
Abstract
Reactive oxygen species (ROS) are deadly weapons used by phagocytes and other cell types, such as lung epithelial cells, against pathogens. ROS can kill pathogens directly by causing oxidative damage to biocompounds or indirectly by stimulating pathogen elimination by various nonoxidative mechanisms, including pattern recognition receptors signaling, autophagy, neutrophil extracellular trap formation, and T-lymphocyte responses. Thus, one should expect that the inhibition of ROS production promote infection. Increasing evidences support that in certain particular infections, antioxidants decrease and prooxidants increase pathogen burden. In this study, we review the classic infections that are controlled by ROS and the cases in which ROS appear as promoters of infection, challenging the paradigm. We discuss the possible mechanisms by which ROS could promote particular infections. These mechanisms are still not completely clear but include the metabolic effects of ROS on pathogen physiology, ROS-induced damage to the immune system, and ROS-induced activation of immune defense mechanisms that are subsequently hijacked by particular pathogens to act against more effective microbicidal mechanisms of the immune system. The effective use of antioxidants as therapeutic agents against certain infections is a realistic possibility that is beginning to be applied against viruses. Antioxid. Redox Signal. 20, 1000–1037.
I. Introduction
R
Phagocytes reside within tissues or are recruited by inflammatory processes. Phagocytes recognize microbes through many molecular patterns displayed by them and try to engulf them. Once a microbe is phagocytosed, the nature of the molecules recognized on microbe's surface dictates the treatment enacted within the phagosome. Respiratory burst, a process by which NADPH oxidase (NADPH oxidase 2 [NOX2]) generates ROS in response to microbe recognition, is a possible outcome of this process and helps to get rid of many microbes. For instance, β-glucan on the surface of the fungi can engage dectin-1 on the surface of phagocyte (69). Once fungi are phagocytosed, NOX2 is promptly assembled at the phagosome membrane and high amounts of superoxide (O2 −•) are discharged into the phagosome. The recognition of Escherichia coli generates comparatively smaller amounts of ROS than the recognition of Listeria monocytogenes, and these ROS are less promptly released into the phagosome (347). However, the recognition and phagocytosis of Leishmania spp. is well studied, and, except when recognition is mediated by Fc receptor (FcR), virulent parasites do not usually trigger a severe respiratory burst (325) (see section V[D]). Thus, microbes face different fates after phagocytosis dependent on the molecules presented at their surfaces and how they are targeted by innate immune receptors.
Once a pathogen is phagocytosed, it must subvert the respiratory burst, withstand its oxidative power, or escape the phagosome to survive. Microbe recognition sets the immune system in motion, and ROS are produced not only in the phagocyte respiratory burst but also in other cell compartments, such as mitochondria, as intermediaries in many signal transduction pathways, such as leukocyte pattern recognition receptor (PRR) signaling. The generation of ROS is a prerequisite to the formation of neutrophil extracellular traps (NETs) (28); is actively involved in phagolysosomal formation and enzymatic degradation (281); autophagy (118, 119, 298) and ROS inhibition of mammalian target of rapamycin (mTOR) kinase (171, 323); chemoattraction and inflammation (224, 357); cell death of infection reservoirs (9); antigenic presentation, T-helper (Th) polarization, and lymphocyte proliferation (43, 85, 181, 203, 318, 337); iron redistribution among tissues (198) and cell compartment availability of iron (36, 201, 358); and foam cell formation (1, 316). Many of these mechanisms promote microbe clearance, whereas others can potentially contribute to microbe persistence.
In this review, we first provide a brief summary of antioxidants and their mode of action as a tool to understand their use in infection. Next, we discuss the mechanisms by which ROS directly kill microbes or interfere with the immune response. In addition, we discuss the role of ROS in pathogenic viral, bacterial, and protozoan infections, and we highlight to the cases in which ROS production seems to favor infection instead of combating it. The evidence supporting a role for oxidative stress in fueling certain infections is compelling but remains largely unnoticed in the literature to date.
II. Antioxidants and Their Mode of Action
Antioxidants are molecules that act to deplete ROS. Molecules that inhibit ROS-generating pathways, molecules that directly scavenge ROS, and molecules that interfere with ROS degrading pathways can act as antioxidants. Common antioxidants are ROS-scavengers, NOX2-inhibitors, inhibitors of various ROS-generating pathways, and nuclear factor (erythroid-derived 2)-like 2 (NRF2)-activators, which are a class of compounds that induce the expression of antioxidant enzymes, thus classified as indirect antioxidants. ROS-scavengers, such as N-acetyl-cysteine (NAC), which replenishes glutathione, have been by far the most studied class of antioxidants and became accessible at pharmacies, but recently, NRF2-activators raised much interest. Many NRF2-activators are considered “nutraceuticals,” molecules naturally found in foods to which healthy effects have been ascribed: resveratrol (wine), pterostilbene (blueberry), sulforaphane (broccolis), curcumin (turmeric), cafestol (coffee), quercetin (red onion), epigallocatechin-3-gallate (green tea), and carnosol (rosemary) (13). The food conservation additive tert-butylhydroquinone is also a potent NRF2 activator (13). Cobalt-protoporphyrin (CoPP) is a drug largely used in experimental research that is capable of inducing heme-oxygenase 1 (HO-1) expression through NRF2 activation (295). NOX2 inhibitors, however, have attracted less attention, most likely due to their less specific effects on NOX family proteins. Apocynin, the most studied NOX2 inhibitor, derives from vanillin and is nontoxic but has no current use in clinics (313).
The housekeeping production of ROS is generally neutralized by constitutive antioxidant defenses. Oxidative stress ensues when ROS production overwhelms antioxidant defenses. The oxidative hit then promotes the dissociation of kelch-like ECH-associated protein (Keap) from NRF2, allowing NRF2 to translocate to the nucleus where it activates cytoprotective and antioxidant defenses by turning on the transcription of genes that contain antioxidant response element (ARE) motifs in promoters (335). A subject recently reviewed in the literature is the ability of NRF2 to interact with many other transcription factors (27). The indirect antioxidants that act by activating NRF2-dependent mechanisms are usually oxidants that promote transient surges of ROS production and some of them can even act as pro-oxidants in large concentrations. Most NRF2 activators fit into the general definition of “hormetic” agents: they induce a low level of stress that activates the antioxidant defenses, and the general outcome is beneficial to the organism (27).
The NRF2-target genes include the following phase II enzymes: HO-1, NAD(P)H quinione oxidoreductase 1, glutathione peroxidase, glutamate cysteine ligase, and glutathione S-transferases. However, not all genes under NRF2 control are enzymes of direct antioxidant action. For instance, H-ferritin and ferroportin (FPN)-1, proteins that regulate the labile iron pool, contain ARE motifs in their promoters (112, 186) but are only indirectly linked to redox regulation. NRF2 acts on tissue regeneration, DNA repair, and lipid metabolism genes (335). Some promoters that contain ARE motifs, such as that of CD36, also contain peroxisome proliferator-activated receptor (PPAR)-γ-controlled PPARE motifs and can be operated by both factors at a time (233). Recently, the macrophage phenotype MHem was described to result from a genetic program commanded by activation transcription factor (ATF)-1 and NRF2 simultaneously and to be capable of countering foam cell formation (24). This indicates that an NRF2-dependent event does not necessarily activate antioxidant defenses.
NRF2 activation orchestrates a tolerant response that can protect tissues from damage caused by inflammation, infective, and oxidative insult (335). The immune system is specialized to provide resistance against infection, but body tissues directly deal with pathogens and defend themselves by a myriad of mechanisms. Thus, it follows that when tissue tolerance protects body tissues or the immune system itself against infection-induced damage (192), it can potentially affect resistance.
III. ROS Can Promote Pathogen Elimination by Direct Oxidative Damage or by a Variety of Innate and Adaptive Mechanisms
When a microbe is recognized by phagocytes and engulfed, it triggers a process, named respiratory burst, in which phagocytes elevate their oxygen consumption. The enzyme NADPH-oxidase (NOX2) is pivotal to the respiratory burst and attaches to the phagosomal membrane during phagocytosis (353). Particular stimuli such as phorbol-12-myristate-13-acetate (PMA) stimulation can promote NOX2 attachment to the cell membrane. NOX2-derived ROS promote oxidative (Fig. 1) and nonoxidative (Figs. 2 –12) mechanisms of microbe elimination.
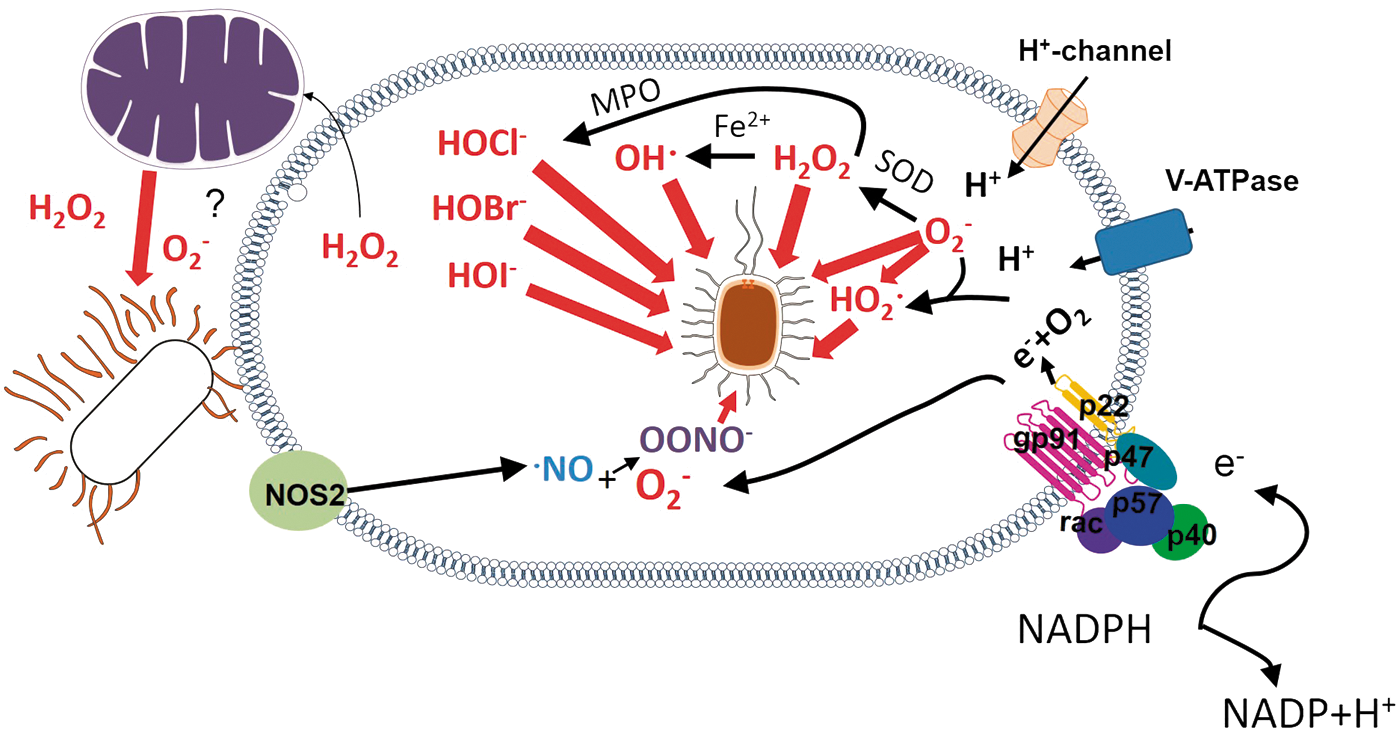
NOX2 is a heme-binding protein composed by the subunits gp91phox, p22phox, p67phox, p47phox, p40phox, and Rac1 or 2 (depending on the cell type). The subunit gp91phox forms a transmembrane channel bound to p22phox through which electrons from NADPH are ultimately transferred to oxygen in the phagosome to generate O2 −• at micromolar concentrations (353).
Activation of Rac is a critical event for NOX2 assembly at the phagosome membrane. The protein Rac, a member of the Rho family that is usually bound to GDP, is activated by the replacement of GDP with GTP. The GTP-bound Rac and other NOX2 cytosolic components are then recruited to the membrane. The process is further regulated by the following: (i) proteins that help in the replacement of GDP with GTP (guanine nucleotide exchange proteins); (ii) proteins that accelerate the hydrolysis of GTP to GDP and contribute to detaching it from the membrane (GTPase-activating proteins); (iii) proteins that bind to GDP-bound Rac and prevent the interaction between Rac and guanine nucleotide exchange proteins (GDI or GDP dissociation inhibitor family) to prevent Rac activation; (iv) proteins that dissociate the GDI from GDP-bound Rac (GDI dissociation factors) contributing to Rac activation.
Gp91phox and p22phox are stored in intracellular granules, whereas p67phox, p47phox, and p40phox subunits are cytosolic and form a ternary complex (170). On phagocytic stimulation, granules fuse to the phagosome, whereas Rac and the ternary complex bind to the phagosome by independent mechanisms that originate in pathogen recognition. Activated Rac binds to gp91phox. The phosphorylation that p47phox undergoes during phagocytic stimulation allows the interaction of the ternary complex with p22phox-gp91phox. Activated Rac then binds to p67phox leading to NOX2 assembly on the phagosome. The signal pathway from pathogen recognition to Rac activation and its relation to the amount of oxidative attack the microbe will undergo inside the phagosome, if any, are still to be determined.
The host susceptibility to various pathogens in the absence of NOX2-derived ROS production was first observed in chronic granulomatous disease (CGD) patients, which lack a functional NOX2 protein (15, 72). Mice deficient in ROS production have also been shown to be more susceptible to a number of infections. Opportunistic infections such as Salmonella enterica serovar Typhimurium (Salmonella typhimurium), Staphylococcus aureus, Serratia marcescens, and Aspergillus spp. accompany the lack of a functional NOX2 protein. Surprisingly, however, there are scarce demonstrations that the use of pharmacological antioxidants (direct or indirect) can increase pathogen burden, such as in hearts from Trypanosoma cruzi chronically infected mice treated with apocynin (64).In contrast, the antioxidant resveratrol reduced S. marcescens burden in mice (177), and antioxidants NAC and glutathione dramatically increased neutrophil killing of S. aureus in vitro, although they did not kill the bacteria directly (227). The reasons for the scarcity of data indicating that antioxidants can increase pathogen burden and the presence of contradictory data are unknown.
Phagocytosis is tightly coupled to the respiratory burst in phagocytes. As soon as the microbe is engulfed, the delivery of high amounts of ROS inside the phagosome prevents the microbe from escaping to the cytosol. Knowledge of the role of CARD9 in this process is emerging. CARD9-deficient mice are susceptible to L. monocytogenes and fungal infection (90, 106, 117). Wu et al. found that CARD9−/− and wild-type macrophages were able to clear E. coli, but the clearance was faster by wild-type macrophages (347). Nevertheless, CARD9−/− macrophages were unable to clear L. monocytogenes. Knowing that E. coli does not escape the phagosome, whereas L. monocytogenes escapes it 30 min after being engulfed (216), the authors hypothesized that CARD9 was involved in the early ROS surge that kills the bacteria before it can escape from the phagosome. Through further study, researchers found that CARD9 binds to LyGDI, a protein that inhibits the activation of Rac, to unleash Rac to be activated by guanine nucleotide exchange proteins, which leads to the early surge in ROS promoted by NOX2 assembly at the phagosomal membrane (347). Pathogens that take time to escape to the cytosol after phagocytosis most likely undermine CARD9-mediated assembly of NOX2 at the phagosomal membrane or are able to withstand the oxidative stress inside the phagosome.
A. Direct oxidative damage to microbes
Two major lines of evidence support the notion that microbes can be directly killed by ROS. A deficiency in a pathogen's antioxidant machinery can turn a highly virulent pathogen into a ROS-susceptible pathogen, indicating that ROS directly damages microbes (174, 228, 311). Additionally, the oxidative damage caused by ROS, such as lipid peroxidation, DNA strand breakage, base oxidation and deamination, and oxidation of methionine residues (255), can be directly tracked in microbes exposed to respiratory burst. The direct oxidative damage to microbes is shown in Figure 1.
Whether O2 −• itself can kill microbes is a question still to be resolved (302). O2 −• does not diffuse across membranes efficiently and is rapidly dismutated to hydrogen peroxide (H2O2) by Cu/Zn-superoxide dismutase (SOD) (267). However, H2O2 can diffuse more freely, although it depends on the presence of aquaporin-3 to pass through membranes (196). H2O2 causes direct oxidative damage to many pathogens. It is also a substrate to a metal-catalyzed oxidation that produces hydroxyl radicals (OH·) and a myeloperoxidase (MPO)-catalyzed reaction that produces halogenated acids, such as hypochlorous acid (HOCl−), hypobromous (HOBr−), and hypoiodous (HOI−) acids, which are highly reactive ROS (220). Still, O2 −• can also be protonated in the low pH of phagolysosomes to form the highly reactive HO2 • species (144).
Although the production of MPO-derived halogenated acids is considered an important oxidative killing mechanism, since MPO-deficient mice and MPO-deficient neutrophils display a decreased capacity to clear a number of pathogens in high doses (8), MPO-deficient individuals do not present particular susceptibilities to infection. A full review on the subject of microbe susceptibility to MPO has been published recently (345).
In the presence of nitric oxide (NO) generated by the enzyme inducible nitric oxide synthase (iNOS), O2 −• is consumed to produce peroxynitrite (OONO−) that can be protonated and degraded to form OH· and nitrogen dioxide (·NO2), which are more reactive products (220). However, the expression of iNOS and the ROS/NO ratio that drives the production of a given species depend on the nature of the stimulus recognized by the phagocyte. E. coli, for example, is chlorinated but not nitrated in human neutrophils, suggesting that it is exposed to HOCl− but not to OONO− (277).
Thus, O2 −•, although not highly toxic by itself, is a precursor to other ROS, such as HOCl−, OH·, and H2O2, and is combined with NO to form OONO− and ·NO2, which are highly reactive molecules (103).
NOX2-derived ROS production is the main, but not the only, source of oxidative attack on invading organisms (15, 72). Toll-like receptor (TLR) stimulation, for instance, can trigger NOX2 and subsequent mitochondrial-derived ROS production (343). In this case, mitochondria were juxtaposed to the phagocytic vacuoles, and the use of both inhibitors of mitochondrial ROS generation and mitochondrial-targeted catalase expression inhibited the killing of Salmonella typhimurium, demonstrating that mitochondrial ROS can be bactericidal (343). Dual oxidases (DUOXs) are Ca2+-activated NOXs and operate as H2O2-generators in various tissues. ROS production by DUOX2 is required for the killing of L. monocytogenes induced by Nod2 stimulation in intestinal cells (173), indicating that ROS can also promote the elimination of cytoplasmic pathogens, at least in certain circumstances. However, whether mitochondrial and DUOX2-derived ROS kill by direct oxidative damage is still to be fully demonstrated.
Microbes can be eliminated by ROS produced by various nonphagocyte cell types, such as intestinal epithelium (as mentioned above) and lungs, but the subject has not been thoroughly exploited. In airways, DUOX produces H2O2 in response to bacterial infection, and lactoperoxidase acts analogously to phagocyte MPO, converting H2O2 to bactericidal OSCN− and leading to bacterial killing (51).
As the production of ROS is a microbicidal mechanism, it is not surprising that many pathogens downregulate the expression or interfere with the activity of NOX2 or downstream effectors. This subject was reviewed elsewhere (160, 308).
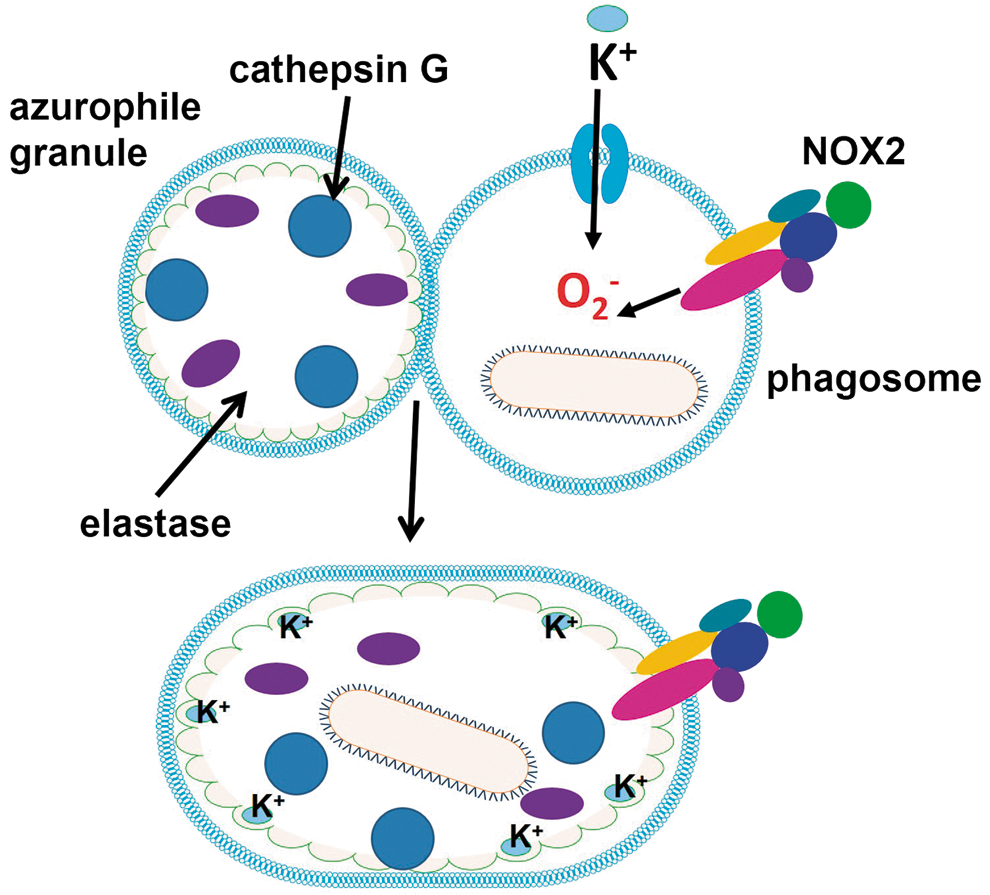
B. O2 −• promotes proteolytic elimination of microorganisms indirectly
Although ROS are thought to kill some microbes directly by causing oxidative damage to biocompounds, an alternative mechanism by which the eletrogenic activity of NOX2 is responsible for ROS-promoted elimination of pathogens in neutrophils has been proposed (Fig. 2). O2 −• production promotes an increase in K+ concentration in phagosome to compensate for negative charges. Cytoskeleton proteins initially constrain the swelling of the vacuole allowing hypertonicity to develop. The concentration of K+ promotes the release of contents from insoluble granule stores, such as elastase and cathepsin G (bound to an anionic sulphated proteoglycan matrix) enzymes, which in turn kill microbes by proteolytic attack (266). Depending on the cell type, the negative charge of O2 −• can be compensated by H+ ions that are pumped by vacuolar-H+-ATPase into the phagosome, indicating that phagosomal acidification and ROS generation can interfere with each other (128, 293). The activation of NOX2 creates a hypertonic, K+ rich, and less acidic environment, which allows the proteases to be activated.
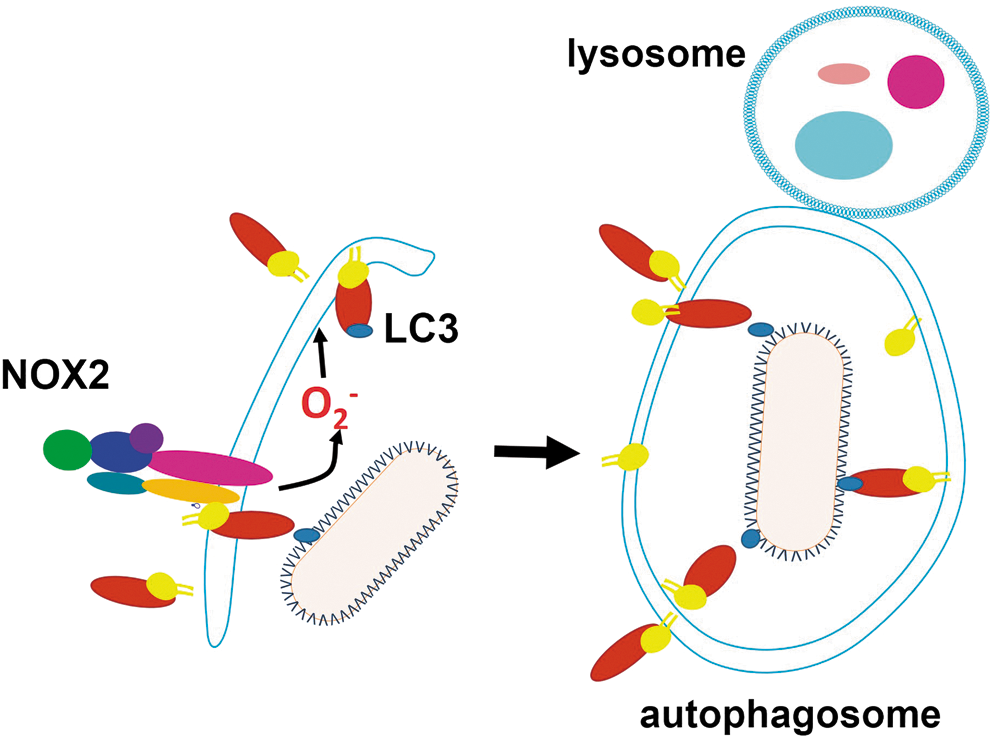
C. ROS promote autophagy
Autophagy is a mechanism of defense against intracellular pathogens such as Mycobacterium tuberculosis (99), Group A Streptococcus (219), Salmonella typhimurium (118), and L. monocytogenes (258). NOX2 has been identified as a factor promoting autophagy [reviewed in (119)] (Fig. 3). In neutrophil infection by Salmonella, NOX2-generated ROS are involved in the recruitment of microtubule associated protein 1A/1B light chain 3 (LC3) to the phagosome, promoting antibacterial autophagy (118). A similar process occurs in intestinal epithelial cells with ROS from other sources. ROS also contribute to the elimination of M. tuberculosis by autophagy (298). The participation of ROS in autophagy is depicted in Figure 3. However, some pathogens sabotage autophagy and take advantage of it to favor infection: Porphyromonas gingivalis (66), Brucella abortus (310), Coxiella burnetii (100), Chlamydia (354), S. aureus (291), Toxoplasma gondii (338), and T. cruzi (275). Whether ROS-promoted autophagy affects pathogen burden in any of these infections remains to be determined.
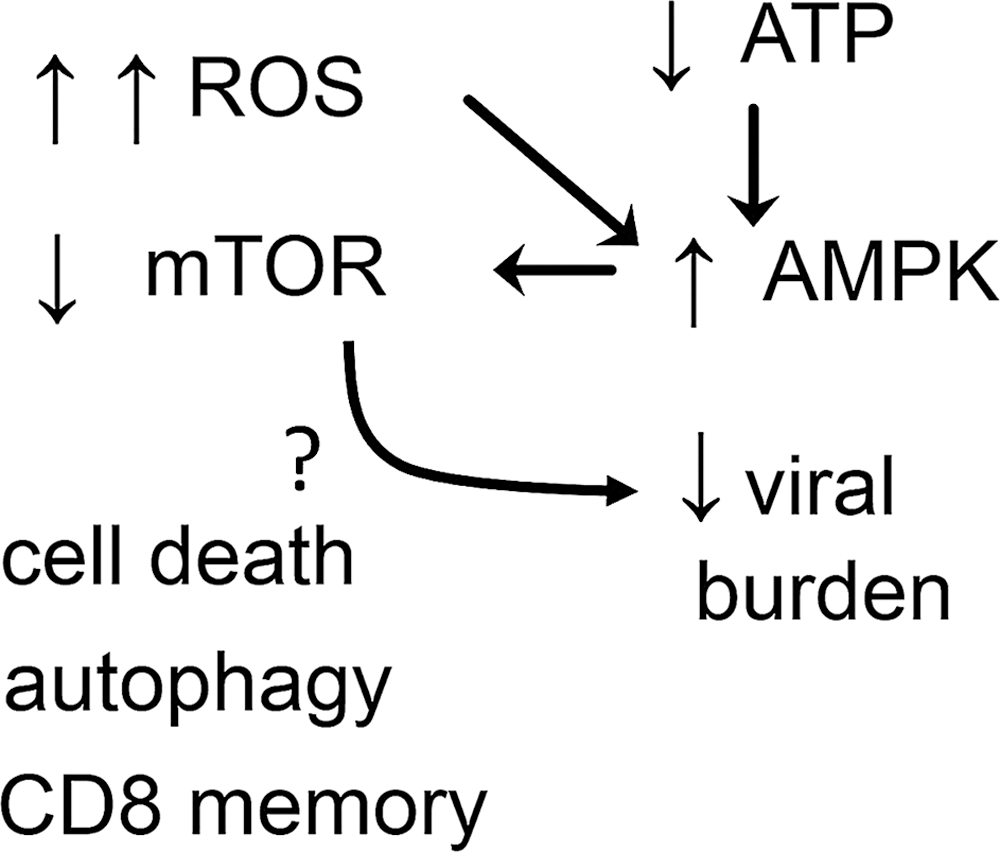
D. ROS inhibit mTOR kinase, triggering an antiviral response
The activation of the mTOR is a sensor of nutrient status that controls cell growth and metabolism. AMP-activated kinase (AMPK) keeps mTOR repressed, and AMPK is activated by low ATP levels, a phenomenon that signals low nutrient status (340). High amounts of ROS during long timespans inhibit the mTOR pathway (171). Recent experiments demonstrated that ROS inhibit human cytomegalovirus (HCMV) infection by inhibiting mTOR (323) (for more details, see viruses infections combated by ROS in section IV[B]). The inhibition of mTOR can represent an effective mechanism against viral infections (214). The potentially microbicidal mechanisms downstream of mTOR inhibition are autophagy, programmed cell death, and potentiation of CD8 cell memory formation (340) (Fig. 4). Nevertheless, the production of type I interferon (IFN) by plasmacytoid cells depends on mTOR (53). It is not known how mTOR inhibition induced by ROS acts to reduce HCMV infection.
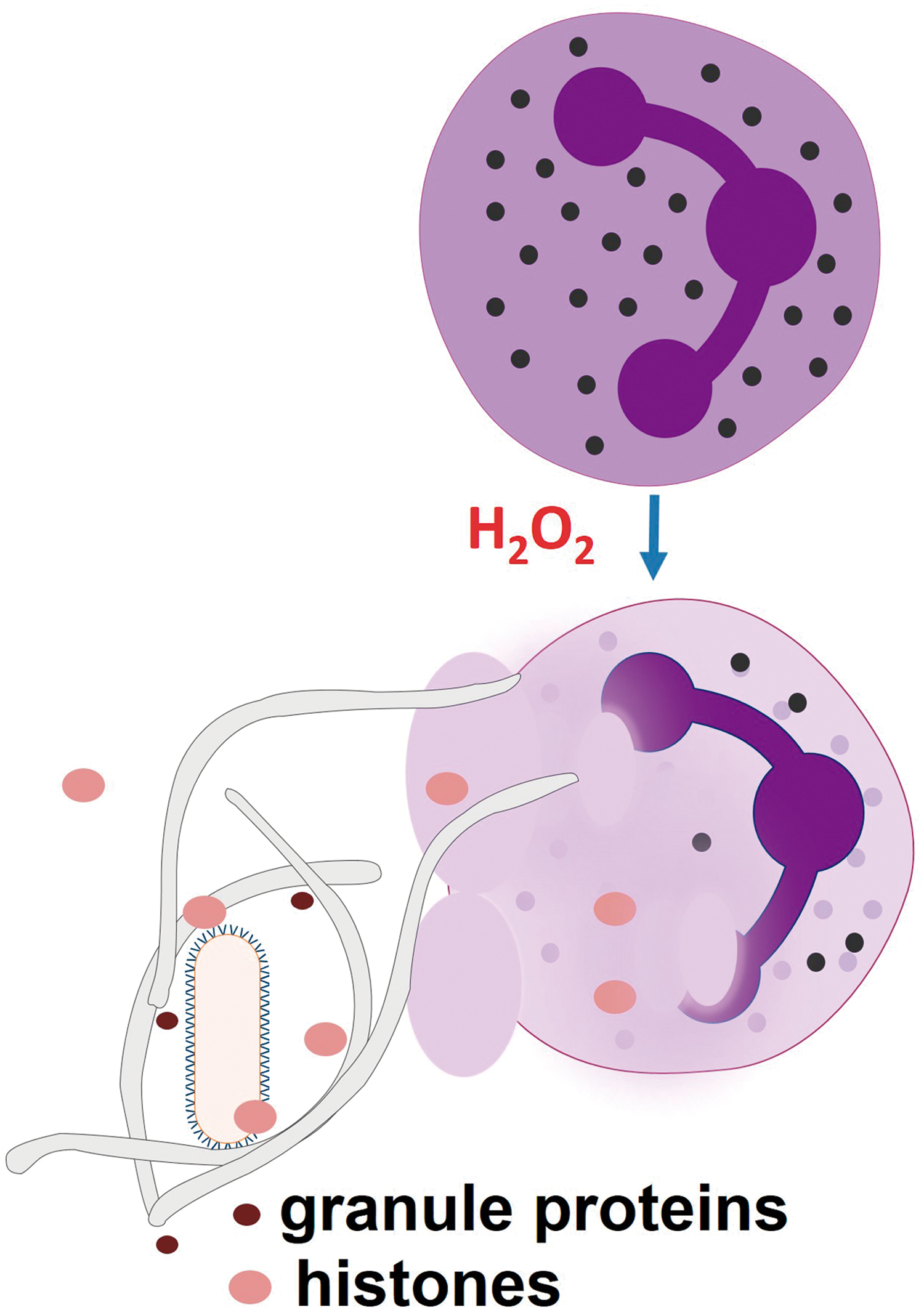
E. ROS promote NETosis
Neutrophils extrude NETs on activation with bacteria, PMA, or interleukin (IL)-8 (95). NETs are formed by chromatin and associated proteins, whereas the neutrophil dies. NETs entrap pathogens and are thought to kill them by delivering bactericidal and fungicidal proteins, such as cathelicidin LL-37, defensins, bactericidal /permeability increasing protein, lactoferrin, MPO, proteinase 3, and elastase. Histones can also kill many pathogens. The ras-related C3 botulinum toxin substrate 2 (Rac2) subunit from NOX2 is required for NET release, and H2O2 can rescue NET release in Rac2-deficient cells (172). Additionally, MPO helps efficient NET formation (240) (Fig. 5). Gene therapy with NOX2 restores the NET formation and controls aspergillosis in cells from CGD patients (18), indicating that NOX2-generated ROS participate in NETs. The NET release mechanism is dependent on a Raf-MEK-ERK pathway of NOX2 activation that is opposed to apoptosis (102). ROS-dependent NET formation was also observed in conditions in which neutrophils remained viable (359). Antioxidants inhibit NET formation (172). Macrophages and monocytes can also release ETs, but their role in the microbicidal activity of macrophages remains unknown.
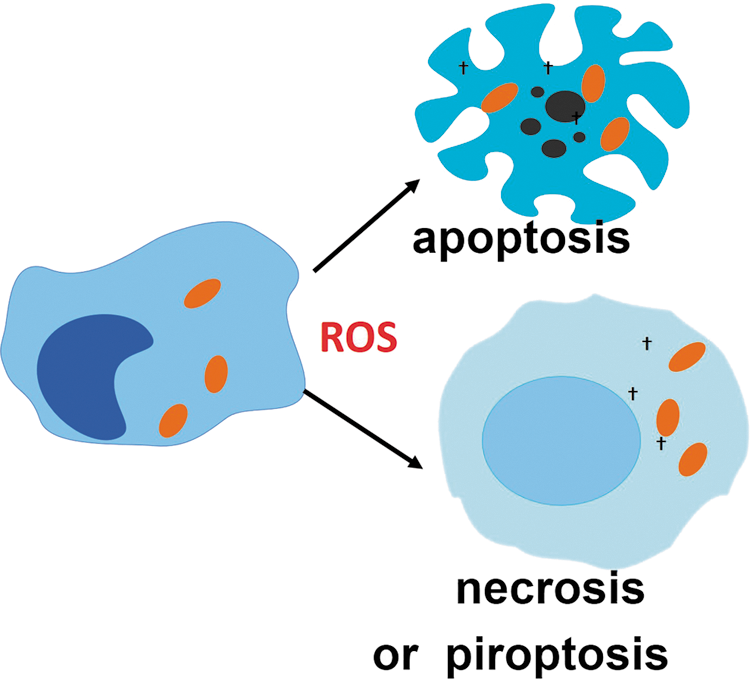
F. ROS promote cell death of infected reservoirs
Death of infected cells limits the spread of infection by preventing pathogen replication and/or differentiation into a cell-invasive phenotype [reviewed in (9, 159)]. In some particular cases, however, it can aid infection by facilitating the exit of the pathogen from the infected cell or killing phagocytes that could otherwise have acted to eliminate the pathogen. Unbalanced ROS production promotes cell death by various mechanisms including necroptosis, apoptosis, and pyroptosis (38, 42, 78, 161, 329), which can contribute to the demise of infection reservoirs (Fig. 6). Recently, ROS production induced by tumor necrosis factor (TNF) has been shown to participate in both M. tuberculosis elimination and promotion of pathogen infection, depending on the amounts produced by macrophage (269). In this work, low amounts of ROS were shown to be microbicidal and to contribute to controlling M. tuberculosis growth, whereas high amounts induced necroptosis and released bacteria in the surrounding, which favors bacterial extracellular proliferation (exuberant and associated with bacterial cording). The pros and cons of ROS-induced cell death in infection have been reviewed recently (42).
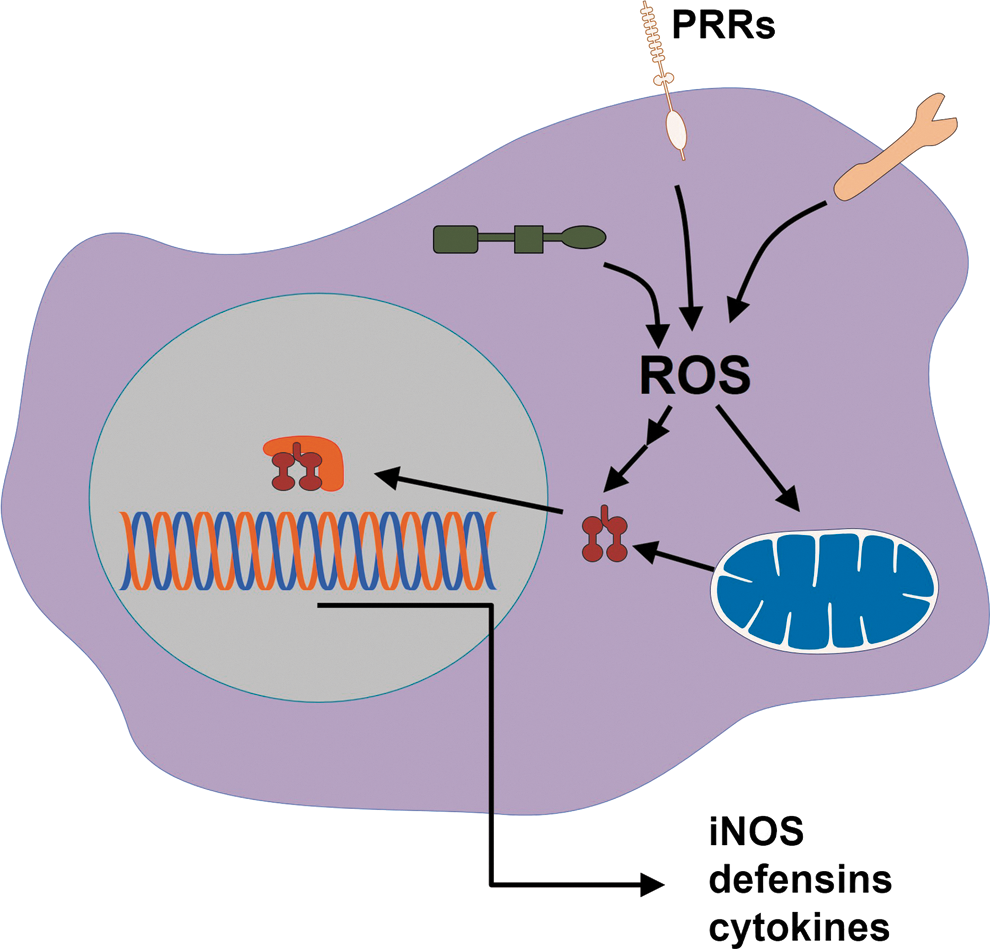
G. PRRs use ROS as signaling intermediaries in inflammation
PRR signaling orchestrates several mechanisms of pathogen elimination, and ROS production can interfere with it. The redox signaling downstream of PRRs, such as toll-like (TLR), nod-like (NLR), C-lectin like, and RIG-like receptors (RLR), controls the expression of genes involved in microbicidal activities (153). In general, it does so by reversible oxidation of cysteine residues in proteins in the NF-κB, AP1, MAPK, and PI3K pathways (321). Thus, it is expected that interference with ROS production can promote pathogen elimination by enhancing redox signaling. TLR-stimulated production of pro-inflammatory cytokines can be enhanced by oxidative stress (74), and TLR-deficient cells do not respond to such stimulation (145, 242, 350). In fact, the literature presents good demonstrations that ROS-enhanced PRR signaling can contribute to pathogen elimination (282, 317, 352).
Inflammasome is a multiprotein complex that senses danger signals and pathogens. The most well-studied inflammasome is the one formed by the NLR NACHT, LRR and PYD domains-containing protein 3 (NALP3). ROS are produced in response to NALP3 activators in Aspergillus fumigatus (284), Candida albicans (91), and influenza A (3) infections, but much controversy surrounds the role of ROS in inflammasomes. There are studies showing that ROS are required for mature IL-1β production induced via P2X7 receptors (206), and antioxidants were shown to prevent pro-IL-1β synthesis or NALP3 expression (67). However, the activation of NRF2, a gene that controls antioxidant defenses (see section III[B]), was shown to be required for cholesterol crystals to activate the inflammasome (80). Rubartelli et al. reviewed the subject and proposed that the activation of antioxidant defenses, which is the usual response to an oxidative hit such as that caused by TLR ligands, is the key to reconcile contradictory studies in favor and against ROS as promoters of inflammasome activation (278). They argue that the greater the activation of Nrf2 by an oxidative insult, the greater the inflammasome response.
H. ROS are chemoattractors to phagocytes
H2O2 from epithelial cells is produced very quickly after tissue injury and appears to be an initial signal that causes neutrophil recruitment to the wound (the region where epithelial tissue was injured) (224). The Src-family kinase Lyn is a sensor of H2O2 and its inhibition impairs neutrophil recruitment to the wound (357). Much is still to be learned about how leukocytes react to oxidative stress through ROS-sensors, but neutrophil recruitment by H2O2 most likely contributes to rapid pathogen clearance after tissue injury.
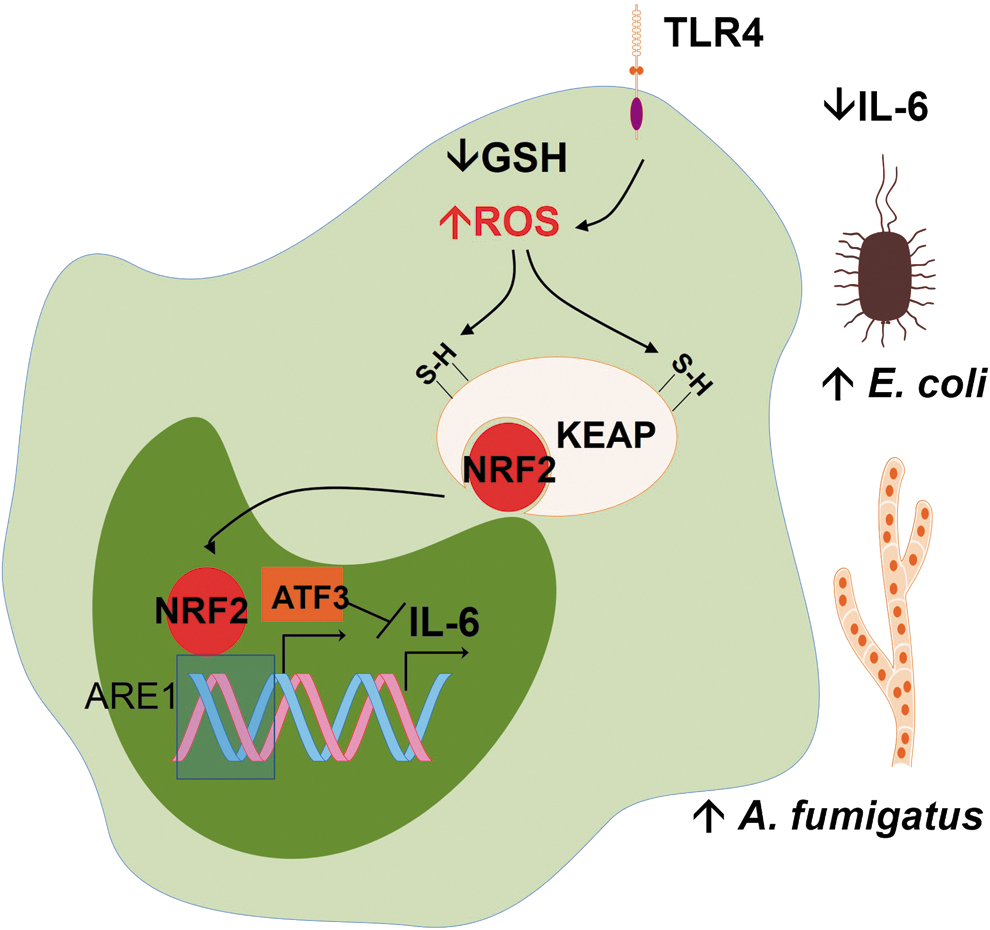
I. ROS can activate NRF2-target genes, a part of the antioxidant defense response that interferes with innate immunity
In section II, we discussed in detail how ROS activate antioxidant defenses through transcription factor NRF2. For now, we should assume that NRF2 is a transcription factor that orchestrates the antioxidant response to oxidative stress and is the target of many antioxidants, the so-called indirect antioxidants. Oxidative stress in macrophages leads to NRF2-dependent ATF3 (a negative regulator of TLR4) superexpression during sepsis. ATF3 superexpression protects against endotoxic shock but increases susceptibility to fungal and bacterial infections by inhibiting IL-6 production (114) (Fig. 8). In this work, ROS production was increased in reduced glutathione (GSH)-depleted monocytes. The liver load of A. fumigatus and the E. coli bacteremia were increased in response to GSH depletion, challenging the overall notion that antioxidant conditions are detrimental to defenses against fungi and bacteria. Thus, it is possible that oxidative stress can fuel infection in a number of cases by ultimately turning off the production of IL-6.
Another NRF2-target gene that participates in innate immunity is CD36 (58, 123). The molecule CD36 is a scavenger receptor best known for its capacity to bind to oxidized low density lipoprotein (LDL) and mediate its internalization, which is part of the atherosclerotic foam cell formation. CD36 also recognizes some microbes and is able to enhance the phagocytosis of Plasmodium falciparum (233). Nevertheless, the NRF2-dependence of CD36 expression is somewhat controversial, as it seems to depend simultaneously on PPARγ (252) and also on monocyte/macrophage differentiation at least in some instances (149). Still, NRF2 controls genes involved in iron and lipid metabolism, described below.
J. ROS interfere with iron storage and tissue mobilization, influencing iron availability to pathogens
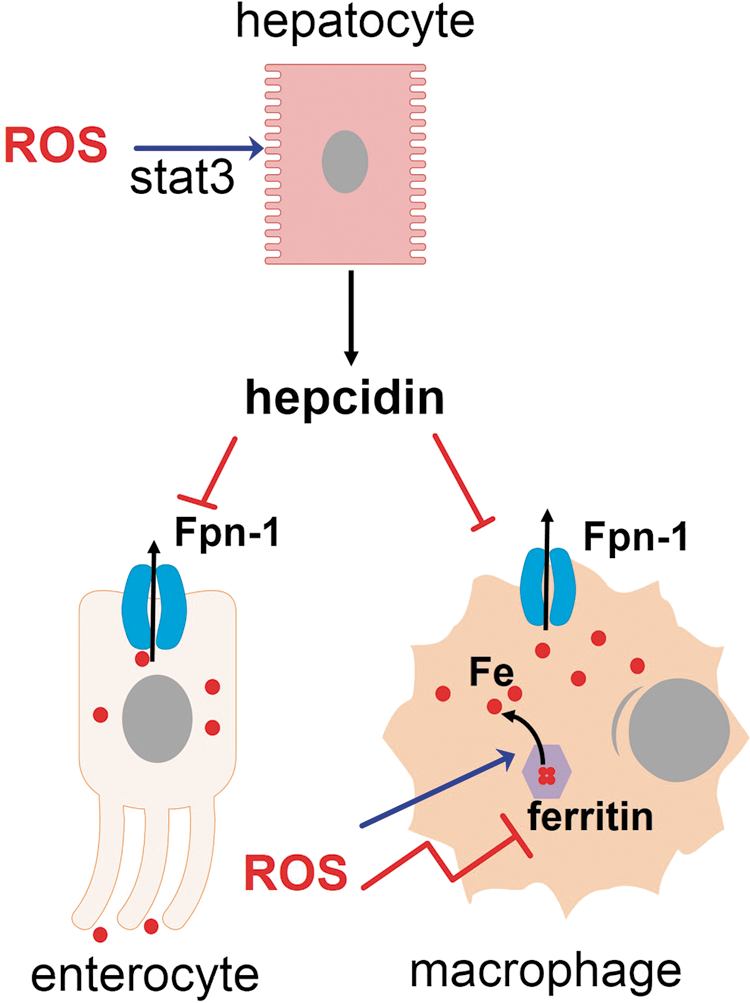
The levels of iron are tightly regulated in the body. Iron is preferentially transported and stored in its redox-inactive form because its free form, the so-called labile iron, can generate cell-damaging ROS (26). Iron is a required nutrient for pathogens and its sequestration away from pathogen reach is part of innate defenses (132). ROS stimulate secretion of hepcidin, a hormone that commands mobilization of iron from the liver to macrophages (198). Ironically, oxidative stress releases iron from intracellular storages (36, 201, 358), thus propagating ROS generation and offering some cytosolic pathogens an opportunity to acquire this nutrient. The outcome of an infection, in certain instances, can presumably depend on the capacity of a given pathogen to withstand oxidative damage, its need for iron, and iron availability in its niche (Fig. 9).
The NRF2-target gene Hmox1 codes for HO-1 (for HO-1 as part of NRF2-orchestrated antioxidant defenses, see section III). HO-1 is an enzyme that has antioxidant capacities, not only because it degrades the prooxidant group heme but also because it leads to biliverdin production, an antioxidant molecule, which is next converted by biliverdin reductase to bilirubin, an even more potent antioxidant (87). Some microorganisms use heme as a source of iron and therefore can be directly affected by the reduced heme availability in the presence of greatly increased HO-1 expression. S. aureus, for instance, takes up hemoglobin and degrades its heme group to produce the free iron it needs (300). HO-1 overexpression produces iron overload in macrophages, but most of this iron is ferritin-bound, due to increased expression of ferritin that comes along with HO-1 overexpression. In fact, HO-1-overexpressing cells have 25% of the free iron usually found in the original cell lineage (162). However, tissue iron is concentrated in splenic and hepatic macrophages in wild-type mice, but in the genetic absence of HO-1 expression, iron leaves these macrophages and can be found in kidneys and hepatocytes (150). The reasons for such redistribution are unknown, but most likely linked to the active heme degrading function in macrophages, since macrophages from HO-1−/− mice die after erythrophagocytosis.
Iron availability to pathogens can greatly influence the outcome of infection. The mononuclear phagocyte system and the liver constitute the main iron reservoirs in the body. A number of hormones and cytokines orchestrate the iron mobilization between those reservoirs.
Hepcidin is mainly secreted by hepatocytes in response to macrophage IL-6 and bone morphogenetic proteins agonists and binds to FPN, a transmembrane protein that exports iron and is expressed in macrophages, hepatocytes, and enterocytes (68). The interaction between hepcidin and FPN produces the degradation of the later. In enterocytes, FPN degradation reduces iron absorption, reducing the flow of iron into the plasma. Plasma iron-binding proteins, which have a general bacteriostatic effect (except for transferrin, targeted by some microorganisms), are upregulated on infection and efficiently sequester iron from extracellular pathogens. These proteins include ferritin (which besides being a major intracellular iron storage, is also present in low amounts in the plasma), lactoferrin, haptoglobin (hemoglobin-binding transporter), and hemopexin (heme-binding transporter). In macrophages, FPN degradation produces iron sequestration inside these cells, so hepcidin provides an opportunity for intracellular microorganisms residing within macrophages (68). The net result of hepcidin action is the mobilization of iron from hepatocytes to macrophages, and the increase in ferritin expression within macrophages. In P. berguei infection, iron mobilization by hepcidin results in decreased liver infection, due to decreased iron availability (253). However, nontyphoidal Salmonella grows within macrophages and its growth is reduced by high levels of FPN expression, whereas hepcidin reverses this effect (48). A similar phenomenon occurs in human immunodeficiency virus (HIV) infection (349). Decreased burden as a consequence of increased FPN expression occurs in M. tuberculosis (131) and also in T. cruzi-infected macrophages (235). Therefore, it should be expected that pathogens that live in the liver and those that live in macrophages exploit the hepcidin-FPN axis to allow maximal iron availability.
In addition, depending on the cell compartment in which the pathogen lives, iron availability can vary. Intracellular pathogens that live inside a phagosome (such as Leishmania, Mycobacteria, and Salmonella) can be subjected to iron starvation mediated by natural resistance-associated macrophage protein (NRAMP)-1, a protein that pumps iron out of the phagosome (132). However, pathogens that live in the cytosol can directly scavenge iron-laden ferritin, as is performed by Neisseria meningiditis (164), or feed on the labile iron pool (non-bound to ferritin), as is performed by Leishmania donovani (61). L. amazonensis expresses an iron transporter called LIT1 and depends greatly on iron availability inside the phagolysosome to express the iron superoxide dismutase (FeSOD), to differentiate into amastigotes, and to grow efficiently inside macrophages (200). Contrary to expectations, L. major can be eliminated in certain conditions of systemic iron delivery, apparently due to increased NF-κB activation caused by increased ROS (19).
M2 macrophages release iron, whereas M1 macrophages exhibit iron retention (34). The generation of NO by macrophages is dependent on the labile iron pool. The activity of iNOS has previously been associated with iron content inside macrophages (341). This finding was confirmed in Mycobacterium-infected macrophages because the upregulation of FPN reduced bacterial burden in its early stages and also decreased NO production in response to macrophage stimulation by lipopolysaccharide (LPS) (131).
Oxidative stress and iron metabolism are closely related. The intricate relations between ROS and iron had been the subject of recent studies. It is well known that excess free iron generates oxidative stress, whereas iron sequestration by ferritin precludes it (26). ROS are in fact able to release iron from iron-containing proteins (201), inhibit ferritin synthesis, increase iron uptake (36), and propagate ROS formation. Inside the cell, nonferritin bound iron complexes with small molecules in different cell compartments that comprise the labile iron pool. In the cytosol, most labile iron forms a complex with glutathione, a molecule involved in antioxidant defenses of the cell (111), and high levels of glutathione are limiting for iron metabolism (154). A short exposure to redox-active agents produces parallel increases in ROS and the labile iron pool (25), an effect that seems to be the net result of a number of phenomena. H2O2 increases the degradation of
Loading of macrophages with saturated Fe-ferritin produces lysosomal membrane permeabilization (a condition associated with cell damage) when cells are subsequently exposed to H2O2. Incubation of macrophages with apo-ferritin prevented lysosomal membrane permeabilization, suggesting that the progressive chelation of free iron by ferritin represents a defense against oxidative stress by preventing lysosomal membrane permeabilization and cell damage (157). In fact, certain microorganisms exploit the redox balance of the cell by inducing the expression of ferritin, as is observed with Chlamydia trichomatis, which reduces ROS-induced apoptosis to preserve the host cell (330). Moreover, NRF2 activators, the so-called indirect antioxidants, increase ferritin and FPN expression (112, 186) since both genes have ARE consensus in their promoters, in agreement with the general role of NRF2 in tissue protection against damage. The outcome of oxidative stress actions on iron availability to each microorganism will depend on the tissue the microorganism infects and/or the intracellular compartment it lives in, and the iron availability can greatly impact the infection. The case for T. cruzi, a cytosolic infection in which oxidative stress acts to increase the labile iron pool in macrophages and to favor parasite growth (235), is discussed in detail in a section below (V[D]).
K. ROS interfere with lipid metabolism and foam cell formation
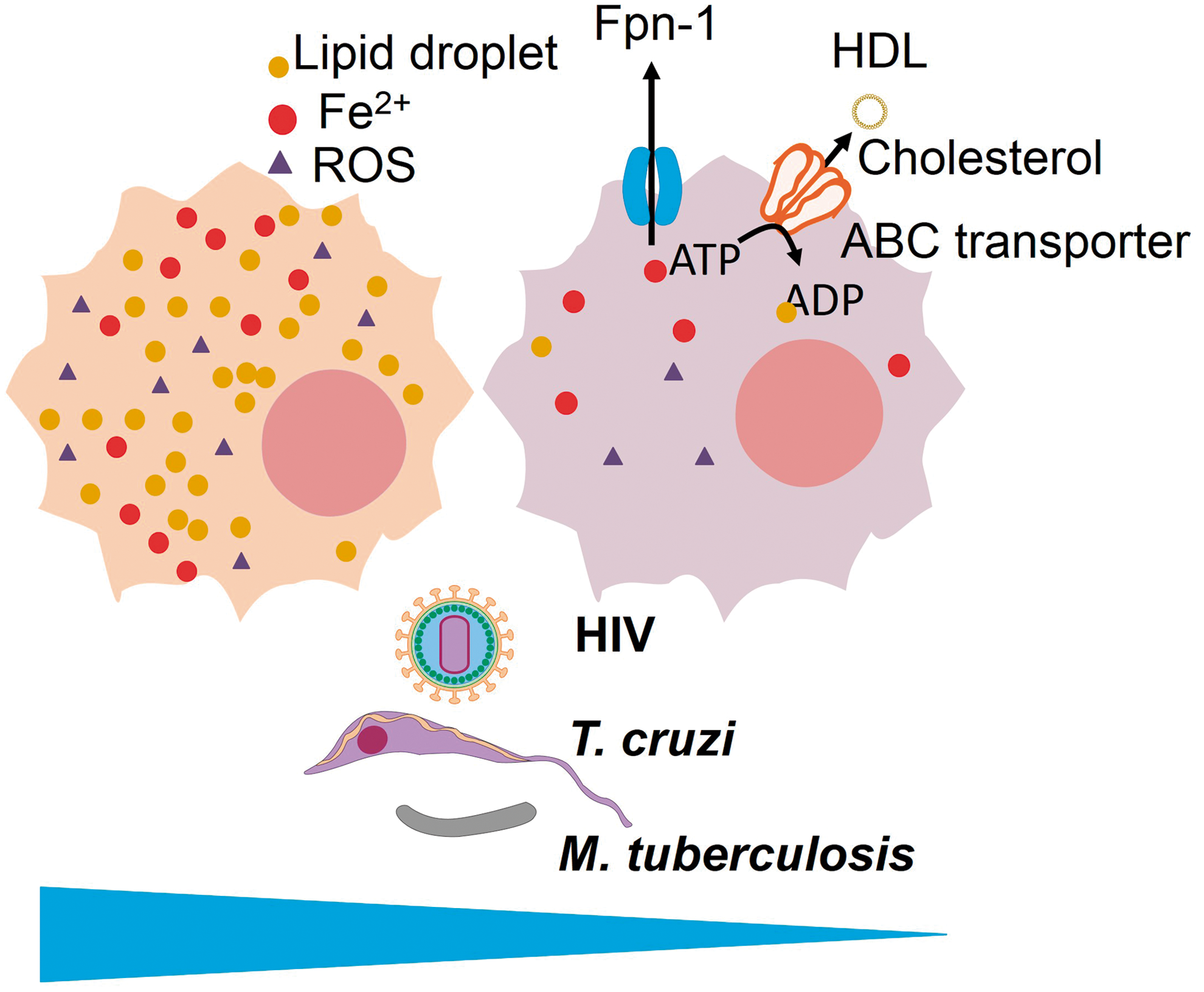
Atherosclerosis and oxidative stress are closely associated. Macrophage accumulation in the vascular wall occurs in atherosclerotic lesions and contributes to plaque formation. On uptake of oxidized lipids through scavenger receptors, macrophages turn into foam cells, a phenotype specialized in lipid accumulation within cytosolic droplets (1, 316). Several pathogens induce foam cell formation, and some pathogens seem to benefit from this formation to maintain their optimal growth, whereas it appears to be counterproductive to others. Some of these pathogens, such as Actinobacillus actionomycetemcomitans, P. gingivalis, Chlamydia pneumonia, HIV, have even been observed in clinical specimens taken from atherosclerotic plaques (151, 155, 202, 209). Foam cell formation can be stimulated by the inhibition of cholesterol efflux through ABCA1 and ABCG1 to high-density lipoprotein (HDL) (360), and recent studies have demonstrated that ROS is a major factor inhibiting cholesterol efflux through ABC transporters, favoring foam cell formation (46, 76, 319). In fact, the oxidative stress and lipid peroxidation produced by iron ascorbate in macrophages (through Fenton reaction) inhibit cholesterol efflux through ABC transporters (183). Therefore, it is highly likely that ROS can influence the outcome of macrophage infections through the inhibition of cholesterol efflux and the generation of the seven cell phenotype.
Several pathogens (both those that are harbored inside macrophages and not) induce foam cell formation: HIV (209), Streptococcus sanguinis (232), Porphyromonas gingivalis (259), M. leprae (55), M. tuberculosis (299), C. pneumoniae (37), T. cruzi (60), L. amazonensis (249), T. gondii (45), P. falciparum (124), and P. berghei (273), to name a few. HIV (209) and C. pneumonia (155) have been observed infecting foam cells in atherosclerotic plaques, indicating that infection and foam cell formation co-exist in the highly oxidant environment of atherosclerotic plaque. HIV Nef is responsible for foam cell formation (209), whereas C. pneumoniae LPS is capable of inhibiting cholesterol efflux and turning macrophages into foam cells (137). In fact, bacterial LPS can induce foam cell formation (40), and other TLR ligands from many different bacteria also appear capable of inducing foam cells (223). M. tuberculosis diverts the cell metabolism toward ketone body synthesis, contributing to the formation of foam cells (299). Studies on L. major-infected macrophages have revealed that the microbe downregulates the expression of ABCA1, producing lipid accumulation inside the cell (262). In some of these infections, a role for foam cell generation in pathogen survival and growth has been proposed. In other infections, the opposite holds true.
Some pathogens reduce cholesterol efflux to HDL and alter cholesterol homeostasis in favor of foam cell formation, such as C. pneumoniae (via the JNK-PPARγ-ABCA1 pathway) (176), HIV (the HIV protein Nef downregulates ABCA1 expression) (209), and M. tuberculosis (299). In HIV infection, there is evidence that the inhibition of cholesterol efflux contributes to virus persistence at least in CD4 T cells because liver X receptors (LXRs) agonists, which promote ABCA1 expression, reduce viral load, an effect that can be reversed by cholesterol replenishment (130). In macrophages, the stimulation of cholesterol efflux reduces the productivity of the virions, reducing infectivity (209), which suggests that foam cell formation is associated with HIV growth. In patients, a decreased HDL level is observed along with increased ABCA1 and paralleling HIV load, a finding interpreted by authors as being compensatory to the dysfunctional cholesterol efflux (73). Thus, in HIV infection, the evidence points to foam cell formation as a factor contributing to enhancing infection. In C. pneumoniae infection, however, the opposite situation occurs: established foam cells can normally uptake bacteria, but this gives rise to smaller bacterial burdens (21). In agreement with these data, in the closely related C. trachomatis, the growth is also impaired along with the inhibition of cholesterol efflux through ABCA1 (54), indicating that the generation of foam cells represents an innate mechanism of defense against this pathogen. M. tuberculosis, however, depends on the generation of foam cells for its survival (244, 299). In T. cruzi-infected cells, the formation of foam cells is associated with the prostaglandin E2 synthesis that favors macrophage deactivation of microbicidal actions and parasite growth (59).
L. monocytogenes growth is increased in LXRα−/− (a nuclear receptor that controls ABCA1 expression) macrophages, but this phenomenon does not appear to be related to cholesterol efflux, as authors of one study examining the issue claim that both LXRα and LXRβ participate redundantly in cholesterol efflux, and they found a role for the former but not for the latter in affecting the bacterial burden (134). Nevertheless, LXRα−/−LXRβ−/− macrophages presented additional susceptibility that demanded further explanation. The recent finding that ABCA1−/− macrophages have decreased L. monocytogenes burden (364) suggests instead a role of foam cell formation as an innate defense mechanism. It appears that foam cells can be either more susceptible or more resistant than macrophages, depending on the pathogen considered. ROS promotes foam cell formation, and it is possible that ROS promotes infection when a pathogen grows better in foam cells and vice versa.
The interplay between lipid accumulation within macrophage foam cells, oxidative stress, and labile iron pool has been the subject of some recent works. In general, oxidative stress reduces the cholesterol efflux through ABCA1 and ABCG1 transporters (46), contributing to the preservation of the foam cell functional phenotype, whereas antioxidants increase cholesterol efflux and prevent foam cell formation (31, 239, 363). In agreement with these data, the exposure of macrophages to haptoglobin:hemoglobin complexes in vitro for 7 days increases FPN expression, decreases labile iron, and increases ABC cholesterol efflux transporters (ABCA1 and ABCG1) expression by reducing ROS generation and turns macrophages into a phenotype resistant to cholesterol loading. The degradation of FPN by hepcidin reverses the foam cell-resistant phenotype, reduces cholesterol efflux, and increases labile iron and ROS generation. The incubation with SOD significantly increased ABCA1 and ABCG1 expression, which is in perfect agreement with the idea that oxidative stress inhibits cholesterol efflux to HDL (76). In addition, the pharmacological suppression of hepcidin (see section III[J]) reduces the labile iron pool in macrophages, increases FPN expression, enhances macrophage cholesterol efflux, increases ABCA1 expression, decreases H2O2 generation, decreases foam cell formation, and atherosclerotic fatty streaks (283). Paralleling these results, in one study, incubation with heme induced a macrophage phenotype resistant to cholesterol loading due to LXRα and ABCA1 expression (24). This new macrophage phenotype is apparently the same in three different works, and J. J. Boyle proposed calling it MHem (23). Together, these data indicate that cholesterol accumulation in lipid droplets, foam cell formation, and atherosclerosis are in fact related to a macrophage phenotype that is characterized by iron retention and high ROS generation. Whether this phenotype MHem is present in foam cell-inducing infections and plays a role in their infectivity or pathogenicity remains unclear.
As shown in this section and in the previous section, HIV, T. cruzi, and M. tuberculosis benefit from foam cell formation (59, 209, 299) and grow less actively when FPN expression increases iron efflux (131, 235, 349) (Fig. 10). Whether this correlation is related to the MHem versus foam cell phenotype remains to be investigated.
The activation of the Nrf2 gene results in a plethora of mechanisms specialized in lipid metabolism, as detected by proteomic studies (143), and although the effects of Nrf2 on atherosclerosis have been the subject of much debate (279), it appears that NRF2-activators increase cholesterol efflux and prevent foam cell formation (152, 239). Whether ROS determine the outcome of macrophage infections through cholesterol efflux and foam cell formation remains to be established.
L. ROS influence phagosomal proteolysis through cathepsin inactivation
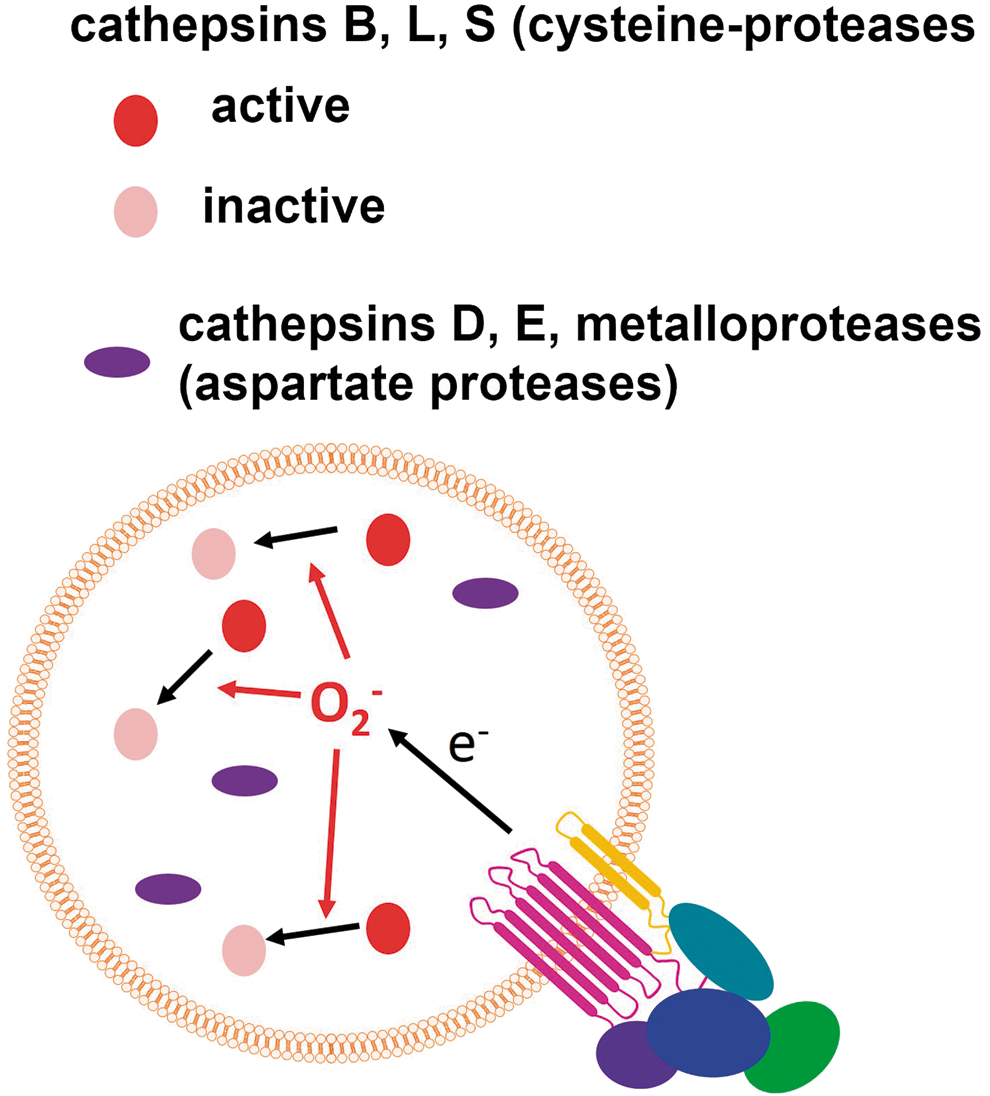
Phagosomal proteolysis is controlled by NOX2-derived ROS, which produce the inactivation of specific cathepsins by oxidation of the catalytic cysteine residues inside macrophage phagosomes (Fig. 11), whereas antioxidants such as resveratrol, quercetin, and diphenylene iodonium (DPI) increase the bulk proteolysis within phagosomes (281). The cathepsin-specific inactivation produced by NOX2 can potentially produce a particular pattern of proteolysis with the emphasis on some antigenic properties during infection.
M. ROS interfere with protein immunogenicity, antigenic presentation, Th polarization, and co-stimulation by dendritic cells
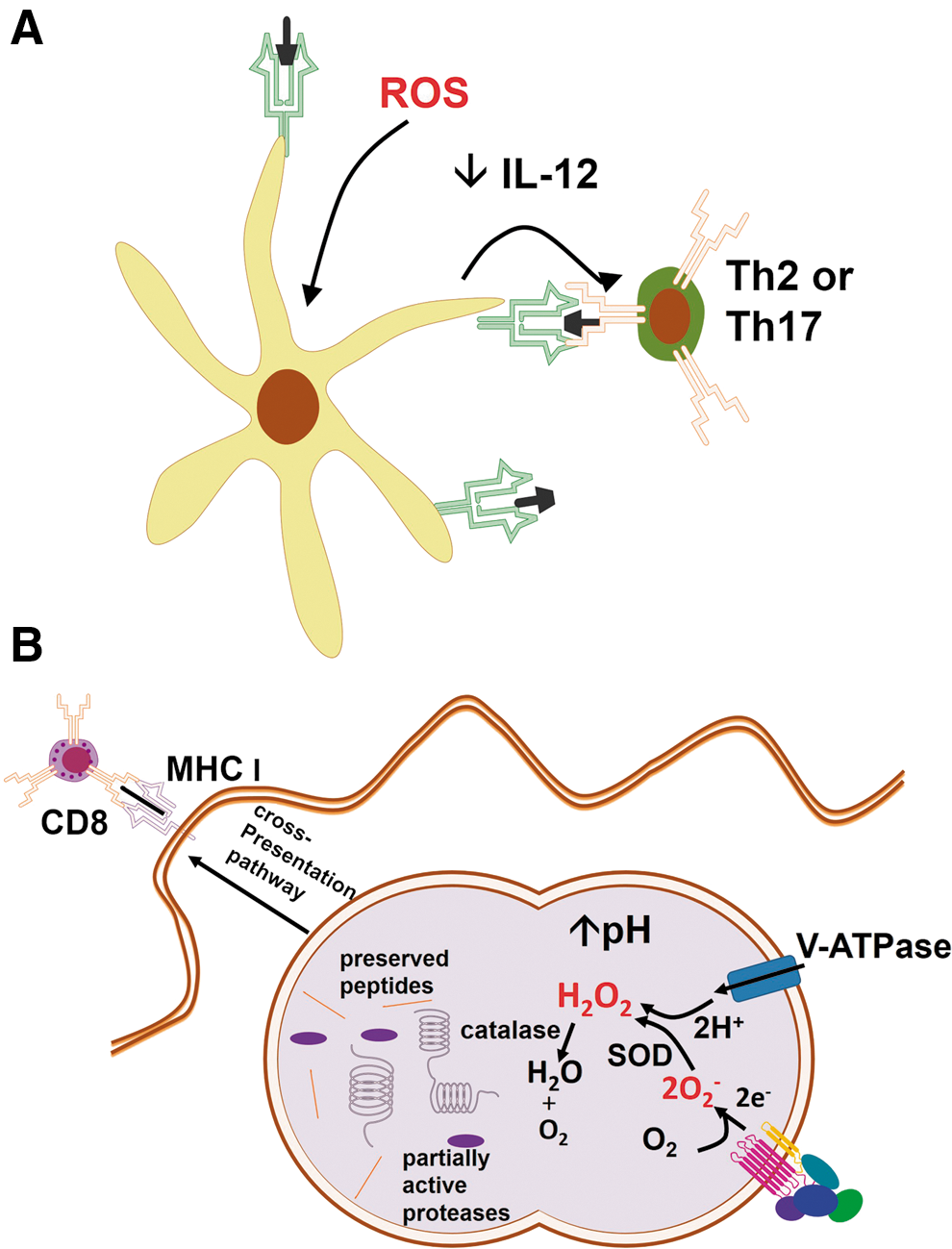
Dendritic cells (DCs) are responsible for the antigen capture, processing, and presentation to T cells that promote T-cell activation and influence the achievement of particular T-cell effector functions. NOX2-derived ROS is required for antigen processing by DCs and can interfere with adaptive immunity (181), increasing immunogenicity of proteins, inducing physiologic changes in DC, or influencing polarization of lymphocytes during antigen-specific responses [reviewed by (148)]. In general, antioxidants produce Th1-polarized responses, whereas the oxidative stress induced by exposure to prooxidants, oxidized proteins, or exhaustion of antioxidant defenses, generates Th2 (203, 243) or Th17 responses (336, 337) (Fig. 12A). An interesting picture has emerged from a work in which responses to oxidative stress generated by diesel particles interfered with the ability of TLR agonists to induce the expression of maturation receptors in DC, thus programming DCs to instruct Th2 responses (43). Oxidative stress increases influenza virus titers in association with increased polarization of Th2 responses (85). The generation of ROS stimulated by papain was shown to orchestrate Th2 responses by generating TLR4-induced lymphopoietin secretion, directly inhibiting IL-12 secretion by DCs, and promoting DC–basophil interaction in lymph nodes (318). It is possible that oxidative stress acts through the activation of NRF2, as Nrf2 −/− DC fail to react to diesel particles by programming cells to Th2 responses (43), and NRF2 activation is known to directly induce Th2 polarization (271).
ROS interference in adaptive immunity can potentially alter resistance to pathogens, and antigen processing and presentation are subject to ROS regulation. ROS and pH are critical to antigen degradation within DC phagosomes, as reviewed elsewhere (148). The consumption of V-ATPase-pumped H+ by NOX2-derived O2 − increases pH in DC phagosomes, reducing proteolysis and allowing antigen preservation for presentation (Fig. 12B). In fact, ovalbumin presentation to CD4+ cells is impaired by DPI, an inhibitor of flavoproteins that decreases NOX2-derived ROS (189). The MHC class I presentation may be compromised under oxidative stress, as demonstrated in macrophage T. cruzi infection, in which ROS inhibit protein tyrosine phosphatase activity, thereby inhibiting IFNγ-mediated immunoproteasome synthesis and causing MHC class I downregulation (17). In various models, ROS have been demonstrated to interfere with antigenic cross-presentation to CD8 cells (148). Recently, NOX2-inhibitors were shown to decrease the antigenic cross-presentation by DC to CD8 T cells due to defective autophagy and alkalization of phagosomes, in a model in which NOX2 controls autophagy of Aspergillus conidia (178). It is possible that the susceptibility of NOX2-deficient mice as well as of CGD patients to some of the pathogens depend on the adaptive CD8 response instead of on the microbicidal phagocyte response.
IV. Pathogens That ROS Contribute to Eliminating
A. Bacterial infections combated by ROS
Most bacterial infections are at least partially susceptible to ROS produced by phagocytes. In CGD, there is susceptibility to fungal (Aspergillus, Candida) and to particular bacterial infections, indicating that the immune system relies on NOX2-derived ROS production to clear these pathogens. The main bacteria that prosper in the absence of NOX2 are S. aureus, L. monocytogenes, Francisella tularensis, S. typhimurium, and S. marcescens (15, 303). It has been previously proposed that CGD patients are susceptible to catalase-positive microbes but exhibit normal killing of noncatalase microbes through the production of HOCl− using the H2O2 produced by microbes themselves in the phagosomes (168, 339).
The most frequently isolated bacterial pathogen from CGD patients is S. aureus (303). S. aureus activates oxidative and nonoxidative microbicidal mechanisms in neutrophils (110). NOX2 (gp91phox) hemizygous mice infected with S. aureus develop abscesses in the abdominal cavity, different from wild-type mice, and have impaired bacterial clearance in peritoneum but can ultimately clear the infection (251). NOX2 (p47phox)−/− mice kill 10-fold fewer S. aureus than wild-type mice (125). These results indicate that ROS production is effective against S. aureus infection, but in its absence, other less effective immune mechanisms undertake clearance and ultimately eliminate the infection.
L. monocytogenes grow unrestricted in NOX2 (gp91phox)−/− mice, and the mice develop high splenic and hepatic bacterial burdens, along with an increased number of microabscesses (65). The enhanced susceptibility of gp91phox−/− mice can be detected from 2 days on, the time when lesions are caused by neutrophils in the liver (65). Nevertheless, by day 6 postinfection, the mice are able to control the infection, most likely due to the establishment of an adaptive response. NOX2 (gp91phox)−/− mice have been shown by another group to resist L. monocytogenes infection with a splenic bacterial load equal to that of wild-type mice, even though their macrophages displayed no ability to kill L. monocytogenes in vitro (297). Another NOX2-deficient murine model, p47phox−/− mice, is relatively resistant to infection (70). The differences between these two CGD murine models were exploited recently (356). The authors observed that the macrophages from the p47phox−/− mice have an increased STAT6 phosphorylation in response to IL-4 compared with gp91phox-/- or wild-type mice, producing the alternatively activated macrophage phenotype (high expression of Ym1 and FIZZ1). How precisely the alternatively activated macrophage phenotype produces resistance to L. monocytogenes remains to be elucidated, although it is known that the cells hypersecrete IL-1α, and this secretion is capable of increasing resistance to the bacteria.
Activated macrophages retain L. monocytogenes in vacuoles for microbicidal purposes. In nonactivated macrophages, L. monocytogenes escapes from macrophages vacuoles within 30 min and incubation with the iNOS inhibitor NG-Monomethyl-
F. tularensis is a facultative intracellular pathogen that causes intense oxidative stress (250). NOX2 (gp91phox)−/− mice are more susceptible to infection with F. tularensis, developing significantly higher bacterial burdens in lungs and spleens and dying 1 day sooner than wild-type mice (156). Nevertheless, the differences observed in existing studies were small, and the authors have proposed that NOX2 plays a minor role in the host defense to F. tularensis. Neutrophils are recruited to the lungs as soon as mice are infected, but the depletion of neutrophils with monoclonal antibodies did not alter bacterial burden (156). A live vaccine strain opsonized by serum was phagocytosed by neutrophils, but not destroyed (191). In this work, F. tularensis was observed to impair neutrophil activation and disrupt the assembly of NOX2 in the phagosome membrane, preventing ROS production. The genes involved in this process have been characterized (292). In another work, the oxidative stress induced by the infection of macrophages with F. tularensis was dissected. The genes leading to the depletion of glutathione were upregulated, but the incubation with NAC did not affect the bacterial burden (7). Taken together, the evidence supports rather limited importance of NOX2-mediated respiratory burst in the killing of F. tularensis.
S. marcescens is one of the most prevalent pathogens found as an opportunistic infection in CGD (15), but no studies concerning how human phagocyte ROS kill S. marcescens or mouse CGD models infected with S. marcescens have been published.
In one study, M. avis, which is pathogenic to humans and mice, was lethal to 40% of NOX2 (gp91phox)−/− mice by 4–6 weeks postinfection (81). These mice displayed a much higher bacterial load in the lungs and their macrophages developed increased loads when infected in vitro with M. avis. These data are consistent with a case of human CGD that was first manifested by the presentation of M. avis infection (231), indicating that NOX2-derived ROS are an important mechanism of elimination in this case.
One of the most striking cases of bacterial susceptibility to NOX2-derived ROS is Acinetobacter baumannii infection in mice. These bacteria are closely related to Pseudomonas aeruginosa and cause a nosocomial pulmonary infection. Multiple drug resistance contributes to its emerging status. Intranasal infection with A. baumannii kills 100% of NOX2 (gp91phox)−/− mice in 48 h but does not kill NOS2−/− or wild-type mice (261). The bacterial counts are 1000-fold increased in the lungs and 10-fold in the spleens of infected gp91phox−/− mice compared with wild-type mice. To our knowledge, no cases of A. baumannii infections have been reported in CGD patients.
B. Viral infections combated by ROS
There is a paucity of data concerning the role of phagocyte ROS in mediating virucidal activity, but in general, findings support a role for ROS in apoptosis induction as a first line of defense against infection reservoirs (301). However, in betanodavirus (nervous necrosis virus) infection, ROS have been shown to induce apoptosis of the host cells, but catalase overexpression still reduced viral titers at the early stages of infection, most likely because apoptosis helps in the spread of infection in early stages (44).
In 1982, Rager-Zisman et al. used a variant of the J774 monocyte cell line defective in oxidative metabolism to evaluate the contribution of respiratory burst to vesicular stomatitis virus (VSV) clearance (263). PMA-induced oxidative burst significantly reduced viral burden when small amounts of viral particles were used to infect cells. No reduction of viral burden was observed on stimulation of oxidative metabolism-deficient cells and catalase was able to inhibit PMA effects on parental cells, indicating that respiratory burst was indeed responsible for the reduction of viral burden. Free virus suspended in H2O2 solutions were killed only when a limited amount of viral particles were used, suggesting that there is an amount of H2O2 required to inactivate each virus and leading the authors to speculate that it most likely acts on the small extracellular input virus rather than on intracellular viral progeny. In 1992, VSV-infected cells were transfected with CuZnSOD to evaluate whether their sensitivity to type IFN I-antiviral state depended on ROS generation. In fact, the more gene copies of CuZnSOD used in transfection, the more cells became resistant to induction of antiviral state and susceptible to infection, indicating that O2 −• participates in determining the sensitivity to type I IFN (120). In this study, paraquat, a drug that induces oxidative stress, reduced viral burden, confirming the role of ROS in reducing viral burden. VSV-infected VERO cells were spared from cell death by antioxidants, which offer a clue to explain the decreased viral burden (268). Epithelial cells deficient in autophagy (Atg5−/−) were observed to be resistant to VSV infection, apparently due to increased RLR signaling (317). Such increased RLR signaling was produced by the deficient autophagy of mitochondria, the main sources of ROS in nonphagocytic cells, generating increased ROS signaling and increasing viral clearance via effector mechanisms downstream of RLR signaling (see section III[G]).
HCMV induces ROS production as soon as it invades a cell (307). These ROS would normally contribute to the effective activation of NF-κB, but the virus has ways to overcome this barrier. Recent experiments have shed light on the subject. HCMV induces non-NRF2-mediated induction of the antioxidant enzymes, SOD, GLCG, and GPX-1 (323). The addition of buthionine sulfoximine (BSO) to HCMV-infected cells, a drug capable of depleting antioxidant GSH, produced unbalanced ROS production that inhibited mTOR and decreased viral growth. Thus, as a result of antioxidant enzymes induction, the levels of ROS are reduced, and H2O2 does not reach the levels necessary to inhibit mTOR. The inhibition of mTOR, a nutrient sensor, halts cell growth and represents an important effector mechanism against viruses (see section III[D]).
Chikungunya virus (CHIKV) is an arbovirus transmitted by mosquitoes. Autophagosome induction in infected fibroblasts is reduced in infected NAC-treated cells, indicating the mediation by ROS (135). Moreover, the oxidative stress caused by CHIKV infection inhibits mTOR (mTORC1, linked to autophagy). In this model, autophagy has been observed to be prosurvival, inhibiting the apoptosis of the host cell, whereas the blockage of autophagy results in massive apoptosis (Z-vad-sensitive). The inhibition of apoptosis decreases the numbers of infected cells, apparently due to the apoptosis-mediated propagation of infection in cell culture, most likely achieved by the enhancement of virus release from apoptotic cells. Although presumably ROS production should inhibit the viral burden as a result of inhibition of both apoptosis and mTOR activation, in the study, the authors focused on the linkage of autophagy, apoptosis and viral propagation and did not directly study how ROS affected viral burden.
In another study, the growth of herpes simplex virus 1 (HSV1) in corneal cells was inhibited by granulocytes, which released H2O2, and the amounts of H2O2 equivalent to that measured were sufficient to cause viral inactivation (109). HSV1 was observed to be somewhat resistant to inactivation by H2O2, and this resistance was mediated by the presence of its viral catalase since catalase inhibition increases its susceptibility (222). These data indicate that the virus is adapted to the oxidative environment where it grows. However, treatment with sulforaphane, an efficient NRF2 activator, did not alter HSV1 burden, despite causing great a reduction in the production of ROS by neutrophils and macrophages infiltrating the central nervous system (288).
Porcine reproductive and respiratory syndrome arterivirus stimulates ROS generation in infected cells, promoting the activation of NF-κB (166). Despite the possible role in pathology, in this case, ROS production and the activation of NF-κB do not seem to be related to viral reproduction, as treatment with antioxidants greatly reduces NF-κB activation but does not alter viral titers.
V. When the Paradigm Fails Us: Pathogens That Thrive on Oxidative Stress
Intriguingly, NRF2 activation results in the reduction of viral burden in respiratory syncytial virus (RSV) infection (49), whereas Nrf2 −/− mice are more susceptible to P. aeruginosa (265). The expression of HO-1, a NRF2-activated heme-degrading enzyme that has many antioxidant properties, has also been shown to reduce pathogen burden in hepatitis B (256), hepatitis C (167), enterovirus-71 (324), and HIV (62) infections, to mediate macrophage resistance to S. typhimurium (361) and T. cruzi (235), and to enhance the bacterial clearance of Enterococcus faecalis (50). Whether NRF2-target genes participate directly in innate immunity against pathogens or act by protecting tissues from infection-induced damage and allowing them to respond to pathogens remains to be elucidated. However, ROS scavengers or NOX2 inhibitors also contribute to the control of the burden of some pathogens, and NOX2-deficiency is associated with decreased burden in some cases (84, 188, 235), indicating that the production of ROS is counterproductive against some pathogens.
In the following sections, we dissect infection cases in which oxidative stress appears to enhance a pathogen's growth and antioxidants inhibit infection. Cell death can be a drawback in studies with prooxidants, as they tend to increase the number of dead cells, and the uptake of pathogens released from dead cells by live cells can potentially distort the results (44, 135), and therefore, most of these studies are based only on antioxidants. In addition, one must keep in mind that prooxidants may later turn into functional antioxidants, as they stimulate NRF2-target genes. Heme, for example, is capable of acting as both a prooxidant, as it can induce oxidative stress, and an antioxidant, as it induces HO-1 expression with delayed kinetics.
A. Bacterial infections in which the participation of ROS in clearance is dubious, controversial, or irrelevant
CGD patients have increased susceptibility to M. tuberculosis and also to environmental mycobacteria (32, 158, 165). Human neutrophils are rather inefficient at eliminating M. tuberculosis (52), and neutrophil respiratory burst, besides apparently being dispensable as a mycobactericidal mechanism (133), produces neutrophil necrotic death and allows bacterial escape (52). However, the respiratory burst contributes to the elimination of M. tuberculosis in macrophages, as shown recently in patients with a NOX2 (gp91phox)-deficiency (CGD) that only affects macrophages (33). In mice, the inhibition of TNF-induced macrophage apoptosis is achieved by M. tuberculosis through the inhibition of NOX2-derived ROS by its type I NADH dehydrogenase (197), a source of ROS that seems to be involved in the redox signaling pathway to apoptosis downstream of TNF. These data indicates that ROS in this case participate in a second line of defense beyond the initial respiratory burst, which is the induction of host cell apoptosis that favors the elimination of infection reservoirs, but the bacteria manages to escape this innate mechanism of control.
M. tuberculosis is relatively resistant to ROS, but NOX2-derived ROS mediates the TLR2 inflammatory responses that contribute to clearing M. tuberculosis through the enhancement of vitamin-D receptor-induced cathelicidin expression in macrophages (352). However, the M. tuberculosis gene Eis reduces ROS production, downstream autophagy and pro-inflammatory cytokine production, but still, does not alter bacterial burden (298). Treating M. tuberculosis-infected guinea pigs with NAC resulted in decreased splenic bacterial counts (236), indicating that the role of oxidative stress in tuberculosis deserves further consideration. In fact, GSH is toxic to M. tuberculosis and GSH depletion increases the bacterial numbers inside macrophages (332, 333), but it remains unknown whether GSH disturbs the redox balance of the bacteria. At least some effects of GSH appear to be related to the stimulation of the NK activity (92). The effects of GSH on M. tuberculosis infection are reviewed elsewhere (207).
Streptococcus pneumoniae is not an opportunistic infection in CGD (169). In 2003, Schaper et al. observed that NOX2 (p47phox)−/− mice have an increased bacterial burden in S. pneumonia-induced meningitis, but NOX2 (gp91phox)−/− mice did not differ from wild-type mice (289). Confirming these findings in gp91phox−/− mice, the administration of a low inoculum resulted in bacterial clearance similar to wild-type controls (184). In 2008, examining gp91phox-/- mice subjected to high inoculum pneumonitis, Marriot et al. observed a surprisingly decreased mortality and decreased bacterial burden in the lungs and blood (185). The enhanced recruitment and activation of neutrophils were observed in gp91phox−/− mice, and the authors concluded that gp91phox is fundamental for control inflammation in this infection but not for microbial clearance. The role of ROS in clearance of S. pneumoniae remains to be fully unraveled.
The killing of P. aeruginosa by neutrophils is normal in CGD patients (306). In this work, the authors found evidence that CGD is associated with P. cepacia, but not with P. aeruginosa infection. These results contrast with NOX2 (p47phox)-deficient mice, which present deficient P. aeruginosa clearance in the lungs and by macrophages, due to decreased TLR4-induced NF-κB (282). In fact, p47phox−/− mice have additional macrophage deficiencies (175), and it is important to note that these results have been confirmed in a NOX2 (gp91phox)-deficient model. Accordingly, mice deficient in Vav, a guanine nucleotide exchange factor involved in Rac activation, display increased mortality, deficient oxidative burst, and deficient clearance of P. aeruginosa by neutrophils, but the authors observed that this protein has additional signaling functions to those predicted by its Rac-activating role, and the mechanism by which Vav deficiency promotes infection requires further research to clarify (88). In addition, when studying the response of mice deficient in vitamin D3-upregulated protein-1 to P. aeruginosa bacteremic shock, the authors observed the mice to be resistant, to have increased bacterial clearance, and to produce increased amounts of ROS (248). Treatment with NAC reversed ROS production and resistance, demonstrating that, in this case, ROS was responsible for increased bacterial clearance. Moreover, in cystic fibrosis conductance regulator molecule-deficient mice, the reduced clearance of P. aeruginosa by alveolar macrophages has been associated with the failure to cluster gp91phox in ceramide-enriched membrane platforms, to release ROS, and to acidify vesicles. The inhibition of ROS production by the NOX2 inhibitor apocynin or incubation with ROS-degrading enzymes (SOD and catalase) of P. aeruginosa-infected macrophages results in increased infection (362). Taken together, these data indicate that ROS contribute to the elimination of P. aeruginosa, at least in mice.
Some studies have not found a role for ROS in P. aeruginosa clearance, or even supported a role for ROS in enhancing infection. Pyocyanin, a redox toxin from P. aeruginosa that induces ROS and depletes GSH in neutrophils, does not alter killing by neutrophils (210). A recent study analyzed the role of MUNC13-4 in P. aeruginosa infection. The molecule is necessary for the regulation of p22phox trafficking to the plasma membrane and extracellular ROS production by neutrophils. Phagosomal maturation after phagocytosis of P. aeruginosa was dramatically impaired in MUNC13-4−/− neutrophils, a stimulation that triggers exclusively intracellular ROS production, but the assembly of p22phox in phagosomes did not depend on MUNC13-4. However, the fusion of phagosomes to azurophilic granules depended on MUNC13-4, as well as the killing of P. aeruginosa. To determine which of these effects was required for MUNC13-4 killing of P. aeruginosa, the authors used gp91phox−/− neutrophils in killing assays and observed that NOX2 is totally dispensable for the neutrophil killing of P. aeruginosa (204).
In a different study, treatment with the NRF2-activator sulforaphane produced the enhanced clearance of bacteria (P. aeruginosa and Haemophilus influenza) by the macrophages from chronic obstructive pulmonary disease patients, an effect that could be reversed by siRNA against NRF2 (107). Sulforaphane also enhanced bacterial clearance in the lungs of wild-type mice but failed to do so in Nrf2 −/− mice. NAC failed to enhance bacterial clearance, indicating that this is an NRF2-mediated effect rather than a general antioxidant effect, but the failure of NAC to perform appropriately in vivo is a rather common finding and should not be taken as a conclusive evidence of a nonantioxidant effect. Nrf2 −/− mice displayed reduced bacterial clearance on exposure to cigarette smoke, an effect that could be credited to the reduced phagocytosis by alveolar macrophages ex vivo under oxidative conditions. In another work, Nrf2 −/− mice succumbed to P. aeruginosa infection provoked after hyperoxia insult and had increased bacterial burden compared with wild-type mice (265). The reduced capacity to clear bacteria could be verified as early as 4 h after infection in Nrf2−/− mice. After 48 h, hyperoxia-exposed infected Nrf2 −/− mice presented increases in IL-6 and IL-1 production and dramatically reduced the expression of phagocytosis receptors in alveolar macrophages compared with wild-type cells. Supplementation with GSH restored the ability of Nrf2 −/− cells to clear the bacterial burden and suppressed the increases in cytokine expression, indicating that in this case, the antioxidant effects of NRF2 mediate the protective effect against P. aeruginosa. Contradictory evidence favoring or refuting a role for ROS in bacterial clearance are most likely due the various cell mechanisms controlled by ROS or due to the differences between mice and humans. These differences need to be dissected and reconciled.
Salmonella is the second most prevalent bacterial infection in CGD (208). Nontyphoidal Salmonella is a common infection causing bacteremia in CGD patients (86), and Salmonella typhimurium has been observed, as an initial manifestation of CGD, to cause bilateral pneumonia and sepsis in association with Pneumocystis jiroveci (303). S. typhimurium is believed to be easily killed by NOX2-derived ROS, despite avoiding NOX2 assembly and remaining in phagocyte vacuoles that do not co-localize with gp91phox (82) since NOX2 (gp91phox−/−)-deficient mice presented increased mortality (297), increased bacterial loads in spleens, livers, and macrophages (187), and their macrophages present less efficient killing of S. typhimurium in vitro (297). These experiments were performed with gp91phox−/− mice in an Nramp1s background to allow for susceptibility to Salmonella and relying on the exclusive and noninteractive effects of the two phenotypes. The susceptibility of Salmonella to killing by ROS was confirmed by the increase in bacterial load observed in mice treated with SOD (326). The survival of S. typhimurium inside peritoneal macrophages from gp91phox−/− mice was greatly increased, and the main killing effects of gp91phox-dependent respiratory burst lasted for less than 5 h since wild-type mice and even NOS2−/− mice cleared a percentage of the bacteria within this interval, which was different from gp91phox−/− and gp91phox−/− NOS2−/− mice (331). Salmonella variants susceptible to oxidative burst (29) or with low capacity to live inside macrophages (75) were identified as avirulent in vivo. In NOX2 (p47phox)−/− mice, an oxidative burst-sensitive mutant regained virulence (328). The restoration of the virulence of a Salmonella variant unable to repair damaged DNA (thereby sensitive to oxidative damage) in gp91phox−/− and gp91phox−/−NOS2−/− mice suggests a role for direct oxidative damage as a mechanism of bacterial killing (297). Taken together, these data appear to draw a clear picture in which Salmonella depends greatly on respiratory burst evasion mechanisms to establish infection, and its growth is favored by the lack of NOX2. Salmonella susceptibility to ROS is reviewed elsewhere (72).
However, Salmonella resists direct oxidative damage, as it is capable of rapidly fixing ROS-induced DNA damage and can adapt to grow normally in highly oxidative environment (250 μM H2O2) within 24 h (182). Surprisingly, Salmonella has been shown to benefit from neutrophil transmigration and ROS production in inflamed guts (312) due to the generation of a luminal oxidized electron acceptor that confers a metabolic advantage and allows Salmonella to outcompete anaerobic microbiota (344). The contrast between Salmonella resistance and enhanced growth in the oxidative environment and its susceptibility to NOX2 activity in macrophages demand further explanation. It is possible that a percentage of Salmonella resists oxidative damage while NOX2-derived ROS promotes Salmonella elimination by nonoxidative mechanisms, such as autophagy (118).
B. Viral infections that thrive in oxidative environments
It is widely accepted that the ROS produced by phagocytes during respiratory burst contribute to the elimination of pathogens. Whereas NOX2-generated ROS can promote direct elimination of microbes engulfed by phagocytes, such as bacteria, fungi, and protozoa, the virus case is more complicated. Viral infections are more frequently inhibited by antioxidants (Table 1), a subject previously examined by Fraternale et al. (79).
CoPP, cobalt-protoporphyrin; GCLC, glutamate cysteine ligase catalytic subunit; GSH, reduced glutathione; GST, glutathione S-transferase; H2O2, hydrogen peroxide; HBV, human hepatitis B virus; HCV, human hepatitis C virus; HIV, human immunodeficiency virus; HO-1, heme oxygenase 1; IFN, interferon; IL, interleukin; KSHV, Kaposi's sarcoma-associated herpesvirus; NAC, N-acetyl-cysteine; NOX2, NADPH oxidase 2; NRF2, nuclear factor (erythroid-derived 2)-like 2; pCV2, porcine circovirus type 2; PMA, phorbol-12-myristate-13-acetate; ROS, reactive oxygen species; RSV, respiratory syncytial virus.
Viruses are usually targeted by autophagy but are barely detected by PRRs on the surface of phagocytes for engulfment and subsequent exposure to ROS and reactive nitrogen species (RNS). Rather, dying virus-infected cells (2) or Ig-opsonized viruses are engulfed (104). Endosomal TLRs, sensors of viral infections, do not augment ROS production, which is different from cell surface TLRs (343). Nevertheless, other sensors of viral infections, such as cytosolic NLRP and RLR, employ ROS as part of cell signaling. ROS can thus potentially influence the elimination of viruses by nonoxidative routes, such as by increasing PRR signaling. Virus detection by intracellular PRRs trigger the production of inflammatory cytokines, type I IFN, autophagy, unfolded protein response, apoptosis, necroptosis, and the onset of an adaptive response that targets virus reservoirs. ROS can also potentially interfere with all these processes. Moreover, many viruses have developed an evasion strategy that depends on NF-κB to allow their growth, such as influenza (225) and HIV (217). In this case, ROS enhance NF-κB activation and thus increase viral burden.
Kaposi's sarcoma-associated herpesvirus (KSHV) enters cells by binding to the NRF2-target cysteine glutathione exchanger (xCT), a cystine/glutamate exchanger that contributes to reducing oxidative stress by importing cysteine to replenish glutathione. Viral miRNAs upregulate the macrophage production of RNS, but RNS-induced apoptosis is prevented by concomitant upregulation of xCT, which is also induced by viral miRNAs in infected cells (260). In this case, the inhibition of RNS generation reduces viral burden, revealing the pathogen's stratagem. Incubation with H2O2 produces KSHV reactivation from latency, whereas ROS-scavenging NAC, glutathione, and peroxide-degrading catalase inhibit KSHV replication in culture, and treatment with NAC in vivo prolongs mice lifespan and reduces their viral load (355). It remains to be demonstrated how ROS and RNS generation contribute to promoting KSHV infection.
When acquired immunodeficiency syndrome (AIDS) became an epidemic, it was soon discovered that NAC could reduce viral burden in vitro (274), and Stanford Hospital in San Francisco observed that ROS-scavenging NAC could prolong patient survival (126, 127). Evidence was found in favor of NAC-induced reduction of viral burden, and the effects of oxidative stress in promoting HIV infection soon became clear (14, 274, 309). It was also observed that PMA promoted viral replication, which could be blocked with NAC (309). In fact, HIV infection leads to chronic oxidative stress, and the induction of HO-1 by heme can produce >90% reduction in the viral DNA found in infected human monocytes, a process reversed by the inhibitor of HO-1 activity tin-protoporphyrin (SnPP) (62). In a humanized mouse model of HIV infection using Hu-NOD-Scid mice, heme was capable of reducing viral burden. The inhibition of NF-κB activation is a suspected mechanism in this case, as hemin-induced HO-1 inhibits NF-κB, and NF-κB activation is a known mechanism promoting infection. Although HIV replication is enhanced by NF-κB activators, treatment with LPS, a TLR4 ligand that activates NF-κB, is capable of inhibiting infection through the induction of HO-1 (63).
Influenza A virus causes oxidative stress, and its inhibition by antioxidants effectively reduces viral replication. ROS-scavenging NAC reduces infection by influenza A (H3N2, H5N1, but less effective against H1N1) (84, 188) and B viruses (188) and is used in clinics to inhibit mucin secretion and reduce viral load. The inhibition of NOX2 by apocynin has been shown to reduce viral titers of influenza A H3N2 (X31) and H1N1 (PR8) in the lungs (334), despite the participation of ROS in RIG-I-elicited antiviral effects (304). Mice deficient in the NOX2 subunit gp91phox present reduced viral titers of influenza A H3N2 (X31) compared with wild-type mice and increased amounts of IL-1β in the lungs (334), whereas mice deficient in NOX2 p47phox present reduced H5N1 burden (122). The effects of NOX2 inhibition include the preservation of CD8+ producing IL-2, IFNγ, and IL-12 (334), but it still remains to be demonstrated how NOX2 inhibition acts to inhibit influenza A infection. In addition, the induction of NRF2 activation (using sulforaphane and epigallocatechin gallate [EGCG]) can reduce influenza A H3N2 (Bangkok/1/79) titer, whereas NRF2 inhibition (via lentivirus-containing shRNA NRF2) has been shown to increase epithelial viral burden (140). EGCG, the NRF2-activator, found in green tea, has been shown to be effective against H1N1, H3N2, and B virus. In these cases, the NRF2-activators induced type I IFN, RIG-I, and MxA molecules involved in virucidal mechanisms and potential mediators of their effects. However, NRF2 overexpression induced by an adenovirus containing Nrf2 gene (AdNRF2) infection reduced viral burden in epithelial and alveolar macrophages but did not increase IFNλ levels (147). However, in another study, HO-1 overexpression resulting from AdHO-1 infection did not reduce H1N1 PR8 titers (108).
RSV infection generates oxidative stress (41). Treatment with NAC reduces viral burden in vivo (188). The administration of aerosols containing SOD reduced the pulmonary RSV load in rats (348), whereas synthetic catalytic scavengers that possess SOD, catalase, and glutathione-peroxidase activity reduced viral replication in human epithelial pulmonary cell (115). Similarly, Nrf2 −/− mice present increased RSV replication compared with wild-type mice, which can be verified as soon as 1 day after infection (49). Moreover, wild-type mice, but not Nrf2 −/− mice, reacted to treatment with NRF2-activator sulforaphane with reduced viral burden in lungs. Taken together, these results indicate that ROS promote RSV infection. The mechanisms by which ROS can do so are discussed in detail elsewhere (83).
Porcine circovirus type 2 (PCV2) replication inside the kidney cells is inhibited by treatment with NAC, whereas GSH depletion with BSO increases PCV2 replication (47). ROS-induced NF-κB activation appears to be involved in promoting this infection.
Enterovirus-71 induces oxidative stress by increasing NOX2-mediated ROS production. Antioxidants such as the NRF2 activators EGCG and GCG have been shown to decrease the viral burden of infected VERO cells (113). Treatment of cell cultures with NAC, apocynin, or siRNA to the p47phox subunit of NOX2 reduces viral burden, and the overexpression of HO-1 (by prior infection with Ad-HO-1) suppresses ROS production and also enterovirus-71 replication (324). The inhibitory effect of Ad-HO-1 on virus replication can be reversed by the inhibitor of HO-1 activity SnPP. The HO-1 metabolite CO is able to inhibit ROS generation and to mimic the effects of HO-1 on the inhibition of virus replication. These results are highly indicative that oxidative stress promotes infection with enterovirus 71.
Chronic viral hepatitis C (human hepatitis C virus [HCV]) is associated with high oxidative stress and reduced expression of NRF2-target genes (39, 342). However, in one study, no positive effects were observed when NAC was added to the conventional IFNα treatment (89). The treatment of hepatocytes with inducers of HO-1 expression such as CoPP or heme decreases HCV replication, as wells as genetically induces HO-1 overexpression (366) or repression of Bach with miRNA (116, 296). A similar situation was observed in human hepatitis B virus (HBV)-infected mice, in which CoPP-induced HO-1 expression reduced viral replication in livers, which could be verified as early as 5 days after infection (256). In hepatocytes, the inhibition of HBV replication with CoPP was reversed by HO-1-specific siRNA and treatment with CoPP promoted HBC core protein degradation in hepatocytes within 6 h (256). As the product of heme degradation billiverdin induces the upregulation of type I IFN and proteins involved in the antiviral state, it is possible that the induction of HO-1 reduces viral burden by increasing type I IFN production. Recently, the inhibition of HCV protein NS3/4A by billiverdin or heme was shown to reverse the blockade of TLR3 and RIGI signaling caused by infection and to restore the type I IFN production in infected hepatocytes (98, 167, 365). The role of HO-1 and heme in viral hepatitis was recently reviewed elsewhere (290), and much of their effects appear to be directly antiviral and unrelated to antioxidant capacities. Nevertheless, it has been shown that H2O2 also increases HCV replication in hepatocytes (10–100 μM), except when incubated at a high and nonphysiological concentration (1 mM) (167). Silybin, the active principle of silymarin, an antioxidant and hepatoprotective plant used since the ancient Greeks, dramatically reduces the viral load in HCV patients (280), indicating that antioxidants in general act on HCV replication.
Lymphocytic choriomeningitis virus (LCMV) is eliminated in mice by perforin and IFNγ-producing CD8 cells. In NOX2 (p47phox)-deficient mice, disease development is attenuated, a phenomenon associated with reduced viral burden, increased numbers of antigen-specific CD8 cells, and increased IFNγ production, starting 8 days after the onset of infection (163). The evidence indicate that CD8 cells and their IFNγ production are sensitive to ROS, as depletion of GSH using BSO is capable of preventing the increase in virus-specific T-cell frequencies and the fraction of these cells that produces IFNγ. In fact, the binding and growth of LCMV in peritoneal macrophages also depends on the ROS generated upon infection (195), indicating that multiple effects of ROS contributes to favoring LCMV infection.
C. Bacterial infections that thrive in oxidative environments
Certain bacterial infections can be promoted by oxidative stress (Table 2). Studying P. gingivalis mutants deficient in rubrerythrin, a molecule involved in oxidative burst defense, Mydel et al. observed that rubrerythrin ensured bacteria proliferation. Surprisingly, NOX2 (gp91phox)-deficient mice were observed to be highly resistant to both wild-type and rubrerythrin-deficient bacteria (215). The authors concluded that the host's oxidative burst enhanced the survival of P. gingivalis and observed evidence that the compensatory increase in NO production was responsible for the resistance of NOX2-deficient mice to infection.
UDCA, ursodeoxycholic acid.
The first clear demonstration that ROS could directly promote a bacterial infection was made in M. abscessus-infected macrophages (230). The incubation of infected THP-1 macrophages with H2O2 or PMA, a respiratory burst inducer, was capable of increasing the growth of M. abscessus, whereas ROS-scavenger antioxidants inhibited the pathogen growth during 4–7 days of infection. The authors had previously demonstrated that the antioxidant MnTE-2-PyP reduces burden in infected macrophages, a phenomenon associated with rescue from infection-induced cell death and promotion of phagosome–lysosome fusion (229). To date, there are no clues to the mechanism by which oxidative stress promotes M. abscessus growth, but it is possible that it acts by preventing phagosome–lysosome fusion, as preventing fusion appears to be an important step in establishing infection.
Helicobacter pylori is known to induce severe oxidative stress and to feed on damaged tissue. Anecdotal evidence led researchers to conduct studies with sulforaphane-rich broccoli sprouts in H. pylori-infected mice and humans. Sulforaphane is a well-known NRF2 activator. Broccoli reduced bacterial colonization, an effect that could not be obtained in Nrf2 −/− mice (351), indicating that sulforaphane acts through NRF2 activation. In fact, the transfection of gastric epithelial cells with the HspB protein from H. pylori increased Keap expression and reduced the expression of the phase II detoxifying enzymes HO-1, SOD1, and NQO1, a mechanism that appears to be involved in the decrease in antioxidant defenses that follows infection (30). The mechanisms by which NRF2 activation reduces H. pylori colonization remain to be clarified. In addition, ROS-scavenging ursodeoxycholic acid (UDCA) reduced H. pylori bacterial colonization (322), whereas NAC helped to efficiently eradicate infection when associated with antibiotics, an effect that could not be attained in oxidative stress-exposed smokers (97). However, those studies are difficult to interpret, as sulforaphane, UDCA and NAC have direct bactericidal effects, although at higher doses (241, 351). The bacterial load of NOS2−/− gp91phox−/− or gp91phox−/− mice infected with H. pylori was far smaller than that of wild-type or NOS2−/− mice, indicating that indeed ROS are counterproductive against these bacteria, and gp91phox−/− mice also had greatly decreased H. felis burden (20). An increase in gastric mononuclear inflammation was associated with the absence of gp91phox expression, but whether it is related to the reduced bacterial load remains unknown. In another work, no significant decrease in H. pylori burden was observed in NOX2 (gp91phox)−/− mice, although the results were suggestive of a moderate decrease (139).
Bacillus anthracis infect macrophages, a critical step for their propagation, as spores germinate within the cell (94). B. anthracis is highly cytolytic to macrophages in vivo. The cytolytic toxin has been identified previously and is known to be responsible for the sudden shock that occurs during infection. Within macrophages, the toxin induces a lethal oxidative burst that kills the cells in hours (105, 287). Therefore, B. anthracis spores must germinate in the oxidative environment induced in macrophages. In fact, it appears that spores from B. anthracis are highly protected against oxidative stress by various SODs (57) and are stimulated to germinate by O2 − exposure (12). B. anthracis is therefore able not only to cope with oxidative stress but also is able to thrive on its generation.
Transgenic mice overexpressing HO-1 present increased survival in the cecal ligation and puncture (CLP) model of polymicrobial sepsis and also decreased bacterial burden, whereas Hmox1 −/− mice do not survive CLP and have higher bacterial burdens than wild-type mice (50). When instead of CLP, a bacterial fibrin clot was placed in their abdominal cavities, HO-1-overexpressing mice presented improved survival and phagocytosis of E. faecalis but not E. coli. These results indicate that HO-1 enhances clearance of particular bacterial species. In addition, when CLP was performed in Nrf2 −/− mice, bacteremia was increased, whereas in Keap1 −/− mice, which present increased activation of NRF2, bacteremia was reduced, a phenomenon associated with altered phagocytosis of bacteria (146).
Infection with the protozoan T. gondii increases enterococci and E. coli counts in the ileum, whereas treatment with NRF2 activators such as curcumin and resveratrol reverse this increase in ileum counts (16). No direct bactericidal activity has been observed that could be ascribed to these compounds, but inflammation is greatly reduced and the intestinal tissue is well preserved after treatment, suggesting that changes in intestinal flora are caused by their interactions with tissues protected from oxidative and inflammatory damage by the NRF2 activators (192).
Recently, HO-1-inducer CoPP was observed to be capable of reducing the numbers of live Salmonella in the lamina propria, mesenteric lymph note, spleen, and liver, whereas mice deficient in the expression of HO-1 in macrophages presented impaired clearance of E. fecalis, E. coli, and S. typhimurium (234). The by-product of HO-1 activity CO mimicked the effects of HO-1, suggesting that in this case, the effects of HO-1 were dependent on CO production. Antioxidant studies are still required to test whether the inhibition of ROS production is involved in the phenomenon. No differences in intestinal permeability were observed among nontreated CO-releasing molecule or CoPP-treated mice, indicating that the effects of HO-1/CO are not due to the protection of the epithelial intestinal barrier against damage.
D. The role of ROS in clearance or promotion of infection by protozoan parasites of medical importance
Whereas in bacterial and viral infections, it is clear that certain pathogens thrive in oxidative environment and others are eliminated by ROS, the situation with protozoans is a bit more complex. Parasites have complex genomes, life cycles, and evasion strategies from the mammalian immune system, and ROS may have different effects depending on the infected cell type or compartment the parasite occupies. The evidence that some protozoan infections can be fueled by oxidative stress is summarized in Table 3.
BSO, buthionine sulfoximine; LPS, lipopolysaccharide; NO, nitric oxide; SOD, superoxide dismutase.
Treatment with NAC reduces the parasitism in footpads of BALB/c mice infected with L. major (270, 276), whereas treatment with BSO, a drug that inhibits the synthesis of glutathione, increases the burden in the footpads of C57BL/6 mice infected with L. major (56). None of these works established whether innate or adaptive immunity was involved in the effect or when treatment started to result in altered burden, but BSO effects were verified at the second week postinfection and an increase in the frequency of IFNγ-secreting lymphocytes was observed in the draining popliteal lymph nodes of NAC-treated mice (56). In another work, however, macrophages were treated with BSO for 24 h, infected with L. major and then activated with LPS/IFNγ (276). Prior BSO treatment resulted in increased macrophage parasite burden. The authors credited the result to the decrease in NO production in BSO-treated macrophages. The NRF2-activator resveratrol, although directly leishmanicidal, reduces the macrophage L. major burden at much lower doses than it does acting directly on the parasite (138), indicating that the antioxidant effects stimulate leishmanicidal macrophage mechanisms. In different study, NOX2 (gp91phox)−/− female homozygous mice had smaller acute infection lesions than wild-type controls, although in macrophages and neutrophils, no role for gp91phox was observed in the killing of L. major (22). Later in the course of infection (from 127 dpi on), L. major burden did not decrease in skin, popliteal lymph nodes, and spleens from gp91phox−/− mice as it did in wild-type mice, and the burden was greatly elevated in spleen, indicating visceralization of leishmaniasis for unknown reasons (22). Taken together, these results suggest that NOX2-derived ROS does not contribute to killing L. major inside macrophages but does interfere with the establishment of later immune responses. A full review on the subject of Leishmania susceptibility to ROS has recently been published elsewhere (327).
Not all infections with Leishmania respond to antioxidants with reduced burden. In L. amazonensis-infected macrophages, diethyldithiocarbamate, an inhibitor of SOD activity, promotes parasite destruction, and NAC reverses this effect (141). However, these findings contrast with the reduced parasite burden observed in the footpads from L. amazonensis-infected mice treated with NAC (205). It is possible, however, that NAC does not act on macrophages to reduce footpad parasite loads but instead on adaptive immunity. Recently, studying the iron uptake by L. amazonensis, Mittra et al. observed iron uptake to be increased after promastigotes were subjected to the iron-deficient medium, a compensatory mechanism dependent on the upregulation of iron transporter LIT1 and which promoted differentiation into amastigotes (200). The increased iron uptake allowed the synthesis of FeSOD, an amastigote antioxidant enzyme that depends greatly on iron availability. When macrophages were infected with L. amazonensis promastigotes subjected to iron depletion or to O2 −• induction by menadione, the parasite displayed increased cell invasion and growth. A similar situation occurred in vivo: O2 −• induction or iron stress increased the parasite burden in footpads. The ROS-enhancing effect on L. amazonensis infection appears to be related to the differentiation into amastigotes, as controls prepared with axenic amastigotes produced similar results.
Leishmania spp. have evolved a complex lipophosphoglycan that allows them to carefully avoid binding to mannose receptors, which triggers respiratory burst and other microbicidal mechanisms in phagocytes (325). Binding to FcR induces intense respiratory burst upon phagocytosis (325), but L. major amastigotes are coated with IgG1 in the footpads of infected mice and bind to macrophages through IgG1 (101). How L. major uses FcR to enter the macrophages and still survive respiratory burst remains unknown. In fact, L. major parasites trigger intense ROS production upon macrophage infection, different from L. amazonensis, which proceeds through a relatively silent cell invasion (4). FcR most likely represent an entry portal to the macrophages at least in L. amazonensis or L. pifanoi, as FcR-deficient mice present smaller cutaneous lesions (142).
L. guyanensis and L. brasiliensis do not cause large lesions in BALB/c mice. J. Souza-Franco et al. observed that the infection of BALB/c macrophages with L. guyanensis triggers an intense respiratory burst able to kill parasites (305).
Human infection with L. chagasi induces the upregulation of HO-1, and treatment with antileishmanial downregulates it (179). L. chagasi apparently benefits from HO-1 upregulation, as induction of HO-1 by treatment with CoPP increases parasite burden in macrophages, and macrophages from HO-1 knockouts infected in vitro have decreased burden and do not respond to treatment with CoPP. Antioxidants such as NAC were not tested in this infection and the role of NOX2 was also not assessed, but L. chagasi has enzymes that protects it against RNS and enhances its survival within macrophages.
Mice deficient in NOX2 (gp91phox) and infected with L. donovani presented increased peak parasite burden and delayed control of infection but were still, ultimately, able to control the infection (213). L. pifanoi interferes with NOX2 recruitment to phagosomes, and only a premature 65 kDa form of gp91phox is found in vacuoles containing amastigotes, which is different from 91 and 65 kDa-containing zymosan vacuoles (245). The maturation of gp91phox to its full-weight form of 95 kDa is known to depend on the availability of heme. L. pifanoi also upregulates HO-1, and the authors speculated that HO-1 overexpression could interfere with NOX2, as it promotes heme degradation. In fact, the HO-1 inducer CoPP reduces O2 −• production (245). Nevertheless, they did not demonstrate the outcome of infection in macrophages treated with the HO-1 inducer CoPP or the parasite burden in gp91phox-knockout macrophages. Therefore, we can only speculate on the role of ROS on L. pifanoi infection.
NOX2 (gp91phox)-deficient mice do not display any differences in parasite burden when infected with P. berghei ANKA, P. yoelli, or P. chabaudi (254), indicating that phagocyte respiratory burst does not participate in Plasmodium clearance. Nevertheless, in P. berghei ANKA infection, although HO-1−/− mice did not differ from controls in parasite burden, the NRF2/HO-1-inducer CoPP did reduce parasitemia in an early stage of infection, an effect that was lost after the drug was discontinued (237). The authors considered that perhaps CoPP interfered with the ability of the parasites to polymerize heme, decreasing parasitemia, but no functional data support this assumption. In the liver, HO-1 expression promoted infection by P. berghei and yoelii, whereas NAC did not alter parasitism, indicating that HO-1 effects are not related to its antioxidant effects (71).
CD36, a receptor for the uptake of oxidized LDL, is a NRF2-target gene that also mediates phagocytosis and clearance of Plasmodium by macrophages (199, 233, 294). CD36 does not have known antioxidant actions, although its expression can be an NRF2-dependent event triggered by an oxidative hit or other kind of stimulus. Thus, although some results might be a bit misleading, we consider that there is no strong evidence in the literature to support a role for ROS in promoting Plasmodium infection.
P. vivax induces oxidative stress in the gut of Anopheles aquasalis, a mosquito vector of malaria. Gene silencing of catalase in the A. aquasalis midgut epithelium increases the percentage of infected insects. A decrease has been observed in the load of potential bacterial competitors, which could be responsible for the association between increased ROS and increased P. vivax infection in the midgut (11). The role of ROS in mammalian infection by P. vivax awaits investigation.
T. cruzi induces oxidative stress (193), which causes much of the cardiac damage associated with Chagas disease (342). T. cruzi infection activates macrophages to respiratory burst, as shown by the decreased production of ROS by macrophages from NOX2 knockout mice (235), whereas in cardiomyocytes, T. cruzi induces mitochondrial ROS (342). Details about the molecular pathway that elicits ROS production in these cases remain unknown. The parasite is well equipped to withstand oxidative insults (246, 247), and the epimastigote forms even proliferate more actively in the presence of H2O2 (77, 226) and heme (226), which probably mimic the highly oxidative environment the epimastigotes face within the insect gut. Nevertheless, a role for ROS in T. cruzi killing was established by early studies. The first evidence came from studies of exhausted respiratory burst in macrophages, in which T. cruzi growth increased, suggesting that ROS are protective against infection (212). This finding was supported by the association between macrophage trypanocidal activity and the production of H2O2 (221), even though others claimed that they observed no evidence that macrophage-derived ROS contribute to T. cruzi killing (190, 211). Arguing against a role for ROS in T. cruzi elimination, another study demonstrated that the amastigote burden was significantly decreased when preinfected macrophages were treated with SOD and catalase during stimulation with IFNγ (194), although the decrease was small. These authors interpreted this finding as a possible consequence of the increased NO production, as ROS inhibition decreases OONO− formation and increases NO availability. They also observed that SOD and catalase did not affect parasite burden when the macrophages were activated before infection and enzymatic treatment. In another work, the percentage of Y-strain-infected cells appeared to be reduced when nonstimulated inflammatory macrophages were infected and then incubated with SOD or catalase, whereas increased percentages were demonstrated when they were incubated with PMA 1.5–3 h before infection (190). An even more clear picture emerged when the percentages of infected cells were compared among PMA (15%) and SOD (1%) treated cells, but the finding was most likely overlooked due to the bad sensitivity of the assay (190).
Recently, we have demonstrated that in elicited macrophages infected in vitro with T. cruzi, incubation with antioxidants (NAC, apocynin, billiverdin), permeant ROS-degrading enzymes (SOD-polyethylene glycol [PEG], catalase-PEG), and transfection of NRF2 or HO-1, reduced parasite burden, whereas incubation with SnPP, an inhibitor of HO-1 activity, or prooxidants (H2O2 or respiratory-burst activator PMA) increased parasitism (235). Consistently, we observed that infected NOX2 (gp91phox)-deficient mice have a decreased macrophage parasite burden and that elicited macrophages from gp91phox−/−-mice subjected to in vitro infection have reduced parasitism. In elicited macrophages infected in vitro before treatment, NRF2 activators, including resveratrol, sulforaphane, pterostilbene, and oltipraz, reduced parasitism (235), whereas in another work, the NRF2 activator curcumin reduced parasitism of infected fibroblasts (218). The mechanisms by which NRF2 activators operate remain to be fully elucidated. The classic mechanisms of parasite elimination such as NO production are not involved in CoPP-mediated reduction of parasitemia or parasitism, as well as the production of type I IFN, or adaptive immunity (235). The labile iron pool is reduced by treatment of infected cells with antioxidants, and the reduction of labile iron pool achieved by various strategies can reduce parasitism (235), indicating that iron sequestration is most likely involved in the burden-reducing effects of antioxidants.
In another work, however, performed with the T. cruzi CL-Brenner clone, apocynin did not alter parasite burden when incubated with infected nonstimulated macrophages and even blocked the killing of T. cruzi in IFNγ/LPS prestimulated macrophages (5). A role for NOX2-derived ROS in TLR4 signaling was not tested in this model. Evidence for OONO− as a mediator of T. cruzi killing has been observed in IFNγ/LPS preactivated macrophages (5, 6). Protein oxidation in parasites has been detected soon after phagocytosis mainly in IFNγ/LPS stimulated macrophages, and NOX2 assembly was confirmed in amastigote-containing vacuoles (5). Parasites overexpressing T. cruzi cytosolic tryparedoxin peroxidase (TcCPX), an enzyme that degrades OONO−, presented increased survival in short-term phagocytosis assays (5), but the parasite burden in nonstimulated macrophages did not differ between the wild-type and TcCPX-overexpressing parasites (247). These findings indicate that OONO− participates in T. cruzi killing. The reasons for the contradictory results concerning the role of ROS in T. cruzi infection await further studies.
The main capacity to induce ROS production in macrophages has been ascribed to T. cruzi cruzipain (96). Cruzipain enhances the oxidative burst by inducing the expression of p47phox and gp91 and enhancing the co-localization of NADPH oxidase enzyme subunits. As cruzipain is known to increase susceptibility of macrophages to T. cruzi infection (314, 315), it remains to be demonstrated if it does so by increasing ROS production by macrophages.
Slamf1-deficient mice were recently described as resistant to T. cruzi Y strain infection (35). As Slamf1 is a molecule expressed in myeloid cells that promote NOX2 activation (180), it is possible that the promotion of T. cruzi infection by ROS underlies these effects.
The effects of antioxidants in vivo are a bit more complex. The HO-1 inducer CoPP administered to T. cruzi Y-strain C57BL/6-infected mice reduces parasitism, whereas the inhibitor of HO-1 activity SnPP increases parasitism (235). The prooxidant paraquat also increases peak parasitemia and mortality in T. cruzi acutely infected mice, indicating that oxidative stress fuels T. cruzi infection in vivo (235). However, we failed to reduce parasitemia by treating mice with NAC in vivo, and only slightly decreased parasitemia by treating with apocynin (our unpublished results), whereas other researchers have succeeded by treating mice with NAC or glutathione (93). In mice infected with the Sylvio strain and treated with the antioxidant phenyl-alpha-tert-butyl-nitrone at the acute or chronic stages, heart parasite burden did not change (342), but conversely, in mice similarly infected and treated with apocynin, parasite burden was observed to be increased in heart and blood (64), indicating that NOX2 is required to control parasitemia in this model. Treatment with NRF2 activators, such as resveratrol, pterostilbene (235), melatonin (285), 15ΔPGJ2 (272), and curcumin (218) reduced T. cruzi parasite burden in mice. The reversal of CoPP's effects on parasitemia by peroral iron sulfate indicated that iron mobilization is involved in the effects of antioxidants on T. cruzi infection (235).
NOX2 (gp91phox−/−)-deficient mice infected with T. cruzi Y strain were examined by us and by Santiago et al. (286). Although we observed a significant decrease in parasitemia (235), they showed a nonsignificant trend toward decreased parasite burden in the gp91phox−/− mice. Similar to Santiago et al., we observed increased parasite burden at some timepoints postinfection in hearts and spleens by quantitative polymerase chain reaction (our unpublished results), and we believe that the lack of gp91phox expression causes distinct effects depending on the tissue and interval postinfection. Both groups observed that T. cruzi-infected gp91phox−/− mice die during acute infection, despite the early control of parasitemia. Their deaths were credited by Santiago et al. for the arterial pressure drop caused by NO overexpression, but an iNOS inhibitor, despite being capable of reversing the drop in arterial pressure, did not prevent the deaths of the gp91phox−/− mice (286). However, we observed that the treatment of gp91phox−/− mice with the HO-1-inducer CoPP produced a further decrease in their parasitemia and totally prevented their deaths. It is not clear to us whether treatment with CoPP spared the lives of infected gp91phox−/− by reducing parasitism in critical tissues or by reducing non-NOX2 generated ROS, and the phenomenon awaits a mechanistic explanation. There are various obscure points in these studies and the role of NOX2 deserves further consideration in T. cruzi infection.
VI. What Do Pathogens That Thrive on Oxidative Stress Have in Common?
Pathogens differ in their escape strategy within phagocytes (121). Some bacteria persist in the phagosome but inhibit their maturation to a fully microbicidal organelle, such as Legionella pneumophila and C. burnetii. M. tuberculosis and L. monocytogenes, not only arrest phagosomal maturation, but also ultimately escape to the cytosol. Listeria can alternatively live within large modified vacuoles (spacious Listeria-containing phagosomes). T. cruzi trypomastigotes induce their phagocytosis, escape phagosomes and change into amastigotes to grow in the cytoplasm. Most viruses invade cells and go directly to their cytoplasm, although some of them stimulate phagocytosis and use the phagosomal vacuole to enter the cell. Whether a pathogen will survive ROS production, live with it, or thrive on it, will probably depend on the cell type that harbors them, the location they reach within the cell, and their susceptibility to particular ROS-promoted mechanisms of microbe elimination.
It is noteworthy that none of the pathogens shown to thrive on oxidative stress spends most of their lifespan inside a mature phagosome, and it is probably an indication that pathogens that benefit from ROS production are not fully exposed to direct oxidative damage.
The literature is full of examples of immune mechanisms sabotaged by pathogens. Most intracellular pathogens succumb to type I IFN-elicited mechanisms of microbe elimination. L. monocytogenes, however, benefits from type I IFN secretion (10), most likely because it desensitizes the cell to the bactericidal mechanisms induced by IFNγ, which are effective against the bacteria (264). In this case, the innate mechanism of protection mediated by IFNγ has been somehow victimized by the pathogen, which turns type I IFN production against it, using the strength of the opponent against himself. It is like an “aikido” blow (Japanese martial art performed by redirecting the force of the attacker rather than opposing it head-on), to get rid of the much more efficient microbicidal mechanism commanded by IFNγ. The production of type I IFN was also stimulated by infection with M. leprae, turning on IL-10 production and downregulation of IFNγ, which in turn inhibits vitamin D-CAMP (cathelicidin)/DEFB4 (beta-defensin) antimicrobial pathway (320). Similarly, in M. tuberculosis infection, PKR activation, part of a microbicidal mechanism that engenders the activation of eIF2α, turns against the host, sensitizing macrophages to enhanced IFNγ-activation and subsequent apoptosis (346). It is possible that an analogous situation occurs with ROS-induced mechanisms of pathogen elimination, such that some pathogens, which are shielded by their own antioxidants, benefit from ROS production as it turns against effective killing mechanisms (Fig. 5). Iron sequestration could be one of these mechanisms, as oxidative stress determines the release of iron from ferritin in T. cruzi infection (235).
Alternatively, it is possible that some pathogens indulge ROS as an indirect metabolic supplement. T. cruzi epimastigotes multiply more actively in axenic cultures supplied with H2O2 (77, 226), for unknown reasons. Salmonella typhimurium grows more actively in inflamed intestinal lumen when ROS is actively produced (312). In this case, intestinal ROS converts luminal thiosulfate to tetrathionate, an electron acceptor that provides a metabolic advantage to Salmonella (344), as demonstrated by the outgrowth of wild-type bacteria over mutants unable to use tetrathionate in normal but not in gp91phox−/− mice.
In Caenorhabditis elegans, the worm NRF2-homolog SKN-1 regulates the responses to xenobiotics and oxidative stress (129). Recently, SKN-1-deficient C. elegans was shown to be susceptible to P. aeruginosa and E. faecalis (238), in a case reminiscent of the role of NRF2-target genes in enhancing immunity against certain bacteria (50, 107, 265). The conserved participation of NRF2-target genes in innate immunity appears to indicate a common microbicidal mechanism underlying most of these cases described here that has yet to be identified.
VII. Concluding Remarks
Oxidative attack is a major weapon used by phagocytes and other cell types against invading pathogens and ROS production can interfere with microbe elimination through a myriad of mechanisms. Although the current paradigm predicts that ROS production contributes to elimination of pathogens, it is becoming clear that for viruses, bacteria, and protozoans, ROS production can contribute to increasing pathogen burden. These new findings open an avenue to the use of antioxidants against particular infections.
Footnotes
Acknowledgments
The authors received financial support from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Pesquisa (CNPq), Fundaçao de Amparo à Pesquisa do Rio de Janeiro (FAPERJ) and INCTDengue. “Figures were made with the help of Motifolio (