
Abstract
Chronic obstructive pulmonary disease (COPD) and lung cancer are frequently caused by tobacco smoking. However, these diseases present opposite phenotypes involving redox signaling at the cellular level. While COPD is characterized by excessive airway epithelial cell death and lung injury, lung cancer is caused by uncontrolled epithelial cell proliferation. Notably, epidemiological studies have demonstrated that lung cancer incidence is significantly higher in patients who have preexisting emphysema/lung injury. However, the molecular link and common cell signaling events underlying lung injury diseases and lung cancer are poorly understood. This review focuses on studies of molecular mechanism(s) underlying smoking-related lung injury (COPD) and lung cancer. Specifically, the role of the ceramide-generating machinery during cigarette smoke-induced oxidative stress leading to both apoptosis and proliferation of lung epithelial cells is emphasized. Over recent years, it has been established that ceramide is a sphingolipid playing a major role in lung epithelia structure/function leading to lung injury in chronic pulmonary diseases. However, new and unexpected findings draw attention to its potential role in lung development, cell proliferation, and tumorigenesis. To address this dichotomy in detail, evidence is presented regarding several protein targets, including Src, p38 mitogen-activated protein kinase, and neutral sphingomyelinase 2, the major sphingomyelinase that controls ceramide generation during oxidative stress. Furthermore, their roles are presented not only in apoptosis and lung injury but also in enhancing cell proliferation, lung cancer development, and resistance to epidermal growth factor receptor-targeted therapy for treating lung cancer. Antioxid. Redox Signal. 21, 2149–2174.
III. The Role of Ceramide in Oxidative Stress-Induced Lung Epithelial Apoptosis
V. Smoking and Lung Diseases: The Enigmatic Association Between Lung Injury and Lung Cancer
VI. The Dichotomous Response of Airway Epithelial Cells to CS Oxidants: A Critical Role for Src
VII. The Unexpected Role of Ceramide in Cell Proliferation and Tumorigenesis
IX. Updates on Novel Unresolved Complexities in the Ceramide-Generating Machinery
I. Introduction
C
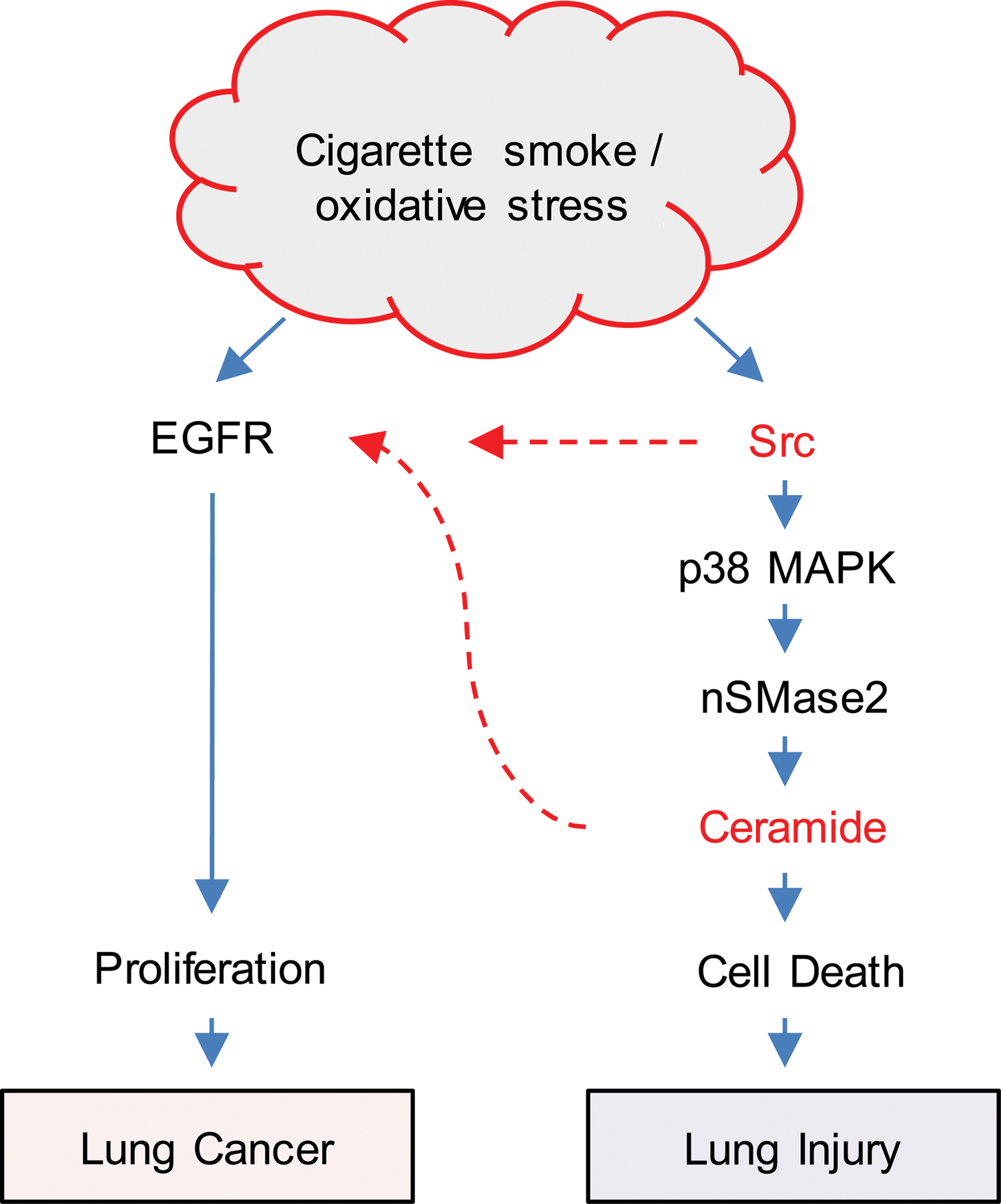
Despite sharing a common etiology of tobacco smoke exposure, COPD (which includes chronic bronchitis in the main airways and emphysema in the parenchymal tissue) and lung cancer differ at the cellular level. Epithelial cell death, which results in tissue destruction and injury, is one of the main features of COPD, whereas lung cancer is characterized by uncontrolled clonal growth of the lung epithelial cells. Furthermore, many epidemiological studies over the years have established increased lung cancer risk and incidence in COPD patients (79). Intriguingly, smoking cessation does not significantly reduce the risk of developing lung cancer, which may be explained by persistent lung oxidative stress and inflammation even after smoking cessation (7). For example, a large prospective trial of nonsmokers demonstrated that lung cancer incidence is common in patients with nonsmoking COPD (211), emphasizing that oxidative stress and chronic inflammation, which are the characteristics of all patients with COPD, could be common determinants in the association between COPD and lung cancer. In addition, mild-to-moderate disease stages of COPD patients (GOLD 1 and 2 standards, respectively) present even higher risk for developing lung cancer than severe (GOLD 4) ones (44). While longer survival of early-stage COPD patients can explain this phenomenon, it is unlikely to be the only reason. That withstand, this association between COPD and lung cancer is not surprising because cancer has been long considered a chronic “wound that does not heal,” which occurs in conditions such as chronic oxidative stress and inflammation mirrored in all patients with COPD (52). Therefore, injury-initiated repair mechanisms could represent an opportunity for cancer to develop. However, the molecular mechanism(s) and cell signaling pathways underlying these clinical observations are still poorly understood (Fig. 1).
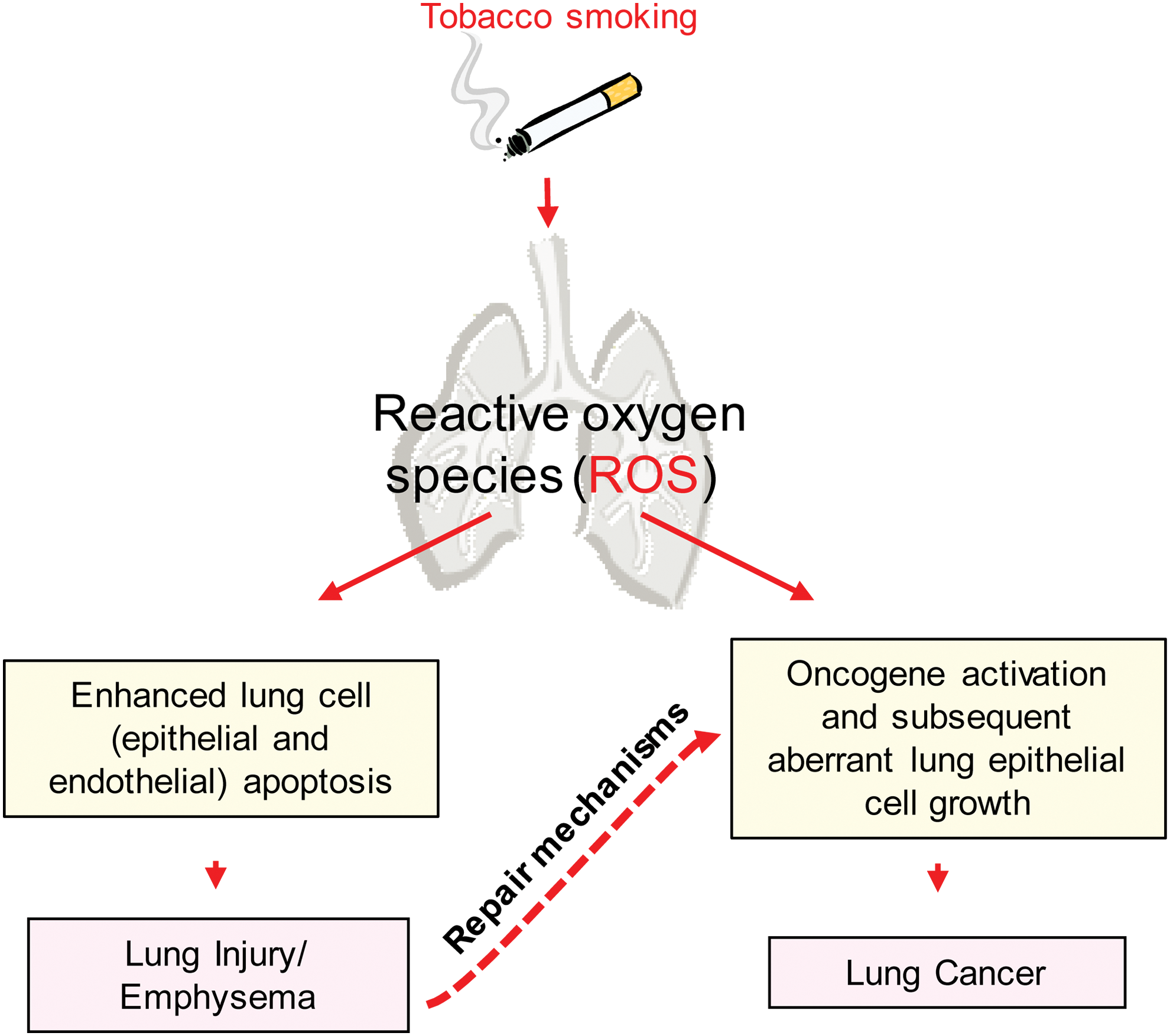
An increasing number of studies demonstrate that increased oxidant burden in smokers contributes to lung injury through many biological actions, including epithelial injury via apoptosis (79). Moreover, markers of oxidative stress (pathological accumulation of reactive oxygen species [ROS]), such as hydrogen peroxide (H2O2), are elevated in the breath and serum of COPD patients (200) and documented to be present in all stages of COPD (62). At the same time, exactly how oxidative stress incites COPD association with lung cancer is poorly understood at the molecular level, despite a role for oxidative stress having widely been proposed in cancer initiation and promotion (65, 89, 143). Such molecular underpinning could possibly be found at both genetic and epigenetic levels, and therefore, further studies are needed in these directions (3). For example, Malhotra et al. found that a reduction in the activity of the transcription factor nuclear factor (erythroid-derived 2)-like 2 (Nrf2) reduces the expression of antioxidants, such as heme oxygenase-1 (HO1) and glutamate–cysteine ligase in COPD patients, thus increasing oxidative stress markers (140). Similar findings were demonstrated in vitro by Goven et al. (82) and ex vivo (83) in lung biopsies from emphysema patients. The downregulation of Nrf2-dependent genes that are involved in the detoxification of CS constituents could lead to enhanced carcinogenic potential and also increase the metastasis of lung cancers (24).
Notably, most molecular studies in airway epithelial cells center on the mechanism(s) of either cell death or proliferation (76, 118, 119, 176, 177). However, cell death and hyperplasia of airway epithelial cells as well as infiltration of inflammatory cells occur simultaneously during lung injury and repair, as documented by animal- and cell-level studies (45, 53, 96, 115, 147, 164, 179, 200, 235). Thus, the mechanisms of cell death and proliferation in the lung constitute two sides of the same coin (Fig. 1). Since these two events are intimately related with each other (50), the scope of this review is not only to present evidence underlying cell death (lung injury) and cell proliferation (lung cancer) during CS-induced oxidative stress but also to discuss a possible molecular interplay between the two pathological conditions. Studies by Goldkorn et al. demonstrated that the oxidative stress component of CS, an equivalence of 200–600 μM H2O2 generated in situ per cigarette, is the driving force behind both smoke-induced cell death (via ceramide generation) and smoke-induced proliferation (via epidermal growth factor receptor [EGFR] activation) (77, 80, 118, 119, 133, 134). These studies are discussed herein together with novel concepts regarding the dichotomous roles of Src in regulating both the ceramide-generating machinery and aberrant EGFR signaling in the pathology of airway epithelial cells exposed to CS-induced accumulation of ROS (CS/oxidative stress). The goal is to provide breath in a challenging and mostly undefined research field that will lead to a better understanding of the molecular connections between smoking-related lung injury and lung cancer.
II. Oxidative Stress and Pulmonary Disease
Oxidative stress reflects an imbalance between the systemic manifestation of ROS and a biological system's ability to readily detoxify the reactive intermediates and to repair the resulting damage. ROS are a group of ubiquitous molecules that include species, such as superoxide anion (O2 −), H2O2, and hydroxyl radicals (•OH). Given that ROS are involved in multiple biologic processes and signal cascades that include normal tissue homeostasis (101, 192), changes to local and global ROS levels directly contribute to the development of many human diseases. In particular, this review focuses on lung diseases since the respiratory system is constantly exposed to gaseous oxygen and ROS, which are quelled by nonenzymatic and enzymatic antioxidant defenses, including glutathione (GSH), superoxide dismutase, and catalase (91). However, when the lung is chronically exposed to oxidants such as ozone (O3) (154) or to those in tobacco smoke, these mechanisms become overwhelmed, leading to the development of pulmonary diseases (144, 235).
Initially, the involvement of ROS in disease was explained with simplistic chemistry in which critical cell proteins and lipids were randomly oxidized and became inactive for their roles in normal cell function (90). Accordingly, antioxidants such as GSH or α-tocopherol were viewed simply as free radical scavengers. More recently, however, ROS have been recognized to function as signaling molecules. Several studies demonstrate the various roles for ROS in the pathogenesis of pulmonary diseases, such as acute respiratory distress syndrome, COPD, asthma, interstitial pulmonary fibrosis (9, 90, 101, 173 –175), and lung cancer (58, 61, 119). Among these diseases, both COPD and lung cancer share a common etiology rooted in exposure to tobacco smoke (80%–90% of the cases), a major source of oxidative stress (as described in Sections III and IV) (Fig. 1).
A. Smoking, oxidative stress, and inflammation
Despite the well-documented dangers of smoking, millions of people continue to smoke cigarettes daily, each puff containing roughly 5000 toxic compounds in addition to over 1015 free radicals, such as H2O2 and hydroxyl and organic radicals in the gaseous phase (30). Importantly, toxic oxidants in tobacco smoke have a detrimental effect on epithelial cells lining the airway, causing both oxidative stress and inflammation, which contribute to several lung pathological conditions, including COPD (79, 184).
COPD is a lung disease endemic among smokers, with little to no therapeutic options. Such lung injury diseases adversely affect more than 16 million people in the United States, with another 50 million estimated to be at risk (2, 155). Up to 85% of COPD cases are directly attributed to smoking and smokers are 10 times more likely to develop COPD than nonsmokers. Importantly, these diseases have no treatments that effectively restore function because they involve destructive and lasting enlargement of distal airspaces and alveolar walls (1, 54, 169), which eventually leads to impaired oxygenation of the blood.
The pathogenesis of emphysema has historically been attributed to protease-antiprotease imbalance resulting from chronic lung inflammation (210). Initially, genetic deficiency in α-1 antitrypsin, a major elastase inhibitor released by neutrophils, was attributed to hastened development of emphysema and led to the hypothesis that levels and activity of proteases and antiproteases play a critical role in emphysema pathogenesis (139). Since then, several studies have shown that chronic exposure to inhaled irritants (the most common being tobacco smoke) leads to chronic inflammation in the lung, during which recruited inflammatory cells release various proteases that destroy the extracellular matrix and lead to the loss of alveolar units (152). Specifically, exposure to tobacco smoke has been shown to increase gene expression and release of cytokines, such as interleukin-8 and tumor necrosis factor-α (TNF-α), in both human airway epithelial (HAE) cells and murine models (31, 32, 148). Subsequent recruitment of neutrophils to the lung leads to localized inflammation, during which apoptosis of irreparable cells and proliferation/differentiation of progenitor cells drive tissue repair (97). At the same time, recruited neutrophils and resident alveolar macrophages secrete matrix metalloproteases (MMPs), such as MMP-2 and MMP-9, and serine proteases, which act to breakdown the extracellular matrix to generate niches for newly dividing cells (207). Importantly, these normal processes become usurped during chronic exposure to tobacco smoke, contributing to pathological alveolar and overall lung destruction. However, the concept of inflammation as the initiator of the cascade of events in lung destruction in diseases such as COPD has recently been challenged. Instead, inflammation could represent the result of a long-standing destructive process in the lung and by itself might be a source of additional injury.
B. Smoking and emphysema/lung injury: the role of apoptosis in the disease development
Recent studies provide a very strong case for cell death having a major role in lung injury in several smoking-related pulmonary diseases. In simple terms, the loss of cells by augmented apoptosis would be expected to be involved, or perhaps, initiate the overall tissue destruction responsible for smoking-induced lung injury (45, 54, 96, 115, 117, 164, 191, 200, 235). This “cell death” hypothesis arose in part due to the observations that smokers with emphysema have increased levels of apoptotic alveolar cells compared to smokers without emphysema (116). Subsequent studies demonstrated that experimental induction of either vascular endothelial cell apoptosis or pulmonary epithelial cell apoptosis in animal models resulted in airspace enlargement (45, 75, 169). For instance, when mice were given a single intracellular injection of active caspase-3 (executioner of apoptosis), the treatment resulted in alveolar epithelial apoptosis followed by alveolar wall damage and airspace enlargement (8, 209). Loss of cells by elevated apoptosis is thus a crucial initiator of the overall tissue damage in COPD (Fig. 1), which could explain the unique nature of alveolar septal damage in emphysema when compared with other lung diseases that are also characterized by inflammation and elevated matrix proteolysis. The irreversible pattern of the disease even when the patient stops smoking (209) could well be accredited to the frequent interface between apoptosis, inflammation, oxidative stress, and matrix proteolysis.
Given the critical role of apoptosis in the pathogenesis of emphysema/lung injury, ceramide, a sphingolipid molecule that serves as a second messenger that modulates cell apoptosis (22, 77, 80, 92, 130, 178), oxidative stress (130), and proteolysis (180), was proposed to be deregulated during the pathogenesis of lung injury diseases, such as COPD, causing induction of alveolar destruction via apoptosis of epithelial and endothelial cells (45, 59). Therefore, the elucidation of the molecular mechanism(s) that modulate the ceramide-generating machinery in the lungs is essential and could lead to the development of improved mechanism-specific drugs for the treatment or prevention of pulmonary injury diseases.
III. The Role of Ceramide in Oxidative Stress-Induced Lung Epithelial Apoptosis
As mentioned before, epithelial cells line both the lung main airways and the terminal alveoli and thus represent the lung's first line of defense—they are extensively exposed to reactive oxidants, such as those present in CS. As introduced earlier, cell signaling events that are initiated in HAE cells during CS exposure lead to either apoptosis (hallmark of injury) or proliferation (hallmark of cancer), as extensively explored both in vitro and in vivo (59, 119, 134). Notably, different stimuli in vitro acting at diverse sites to activate ceramide accumulation trigger apoptosis (178). Specifically, exogenous C6-ceramide was sufficient to induce apoptosis in human lung epithelial cells, supporting the concept that elevated ceramide levels, per se, is sufficient to initiate the apoptotic cascade in lung epithelial cells and that ceramide accumulation is a causative signal for apoptosis induction (178) and not just an outcome of epithelial cell death.
The initial studies addressing oxidative stress-induced lung injury investigated whether HAE cells undergo apoptosis when exposed to H2O2, concentrations of about 50–250 μM, and consequently whether this process is mediated by ceramide generation (22, 77). It was shown that either the administration of exogenous H2O2 or the enhancement of endogenously generated H2O2 was effective in depleting cellular GSH and initiating ceramide generation and apoptosis events (178). Both a dose-dependent relationship (50–250 μM H2O2) and a time-dependent relationship were observed between H2O2 exposure and increased cellular ceramide levels and subsequent apoptosis. Interestingly, even a short 1-h exposure of cells to 250 μM H2O2, followed by growth in regular medium, was sufficient to induce apoptosis (130).
As a ubiquitous antioxidant, GSH, a tripeptide (L-γ-glutamyl-L-cysteinyl-glycine) containing a sulfhydryl group that enables it to be a key intracellular reducing agent, is an essential component of the antioxidant defense mechanism in oxidant-induced lung injury. Notably, both lung epithelial cells and the epithelial lining fluid contain high concentrations of GSH (130). Importantly, Lavrentiadou et al. demonstrated that low GSH levels were essential for ceramide generation, whereas high GSH levels inhibited the production of ceramide in HAE cells (in vitro), which showed that GSH plays a critical role in preventing lung epithelial cell death (130). This demonstrated that the events that control the fate of the cells occur within this hour, during the time shown that GSH is depleted and ceramide is generated (130). This initial model for H2O2-induced cellular oxidative stress and ceramide generation consisted of direct exposures of airway epithelial cells to H2O2 and GSH or N-acetyl cysteine (NAC), a cell-permeable precursor of GSH. Moreover, an increase of intracellular H2O2, mediated by inhibition of catalase by aminotriazole, also induced ceramide generation and apoptosis (118, 130, 178). Although these early studies undertook a reductionist, simplified approach, both scenarios were relevant to the lung epithelium. H2O2 is a ubiquitous molecule that is freely miscible and able to cross cell membranes readily and is present in several air pollutants, including the tobacco smoke (119). It is detected in exhaled air of humans (222), and the amounts of exhaled H2O2 appear greater in subjects with pulmonary disease (205) and in smokers.
Later, in vitro studies showed that CS exposure regulates both growth and death of airway epithelial cells via its H2O2 component (119, 130): CS exposure could generate between 100 and 800 μM H2O2 in situ, as measured in the media of cultured HAE cells, a factor that was dependent on the length of exposure and the amount of combusted tobacco. Furthermore, when the CS-generated H2O2 was quenched by antioxidants, such as GSH or NAC, no enhanced ceramide generation was observed (119). Since then, it has been shown that exposure to oxidative stress generated by either CS exposure or direct administration of H2O2 leads to an increase of ceramide level before caspase-3-dependent apoptosis in airway epithelial cells, mimicking the dominant apoptotic process of emphysema pathogenesis (178). Following this discovery, the potential enzymatic regulators controlling this phenomenon were investigated.
A. Neutral sphingomyelinase 2-induced ceramide generation as a specific target in CS-induced lung injury
Ceramide generation through sphingomyelin hydrolysis is known to have a key role in various types of cell stress induction. While several sphingomyelinases (SMases) may be able to accomplish such ahydrolysis, only some SMases may have unique pathological importance (20, 21, 34, 59, 60, 77, 79, 158, 178, 182, 212, 224). Initial in vitro studies showed that an SMase(s) operating at neutral pH was responsible for ceramide generation in HAE cells exposed to oxidative stress (133). Therefore, a considerable effort of purification and cloning of a putative protein from the lungs of nonhuman primates and HAE cells led to the independent isolation and characterization of a novel SMase, neutral sphingomyelinase 2 (nSMase2; SMPD3 gene), as the only accountable source for ceramide generation as a result of oxidative stress induction (133).
Subsequently, it was shown that nSMase2 was activated in HAE cells (in vitro) not only by H2O2-generated oxidative stress but also in response to CS exposure and subsequent oxidative stress (133, 134). Moreover, silencing of nSMase2 expression using siRNA fully abolished ceramide production and the downstream apoptotic outcomes in response to oxidative stress. At the same time, mice heterozygous for nSMase2 (+/−) demonstrated significantly reduced lung ceramide generation when exposed to CS. Intriguingly, nSMase2 also is overexpressed in human emphysema patients (smokers) and rodents chronically exposed to CS. These studies clearly implicated nSMase2 activation and the subsequent ceramide generation in lung airway epithelial injury during CS-induced oxidative stress (59) (Fig. 2).
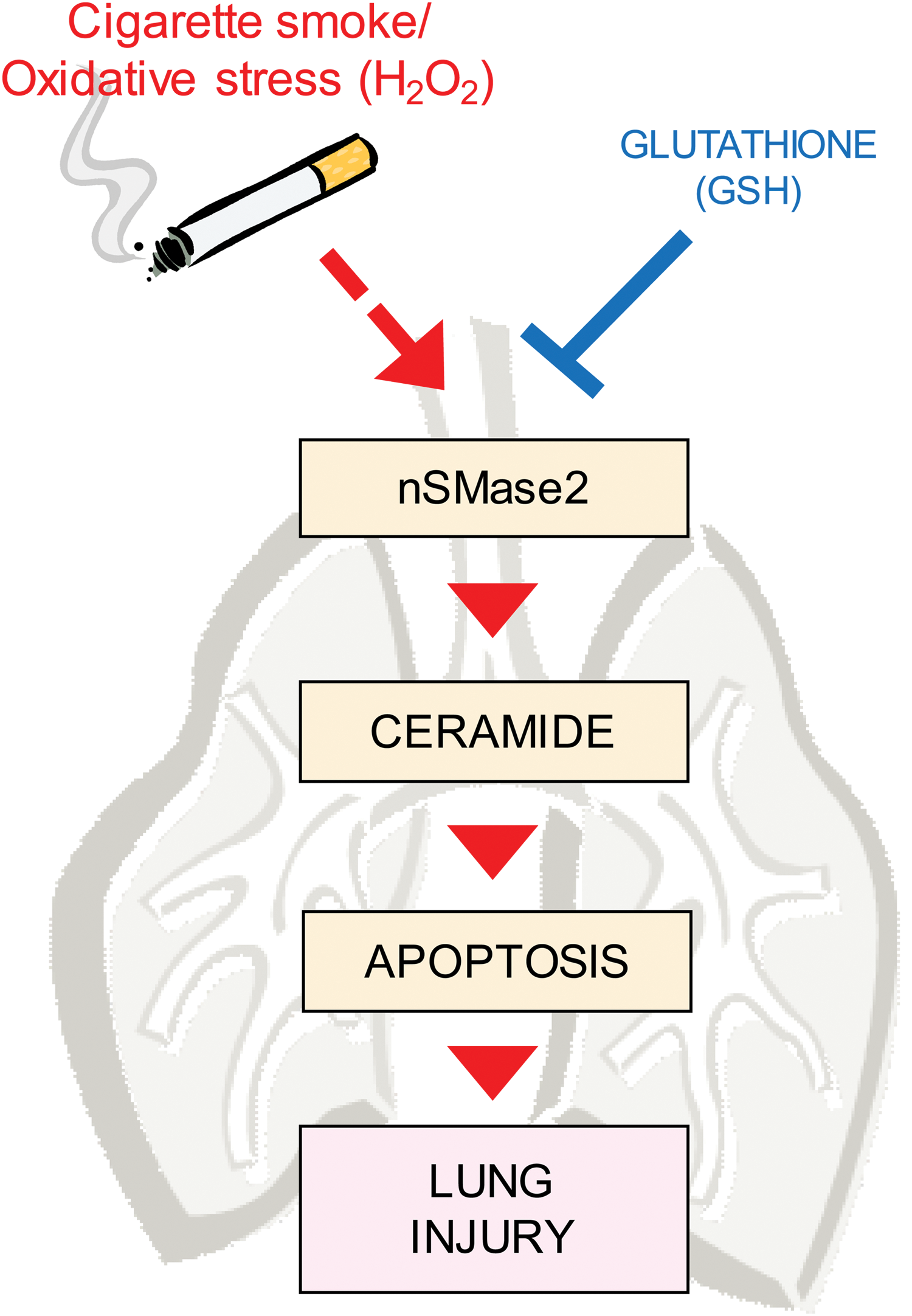
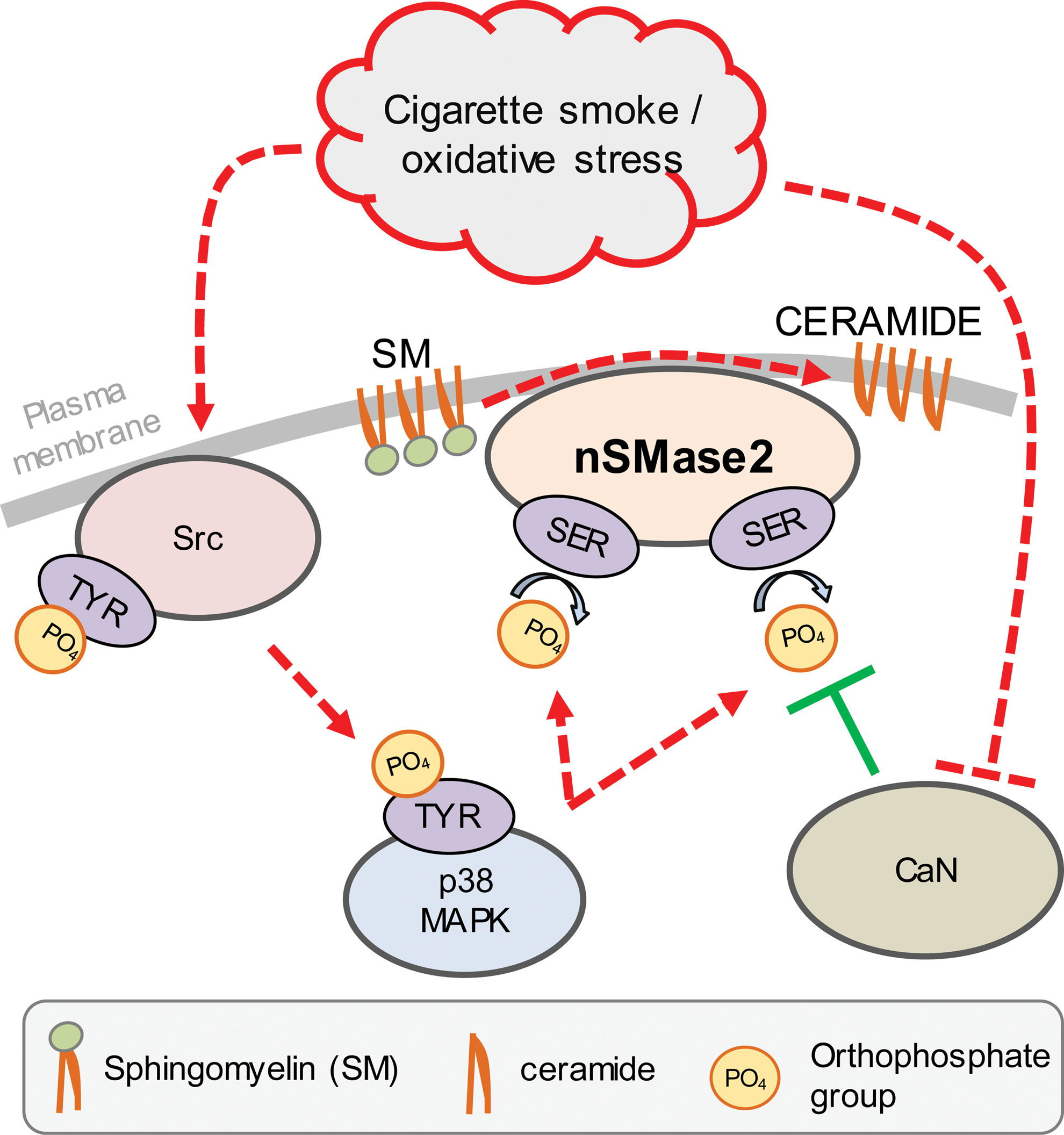
The molecular mechanism that controls nSMase2 function in response to oxidative stress has been recently unraveled by showing that nSMase2 is regulated by an intricate phosphorylation-based machinery (60). Specifically, nSMase2 phosphorylation on specific serines causes both its activation and protein stabilization upon exposure of HAE cells to oxidative stress (56). In addition, some of the protein regulators of nSMase2 phosphorylation were identified. For example, calcineurin (CaN) phosphatase was shown to bind directly to nSMase2 (60). However, during H2O2-induced oxidative stress, CaN becomes inhibited by oxidation of its catalytic cysteine and is degraded, increasing nSMase2 phosphorylation and activity (56, 60).
B. The surprising role of Src in controlling nSMase2/ceramide generation
The nonreceptor tyrosine kinase (RTK) Src is the first identified proto-oncogene, known for both its overexpression and overactivation in various cancers, including non-small cell lung cancer (NSCLC) (73, 202, 236). In contrast, its role in injury-related pulmonary diseases remains unexplored. Several studies demonstrate that Src is activated by CS exposure in an oxidative stress-dependent manner (57, 118, 119), under the conditions for which nSMase2-dependent ceramide generation and apoptosis induction were observed (133, 134).Therefore, it has been recently investigated whether Src activity mediates nSMase2 function and ceramide upregulation during CS-induced oxidative stress of HAE cells.
Surprisingly, it was found that Src indeed upregulates nSMase2 activity and ceramide generation, via the intermediate p38 mitogen-activated protein kinase (MAPK), during oxidative stress exposure of HAE cells (29). Specifically, it was found upon direct H2O2 administration or CS-induced ROS generation (CS/oxidative stress) that inhibition of either Src or p38 MAPK suppresses nSMase2 activity, nSMase2 phosphorylation, and subsequent ceramide generation (29). Importantly, the oxidative stress-dependent activation of p38 MAPK occurs downstream of Src (29) (Fig. 3). It was previously shown by Clarke et al. (35) that p38 MAPK had some role in controlling nSMase2 function. In addition, Schweitzer et al. also proposed a role for p38 MAPK as an upstream regulator of nSMase2 and ceramide-dependent disruption of the lung epithelial–endothelial barrier (190). The studies by Chung et al. (29) add to these findings by demonstrating the unique role of Src in driving ceramide generation/accumulation during CS-induced oxidative stress. At the same time, a recent publication from Cinq-Frais et al. claims that Src can also be activated downstream of nSMase2 and ceramide generation (33) in response to oxidative stress, which implicates the potential existence of a feedback loop that potentiates these signaling events.
Most importantly, as reported by Filosto et al. (59) and Petrache et al. (169), ceramide accumulation in alveolar cells is a dominant driver of smoking-related emphysema pathogenesis. Within this context, a recent article by Geraghty et al. (72) presented that Src inhibition in smoke-exposed mice prevented airspace enlargement in their lungs. Moreover, the same study showed that Src inhibition in HAE cells prevented CS-induced signaling of EGFR, whose aberrant activation was suggested to be Src-related (57). This is very relevant to lung cancer development (as further described in Section VI). Thus, the potential involvement of Src, a known proto-oncogene, in the development of emphysema presents far reaching implications in the age-old conundrum of why smokers (active and former) are prone to both COPD and lung cancer development.
IV. Oxidative Stress and Lung Cancer
Clinical epidemiology and anecdotal accounts have long revealed an intimate relationship between mainstream tobacco smoke exposure and development of lung cancer. Despite exhaustive preclinical and clinical studies investigating drivers of this disease, the prognosis of metastatic lung cancer still remains very poor with the 5-year survival rate hovering between 5% and 15% (110). However, recent progress has increased the understanding of key biological pathways in lung carcinogenesis (37, 86, 100, 187).
Localized chronic inflammation in the lung is observed in countless NSCLC patients. As discussed earlier in section II, the chronic nature of smoking-related inflammation in the lung results in remodeling of the extracellular matrix by protease release/activation, which not only encourages vascularization of the tissue but also promotes repair mechanisms, including epithelial cell proliferation—characteristics that are common and critical for various cancers (204).
At the same time, there are several classical and novel proto-oncogenes and onco-suppressors characterized in lung cancer, for which systematic understanding requires the reading of the referenced articles (48, 86, 220). Within this section, we will specifically focus on two players: the EGFR and Src, which are highly activated in the lungs of COPD (70) and NSCLC patients (236) with smoking history and which become activated by CS in vitro and in vivo.
A. Stress-driven endocytosis of tyrosine-phosphorylated EGFR leads to tumorigenesis: the critical role of oxidative stress
The airway epithelium and the plasma membrane of its cells serve as the first barrier against native gaseous oxidants and particulate matter, species that are in extreme high levels in tobacco smoke. Although such exposure to tobacco smoke can initiate apoptotic pathways and trigger lung injury, as discussed above, it can also drive cell proliferation by its simultaneous stimulation of EGFR in airway epithelial cells (80, 177).
Seminal studies over the years demonstrated that the EGFR plays a prominent role in lung cancer development. Importantly, past reports have shown that cigarette smoking not only augments EGFR expression in human bronchial epithelium (12, 159, 171) but also abnormally activates the receptor kinase (119) (Fig. 4). EGFR activation is thought to promote malignancy through its role in proliferation, angiogenesis, metastasis, and apoptosis inhibition. Although EGFR is expressed in all cells of epithelial origin, a higher level of EGFR expression has been reported in squamous cell carcinoma (50%–80%) (99, 100), which occurs more frequently in smokers and men (233). Moreover, EGFR overexpression is observed in tumors from more than 60% of patients with metastatic NSCLC and is correlated with poor prognosis (195). Although smoking has been established as the most important preventable cause of lung cancer, it has been recently demonstrated that somatic mutations of the EGFR are responsible for ∼10%–25% of all nonsmoking-related lung cancer cases (153, 165). These findings have since provided rationale for the development of EGFR-targeted and now Food and Drug Administration (FDA)-approved cancer therapeutics known as tyrosine kinase inhibitors (TKIs).
More than 3 decades of research have established a fairly defined mechanism of EGFR activation, internalization, and subsequent downregulation upon ligand (EGF) binding to the receptor extracellular domain. However, much less is known about the ligand-independent mechanisms of EGFR activation and trafficking. These “alternative” mechanisms can generate a hallmark of severe human pathological conditions, particularly in cancer, and thus defining them is extremely relevant to the mission of understanding and fighting against EGFR-driven tumorigenesis.
1. Canonical EGFR activation, intracellular trafficking, and degradation
The EGFR (ErbB1) is a member of the ErbB family of RTKs, which also includes ErbB2, ErbB3, and ErbB4. These receptors have a vital role in normal cellular processes, such as cell division, differentiation, and migration, and their overexpression or deregulation has been linked to a variety of human cancers (189, 231, 232). Hence, their activation, particularly that of the EGFR, has been a subject of intense studies.
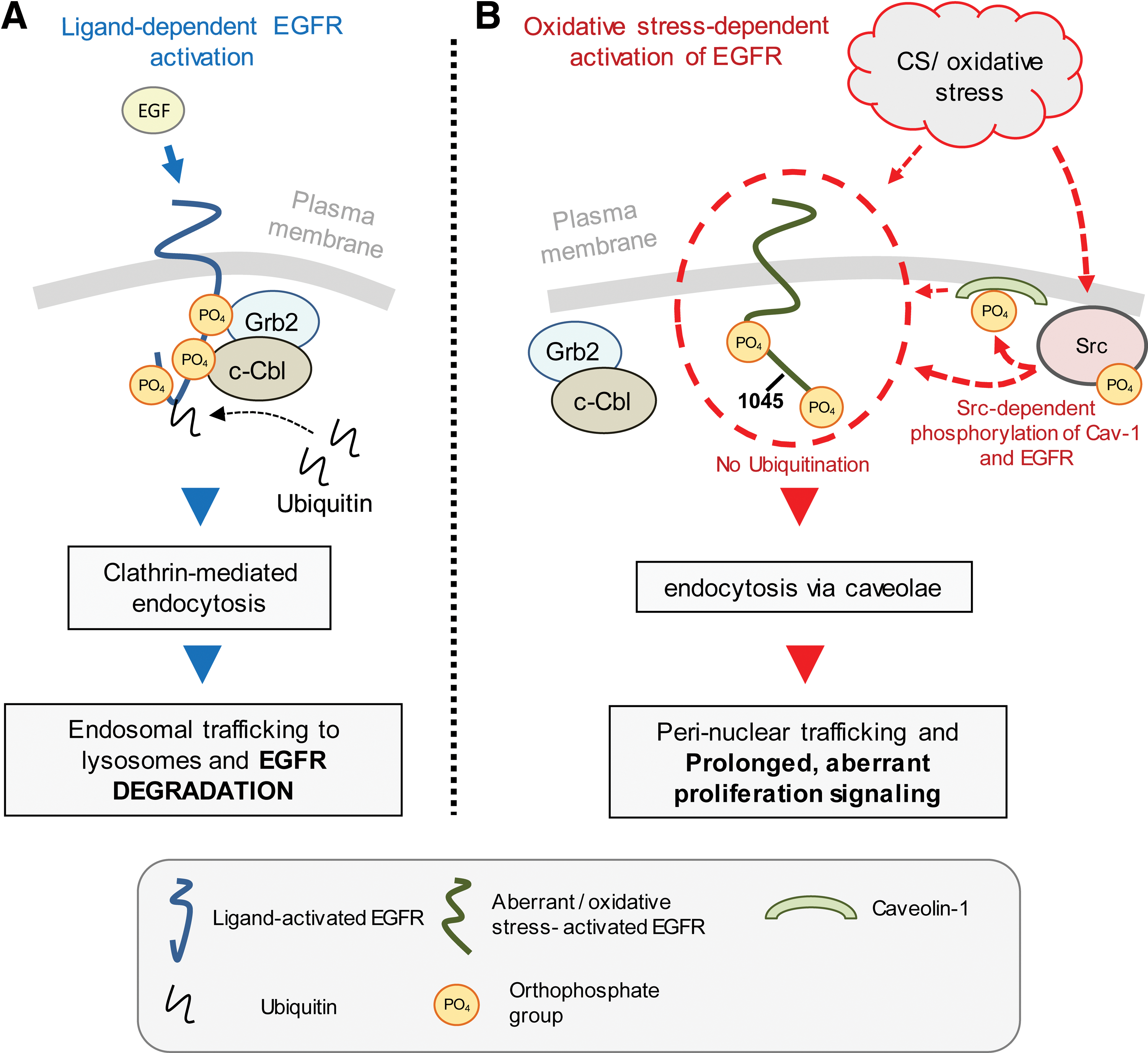
The model of EGFR activation has been established wherein ligand binding induces receptor dimerization, leading to the activation of its intrinsic tyrosine kinase activity, autophosphorylation, and subsequent phosphorylation of downstream signaling molecules (188, 189) involved in cellular survival and proliferation. Upon EGF binding, the EGFRs are rapidly internalized from the cell surface through numerous pathways, including clathrin-coated pits (219). Therefore, to control cellular growth and tumorigenesis, the activation of the EGFR has to be tightly regulated in a process that includes degradation of the receptor. Indeed, the inability of the EGFR to be downregulated via clathrin-mediated endocytosis and degradation has been linked to its oncogenicity (64).
Although the exact mechanism by which the EGFRs are recruited into the clathrin-coated pits is under investigation, the general paradigm of EGFR internalization is as follows. The EGF-activated EGFRs recruit growth factor receptor-bound protein 2 (Grb2), which plays a critical role in the binding of adaptor protein 2 complex (AP-2) (103, 111). AP-2 subsequently recruits clathrin (cytosolic scaffold protein that mediates vesicle formation) to the plasma membrane, which polymerizes to form a clathrin coat that provides the framework required for vesicular budding into the cell and eventual internalization of the activated EGFRs (170).
Internalized RTKs, such as the EGFR, undergo a general modulatory phase consisting of a two-step endocytosis process (86, 142, 219, 221): (1) A fast ligand-dependent internalization step removes activated receptors from the cell membrane and segregates them in endosomes, followed by either (2) sorting of internalized RTK molecules to lysosomes, where they go through degradation, or recycling back to the plasma membrane. Notably, EGFR classified for lysosomal degradation requires the recruitment of cellular-casitas B-lineage lymphoma (c-Cbl), which controls EGFR ubiquitination (132). Expectedly, molecular studies impeding clathrin-mediated endocytosis resulted in no subsequent lysosomal degradation of the ligand-activated EGFR (106, 160, 188). This could maintain prolonged downstream activation of prosurvival and proliferation molecules, such as Akt and extracellular regulated protein kinase (ERK1/2) (71), thereby enhancing cell proliferation and tumorigenesis (42).
2. CS produces H2O2-induced oxidative stress that aberrantly activates EGFR
Since H2O2 is known to be a major component of the gas phase of CS (157), a study was undertaken by Khan et al (119) to determine whether CS has a similar effect as H2O2 on EGFR activation in HAE cells, which included primary immortalized and transformed cells.
First, it was demonstrated that CS could generate comparable amounts of H2O2 that were generated in previous experiments. These earlier studies used artificially produced H2O2 by complementing the cells with high glucose media and glucose oxidase (GO) (118, 176, 177). Next, it was not only shown that CS can activate the EGFR in a dose- and time-dependent manner but also that it aberrantly phosphorylated the receptor with a pattern of phosphorylation sites that was different from that induced by the ligand, EGF, but identical to the phosphorylation pattern induced by GO (namely H2O2). Moreover, the EGFR was not activated and very little or no H2O2 could be detected in cell culture medium exposed to CS in the presence of GSH (119), an antioxidant that targets reactive radicals, including ROS, such as H2O2 (91). This demonstrates that the oxidative stress component of CS was necessary for the activation of EGFR in HAE cells exposed to CS. Notably, Ganesan et al. recently presented an aberrantly activated EGFR receptor in COPD patients (70). Moreover, Geraghty et al. demonstrated in mice exposed to CS that the proto-oncogene Src promoted the EGFR activation in the airway epithelium, thereby enhancing airway inflammation and lung tissue destruction (72). These findings attempted to provide an acceptable mechanism connecting the molecular origins of COPD and lung cancer.
Several in vitro reports demonstrated the inactivation of protein tyrosine phosphatases (PTPs) by H2O2 and thus suggested that this was the cause responsible for EGFR-enhanced tyrosine phosphorylation under oxidative stress (46, 69, 76, 131, 227). For example, data published by Xu et al. indicated that H2O2 induced phosphorylation of EGFR on Y1068 while PTPs activity was reduced to ∼70% of control. Although PTPs inactivation certainly contributes to the enhanced phosphorylation of EGFR upon H2O2 exposure, Khan et al. and Reynolds et al. showed that H2O2 could not induce phosphorylation of a kinase-dead EGFR (118, 181), suggesting that the kinase activity of the receptor is also required for its activation/phosphorylation by H2O2. In addition, the sole inactivation of PTPs cannot explain the change in the pattern of EGFR tyrosine phosphorylation. It was reported that under both CS-induced H2O2 and direct H2O2 exposure, the display of EGFR phosphorylation sites differs from that induced by EGF binding to the receptor. Particularly, EGFR tyrosine (Tyr) Y845 is robustly phosphorylated under H2O2 or CS exposures, whereas Y1045 phosphorylation is totally absent. This ultimately results in an active EGFR that is unable to undergo normal downregulation, demonstrating impaired trafficking and degradation. More explicitly, upon H2O2-induced oxidative stress, the aberrantly activated EGFR is unable to bind c-Cbl and thus is neither ubiquitinated nor targeted to lysosomes for degradation. Instead, EGFR is strongly associated with the phosphorylated caveolin-1 (Cav-1) in an Src-dependent manner and thus is recruited into caveolae and not into clathrin-coated pits (76, 118, 119, 176, 177). Such oxidative stress-dependent impairment of the EGFR clathrin-mediated endocytosis and lysosomal degradation (106, 160, 188) results in prolonged downstream activation of prosurvival and proliferative signals, such as of Akt and ERK1/2 (71). This coincided with enhanced cell proliferation (215) and was shown to facilitate tumor promotion processes in the epithelial cell line T51B (105) as well as to mediate tumor promotion in other nonneoplastic rat liver epithelial cells (42).
Intriguingly, a previous report by Bae et al. indicated that the ligand, EGF-induced EGFR phosphorylation, required the presence of intracellular H2O2. Overexpression of catalase could quench the receptor canonical activation/phosphorylation by EGF. However, the H2O2 generation itself appeared to be EGFR kinase dependent because it was abolished in the presence of a kinase-dead EGFR (10). Conversely, Ushio-Fukai et al. (214) showed no effect of the antioxidant, NAC, on EGF-stimulated activation of EGFR. These authors also claimed that angiotensin II-dependent transactivation of EGFR was the one dependent on ROS generation and was indeed abolished in the presence of the antioxidant NAC (214). The use of different cell types can partially explain the discrepancies in the findings of various studies carried out by different groups; however, one can also speculate that while a local and transient generation of H2O2 has a physiological role in the ligand-initiated signaling, a pathological imbalance in the cell redox potential due to enhanced H2O2 accumulation can lead to an aberrant EGFR activation accompanied by an abnormal downstream signaling. In line with this idea, Reynolds et al. (181) also demonstrated a H2O2-dependent inactivation of PTPs as one of the mechanisms inducing EGFR lateral mobility at the plasma membrane and subsequent amplification of the receptor phosphorylation and signaling (181). In addition, the same study suggested that the H2O2 produced physiologically in response to EGF stimulation may have a different cellular localization compared to the H2O2 generated pathologically by environmental causes. The first was described as spatially constrained to a layer below the plasma membrane (181), whereas the latter, the exogenously administrated H2O2, is also found in the endomembranes, which could be one of the reasons for its aberrant pathological effects. Of note is that the “physiological” H2O2 was never observed in the endomembranes.
Altogether, the above findings indicate that the exposure of cell to pathological H2O2/oxidative stress could induce nonligand-bound EGFR phosphorylation based on both EGFR aberrant kinase activation and PTPs inhibition. Moreover, such oxidative stress-driven EGFR phosphorylation/signaling is not restricted to the plasma membrane compartment.
As discussed, H2O2 can affect both the EGFR kinase itself and other EGFR regulators, including the PTPs. Therefore, to gain more insight into H2O2-induced EGFR signaling and hyperplasic responses, the role of the E3 ligase c-Cbl, as a possible link between oxidative stress, EGFR signaling, and tumorigenesis, was further examined.
3. The lack of c-Cbl binding to EGFR causes prolonged proliferation signaling under oxidative stress
It was well established that upon EGF stimulation of cells, c-Cbl binds directly to the EGFR via Tyr-1045 (176) and indirectly through the SH3 domain of Grb2 (66). c-Cbl binding and its consequential phosphorylation results in the activation of the E3 ligase activity of c-Cbl, recruitment of the ubiquitin-conjugating enzyme Ubc-H7 (234), and EGFR ubiquitination.
On the other hand, upon mapping the EGFR phosphorylation sites, it was found that phosphorylation at Tyr-1045, the docking site for c-Cbl (132), was abrogated under oxidative stress, and thus, the receptor could not be ubiquitinated and degraded. Therefore, it was suggested that this deficiency might have a key role in linking oxidative stress, the EGFR, and tumorigenesis by conferring prolonged receptor signaling (177).
To gain a better understanding of how a receptor lacking c-Cbl binding may lead to tumorigenesis, the mutant EGFR (Tyr-1045 to Phe) was ectopically expressed in CHO cells to determine whether the lack of phosphorylation at this site is indeed the only cause for prolonged EGFR retention at the membrane under oxidative stress. However, additional findings suggested that the inability of the EGFR to bind c-Cbl under oxidative stress is not solely due to its abrogated Tyr-1045 phosphorylation because the Y1045F mutant was still able to bind c-Cbl, probably indirectly via Grb2 (176).
Indeed, other studies suggested that c-Cbl is recruited to the activated EGFR through both direct and indirect binding (112, 218). It has been shown that the direct interaction of c-Cbl with EGFR is mediated through the phosphorylated Tyr-1045 on EGFR (132), whereas the indirect c-Cbl-EGFR interaction is maintained via SH2/SH3 Grb2 domains (67, 111). Consistently, Huang and Sorkin (104) reported that knockdown of Grb2 by RNA-interference inhibited clathrin-mediated endocytosis of the EGFR after EGF exposure of the cells because of the impaired recruitment of the c-Cbl RING domain to EGFR. Of note is that the two binding sites of Grb2 to the EGFR, Tyr-1068 and Tyr-1086, are phosphorylated under H2O2 exposure, but Grb2, which is still bound to c-Cbl, does not bind the EGFR. Furthermore, Shc, whose phosphorylation and binding to Grb2 had been shown to be responsible for Grb2 recruitment to the EGFR (111), is still phosphorylated under H2O2 exposure and is still able to bind to the EGFR. Therefore, most possibly Grb2 fails to bind to the EGFR under oxidative stress exposure due to conformational changes in the EGFR. Consistently, Goldkorn and colleagues (58, 61) have shown that oxidative stress induces a novel active kinase conformation of the receptor, which is described in detail below in section IV.A.5.
Undoubtedly, the studies with the Y1045F mutant (MT) EGFR (176) led to a better understanding of why c-Cbl fails to bind to the EGFR under oxidative stress, which is due to both the abrogation of phosphorylation of Tyr-1045 and the lack of Grb2 binding, thereby resulting in a receptor unable to undergo normal internalization through the early endosomes and subsequent downregulation (176) (Fig. 4).
When is c-Cbl actually required for EGFR sorting? Early on, several studies led to some confusion about when and where c-Cbl is required for EGFR internalization, as well as the role of EGFR Tyr-1045 phosphorylation in c-Cbl recruitment. For example, Jiang and Sorkin (112) showed data suggesting that the Y1045F EGFR MT was internalized despite its inability to undergo ubiquitination, whereas Mosesson et al. (151) demonstrated that the Y1045F MT is internalization-resistant. At the same time, Duan et al. (51) showed in c-Cbl−/− mouse embryonic fibroblasts that the EGFR could still be internalized following EGF stimulation.
To gain a better understanding of the consequences of EGFR inability to recruit c-Cbl under oxidative stress, the exact cellular compartment where c-Cbl-mediated ubiquitination is necessary for EGFR sorting was investigated. In particular, Ravid et al. showed (176) that the administration of PP1 (an Src family kinase [SFK] inhibitor) to A549 cells blocked the EGF-induced phosphorylation of c-Cbl (but not of the EGFR) and the ubiquitination of the EGFR, without inhibiting the internalization of the receptor into early endosomes. Notably, an efficient binding of c-Cbl to the EGFR was observed upon EGF stimulation in the presence of PP1 (in both wild-type [WT] and Y1045F MT receptors), which implies that the tyrosine phosphorylation of c-Cbl is not required for its binding to the EGFR or for its role in the entry of EGFR into the early endosomes. At extended time points of cells treated with PP1 and EGF, the EGFR remained associated with EEA1, an early endosome marker. Only after PP1 removal and the subsequent recovery of c-Cbl phosphorylation and EGFR ubiquitination, EGFR could exit from the early endosomes. Therefore, it was concluded that the c-Cbl-dependent ubiquitination is required for the exit of EGFR from the early endosomes.
In summary, the major findings of that study (176) were that c-Cbl binding to the EGFR is sufficient to enhance receptor internalization, whereas the E3 ligase activity of c-Cbl and EGFR ubiquitination are required for EGFR trafficking out of the early endosomes and eventual transport to the lysosome for the degradation of the receptor. In addition, that study also suggested that the EGFR lack of Tyr-1045 phosphorylation during exposure to oxidative stress is not the only reason for the receptor inability to enter the late endosomes. Rather, the role of Grb2 in the recruitment of c-Cbl as an adaptor added an additional level of complexity to the mechanism of EGFR sorting under oxidative stress.
Importantly, the degree of intricacy was further expanded by other studies that demonstrated that several proteins with ubiquitin interaction domains are in fact required for EGFR transfer from the early endosomes to other vesicular bodies for degradation (11, 213). For example, the proteins Hrs and Tsg 101 were shown to be involved in a large sorting complex that is “somehow” responsible for coupling EGFR transfer between early and late endosomes. Although the detailed mechanism has yet to be identified, one can imagine a situation where this large protein sorting complex binds to the ubiquitinated EGFR, thus allowing its transfer into the late endosomes.
4. EGFR perinuclear sorting under oxidative stress
In light of the above studies, the next question was how the EGFR is specifically sorted under oxidative stress. Another study (118) suggested that, under oxidative stress, EGFR is able to undergo clathrin-independent endocytosis and is sorted to a perinuclear compartment, where it is not degraded and remains active. The mechanism of this trafficking involves Src activation by H2O2-induced oxidative stress, which subsequently phosphorylates Cav-1 at Tyr-14 and triggers the caveolar endocytosis of EGFR (Fig. 4).
Cav-1 and Cav-2 heterooligomerize and form caveolae, which are integrated into the lipid rafts (63, 68, 128, 156, 172). It has been suggested that Cav-1 can function in caveolae in a manner analogous to the way clathrin adaptors draw membrane receptors to coated pits and/or drive membrane invagination and budding (172). Cav-1 is known to interact directly with many signaling molecules through its caveolin-scaffolding domain at residues 82–101 (38, 107). Indeed, EGFR has been reported to interact with the caveolin-scaffolding domain through a caveolin-binding sequence motif located in the intracellular kinase domain (residues 898–905) of the receptor (39, 107). To elucidate the mechanism by which EGFR is being trafficked to the perinuclear compartment under oxidative stress, the involvement of Cav-1 was investigated.
Expressing a WT Cav-1 or a Tyr-14 MT (Y14A) Cav-1 in a cell culture model, it was demonstrated that EGFR is constitutively associated with Cav-1. However, Cav-1 is phosphorylated on Y14 only in the presence of H2O2 and is subsequently accumulated together with EGFR in the perinuclear compartment. When either Src was inhibited or when the Y14A Cav-1 MT was overexpressed, the accumulation of EGFR was not observed at that unusual site (118).
Despite some reports that several SFKs are involved in oxidative stress-induced phosphorylation of Cav-1 (19, 185), the observation that EGFR Tyr-845, a c-Src target (16), is robustly phosphorylated under oxidative stress suggests that c-Src involvement is not coincidental. Consistently, Dittmann et al. have recently described a radiation-induced mode of EGFR trafficking to the nucleus, and the specific knockdown of c-Src (by siRNA) blocked EGFR phosphorylation at Y845 as well as the phosphorylation of Cav-1 at Y14 and resulted in the blockade of EGFR transport into the nucleus (49). It should also be noted that Sanguinetti and Mastick (186) have reported that c-Abl (Abelson murine leukemia viral oncogene homolog) kinase expression is required for oxidative stress-induced phosphorylation of Cav-1, although its eventual role in the perinuclear sorting of EGFR remains to be determined.
Although the above study (118) demonstrated that Src-mediated phosphorylation of Cav-1 Tyr-14 was necessary for caveolar endocytosis of EGFR under oxidative stress, the exact role of Cav-1 remains to be determined. Pelkmans et al. (167) suggested that the manner in which caveolar cargo is taken up and released depends on how the cargo interacts with Cav-1 as well as with other caveolar proteins and on how the cargo is influenced by compartment-specific signals, such a pH changes. The model thus far (118) indicates that H2O2 activation of EGFR results in aberrant receptor phosphorylation that precludes it from being sorted through clathrin-coated pits for eventual lysosomal degradation (176, 177). Concomitantly, H2O2 (but not EGF) activates c-Src, which in turn phosphorylates Cav-1 at Y14 (a proposed trigger for caveolar endocytosis) and also dynamin-2 (at Y231/Y597), which is thought to localize at the neck of caveolae such that the stimulation of its GTPase activity leads to vesicle fission (161, 193, 194). Through this route of caveolar endocytosis, H2O2-activated EGFR is trafficked to a perinuclear region where continued receptor signaling is identified via Tyr-1173 phosphorylation, potentially contributing to prolonged proliferative signaling (61, 118) and tumorigenesis.
5. An aberrant activated conformation of EGFR under oxidative stress underlies lung cancer resistance to targeted therapy
The well spent effort to better understand the mechanisms of EGFR kinase activation was aimed to develop specific inhibitors, such as the TKIs. To date, several TKIs, erlotinib (Tarceva™) and gefitinib (Iressa™), are approved by the FDA for the treatment of metastatic lung cancer driven by the expression of oncogenic mutant EGFR while several second-generation TKIs are in clinical trials (e.g BIBW2992) or in various phases of drug development (55, 87, 217, 225).
Importantly, clinical use of TKIs is effective in a subset of lung cancer patients who express oncogenic, but also TKI-sensitive, EGFR mutations—exon 19 deletion (Δ746–750) and exon 21 single-point substitution mutation (L858R) (153, 165). Despite being first identified by its prevalence in NSCLC patients who were women of Southeast Asian origin and nonsmokers, it is important to note that the expression of the mutant EGFRs itself confers sensitivity to TKI therapy.
Interestingly, however, D'Angelo et al. have recently found that a significant number (40%) of those EGFR mutations (L858R and Δ746–750) are actually found in adenocarcinoma from both men and women who are current and former smokers (41). Although it is generally accepted that patients who are smokers do not benefit from TKI therapy, we still do not know whether lung cancer patients who carry the “TKI-sensitive” EGFR mutations and are former/current smokers would benefit from a TKI therapy (in comparison to those who are never-smokers). This is because those patients (harboring TKI-sensitive EGFR mutations and being smokers) were not rigorously screened and tested in any clinical trial assessing TKI effectiveness. Moreover, a recent anecdotal observation illustrated how patients responsive to TKI therapy acquire resistance to TKIs if they began smoking (Lara PN, personal communication, 2012).
Consistently, recent cellular studies on smoking-induced redox signaling and its impact on epithelial cell proliferation and tumorigenesis demonstrated that smoking-induced oxidative stress can indeed affect the sensitivity to TKIs therapy of both WT and oncogenic mutant EGFR overexpressing NSCLC cells (57, 58).
As described earlier, CS/oxidative stress activation of EGFR was found to be ligand-independent, did not induce “classical” receptor dimerization and was not inhibited by the TKIs AG1478, erlotinib (Tarceva) and gefitinib (Iressa) (58, 61). Thus, an unprecedented activated state of EGFR was described under oxidative stress. This activation mechanism was also shown to be temperature-dependent, suggesting the simultaneous involvement of the cell membrane state of fluidity (61).
Supporting these findings, an in vitro study by Burdick et al. presented only a partial reduction of EGFR phosphorylation by the TKI AG1478 following the exposure of breast cancer cells to H2O2 (18). Conversely, a previous report from Takeyama et al. claimed that the TKI AG1478 is effective against EGFR activation induced by CS (206). However, the technical conditions utilized by Takeyama et al. significantly differed from those used in the studies of Filosto et al. (57, 58). Takeyama's study used CS condensate/solution, whereas Filosto's study employed direct exposure of cells to the gas phase of CS (CS puffs) (58). In addition, Takeyama's study involved high concentrations (10 μM) of the TKI AG1478 without presenting any dose–response curves and without any data on the consequential EGFR phosphorylation. However, Takeyama's conclusions are supported, to some extent, by Hirota et al. studies (98). This group could observe a redox-sensitive (NAC-inhibited) transactivation of EGFR by TNF-α stimulation of NIH3T3 cells, which could be inhibited by 50–250 nM AG1478 TKI (98). Yet, these studies not only used fibroblasts (as opposed to lung epithelial cells) but also never exposed these cells directly to CS/H2O2 or to CS/H2O2 in the presence and absence of AG1478.
As concluded before (section IV.A.2), diverse outcomes could be typically expected in studies, carried out in different cells. Another important factor that may contribute to disparate results in EGFR signaling outcomes is the slightly dissimilar experimental conditions, which may lead to distinct cellular localization of the receptor. For example, the responses of EGFR under physiological concentrations of H2O2, which are strictly localized to the plasma membrane, are expected to significantly differ from responses under pathological higher concentration of H2O2 exposures, which could be diffused throughout the cell.
Recent data from Chung et al. (27), as well as from studies by Bublil et al. (17), demonstrated that EGFR does not need an extracellular ligand to form dimers. EGFR continuously changes from a monomer to a dimer state, where the interactions between the intracellular domains of the receptor are as important as those of the extracellular regions in forming dimers. These recent findings underlie the need for better understanding the mechanisms involved in ligand-independent activation of EGFR and the physiological relevance of such mechanisms.
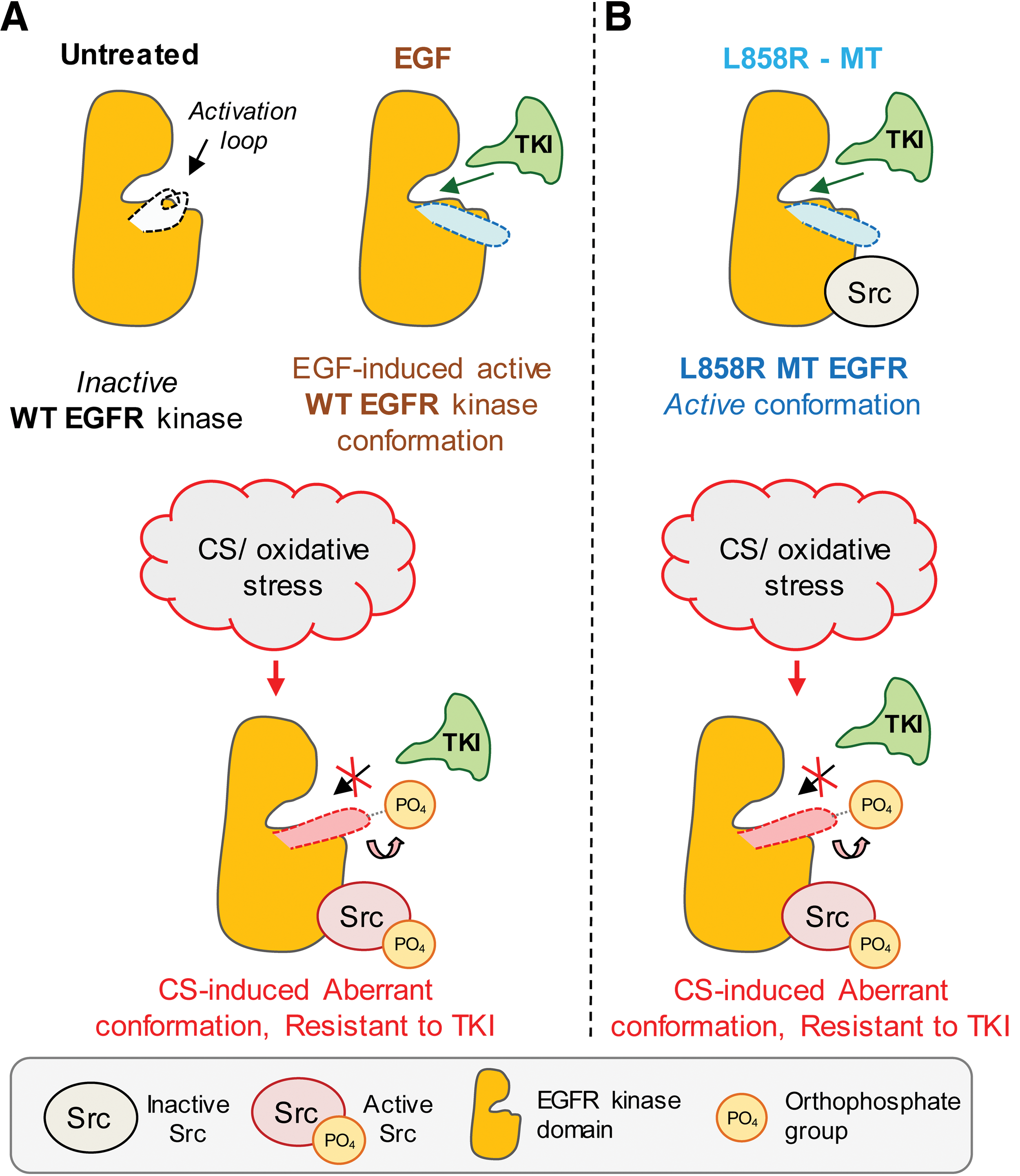
Very important indication that EGFR exposed to oxidative stress undergoes a unique conformational change was supported by the finding that under such conditions, EGFR could strongly bind c-Src. Moreover, the interaction between EGFR and c-Src was not dependent on the activation of c-Src because it persisted even in the presence of the c-Src kinase inhibitor, PP1 (57, 58, 61). Additionally, Kim et al. reported that under physiological conditions, c-Src could stably interact with ErbB2, but not with EGFR (ErbB1), because of the structural differences between ErbB1 and ErbB2 in the kinase domains of the two receptors (120). Consistently, studies with the L858R EGFR MT also demonstrated that this MT could bind c-Src, whereas the WT EGFR under physiological conditions could not. The L858R EGFR MT was crystallized and shown to possess a protein conformation that differs from that of the WT EGFR at the level of the kinase domain, carrying a constitutively open “activating-loop” (237). Interestingly, the L858R EGFR MT was also shown to have a similar functional phenotype to that of the WT EGFR exposed to oxidative stress, previously described by Goldkorn and colleagues (118, 176, 177). These unique features included prolonged phosphorylation/activation, lack of Y1045 phosphorylation followed by lack of ubiquitination, impaired trafficking and degradation, and constitutive interaction with c-Src, without any ligand stimulation (25, 26, 163, 196) (Fig. 5). This further supported the idea that H2O2 induces a conformational change in the intracellular kinase domain of EGFR. Accordingly, dimerization of the extracellular domain could not be captured by the EDAC [1-ethyl-3- (3-dimethylaminopropyl) carbodiimide] cross-linker neither for the WT EGFR under oxidative stress (58, 61) nor for the L858R MT.
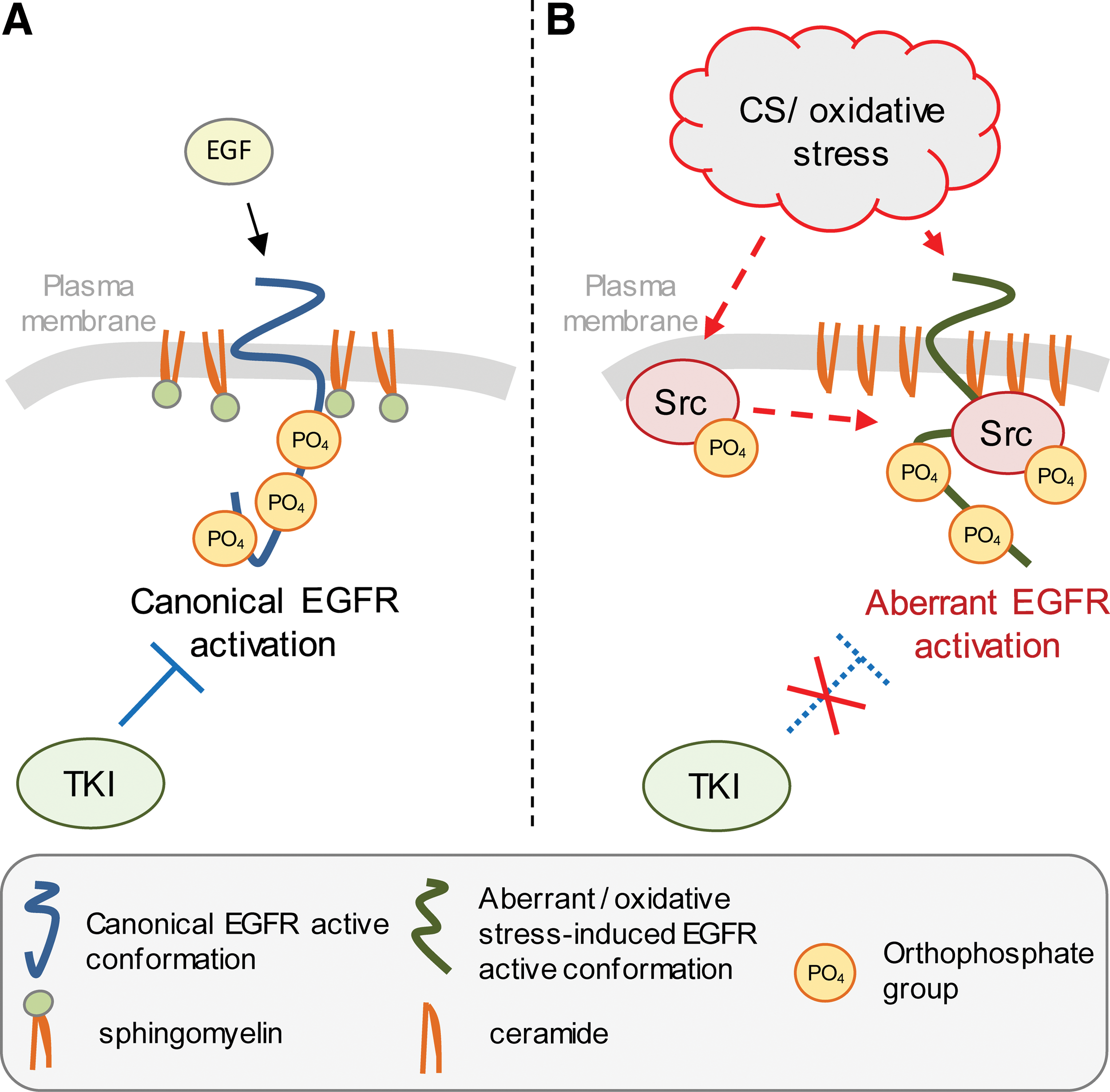
Intriguingly, although WT EGFR under CS/oxidative stress appears to acquire a novel activated conformation, that conformation seemed to be different from that of the L858R EGFR MT, which is known to be sensitive to TKIs (229). This was shown by employing a novel anti-EGFR antibody (α4-2 mAb) (58, 61) that is susceptible to the conformational changes induced by EGF binding to the receptor (108) or by the somatic mutation L858R. It was proven that CS/H2O2-generated oxidative stress induced a unique active conformation of the receptor (58, 61), which is different from that of both the EGF-induced WT EGFR and the L858R EGFR MT (Fig. 6). Furthermore, this distinctive conformation/activation mechanism appeared to be also temperature-dependent, suggesting the simultaneous involvement of changes in the membrane structure (61), as further discussed later on in section VII.E.
The above findings have important clinical implications in that the oxidative stress-activated EGFR cannot be inhibited by TKIs in NSCLC and may explain the ineffectiveness of these drugs in NSCLC patients with smoking history (58). Furthermore, aberrant EGFR activation has been recently shown by others to occur in the lungs of COPD patients (70, 223), who present chronic pulmonary oxidative stress (175), and, as mentioned above, have high risk for developing lung cancer (103). In addition, and very importantly, the proto-oncogene Src and its activation has been recently implicated in supporting the TKI-resistant conformer of EGFR (57), as described and reviewed in detail in section VI.
V. Smoking and Lung Diseases: The Enigmatic Association Between Lung Injury and Lung Cancer
Several prospective clinical studies have shown that lung cancer incidence is significantly increased in COPD patient populations (103). For instance, de Torres et al. reported that in their 5-year prospective cohort of former and current smokers, individuals with emphysema were more likely to develop lung cancer than those without emphysema (43): even the existence of some low-grade emphysema without noticeable airflow obstruction is associated with significantly elevated risk of lung cancer. Individuals with both emphysema and airway obstruction were at even higher risks than those with neither condition (43). Since the identification of mechanisms linking COPD and lung cancer has long been hampered by the heterogeneity of the disorders, it is still not known whether emphysema confers a local–regional risk for lung cancer or a global one (103).
Therefore, the clinical data may require a novel mechanistic understanding of how apoptosis-dependent lung injury may lead to cell proliferation-dependent lung cancer. Indeed, albeit originated by opposing cellular phenotypes (programmed cell death vs. cell proliferation), the two diseases may actually share some common cell signaling events. Numerous studies address these questions at various levels, such as genetic susceptibility, extracellular matrix/proteases, and inflammatory cells in the field of injury (103). Within this context, we herein describe in detail the novel example of the role of Src and the ceramide-generating machinery induced by CS-generated oxidative stress, as a possible signaling complex that regulates both epithelial lung injury and lung cancer as well.
VI. The Dichotomous Response of Airway Epithelial Cells to CS Oxidants: A Critical Role for Src
During CS-induced oxidative stress, Src plays a dichotomous role in airway epithelial cells (79). On one hand, ceramide, a “proapoptotic” sphingolipid, is generated by the induction of nSMase2 activity downstream of Src (29). On the other hand, the EGFR becomes aberrantly activated with the support from Src, promoting proliferative signaling and resistance to cancer therapy.
Src, the first proto-oncogene described, has previously been observed to be redox-sensitive, becoming activated by exogenous H2O2 (61) and CS (58).Owing to this shared stimuli between Src activation and CS-induced ceramide generation in HAE cells, unanticipated evidence was recently provided, demonstrating that Src activation upon CS/oxidative stress exposure controls nSMase2 function and subsequent ceramide generation at the plasma membrane of HAE cells (29), an accepted hallmark of apoptosis induction and lung injury (56, 59, 60, 114, 190).
At the same time, Src is a known key participant in the EGFR family pathway. Phosphorylation of several tyrosine residues within the EGFR has been shown to be increased following Src overexpression both in vitro and in vivo, indicating that Src is needed for full biological response following EGF stimulus (137, 208). Furthermore, using chimeric EGFR/ErbB2 receptors (137, 141), it was demonstrated that Src specifically binds to ErbB2, but not to WT EGFR or other ErbB family members. However, mutant EGFRs isolated from lung adenocarcinomas have the capacity to bind to Src, and these EGFR mutants require Src kinase activity for transformation (25, 141).
Recent in vitro studies of NCLSC cells exposed to CS provided some initial explanation to the dual role that Src may play under CS-induced oxidative stress. It was shown that the oxidative stress not only induced robust phosphorylation/activation of Src that subsequently activated nSMase2 to generate ceramide but also led the activated Src to strongly bind to the aberrantly CS-activated EGFR (58). Src mutants and inhibitors were used to analyze the role of Src in generating resistance to TKIs in EGFR-overexpressing NSCLC cells exposed to CS (57). The findings demonstrated that oxidative stress-activated Src is critical for the TKI-resistant phenotype observed in both the WT- and L858R MT EGFR-expressing cells during CS exposure, supporting the idea that targeting of Src during TKI treatment may overcome smoking/oxidative stress-related aberrant EGFR activation and resistance to therapy in NSCLC (Figs. 5 and 6).
The unprecedented, dual function suggested for Src and its effect on ceramide generation thus provided a potential mechanistic link between smoking-induced lung injury and lung cancer. Such a role is corroborated in that the oxidative stress-induced active conformation of EGFR could be stabilized in ceramide-enriched cellular compartments (61), as discussed in Section VII.
VII. The Unexpected Role of Ceramide in Cell Proliferation and Tumorigenesis
A. Cell membrane ceramide-enriched signaling platform and stabilization of aberrantly activated EGFR
Within the investigation on the mechanism linking redox signaling and lung epithelial cell proliferation, it was recently proposed that the oxidative stress-dependent activation of EGFR might be affected by changes in membrane structure/fluidity (61). Consistently, it was shown in HAE cells that such EGFR activation is dependent on temperature and on membrane cholesterol/ceramide ratio (61). Moreover, the TKI AG1478 could not inhibit EGFR activation during oxidative stress in living cells, whereas it did inhibit it in a crude cell membrane fraction (broken cells), where the proper membrane structure was altered (61). Thus, it was suggested that the membrane lipid constituents may either induce or stabilize the oxidative stress-dependent active conformation of EGFR (58), a theory encouraged by several interesting observations involving the EGFR and the membrane lipid components.
B. Membrane ordered lipid domains
Recently, different groups showed that sphingolipids in cell membranes are not homogenously distributed but are instead compartmentalized into specific domains mediated by interactions between sphingolipids and cholesterol (238). Specifically, sphingolipids and cholesterol are strongly bound with each other because of complementary amphoteric interactions (122), resulting in high local concentration of sphingolipids- and cholesterol-enriched membrane domains known as membrane rafts or lipid rafts. It is believed that cholesterol and some of its precursors not only cooperate with sphingolipids in these membrane rafts but also stabilize the structure of rafts by packing open spaces between the massive sphingolipids (122, 201, 226, 238). It is generally accepted that membrane microdomains exist and facilitate the segregation of specific lipids and proteins, thereby initiating specific signaling events. However, the actual lipid composition and size of these membrane microdomains, or rafts, are still very debatable (162). The major problem is that the postulated diameters of these membrane rafts are in the nanometer order, and therefore, it is difficult to accurately visualize these distinct morphological structures. Thus, conclusions on their structure and functions were mainly drawn based on biochemical experiments that utilized detergent extraction of membranes followed by the isolation of the detergent-resistant membranes (DRMs) on sucrose density gradients. However, such methods inevitably induce various artifacts, confounding results, and interpretation. Moreover, it has been shown recently that there are DRMs that are “nonbuoyant” on sucrose density gradient. These DRMs were defined as “heavy” rafts, as opposed to the “light” or buoyant rafts. However, the specific lipid differences between “heavy” and “light” rafts have not been characterized, yet (162).
C. Ceramide selectively displaces cholesterol from ordered lipid domains (rafts)
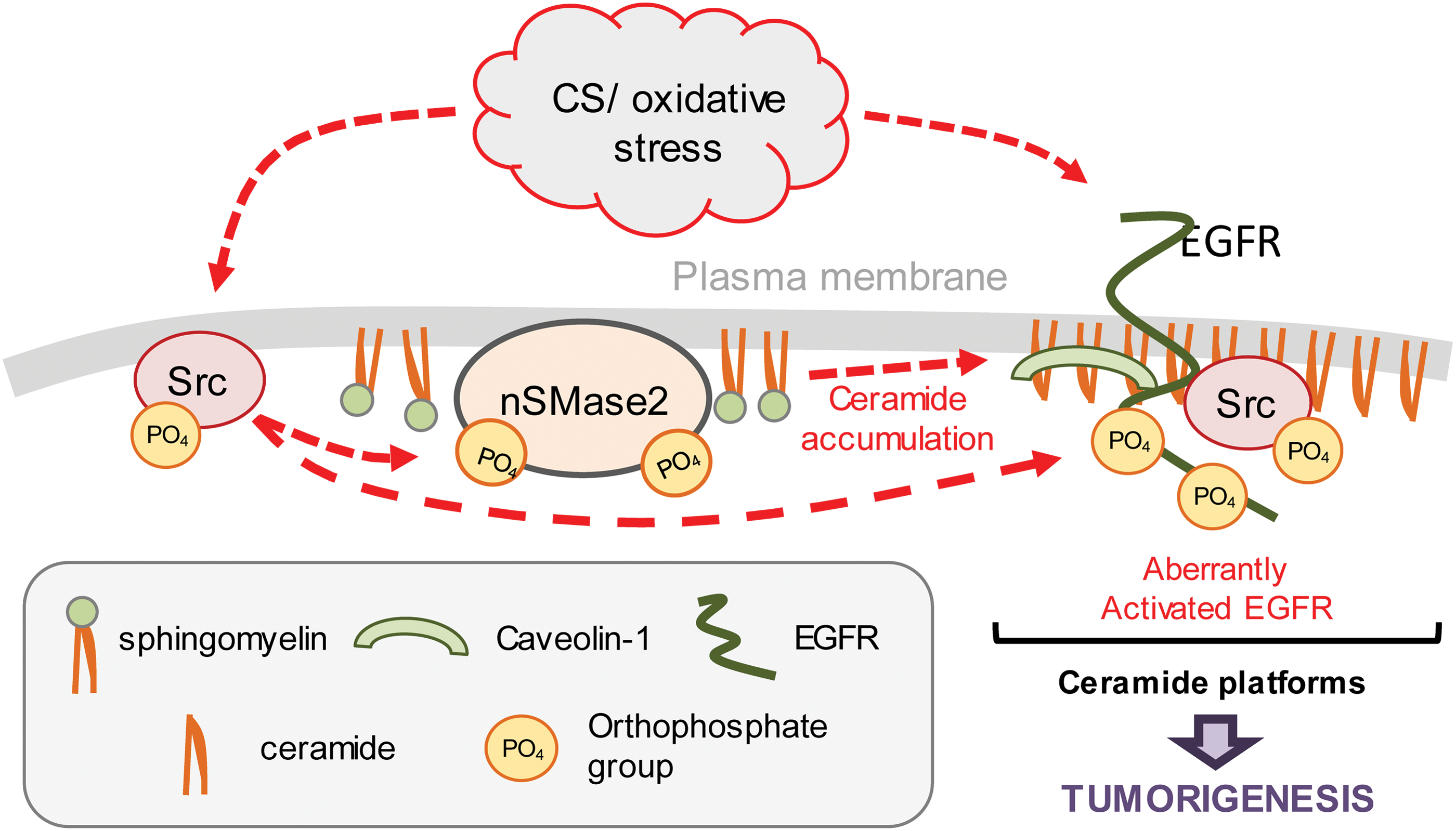
Studies of the composition of artificial membranes demonstrated that the generation of ceramide by sphingomyelin hydrolysis can specifically displace cholesterol from the membrane rafts (145). Therefore, ceramide generation could change composition and structure of the rafts. Furthermore, it has been proposed that ceramide-enriched membrane domains are predisposed to fuse with each other, raising larger membrane domains, the “ceramide-enriched membrane platforms,” whose diameters could reach the order of micrometers (36, 40, 81, 88, 145, 146, 199). These ceramide platforms possess different biophysical properties compared to the “conventional” lipid raft (phospholipid- and cholesterol-enriched ones) and may control important cell functions, such as protein sorting and redistribution of membrane receptors, including the EGFR. Consistently, ceramide-enriched membrane platforms were shown to capture and cluster receptor proteins (40, 84, 88). Therefore, since lipid rafts are thought to serve as signaling platforms for many membrane receptors, including the EGFR (94), changes in membrane structure and fluidity driven by ceramide generation from sphingomyelin breakdown are likely to significantly impact the phenotype and cell fate of airway epithelial cells.
D. Ceramide/cholesterol ratio affects EGFR and Src
Lambert et al. reported that the disruption of cholesterol-enriched lipid rafts caused ligand-independent activation of EGFR (126). One potential explanation was provided by Wang et al. (216). This study suggested that a population of EGFRs is strongly associated with lipid rafts and their disruption by a cholesterol sequestering agent, such as MβCD, would cause the relocalization of EGFR in nonraft portions of the plasma membrane where it could be activated due to its release from raft-associated inhibiting factors (216).
At the same time, Filosto et al. found that no changes of total cholesterol levels could be observed in HAE cells exposed to oxidative stress (61). Furthermore, cholesterol uptake in the plasma membrane could inhibit such oxidative stress-induced activation of EGFR (61). Therefore, it was proposed that membrane ceramide generated during exposure to oxidative stress may displace cholesterol from the rafts, causing the EGFR to transfer to rigid ceramide-enriched platforms, where an aberrant EGFR signaling occurs (61). Specifically, it was also shown that the aberrant binding between EGFR and active Src due to CS exposure was responsible for the TKI resistance in lung cancer patients (57). This interaction was not transient (i.e., in a rapid on/off equilibrium) but, instead, was stable. Thermodynamically, this could happen only if other supporting changes occur simultaneously. Therefore, it was suggested that alteration in the plasma membrane composition and fluidity, specifically the generation of ceramide from sphingomyelin by nSMase2 activation observed during CS exposure, supports and stabilizes the abnormal active structures and interaction between EGFR and Src, as well as their aberrant functions.
E. Ceramide, EGFR, and Src
It was recently demonstrated that during oxidative stress, activated EGFR and activated Src not only strongly bind each other but also colocalize within ceramide-enriched membrane regions (61). Specifically, at early time points of oxidative stress exposure (15 min), active EGFR and elevated ceramide colocalize primarily in the plasma membrane of the cells, while at later time points (30 min), such colocalization is detected mainly in the perinucleus. Consistent with this last observation, it was previously shown that EGFR activated by oxidative stress, unlike the EGF-stimulated receptor, does not undergo clathrin-dependent internalization and subsequent lysosomal degradation. Instead, EGFR remains active and traffics via caveolae to the perinucleus because of strong binding to phosphorylated Cav-1 (118, 119), which is a known substrate of Src. Intriguingly, others have shown in vitro that ceramide generation at the plasma membrane promotes the recruitment of Cav-1 into caveolae (125, 230). Moreover, the oxidative stress-activated EGFR colocalized with the early endosome marker EEA-1 (118, 176) and the recycling endosome marker Rab-11 (unpublished observation). In summary, when airway epithelial cells are exposed to oxidative stress, ceramide generation could support the aberrant activation of EGFR and prompt the caveolin-dependent trafficking of EGFR to the cell perinucleus; this results in sustained EGFR signaling, which is a feature of lung cancer (119). However, whether the change in EGFR conformation under oxidative stress occurs as a result of simultaneous alterations in the membrane structure or happens independently and is only being stabilized by the simultaneous membrane changes requires additional studies.
F. The link between ceramide and Src underlies their dual roles in apoptosis and proliferation
Although it is believed that the generation of ceramide acts as a direct stimulus for apoptosis during cellular stress, it is similarly plausible that ceramide induces proliferation signaling and may thus promote tumorigenesis. Ceramide generation at the cell membrane triggers the formation of signaling platforms that can affect a number of membrane-associated proteins, including Src and EGFR (61), which in turn can initiate prosurvival/proliferation signaling cascades (Figs. 7 and 8).
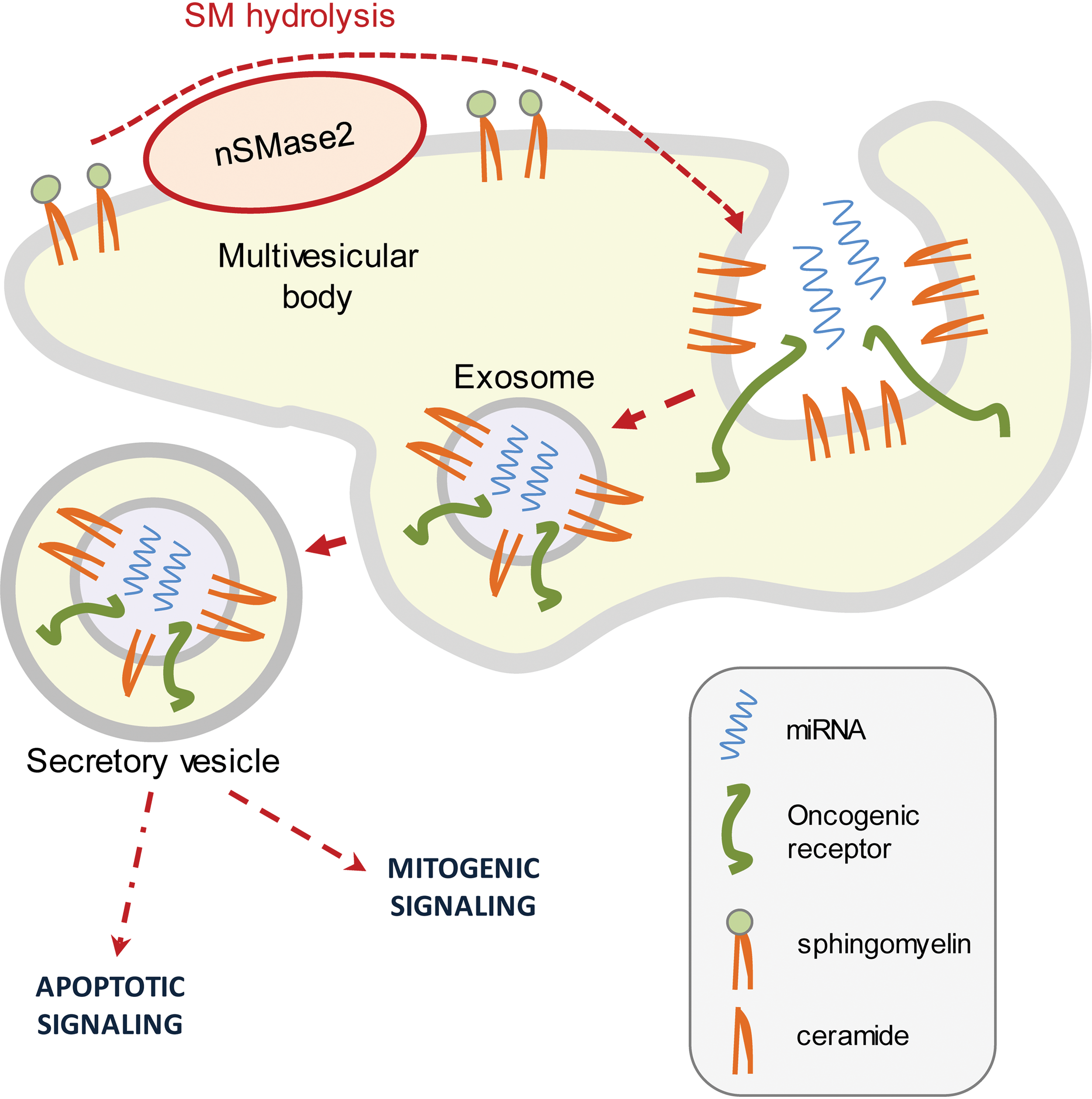
Consistent with this dual role of ceramide, it has been recently shown in vitro that nSMase2 function and subsequent ceramide generation under TNF-α- or oxidized low-density lipoprotein-induced cell stress can be mechanistically linked to both apoptosis initiation and cell proliferation (47). An additional recent publication claims that the loss of ceramide transfer protein (CERT) augments EGFR signaling in breast cancer via its regulation of a cellular pool of sphingomyelin (SM). Reduction of CERT expression led to increased ceramide and reduced SM levels, thereby enhancing tumorigenesis (94). Intriguingly, a recent article by Poirier et al. reports that nSMase2 activity and ceramide level are critical for lung development and function: systemic knockout of nSMase2 catalytic function (fro/fro mice) caused reduced ceramide levels and abnormalities in the lungs, which appear to develop with congenital emphysema-like defects (enlarged alveoli and increased compliance of the respiratory system) (170); this was counterintuitive, based on the commonly accepted role of nSMase2 function and ceramide upregulation in inducing lung injury and emphysema (59, 169). Therefore, it further proves that the role of ceramide in the lung as a mere detrimental lipid needs to be reevaluated. Notably, nSMase2 and ceramide generation could be instrumental in the spreading of oncogenic proteins and microRNAs (miRNAs) via exosomal generation, as further described in section VIII.
VIII. Propagation of Oncogenes and miRNA via nSMase2-Dependent Ceramide Generation and Exosome Secretion
Cell intercommunication has been conventionally described to occur via gradients of soluble molecules, which are recognized by cell-associated receptors. However, membrane nanovesicles coined exosomes have recently been identified as a new vehicle for cellular communication between cells that fuse with receiving cells in the neighboring area (4, 15, 198). Exosomes can be viewed as “units” of information, containing a number of biologically active protein and RNA species, including oncogenic receptors and miRNAs, short noncoding RNAs that modulate gene expression (121, 135). Al-Nedawi et al. reported that EGFRvIII, a truncated and oncogenic form of EGFR, can be transferred via secretory exosomes from human brain glioma cells into neighboring cells lacking EGFRvIII (4). Moreover, a recent publication by Kosaka et al. demonstrated intercellular communication by miRNAs transfer (124). They showed that miRNAs could be secreted from donor cells in exosomes and thereby be incorporated in receiving cells while staying functional (124). Consistently, Pegtel et al. also found that miRNAs released from cells infected with Epstein–Barr virus were transferred to and affected recipient cells (166). Importantly, Kosaka et al. (124) further demonstrated that nSMase2 function and ceramide generation control the formation and secretion of miRNA containing exosomes, underlining a novel fundamental path for the ceramide-generating machinery in regulating cell communication, gene expression, and cell fate.
At the present time, there are no reports on exosomal oncoprotein transfers in the lung. However, many studies, including studies on lung cancer, described the tumorigenic role of exosomes in that they can alter the gene expression of receiving cells by mediating transfer of functional miRNAs. Some miRNAs are deregulated in lung cancer and are associated with clinical outcomes. For example, low expression of let-7a miRNA and high expression of miR-155 miRNA were linked with poor clinical outcome (228). The let-7 miRNA has been widely reported to be differentially lower expressed in lung cancers, including CS-induced lung cancer (109); at the same time, let-7 miRNA can attenuate oxidant-induced injury through increasing the levels of HO1 (102), one of the key ubiquitous antioxidant enzymes. Thus, changes in miRNAs, including let-7 miRNA, could participate in the molecular linkage between injury and cancer development.
Notably, many miRNAs are overexpressed in lung cancer and can modulate cancerous cell growth via affecting EGFR function (136). In addition, miRNA-containing exosomes could be collected from the plasma of tumor patients, including lung cancer patients, thereby potentially serving as prognostic biomarkers (5, 23, 149). Thus, ceramide-dependent secretion of exosomes can control the transfer of oncoproteins and onco-miRNAs between the subset of cell populations, contributing to tumorigenesis. Consistently, Kosaka et al. demonstrated in vivo (mice) that the nSMase2-dependent exosomal transfer of angiogenic miRNAs regulates cancer cell metastasis (123).
On the other hand, exosome release by nSMase2-dependent mechanism as well as exosome uptake in recipient cells has been recently associated with cytotoxic effects in NSCLC cells (74). This demonstrates that such a nanovesicle-mediated machinery can also have proapoptotic effects (Fig. 9). Therefore, whether cells engage in proliferation or apoptosis in response to exosome-dependent signaling will likely depend on the stimuli induced, as well as on processes of adaptation to stress.
IX. Updates on Novel Unresolved Complexities in the Ceramide-Generating Machinery
Additional complexity of this underinvestigated field stems from recent studies showing that suppression or stimulation of tumor growth by ceramide could be observed and could be dependent on the length of the fatty acid chain of ceramide (93). For instance, as recently reviewed by Saddoughi and Ogretmen, C(16)-ceramide was implicated in cancer cell proliferation, whereas C(18)-ceramide mediated cancer cell death (183). Moreover, this could lead to different subcellular localization and therefore to different downstream targets (203), all of which could be also cell-type specific.
In addition, several enzyme families are already known to be involved in ceramide generation (93). However, at the present time, very little is known on their regulation under various stress stimuli/conditions. Recent studies are demonstrating the critical role of ceramide in modulating cell proliferation as a metabolite within the greater sphingolipid machinery. This allows the conversion of what is once a proapoptotic sphingolipid to ones that have proliferative signaling properties (78, 183). For example, Mitra et al. investigated ceramide kinase and its critical role in converting ceramide to ceramide-1-phosphate (C1P), a sphingolipid metabolite demonstrated to have threshold-like effects on cell proliferation (150). In A549 lung adenocarcinoma cells, low levels of C1P drove cell proliferation, whereas high levels of C1P led to apoptosis, possibly due to the conversion of C1P to ceramide (150).
At the same time, a recent publication by Petrache et al. presents that the de novo ceramide synthesis plays a critical role in maintaining the lung structure and function (in vitro and in vivo). Using a murine knockout model, it was shown that downregulation of ceramide synthase 2 resulted in a compensatory increase of ceramide synthase 5 activity. This caused the accumulation of ceramide species that harbor 16 carbon atoms in the fatty acid chain (C16-ceramide) and enlargement of lung parenchymal spaces (resembling emphysematous changes) (168). Another recent publication by Sultan et al. (203) presented in vitro that H2O2 could potentiate the de novo ceramide pathway by cotreatment of lung cancer cells with “short-chain” C6-ceramide, resulting in C16-ceramide accumulation and inhibition of the proto-oncogene c-Myc (myelocytomatosis viral oncogene homolog) (203).
Importantly, such examples include interesting cell- and tissue-fate observations tied to specific ceramide/sphingolipid species, indicating that we are only “scratching the surface of the iceberg” containing the puzzle of the ceramide-generating machinery.
X. Therapeutic Perspectives
Although specific inhibitors of nSMase2 are still not available, a few options already exist in the drug market to target Src and p38 MAPK. In a phase II clinical trial, Laurie et al. suggested that saracatinib, an orally available inhibitor of SFKs, may benefit a subset of NSCLC patients, which is currently molecularly undetermined; thus, further use of saracatinib in populations preselected for target mutations and smoking status should be considered (129). For instance, saracatinib could be functional as a combinatory therapy with EGFR TKIs, erlotinib or gefinitib, in patients who are smokers or former smokers and harboring activating EGFR mutations. Conversely, a small phase II clinical trial presented by Johnson et al. found that dasatinib, an Src-specific inhibitor, has little-to-no efficacy in patients with EGFR-mutant lung adenocarcinoma who acquired resistance to erlotinib and gefinitib; however, this trial included mostly never-smokers (113). To the best of our knowledge, no clinical trial has evaluated so far the use of SFK inhibitors in controlling COPD. On the other hand, p38 MAPK inhibition has recently been used in COPD patients with promising outcomes. For instance, Singh et al. found in a small cohort of randomized patients that SB-681323, a potent p38 MAPK inhibitor, strongly suppressed inflammation in COPD (197). Consistently, MacNee et al. presented that PH-797804 (another orally available p38 MAPK inhibitor) improved lung function parameters in a randomized cohort of 230 COPD patients (138). To date, the use of p38 MAPK inhibitors in lung cancer trials has not been assessed, although a potential role for p38 MAPK in lung tumorigenesis and resistance to therapy has been observed in vitro in NSCLC (28, 85).
In summary, despite the rising attention to the nexus between COPD and lung cancer, it is still important and necessary to additionally study the molecular pathways underlying both lung injury and lung cancer, which urge novel drug discovery and clinical trials.
XI. Concluding Remarks
In this review, several years of studies investigating redox signaling are described. Specific signaling pathways that lead to cell death and to cell proliferation/tumorigenesis were demonstrated in lung airway epithelial cells exposed to CS. In search for common molecular pathways and targets in lung injury and lung cancer, ceramide, a lipid metabolite, was highlighted as a tentative target. This canonically proapoptotic sphingolipid, which already had been described to have a role in lung injury (59), was suggested to also have potential roles in promoting cell proliferation and lung tumorigenesis. To provide the specific context for such a dichotomous role of ceramide, the review focuses on CS-induced oxidative stress and its effects on the airway epithelium.
On one hand, ceramide is generated by the activation of the axis Src-p38 MAPK-nSMase2 during oxidative stress, eventually leading to apoptosis, which is the dominant pathway in lung injury/COPD. On the other hand, the same stimulus aberrantly activates the EGFR, leading to prolonged proliferative signaling as observed in NSCLC. Interestingly, recent studies provide evidence that these two CS-activated pathways may actually converge and interact. Specifically, it was recently found in lung epithelial cells exposed to CS/oxidative stress that EGFR and Src are bound together and preferentially colocalize in ceramide-enriched regions of the plasma membrane, suggesting that nSMase2/ceramide may directly affect the aberrant EGFR/Src activation and mutual interaction, leading to enhanced proliferation/tumorigenic signaling (61).
Moreover, new findings show that CS-induced oxidative stress exposure induces resistance to EGFR-targeted (TKI) therapy, solely through post-translational molecular changes. These molecular alterations consist of an aberrant EGFR phosphorylation pattern accompanied by an aberrant conformation, which is supported by Src binding and activity (57, 58). Intriguingly, these structural anomalies of the oxidative stress-activated EGFR could be supported by the formation of ceramide-enriched microdomains generated in the plasma membrane of lung epithelial cells, which is dependent on Src and p38 MAPK function (29).
Due to the plethora of observations reported herein, the ceramide-generating machinery presents a novel array of target proteins, such as Src, p38 MAPK, and nSMase2, which may serve in mediating smoking-induced lung injury and lung cancer.
Footnotes
Acknowledgments
This work was supported by grants from National Institutes of Health (HL-66189 to T.G.) and from the Tobacco-Related Disease Research Program (TRDRP) (17RT-0131 and 22RT-0090 to T.G. and 20FT-0087 to S.F.).