
Abstract
Introduction
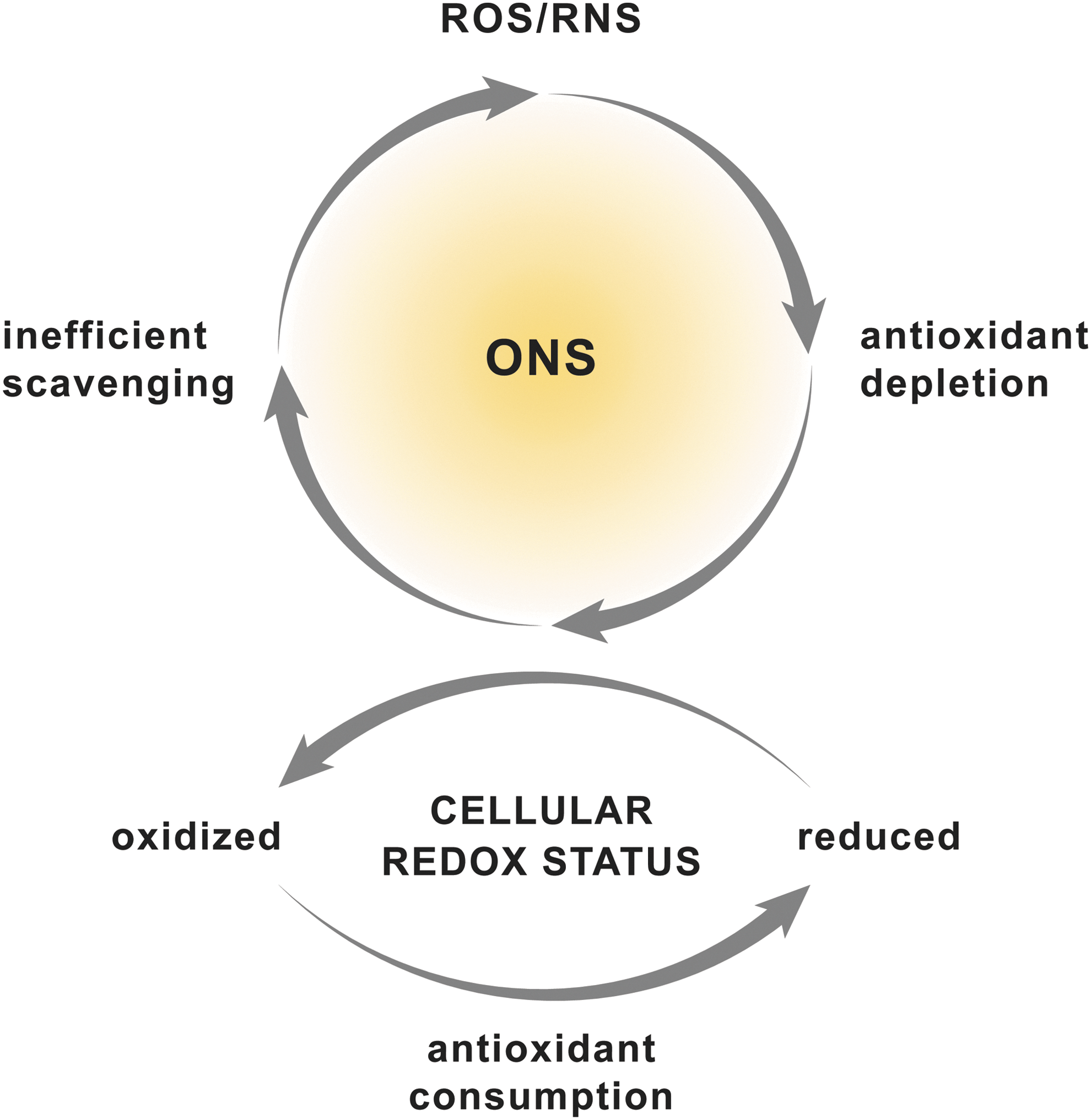
O
The liver is particularly prone to ONS due to its high metabolic rate and because hepatocytes are rich in ROS/RNS-producing mitochondria (section “Mitochondria”), cytochrome P450 (CYP) enzymes (section “Extramitochondrial FA oxidation”), and inducible nitric oxide synthase (iNOS, section “Reactive nitrogen species”) (92). Consequently, many hepatopathologies are associated with ONS, including viral hepatitis (112), alcoholic and non-alcoholic fatty liver disease (NAFLD) (121), cholestasis (33), and ischemia-reperfusion (IR) injury, the inevitable side effect of liver surgery performed under vascular exclusion (warm IR, e.g., liver resection) or liver transplantation (cold IR) (94).
In particular, much attention has been focused on the role of ONS in the interplay between NAFLD and IR, as mounting evidence points to an exacerbation of IR injury in fatty livers (208). Patients with NAFLD who undergo a liver resection have an increased risk of postoperative morbidity and mortality (131, 170). In the transplantation setting, steatotic grafts are commonly considered ineligible for transplantation because of the high risk of primary malfunction (128).
While the ONS-mediated processes that dictate NAFLD (177) and IR injury (205) are well documented, an integrative overview of the role of ROS/RNS in the interplay between these conditions is lacking. Nevertheless, a substantial amount of clinical evidence implies that the development of modalities to preserve fatty livers during IR bears great necessity (208). Consequently, this review provides an in-depth account of how the hepatocellular formation of ROS/RNS and the resulting ONS exacerbate the extent of IR injury in fatty livers.
Although the immune response and the extracellular component of hepatic ONS are critical contributors to both conditions, they will be only briefly addressed because these topics have recently been reviewed in the context of NAFLD (14) and IR injury (206).
Non-alcoholic fatty liver disease
NAFLD is characterized by the presence of vesicular fat in hepatocytes (i.e., steatosis) in the absence of excessive alcohol consumption and constitutes the common denominator in non-alcoholic fatty liver (NAFL), non-alcoholic steatohepatitis (NASH), as well as cryptogenic- or NASH cirrhosis (26). At a cellular level, the key feature of NAFLD is the vesicular accumulation of triglycerides (19) (Fig. 2), the ‘first hit’ in the putative two-hit theory (40). From here on, several ‘second hits’ evoke and amplify an inflammatory response that drives the gradual progression from NAFL to NASH (47) (Fig. 2, inset).
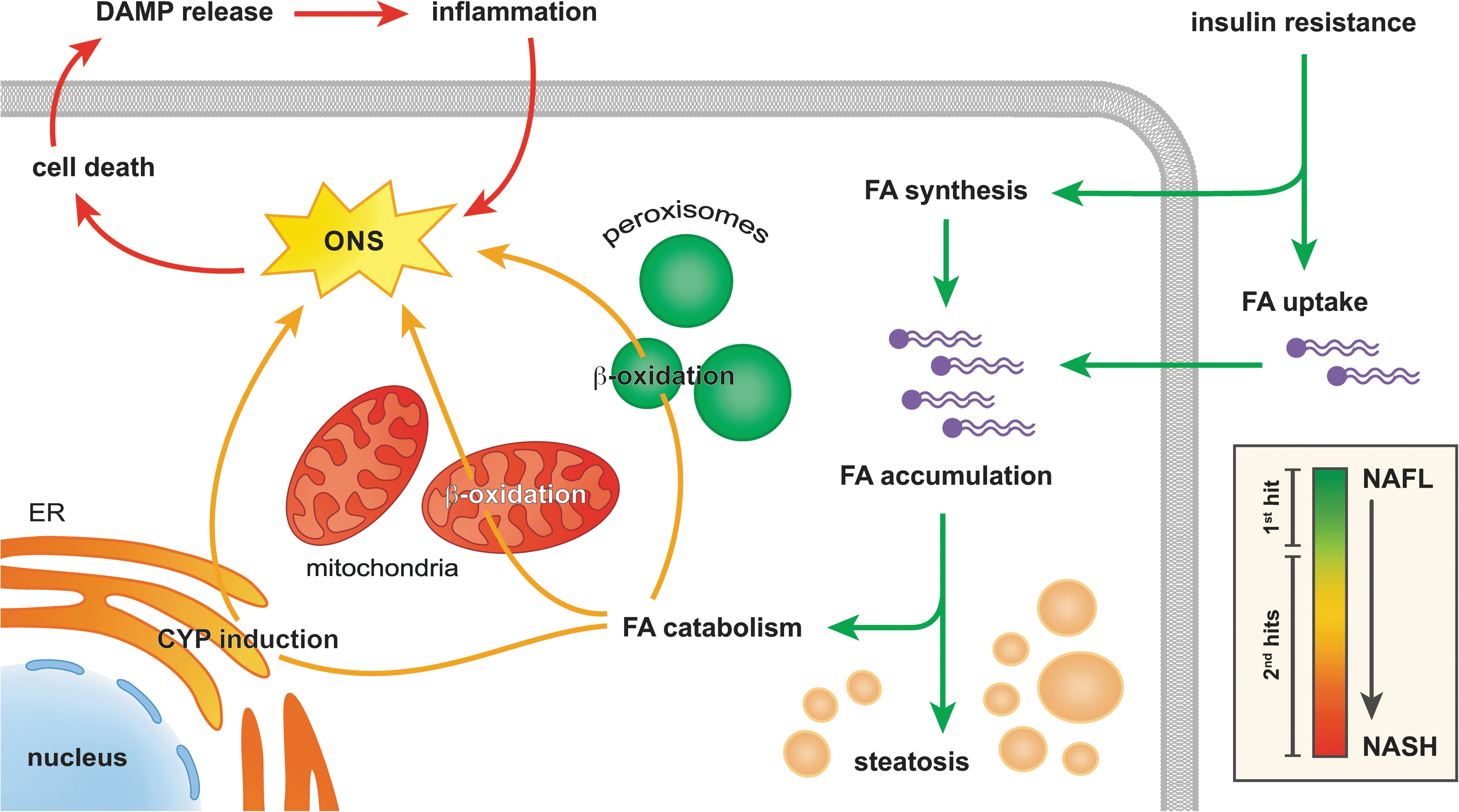
The accumulation of fat in NAFLD is a direct result of insulin resistance, which explains the strong correlation between NAFLD and the metabolic syndrome (i.e., obesity, type-2 diabetes, and hyperlipidemia) (9). The causal factors for insulin resistance in metabolic syndrome-associated conditions such as NAFLD are increased levels of peripheral adipose tissue-derived tumor necrosis factor alpha (TNF-α) and hyperlipidemia (217), which impede (hepatic) insulin signaling by inhibiting insulin receptor substrate protein-1 through the activation of c-Jun N-terminal kinase (JNK) (83, 185, 217) (sections “Tumor necrosis factor alpha” and “Unfolded protein response”). Considering that JNK can be activated by ONS (179) and that activated JNK prompts the mitochondrial formation of ROS (207) (section “Tumor necrosis factor alpha”), ONS is both a cause and a consequence of insulin resistance and NAFLD (88, 217). Nevertheless, the finding that selective hepatocellular ablation of JNK actually induced insulin resistance and hepatic steatosis (178) indicates that other pathways might be involved as well.
In the liver, insulin resistance on the one hand increases the uptake of fatty acids (FAs) from the blood and on the other hand augments intracellular FA synthesis (9) (Fig. 2, green arrows). Hepatocytes respond to the consequent excess in intracellular FAs by storing them as triglycerides and by boosting their catabolism through mitochondrial and peroxisomal β-oxidation as well as induction of CYP enzymes in the endoplasmic reticulum (ER) (169) (Fig. 2, yellow arrows). Although the catabolism of FAs via these pathways is instrumental in the removal of fat, it also augments the generation of ROS/RNS, i.e., superoxide anion (O2 •−) and its derivatives (154) (section “ROS/RNS and Their Chemical Properties in the Context of NAFLD and IR”). The consequent ONS sensitizes hepatocytes to cell death, which constitutes a key ‘second hit’ in the development of NASH (47) (Fig. 2, red arrows).
The main consequence of ONS-induced cell death is the release of damage-associated molecular patterns (DAMPs), which are endogenous self-antigens that alert the immune system of (impending) cell death once released into the extracellular environment (104). Hepatocyte-derived DAMPs trigger parenchymal inflammation by binding to Toll-like receptor 4 on the surface of Kupffer cells (KCs), the liver's resident macrophages (35). In response, KCs not only release cytokines such as interleukin 1β (IL-1β) (35) and TNF-α (199), but also mobilize NADPH oxidase-2 to form O2 •− and upregulate iNOS to generate nitric oxide (•NO) (191, 205). However, genetic ablation of gp91phox (41) or p47phox (136), which prevents the formation of active NADPH oxidase 2, did not affect the extent of lipid peroxidation, hepatic injury, and inflammation in mice that were fed a methionine- and choline-deficient diet (41, 136). An increase in TNF-α mRNA was found mainly in hepatocytes (rather than KCs) of the gp91phox knock-out animals (41). These findings indicate that NASH develops in the absence of NADPH oxidase 2-derived O2 •−, yet not through KC-induced extracellular ONS or cytokine production but by a compensatory mechanism that is based on TNF-α signaling by parenchymal cells. Moreover, it is probable that the NADPH oxidase 2-dependent production of O2 •− by KCs mainly functions to stimulate cytokine production in these cells through activation of the transcription factor nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) (41) rather than contributing to ONS per se.
The pro-inflammatory state invoked by hepatocellular ONS and consequent DAMP release is further aggravated by the increased levels of gut-derived endotoxins (e.g., lipopolysaccharide) that circulate in obese and insulin-resistant mice (21), which also leads to Toll-like receptor 4-dependent KC activation (139). Guided by these KC-derived molecular cues (e.g., TNF-α), neutrophils accumulate in the liver and increase the state of inflammation and ONS (14). Neutrophils not only co-express NADPH oxidase 2 and iNOS (205), but in addition release the enzyme myeloperoxidase that catalyzes the conversion of hydrogen peroxide (H2O2) to strong oxidants such as hypochlorous acid (HOCl) (60). Accordingly, plasma myeloperoxidase levels are increased in patients who suffer from NASH (173), while myeloperoxidase-deficient mice are less susceptible to develop high-fat diet-induced NASH and corollary liver injury (172). Lastly, extracellular ONS, hepatocyte apoptosis [due to the actions of TNF-α (233), section “Tumor necrosis factor alpha”], and lipopolysaccharide collectively activate stellate cells, which leads to collagen deposition and eventually fibrosis (55). The histological features of these processes, namely leukocyte infiltration, necrosis, and fibrosis (19) are clinically used to distinguish between NAFL and NASH (19).
In sum, TNF-α and activated JNK establish a detrimental self-propagating cycle between the liver, the innate immune system, peripheral adipose tissue, and the gut that entails fat accumulation, insulin resistance, ONS, and inflammation (85). In this complex network of metabolic and inflammatory factors, ONS occupies a central role and contributes to both the development and the progression of NAFLD (9). Of note, many of these factors are highly prevalent in Western societies because of their close association with the metabolic syndrome (52). Furthermore, the ongoing adoption of the Western sedentary and nutritional lifestyle in Asian countries significantly adds to the worldwide population that is at risk for developing NAFLD (46). As a result, the prevalence of NAFLD and NASH is currently estimated at 17–45% and 5–17% (9, 46), respectively, which renders NAFLD the most common liver disease worldwide. The number of fatty livers encountered in the surgical setting is therefore expected to follow this trend.
IR injury
Hepatic IR is an iatrogenic condition that results from the temporary withdrawal of blood supply to the liver to deter excessive bleeding during liver resection or allow for transplantation of the organ (159). The pathophysiology of IR injury can be divided into three phases that encompass ROS/RNS production (205). The hyperacute phase marks the first 30 min of reperfusion and is characterized by intracellular ONS (205), which mainly results from excessive mitochondrial formation of O2 •− and derivative ROS/RNS (140). This inflicts cell death to a limited extent (70), yet enough to activate KCs, which initiate the second phase (acute phase, 30 min–6 h of reperfusion) (205). During this phase, activated KCs produce and release ROS/RNS as well as pro-inflammatory mediators (e.g., TNF-α) (206). In doing so, KCs incite the most detrimental third phase (chronic phase, >6 h reperfusion) (205), in which leukocytes migrate to the liver (92) and produce large amounts of extracellular ROS/RNS as well as proteases that collectively induce microcirculatory defects and inflict parenchymal necrosis (205).
Clinically, IR injury is characterized by a steep increase in serum transaminases and decreased liver function (66), both resulting from hepatocellular damage, which in most severe cases culminates in acute liver failure (153). Thus, IR injury is a potentially hazardous side effect of life-saving surgical procedures, particularly in fatty livers (208).
ROS/RNS and Their Chemical Properties in the Context of NAFLD and IR
In order to fully appreciate the detrimental effects of ROS/RNS in IR-subjected fatty livers it is important to define the chemical properties of the individual species. Such an appraisal is imperative to assess which ROS/RNS are most likely involved in the resulting hepatopathology and which factors dictate their effects.
Reactive oxygen species
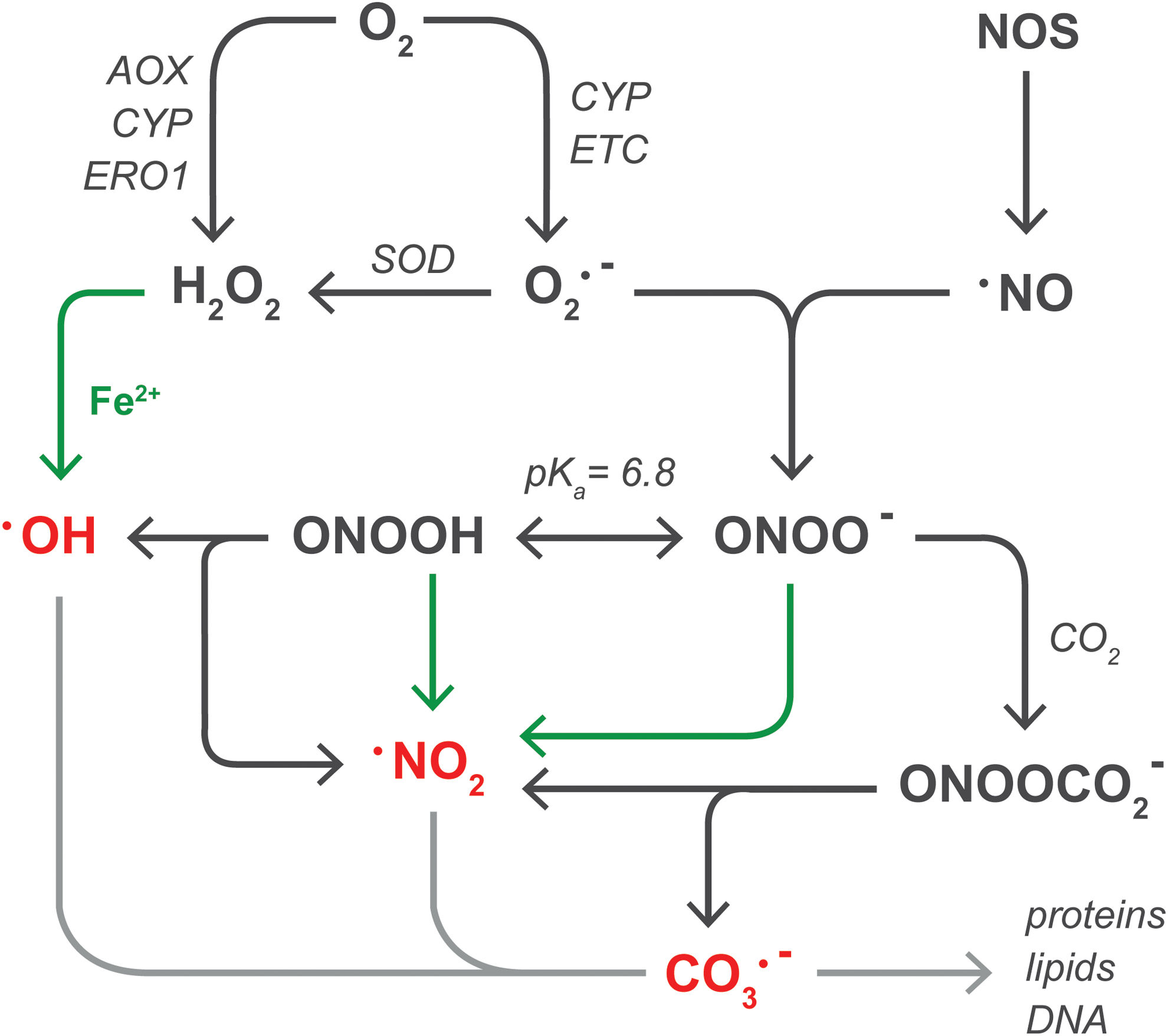
All ROS/RNS are directly or indirectly derived from O2 •−, the product of the one-electron reduction of O2 (Fig. 3). The two main sources of O2 •− in steatotic hepatocytes are the mitochondrial electron transport chain (ETC) (141) (section “Mitochondria”) and CYP enzymes (227) (section “Other sources of ROS/RNS in NAFLD”). Although NADPH oxidase 4 is moderately expressed in human liver tissue (28), the finding that NADPH oxidase 4-deficient mice are more susceptible to high-fat diet-induced obesity and consequent hepatic steatosis compared with wild-type controls excludes this enzyme as a source of O2 •− in NAFLD (117). O2 •− has an estimated biological half life (t 1/2) of ∼10−6 s (63) and swiftly dismutates into the non-radical oxidant H2O2 in a reaction that is catalyzed by three forms of superoxide dismutase (SOD): mitochondrial manganese-SOD (MnSOD), cytosolic copper-zinc-SOD, and extracellular SOD (230). In hepatocytes, H2O2 is directly formed during the two-electron reduction of O2 by peroxisomal fatty acyl-CoA oxidase (168) (AOX) as well as microsomal CYP enzymes (227) and ER oxidoreductin 1 (68) (ERO1, section “Other sources of ROS/RNS in NAFLD”). Compared with O2 •−, H2O2 has a longer lifespan [t 1/2≈10−5 s (63)] that, in combination with its membrane-transgressing capacity, enables it to react more distally from its production site (11). H2O2 is scavenged by peroxiredoxins [1–6] (221), glutathione peroxidases [1–4, 7, 8] (84), and catalase (84), but will participate in metal-catalyzed reactions (MCR) in the presence of transition metals such as ferrous iron (Fe2+) (215). Specifically, the reaction between H2O2 and Fe2+, known as the Fenton reaction (49), leads to the formation of highly reactive hydroxyl radicals (•OH) and Fe3+(215). Thereafter, Fe3+ can be reduced to Fe2+ by O2 •−, establishing an overall reaction that is known as the Haber-Weiss cycle (215). Due to its reactivity [t 1/2≈10−15 s (63)], •OH has a very limited diffusion distance.
The labile iron pool
The role of Fe2+ in ONS deserves close attention, as free Fe2+ associates with amino acids (section “Proteins”) as well as the DNA backbone and, as such, catalyzes redox reactions with H2O2 and peroxynitrite (next section) in a site-specific manner (6, 192). Moreover, Fe2+ directly oxidizes biomolecules in the presence of O2 (89), whereas its non-oxidizing ferric form (Fe3+) is easily reduced to Fe2+ by the antioxidants ascorbic acid and glutathione (GSH) (146). Accordingly, the amount of freely available intracellular iron (i.e., Fe2+ and Fe3+), referred to as the labile iron pool (LIP) (97), is strictly controlled by the storage of iron in ferritin as Fe3+, which prevents its participation in MCR (216). Nevertheless, O2 •− has the ability to liberate Fe2+ from iron-sulfur clusters in metalloproteins such as mitochondrial aconitase (142), attesting to the ability of ROS/RNS to augment the LIP. Corroboratively, aconitase activity was significantly hampered in liver mitochondria that were subjected to in vitro anoxia-reoxygenation, an effect that could be partially prevented by decreasing the mitochondrial formation of O2 •− (188). Similarly, the administration of a lipid-soluble iron chelator (des ferriexochelin 772SM) (7) and overexpression of the ferritin heavy chain (15) reduced ONS and liver damage, respectively, in rat models of cold IR. Moreover, hepatic iron overload reportedly promotes disease progression in human NAFLD (203), altogether underscoring the significant role of the LIP in ONS. It should be noted that the results of the latter study are to be interpreted with caution, because the histological method that was employed to detect intracellular iron [i.e., Perls' Prussian Blue (115)] involves degradation of ferritin and subsequent selective staining of Fe3+, but not Fe2+(218), which implies that this method does not necessarily provide information on the LIP (97).
The hepatic iron accumulation that is associated with NAFLD could be, at least in part, an effect of ROS formation. In that regard, the expression of transferrin receptor 1, which dictates hepatocellular iron uptake, and ferritin, which determines the amount of free iron (i.e., the LIP), is regulated post-transcriptionally via the binding of iron regulatory proteins to iron-responsive elements in ferritin as well as transferrin receptor 1 mRNA (214). In particular, iron regulatory protein 1 (IRP1) only binds to iron responsive elements following a structural rearrangement that entails the loss of its iron-sulfur cluster and consequent exposure of a cysteine-rich mRNA-binding site. This conformational change, which coincides with activation of the protein, is induced not only by a reduction in cellular iron levels but also by oxidative stress (214). More specifically, exposure of HepG2 cells to persistently low levels of extracellular H2O2 (to mimic a state of chronic inflammation such as NAFLD) activated IRP1 and augmented the LIP (8). Moreover, rats that were fed a high-fat diet showed enhanced IRP1 activity, increased transferritin receptor 1 expression, decreased ferritin levels, and an increase in hepatic total iron content (132). These findings indicate that the inflammatory aspect of NALFD could contribute to hepatocellular iron accumulation as well as augmentation of the LIP and hence ONS. Accordingly, leukocyte influx as well as increased hepatic TNF-α mRNA, malondialdehyde (MDA, a lipid peroxidation end product), and 3-nitrotyrosine (a protein nitration marker) levels were also observed in these high-fat diet-fed rats (132).
Notwithstanding the apparent relationship between inflammation, ONS, and the LIP, the cysteine residues on activated IRP1 are preferential targets for oxidation (section “Proteins”), which ameliorates their mRNA-binding capacity (214). Severe ONS could therefore revoke the effects mentioned earlier inasmuch as the inactivation of IRP1 increases ferritin levels and decreases transferrin receptor 1 expression. The finding that pretreatment with N-acetyl cysteine prevented IRP1 inactivation during early reperfusion in a rat model of warm IR substantiates this notion (195). Consequently, additional studies that investigate iron metabolism and the LIP in NAFLD as well as IR injury are required, as hepatic iron kinetics likely govern the ONS-mediated interplay between both conditions.
Reactive nitrogen species
In addition to ROS, O2 •− is a precursor of RNS such as peroxynitrite, the common denominator for peroxynitrite anion (ONOO−) and peroxynitrous acid (194) [ONOOH, pKa=6.8 (165) and t 1/2<0.1 s (166)]. Peroxynitrite is formed in the diffusion-controlled reaction between O2 •− and •NO [t 1/2=1−30 s (90)] (102), which is synthesized by NOS enzymes such as iNOS (61). Its membrane-transgressing ability and long lifetime enable •NO to act on neighboring cells (13), which implies that •NO formed in KCs can diffuse into hepatocytes. iNOS is induced in KCs (211) as well as in hepatocytes (61) in response to inflammatory stimuli such as IL-1β and TNF-α (71), both of which are released during NAFLD (198) and IR (92). Accordingly, hepatic iNOS mRNA and protein levels are increased compared to controls in rodent models of NAFLD (122, 190) and cold IR (82), respectively. Along these lines, iNOS knock-out mice that were fed a high-fructose (i.e., NAFLD-inducing) diet exhibited reduced hepatic TNF-α mRNA, 3-nitrotyrosine, and 4-hydroxynonenal (4-HNE, a lipid peroxidation end product) levels compared with wild-type controls (190). Moreover, the administration of an iNOS inhibitor [N-(1-naphtyl)ethylendiamine dihydrochloride] markedly reduced liver damage, improved regeneration, and increased overall survival in a rat model of cold IR with a concomitant reduction in RNS formation (82).
Although some have claimed that a specific NOS isoform is present in mitochondria, its existence has not yet transcended from dispute (17). Alternatively, hypoxic rat liver mitochondria generate •NO from nitrite (24), although •NO formed under hypoxic conditions was shown to alleviate rather than aggravate mouse liver IR injury in vivo by transiently reducing mitochondrial O2 •− formation during early reperfusion (188) (section “Mitochondria”). Moreover, due to the stability and diffusibility of •NO, mitochondrial •NO production is not a prerequisite for mitochondrial RNS formation (164). Instead, the limited diffusion properties of O2 •− (2) dictate that the formation of peroxynitrite will predominantly occur in the vicinity of O2 •− generation. Since the reaction between O2 •− and •NO directly competes with that of O2 •− and SODs (20), the generation of peroxynitrite also depends on the availability of SODs. Thus, when SODs become inactivated, as occurs in NAFLD (138, 209) (section “Proteins”), and iNOS is simultaneously induced, which takes place in NAFLD (122) as well as IR (82), the formation of peroxynitrite and derivative radicals (next paragraph) will increase.
Similar to H2O2, peroxynitrite participates in MCR, resulting in the formation of nitrogen dioxide (•NO2) (6). Peroxynitrite is scavenged by peroxiredoxins and glutathione peroxidases (194), but the enzymatic inactivation of ONOO− directly competes with its reactivity toward CO2, which generates •NO2 and a carbonate radical anion (CO3 •−) with a ∼35% yield (32, 194). ONOOH, on the other hand, can homolyze into •OH and •NO2 (∼30% yield) (31), but this reaction is slow [k=0.8 s−1] (31, 194) and will therefore predominantly occur in lipophilic compartments (e.g., membranes) where less competing reactions prevail (194). Insofar as the lipid compartment is increased in NAFLD (162), the formation of •OH and •NO2 from ONOOH homolysis might be favored.
The effects of •NO2 [t 1/2≈10−6 s (51)] and CO3 •− [t 1/2≈10−6 s (130)] have only recently received attention as effectors of the cytotoxicity that was initially ascribed to peroxynitrite (12). Both species can directly oxidize biomolecules and are scavenged by ascorbic acid and GSH (12), which is likely to be impaired in NAFLD because of GSH (209) and ascorbic acid depletion (116) as well as in IR due to a transient reduction in GSH levels (103). Furthermore, due to its lipophilicity, •NO2 is an initiator of lipid peroxidation, a process that can be inhibited by α-tocopherol (189) (section, “Lipids”).
In summary, two scenarios can be distinguished in which ROS/RNS are most damaging towards hepatocytes: (i) the combination of H2O2 or peroxynitrite and Fe2+ that induces site-specific, MCR-mediated generation of •OH or •NO2, respectively, and (ii) the formation of peroxynitrite from •NO and O2 •−, which gives rise to •NO2, •OH, and/or CO3 •− as derivative radicals. Notably, either route applies to NAFLD and IR since both conditions are characterized by excessive formation of O2 •− and •NO. Moreover, the formation of peroxynitrite and its derivative radicals is specifically favored in NAFLD due to the depletion of SOD and GSH, respectively. Lastly, experimental evidence suggests a catalytic role for iron in NAFLD and IR, although studies that explicitly address the LIP in both conditions are currently lacking.
Molecular Targets of ROS/RNS in NAFLD and IR
The biological targets of ROS/RNS are generally divided into lipids, proteins, and DNA. Understanding the reaction mechanisms that underlie their damaging effects is essential to interpret the events observed at a cellular level. Considering that (per)oxidation of proteins and lipids has the most relevant effects with respect to the interplay between NAFLD and IR, the reactions between ROS/RNS and these biomolecules will be addressed next with specific focus on the implications for both conditions.
Lipids
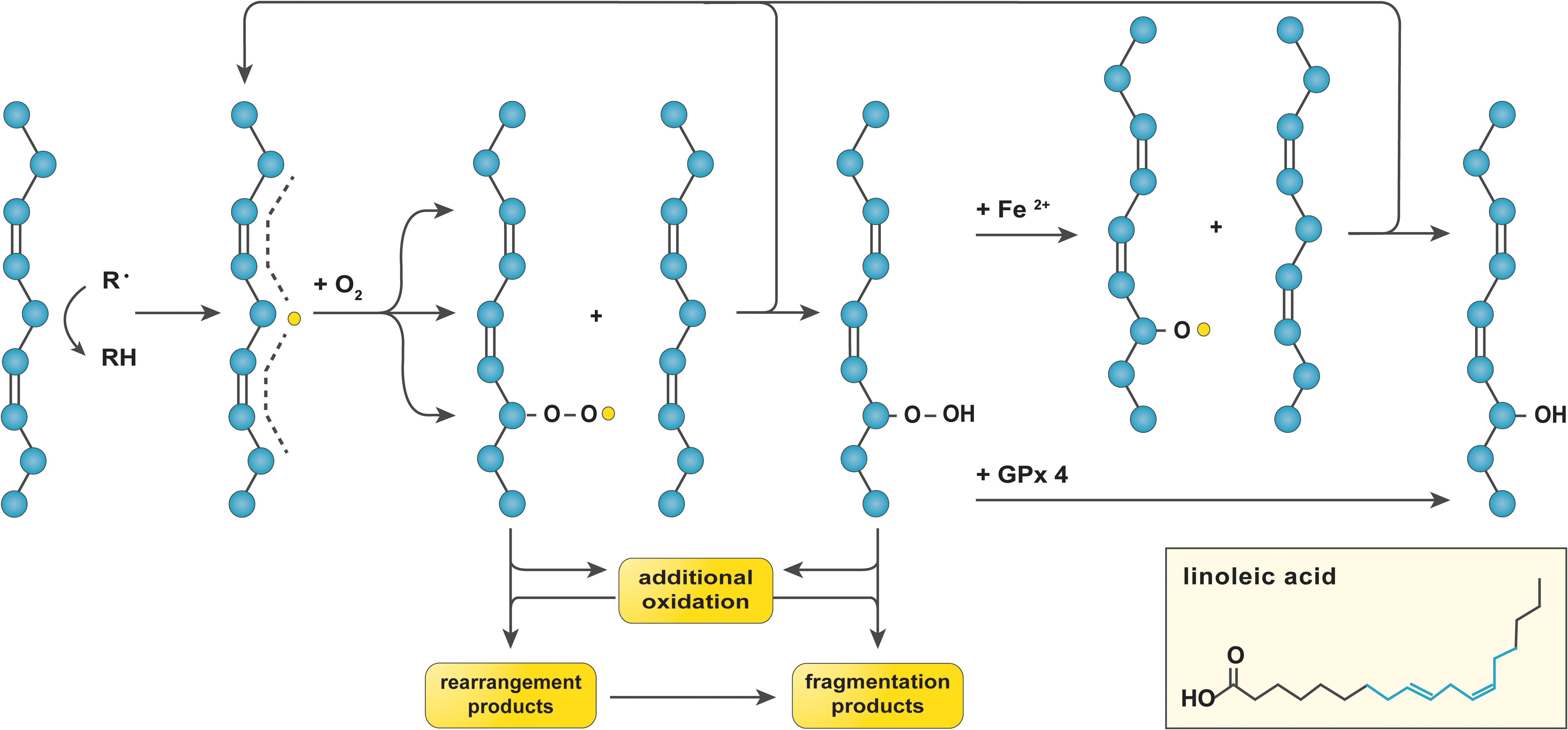
Lipid peroxidation is prevalent in NAFLD, not only due to the abundance of substrate (i.e., FAs) (162) but also because of the nature of the reaction, which is radical chain propagating (226). The susceptibility of an FA to oxidation is proportional to the number of double bonds in its aliphatic tail inasmuch as hydrocarbons that are flanked by two alkenes (i.e., doubly allylic sites) have a ∼20% lower bond dissociation energy compared to those in saturated C–C bonds (226). Accordingly, the polyunsaturated fatty acids (PUFAs, ≥2 double bonds) linoleic acid (LA, ω-6, 1 doubly allylic site), arachidonic acid (AA, ω-6, 3 doubly allylic sites), eicosapentanoic acid (EPA, ω-3, 4 doubly allylic sites), and docosahexanoic acid (DHA, ω-3, 5 doubly allylic sites) are most sensitive to oxidation (226).
Given their lipophilicity, •OH (149) and •NO2 (160) from MCR and/or ONOOH homolysis generally initiate lipid peroxidation (Fig. 4). The thus formed lipid radical (L
NS indicates no significant difference versus control, + indicates a significant increase versus control, + + indicates a significant increase versus control and NAFL, − indicates a significant decrease versus control, and − − indicates a significant decrease versus control and NAFL. ND indicates that no data were obtained in the respective study.
4-HNE, 4-hydroxynonenal; GSH, reduced glutathione; MDA, malondialdehyde; NAFL, non-alcoholic fatty liver; NASH, non-alcoholic steatohepatitis; SOD, superoxide dismutase.
Another contributory factor to the aggravation of IR injury in NAFLD is an increased ω-6/ω-3 FA ratio (4). The disrupted ω-6/ω-3 balance in fatty livers could be in part explained by the reaction rates of LA, AA, EPA, and DHA toward L–OO
Consequently, the presence of antioxidants in the lipid compartment is particularly important. The major free radical scavengers in the lipid compartment are α-tocopherol (44) and ubiquinol (also referred to as coenzyme Q10 [UQH2]); the reduced form of ubiquinone (UQ), which is also a component of the ETC (43) (Fig. 5). These antioxidants work in concert to protect membranes against oxidative modification (43), the clinical implications of which are reflected by the beneficial effects of α-tocopherol supplementation on NASH disease activity (determined histologically) found in a randomized controlled trial (183).
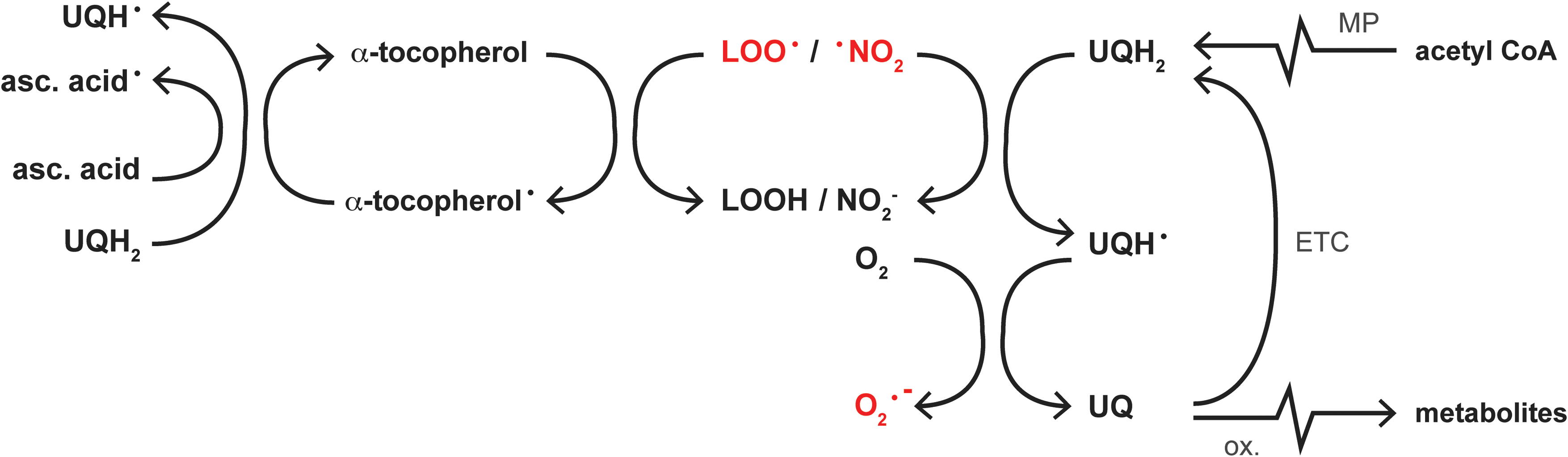
Upon oxidation, the regeneration of α-tocopherol relies on the reducing capacity of ascorbic acid or UQH2 (43), whereas that of UQ depends on the reductive potential of the ETC (126) or de novo synthesis from acetyl CoA via the mevalonate pathway (43). However, hepatic ascorbic acid levels become depleted in rats fed a high-fat diet (116), whereas the decline in UQ(H2) plasma levels in human NAFLD (225) indicates a concomitant reduction in hepatic UQ(H2) content. Inasmuch as UQ is easily (auto-)oxidized into inactive metabolites (38), excessive lipid peroxidation in NAFLD could be responsible for a reduction in intracellular UQ(H2) levels (53), altogether debilitating the antioxidant defense machinery in membranes.
In sum, excessive lipid peroxidation, a disrupted ω-6/ω-3 FA ratio, and a reduced antioxidative capacity contribute to the marked damage in steatotic livers compared with healthy livers after IR (147, 228).
Proteins
Oxidation of proteins irreversibly impairs their structure and function. Generally, cells possess an effective machinery to cope with oxidized proteins, namely the 20S-proteasome (39). However, when the amount of oxidatively modified proteins becomes overwhelming, the extent of protein fragmentation and cross-linking increases (39). This eventually leads to the formation of large protein aggregates, such as the Mallory bodies that are characteristic of NASH (229), which are cytotoxic and resistant to proteolytic digestion (39). The formation of protein aggregates in NAFLD is furthermore promoted by 4-HNE-mediated inhibition of the 20S-proteasome (50).
Proteins are irreversibly modified via three mechanisms (Fig. 6, mechanisms 1–3). First, the protein backbone is a target for oxidation by •OH via proton abstraction (59) (Fig. 6, mechanism 1), which eventually results in cleavage of the protein backbone into carbonylated (R=O) fragments (192). Second, MCR takes place at amino acids that chelate transition metals (i.e., lysine, arginine, proline, threonine, histidine, leucine, and cysteine) (192) (Fig. 6, mechanism 2), which, in the presence of H2O2 or peroxynitrite, results in carbonylation (192) or nitration (R–NO2) (6), respectively, of the transition metal-complexed amino acid. Third, the aromatic moieties of tryptophan, tyrosine, phenylalanine, and histidine are preferred targets of peroxynitrite-derived radicals (6) (Fig. 6, mechanism 3), thereby generating hydroxylated (R–OH) or nitrated residues such as 3-nitrotyrosine (6). Of note, 3-nitrotyrosine is a reliable biomarker for the formation of RNS such as peroxynitrite-derived •NO2 in vivo (210).
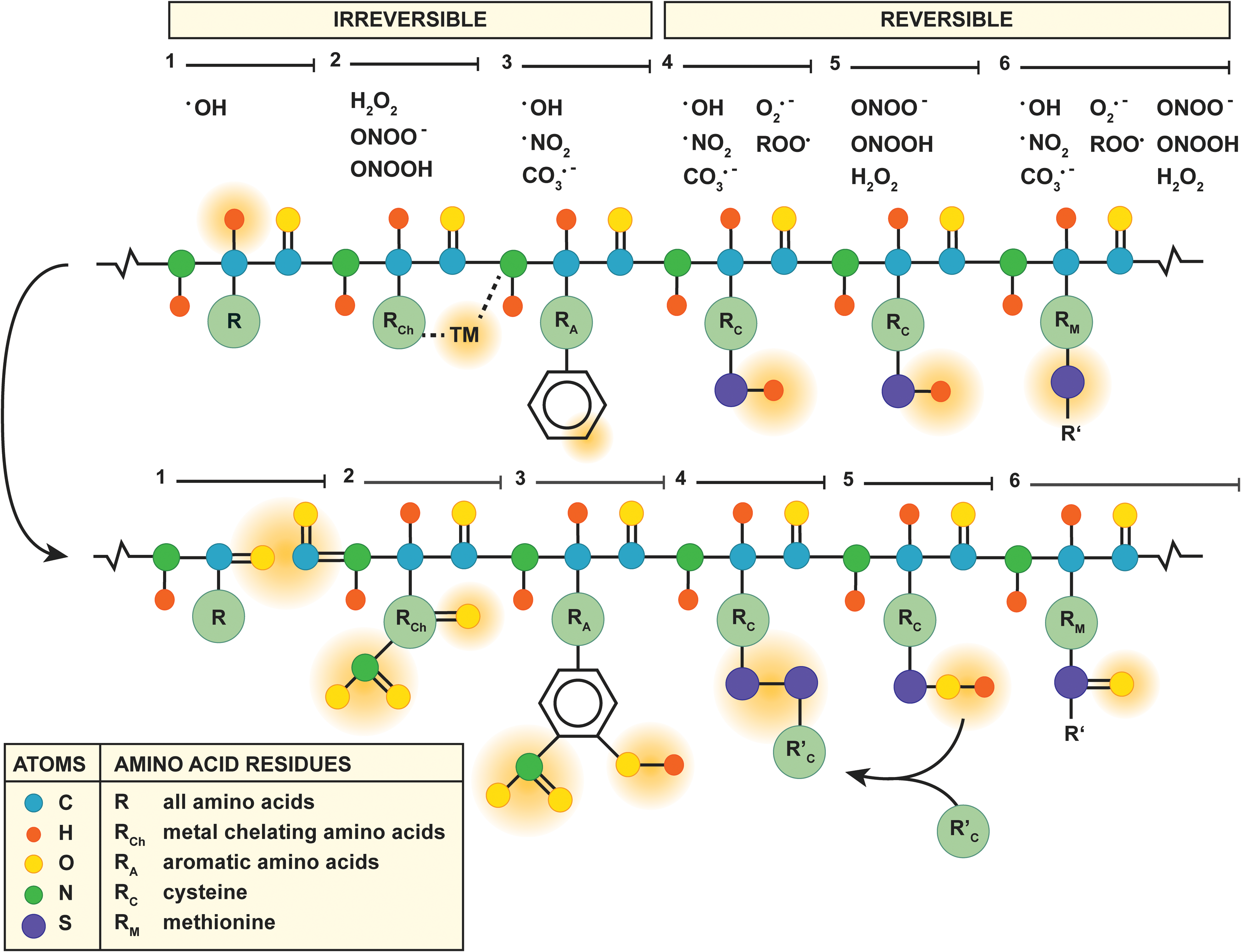
Accordingly, elevated levels of 3-nitrotyrosine and protein carbonyls have been measured in liver samples of NAFLD patients (Table 1) as well as in a mouse model of IR (140). The consequences hereof likely contribute to the susceptibility of steatotic hepatocytes to IR injury, especially when these modifications affect antioxidant enzymes. Tyrosine nitration and histidine oxidation, for instance, are key events in the peroxynitrite-induced inactivation of MnSOD (193) and copper-zinc-SOD (5), respectively. The functional loss of these enzymes was found to contribute to the reduced antioxidative capacity in NAFLD livers (209), which is likely to affect cell viability during IR (section “Pathophysiological implications of ROS/RNS in NAFLD and IR”). Moreover, reduced MnSOD activity was observed in two mouse models of NAFLD, despite a significant increase in MnSOD mRNA levels (138), attesting to the fact that post-transcriptional modifications such as oxidation are responsible for the loss of MnSOD activity in NAFLD.
Contrary to the mechanisms of irreversible protein modification mentioned earlier, oxidation of cysteine and methionine is reversible and, as such, constitutes the main redox mechanism in numerous antioxidant enzymes (192) (Fig. 6, mechanisms 4–6). Oxidation of the cysteine thiol/thiolate (R–SH/R–S−, Fig. 6, mechanism 4) by free radicals, for instance, accounts for the direct antioxidative properties of GSH, in which the formed thiyl radical (R–S
Alternatively, H2O2 and peroxynitrite oxidize cysteine thiols via the formation of an intermediate sulfenic acid (100) (R–SOH, Fig. 6, mechanism 5). In particular, the reaction of protein sulfenic acids or nitrosothiols [R–S–NO, from reaction with •NO (54)] with GSH (77), known as S-glutathionylation (R–S–SG), constitutes an important process in the modulation of TNF-α signaling during ONS (157, 174) (section “Tumor necrosis factor alpha”). Moreover, sulfenic acid formation is the working mechanism of H2O2 and peroxynitrite scavenging by peroxiredoxins (221). Peroxiredoxin-4-overexpressing mice that were fed a high-fructose diet showed reduced levels of hepatic 4-HNE, serum MDA, and hepatic TNF-α mRNA levels compared with their wild-type littermates (143). Collectively, these findings highlight the importance of non-radical oxidant-scavenging enzymes in regard to ONS and inflammation in the context of NAFLD.
However, the catalytic site of peroxiredoxins can be inactivated by overoxidation into sulfinic (R–SO2H) and ultimately sulfonic acids (R–SO3H) (100) that reduces their antioxidative capacity (223), although the sulfinic acids on peroxiredoxin 1–4 can still be regenerated by sulfiredoxin (220). Overoxidation and subsequent recuperation of peroxiredoxins was shown to occur in HeLa cells that were incubated with tert-butyl hydroperoxide (an inducer of acute oxidative stress), but complete recovery was slow and depended on both regeneration and de novo synthesis (30). Accordingly, the non-radical oxidant-scavenging capacity of hepatocytes could be affected by overoxidation of peroxiredoxins during IR, particularly under conditions of steatosis due to the prevalent ONS.
Another amino acid prone to oxidation is methionine, which contains a thiol group that can be oxidized into a methionine sulfoxide (R–S=O) by various ROS/RNS (111) (Fig. 6, mechanism 6). Although reversible, methionine sulfoxide formation is not benign per se as the oxidation of methionine-80 in cytochrome c activates its peroxidase activity (197), which constitutes a key event in ONS-induced cell death (96) (section “Mitochondria”). The detrimental effects that emanate from cytochrome c oxidation at methionine-80 underscore the importance of the mitochondrial antioxidative machinery. Not only are mitochondrial antioxidants critical in preventing cell death during IR (140) (section “Mitochondria”), they are also responsible for the detoxification of ETC-derived ROS/RNS and therefore for deterring their diffusion into the cytosol (77). However, the mitochondrial antioxidative defense system consists of a variety of antioxidants, including MnSOD (99), GSH (124), and peroxiredoxin-3 (221), which can be (over)oxidized by ROS/RNS as addressed earlier. Moreover, the mitochondrial antioxidative capacity strongly depends on the cumulative actions of the individual antioxidants (138). For instance, SOD-overexpressing mice that were fed a methionine- and choline-deficient diet were more susceptible to NASH because of mitochondrial GSH depletion and a consequent rise in H2O2 levels, despite a reduction in peroxynitrite formation (138). Thus, the modulation of a single component of the mitochondrial antioxidative defense system can exert inverse and undesirable effects on its overall function.
Conclusively, the ONS in NAFLD likely contributes to attenuation of the overall cellular and particularly mitochondrial antioxidative capacity by (irreversibly) oxidizing critical amino acid residues in antioxidants, which renders steatotic hepatocytes more amenable to IR damage.
Pathophysiological Implications of ROS/RNS in NAFLD and IR
At a cellular level, there are two pivotal sites at which the interplay between NAFLD and IR takes place: the mitochondria and TNF-α signaling, both of which are strongly affected by ONS. In addition, two other sources of ROS/RNS, namely extramitochondrial FA oxidation and the unfolded protein response (UPR), have been described for NAFLD that are not directly influenced by IR but likely contribute to the ONS that sensitizes fatty livers to IR. Therefore, these will also be addressed.
Mitochondria
Under physiological conditions, mitochondria consume more than 90% of cellular O2 in order to generate ATP via oxidative phosphorylation (137). During oxidative phosphorylation, electrons derived from the oxidation of NADH and FADH2 are carried along complex I–IV of the ETC to eventually reduce O2 to H2O (137). The transport of electrons enables the movement of protons from the mitochondrial matrix across the inner mitochondrial membrane into the intermembrane space, thereby establishing an inward protonmotive force (Δp) that consists of a pH component (i.e., the transmembrane pH gradient, ΔpH) and an electrical component (i.e., the membrane potential, ΔΨ) (105). The Δp-driven return of protons through ATP synthase provides the energy required to phosphorylate ADP to ATP (137). However, a small fraction of the intramitochondrial oxygen is reduced to O2 •−, predominantly by complex I, which is favored at a high NADH/NAD+ ratio as well as a large Δp (141).
As mentioned in the introduction, mitochondria constitute an important source of hepatocellular ROS/RNS in NAFLD (154). The basis hereof lies in the large amount of free fatty acids that are transported into the mitochondria to undergo β-oxidation (154). This generates acetyl-CoA, which enters the tricarboxylic acid cycle to reduce NAD+ and FAD (135). Subsequently, as the NADH/NAD+ ratio and Δp increase, so does the production of O2 •− (141).
In NAFLD, ROS/RNS formation is further enhanced by the accumulation of nonesterified cholesterol in the mitochondrial membranes, which reduces their fluidity and hinders the transport of GSH into the mitochondrial matrix (118). Moreover, 4-HNE can form adducts with histidine residues in the catalytic center of complex IV, significantly reducing its activity (29). Accordingly, when complex IV is inhibited, so is electron transport in the preceding complexes, leading to an increase in the NADH/NAD+ ratio and hence O2 •− formation (141). Although the activity of purified complex IV can be preserved in the presence of 4-HNE when GSH is added (29), the mitochondrial GSH depletion that concurs with NAFLD (118, 187) likely promotes the 4-HNE-dependent inactivation of complex IV. Thus, ONS and mitochondrial GSH depletion further contribute to the generation of (mitochondrial) ROS/RNS in NAFLD.
Mitochondria respond to this metabolic stress by expressing mitochondrial uncoupling protein 2 (UCP2) (145). While UCP2 is usually undetectable in hepatocytes, it is expressed in response to high levels of nonesterified PUFAs via activation of the nuclear receptor peroxisome proliferator-activated receptor α (PPAR-α) (95, 145). UCP2 establishes a proton leak over the inner mitochondrial membrane that lowers Δp and hence the formation of O2 •− (141), which implies that UCP2 induction alleviates mitochondrial ONS. Along these lines, the degree of proton conductance through UCP2 is directly and proportionally controlled by the amount of O2 •− generation in mitochondria via the formation of 4-HNE (142). These findings indicate that UCP2 activity increases parallel to mitochondrial O2 •− formation in NAFLD and, as such, aims to prevent excessive ONS. Corroboratively, UCP2 mRNA and protein expression was observed in hepatocytes of steatotic ob/ob mice in contrast to healthy controls, in which UCP2 mRNA was undetectable (27).
Although intended as an antioxidative mechanism, the expression of UCP2 in steatotic hepatocytes affects mitochondrial energy metabolism. Since ETC uncoupling occurs at the expense of Δp, the mitochondrial ATP synthesizing capacity becomes impaired (137), which explains the low basal ATP levels that were measured in ob/ob mice (27). Moreover, basal ATP levels inversely correlate to body mass index in NASH patients (34) as well as in subjects that are clinically unlikely to suffer from NASH (34, 144), indicating that UCP2 expression might already take place in earlier stages of NAFLD.
Although the negative effects of UCP2 expression on ATP generation are usually quiescent (27, 45), they surface when energy demands increase. For instance, NASH patients who were subjected to a fructose (i.e., ATP-depleting) challenge showed significantly a reduced ATP repleting capacity (34). The negative effect of UCP2 on the mitochondrial ATP-generating capacity becomes even more pronounced when the liver is subjected to IR. During ischemia, the lack of O2 forces hepatocytes to switch to anaerobic glycolysis to generate ATP (222). However, due to the limited ATP yield from anaerobic respiration compared with oxidative phosphorylation, cellular ATP/ADP stores gradually become depleted (103) while cessation of ETC activity increases the NADH/NAD+ ratio (103). The high NADH/NAD+ ratio subsequently fuels the acute burst in complex I-driven mitochondrial O2 •− formation (141) on reoxygenation (i.e., reperfusion) (22).
In line with the previous findings, transient S-nitrosylation (i.e., inactivation) of complex I during the hyperacute phase of reperfusion, resulting from the intramitochondrial formation of •NO from exogenously administered nitrite during hypoxia, significantly attenuated parenchymal damage in a mouse model of IR (188). This beneficial effect was attributed to a reduction in mitochondrial H2O2 formation and improved ATP synthesis during early reperfusion, conclusions that were based on observations made in nitrite- and vehicle-treated rat liver mitochondria that were subjected to in vitro anoxia-reoxygenation (188). In addition, compared with healthy hepatocytes, steatotic hepatocytes are challenged in neutralizing the high levels of ROS/RNS and in restoring ATP reserves during early reperfusion because of mitochondrial antioxidant depletion and ETC uncoupling (187), which has two major consequences.
First, ATP depletion impairs the function of ion transporters in the plasma membrane, including the Na+/K+ ATPase, which leads to an increase in cytosolic [Na+] during ischemia (23). In response, the Na+/Ca2+ antiporter starts working in reverse, reducing intracellular [Na+] at the expense of a rise in [Ca2+] (23). This Ca2+ freely enters the mitochondria during repolarization (i.e., reperfusion), where it induces mitochondrial permeability transition (MPT) (101, 110). MPT encompasses the formation of membrane pores that enable free movement of small solutes between the mitochondrial matrix and the cytosol (110). During MPT, cytochrome c translocates from the inner mitochondrial membrane to the cytosol, where it activates caspase 9 to initiate apoptosis (150). ATP/ADP depletion (72) and ONS (148) greatly lower the threshold for MPT. Specifically, ROS/RNS oxidize vicinal cysteine residues on the adenine nucleotide translocator, one of the MPT pore components, which augments its sensitivity to Ca2+ (129). Moreover, peroxynitrite-dependent tyrosine nitration was shown to directly promote oligomerization and channel activity of the voltage-dependent anion channel protein, another MPT pore component, in cardiac IR (224). In addition, a complex I-dependent reduction in mitochondrial ROS formation (described earlier) elicited protection against Ca2+-induced MPT in isolated rat liver mitochondria that were subjected to in vitro anoxia-reoxygenation (188), collectively attesting to the role of ROS/RNS in MPT induction.
In addition to MPT induction, ROS/RNS play a pivotal role in the release of cytochrome c by switching on its peroxidase activity (197) (section “Proteins”) and enabling the subsequent peroxidation of cardiolipin (96), a phospholipid with LA side chains that anchors cytochrome c into the inner mitochondrial membrane (96, 150). The formation of cardiolipin-hydroperoxide (CL-OOH) is essential for the release of cytochrome c from mitochondria, because it disrupts the hydrophobic cardiolipin–cytochrome c interaction (150) and destabilizes the lipid bilayer, thereby promoting Bax-dependent permeabilization of the outer mitochondrial membrane (123) (section “Tumor necrosis factor alpha”). Taken altogether, the prevailing ONS in steatotic hepatocytes likely predisposes mitochondria to MPT and cytochrome c release during IR, causing mitochondria to initiate cell death programs.
Second, the cytochrome c-dependent activation of caspase 9 and subsequent apoptosis can only take place when ATP levels are at least 15–20% of physiological baseline (93). When the energy status drops below these levels, hepatocytes are unable to maintain cellular ion homeostasis, which leads to osmotic swelling and ultimately oncotic necrosis (93). In addition, when ATP levels become insufficient during apoptotic signaling, the cell averts to secondary necrosis (93). Given the low basal ATP levels (27, 45) and uncoupling of the ETC (27, 187) in steatotic hepatocytes, the cells are more likely to undergo (secondary) necrosis than healthy cells following MPT (45). This is particularly important in the context of IR injury, as necrotic cells are highly immunogenic (104). As a consequence, KC activation and leukocyte chemotaxis will be more pronounced, giving rise to the extensive tissue damage observed in various mouse models of fatty liver IR (42, 118). In that regard, UCP2-dependent ATP depletion in particular has been correlated to the poor response of NASH-affected rat (187) and mouse (45) livers to IR. However, the role of this phenomenon in the more prevalent NAFL requires more research.
Taken altogether, the metabolic perturbations in NAFLD negatively influence hepatocyte viability during IR through mitochondrial pathways. First, prevalent mitochondrial ONS favors MPT induction and cytochrome c release during IR. Second, since UCP2 expression impairs ATP production, cells are more likely to undergo necrosis than apoptosis following MPT. The consequent exacerbation of DAMP release during reperfusion likely augments the severity of the inflammatory response and corollary parenchymal damage in steatotic versus healthy livers.
Tumor necrosis factor alpha
TNF-α is a pleiotropic cytokine that is released by a plethora of (liver) cell types in response to inflammatory stimuli (206). In addition to its ability to elicit (hepatic) insulin resistance (3), TNF-α is a strong chemoattractant for leukocytes (32) and a potent iNOS inducer (71). Via its pro-inflammatory effects, TNF-α not only mediates the progression of NAFL to NASH (199) but also significantly contributes to IR injury (32, 200). Nevertheless, TNF-α has a dual effect on hepatocytes because it simultaneously activates a multi-branched death signaling cascade as well as a proliferative ‘survival’ pathway (78). In addition, it has recently been described that TNF-α also induces a novel, programmed type of necrosis (204). In fatty livers, the prevailing ONS likely directs hepatocytes toward TNF-α-mediated cell death rather than survival during IR.
Shortly after binding TNF-α, the cytosolic domain of the TNF-α receptor-1 recruits TNF-α receptor-associated protein with death domain (TRADD), TNF-α receptor-associated factor 2, and receptor-interacting kinase 1 (RIP1) to form the plasma membrane-bound complex 1 (134) (Fig. 7). Complex 1 initiates the survival signaling pathway, which depends on the activation of NF-κB (134), as well as two branches of the trinomial death signaling cascade that are activated by JNK phosphorylation (i.e., activation) (78) and ceramide formation (58). Thereafter, part of complex I dissociates from the cytosolic receptor domain to form complex 2, which mediates the third branch of the death signaling cascade (134). Whether hepatocytes will ultimately proliferate or die following stimulation with TNF-α is strongly influenced by ONS (78).
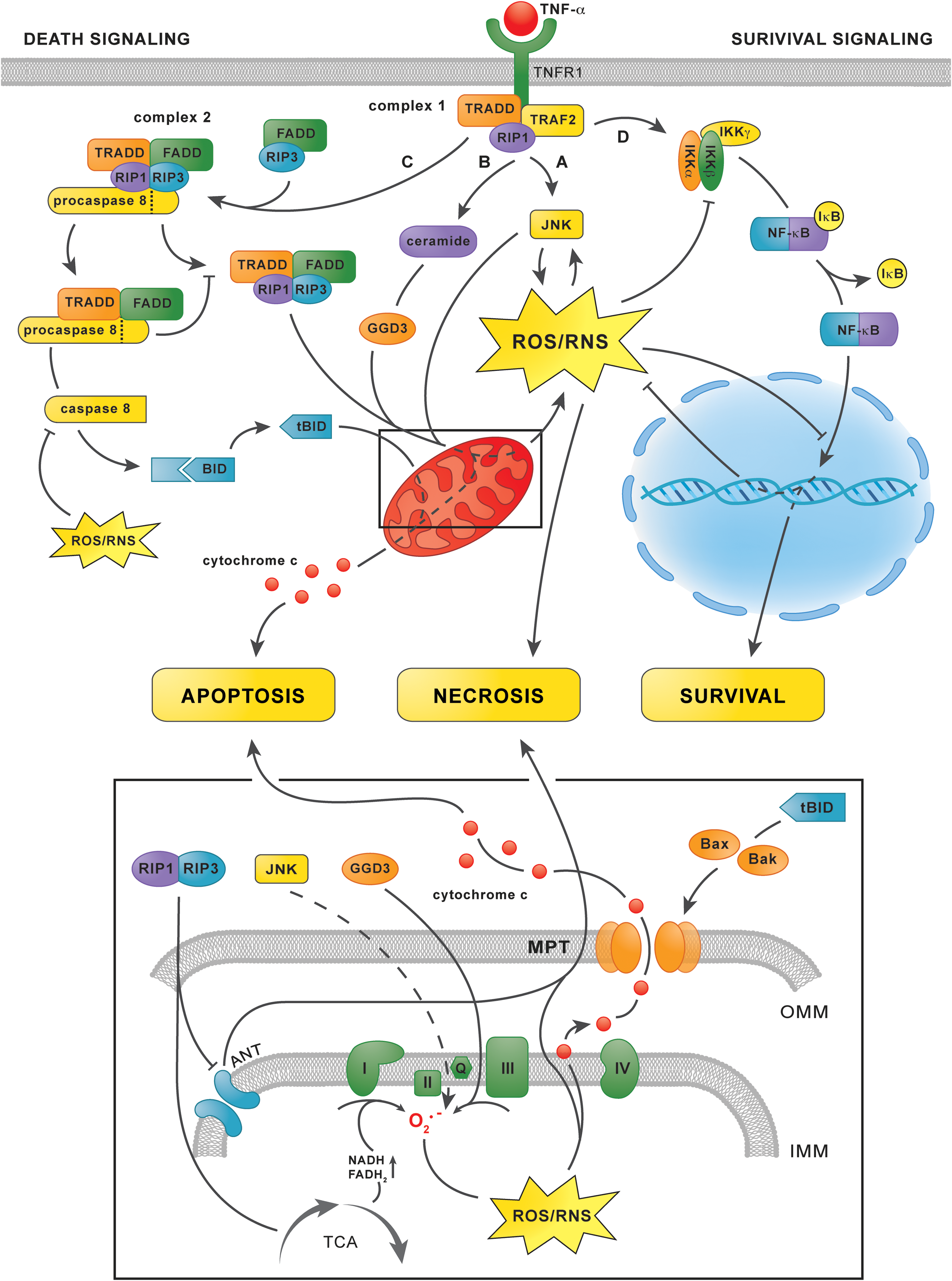
The first branch of the death signaling cascade is initiated by activation of the mitogen-activated protein kinase cascade by complex 1, which consists of apoptosis signal-regulating kinase 1 (ASK1), mitogen-activated protein kinase kinase 4/7, and finally phorsphorylation of JNK (78) (Fig. 7, pathway A). Phosphorylated JNK causes ONS by inducing mitochondrial O2 •− formation via an unknown mechanism (75, 207) as well as via degradation of ferritin, which augments the LIP (10). Moreover, ROS/RNS sustain JNK phosphorylation by directly activating ASK1 (179). Specifically, oxidation of thioredoxins that constitutively bind to ASK1 enables their dissociation, which enables ASK1 phosphorylation and downstream JNK phosphorylation (179). The existence of this positive feedback loop enables sustained JNK activity, ONS, and ultimately necrosis (98). Accordingly, JNK-dependent ROS/RNS formation and MPT induction were observed in primary hepatocytes in which mitochondrial GSH had been depleted (75), such as fatty hepatocytes (187), as well as in fibroblasts in which NF-κB was selectively inhibited (180, 207)—an ONS-dependent effect that is likely to occur in NAFLD (discussed next). Moreover, selective inhibition of JNK significantly reduced liver damage in rat models of warm (200) and cold IR (201). These findings point to phosphorylated JNK as an important mediator of IR injury, an effect that is likely to be even more prominent in NAFLD because of the prevalent ONS.
In addition, complex 1 initiates the second branch of the death signaling cascade via the activation of acid sphingomyelinase, which hydrolyzes the membrane sphingophospholipid sphingomyelin to form ceramide (58) (Fig. 7, pathway B). The subsequent accumulation of ceramide accelerates the synthesis of ganglioside GD3 in the ER, which enters mitochondria because of their physical continuity with the ER (175), where it promotes O2 •− formation at complex III (69). Ceramide accumulation could therefore contribute to ONS in NAFLD when TNF-α is involved, although conclusive evidence is unavailable at this point (152). Nevertheless, inhibition of acid sphingomyelinase by imipramine led to a reduction in parenchymal damage in a mouse model of IR, suggesting that ceramide accumulation contributes to TNF-α-dependent ROS/RNS formation and cell death in IR (119).
At a later time point (>2 h) after TNF-α stimulation, the third branch of the death signaling cascade is activated (134) (Fig. 7, pathway C). Complex 1-derived TRADD and RIP1 bind Fas-associated protein with death domain, procaspase 8, and RIP3, which leads to the formation of complex 2 (134). Complex 2 subsequently releases activated caspase 8, which silences the RIP1/RIP3 complex and cleaves the Bcl-2 protein Bid into tBid (234). Thereafter, tBid induces mitochondrial permeabilization via oligomerization of its Bcl-2 family members Bak and Bax in the outer mitochondrial membrane (56), a process that is facilitated by CL-OOH (123), which enables the release of cytochrome c and the initiation of apoptosis (56). At this point, the three branches of the death signaling cascade converge, as the JNK/ceramide-mediated mitochondrial formation of ROS/RNS is necessary for the formation of CL-OOH and the detachment of cytochrome c from the inner mitochondrial membrane, (i.e., events that are a prerequisite for the initiation of apoptosis) (150) (section “Mitochondria”).
However, when caspase 8 is inactivated in fibroblasts, the RIP1/RIP3 complex triggers MPT-dependent necrosis rather than apoptosis via upregulation of the tricarboxylic acid cycle (231), resulting in an increased NADH/NAD+ ratio and hence, mitochondrial ROS/RNS formation (141), as well as ATP depletion due to inhibition of the adenine nucleotide translocator (196). Considering that the active site of caspase 8 is a cysteine that is susceptible to reversible oxidation and subsequent inactivation by H2O2 in vitro (16), and possibly also peroxynitrite (100), the activity of caspase 8 is probably directly dependent on the cytosolic redox status. Therefore, severely oxidatively/nitrosatively stressed cells could be more likely to undergo necrosis than apoptosis when stimulated by TNF-α because of oxidative caspase 8 inactivation.
As previously mentioned, complex 1 activates a survival pathway parallel to the death signaling cascade (78) (Fig. 7, pathway D). The survival pathway is initiated by activation of the IκB kinase complex, which subsequently phosphorylates nuclear factor of κ light polypeptide gene enhancer in B cells inhibitor α (IκB-α) (62). Phosphorylation of IκB-α enables its dissociation from NF-κB, which thereafter translocates to the nucleus to initiate the transcription of anti-apoptotic and antioxidative proteins such as MnSOD (219), the ferritin heavy chain (156), and cellular FLICE-like inhibitory protein (c-FLIP) (133). When sufficiently activated, the effects of NF-κB impede the death signaling cascade so effectively that, without its inhibition, the transcription of antioxidative (e.g., MnSOD and ferritin) and anti-apoptotic proteins (e.g., c-FLIP) swiftly abrogates the effects of phosphorylated JNK and complex 2, respectively (133).
The activity of NF-κB is, however, inhibited by S-glutathionylation of a specific cysteine on the β-subunit of the IκB kinase complex, which impairs IκB-α phosphorylation (174) and hence its dissociation from NF-κB, as well as a cysteine on the p50 subunit of NF-κB itself, which inhibits NF-κB binding to DNA and subsequent transcription of target genes (157). Accordingly, primary hepatocytes that are incubated with ONS-inducing agents such as glucose oxidase, which generates H2O2, antimycin, which inhibits the ETC at complex III, or diamide, which depletes GSH, undergo either apoptosis or necrosis after stimulation with TNF-α, depending on the extent to which the stressors were applied (76). Inhibition of IκB-α phosphorylation and suppression of NF-κB translocation to the nucleus were proposed as causative factors of the observed effects (76). As mentioned earlier, cytosolic ONS occurs in NAFLD as a direct effect of extramitochondrial FA oxidation (1) (section “Extramitochondrial FA oxidation”) and as an indirect effect of mitochondrial GSH depletion (187), which facilitates efflux of ROS/RNS into the cytosol (77). Thus, the viability of healthy hepatocytes is not affected by TNF-α because of adequate activation and unimpaired NF-κB signaling (180), whereas oxidatively/nitrosatively stressed steatotic hepatocytes that are stimulated with TNF-α have a predilection for apoptosis (233) because of NF-κB inhibition (Fig. 8). Accordingly, apoptosis is a prominent feature of NASH (48). Moreover, when acute intensification of ONS occurs in combination with severe ATP depletion, as for instance during IR (103), additional modulation of the death signaling cascade in the form of caspase 8 inactivation and sustained JNK phosphorylation likely directs steatotic hepatocytes toward necrotic rather than apoptotic cell death following stimulation with TNF-α (Fig. 8). Considering that TNF-α is profusely secreted during IR (200), pervasive parenchymal necrosis is likely to occur in IR-subjected fatty livers.
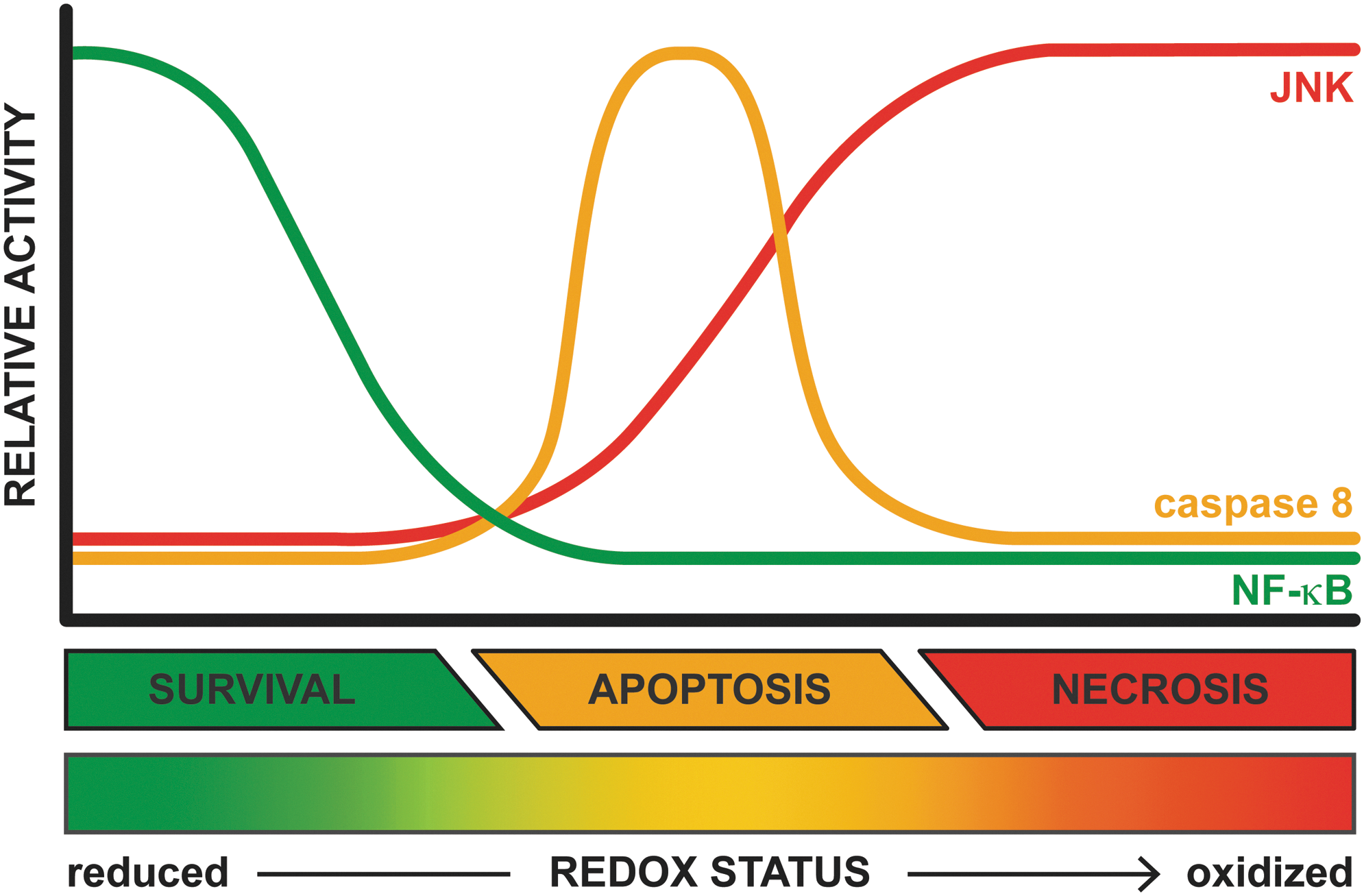
Taken altogether, TNF-α is an important factor in the interplay between NAFLD and IR in which ONS sensitizes hepatocytes to cell death rather than proliferation upon stimulation with TNF-α. TNF-α is a confirmed inducer of apoptosis in NAFLD, whereas the preferred mode of cell death shifts toward necrosis when fatty livers are subjected to severe ONS in combination with high levels of TNF-α and depletion of intracellular energy stores, all of which are cardinal features of IR.
Other sources of ROS/RNS in NAFLD
Extramitochondrial FA oxidation
Mitochondria are supported in the oxidation of FAs by peroxisomal β- and microsomal ω-oxidation; pathways that are controlled by PPAR-α (169) (Fig. 9). Peroxisomal β-oxidation is mainly responsible for shortening very long chain FAs that cannot be directly processed by mitochondrial β-oxidation (169). Microsomal ω-oxidation, located in the ER and usually a minor contributor to FA oxidation, involves ω-hydroxylation of medium and long chain FAs (169). This reaction, which is catalyzed by CYP4A enzymes, generates dicarboxylic acids that are further metabolized by peroxisomal β-oxidation (168, 169). In addition, ketone-containing catabolites produced through mitochondrial β-oxidation as well as the FAs AA and lauric acid are processed by another member of the CYP family, namely CYP2E1 (114). In contrast to the other pathways, CYP2E1 is not regulated by PPAR-α but is induced by several other NAFLD-related phenomena such as increased levels of saturated FAs, insulin resistance, ketosis, and obesity (114, 176).
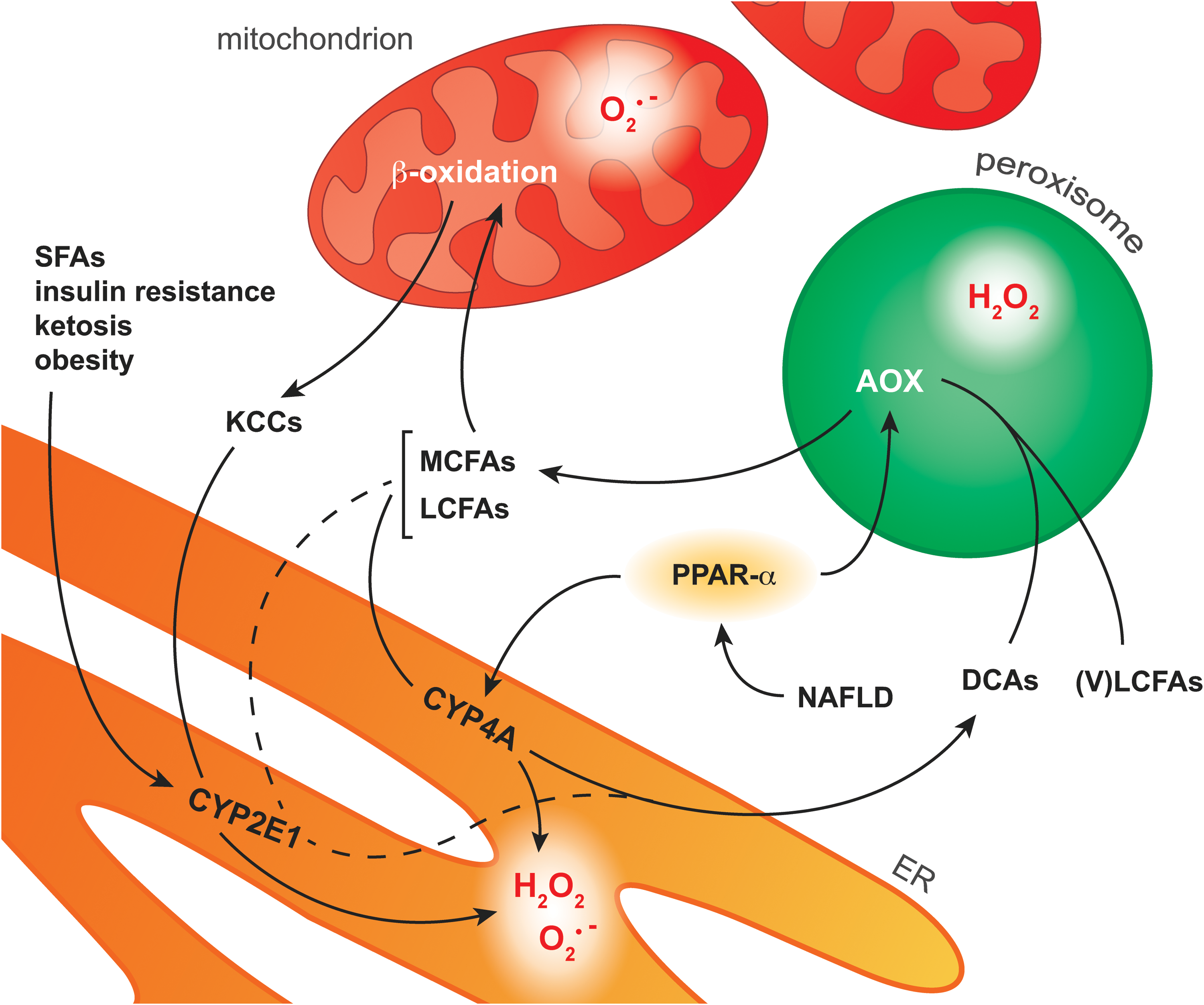
Similar to the mitochondrial pathway, extramitochondrial FA oxidation also generates ROS as byproducts. All CYP enzymes possess a heme center that catalyzes the mono-oxygenation of substrate(s) using O2 and electrons derived from the oxidation of NADPH (227). However, at different stages of the catalytic cycle, oxygen can be prematurely released from the reduced complex, thereby generating O2 •− or H2O2 (227). It is likely that peroxynitrite is also formed when iNOS is concomitantly induced (section “ROS/RNS and their chemical properties in the context of NAFLD and IR2”). Insofar as CYP2E1 is the most inefficient or ‘leaky’ CYP isoform, it generally exhibits the largest ROS-generating potential (64). In addition to the microsomal pathway, H2O2 is formed in the first step of peroxisomal β-oxidation, which is catalyzed by the FAD-containing enzyme AOX (168). In this reaction, AOX oxidizes fatty acyl CoA to enoyl CoA, by which the FAD subunit of AOX is reduced to FADH2. To regain catalytic activity, FADH2 is oxidized to FAD in the presence O2, reducing O2 to H2O2 (106).
Although all extramitochondrial pathways constitute a source of ROS and possibly RNS, much attention has been focused on CYP2E1 as a contributor to ONS in NAFLD (176). Not only could the ability of microsomes to oxidize lipids be strongly inhibited by a specific CYP2E1 antiserum (108), but CYP2E1-null mice fed a high-fat diet also showed significantly lower levels of MDA and protein carbonyls as well as reduced JNK phosphorylation compared with wild-type controls (1). The increased activity of CYP2E1 in NAFLD has also been confirmed in patients. CYP2E1 protein levels are increased in NASH-affected livers compared with matched controls (25). In addition, an increased CYP2E1 to total CYP protein ratio was reported for patients with NAFL (209), collectively attesting to the fact that CYP2E1 contributes to the ONS observed in NAFLD (176).
Furthermore, PPAR-α activation by non-esterified PUFAs is expected to contribute to ONS via the induction of CYP4A enzymes and peroxisomal β-oxidation (168) (Fig. 9). However, a PPAR-α agonist (Wy-14,634) actually attenuated fibrosis in mice that were fed a methionine- and choline-deficient diet, despite increased CYP4A expression (91). Moreover, mice nullizygous for AOX, the H2O2-generating enzyme in peroxisomal β-oxidation, spontaneously develop severe steatohepatitis due to the inability to metabolize very long chain FAs, which accumulate, and dicarboxylic acids (80), which damage mitochondria (168). The ability of PPAR-α to increase FA metabolism (169), which prevents dicarboxylic acid-dependent mitochondrial dysfunction as well as the accumulation of peroxidizable PUFAs (168), and to induce UCP2 expression, which reduces mitochondrial ROS/RNS formation (section “Mitochondria”), actually curtails ONS despite the concomitant upregulation of ROS-producing enzymes (91).
Collectively, these findings indicate that peroxisomal β-oxidation and CYP4A are sources of ONS in NAFLD. However, the beneficial effects of inhibiting these ROS-producing pathways are outweighed by the corollary accumulation of harmful substrates (e.g., dicarboxylic acids). Moreover, ample evidence points to PPAR-α-independent CYP2E1 as a significant contributor to cytosolic ONS in NAFLD.
Unfolded protein response
Another potential source of ROS/RNS in NAFLD is the UPR, which is triggered by proteins that are incorrectly assembled and/or processed by the ER, generally resulting from the inability of the ER to deal with perturbations in cellular homeostasis (i.e., ER stress) (86). In NAFLD, ER stress results from elevated levels of saturated FAs (213) and ONS (81), which is underpinned by the causal relationship between obesity and ER stress (151). The ER has three sensors that initiate the UPR: inositol-requiring enzyme 1α, activating transcription factor 6, and double-stranded RNA-dependent protein kinase-like ER kinase (PERK) (232). Of these three, increased phosphorylation of the PERK-regulated transcription factor eukaryotic initiation factor 2 α (eIF2α) has been observed in both NAFL- and NASH-affected patients (163), confirming that the UPR is activated in fatty livers.
The UPR likely contributes to ONS in NAFLD via three routes. First, the redox-active enzymes protein disulfide isomerase (PDI) and ERO1 constitute a source of ROS. PDI and ERO1 regulate protein tertiary structure by catalyzing the formation of intramolecular disulfide bridges (65). In the terminal step of this redox process, the FADH2 subunit on ERO1 reduces O2 to H2O2 and thus regains its oxidizing potential (68). When the ER protein folding demand rises during ER stress, ERO1 and PDI enter a perpetuating cycle of what has been described as ‘futile refolding attempts’ (181), thereby amplifying H2O2 formation (81). Second, ER stress leads to Ca2+ translocation from the ER to the mitochondrial matrix (65), causing increased mitochondrial ROS/RNS formation due to Ca2+-dependent upregulation of the tricarboxylic acid cycle (18), which can induce MPT when the rise in intramitochondrial Ca2+ is sizeable enough (155). Third, inositol-requiring enzyme 1α activates JNK (202), as has been confirmed in NASH-affected human livers (163). Given the fact that activated JNK stimulates mitochondrial ROS formation (207) these findings point not only to ER stress as a source of ROS/RNS in NAFLD but also as a direct contributor to its development, as phosphorylated JNK also induces insulin resistance (3) (section “Non-alcoholic fatty liver disease”).
Normally, PERK concomitantly mobilizes an antioxidant salvage pathway to curtail its own effects (36, 113). In NAFL- and NASH-affected patients, however, there was no increased action of activating transcription factor 4 (163), which is controlled by the PERK-eIF2α signaling axis and usually replenishes the antioxidant reserves, (e.g., by increasing the cellular GSH pool) (113). Considering that the ER typically consumes the newly formed GSH to reduce the disulfide bonds of incorrectly assembled proteins (37), the failure to upscale GSH synthesis not only impacts the cellular antioxidative capacity but also further increases ER stress and ROS production. Although PERK is able to augment the antioxidative capacity via activation of the antioxidant response gene nuclear factor (erythroid-derived 2)-like 2 (Nrf2) (36), which has been confirmed in NAFLD-affected human livers (79), its beneficial effects are likely insufficient to effectively combat the concomitant ONS (79, 107).
In conclusion, the UPR contributes to cytosolic as well as mitochondrial ROS/RNS production, while the concurrent induction of antioxidative defenses either fails or is inadequate in countering ONS.
Concluding Remarks
The exuberant formation of ROS/RNS is a cardinal feature of NAFLD as well as IR and, although the underlying pathological processes differ, several ROS/RNS-mediated effects are common in both conditions. At the molecular level, H2O2 and peroxynitrite are important sources of toxic derivative radicals, particularly in the presence of free Fe2+, which initiate the (per)oxidation of lipids and proteins that leads to depletion of the intracellular antioxidative capacity, functional impairment of biomolecules, and the formation of toxic metabolites.
At a cellular level, ONS sensitizes steatotic hepatocytes to apoptotic and necrotic cell death. UCP2-dependent ATP depletion and ONS render the mitochondria of fatty hepatocytes prone to MPT, which becomes especially relevant during IR. Consequently, DAMP release, KC activation, and leukocyte chemotaxis will be more prolific, prompting severe extracellular ONS and TNF-α release that ultimately culminate in widespread parenchymal necrosis.
ROS/RNS predispose the fatty liver to more severe IR injury via a plethora of damaging effects. Notably, these effects emanate from specific (bio)chemical mechanisms that can serve as targets for interventions that are aimed at reducing ONS-mediated liver injury. Much research so far has been devoted to the role of ONS and mitochondrial uncoupling in either NAFLD or IR, whereas only a few papers address the interplay between both conditions (42, 45, 118, 127, 147, 187). Therefore, more research on this topic is warranted given the increasing prevalence of NAFLD and corollary rise in surgical interventions in patients with fatty livers.
Footnotes
Author Disclosure Statement
R.F.v.G. is supported by a PhD Scholarship from the Academic Medical Center (University of Amsterdam), and M.H. is supported by grants from the Dutch Anti-Cancer Foundation (Stichting Nationaal Fonds Tegen Kanker) in Amsterdam, the Phospholipid Research Center in Heidelberg, the Nijbakker-Morra Foundation in Leiden, and Stichting Technologische Wetenschap (STW). T.M.v.G. and M.J.R. state that no competing financial interests exist.