
Abstract
The protein tyrosine phosphatase (PTP) CD45 is critical in regulating the earliest steps in T-cell-receptor signaling but, similar to all PTPs, it is susceptible to oxidative inactivation. Given the widely reported effects of oxidant damage associated with rheumatoid arthritis (RA), we examined whether CD45 phosphatase activity was altered in CD4+ T cells from RA patients and related this to CD4+ T-cell function and redox status. CD45 phosphatase specific activity in T cells from RA peripheral blood (PB) and synovial fluid was 56% and 59% lower than in healthy control (HC) PB, respectively. In contrast, CD45 activity in T cells from disease controls (DSC) was not significantly different from HC. Both reduced glutathione (GSH) (p<0.001) and oxidized glutathione (GSSG) (p<0.05) were significantly lower in RA PB T cells compared with HC PB T cells. Treatment of RA PB T cells with the GSH precursor N-acetyl cysteine increased CD45 phosphatase activity and proliferation, while it decreased Lck kinase phosphorylation, which is regulated by CD45. Our observations lead to the hypothesis that the largely reversible oxidative inactivation of the CD45 phosphatase may underlie the decreased signaling efficiency and functional responsiveness which are characteristic of RA PB CD4+ T cells. Antioxid. Redox Signal. 19, 2280–2285.
Introduction
R
We have described an acquired signaling dysfunction in chronic inflammatory arthritis that occurs through oxidative inactivation of the CD45 phosphatase. This dysfunction may substitute for or reinforce genetically determined immune aberrations that may together initiate and perpetuate chronic inflammatory disease. Oxidative stress and immune cell dysfunction are two long-recognized features of rheumatoid arthritis. We have provided a novel mechanism, the oxidative inactivation of CD45, that unifies these features and provides a rationale for new therapies of chronic inflammatory arthritis through antioxidant supplementation.
In RA patients, reactive oxygen intermediates (ROI) are abundant in the SF and PB, and glutathione (GSH) antioxidant protection within cells may be impaired (2). This may not only result from the inflammatory processes, but it could also result from dietary insufficiency in antioxidant vitamin C, which has been linked to the occurrence of inflammatory arthritis (3). Cigarette smoking, which also depletes blood levels of reduced GSH, is also an important risk factor for RA. The altered redox state in RA may promote alterations in TCR signaling in RA (2), a process that may involve displacement of LAT from the membrane. The protein tyrosine phosphatase (PTP) CD45, which is the most abundant PTP in leucocytes, regulates the very first stages of the TCR signaling cascade and, in systemic lupus erythematosus patients (9), its activity is decreased, probably because of the sensitivity to oxidation of the cysteine at the active site of all PTPs (7). Given this, we set out to investigate the activity of CD45 phosphatase in RA T cells and to relate this to both the redox status of the cells and their functional responses.
Proliferation Responses of RA PB T Cells Are Decreased
RA PB CD4+ T cells display a reduction in the response of the cells to activation through the TCR (1), and so, we initially set out to confirm these findings in the RA patients investigated in this study (PB taken from seven patients in Table 1). After stimulation with anti-CD3/anti-CD28, there was a significant reduction in the proliferation of the cells from the RA patients compared with the HC (Fig. 1A).
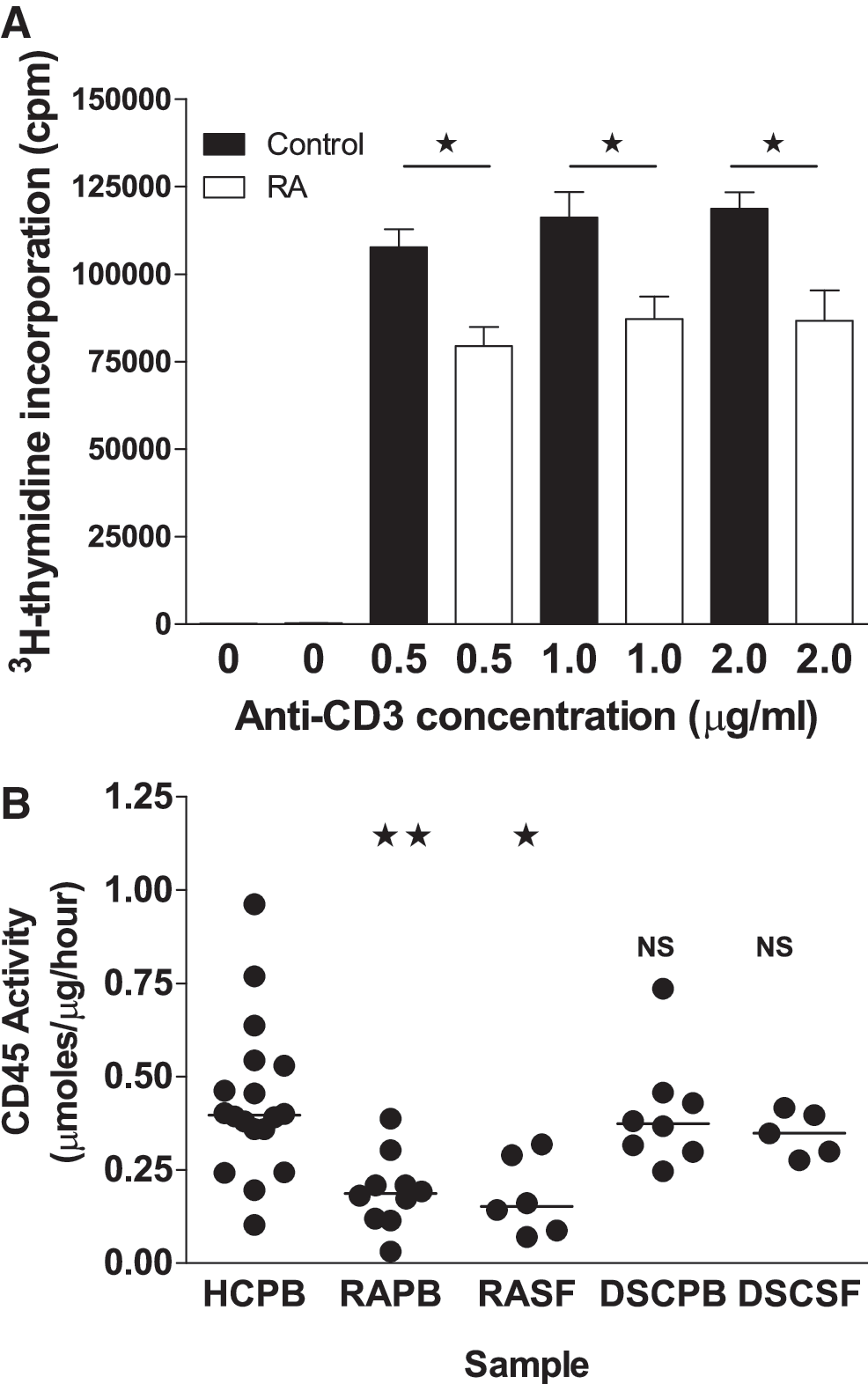
Seven sero-positive RA patient samples were used for proliferation responses and CD45 enhancement assays using N-acetyl cysteine. Eleven sero-positive RA samples and 8 DSC were used for CD45-specific activity and GSH measurements. All assays on patient samples were done in parallel with an age- and sex-matched HC sample.
RA, rheumatoid arthritis; DSC, disease control; GSH, glutathione; ESR, erythrocyte sedimentation rate; CRP, C-reactive protein.
CD45 phosphatase activity is decreased in RA but not in disease control patients
Phosphatase activity of CD45 was then assessed in both RA PB and RA SF, and this was compared with that of HC PB CD4+ T cells (Fig. 1B). The CD45 activity in RA CD4+ T cells was 56% lower in PB (0.19±0.03 μmoles/μg/h; mean±SEM CD45 activity; p<0.02) and 59% lower in SF (0.18±0.04 μmoles/μg/h; mean±SEM CD45 activity; p<0.05) than in HC (0.43±0.05 μmoles/μg/h; mean±SEM CD45 activity). This was restricted to RA patients, as there was no significant difference in the activity of CD45 from the PB (0.40±0.05 μmoles/μg/h; mean±SEM CD45 activity) and SF (0.35±0.03 μmoles/μg/h; mean±SEM CD45 activity) CD4+ T cells of disease control (DSC) patients (Fig. 1, last two columns). Furthermore, the CD45 from the DSC PB and SF CD4+ T cells was significantly more active than the RA PB and SF CD4+ T cell CD45 (PB p<0.02 and SF p<0.05). Our observation that the phosphatase activity of CD45 isolated from RA PB and SF CD4+ T cells is decreased, when compared with HC PB CD4+ T cells, may lead to changes in the activity of Src kinases and in downstream calcium signaling. Interestingly, this decreased activity was restricted to RA patients, which is consistent with previous studies in which calcium signaling depression was not seen in DSC groups comprising just ankylosing spondylitis and osteoarthritis patients (1). The absence of any significant change in CD45 activity in the rheumatoid factor sero-negative DSC group suggests that inflammation alone is not the sole cause of the changes we have seen in RA.
Antioxidant defense mechanisms of RA CD4+ T cells and fluids are depressed
Levels of both GSH and oxidized glutathione (GSSG) were significantly lower in both the RA serum and the RA PB CD4+ T cells than in their matched HC serum and PB CD4+ T cells (Fig. 2A, B). SF CD4+ T cell levels of GSH were even lower than both HC CD4+ T cell and RA PB CD4+ T cell levels. GSH in CD4+ T cells from DSC patients was not significantly different from either the HC or RA samples. DSC GSH was clearly closer to HC levels (HC PB 10.28±1.90; DSC PB 9.276±1.46; RA PB 6.64±1.42 μM). The DSC PB CD4+ T cell samples showed no difference in their reduction capacity compared with HC samples but were significantly higher than RA PB CD4+ T cells. Despite this, RA patients maintained reduction potentials, (dependent on GSH and GSSG concentrations), at levels similar to those in HC, demonstrating the maintenance of the normal redox environment, which is vital for cell function and survival (8). The reduction potentials observed in the PB CD4+ T cells of all groups (Fig. 2) are in the normal range, and so, this suggests that their survival is not compromised by redox stress. However, the decreased reduction capacity in RA PB CD4+ T cells suggests that they are less able to withstand the effects of ROI, thus allowing the oxidative inactivation of the CD45 phosphatase.

Boosting reduction capacity in vitro enhances RA T cell function, CD45 phosphatase activity and decreases Lck phosphorylation
Incubation with N-acetyl cysteine (NAC) (100 μM) for 2 h before stimulation significantly increased RA PB CD4+ T cell responses compared with untreated cells from the same patient (Fig. 3A, last two columns). The proliferative responses of the RA preincubated cells were almost equivalent to those of HC cells not treated with NAC (Fig. 3A, first column).

We also measured the relative increase in CD45 phosphatase activity after pre-treatment of RA PB CD4+ T cells and matched HC samples with NAC (Fig. 3B). The increase was significantly greater (p<0.05) in RA PB CD4+ T cell samples (35.8 [14–74]%; median [range]) than that observed with HC PB CD4+ T cells (12.6 [5–70]%; median [range]). The increase in CD45 activity in RA cells correlated with the increase in proliferative responses at 1.0 μg/ml anti-CD3 (p<0.02) (Fig. 3C).
Dephosphorylation of Lck Tyr 505 by CD45 is a priming event in the activation of Lck and subsequent events in downstream TCR signaling. We assessed the levels of Lck phospho-Tyr 505 in cells preincubated with NAC and then activated by cross-linking CD3. In resting cells (Fig. 4 top panels), NAC caused the decrease in the level of phospho Lck as the concentration of NAC increased. In activated cells (Fig. 4 bottom panels), levels of phospho-Lck were higher, particularly in the cells not incubated with NAC. However, as the concentration of NAC increased a distinct population of Lck phospho negative cells appeared. Given that the phosphorylation of tyrosine 505 is tightly regulated by CD45, this demonstrates that the decreased activity of CD45 phosphatase that we have observed in the RA patients (Fig. 1) results in the poor proliferation and responses of the cells (Fig. 3) through altered regulation of Lck phosphorylation.

Since CD45 activity was enhanced by NAC in the RA patients, it suggests that the inactivation was due to a partially reversible oxidation of the CD45 phosphatase active site. However, CD45 phosphatase activity in RA PB CD4+ T cells was not fully restored to the level in HC by NAC (data not shown), suggesting that a degree of irreversible modification may also have occurred. Recent structural studies on the oxidation of PTPs show that the formation of a sulfenyl-amide linkage is the first step in the oxidation (7). While this inactivates the enzyme, it may also protect against further irreversible oxidation to sulfinic and sulfonic forms, and so may explain why much of the oxidation observed was reversible. Enhanced proliferation correlated with the increase in CD45 phosphatase activity, demonstrating that the function of RA PB CD4+ T cells can be significantly enhanced by NAC to a near normal response.
There is considerable evidence of oxidative damage occurring both locally and systemically in RA (2), and so, we suggest that in this environment a decreased CD45 phosphatase activity results due to oxidation. Chronic exposure of blood to what may be generally low levels of oxidants, associated with hypoxic reperfusion injury and systemic inflammation, would mean that the antioxidant defenses will be continually attacked and depleted. This decreased reduction capacity may be particularly important for T cells, which are long-lived. A similar chronic accumulation of oxidative damage may occur in aging. We have demonstrated that CD45 phosphatase activity is decreased in T cells from healthy elderly individuals (4), and the accumulation of oxidative damage in elderly people is known to correlate with a decrease in the plasma GSH levels.
In TCR signaling, the importance of CD45 in controlling early events means that inhibition of its action will supersede any other signaling changes. The potential importance of these early TCR signaling events for the etiology of arthritis was demonstrated in a mutant mouse model (6) in which a point mutation in the TCR-proximal ZAP-70 protein leads to an attenuated CD4 T cell TCR signal, very similar to what we have observed in RA patients. In these animals, a spontaneous persistent arthritis ensued that could be prevented by re-introducing a fully functional ZAP-70 molecule. While in this model thymic selection of autoreactive T cells was shown to occur, the reasons for the development of arthritis remain unclear. However, it suggests that the acquired dysregulation of TCR proximal signaling which we have observed has the potential to allow aberrant autoimmune responses to occur, perhaps by interfering with the regulation of peripheral tolerance, giving rise to a persistent inflammatory arthritis. Low dietary intake of vitamin C (the other major anti-oxidant, in equilibrium with GSH) has been related to the occurrence of polyarthritis (3). Such alterations in the systemic redox balance could enable inactivation of CD45, thus allowing aberrant immune function and initiation and/or perpetuation of inflammatory arthritis.
Notes
Materials and methods
RA and DSC patients
PB was obtained from arthritis patients undergoing treatment in the clinics of the Rheumatology Department of Selly Oak, or City Hospitals, Birmingham (n=18). SFs were collected when available from patients undergoing therapeutic aspirations of knee joints. All RA patients satisfied the 1987 American College of Rheumatology (ACR) criteria for RA. PB (and SF when available) was also taken from arthritis patients not meeting the 1987 ACR criteria for RA (n=8) (Gout; reactive arthritis; psoriatic arthritis; sero-negative arthritis; osteoarthritis). These formed the disease control patient group (DSC). All samples were obtained under the approval of South Birmingham and Sandwell & West Birmingham Hospitals Local Research Ethics Committees. All patient samples were matched with a PB sample collected from age- and sex-matched healthy control (HC) staff members of the University of Birmingham (n=26, mean age of controls=49.42, range 26–54, mean age of patients=57.42, range 18–81) (see Table 1).
Lymphocyte isolation from PB and SF
PB and SF were collected into preservative-free heparin, and CD4+ T cells were isolated by Ficoll–Paque (Amersham Biosciences) density gradient centrifugation and magnetic bead separation.
Incubation of CD4+ T cells with NAC
Purified cells were incubated for 2 h with and without NAC (100 μM) (found to be the optimal concentration for restoring PTP activity—data not shown) in complete medium and were then stimulated as described earlier or used in the assay for CD45 activity (below).
Measurement of CD45 phosphatase activity
CD45 phosphatase specific activity was measured using an antibody capture technique as previously described (5).
Measurement of GSH
Purified CD4+ T cells (5×105) were lysed and de-proteinated in 1.8% perchloric acid. Fresh serum and SF were similarly treated and stored at −80°C. 10 mM dithio-bis (2-nitrobenzoic acid), 0.5 mM HCl, 0.075U GSH reductase, and 1 mM NADPH were added to triplicate samples, and the change in absorbance at 412 nm over 15 min at 37°C was measured and compared with a GSSG standard curve. GSSG was then determined by subtracting the amount of GSH from the total GSH concentration. GSH was measured similarly but in the absence of GSH reductase and NADPH.
Proliferation assays
105 cells CD4+ T cells were activated with anti-CD3 (1 μg/ml) and anti-CD28 (2 μg/ml) immobilized onto 96-well plates using goat anti-mouse IgG (100 ng/ml). After 48 h, 3H-thymidine was added, and cells were harvested at 72 h for counting using an LKB Betaplate reader (LKB-Wallac).
Flow cytometry of phospho-lck
Peripheral blood mononuclear cells (0.5×106) were plated onto 24-well plates in medium (RPMI 1640 with 10% fetal calf serum and gps) (2mls) with and without a range of concentrations of NAC. After 2 h of incubation at 37°C, cells were rapidly cooled on ice, sedimented, and then stimulated at 37°C using prewarmed anti-CD3 (Immunotools) and goat anti-mouse IgG (Sigma) to cross-link receptors. One minute after cross-linking, cells were fixed by the addition of three volumes of Phosflow Fix buffer (BD). This was left for 15 min at 37°C, and then, the cells were permeabilized using Phosflow perm/wash buffer (BD) to check for the presence of phosphorylated tyrosine residue 505 of Lck (BD LcK [Y505] PE) and CD4 (anti-CD4 APC Immunotools), and they were then analyzed by flow cytometry using a Coulter Cyan flow cytometer. Parallel samples with appropriate isotype controls were also analyzed.
Footnotes
Acknowledgments
D.R. and R.B. were funded by the Arthritis Research, the United Kingdom (Grants Y0510 and 19325); E.C. was funded by the Wellcome Trust (grant 089384/Z/09/Z).