
Abstract
Smooth muscle cells (SMCs) are key components within the vasculature. Dependent on the stimulus, SMC can either be in a proliferative (synthetic) or differentiated state. Alterations of SMC phenotype also appear in several disease settings, further contributing to disease progression.
Introduction
E
In the present work, we postulate a strong impact of microRNA (miR)-24 on the regulation of smooth muscle cells (SMCs). Enhanced miR-24 expression is deteriorating SMC biology mainly via inducing apoptosis and autophagy, inhibiting proliferation and reducing contractile marker genes. A factor contributing to the observed phenotype is the target gene heme oxygenase 1. Elevated miR-24 expression (e.g., induced by hypoxia) is also impairing vascular density in an engineered heart tissue model. Thus, miR-24 is an interesting candidate to develop novel miRNA-based methods to modulate tissue vascularization.
Results
Pre-miR screening highlights miR impact on human aortic smooth muscle cell viability
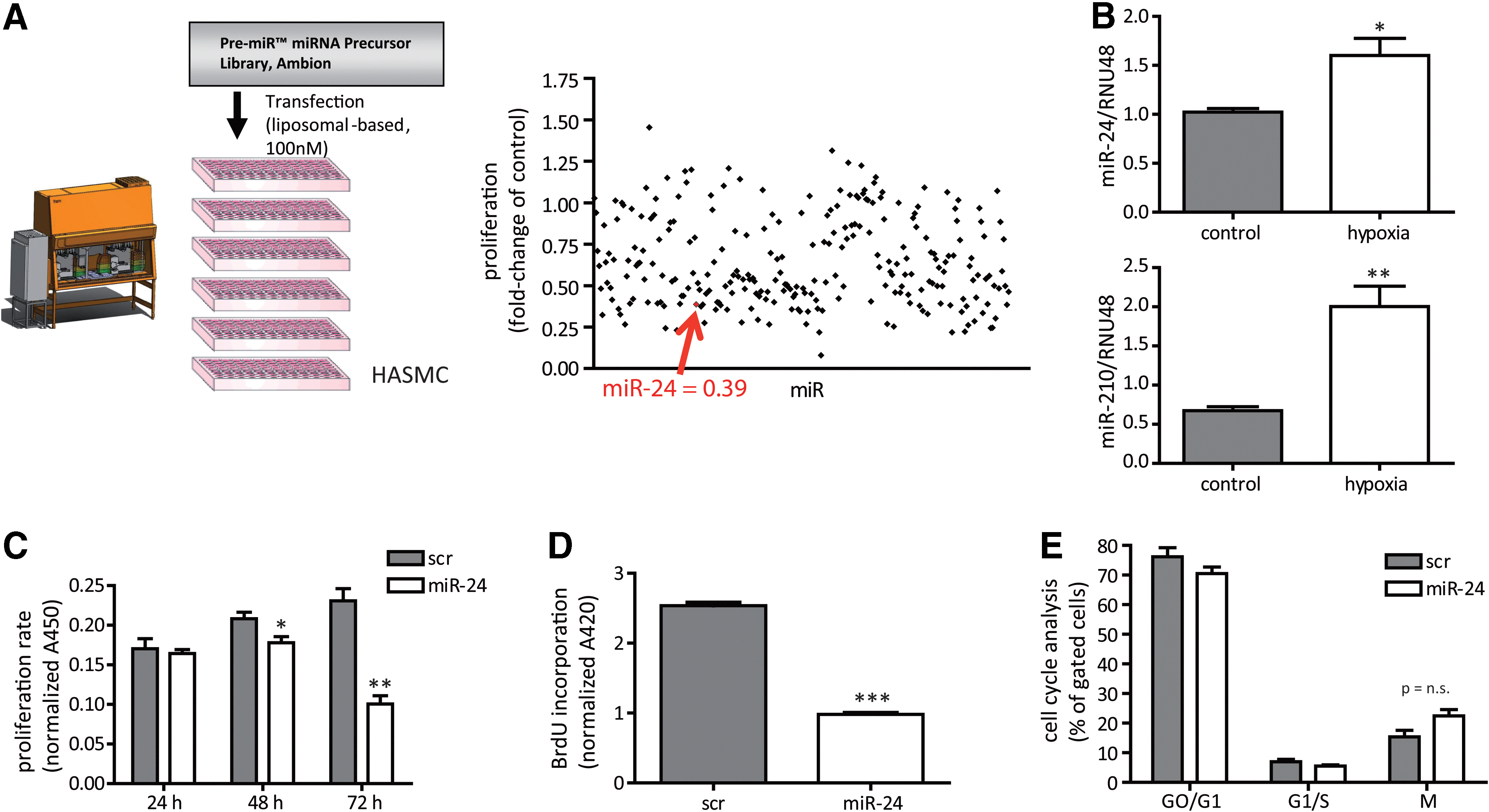
To identify miRNAs that alter human aortic smooth muscle cells (HASMCs), we performed a robotic-assisted high-throughput precursor (pre)-miR screening. Applying a miR library of >250 pre-miRs, liposomal transfection identified several miRs that altered cellular proliferation 72 h after pre-miR transfection (Fig. 1A and Supplementary Table S1; Supplementary Data are available online at
miR-24 impairs HASMC viability
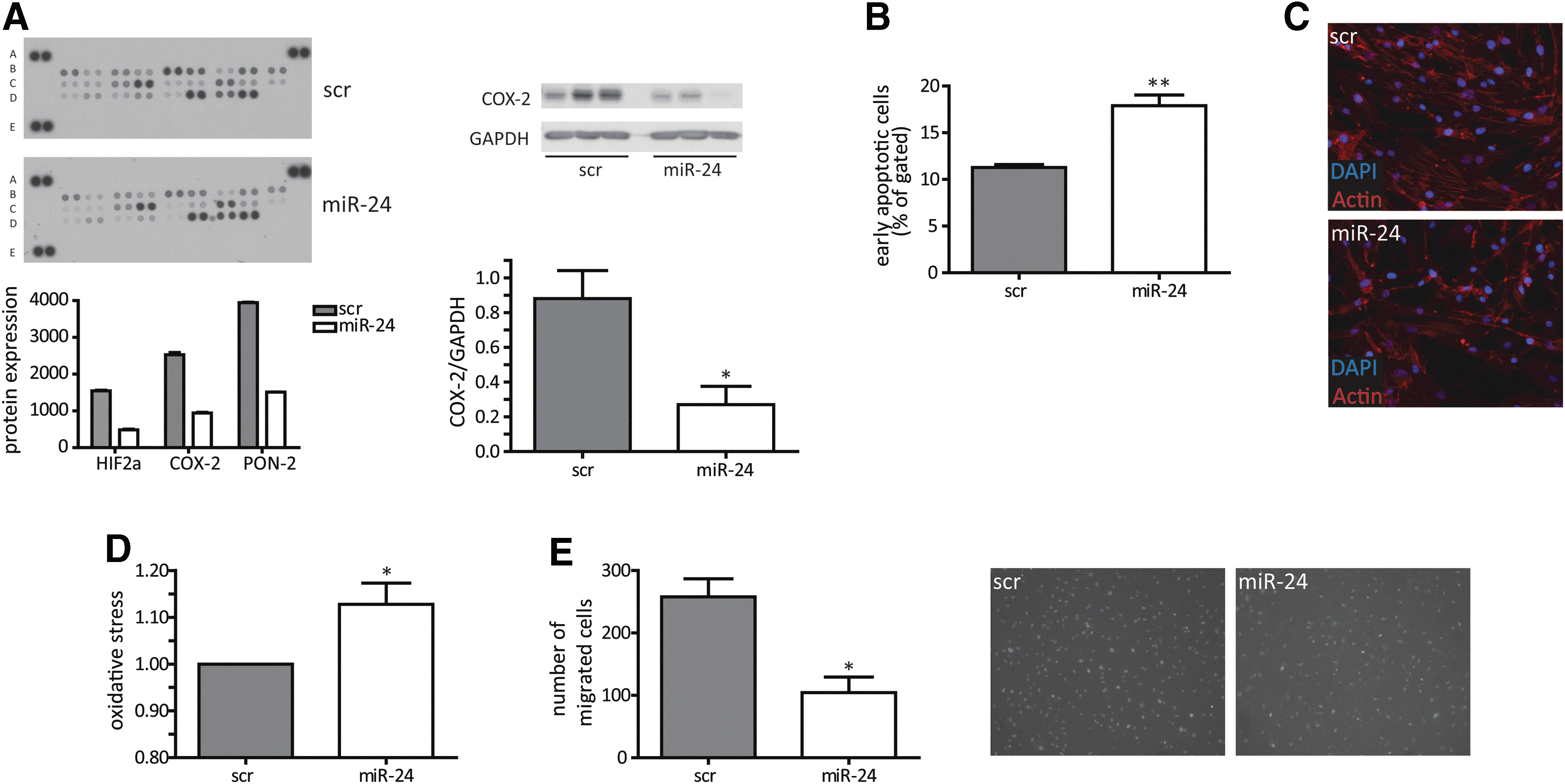
Among theses candidate miRs, miR-24 was chosen for further analysis because of its strong effects on HASMC proliferation and its well-known role being a “hypoxaMir” in vascular cells (10). Indeed, hypoxia significantly increased miR-24 levels in HASMCs (Fig. 1B), a finding that previously has been also reported in endothelial cells (10). Validation experiments confirmed the initial screening results for miR-24 to function as a potent antiproliferative miRNA in time-dependent manner with most prominent effects at 72 h after initial transfection with high efficacy (Fig. 1C; Supplementary Fig. S1). Of interest, the other miR-24 gene cluster members miR-23/27 also impaired proliferative potential of HASMCs (Supplementary Table S1). Next to the proliferation assay based on turnover of WST-1, we additionally checked BrdU incorporation rate, which was also reduced upon miR-24 overexpression (Fig. 1D). We next tested whether miR-24-induced inhibition of HASMC proliferation was mediated through changes in the cell cycle. Surprisingly, cell cycle progression was not changed upon miR-24 transfection to HASMCs (Fig. 1E; Supplementary Fig. S2). Thus, alterations in cell cycle are not likely to be responsible for the observed phenotype. Next, we employed a proteomic-based approach to study the effects of miR-24 on stress response proteins. This approach revealed the repression of several factors important to resist cellular stress, such as hypoxia-inducible factor 2α, cyclooxygenase (COX-2), and paraoxonase-2 (Fig. 2A; Supplementary Table S3), suggesting miR-24 overexpression resulted in decreased resistance to stress. Validation experiments confirmed that miR-24 induced repression of COX-2 (Fig. 2A). In agreement, miR-24 overexpression led to a significant increase in apoptosis levels in HASMCs (Fig. 2B) and to disturbed actin filament appearance (Fig. 2C) suggesting increased cellular stress.
Enhanced miR-24 expression reduces functional capacity of HASMCs
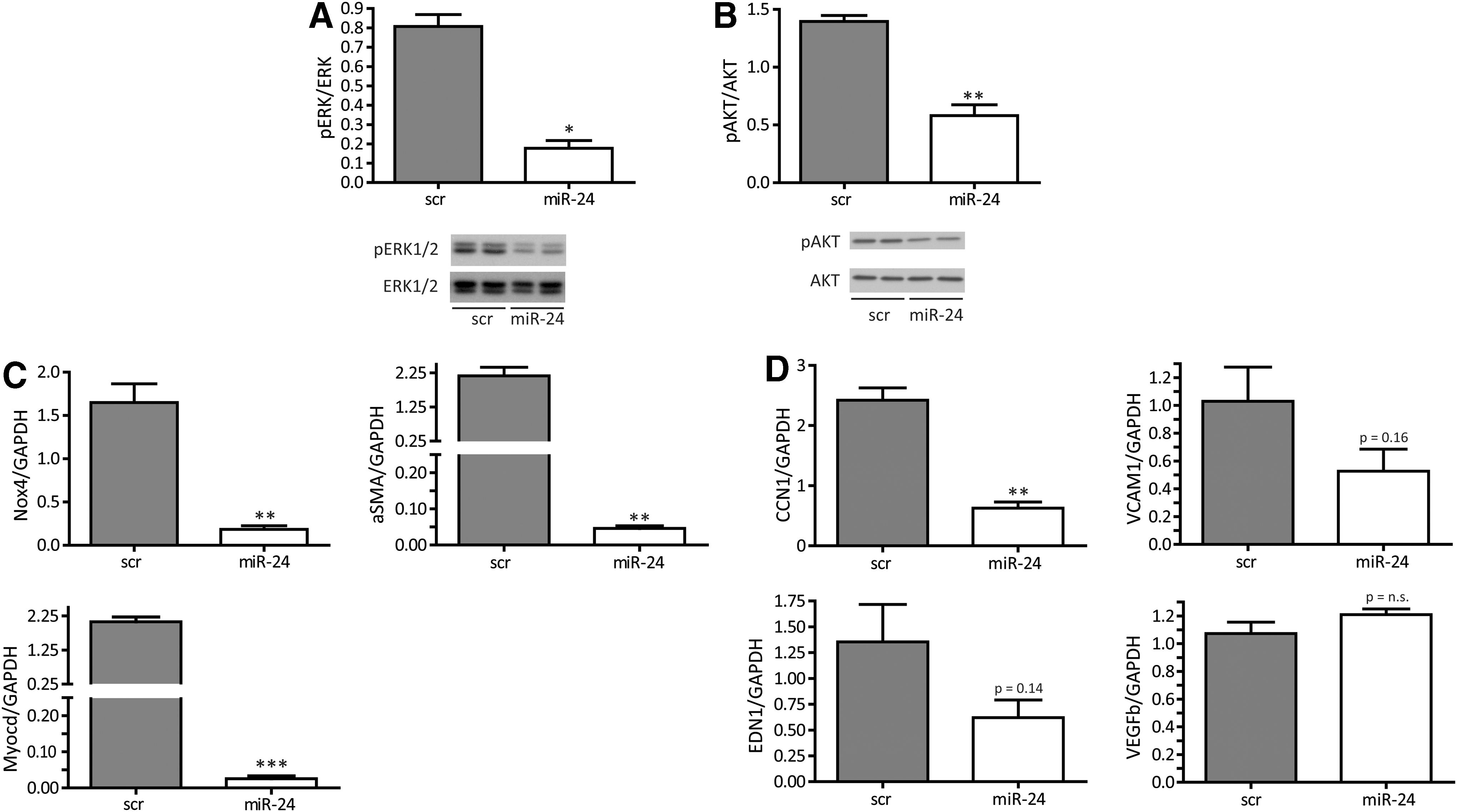
To understand the underlying molecular events leading to miR-24-induced decrease in stress resistance, a number of further studies were performed. To study additional reasons of increased apoptosis and further functional consequences, we employed an assay to study miR-24 effects on the generation of intracellular oxidative stress. Indeed, miR-24 overexpression increased oxidative stress and impaired migratory capacity in a Boyden-chamber assay (Fig. 2D, E). In line, important survival pathways such as extracellular kinase (ERK)-mediated signaling and the Akt pathway were significantly inhibited when miR-24 was overexpressed in HASMCs (Fig. 3A, B). In addition, there was a loss of expression of contractile marker genes such as NOX-4, αSMA, and MYOCD (Fig. 3C) upon miR-24 overexpression being in line with the observed deregulated phenotypic appearance of HASMCs (Fig. 2C). Time-dependent expression patterns of those genes reflected an early response to miR-24 overexpression (data not shown). In addition to contractile marker genes, we validated gene expression of angiogenic genes cysteine-rich angiogenic inducer 61 (CCN1), endothelin 1 (EDN1), vascular cell adhesion molecule 1 (VCAM1), and vascular endothelial growth factor B (VEGFb) (Fig. 3D). Of those, the expression level of proangiogenic CCN1 was most strongly repressed upon enhanced miR-24 expression.
HMOX-1 is a direct target for miR-24
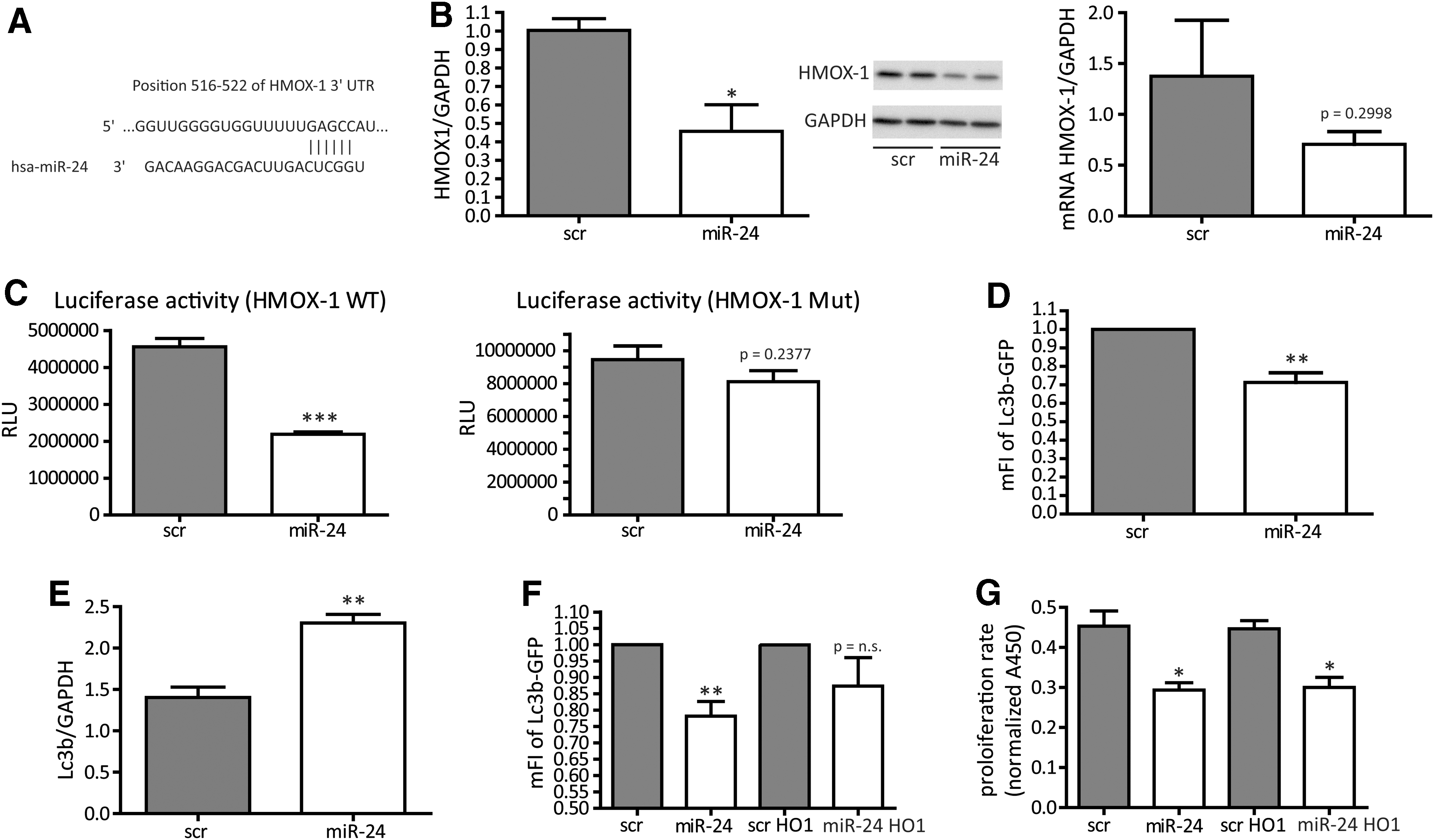
We next wanted to identify a direct miR-24 target that helps to understand the miR-24-induced phenotype of HASMCs, for example, increase of cellular apoptosis and loss of proliferation and contractile marker genes. We thus screened putative miR-24/target gene interactions via the software and miRNA database targetscan (targetscan.org) and identified heme oxygenase-1 (HMOX1, HO-1) to be an interesting candidate as it is an important and well-established cytoprotective factor (25). An indirect search investigating the 3′-UTR region of HO-1 revealed potential miR-24 base pairing (Fig. 4A). We conducted several experiments to validate this single miR-24 binding site to be of relevance in miR-24/HMOX-1 interaction. Indeed, increased miR-24 levels effectively repressed HMOX-1 expression at the protein level and (as a trend) at the mRNA level (Fig. 4B), suggesting translational repression to be the main mechanism of miR-24 in HMOX-1 regulation in human SMCs. In murine cells, miR-24-dependent repression of HMOX-1 was also observed confirming a conserved mechanism (Supplementary Fig. S3). To next validate direct miR-24 binding to HMOX-1 3′-UTR region, luciferase reporter gene constructs were generated. Co-transfection of wild-type reporter construct along with normalizing plasmid and miR-24 precursors significantly lowered luciferase activity indicating miR-24-mediated repression of HMOX-1 (Fig. 4C). When the miR-24 binding site was mutated, however, the repressive effects of miR-24 were abolished further indicating direct miR-24 targeting to the HMOX-1 3′-UTR region (Fig. 4C). As the HMOX-1 enzyme system was suggested to have a role in the regulation of autophagy (3), we next tested the effects of miR-24 on HASMC autophagy. Indeed miR-24 increased basal autophagy (Fig. 4D). The specific inhibition of autophagy by wortmannin, however, had no rescuing effects on the antiproliferative phenotype of miR-24 overexpression (data not shown), suggesting autophagy and apoptosis to have independent effects in the context of miR-24-mediated functions in HASMC. Lc3b, a common marker gene for autophagy induction, is also upregulated upon miR-24 transfection (Fig. 4E). We next tested if the miR-24-mediated effects on HASMC biology were at least in part dependent of HMOX1. For this, we employed viral-based miR-24-resistant HMOX1 overexpression (HMOX1 lacking the miR-24 binding site in 3′-UTR), which indeed partly normalized the increased miR-24-induced autophagy (Fig. 4F). This reflects an important role of the miR-24 target HMOX1 to balance HASMC autophagy. Other miR-24 dependent effects on proliferation rate or oxidative stress were not affected by HMOX-1 overexpression (Fig. 4G and data not shown).
Translation of the in vitro findings into a model of vascularized ex vivo myocardium
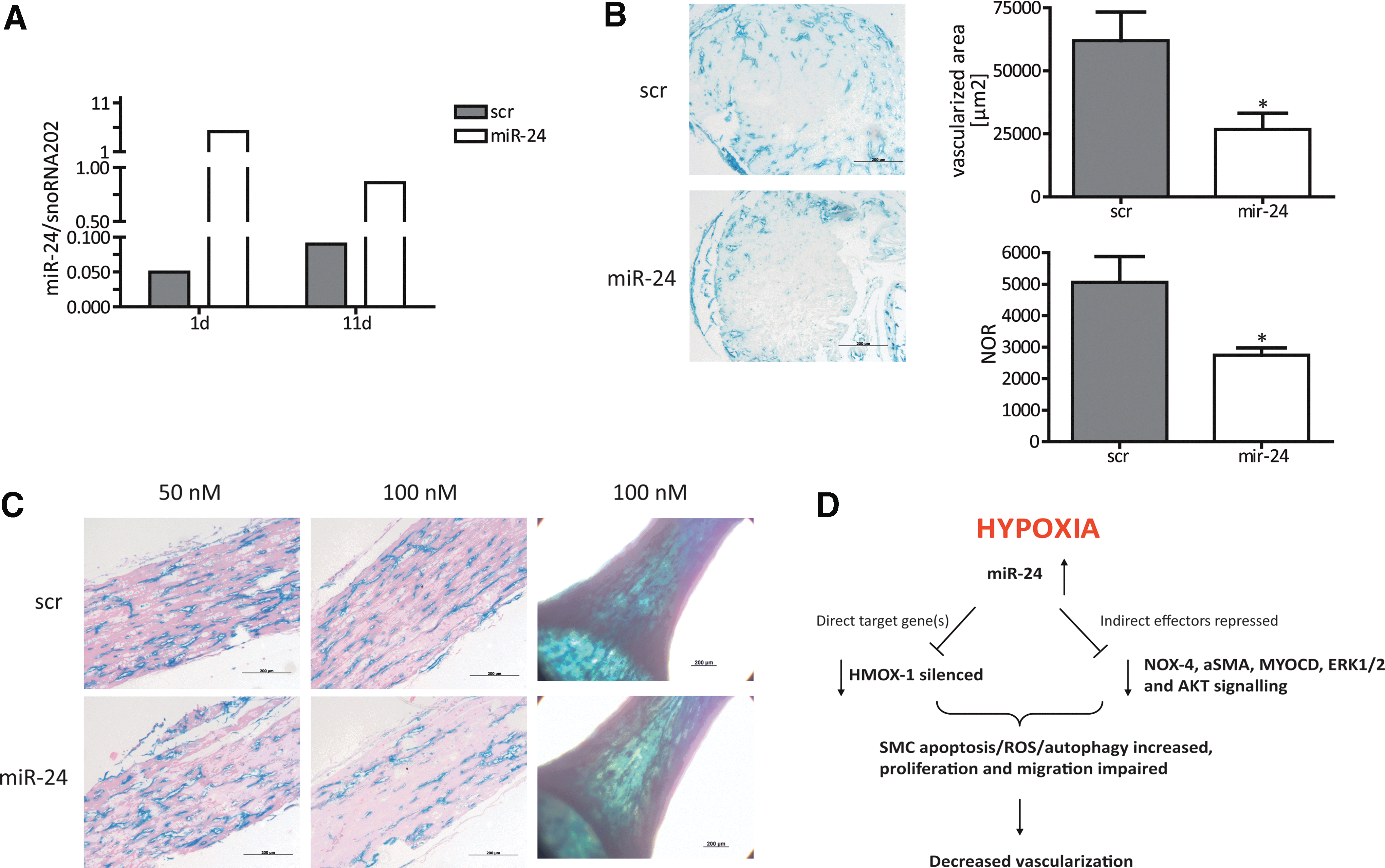
To translate our in vitro findings into an in vivo scenario, we employed the following approach. EHTs represent an excellent opportunity to study the effects of novel substances in a well-organized three-dimensional construct of ex vivo generated vascularized myocardium (15). Our hypothesis was that the detrimental effects of miR-24 on vascular SMCs would affect overall vascularization in such EHTs. For this purpose, we generated EHTs with genetically labelled endothelial cells for the simple and robust identification of vascular structures ex vivo. Heterozygous inducible Cre-positive VE-Cadherin-Cre-ERT2 mice (2, 18) were crossed with homozygous Rosa26R-LacZ reporter mice to specifically label endothelial cells by LacZ gene expression as detailed in the “Materials and Methods” section. To confirm transfection efficiency in EHTs, miR-24 expression levels in miR-24-transfected EHTs were analyzed in comparison to control-transfected EHTs. miR-24 expression level were highest 1 day after transfection (fivefold of the miR-24 mRNA levels in scr EHTs) and decreased to day 4 (twofold) and day 11 (onefold), indicating that a transient effect of miR-24 that was most pronounced in the first days of EHT culture (Fig. 5A). miR-24- or control-transfected VE-CadherinxRosa26R-LacZ EHTs were treated with 5-OH-tamoxifen to induce LacZ expression in endothelial cells, stained with X-gal, fixed, paraffin-embedded, and cross sectioned. Sections were quantified without counterstaining to avoid potential color overlay of the LacZ signal. Native VE-Cadherin EHT showed endothelial cell-specific X-gal+ tubular structures with clearly discernible lumina, primarily located in the border zones of the EHT (Fig. 5B). Less X-gal-positive regions and tube-like structures were detected in miR-24-transfected VE-Cadherin EHTs (46%), and endothelial cell density was 57% lower in miR-24-treated EHTs (100 nM; Fig. 5B). These findings in cross sections were supported by longitudinal histological sections, which showed extensive interconnected endothelial cells forming branched vascular networks in EHTs (Fig. 5C). Again, vascular structure appeared less dense in miR-24-treated EHTs in the whole mount view (Fig. 5C). In addition, EHT showed a loss of smooth muscle actin (ACTA2) expression under miR-24 overexpression conditions (Supplementary Fig. S4). These new data suggest that miR-24 overexpression not only in vitro but also in an ex vivo EHT model impairs vascularization. Whether these effects are mediated via the previously identified detrimental effects of miR-24 on endothelial cells (10) and/or the detrimental effects on SMCs as shown in this report remains to be determined in future studies.
Discussion
Vascular injury (e.g., seen in atherosclerosis or in-stent restenosis) involves the activation of resident, quiescent SMC to a more proliferative status. Thus, a broader knowledge about the involvement of miRNAs in the proliferation of SMCs is highly valuable. In this study, we could identify miR-24 as a strong antiproliferative miR by a robotic-assisted miR screening approach (mechanistic scheme summarized in Fig. 5D). In line with previous results, miR-24 is designated as a “hypoxaMir.” Hypoxia itself is a strong inductor of proliferation in SMC (12). Enhanced miR-24 expression triggers functional SMC defects as well as the loss of contractile potential measured by strong repression of marker genes, which is in agreement to the study from Chan et al. (5). In contrast, we did not observe a parallel induction of a clear synthetic phenotype characterized by elevated proliferation. It is more likely that miR-24 transfection put cells into a more intermediate cellular status with less proliferation and contractile activity. Of note, miR-24 is a clustered miR altogether with miR-23/27 (5). In our screening, we also observed detrimental effects on proliferation rate of the other cluster members, whereas miR-24 effects were strongest. At the transcriptional level, regulatory mechanisms for this miR cluster are less studied and may also be of interest for extensive future analysis. Impairment in proliferation, however, cannot be explained by cell cycle alterations as we did not see any defects in cell cycle progression triggered by miR-24. Of note, endogenous miR-24 repression did not influence the HASMC activity demonstrating the crucial role of balanced miR-24 expression (data not shown). miR-24 is altering the expression level of various cellular stress proteins and induces apoptosis and oxidative stress under basal conditions. Prominent cellular survival pathways (e.g., AKT and ERK) are also effectively repressed via miR-24. This likely contributes to the loss of the migratory potential of SMCs. There are some different previous results about miR-24 in SMC present in the literature. Whereas Chan et al. (5) identified PDGF-BB driving in a miR-24-dependent manner a more synthetic phenotype in SMC, miR-24 has also recently been reported to inhibit SMC migration capacity during vascular injury (20), being in line with our observations. The miR-24-dependent effect on CCN1 expression may also reflect a paracrine function since this factor was reported to be activated in neovascularization processes (14). The identification of cytoprotective HMOX-1 as a direct miR-24 target gene implies high relevance of miR-24-dependent pathways in SMC. Indeed, HMOX-1 could recently be linked to counteract apoptosis and autophagy in renal cells (3). Here, miR-24-dependent repression of SMC HMOX-1 also induces apoptosis and autophagy. In our study, HMOX-1 supplementation could partly rescue the defective autophagy phenotype additionally supporting the impact of miR-24/HMOX-1 axis in SMC. Further functional impairment of SMC function (e.g., proliferation, oxidative stress), however, could not be reverted by HMOX-1 suggesting that other known or unknown miR-24 target genes participate in these processes, for example, p21 protein-activated kinase 4 (PAK4) and c-myc (Supplementary Fig. S5). The identification of further miR-24 downstream effectors is thus an open issue and remains to be determined in future studies. Translation of in vitro findings to an ex vivo model of EHT validated miR-24 effects on vascular cells. Efficient miR-24 modulation was seen in the developing EHT, which finally disturbed the appearance of vascular structures. We think that this is a further evidence for proapoptotic properties of miR-24 in the interplay between endothelial and SMCs. Thus, during vascular injury, for example, miR-24 should be held at a low expression level. Therapeutic intervention may be of use applying antagonistic chemistries (e.g., antagomir or locked nucleic acid chemistry). The efficacy of therapeutic miR-24 targeting should thus be tested in other models of vasculature.
Materials and Methods
Cell culture
HASMCs were purchased from Invitrogen (Gibco) and cultured in SmGM-2 (Lonza) supplemented with human epidermal growth factor, insulin, human fibroblast growth factor-B, gentamicin/amphotericin-B, and 10% fetal bovine serum under standard cell culture conditions (37°C, 5% CO2). In paraformaldehyde (PFA)-fixed cells, actin cytoskeleton was stained applying TRITC-tagged Phalloidin (Sigma; #1951). Hypoxia was considered as an oxygen level of 0.1–0.2% and was performed for 24 h. SV40-transformed endothelial cells were purchased from ATTC (#CRL-2167) and cultivated according to the manufacturer's instructions.
Transfection
Cells were transfected at a confluency of 60–70% 1 day after seeding. Specific miRNA (pre-miR-24; Ambion, Ambion human pre-miR Library #4385830) or control miRNA (pre-miR Negative Control #2; Ambion) at a final concentration of 100 nM and Lipofectamine 2000 (Invitrogen) were incubated separately with Opti-MEM I (Invitrogen) for 5 min, then mixed and incubated for 20 min. Cell medium was replaced for the transfection reaction. After 4 h, the medium was refreshed. Cells were collected and analyzed after 72 h.
Pre-miRNA screening and proliferation assay
HASMCs were seeded in 96-well cell culture plates and transfected with Human Pre-miR Library (Ambion) at a final concentration of 100 nM 1 day after seeding manually or using a robot pipetting device (Bravo System; Agilent). To measure the proliferative capacity in miRNA-modulated cells, a WST-1 proliferation assay (Roche) was applied. Seventy-two hours after transfection, the medium was changed and replaced by WST-1 reagent according to the manufacturer's instructions. Finally, WST-1 absorbance was measured at 450 nm.
Migration assay
For Boyden-chamber migration, cells were incubated for 1 h with 4′,6-diamidino-2-phenylindole, pelleted, and resuspended in SmGM-2 with 0.1% bovine serum albumin. Twenty-five thousand cells per sample were seeded on fibronectin coated trans-well inserts (BD Biosciences), previously placed in wells containing chemoattractants (50 pg/μl VEGF, 100 pg/μl stromal cell-derived factor). Images were captured after 6 and 24 h, and the number of migrated cells was counted using NIS-elements BR software (Nikon).
Autophagy measurement
HASMCs were seeded in 48-well cell culture plates and transfected with miRNA 1 day after splitting. Immediately after transfection, LC3b-GFP construct was added. After 4 h, the medium was refreshed and cells were incubated for another 48 h. Finally, cells were harvested, fixated with 0.5% PFA, and analyzed for GFP fluorescence on Guava FACS Easycyte. Fluorescence-activated cell sorting (FACS) data were analyzed using FlowJo software.
Real-time polymerase chain reaction analysis
Total RNA isolation was performed with TriFast reagent (Peqlab) according to the manufacturer's protocol. For gene expression analysis, RNA was reverse transcribed with iScript Select cDNA Synthesis Kit (Biorad) and Real-time polymerase chain reaction (PCR) analysis was performed using iQ SYBR Green Supermix (Biorad). Primer sequences were as following: NOX4: forward 5′-CCGGCTGCATCAGTCTTAACC-3′, reverse 5′-TCGGCACAGTACAGGCACAA-3′; Acta2: forward 5′-CCTGACTGAGCGTGGCTATT-3′, reverse 5′-GATGAAGGATGGCTGGAACA-3′; Myocd: forward 5′-CTGTTCCTGCAGCTCCAAAT-3′, reverse 5′-GGAGACAAGGGGGTATTGCT-3′; Lc3b: forward 5′-AACAAAGAGTAGAAGATGTCCGAC-3′, reverse 5′-GAACTTTGTTTTATCCAGAACAGG-3′; c-myc: forward 5′-TCAAGAGGCGAACACACAAC-3′, reverse 5′-GGCCTTTTCATTGTTTTCCA-3′; GAPDH: forward 5′-CCAGGCGCCCAATACG-3′, reverse 5′-CCACATCGCTCAGACACCAT-3′. Primers for human CCN1 (CYR61), VCAM1, EDN1, CDK1/2/4/6, PAK4, and VEGFb were purchased from Qiagen as Quantitect primer sets. To analyze miRNA expression, two-step real time-PCR primer sets from Applied Biosystems were used according to the manufacturer's protocol. RNU48 and sno-RNA-202 served as a housekeeping control.
Western blotting
About 10–40 μg of total protein were separated by sodium dodeyl sulfate–polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membrane, and analyzed by western blotting using standard protocols. Antibodies were purchased from: HMOX-1 (R&D Systems; AF3776), ERK (Cell Signaling; #9101/02), AKT (Cell Signaling; #9272/75), COX-2 (Santa Cruz; #1747), ACTA2 (ab5694), and GAPDH (Abcam; ab8245). Luminol reagent was used to visualize the signals on the membrane using X-ray film. The band intensity was calculated by applying ImageJ software.
Proteome profiler
Human Cell Stress Array (R&D Systems; ARY018) was performed with 300 μg of cell lysate pooled from three independent experiments following the instructions of the manufacturer. ImageJ software was applied to calculate signal intensities.
miR target prediction and luciferase reporter gene assay
The miRNA database and target prediction tool TargetScan (
Apoptosis assay
Transfected cells were harvested after 72 h and stained with the Annexin-V-Fluos kit from Roche Diagnostics according to the manufacturers' instructions. FACS analysis was performed on a FACSCalibur (BD Biosciences).
BrdU enzyme linked immunosorbent assay
To measure the proliferative capacity in miR-24-modulated HASMC, a colorimetric BrdU enzyme linked immunosorbent assay kit from Roche (#11647229001) was applied. Standard procedures were performed according to the manufacturers' instructions.
Cell cycle analysis
Analysis of miR-transfected cells was performed applying Guava Cell Cyle Reagent (Millipore) according to the manufacturers' instructions. Subsequent FACS analysis was performed on guava easyCyte (Millipore).
Detection of oxidative stress
To monitor intracellular oxidative stress, cells were stained in culture with CellROX Green Reagent according to the manufacturer's instructions (Invitrogen). Afterward, FACS analysis was performed on guava easyCyte (Millipore).
Mouse model for genetic labeling of endothelial cells
Heterozygous VE-Cadherin-Cre-ERT2 mice [kindly provided by Prof. Florian Limbourg; (2, 18)] were crossed with homozygous Rosa26R-LacZ reporter mice to specifically label endothelial cells by LacZ gene expression. Double transgenic mice were identified by genotyping via PCR from tail DNA using primers R1295: 5′-GCG AAG AGT TTG TCC TCA ACC-3′, R523: 5′-GGA GCG GGA GAA ATG GAT ATG -3′, R26F2: 5′-AAA GTC GCT CTG AGT TGT TAT-3′, CRE1: 5′-GCC TGC ATT ACC GGT CGA TGC AAC GA-3′, and CRE2: 5′-GTG GCA GAT GGC GCG GCA ACA CCA TT-3′. In Rosa26R-LacZ reporter mice, a floxed stop codon is inserted between the promoter sequence and the LacZ gene, inactivating the LacZ gene until Cre recombinase expression is induced. Cre-mediated recombination leads to the excision of the floxed stop codon and LacZ gene expression. During EHT culture, Cre expression was induced by the addition of o-hydroxytamoxifen (1 nM) to the culture medium on day 15 for 48 h. The endothelial cell specificity of the system is obtained by control of the inducible Cre-ERT2 fusion protein by the endothelial cell-specific surface protein VE-Cadherin and was verified in mice (data not shown).
Neonatal mouse cardiomyocytes
Neonatal mice (day 0–1, postnatal) were sacrificed by decapitation. After heart extraction, neonatal heart cells were isolated by a serial DNase/trypsin digestion based on methods to isolate neonatal rat cardiomyocytes (23). The resulting cell population was immediately subjected to EHT generation. Experimental procedures were reviewed and approved by the Ethics Committee, University of Hamburg.
Sylgard posts and Teflon spacers
Sylgard 184 (Dow Corning) was used to fabricate silicone racks with silicone posts for EHT cultivation, and Teflon spacers for casting mold generation were manufactured as previously described (15). Silicone racks were produced by Silitec GmbH & Co. KG.
Generation and culture of EHT
To generate EHTs, a reconstitution mix was prepared on ice as follows (final concentration): unpurified 6.8×106 cells/ml, 5 mg/ml bovine fibrinogen (stock solution: 200 mg/ml fibrinogen plus aprotinin 100 μg/ml in NaCl 0.9%; Sigma F4753), 10% Matrigel (BD Bioscience; 356235). 2×Dulbecco's modified Eagle medium (DMEM) was added to match the volumes of fibrinogen and thrombin stock (100 U/ml; Sigma T7513) to ensure isotonic conditions. Casting molds were prepared as previously described (15). For each EHT, 97 μl reconstitution mix was briefly mixed with 3 μl thrombin and pipetted into the agarose slot. For fibrinogen polymerization, the constructs were placed in a 37°C, 7% CO2 humidified cell culture incubator for 2 h. The racks were transferred to 24-well plates filled with the culture medium. EHTs were kept in a 37°C, 7% CO2 humidified cell culture incubator. The cell culture medium was changed after 48 h and consisted of DMEM (Biochrom F0415), 10% horse serum (Gibco 26050), 2% chick embryo extract, 1% penicillin/streptomycin (Gibco 15140), insulin (10 μg/ml; Sigma I9278), and aprotinin (33 μg/ml; Sigma A1153).
Transfection of murine EHT
Transfection of VE-CadherinxRosaR-26 EHTs was performed with Lipofectamine™ 2000 Reagent (Invitrogen) according to the manufacturer's instructions. The amount of miRNA and transfection agent was adapted to the use for the EHT culture format. After casting, EHTs were immediately placed into the wells including the transfection mix. EHTs were kept in the transfection mix at 37°C and 7% CO2 for 24 h. Subsequently, EHTs were transferred to a fresh plate containing the EHT culture medium.
Histology
For X-gal staining, EHTs (whole-mount) were transferred to a clean dish containing phosphate-buffered saline (PBS, w/o MgCl2, w/o CaCl2; Gibco). The tissue was washed two times with PBS for 10 min. Then, the samples were transferred to a dish containing the prefixation solution (20×PBS [64 mM Na2HPO4, 10 mM KH2PO4, 26 mM KCl, 2.7 M NaCl, pH 7.4], 5% [v/v]; 37% formaldehyde, 5% [v/v]; 25% glutaraldehyde, 0.2% [v/v] in aqua ad injectabilia) for 45 s. The prefixation solution was discarded immediately in two times PBS washing steps for 10–20 min. Staining was performed in the solution (20×PBS [64 mM Na2HPO4, 10 mM KH2PO4, 26 mM KCl, 2.7 M NaCl, pH 7.4], 5% [v/v]; potassium ferricyanide crystalline [500 mM], 1% [v/v]; potassium ferricyanide trihydrate [500 mM], 1% [v/v]; magnesium chloride [1 M], 0.2% [v/v] in aqua ad injectabilia) containing 0.4 mg/ml X-gal (Sigma B4252) at 37°C over night. The X-gal-positive cells could be detected by the blue staining. After staining, samples were washed in PBS for 2–3 min and postfixed with Histofix® (Roth) for 60 min. Paraffin embedding, sectioning (4 μm), and eosin staining was performed according to standard procedures. Photographic images of the slides were taken with a microscope (Zeiss-Axioplan IM-35), and the suitable software (Zeiss-Axiocam) was used to quantify blue regions in the sections.
RNA isolation from EHT
Total RNA was extracted from 1 EHT using TRIzol® (Invitrogen) reagent (300 μl per EHT) according to the manufacturers' instructions. EHTs were homogenized by the use of a TissueLyser® (Qiagen) at a vibration frequency of 30 Hz. RNA concentration was determinded by measuring the absorbance at a wavelength of 260 nm with a spectrophotometer (NanoDrop® ND-1000; PeqLab). Absorbance was also determined at the wavelength of 280 nm, and the ratio A260/A280 was calculated to test for purity. RNA samples were stored at −80°C for further use.
Statistical analysis
For statistical analysis, GrapPad Prism (Version 4) was applied. In case of two groups, unpaired t-test was performed, unless Figure 2D where paired t-test was performed. Error bars in graphs indicate standard error of the mean. Asterisk mean: *p<0.05; **p<0.01; ***p<0.001.
Footnotes
Acknowledgments
We thank Dr. Mortimer Korf-Klingebiel and Prof. Kai Wollert for providing HMOX-1 virus and Prof. Lucie Carrier for providing Lc3b-GFP virus. Histological standard procedures were performed by the HEXT core facility (UKE, Hamburg) mouse pathology (
Author Disclosure Statement
The authors disclose support from the IFB-Tx (BMBF 01EO0802; T.T.) and DFG TH 903/10-1 (T.T.). T.T. and J.F. have filed patents in the field of cardiovascular miRNA diagnostics and therapeutics. The work of A.S. and T.E. was supported by the EU FP7 Angioscaff and BIODESIGN projects and the German Ministry of Research and Education, BMBF (DZHK, German Centre for Cardiovascular Research).
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.