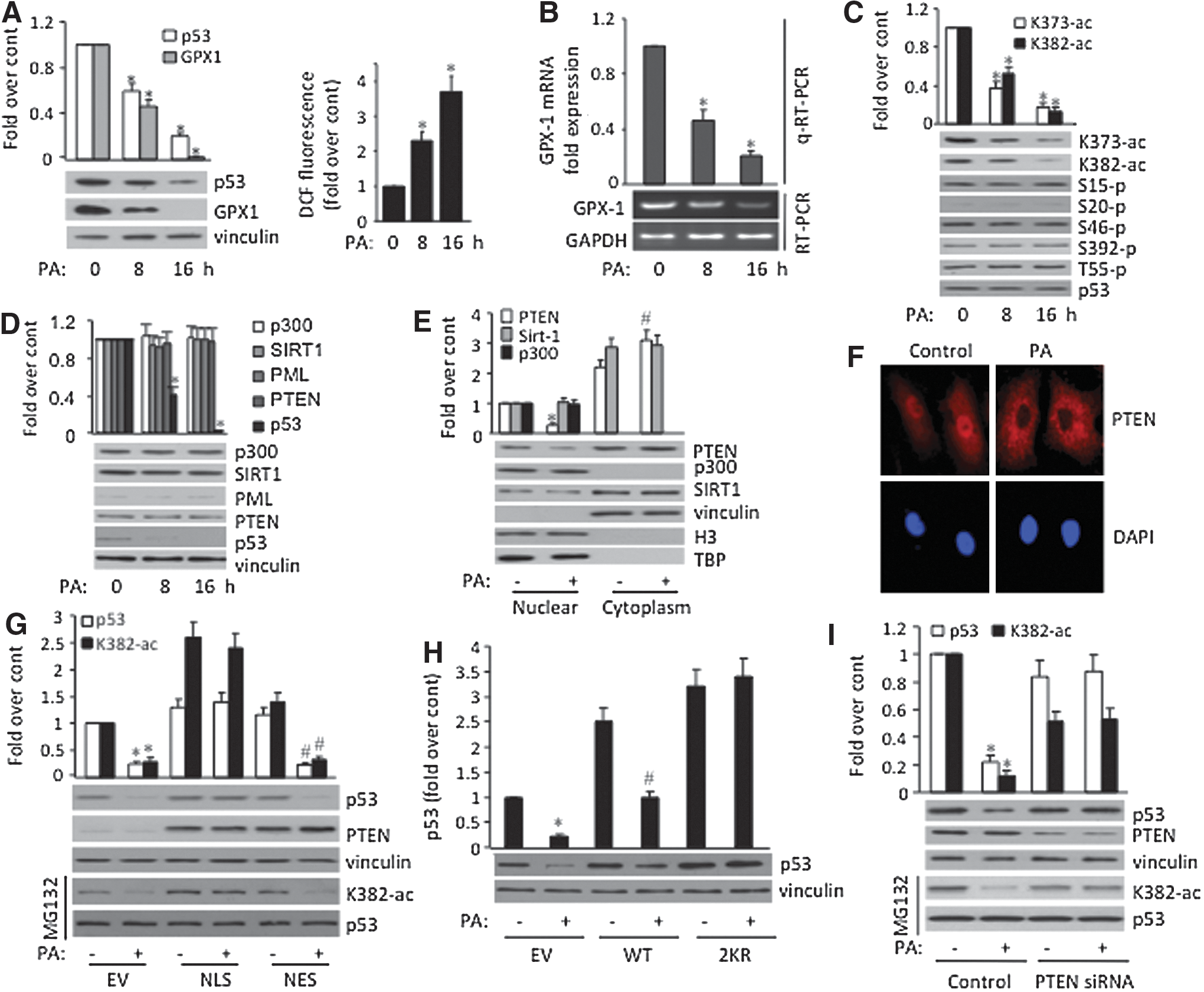
Because posttranslational modifications play an important role in the regulation of p53 activity (4), we assessed p53 posttranslational modifications upon PA exposure. Our data show that exposing cells with PA leads to a significant decrease in p53 acetylation at K373 and K382, whereas other tested modifications are unaffected (Fig. 1C). To identify mechanisms underlying the inhibition of p53 acetylation by PA, we examined levels of several factors that are known to regulate p53 acetylation, including acetyltransferase p300 (19), deacetylase SIRT1 (49), PML (43), and PTEN (26). To our surprise, at total protein levels, none of the proteins tested was affected upon PA treatment (Fig. 1D). Because our previous studies suggested that nuclear PTEN plays a role in maintaining high p53 acetylation (26), we tested whether PA treatment could alter subcellular distribution of PTEN. As shown in Figure 1E, subcellular fractionation indeed reveals reduced nuclear and increased cytoplasmic localization of PTEN upon PA treatment, where subcellular localization of p300 and SIRT1 was not affected. Importantly, immunostaining of PTEN also confirmed that PA treatment leads to PTEN cytoplasmic localization in HUVEC (Fig. 1F).
To verify the role of PTEN subcellular localization in PA-suppressed p53 acetylation, HUVEC were first introduced with PTEN expression plasmids tagged with either nuclear localization signal (NLS) or nucleus exclusion signal (NES). As illustrated in Figure 1G, nuclear-localized PTEN, but not cytoplasmic-localized PTEN, rescued PA-suppressed p53 acetylation and protein downregulation. To ensure that the p53 protein downregulation is mediated through p53 acetylation, ECs were transfected with wild-type (WT) p53 or the double acetylation mutant (K373/382R; 2KR). The assay shows that 2KR but not WT abates PA-induced p53 downregulation (Fig. 1H). Finally, we showed knock down of endogenous PTEN by siRNAs indeed abolished PA-suppressed acetylation and downregulation of endogenous p53 (Fig. 1I). Taken together, our results demonstrate that reduced PTEN nuclear localization and p53 acetylation mediate PA-induced p53 inhibition.
S6K phosphorylates PTEN at S380 and suppresses PTEN monoubiquitination in PA-treated cells
A recent study suggested that the activation of the mTOR/S6K cascade is involved in regulating subcellular localization of PTEN (34). To determine whether mTOR/S6K is responsible for PA-induced PTEN subcellular localization, we treated HUVEC with sodium salicylate (NaSal), a specific inhibitor of S6K, and observed that PA-induced PTEN nuclear export as well as inhibition of p53 acetylation and GPX1 expression were all blocked by the treatment (Fig. 2A). Furthermore, PA treatment activates S6K in the nucleus as indicated by S6K-T389 phosphorylation of nuclear S6K (Fig. 2B). Because PTEN S380 phosphorylation has been suggested to control its subcellular localization, we also examined this phosphorylation upon PA exposure (8, 44). The assay shows that, concurrent with nuclear S6K activation, increased PTEN-S380 phosphorylation in the nucleus was indeed observed after PA exposure (Fig. 2B). Importantly, inhibition of S6K by NaSal abolished the PA-induced PTEN phosphorylation (Fig. 2B). The data imply that, upon PA treatment and activation, S6K may directly phosphorylate PTEN at S380, which regulates PTEN subcellular localization.
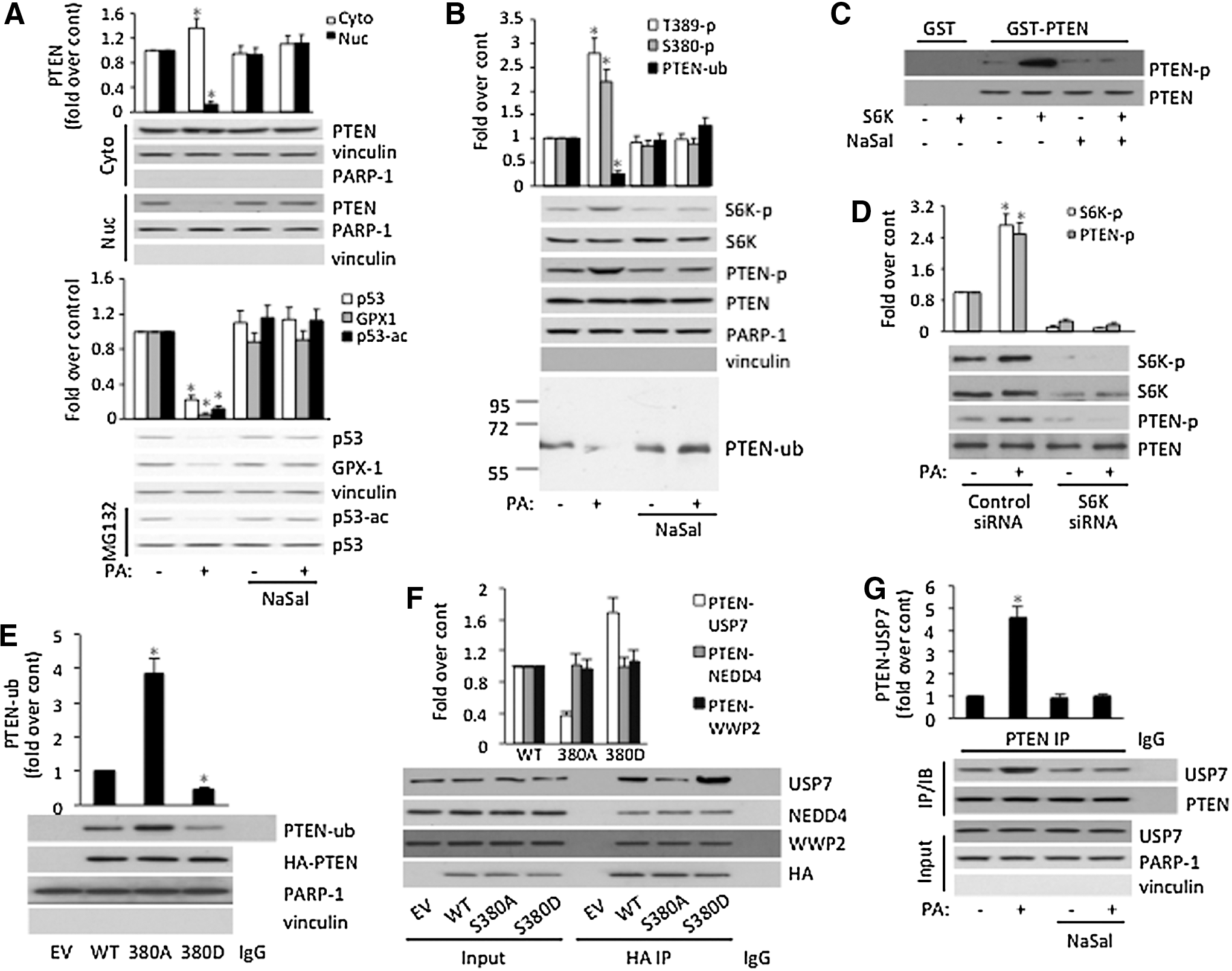
To test this possibility, we performed in vitro phosphorylation assay using purified PTEN and S6K proteins. As shown in Figure 2C, purified recombinant human S6K kinase indeed phosphorylates PTEN at S380 in vitro, and NaSal completely blocks this phosphorylation. To further verify the phosphorylation in vivo, we showed knock down of S6K by S6K-siRNA reduced PTEN phosphorylation after PA exposure (Fig. 2D). Thus, our data, for the first time, suggest that S6K directly phosphorylates PTEN at S380 in response to high FFA.
Because PTEN monoubiquitination is critical for its subcellular localization (54), we tested whether S380 phosphorylation affects PTEN monoubiquitination. As shown in Figure 2B, upon PA treatment and concurrent with appearance of PTEN S380 phosphorylation, PTEN monoubiquitination was reduced significantly. Furthermore, blocking PTEN S380 phosphorylation by NaSal rescued PA-induced reduction, indicating that PA may inhibit PTEN nuclear localization via S380 phosphorylation and monoubiquitination.
To test this directly, HUVEC were transfected with PTEN phosphorylation defect mutant S380A and phosphorylation mimic mutant S380D. As shown in Figure 2E, S380A undergoes more PTEN monoubiquitination than WT protein, whereas S380D shows lower level of PTEN monoubiquitination, suggesting that PTEN S380 phosphorylation suppresses monoubiquitination. Because PTEN ubiquitination is dynamically regulated by its E3 ligase NEDD4-1 (54, 56), WWP2 (35), and deubiquitinase HAUSP/USP7 (50), we tested whether S380 phosphorylation affects the interaction of PTEN with those proteins. As shown in Figure 2F, S380A displays decreased interaction with USP7, whereas S380D shows increased interaction with USP7 than with WT protein. In contrast, the interaction of PTEN with either NEDD4 or WWP2 was not affected by phosphorylation. To ensure PTEN S380 phosphorylation indeed affects endogenous PTEN-USP7 interaction, we showed treating cells with NaSal that inhibits S380 phosphorylation blocked PA-induced PTEN-USP7 interaction in HUVEC (Fig. 2G). As a control, endogenous PTEN-NEDD4 and PTEN-WWP2 interaction was not affected by PA treatment (Supplementary Fig. S2). Together, our data suggest that PA treatment activates S6K, which leads to the phosphorylation of PTEN at S380. Phosphorylation of PTEN in turn enhances PTEN-USP7 interaction, reducing PTEN monoubiquitination, and nuclear localization.
mTOR/S6K mediates PA-impaired PTEN nuclear localization and GPX1 expression
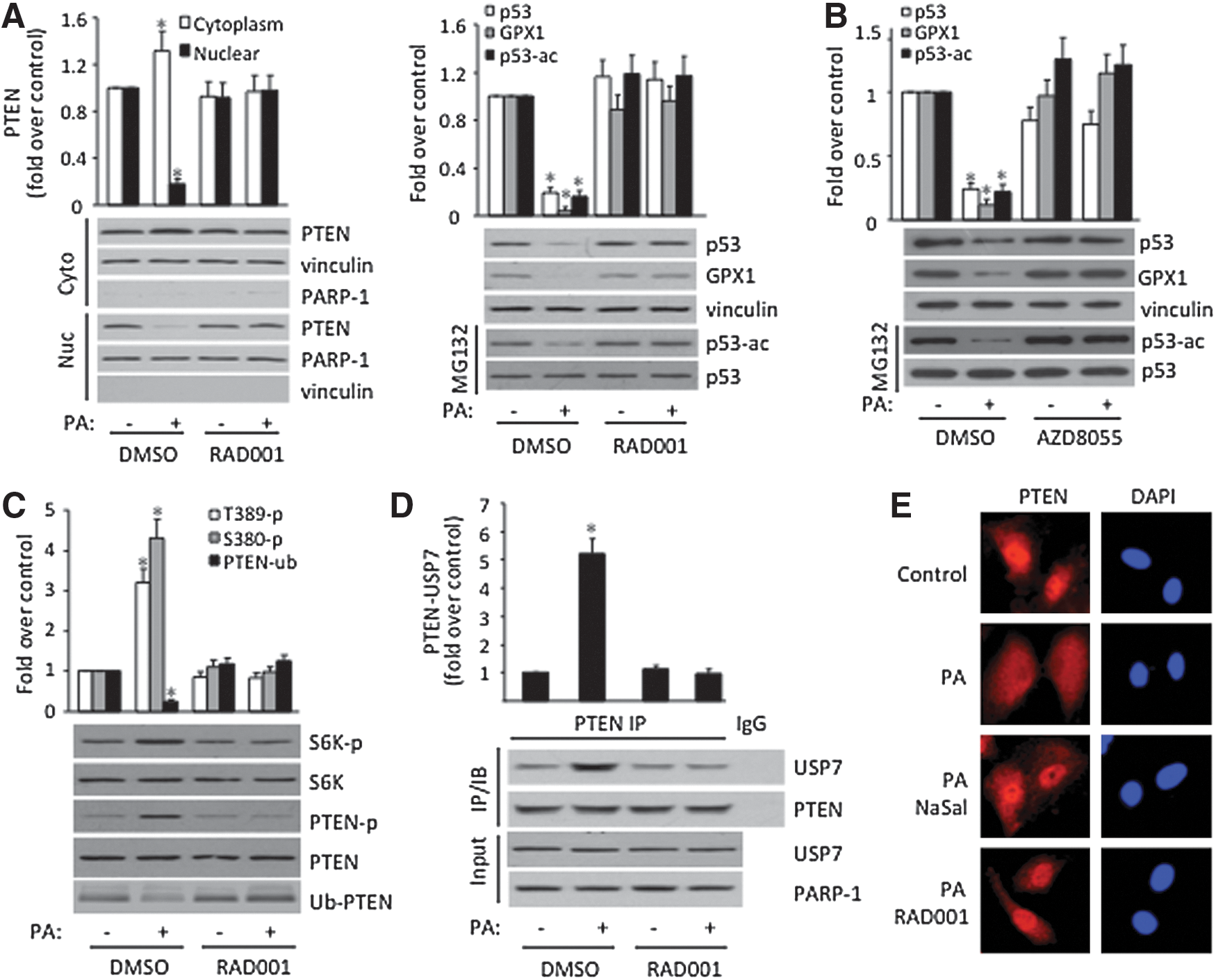
Next, we determined whether mTOR, an upstream kinase of S6K, plays a role in PA-induced PTEN nuclear export. As indicated in Figure 3A, inhibition of mTOR by RAD001, an mTOR inhibitor, abolishes PA-suppressed PTEN nuclear localization, p53 acetylation, and GPX1 expression in ECs. Similar effects were also observed when cells were treated with another mTOR inhibitor AZD8055 (Fig. 3B). These data indicate that, upon FFA treatment, mTOR become activated, which phosphorylates and activates S6K, leading to PTEN phosphorylation. To test this, we showed that PA-induced S6K T389 and PTEN S380 phosphorylation are indeed abolished by RAD001 (Fig. 3C). Importantly, PA-induced PTEN-USP7 interaction (Fig. 3D) and PA-suppressed PTEN monoubiquitination (Fig. 3C) are all eliminated upon RAD001 treatment, supporting the notion that mTOR/S6K mediates PA-impaired PTEN nuclear localization via regulating PTEN phosphorylation and monoubiquitination. Immunostaining with anti-PTEN antibody further confirmed that PA-induced PTEN cytoplasmic localization was blocked by RAD001 and NaSal (Fig. 3E). Finally, to further substantiate the physiological relevance of mTOR/S6K/PTEN pathway in vascular damage, we showed that PA treatment also leads to mTOR/S6K activation, PTEN S380 phosphorylation, and PTEN nuclear export in HAECs (Supplementary Fig. S3).
PA treatment impairs p53 binding to the GPX1 promoter and GPX1 expression
Because p53 acts as a DNA-bound transcription factor that regulates GPX1 (53), to substantiate the role of mTOR/S6K in cellular response to PA, we investigated whether PA treatment impairs p53 binding to the GPX1 promoter by chromatin immunoprecipitation (ChIP) assay. As shown in Figure 4A, PA exposure significantly inhibits p53 binding to GPX1 promoter. Furthermore, this inhibitory effect is blocked by NaSal and RAD001. To further verify the role of S6K in p53 DNA binding to the GPX1 promoter and GPX1 expression, we showed that knock down of S6K by S6K-siRNA also rescued the inhibition of p53 binding to GPX1 promoter (Fig. 4B). Importantly, PA treatment reduces GPX1 mRNA level, which was also rescued by NaSal, RAD001, and S6K-siRNA (Fig. 4C, D). Taken together, our results suggest that, in EC nuclei, mTOR/S6K is activated in response to PA treatment, which leads to PTEN S380 phosphorylation, PTEN deubiquitination, and nuclear export. The absence of PTEN in the nucleus, in turn, reduces p53 acetylation, transcriptional activity, and GPX1 expression.
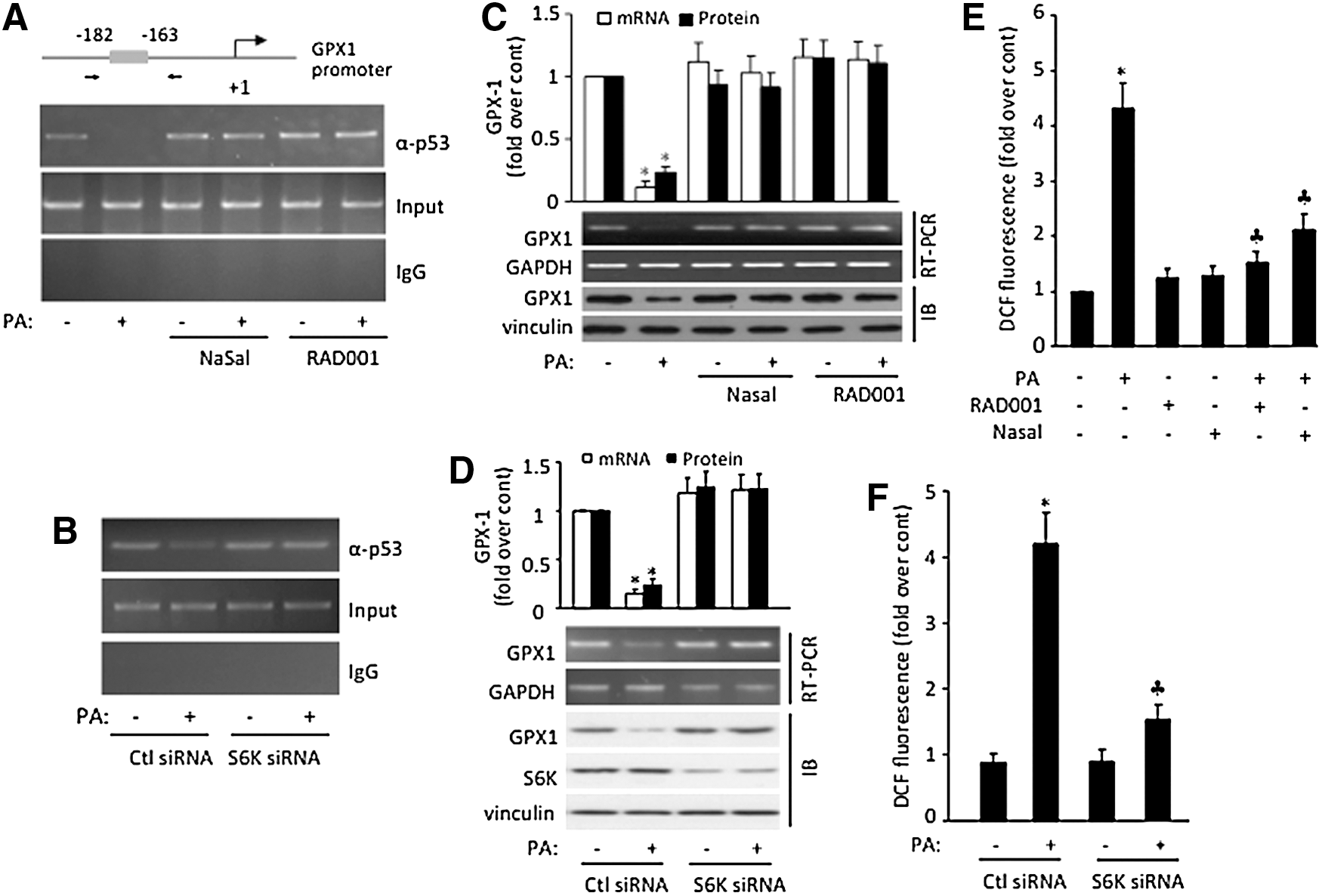
To substantiate the physiological role of mTOR/S6K pathway in cellular response to PA, we show that PA-induced ROS formation was partially blocked by either NaSal or RAD001, whereas treating cells with either RAD001 or NaSal alone has little effect on ROS accumulation (Fig. 4E). Similarly, PA-induced ROS formation was also partially blocked by the introduction of S6K-siRNA (Fig. 4F). Taken together, these results suggest that mTOR/S6K pathway is involved in PA-induced enzymatic antioxidant system impairment and endothelial oxidative stress.
PTEN-mediated GPX1 regulation plays a role in PA-suppressed nitric oxide bioavailability
Reduced nitric oxide (NO) bioavailability and increased ROS formation are major components of endothelial dysfunction (17). In addition, reduction in the NO activity occurs very early in metabolic syndrome, even before any structural changes in the vascular wall (6). To further elucidate physiological relevance of our findings in endothelial dysfunction, we detected intracellular generation of ROS and NO as an index of endothelial function in HAEC. PA exposure significantly increases ROS formation and decreases NO production in HAEC (Fig. 5A). Importantly, this effect was partially reversed by the addition of RAD001 and NaSal, supporting the role of mTOR/S6K pathway in increasing ROS and reducing NO upon PA exposure (Fig. 5A). As a control, the administration of polyethylene glycol catalase (PEG-catalase; a membrane-permeant analog of catalase) to the cell also alleviates PA-induced ROS accumulation. To ensure that the reduction of GPX1 levels indeed plays a role in those events, we showed that GPX1 overexpression partially rescued PA-induced ROS formation and NO reduction (Fig. 5B). Importantly, introduction of PTEN into the nucleus (NLS-PTEN) but not cytoplasm (NES-PTEN) also leads to increased GPX1 expression and partially rescues PA-induced ROS production and NO suppression (Fig. 5C), suggesting mTOR/S6K/PTEN/p53/GPX1 pathway, at least in part, plays an important role in endothelial oxidative damage by PA.
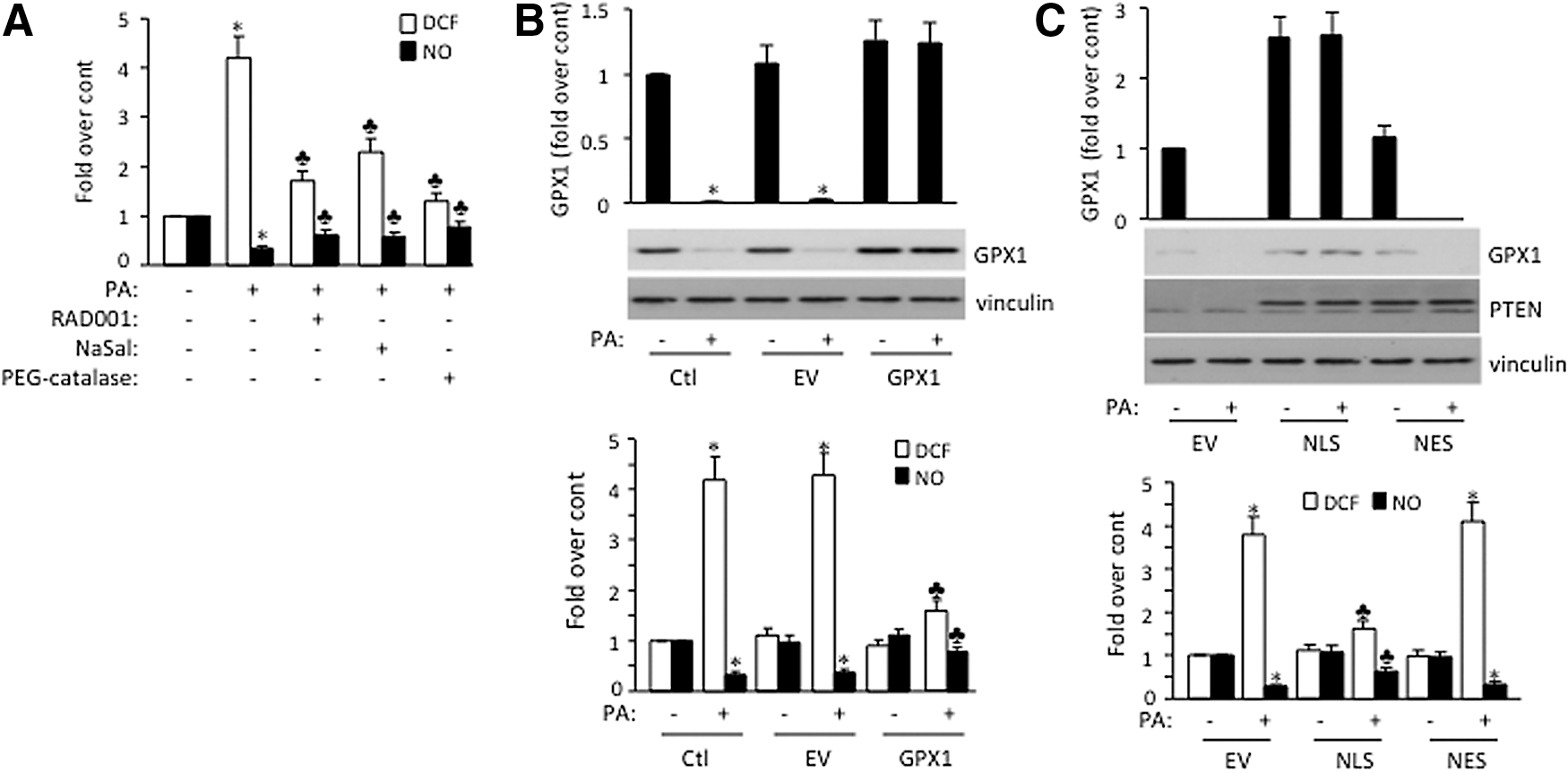
mTOR/S6K is involved in PA-induced aortic oxidative stress and lesions
We next determined if the mTOR/S6K/PTEN/p53/GPX pathway observed in cell culture could modulate vascular disease in the intact animals. C57BL/6 mice are susceptible to atherosclerotic lesion development when fed a separate high-fat diet containing cholesterol and bile acids (HFD atherogenic diet, HFAD). This mouse model closely simulates vascular diseases observed in human metabolic abnormalities (39). To determine whether HFAD feeding induces PTEN nuclear export, we assayed PTEN subcellular localization in aortas after 14 weeks of HFAD feeding and our data revealed reduced nuclear and increased cytoplasmic localization of PTEN compared to those fed a chow diet (Fig. 6A). To assess whether mTOR/S6K is responsible, we treated the mice with RAD001 and NaSal and observed that HFAD-induced PTEN nuclear export was completely blocked in aortas (Fig. 6A). Importantly, HFAD feeding also leads to decreased p53 and GPX1 protein expression in aortas, and this effect can be effectively blocked by RAD001 or NaSal administration (Fig. 6B). Accordingly, ROS formation is significantly increased in aortas of HFAD-fed mice compared to those fed a chow diet, and this inductive effect could be partially blocked by RAD001 or NaSal (Fig. 6C). As shown in Figure 6D, C57BL/6 mice fed with HFAD show a significant lesion in aorta root, which was inhibited by RAD001 (∼57%, *p<0.05) and NaSal (∼40%, *p<0.05). These results are consistent with previous studies in LDL−/− mice, which demonstrated that the mTOR inhibitor RAD001 prevents atherosclerosis despite severe hypercholesterolemia (38). Taken together, these data suggested that mTOR/S6K pathway, at least partially, mediates HFAD-induced oxidative stress and vascular damage via PTEN nuclear export, p53/GPX1 inhibition, and that the mTOR/S6K inhibitors attenuate hyperlipidemic vascular damage in vivo (Fig. 6E).
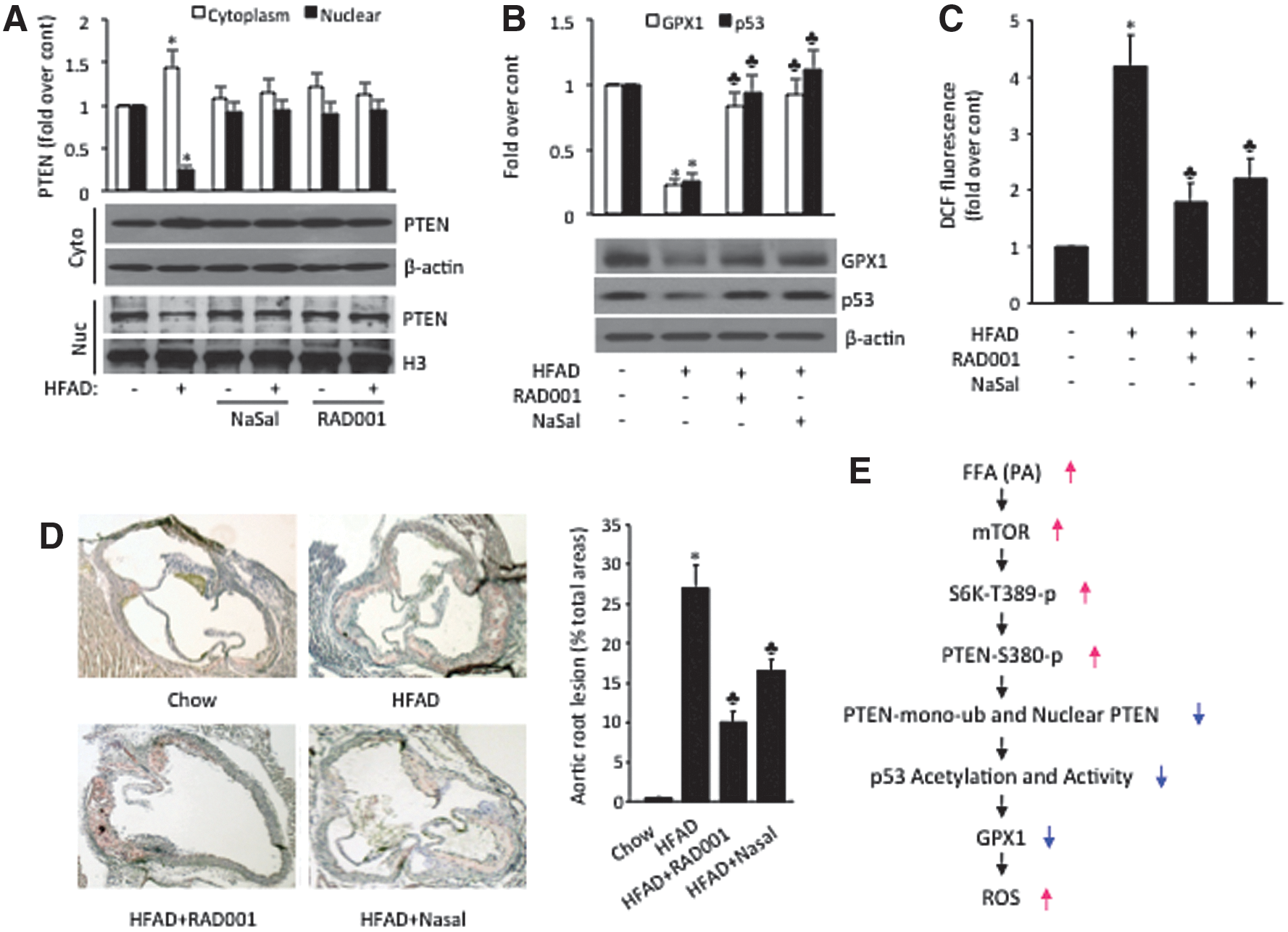
Discussion
Hyperlipidemia is an independent risk factor for the development of cardiovascular diseases. Clinical studies have also shown that inappropriate elevation of plasma FFA levels is associated with the impairment of endothelium-dependent vasodilation, high blood pressure, myocardial infarction, stroke, and sudden death (20). Despite its significance, the precise mechanism underlying FFA-induced endothelial oxidative stress remained elucidated. Here, we show that high FFA level activates mTOR/S6K that directly phosphorylates PTEN at S380 and affects PTEN nuclear function in both HUVEC and HAEC cells (Fig. 6E). The mTOR/S6K signaling is an ancient nutrient-sensing pathway. The importance of this pathway in the pathogenesis of human diseases has been underscored by the wide use of the rapamycins in a number of clinical settings, including, transplantation, restenosis, rheumatoid arthritis, type 2 diabetes, and most recently cancer (37). Our results that S6K phosphorylates and inactivates PTEN under high FFA conditions thus hold therapeutic potential for those diseases, particularly hyperlipidemia-related cardiovascular diseases and cancer. Furthermore, our finding that S380 phosphorylation affects PTEN monoubiquitination via PTEN-USP7 interaction represents the first example of coordinated regulation of PTEN nuclear localization and thus providing a new insight into the regulation of PTEN nuclear localization. Overall, our study defined a novel molecular mechanism by which high level of FFA causes ROS accumulation and oxidative damage through inactivation of nuclear PTEN function. Interestingly, the elevated level of ROS has also been reported to inactivate PTEN either directly by oxidizing its cysteine residue or indirectly by inducing its phosphorylation, resulting in the activation of the PI3K/Akt signaling cascade (21), suggesting the existence of multiple mechanisms for the inactivation of PTEN by ROS in the cell.
Oxidant stress is involved in the vascular effects of cardiovascular risk factors, including hyperlipidemia (1). The addition of PA has been shown to elevate ROS in cultured ECs through PKC-dependent activation of NADPH oxidase (18). Indeed, we have confirmed the role of NADPH oxidase in PA-induced ROS accumulation in HAEC cells (Supplementary Fig. S4). However, inhibition of NADPH oxidase only partially blocks PA-induced ROS accumulation in HAEC (Supplementary Fig. S4), and combination of the inhibition with PEG-catalase, a H2O2 scavenger that mimics GPX1 function, is required for complete blocking, suggesting that both increased upstream production and decreased downstream scavenging contribute PA-induced ROS elevation. Our finding that PA treatment leads to mTOR/S6K activation, PTEN phosphorylation, and inhibition of p53/GPX1, thus provides a new mechanism for decreased ROS scavenging system induced by PA. In any case, the elevated level of ROS and nitrogen species by the overabundance of FFA induces oxidative stress leading to faulty signal transduction and apoptosis of ECs, VSMC, and cardiomyocytes. The role of these species in FFA-mediated apoptotic cell death is relevant to the complications of metabolic syndrome such as cardiovascular disease. In addition, increased concentrations of ROS reduce bioactive nitric oxide (NO•), an important vasoprotective molecule, through chemical inactivation, forming toxic peroxynitrite, which in turn can uncouple eNOS to form a dysfunctional superoxide-generating enzyme that contributes further to oxidative stress leading to impairment of endothelium-dependent vascular relaxation and apoptosis. Endothelial dysfunction observed in conjunction with risk factors for cardiovascular diseases is characterized by enhanced inactivation or reduced synthesis of NO·, alone or in combination. Thus, a sophisticated antioxidant enzyme defense system is crucial for ECs to protect themselves from oxidative damage. GPX1 represents the key antioxidant enzyme in vascular ECs (10). GPX1-deficient mice have endothelial dysfunction and abnormal cardiac function after ischemia/reperfusion injury (11, 12). In addition, GPX1 is implicated in diabetes (15, 24) and cancer development (24). In clinical studies, GPX1 activity measurements have been found to be predictors of cardiovascular risk in coronary artery disease patients (46). Here, we demonstrate that FFA exposure dramatically suppresses GPX1 expression by inhibiting its transcription factor p53 (Fig. 1). Moreover, we show that GPX1 plays an essential role in preserving endothelial function and NO bioavailability (Fig. 5). This may be attributed to the antioxidant actions of GPX1 in removing intracellular H2O2, which can induce eNOS uncoupling (7) and augment superoxide production by stimulating NADPH oxidase activation (29). Together, our results reveal that impairment of antioxidant system GPX1 plays an important role in FFA-induced endothelial oxidative damage. These findings yield insights into the biological importance of p53/GPX1 in mediating the pro-atherosclerotic effects of FFA condition in metabolic syndrome. The characterization of molecular mechanisms involved in the FFA-induced oxidative stress in endothelium should provide an avenue for future use of PTEN, p53, and GPX1 as potential targets of therapy for cardiovascular diseases.
Materials and Methods
Materials, cell culture, and plasmid construction
Antibodies used in this study and their commercial sources are as follows: anti-GPX1, anti-p53-S15-phos, anti-p53-S20-phos, anti-p53-S46-phos, anti-p53-K373Ac, anti-p53-K382Ac, anti-PTEN, anti-PTEN-S380-phos, anti-S6K, anti-S6K-T389-phos, and anti-HAUSP antibodies were from Cell Signaling Technology; anti-p53 (DO1), anti-p53-S392-phos, anti-PML, anti-p300, anti-SIRT1-1, anti-H3, anti-TBP, anti-Nedd4-1, anti-PAPR-1, and anti-ubiquitin antibodies from Santa Cruz Biotechnology; anti-myc (Ab910B) antibody from Abcam, and anti-vinculin antibody from Sigma. Anti-TAF1 and anti-p53-T55-Phos antibodies were home generated. All protein modifications were detected after normalization of protein levels. S6K inhibitor NaSal (10 mM), mTOR inhibitor RAD001 (20 nM), TSA (0.5 μM), leptomycin B (LMB, 15 ng/ml), PA, PEG-catalase (500 U/ml), and bovine serum albumin (BSA) were purchased from Sigma. AZD8055 (150 nM) was purchased from Selleck Chemicals. PA was added into the cell culture medium as PA-BSA complex as described previously (58). Control cells were incubated with fatty acid-free BSA. The proteasome inhibitor MG132 was purchased from A.G Scientific, Inc. S6K and mTOR inhibitors were added to cells 30 min before PA. LMB was added simultaneously with PA. PTEN or S6K siRNA was transfected into cells 24 h before PA.
HAEC and HUVEC were maintained in EGM-2 and EGM-MV (Lonza) at 37°C, and passages 2 to 5 were used for all experiments.
For constructing pcDNA3.1/GPX1, RNA extracted from HUVEC was used to generate GPX1 cDNA using the SuperScript™ One-Step reverse transcription–polymerase chain reaction (RT-PCR) with Platinum Taq kit from Invitrogen. Gene specific primers (5′-ACTGTAGGATCCCAGTTAAAAGGAGGCGCCTGCTGGCCT-3′; 5′-TGACATTCTAGAAGTGGGGAAACTCGCCTTGGTCTGGCA-3′) were designed based on the human GPX1 sequence (GenBank accession number NM_000581.2), including a portion of the 5′UTR and most of the 3′UTR including selenocysteine-insertion sequence (SECIS) motif (2) and incorporated BamHI and XbaI for cloning.
Animals and diets
Female C57BL/6J mice, 6–8 weeks of age, originally were obtained from Jackson Laboratories. This strain is highly susceptible to diet-induced atherosclerosis (39). The animals were maintained in a temperature-controlled room (25°C) on a strict 12-h light–dark cycle and were given free access to food and water. Mice were fed rodent chow pellets for 1 week before initiation of diet studies.
Mice were either fed with rodent chow or with a HFAD known to elicit fatty streak lesions in the aortas of susceptible mice (31, 39, 40). The rodent chow diet contained ∼4% fat (by weight), 24% protein, and 4.5% crude fiber (47). The HFAD, which has been described in detail (41), was a mixture of rodent chow and other ingredients resulting in ∼15% fat (∼30% of calories, primarily cocoa butter) with 1.25% cholesterol and 0.5% sodium cholate. Mice were fed rodent chow or atherogenic diet for 14 weeks, as noted. Two weeks after the start of HFAD feeding, the mice received solvent or RAD001 at 1 mg/kg or NaSal at 2 mg/kg per body weight per day by subcutaneously implanted osmotic minipumps for the study period of 12 weeks. Food intake and body weight were measured once a week. At the end of experiment, the mice were euthanized by CO2 inhalation. Mouse aortas were freshly prepared and immersed in iced Tris buffer (5 mM, pH 7.4) containing the protease inhibitors leupeptin, benzamidine, aprotinin, PMSF, and antipain (10 μg/ml). The tissues were homogenized for 30 s in a Polytron homogenizer. The homogenates were then centrifuged for 10 min at 100 g to remove particulate matter and unbroken cells. Western blot analysis to compare p53 and GPX1 expression in normal and HFAD-fed mice were performed with the corresponding antibodies.
Quantification of atherosclerotic fatty streak lesions was carried out by the evaluation of lesion size in the aortic root as described (41, 47) with modifications (22). Briefly, after the mice were killed, the freshly removed hearts were snap-frozen in the OCT-embedding medium (Miles) and sequentially cut into a total of 60 (6-μm-thick) sections around the aortic sinus as reported previously (41). Every third section throughout the aortic sinus (400 μm) was taken for analysis. The sections were evaluated for fatty streak lesions following lipid staining with oil red O and nuclei staining with hematoxylin. Lesion areas per section were quantified by capturing images directly from an RGB camera attached to a light microscope (Axioscope; Karl Zeiss) and displayed on a Trinitron RGB monitor. Area measurements were analyzed with the use of KS400 software (Karl Zeiss Vision).
Nuclear/cytoplasmic fractionation
Subcellular fractionation was performed as described (5). Briefly, one 100 mm-plate of HUVEC were washed with PBS and resuspended in 350 μl of 1:5 diluted buffer A (50 mM Hepes [pH 7.4], 1 mM EDTA, 10 mM mannitol, 1 mM DTT, 2 μg/ml aprotinin, 2 μg/ml leupeptin, and 1 mM PMSF). After incubation on ice for 10 min, cells were homogenized with 25 G needle for 10 strokes. After brief centrifugation at 6000 g at 4°C, the supernatant (cytoplasmic fraction) was collected. The pellet was washed with buffer A and then resuspended in 350 μl of L-buffer (50 mM Tris [pH 8.0], 120 mM NaCl, 0.5% Nonident P-40, 1 mM DTT, 2 μg/ml aprotinin, 2 μg/ml leupeptin, and 1 mM PMSF). After centrifugation, the supernatant (nuclear fraction) was collected. Nuclear and cytoplasmic fractions were assessed by immunoblotting of histone H3, TBP, or vinculin, respectively. Nuclear and cytoplasmic fractions from fresh aortas were prepared with a NE-PER* Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific).
Immunoprecipitation, immunoblotting, and immunostaining
Whole-cell extract was prepared by lysing cells in lysis buffer containing 20 mM Tris-HCl (pH 7.9), 150 mM NaCl, 0.5% NP-40, 20% glycerol, 2 mM EDTA, 0.5 mM DTT, 2 mg/ml aprotinin, 2 mg/ml leupeptin, and 0.5 mM PMSF. For protein–protein interaction assay, cell lysates were immunoprecipitated with 1 μg anti-PTEN or anti-HA antibody. The amounts of USP7, NEDD4-1 and WWP2 in the immunoprecipitates were determined by anti-HAUSP, anti-NEDD4-1 and anti-WWP2 antibody, respectively. For detecting p53 T55 phosphorylation, the cell lysate was immunoprecipitated with anti-T55-Phos antibody and immunoblotted with anti-p53 (DO-1) antibody. To normalize the p53 protein levels, MG132 (20 μM; Calbiochem) was applied 6 h before cell harvesting. For the PTEN ubiquitination assay, HUVEC were transfected with Myc-ubiquitin and then treated with PA for 16 h in the presence or absence of NaSal or RAD001. The nuclear extract was subjected to immunoprecipitation with anti-myc antibody, followed by immunoblotting with anti-PTEN antibody.
For immunostaining, HUVEC were fixed with 3% formaldehyde for 20 min, permeabilized with 0.5% Triton X-100 for 10 min, and preblocked with PBS containing 3% BSA for at least 1 h. The cells were incubated with anti-PTEN antibody at 4°C overnight and then incubated with the secondary antibody conjugated with Alexa fluor 568 (Invitrogen) for 2 h. After incubation, the cells were mounted with mounting solution containing 1 μg/ml DAPI. Images were obtained with a Nikon E-800 fluorescence microscope.
In vitro phosphorylation assay
In vitro phosphorylation assay for PTEN was performed using bacterially expressed purified PTEN (14) and baculovirus expressed and purified human S6K protein (1-421, T412E active; Millipore) in a phosphorylation buffer containing 20 mM Hepes, pH 7.8, 20 mM MgCl2, 20 mM glycerophsphate, 1 mM DTT, 100 μM ATP, 1 mM EDTA, and 1 mM PMSF. The phosphorylation reaction was resolved by 10% SDS-PAGE, and the phosphorylation of PTEN was assayed by immunoblotting with anti-PTEN-phospho-S380 antibody. The GST-PTEN proteins were detected by immunoblotting with anti-PTEN antibody (Cell Signaling) after stripping.
Detection of ROS
To detect the generation of intracellular ROS, the ROS-sensitive fluorescent indicator 2′,7′-dihydrodichlorofluorescin diacetate (DCFDA; Molecular Probes) was used in HUVEC or HAEC according to the manufacturer's protocol and as described previously (55). Confluent HUVEC in 96-well plates were preincubated with the fluorescence probe DCFDA (10 μM) for 30 min. After removal of the medium from wells, cells were washed three times in PBS, followed by the measurement of fluorescence intensity at 485-nm excitation and 538-nm emission spectra with a fluorescence microplate reader. Data are presented as the fold of increase in DCF fluorescence compared with that in control cells.
Detection of NO release
NO quantification was performed using the NO-specific fluorescent dye 4,5-diaminofluorescein diacetate (DAF-2 DA; Cayman Chemical) as described previously (13). Briefly, HAEC were seeded in 96-well plates (3×104 cells/well), cultured for 24 h, and then serum starved for 2 h in phenol red-free EBM supplemented with L-arginine (100 μM) and uric acid (100 μM). Cells were then loaded with DAF-2DA (5 μM final concentration) for 10 min at 37°C. In some experiments, RAD001, NaSal, or PEG-catalase was added to the media before PA treatment. After being loaded with DAF-2 DA, cells were washed three times with EBM at 37°C and kept in the dark. The fluorescence intensity was measured with a multilabel plate reader Wallac 1420 VICTOR2 (PerkinElmer) using 485 nm as excitation and 535 nm as emission wavelengths. The fluorescence intensities were corrected by subtracting the nonspecific fluorescence in wells without the addition of DAF-2 DA and in wells without cells.
Transfection and siRNA gene silencing
Cell transfection was performed using TransPass HUVEC Transfection Reagent (New England Biolabs) with 1 μg PTEN expression vector (NLS-PTEN or NES-PTEN), 1 μg HA-PTEN (HA-WT-PTEN, HA-S380A-PTEN or HA-S380D-PTEN), 1 μg p53 expression vector (pcDNA3-p53-WT or pcDNA3-p53-2KR), or empty vector. For PTEN and S6K knock down, HUVEC were transfected with human-specific PTEN siRNA (5′-AAGAUCUUGACCAAUGGCUtt-3′), S6K siRNA (5′-GUGCCAAUCAGGUCUUUCU-3′), or scrambled siRNA (Cell Signaling, Inc.) for 48 h using BioT transfection reagent (BiolandScientific LLC) according to the manufacturer's instructions.
ChIP analysis
ChIP analysis was carried out as described previously (25). Primers for PCR, which amplify a putative p53-binding site (−182 to −163) on the GPX1 promote, are as follows: 5′-CCTAACTCAGGAACCTCTGAGAAA-3′ and 5′-CAGGAAAAGGCTGGAGAGTG-3′. The PCR products were electrophoresed by agarose gels and visualized by ethidium bromide.
RT-PCR and real-time quantitative RT-PCR
Total RNA was extracted using the TRIzol reagent (Sigma), and RT-PCR was performed using SuperScript One-Step RT-PCR kit (Invitrogen) according to the manufacturer's protocol. One set of primers was designed to amplify GPX1 mRNA (5′-TCCCTCTGAGGCACCACGGTC-3′ and 5′-TTGGCGTTCTCCTGATGCCCAAAC-3′). Another set of primers for GAPDH (5′-AGGTGAAGGTCGGAGTCAAC-3′ and 5′-GACAAGCTTCCCGTTCTCAG-3′) was used as a control. Real-time quantitative RT-PCR was performed in a 96-well format in the Bio-Rad CFX96 Real Time System (Bio-Rad Laboratories). The real-time PCR mixtures consisted of 2 μl cDNA, 10 μM forwarded primer, 10 μM reversed primer, and 10 μl iQ SYBR Green Supermix (Bio-Rad Laboratories) in a final volume of 20 μl. The thermal profile of the RT-PCR procedure is as follows: 95°C for 2 min; 39 cycles of 95°C for 15 s; 55°C for 15 s; 72°C for 40 s; and 4°C. Dissociation curve consists of 10 s of incubation at 95°C, 5 s of incubation at 65°C, a ramp up to 95°C. Melting curves were used to validate product specificity. All samples were amplified in triplicates from the same total RNA preparation and the mean value was used for further analysis.
Statistical analysis
Values are presented as mean±SD. Differences between experimental groups were determined by one-way analysis of variance (ANOVA), followed by Student t-test as appropriate. All results obtained from the time-course studies were analyzed with repeated-measures ANOVA. Values of P<0.05 were considered statistically significant.
Footnotes
Acknowledgments
We are grateful to Dr. John Shyy for valuable suggestions and helpful discussion. We thank all members of our laboratory for discussions. This work was supported by NIH Grant CA075180 to X.L. and by American Heart Association Award POST3530033 to Y.W.
Author Disclosure Statement
No competing financial interests exist for any of the authors.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.