
Abstract
Introduction
I
It is widely recognized that endothelial dysfunction in the human arterial wall is a major early step in atherogenesis and it is characterized by reduced production of the vasodilator nitric oxide (NO). Exposure of the endothelium to reactive oxygen species (ROS) plays a major role in the pathophysiology of endothelial dysfunction. Dysregulation of vascular homeostasis due to reduced NO bioavailability triggers a cascade of events that ultimately lead to the formation of the atherosclerotic plaque (9). Therefore, evidence suggests that interventions aiming at improving the balance between ROS and NO bioavailability in the vascular endothelium may be beneficial in both primary and secondary prevention.
The Role of Vascular Endothelium in Atherosclerosis
Endothelial dysfunction is widely regarded as one of the key early events in the atherosclerotic process. Endothelial dysfunction is characterized by reduced availability of NO; this is a gaseous molecule that diffuses to the vascular smooth muscle cells (VSMCs), triggers an increase in intracellular cyclic GMP, and induces vasorelaxation. Therefore, it has a major role in the regulation of vascular tone, as it opposes the actions of vasoconstricting factors such as endothelin-1. Moreover, NO is also implicated in the regulation of other processes, such as platelet aggregation and leukocyte adhesion, thus playing an integral role in vascular homeostasis (28).
The major enzymatic source of NO in the vasculature is endothelial nitric oxide synthase (eNOS), which is located predominantly in endothelial cells (ECs). eNOS is a complex homodimer that uses
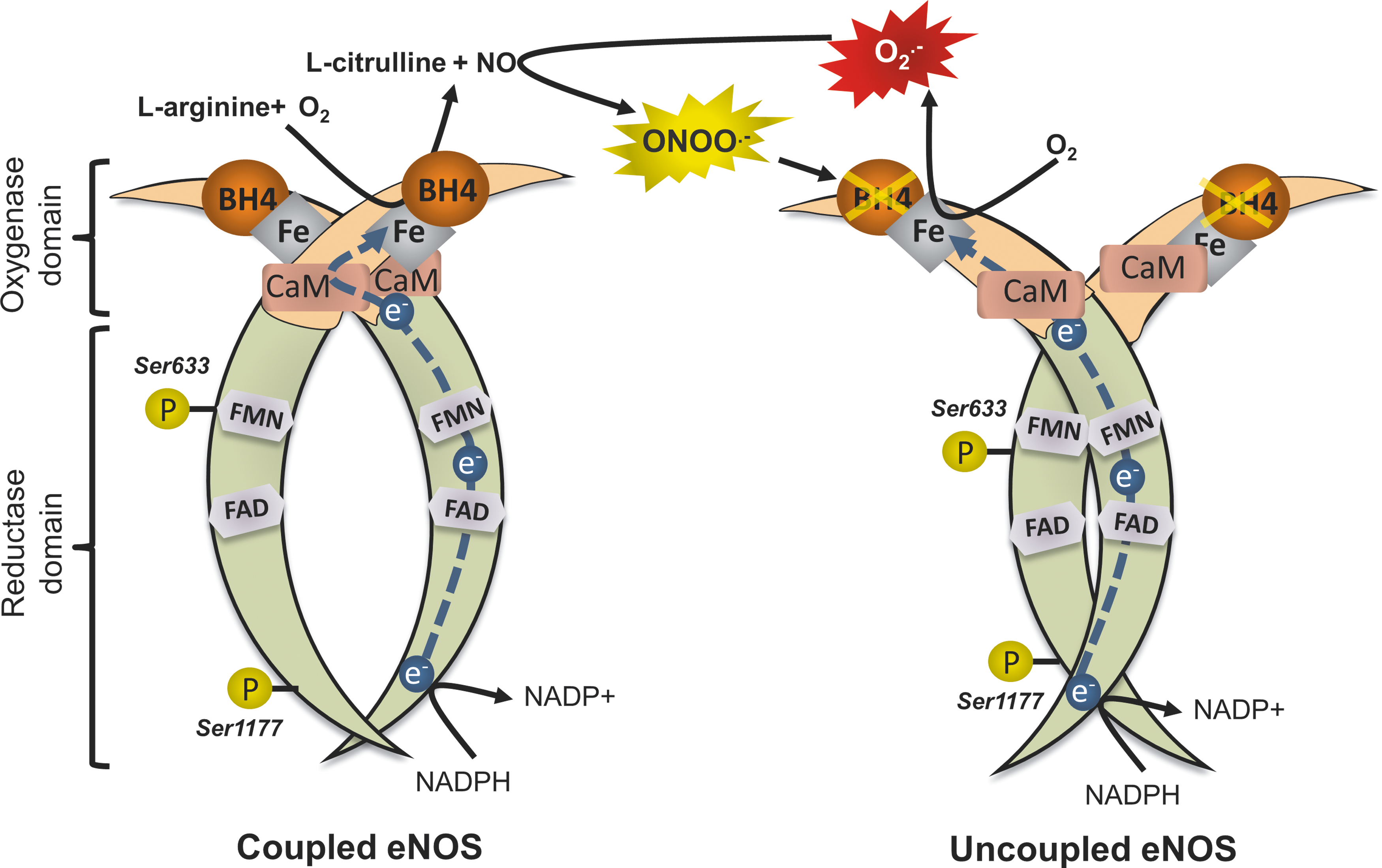
Another mechanism by which eNOS function is negatively affected is through the effects of the endogenous eNOS inhibitor asymmetric dimethylarginine (ADMA). ADMA is produced as a by-product of protein metabolism through methylation of
Measurement of endothelial function has been established as an important predictor of future cardiovascular events, independent of other risk factors. A number of studies have demonstrated an association between impaired flow-mediated dilatation (FMD) of the brachial artery (a noninvasive, ultrasound-based method for determining endothelial function) and increased risk of cardiovascular events in patients undergoing vascular surgery (38), as well as in the absence of obstructive atherosclerotic disease (104). A very recent meta-analysis concluded that FMD of the brachial artery is inversely associated with risk of CVD events, possibly more so in populations already at higher risk based on traditional predisposing factors, suggesting that 1% increase in FMD leads to reduction of cardiovascular risk by 9% (90).
Oxidative Stress in Atherosclerosis
Our current understanding of CVD pathogenesis supports a major role for ROS in atherogenesis (10). Classic atherosclerotic risk factors, such as diabetes mellitus, hypertension, and smoking, have all been linked to elevated production of ROS by the vascular wall (10). A major event in the atherosclerotic process is oxidation of lipoprotein molecules, primarily LDL by ROS, leading to the formation of oxidized LDL (oxLDL). This takes place primarily in the subendothelial space rather than in plasma, which hosts a variety of antioxidant defense mechanisms (37). ROS-mediated uncoupling of eNOS causes dysfunction of the endothelial layer, through the mechanisms presented previously. Binding of oxLDL to the lectin-like oxidized receptor triggers activation of redox-sensitive, proinflammatory transcriptional pathways (such as nuclear factor kappa B [NF-κB]) in ECs. This induces the release of inflammatory cytokines (such as interleukin [IL]-6) and increases the expression of cellular adhesion molecules (such as vascular cellular adhesion molecule 1 [VCAM-1]). As a result, circulating monocytes are attracted to the vascular endothelium, where they adhere due to increased expression of adhesion molecules and infiltrate the subendothelial space. There, they phagocyte oxLDL molecules, acquire macrophage-like characteristics, and eventually transform into foam cells. This further aggravates the inflammatory processes triggered in the vascular wall and creates a vicious circle where more monocytes/macrophages are attracted to the subendothelial space. In addition, ROS and inflammatory cytokines trigger VSMC migration from the media to the intima. The accumulation of lipid-laden foam cells and the parallel migration/proliferation of VSMCs eventually lead to atherosclerotic plaque formation.
Although the presence of increased oxidative stress in atherosclerosis cannot be doubted, actual causality has not been conclusively proven in humans. Several questions have been raised on whether increased ROS generation constitutes an actual cause of atherosclerosis or if it is simply a consequence or a bystander in the process (60). Mechanistic studies in humans are always challenging, given that access to human tissue is limited; therefore, proving a causal relationship is difficult. The oxidative stress hypothesis has been extensively explored in experimental models, and clinical studies have yielded results that are largely consistent with basic studies. Therefore, oxidative stress is now widely accepted as a key component of human CVD (17).
Given the implication of ROS in the pathogenesis of atherosclerosis, a logical therapeutic approach seemed at first to be the administration of dietary antioxidant supplements, such as ascorbic acid (vitamin C), α-tocopherol (vitamin E), and others. These compounds act as exogenous ROS scavengers and their supplementation was considered to be a rational therapeutic attempt at restoring vascular redox balance and preventing CVD. Indeed, since 1994, more than 50 large randomized trials that used antioxidant vitamins in primary and secondary CVD prevention have been published, with disappointing results. The latest meta-analysis, based on a total population of almost 300,000 participants spread across 50 randomized trials, concludes that there is no evidence in favor of antioxidant supplementation to prevent CVD (73).
The disappointing results of the antioxidant supplement trials should not in any case be interpreted as an invalidation of the oxidative stress hypothesis. For example, Patrignani et al. (82) found that supplementation with pharmacological doses of vitamin E up to 1200 mg/day was not associated with any detectable change in urinary 8-iso-PGF2a excretion (a noninvasive marker of oxidant stress in vivo) in healthy cigarette smokers. In contrast, comparable pharmacological doses of vitamin E affect 8-iso-PGF2a in clinical settings of patients with CVD risk factors and inflammation. Interestingly, they showed that there is a linear correlation between the basal rate of 8-iso-PGF2a excretion and the slope of changes in this index of lipid peroxidation as a function of changes in plasma vitamin E associated with short-term dosing with 600 mg/day in different clinical settings. These results are consistent with the hypothesis that the basal rate of lipid peroxidation is a major determinant of the response to vitamin E supplementation.
Aside from methodological issues concerning the design, population, and dosage of antioxidants in the clinical trials, there could also be biological reasons behind the failure of traditional, dietary antioxidant-based strategies to exert any beneficial effect on CVD prevention. All of the compounds tested in trials act as scavengers of ROS in vivo; they do not prevent the formation of ROS by cellular enzymatic systems, but rather eliminate ROS already formed in an attempt to restore redox balance. However, compartmentalization of ROS formation is an important aspect of redox physiology; a large number of enzymatic systems generate intracellular ROS, in distinct cellular compartments or organelles, such as the mitochondria (34). In addition, ROS are not just harmful by-products of cellular metabolism; they are important signaling molecules involved in a plethora of signaling transduction cascades that mediate responses to stress, injury, hypoxia, and so on (64). Indiscriminate, nontargeted scavenging of ROS, as achieved through most antioxidant strategies tested in clinical trials, would not necessarily restore redox balance, due to the inability of these compounds to reach the proper cellular compartment, but also through interference with potentially beneficial ROS-mediated signaling pathways.
Therefore, an effective antioxidant compound should not act as a simple ROS scavenger, but rather it should achieve a targeted inhibition of increased ROS generation by dysregulated ROS-generating enzymatic systems, while at the same time maintaining the beneficial effects of low-level ROS-mediated signaling. For example, Dikalova et al. (34) showed that a mitochondria-targeted O2 − scavenger managed to significantly attenuate angiotensin II-induced hypertension and total O2 − production in an animal model, whereas a nontargeted scavenger did not. Similarly, statins, both through their LDL-lowering actions and through their pleiotropic effects on ROS-generating enzymatic systems in the vascular wall, could constitute an elegant and efficient strategy aimed at restoring redox balance compared to traditional antioxidant supplements.
The Role of Statins in Cardiovascular Medicine: An Effective Lipid-Lowering Strategy
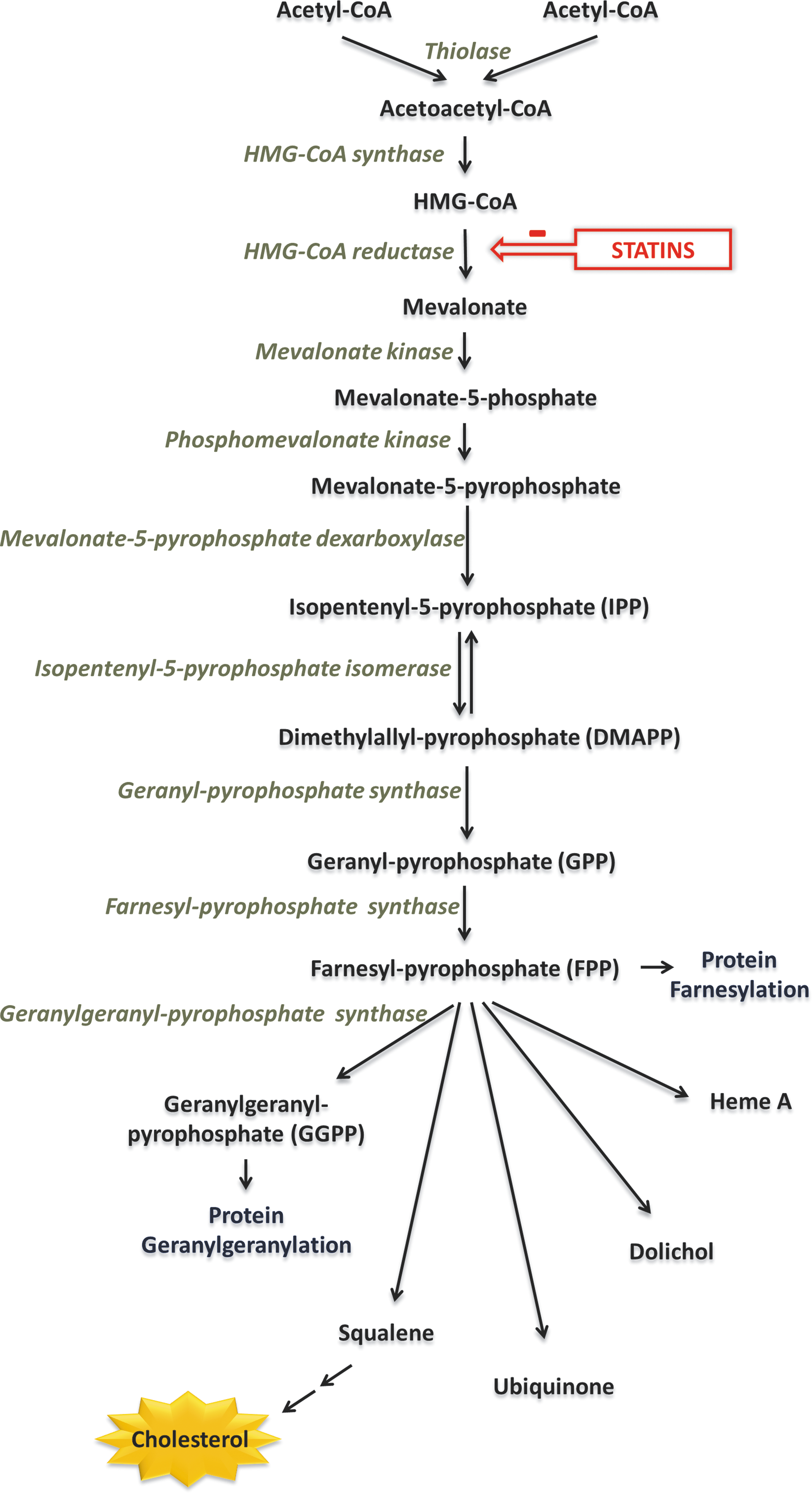
Statins are organic molecules originally derived from fungi. They exert their effects by inhibiting the enzyme 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, by competitively inhibiting binding of substrate to the active site of the enzyme, owing to their similarity in structure to HMG-CoA (47). HMG-CoA reductase is the rate-limiting enzyme in the mevalonate pathway (Fig. 2). This enzyme converts HMG-CoA into mevalonate, which is subsequently phosphorylated and converted into isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP). These molecules serve as precursors to the isoprenoid farnesylpyrophosphate (FPP), which is converted into geranyl-geranyl-pyrophosphate (GGPP) and squalene. In hepatic cells, squalene forms the basis of ergosterol and subsequently cholesterol synthesis. Because a significant percentage of the total cholesterol in the human body is endogenously produced (rather than up-taken by diet), inhibition of HMG-CoA by statins leads to a subsequent reduction in cholesterol and particularly low-density lipoprotein (LDL) biosynthesis (63).
Many large-scale randomized clinical trials have shown that lipid-lowering strategies that include statins have an antiatherogenic potential and suppress the risk of myocardial infarction and stroke. The landmark WOSCOPS trial (West of Scotland Coronary Prevention Study Group) demonstrates a 31% reduction in coronary events in patients with hypercholesterolemia after treatment with pravastatin (40 mg/day) (98). Since then, a number of studies have provided more evidence of beneficial effects for primary prevention; the latest Cochrane Database review concludes that statins reduce vascular events and all-cause mortality in subjects without evidence of prior CVD (106). Similarly, the Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) trial demonstrates significantly reduced coronary and all-cause mortality in patients who received pravastatin (40 mg/day) versus placebo following an ACS (2). In addition, the Post Coronary Artery Bypass Graft (POST-CABG) trial shows a reduction of saphenous vein graft (SVG) obstructive atherosclerotic disease in patients under aggressive lipid-lowering treatment with lovastatin±cholestyramine compared to more conservative treatment (1). As a result of these and other large-scale trials, statins now form an integral part of the medicinal regimen prescribed following a major cardiovascular event; the latest AHA/ACCF guidelines recommend initiation of statin therapy as secondary prevention in the presence of coronary or other atherosclerotic vascular disease (class IA), with target LDL levels below 100 mg/dl aiming for at least a 30% reduction from previous levels (101).
Pleiotropic Effects of Statins: Beyond Lipid Lowering
Most of the early clinical trials focused on dyslipidemic individuals or CVD patients with high LDL cholesterol levels. However, analysis of the LIPID trial uncovers a beneficial effect of statin treatment in CVD patients against new-onset coronary events or need for revascularization, irrespective of their LDL levels (95). Similarly, post hoc analysis of the POST-CABG trial reveals a beneficial effect of intensive statin treatment on SVG disease independently of lipid lowering (35). The concept that statins could potentially have effects beyond their lipid-lowering capabilities is further reinforced by the landmark JUPITER trial, in which normolipemic, apparently healthy subjects with high C-reactive protein (CRP) levels were randomized to rosuvastatin 20 mg/day or placebo (92). The results of this study show that statin treatment reduces CRP levels and the incidence of major cardiovascular events. Statin effects that are independent of LDL lowering have been termed “pleiotropic” and have been the focus of extensive research in the past decade.
The concept of direct pleiotropic effects of statins on vascular endothelial function in humans has also been demonstrated by comparing the effects of different lipid-lowering strategies. In a randomized trial of chronic heart failure patients treated for 4 weeks with simvastatin or ezetimibe (10 mg/day), the statin arm shows markedly improved FMD of the brachial artery compared with the ezetimibe arm, despite the similar reduction of LDL levels in the two study groups (54). Similarly, in dyslipidemic patients without CVD, 40 mg/day simvastatin treatment improves FMD and reduces vascular Rho kinase activity significantly more than simvastatin 10 mg/day plus ezetimibe 10 mg/day (58). Another recent meta-analysis that examines the effects of statin treatment on FMD in diabetic patients concludes that statins confer improvement of endothelial function, but only in patients who did not already present with severe endothelial dysfunction (125); this highlights the role of early prevention and modification of risk factors, in addition to the beneficial effects of statin treatment. Interestingly, sudden discontinuation of simvastatin treatment in patients with coronary artery disease leads to significant reduction in FMD 1 week after halting treatment, an effect not correlated with rebound increase in LDL levels (58). Conversely, in healthy individuals, abrupt halting of statin treatment also leads to a decline in FMD in day 1 after discontinuation, which is restored to baseline levels in 1 week (58). These results highlight both the potency of the endothelium-protecting pleiotropic effects of statins, and the need for proper adherence to treatment.
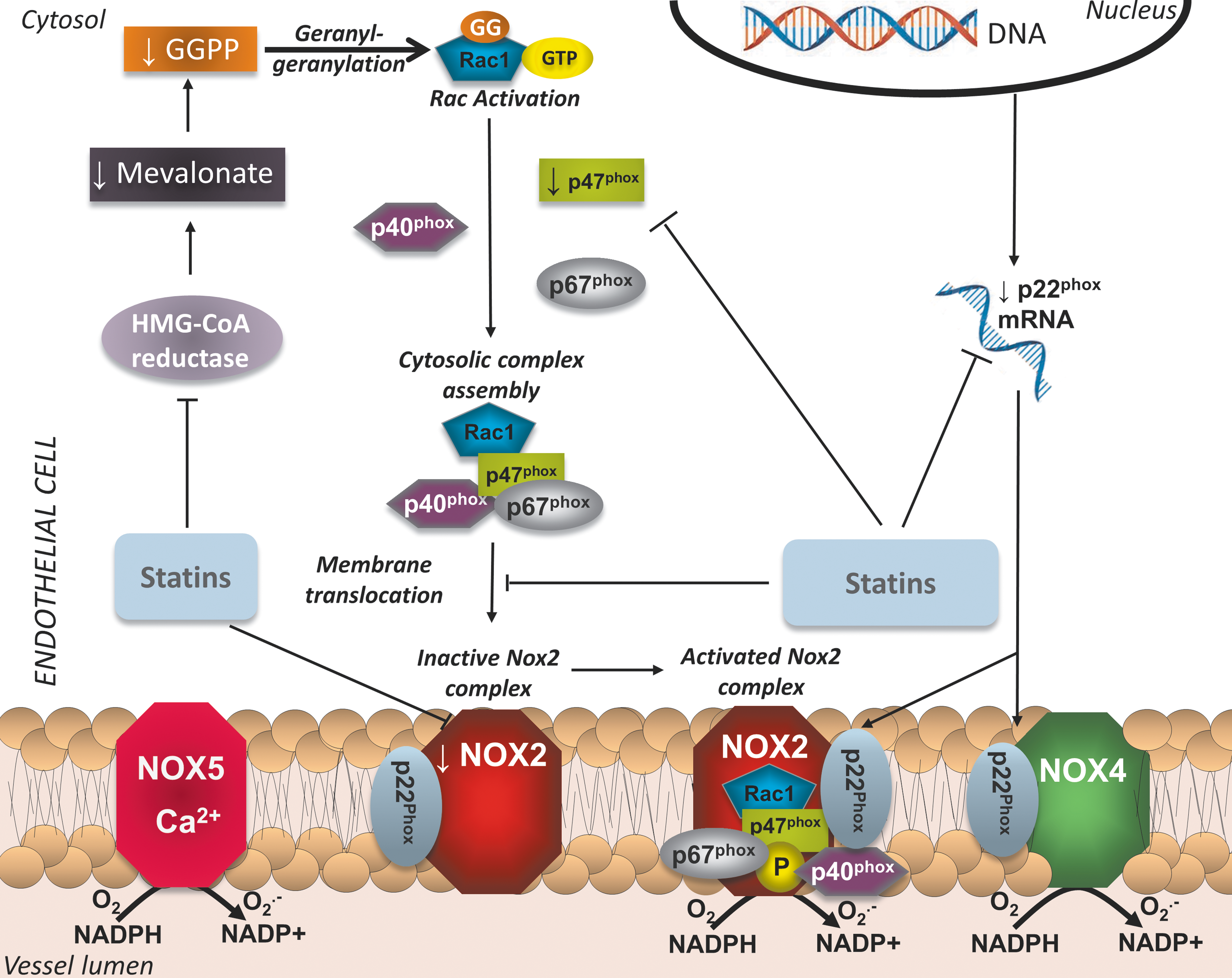
As mentioned before, the isoprenoids FPP and GGPP are intermediate products of the mevalonate pathway, which is inhibited by statins. Protein prenylation (the addition of isoprene units) is an important post-translational modification. The family of small GTP-ases (Ras, Rho, Rac, etc.) is an important cellular target for prenylation. Addition of FPP or GGPP moieties in proteins (farnesylation or geranylgeranylation) is critical for lipid anchoring and activation of Rho and Rac (11). Geranylgeranylation of Rho activates the Rho-associated protein kinase (ROCK); the Rho/ROCK pathway is implicated in the control of a variety of cellular functions, including proliferation, cell migration, redox signaling, and apoptosis (102). In addition, Rac is involved in the activation of the NADPH-oxidase complex and thus plays a major role in regulation of ROS generation, as will be extensively discussed later on.
The mevalonate pathway is active not only in hepatocytes, where it was first described, but it is also important for cellular homeostasis of vascular cells (ECs and VSMCs). Lipophilic statins (such as atorvastatin and simvastatin) can be passively diffused through the lipid bilayer of the cellular membrane and can therefore be uptaken by a large number of cells and not just hepatocytes (which also possess mechanisms of active transport) (50) (Fig. 3). Direct inhibition of small GTP-ase prenylation in vascular cells forms the core of the hypothesis that explains the rapid pleiotropic effects of statins on the vascular wall, which are independent of lipid lowering.
Sources of ROS as Therapeutic Targets in the Vascular Wall: Effects of Statins
Cellular redox imbalance, characterized by increased production and reduced elimination of ROS, is implicated in many human pathophysiological disease states, including CVD. Statins are probably the most effective, currently available strategy that inhibit the pro-oxidant enzymatic systems present in the vasculature and enhance the antioxidant defense mechanisms.
The effects of statins on redox state regulation of ECs have been extensively explored in cell culture and animal models. However, results obtained from experiments performed in such “model systems” do not necessarily reflect human physiology and may not be applicable to human disease. For example, primary cells and even more so cell lines cannot in any way take into account the complex interactions that occur on a tissue level. In addition, a large number of experiments have been performed in human umbilical vein endothelial cells (HUVECs), which exhibit significant functional differences compared with arterial ECs, which are more relevant to the development of atherosclerosis. Moreover, some cell culture studies have used concentrations of statins that are many folds higher than what is thought to be achievable in vivo. Similarly, even though this issue does not exist with animal models that have been treated with statins, there are significant differences in the pathophysiology of atherosclerosis compared with humans, which makes interpretation of the results challenging. However, in the literature, there are a large number of human translational studies, which have yielded similar results with the findings of cell and animal models. The remarkable consistency of the translational study results reinforces the validity of the original findings from basic science studies on the effects of statins on the regulation of redox state in ECs.
Effects of statins on eNOS
It is well documented in the literature that the pleiotropic effects of statins in the vasculature are at least partly mediated through modifications in eNOS expression, activity, and enzymatic coupling (Fig. 4). It has been shown that geranylgeranylation of the GTP-ase Rho leads to downregulation of eNOS in ECs; statin treatment inhibited GGPP formation and therefore led to enhanced eNOS expression, which was reversed by co-incubation with GGPP (56). Increased endogenous LDLs have also been shown to negatively affect eNOS mRNA expression in HUVECs, an effect reversed at the post-transcriptional level after exposure to simvastatin (65). The mechanism by which statins augment eNOS mRNA stability is an increase in eNOS mRNA polyadenylation due to alterations in the actin cytoskeleton, following Rho inhibition (51). In addition, exposure of H2O2-induced senescent HUVECs to statins activates the phosphatidylinositide 3-kinase (PI3K)/Akt pathway and enhances eNOS expression (80). More recently, eNOS mRNA has been identified as a target of the micro-RNA miR-155, which is upregulated by inflammatory stimuli such as tumor necrosis factor alpha (TNFα). Importantly, this effect was attenuated by simvastatin, but the beneficial actions of the statin were abolished by co-incubation with mevalonate or GGPP. Interestingly, they were mimicked by inhibition of RhoA, suggesting that the mevalonate/GGPP/RhoA pathway underlies the observed effects of simvastatin on miR-155 expression (103). The increased expression of eNOS mediated by statins translates into improved endothelial function, as evidenced in animal models of age-related (27) and high-fat-diet-induced (119) endothelial dysfunction.
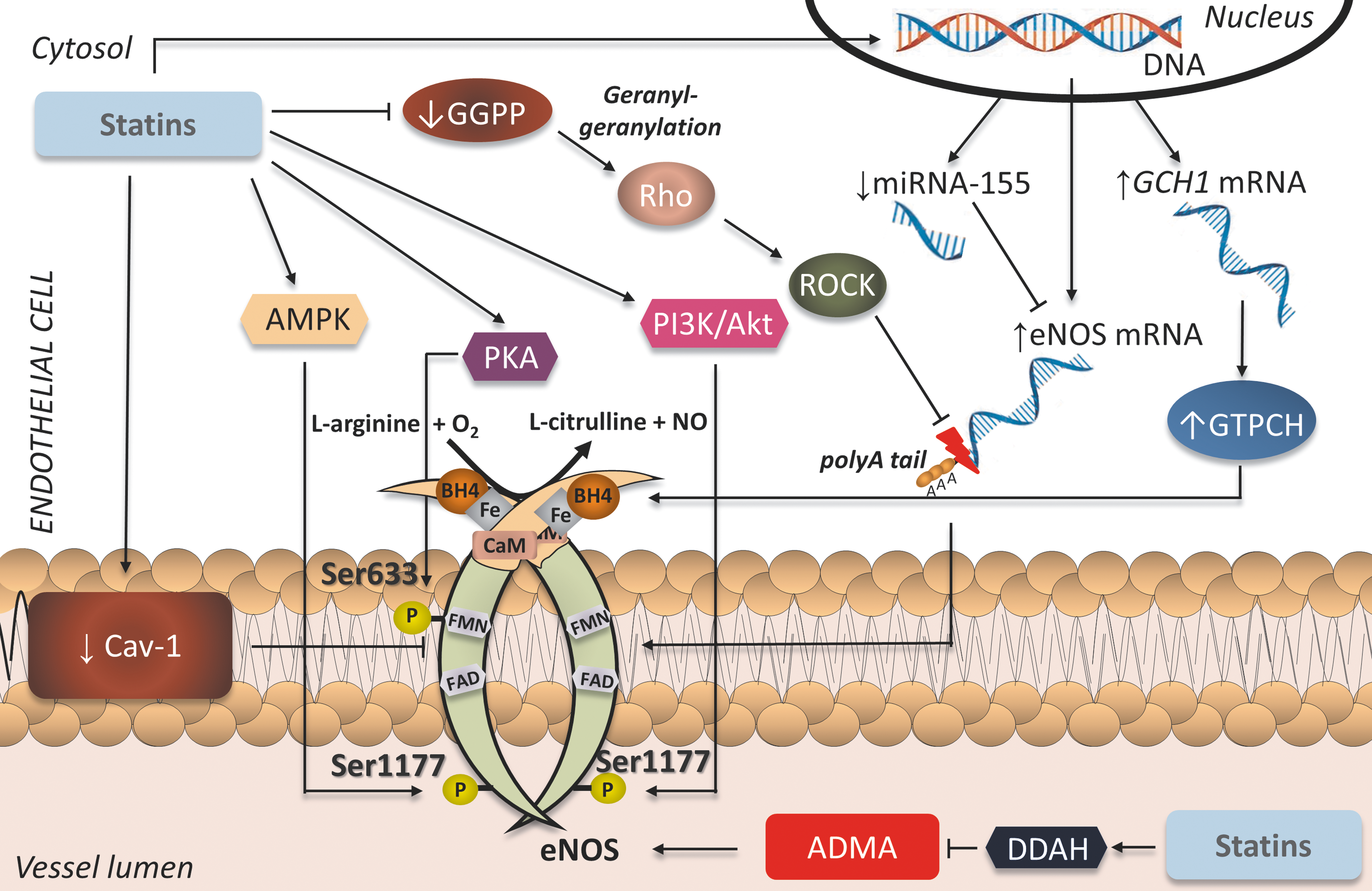
In addition to augmenting eNOS expression on a transcriptional and post-transcriptional level, statins have been shown to enhance eNOS activity at a post-translational level as well. eNOS possesses several activation phosphorylation sites at serine/threonine residues. Exposure of HUVECs to fluvastatin has been demonstrated to increase phosphorylation of eNOS at the activation sites Ser-633 and Ser-1177 through protein kinase A (PKA)- and PI3K/Akt-mediated pathways, respectively (12). Statin-mediated induction of eNOS phosphorylation at Ser-1177 has been shown to rely on heat shock protein-90 recruitment of Akt to the eNOS complex in ECs (18). Atorvastatin has also been shown in ECs to increase eNOS phosphorylation at Ser-633 in an adenosine monophosphate activated protein kinase (AMPK)–mediated way (79). Moreover, ex vivo incubation of rat mesenteric resistance arteries with simvastatin induces rapid AMPK-mediated eNOS phosphorylation at Ser-1177 (93). This leads to improved endothelium-dependent vasorelaxation in myograph chamber vasomotor studies, an effect reversed both by inhibition of eNOS by N-nitro-
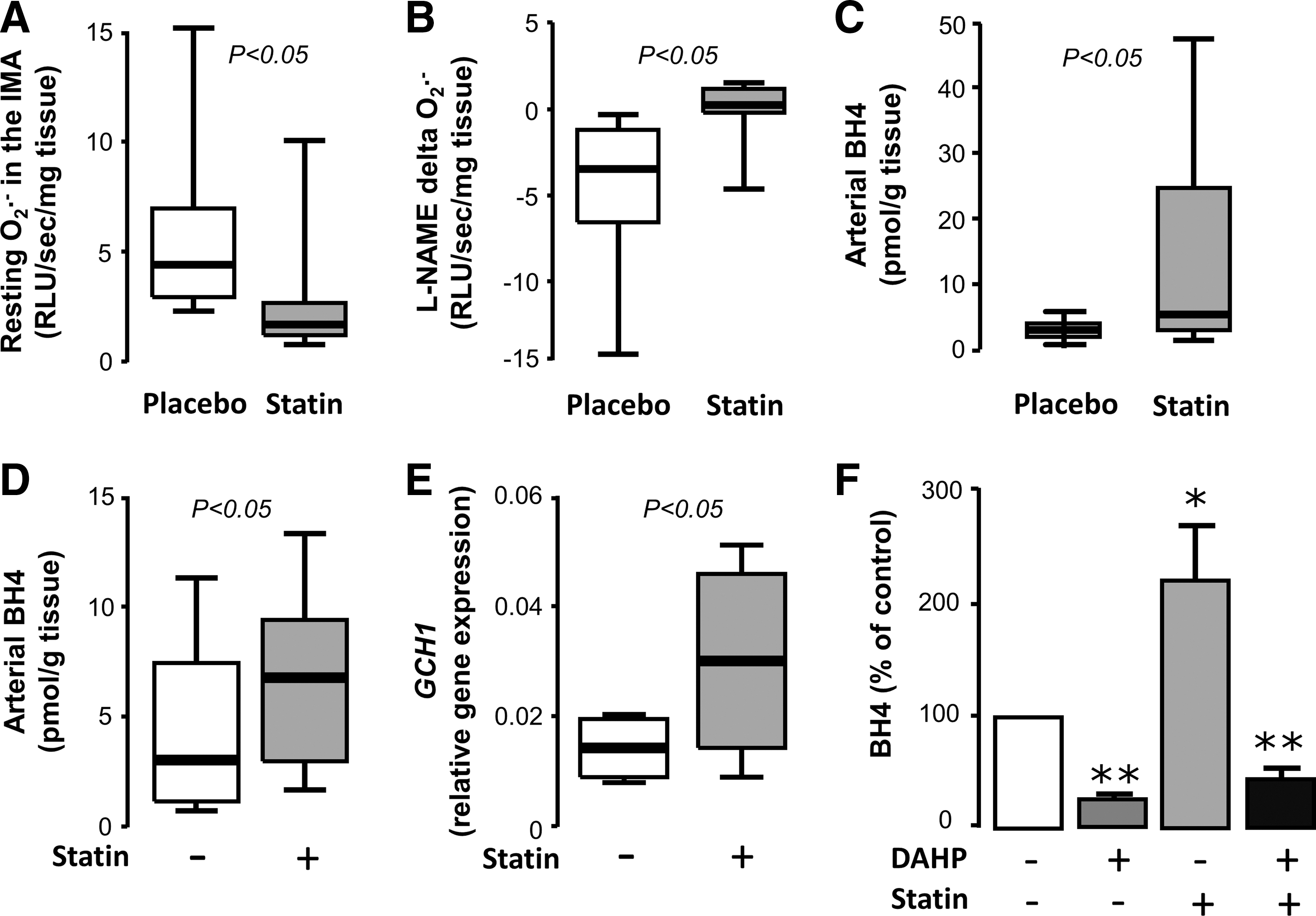
Activation of eNOS does not necessarily translate into improved NO production; if eNOS is largely uncoupled, then its phosphorylation-induced activation leads to increased production of O2 − instead of NO (24). However, statins have been shown to function on multiple levels to increase both eNOS activity and its coupling status. Exposure of HUVECs to cerivastatin or fluvastatin leads to upregulation of GTPCH expression and increases BH4 bioavailability, leading to improvement of eNOS coupling (12, 42). In an animal diabetic model, atorvastatin administration prevents eNOS uncoupling through the same mechanism (118). We have recently demonstrated in a randomized double-blind placebo-controlled clinical trial that 3-day preoperative atorvastatin treatment (40 mg/day) of patients undergoing CABG surgery increases BH4 content and improves eNOS coupling in the human internal mammary artery (IMA) compared with the placebo group (Fig. 5) (5). This is accompanied by improved FMD of the brachial artery. Importantly, these effects are rapid and independent of the LDL-lowering actions of atorvastatin. Ex vivo exposure of human IMAs to atorvastatin leads to upregulation of GCH1 gene, encoding for GTPCH, and a subsequent increase in vascular BH4 and total biopterin levels, effects that are reversed by co-incubation with mevalonate (5).
A possible indirect mechanism by which statins could increase eNOS activity and improve its coupling is enhanced metabolism or interference with the effects of the endogenous eNOS inhibitor ADMA. ECs exposed to ADMA exhibit an increased inflammatory response that is markedly reduced by simvastatin through extracellular receptor kinase pathways (49). Rosuvastatin treatment of spontaneously hypertensive rats has been shown to reduce circulating ADMA levels and reduce vascular oxidative stress, independently of cholesterol lowering (99). Similarly, in a rat model of pulmonary hypertension, rosuvastatin increases DDAH expression and subsequently reduces serum ADMA levels, while also increasing eNOS phosphorylation through the PI3K/Akt pathway (83). However, the results of human studies have been contradictory. Some randomized trials have demonstrated that short-term statin treatment lowers circulating ADMA levels in humans (61, 76, 112), while other studies have failed to find such an effect (36, 74, 85, 109, 121). These discordant results could possibly be attributed to differences in the characteristics of the populations examined. Interestingly, in a small randomized trial, simvastatin failed to improve endothelial function in patients with high ADMA levels, but managed to do so in combination with oral
Effects of statins on NADPH oxidase
NADPH oxidase is the most important source of ROS in the vasculature, both in ECs and VSMCs. It catalyzes the conversion of O2 into O2 •− radicals, by using NADPH as an electron donor. It is a membrane-bound enzymatic complex; the components of which depend on the respective homolog of the membrane subunit present: Nox1–5 and Duox1 or 2. ECs have been shown to possess Nox2, Nox4, and Nox5, whereas Nox1, Nox4, and Nox5 have been detected in VSMCs. Nox2 is the form of the enzyme present in phagocytes; it consists of two membrane-bound subunits, p22phox and gp91phox–Nox2 (which constitute the cytochrome b558 complex), and four cytosolic subunits, p40phox, p47phox, p67phox, and Rac1 or 2, which upon stimulation translocate to flavocytochrome b558 and lead to assembly and activation of the enzymatic complex. The activity of Nox1 and Nox2 can be stimulated by angiotensin II, growth hormones, and cytokines, through p47phox phosphorylation (33), whereas Nox5 is quiescent until Ca-dependent stimulation (40). On the contrary, Nox4 does not require Rac translocation for activation, but is constitutively active (33). In disease states, it is well established that elevated expression and activity of NADPH oxidase isoforms in the vasculature, together with the resulting increased ROS production, contribute to the initiation and maintenance of the atherosclerotic process. However, it should be noted that recent studies have unveiled a distinct, potentially beneficial role for the Rac1-independent Nox4 isoform in the vascular wall, by enhancing vasodilation in a H2O2-mediated way (91), underlying the fact that controlled ROS production in specific cellular compartments may be beneficial for vascular homeostasis, as they serve as signaling molecules that mediate multiple biological processes.
A significant amount of literature has focused on the role of statins in inhibiting NADPH oxidase activity. As mentioned previously, Rac1 activation and translocation to the membrane is necessary for the activation of Nox1 and 2 complexes, which are abundant in vascular cells and are involved in the pathophysiology of atherosclerosis. A crucial step in Rac1 activation is its geranylgeranylation by the GGPP produced through the mevalonate pathway. Given that statins suppress the formation of isoprenoids like GGPP, a large number of studies have sought to determine whether inhibition of HMG-CoA reductase could lead to attenuation of NADPH oxidase activity.
In HUVECs, statin treatment reduces p22phox mRNA and p47phox protein levels by increasing PPARα activity (46). VSMCs exposed to atorvastatin show downregulation of Nox1 expression and Rac1 membrane translocation, resulting in reduced ROS generation (116). Statins are also able to blunt high glucose-induced ROS generation in porcine coronary arteries, by reducing p22phox mRNA levels (25). Withdrawal of cerivastatin in VSMCs induces the translocation of Rac1 to the membrane, with a subsequent activation of NADPH oxidase complexes and increase in O2 − production (16). A mechanism through which NADPH oxidase exerts its effects is colocalization with ceramide and acid sphingomyelinase to form membrane rafts in the cellular membrane; in human coronary artery ECs, statin treatment prevents the oxLDL-induced formation of these membrane rafts and subsequently reduces O2 − generation (117).
The effects of statins on NADPH oxidase have also been examined in animal models that exhibit various CVD states. Statin administration in rats has been shown to suppress NADPH oxidase activity through a mevalonate-dependent prevention of Rac activation, leading to improved vascular NO availability and subsequently improved endothelial function (113). In atherosclerotic rabbits, fluvastatin treatment prevents an increase in p22phox and gp91phox induced by high-fat diet, improving endothelial function and reducing plaque size (68). Similarly, simvastatin treatment suppresses ROS generation and restores endothelial function in diabetic rat coronary arteries (105). In a normocholesterolemic hypertensive rat model, statins reduce gene expression of p22phox and Nox1 (116), as well as angiotensin type I receptors and p22phox protein levels, thereby reducing ROS production and improving endothelial function (115). In a rat model of ischemic stroke, atorvastatin pretreatment prevents increases in ROS generation and NADPH oxidase activity in the penumbra, by reducing gp91phox mRNA levels and p47phox translocation to the membrane (45). On the other hand, withdrawal of statins induces endothelial dysfunction in mice, through a Rac1-mediated activation of NADPH oxidase (110).
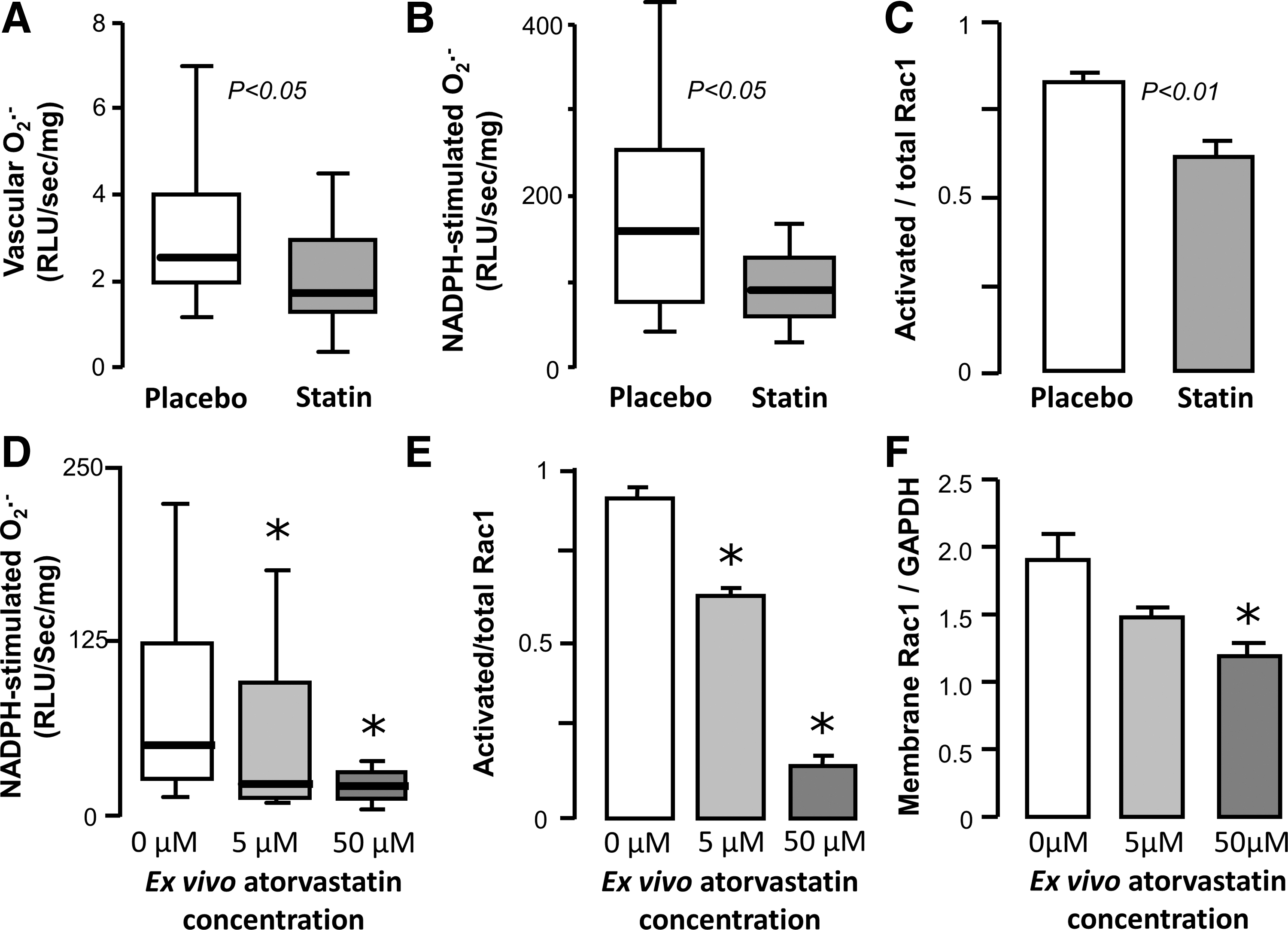
Translational studies in tissues obtained from human subjects have demonstrated the relevance of the aforementioned observations to human physiology as well. Statin treatment has been found to downregulate gp91phox levels in human internal mammary arteries obtained from patients undergoing CABG (94). In a cross-sectional study, statin treatment of hyperlipidemic individuals was found to be associated with reduced circulating levels of gp91phox, as well as markers of systemic oxidative stress (88). In the Endothelial Protection, AT1 blockade and Cholesterol-Dependent Oxidative Stress (EPAS) trial, 49 patients undergoing scheduled CABG were randomized to placebo, pravastatin (40 mg/day), irbesartan (150 mg/day), or both for 4 weeks (69). Statin treatment, as well as combination therapy, significantly increases eNOS and suppresses gp91phox expression in the left IMA, suggesting a synergistic effect on endothelial function, independently of LDL-lowering effects (69). We have recently demonstrated that short-term treatment with atorvastatin rapidly suppresses O2 − generation and NADPH oxidase activity in SVGs from patients undergoing CABG, independently of LDL-lowering effects (Fig. 6) (6). This is also accompanied by a reduction in plasma malonyldialdehyde levels, a marker of systemic oxidative stress. Ex vivo incubation of human SVGs with atorvastatin leads to reduction in p67phox and Rac1 translocation to the membrane, effects that are reversed by coincubation with mevalonate (6). A similar NADPH-oxidase-suppressing mechanism has been observed by our group to be relevant to human myocardium as well (7). Figure 7 summarizes the antioxidant effects of statins through modulation of NADPH oxidase expression and activity in the endothelium.
Effects of statins on other enzymatic systems and pathways in ECs
Prostacyclin (PGI2) is produced in ECs by the enzyme PGI2 synthase. Its precursor molecule is prostaglandin H2, itself synthesized from arachidonic acid through oxidation of fatty acids by cyclooxygenases. PGI2 is an important vasodilator and antithrombotic molecule, counterbalancing the effects of thromboxane A2. It has been recently reported that vascular PGI2 may be involved in the regulation of eNOS expression (122). Importantly, Di Francesco et al. showed that cyclooxygenase-2-dependent PGI2 induced by steady laminar shear stress upregulates heme-oxygenase 1, which halts the effects of TNFα in human ECs (31). Given that fluvastatin has been shown to increase expression of PGI2 synthase and PGI2 in human ECs (100), this pathway may also participate in the vasoprotective effects of statins.
Statin-mediated alterations in the activity of pro-oxidant enzymes could also partially explain the beneficial antithrombotic effects of statins, as evidenced by reduced release of thromboxane A2 from aged rat aortas, in a cyclooxygenase-2-mediated way (27). Release of 8-isoprostane through cyclooxygenase-2 is elevated in spontaneously hypertensive rats; this is abrogated after atorvastatin treatment, leading to an improvement of endothelial function and restoration of vascular redox state (111). Similarly, atorvastatin-induced modifications of the 5-lipoxygenase pathway (which oxidizes fatty acids into fatty acid hydroperoxides) lead to reduction of plaque vulnerability in rabbits (127).
Endogenous Antioxidant Defense Mechanisms: Effects of Statins
Due to constant exposure to ROS generated through various pathways, almost all living cells possess several enzymatic antioxidant defense systems, controlling the final ROS availability. Superoxide dismutase (SOD) is an enzyme that catalyzes the conversion of O2 − to H2O2. Catalase is an enzyme located in peroxisomes, which decomposes H2O2 to water and O2. Glutathione (GSH) is a tripeptide that acts as an important antioxidant limiting the effects of H2O2; this is achieved by reducing sulfhydryl groups in cysteine residues of other proteins, thus protecting those proteins of oxidative damage. This is catalyzed by glutathione-S-transferases, as well as glutathione peroxidases (GSH-Pxs), leading to the formation of glutathione disulfide (GSSG) from two GSH molecules; GSH can be replenished through the effects of glutathione reductase. The GSH/GSSG ratio is widely used as a marker of cellular redox state. Paraoxonase is an enzyme involved in protection against oxidation of the LDL molecule. Other antioxidant enzymatic systems are also present in cells, such as thioredoxins, peroxiredoxins, and so on.
Statins have been shown to exert beneficial effects in the vasculature not only through suppression of pro-oxidant enzyme but also through enhanced levels and activity of endogenous antioxidant systems, in both experimental models and human studies. Simvastatin can partially restore renal levels of all three major cellular antioxidant defense systems (SOD, GSH-Px, and catalase) and reduce the levels of oxidative damage biomarkers in diabetic animals (53, 128). It has also been shown to improve endothelial function by increasing SOD levels in rats chronically treated with the eNOS inhibitor LNAME (86). In addition, the beneficial effects of atorvastatin on plaque size reduction in animals fed an atherogenic diet are potentially mediated by an increase in circulating paraoxonase levels (97).
The effects of statins on catalase levels and activity have been demonstrated in a number of studies. Rat aortic VSMCs and senescent HUVECs show increased gene expression and protein levels of catalase after atorvastatin treatment (116), an effect mediated by the PI3K/Akt pathway (80). In animal models of hypertension and diabetes, statins elevate aortic catalase expression through a sirtuin-1-dependent pathway (80, 116). A statin-induced increase in catalase levels has also been demonstrated in human abdominal aortic aneurysms (87).
Effects of Statins on Redox-Sensitive Transcriptional Pathways
It is well established that a major mechanism through which increased production of ROS promotes atherosclerosis is the triggering of redox-sensitive transcriptional pathways in the vascular endothelium (Fig. 8). These pathways regulate the production of pro-inflammatory, pro-atherogenic cytokines, and cellular components, which enhance oxLDL and macrophage infiltration, as well as VSMC proliferation and migration to the intima. One of the most important redox-sensitive pathways is the NF-κB pathway. NF-κB is a transcription factor that controls the expression of a large number of pro-inflammatory genes. In its inactivated form, NF-κB is located in the cytosol, bound to its inhibitor (IκBα). While H2O2 had been initially proposed as being able to directly activate NF-κB (107), it is now widely accepted that ROS have the ability to indirectly modulate NF-κB function (70). Pro-inflammatory mediators, such as TNFα, have been shown to activate the IκB kinase (IKK), which phosphorylates IkB and leads to its ubiquination and degradation, thus freeing the NF-κB molecule. Consequently, NF-κB translocates to the nucleus and binds to its response elements, initiating the transcription of pro-atherogenic mediators, such as IL-6, TNFα, the adhesion molecules VCAM-1 and intercellular adhesion molecule (ICAM)-1, and others. Another pro-inflammatory transcription factor that can be triggered by ROS is activator protein-1 (AP-1). Like NF-κB, AP-1 controls the expression of genes involved in inflammatory processes, cellular proliferation, and apoptosis, and has a role in the atherosclerotic process.
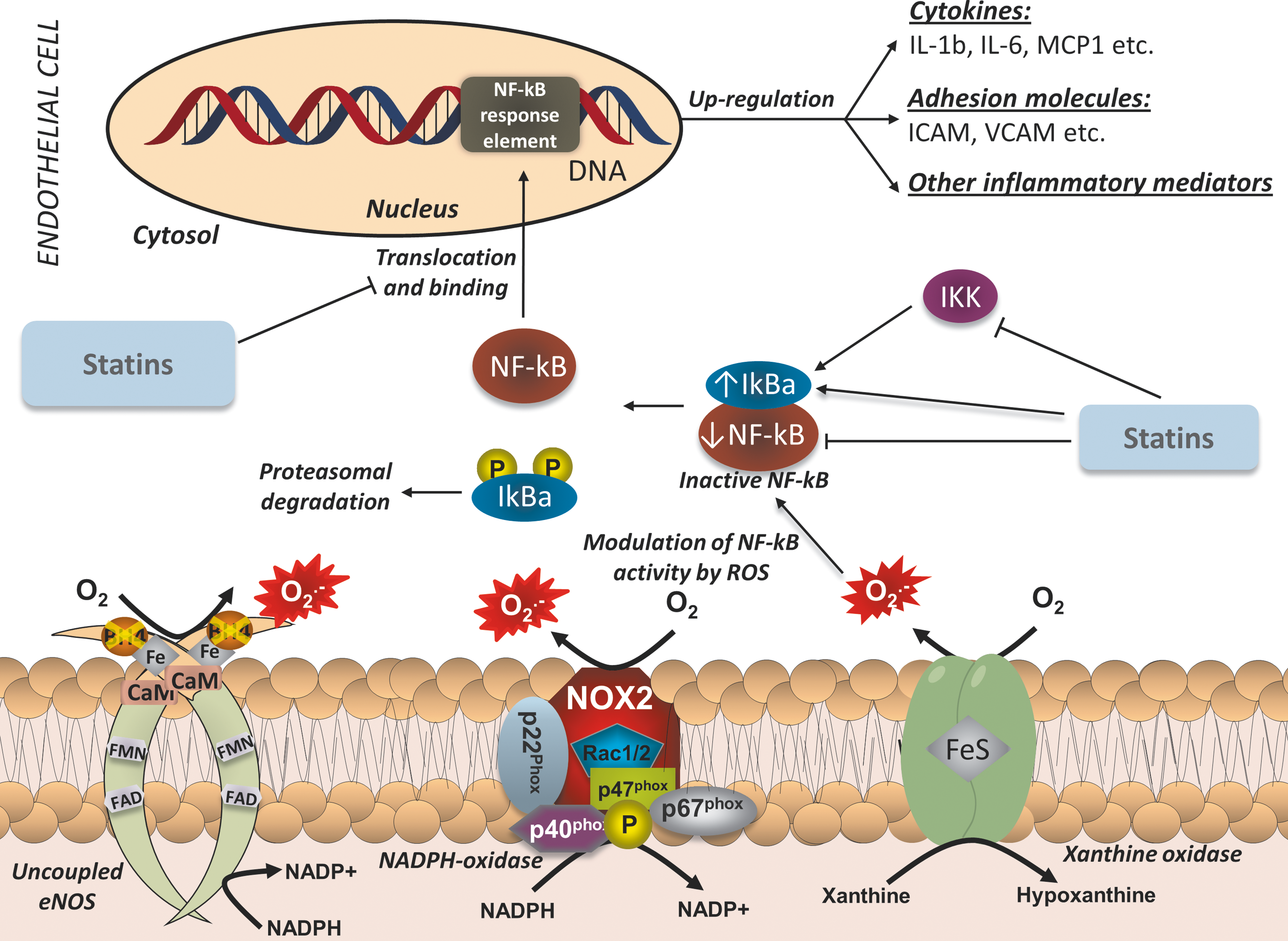
The role of statins as potential inhibitors of NF-κB has been investigated in cell models, where NF-κB activity has been induced through a variety of different stimuli. Exposure of VSMCs to atorvastatin prevents TNFα- and angiotensin II-induced activation of NF-κB and the subsequent upregulation of inflammatory mediators, effects prevented by FPP and GGPP (77). Fluvastatin also appears to attenuate CRP-induced activation of NF-κB both in ECs (114) and VSMCs (41). Both simvastatin and atorvastatin attenuate oxLDL-induced activation of NF-κB in human coronary artery ECs (57). These statin-mediated effects have also been demonstrated in animal models of CVD. Simvastatin treatment reduces NF-κB activity in both the atherosclerotic plaques and circulating mononuclear cells of diet-induced atherosclerotic animals, independently of lipid-lowering effects (43). Similarly, administration of cerivastatin significantly reduces cardiac activation of NF-κB induced by angiotensin II, without affecting cholesterol levels (29). The LDL-independent, anti-inflammatory role of statins has also been extensively explored in cell models where atherosclerosis-like inflammation has been induced by infection with a pathogen. In an EC and a VSMC model of Chlamydia pneumoniae-induced inflammation, cerivastatin significantly attenuates the upregulation of inflammatory mediators, through suppression of NF-κB activation (30, 52). Similarly, in HUVECs infected with cytomegalovirus, exposure to fluvastatin attenuates NF-κB activation, leading to reduced viral replication (89).
Statin interference of the NF-κB pathway can also have a crucial role in preventing monocyte infiltration, foam cell formation, and VSMC proliferation. Simvastatin inhibits in a time- and dose-dependent way the TNFα-induced upregulation of ICAM and VCAM-1 in HUVECs by reducing NF-κB activation; this leads to reduced monocyte–EC interactions (123). Importantly, in vitro exposure of human monocytes to pravastatin abrogates LDL-induced activation of NF-κB and subsequent expression of inflammatory mediators (124). In addition, atorvastatin inhibits IL-18-induced migration of aortic VSMCs through AP-1 and NF-κB inactivation (21).
Various mechanisms have been proposed to explain the observed reduction in NF-κB activity triggered by statins. It has been demonstrated that statins induce upregulation of gene expression of IκBα in ECs as well as downregulation of expression of NF-κB and reduction of its protein-binding capacity in both ECs and VSMCs, thus leading to an overall reduction in NF-κB activity in vascular cells (32). In addition, statins have also been shown to reduce ROS-mediated activation of NF-κB in monocytes through a reduction in IKK activity (78). An IKK-independent pathway that involves inhibition of PI3K/Akt signaling has also been demonstrated in human ECs (44).
The ability of statins to suppress NF-κB signaling has also been shown in clinical studies. In a small, nonrandomized study, 3 months of pravastatin treatment (40 mg/day) before scheduled carotid endarterectomy led to a more stabilized phenotype of the carotid plaque, as evidenced by reduced lipid and inflammatory content, characterized by reduced activation of NF-κB (26). In the human abdominal aortic aneurysm wall, simvastatin suppresses ROS generation and NF-κB activity (87). In a randomized trial, atorvastatin treatment (80 mg/day) 1 month before endarterectomy was associated with reduced NF-κB activation in circulating mononuclear cells and the carotid plaque, coupled with reduced inflammatory gene expression and cell infiltration of the plaque (78).
A major transcriptional pathway that is pivotal to the cellular antioxidant response is that of the transcription factor Nrf2 [Nuclear factor (erythroid-derived 2)-like 2]. In its inactivated form, Nrf2 is located in the cytoplasm, bound to the proteins Cullin 3 and Keap1 (Kelch like-ECH-associated protein 1). Keap1 possesses cysteine residues, which are sensitive to ROS modifications due to their sulfhydryl group (–SH). Oxidation of these –SH groups dissociates the Cullin3/Keap1 complex from Nrf2, which translocates to the nucleus and initiates transcription of genes encoding for endogenous antioxidant defense proteins. Simvastatin has been shown to activate Nrf2 through the PI3K/Akt pathway and subsequently suppress ROS generation in primary mouse embryonic fibroblasts (23). Similarly, fluvastatin significantly increases Nrf2 nuclear translocation in human coronary artery SMCs through the same intracellular pathway, leading to upregulation of antioxidant enzymes and reduction in ROS production (62).
Effects of Statins on Gene Expression Profile of Vascular Cells
Gene expression profiling through gene chip microarray analysis has revolutionized the way researchers explore disease and treatment. By identifying gene targets that are differentially regulated in disease states or after intervention, we can gain substantial insights into the causation and processes underlying pathophysiological conditions. This allows for identification of novel therapeutic targets or improvement of existing treatment strategies. In this context, the effects of statins on the endothelial transcriptome have been the focus of significant research in the last years, in an effort to better understand the mechanisms that underlie the multifaceted pleiotropic actions of these compounds.
Transcriptome analysis of HUVECs exposed to atorvastatin or pravastatin reveals a shift toward a less inflammatory, prothrombotic, and vasoconstricting gene expression profile; this is evidenced by downregulation of IL-8, monocyte chemoattractant protein-1, plasminogen activator inhibitor-1, and endothelin-1, as well as upregulation of thrombomodulin and eNOS (71). Human coronary artery SMCs have also been shown to have significant upregulation of thrombomodulin after statin treatment (72). Two different studies, the first in human pulmonary artery ECs exposed to simvastatin for 16 h (48) and the other in aortas of ApoE-deficient, high-cholesterol-diet mice treated with pravastatin for 6 weeks (59), showed very similar results in relation to changes in gene expression profile; in both studies, statin treatment activated genes related to cytoskeleton organization. This means that both acute and chronic statin treatment led to modifications in cytoskeleton proteins, which could mediate the beneficial effects of statins on endothelial function. A very interesting study reveals that more than 20% of human coronary artery EC genes affected by simvastatin treatment have a currently unknown physiological function (126). The same study shows a significant downregulation of genes involved in proliferation and cell cycle control (126). These results could potentially explain the protective effects of statins against neointimal formation, while also highlighting the fact that a significant portion of the mechanisms that underlie their pleiotropic actions on human endothelium is still incompletely understood.
Adverse Effects of Statin Treatment
Like any other medication, statins are not devoid of adverse effects. The most significant side effects associated with statin treatment are elevation of liver enzymes and muscle-related complaints, which can range from myalgias to myositis and rhabdomyolysis (39). The mechanisms that underlie these effects have been extensively explored, and mostly relate to mitochondrial defects due to depletion of ubiquinone (Coenzyme Q10), the production of which depends on the mevalonate pathway. The appearance of these side effects depends on the genetic predisposition of the individual but is also largely dose dependent (41). Therefore, care should be taken by clinicians when using statin therapy to achieve optimal treatment targets while at the same time avoiding onset of adverse effects.
Conclusions
The use of statins has become a key strategy to reduce cardiovascular risk both in primary and secondary prevention. Further to their lipid-lowering properties, statins also have a number of direct, or pleiotropic, effects on vascular function, suppressing atherogenesis and in some cases may even lead to atherosclerotic plaque regression. Over the last decade, the vascular endothelium has been identified as one of the primary targets for the antiatherogenic/pleiotropic effects of statins. Statins have the ability to restore the physiological balance between NO and ROS production in the vascular endothelium, through a number of LDL-dependent and -independent effects. They increase the expression, activity, and enzymatic coupling of eNOS, the main source of O2 •− radicals in the human vascular endothelium, leading to increased NO biosynthesis. At the same time, they suppress the activity of pro-oxidant enzymes (such as NADPH oxidase, uncoupled eNOS, and others) and enhance the efficiency of endogenous antioxidant defense systems in the vascular endothelium, leading to a net reduction of ROS bioavailability. By restoring the balance between NO and ROS in the vascular endothelium, statins also control the activation of key redox-sensitive transcriptional pathways with a key role in the control of vascular inflammation, and they also prevent the proliferation/migration of VSMCs by suppressing atherogenesis.
Future Perspectives
There are several remaining issues that need to be addressed regarding the biological role of statins as regulators of vascular redox state. Their indirect effects on vascular redox state (e.g., by modifying the biology of perivascular adipose tissue that exerts paracrine effects on the vascular wall) should be further explored in the future. At a clinical level, the beneficial role of statin treatment in vascular disease pathogenesis and especially in atherosclerosis is clear. Statins are now the corn stone of all therapeutic strategies in both primary and secondary cardiovascular prevention (101). Their use is also clearly indicated in ACSs, as they stabilize atherosclerotic plaques and they improve survival (3). However, given the complex biological effects of statins, it is hard to support that these clinical effects are primarily due to their direct or indirect “antioxidant capacity.” Further research is required to examine whether statins are clinically effective in other CVDs with significant involvement of ROS in their pathogenesis. That would expand further their already wide clinical indications.
Finally, the ability of statins to exert their biological effects in the vascular endothelium independently of their primary pharmacological action, which is LDL-lowering effect, could serve as a “pharmacological model” for the development of novel antioxidant strategies that aim to control the intracellular balance between production/elimination of ROS beyond their simple chemical scavenging in the vascular endothelium. These clever strategies should aim to mimic the pleiotropic effects of statins on the vascular endothelium, to achieve the appropriate balanced regulation of vascular redox signaling in the human endothelium.
Footnotes
Acknowledgments
This study was funded by the British Heart Foundation (FS/11/66/28855). C.A. and M.M. also acknowledge support from the BHF Centre of Research Excellence, Oxford (RE/08/004).