
Abstract
Introduction
R
The Nox family of NADPH oxidases comprises seven members, with tissue-specific expression patterns and different modes of action and function. In this article, we will focus on the role of Nox proteins in cellular mechano-transduction with a particular emphasis put on the vascular system. We will elaborate on isoform-specific differences between the individual Nox homologues for this process and also cover the less well studied Nox homologues Nox4 and Nox5.
The Nox Family of NADPH Oxidases
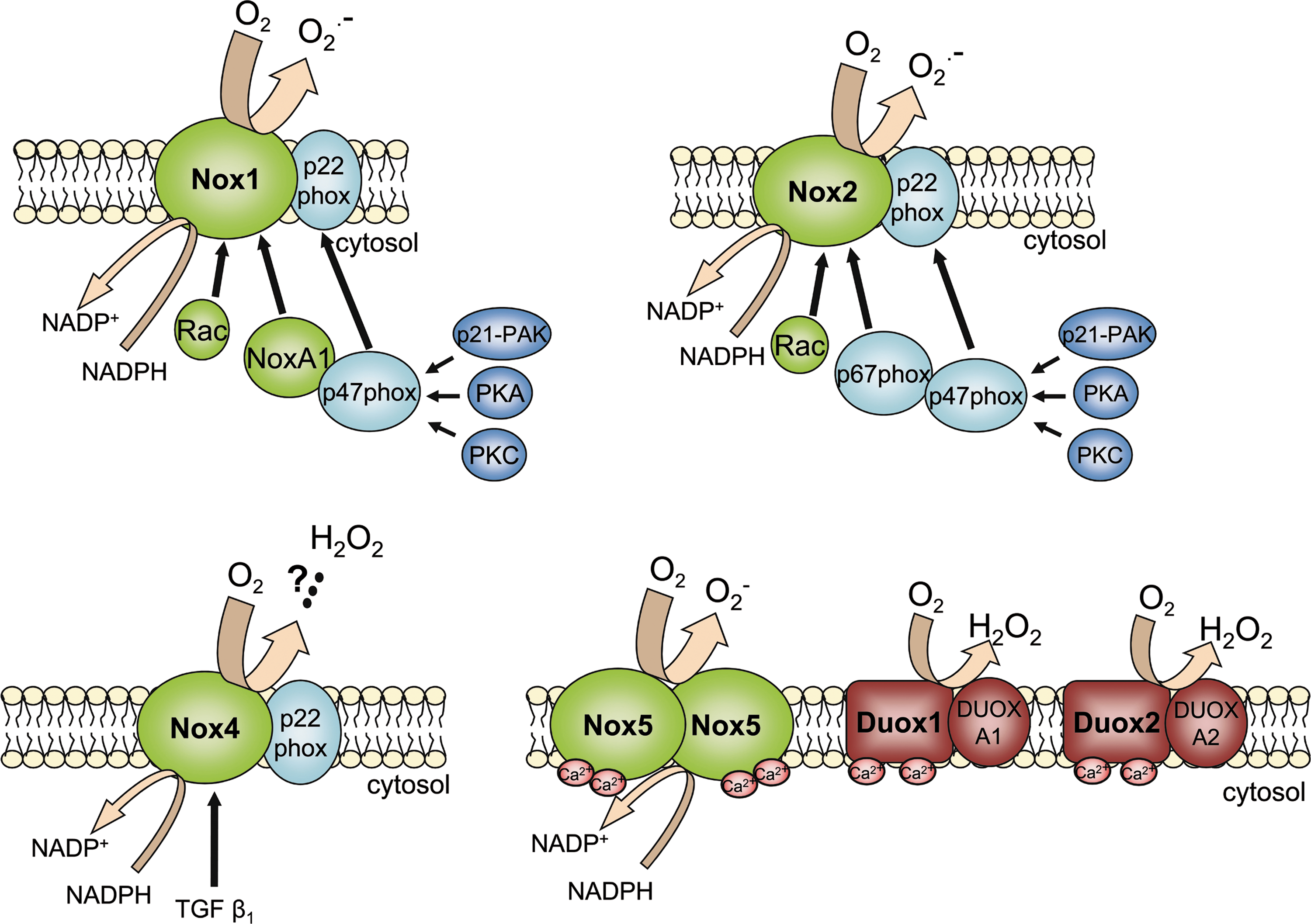
Starting with the discovery of the leukocyte NADPH oxidase, up to now, seven members of the Nox family of NADPH oxidases have been identified (66). Some of them have a rather unique expression pattern und function, whereas others are expressed throughout the body. Nox designates the large catalytic subunit, but with the exception of Nox5, which forms homo-dimers (58), all Nox NADPH oxidases depend on additional interacting proteins (8). With respect to transmembrane proteins, these are p22phox and Duoxa1/2, which facilitate assembly and maturation of the transmembrane Nox and Duox enzymes, respectively. The cytosol contains proteins which are required for the activation of Nox1, Nox2, and to some extent Nox3 (81). Of these, p47phox and Noxo1 are scaffolding proteins, which tether p67phox and Noxa1, respectively, with the plasma membrane and the Nox protein, which is then activated (11). The activation of Nox proteins by this protein-protein interaction is a complex process, controlled by several mechanisms involving small GTPases like Rac1 in nonmyeloid cells and Rac2 in leukocytes and phosphorylation events by serine-threonine-kinases like protein kinase C, AKT or p21-activated protein kinase (PAK) (77). Different to all other Nox proteins, Nox5 and Duox1 as well as Duox2 are directly activated by calcium through EF hands. With respect to activation, Nox4 stands out as it has constitutive activity, at least if overexpressed, and (with the exception of 22phox) is to a large degree independent of accessory proteins, although some interacting proteins like protein disulfide isomerase (52) and Poldip-2 (76) have been reported to be of relevance. Thus, protein expression is the main factor controlling Nox4-mediated ROS production. Nox proteins also differ in the type of ROS released: Whereas Nox1, Nox2, Nox3, and Nox5 mainly produce superoxide anions (O2 −), H2O2 is the prominent product of Nox4, Duox1 and Duox2 (8, 109) (Fig. 1). The mechanisms underlying this potentially important functional difference are incompletely understood. Finally, each NADPH oxidase has a distinct expression pattern. On the mRNA level, Nox1 expression predominates in epithelial cells, Nox2 is highly expressed in leukocytes, Nox3 is largely restricted to the inner ear and Duox enzymes are highly expressed in the thyroid gland (8). Expression data on Nox5 are still sparse as the enzyme is not expressed in rodents (7). Nox4 in contrast, at least on the mRNA level is highly expressed in most differentiated cells, although protein expression is highest and readily detectable in the kidney (82).
Nox Proteins and Mechano-Transduction
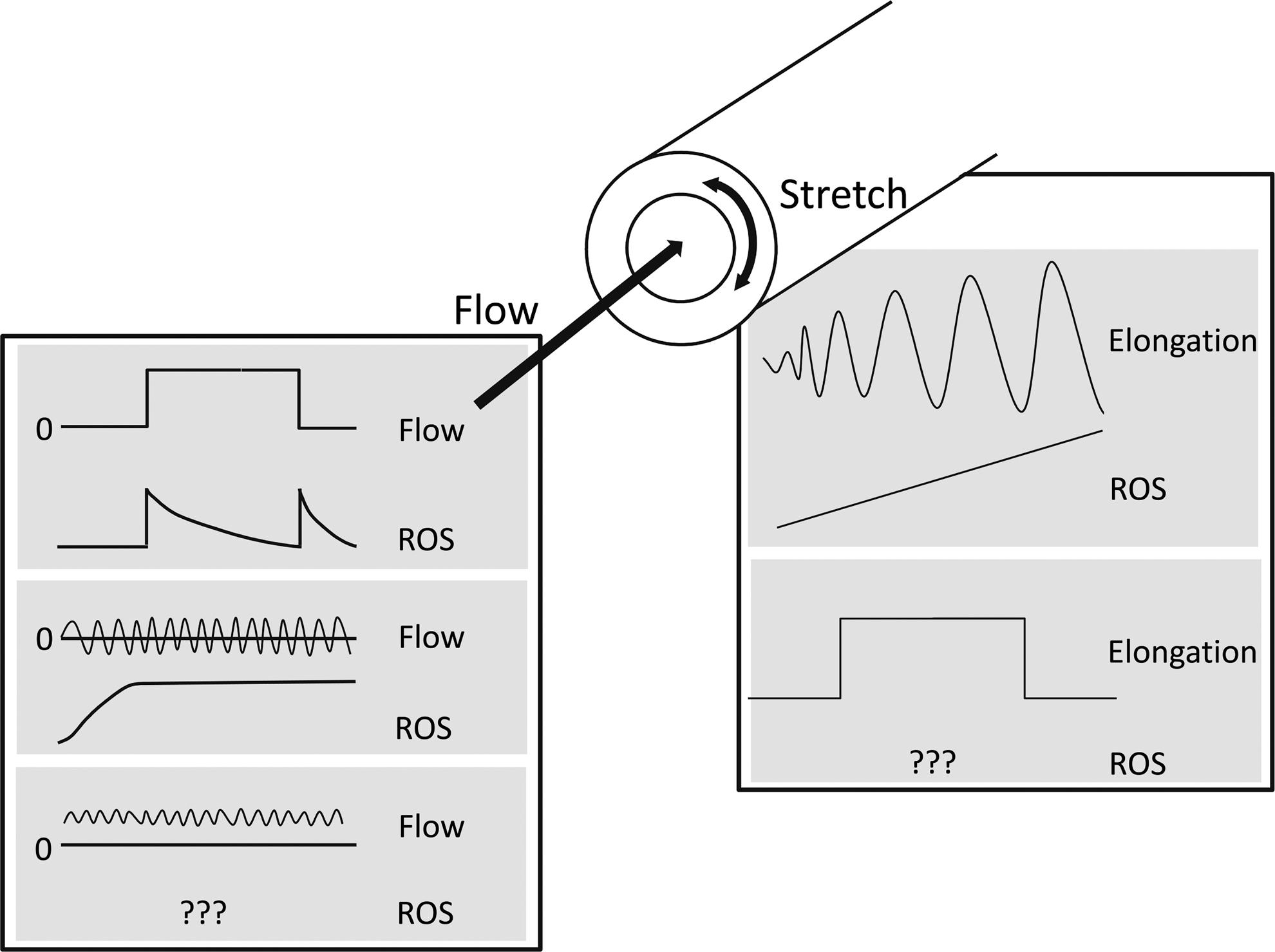
For obvious reasons, most studies on mechano-transduction and Nox proteins focus on the cardiovascular system, and the reaction of cells, the heart and vessels to stretch and strain as well as to different flow patterns. Although hardly systematically researched, the different forms of mechanical stimulation seem to specifically affect individual Nox proteins. This relates to the control of Nox activity but also to Nox expression. In a nutshell, disturbed or oscillatory flow rather increases Nox-dependent ROS production (105), whereas continuous laminar flow at physiological levels has the opposite effect. Cyclic strain increases the cellular ROS production by inducing and activating Nox proteins but little is known how a constant elevation in the stretch level impacts on ROS production. Either, there is a U-shaped relation between stretch intensity and ROS formation or rather a proportional increase of ROS related to relative elongation and frequency of the strain cycles (Fig. 2).
The cellular signal transduction activated in response to physical forces is complex and incompletely characterized (54). In a confluent cellular monolayer, stretch leads to elongation of the whole cell, and to conformational changes at the cell-matrix-interfaces and particular at cell-cell-contacts (68). Shear stress in contrast imposes force on the glycocalyx and the apical cell surface, which results in a displacement of this layer relative to the basal cell membrane and leads to traction forces (65). Finally, increase in pressure not only results in tangential force development but also compression of the cell, which may directly affect integral membrane proteins (93). Depending on specific features of the cell, these effects are further modulated: in flat cells, like in the endothelium, the perinuclear region surmounts the cell and thus, is a site of high force development. The glycocalyx of these cells may also serve to enhance shear forces. Similarly, villous or cilia structures of cells may act as sites of shear stress signaling or even sensing of fluid movement as in the case of the primary cilium (118).
Obviously, there is not just a single mechano-sensor and rather physical forces appear to activate a broad spectrum of signaling cascades (106). Force application to the surface of cells can directly result in ion channel opening (54) but also stimulates integrin signaling, which involves the formation of phosphatidylinositol-triphosphate (PIP3) and the activation of Rac1 (70), two important elements in the activation of Nox1 and Nox2 (11). Also, the reorganization of the cytoskeleton in response to integrin activation may help to tether cytosolic Nox activators to the catalytic subunit of the enzyme. Integrin signaling, in turn, is redox-sensitive and hot spots of ROS-generation directly modify integrins when cells newly attach to a matrix (26). Mechano-stimulation of the cell also activates several G-protein coupled receptors, such as the AT1 or ETA receptor (106), which are well known to activate Nox proteins.
Stretch and Nox-Dependent ROS Production
Stretch is an important cellular stimulus. It promotes the reorganization of matrix structures like bone (103), signals the filling state of hollow organs like the bladder or the intestine (37) and is a direct consequence of passive or active increases in muscle tension (110). Force development imposed on the stretched cell can be static, oscillatory, like in the heart, or a combination of both as present in arteries. The subsequent cellular reaction appears to vary between the patterns of stretch applied but only few studies have taken the technically challenging approach to systematically relate cellular responses to the differential pattern of force application and considered the technical limitations present in all model systems (22, 112).
Since the earliest studies on vascular Nox functions, vascular stretch, as a consequence of an increased tangential wall tension as present in hypertension, has been linked to ROS production (114). Prohypertensive hormones like angiotensin II (46, 66), urotensin II (28), endothelin (73), noradrenaline (9) and aldosterone (34) all induce components of the Nox family and activate the enzyme complex. Also, the direct effect of strain on ROS production was studied. These studies either utilized pressurized vessels ex vivo, aortic constriction or, for cell culture experiments, the Flexcell device. In this apparatus, cells are cultured on flexible silicone elastomer membranes, which are distended in a defined manner by a cone pushed into the membrane from the bottom, cell free-side of the culture plate. With such techniques, it was demonstrated that strain itself also increases ROS production (71) and induces Nox activity and expression (39, 46, 72, 117) (Fig. 3).
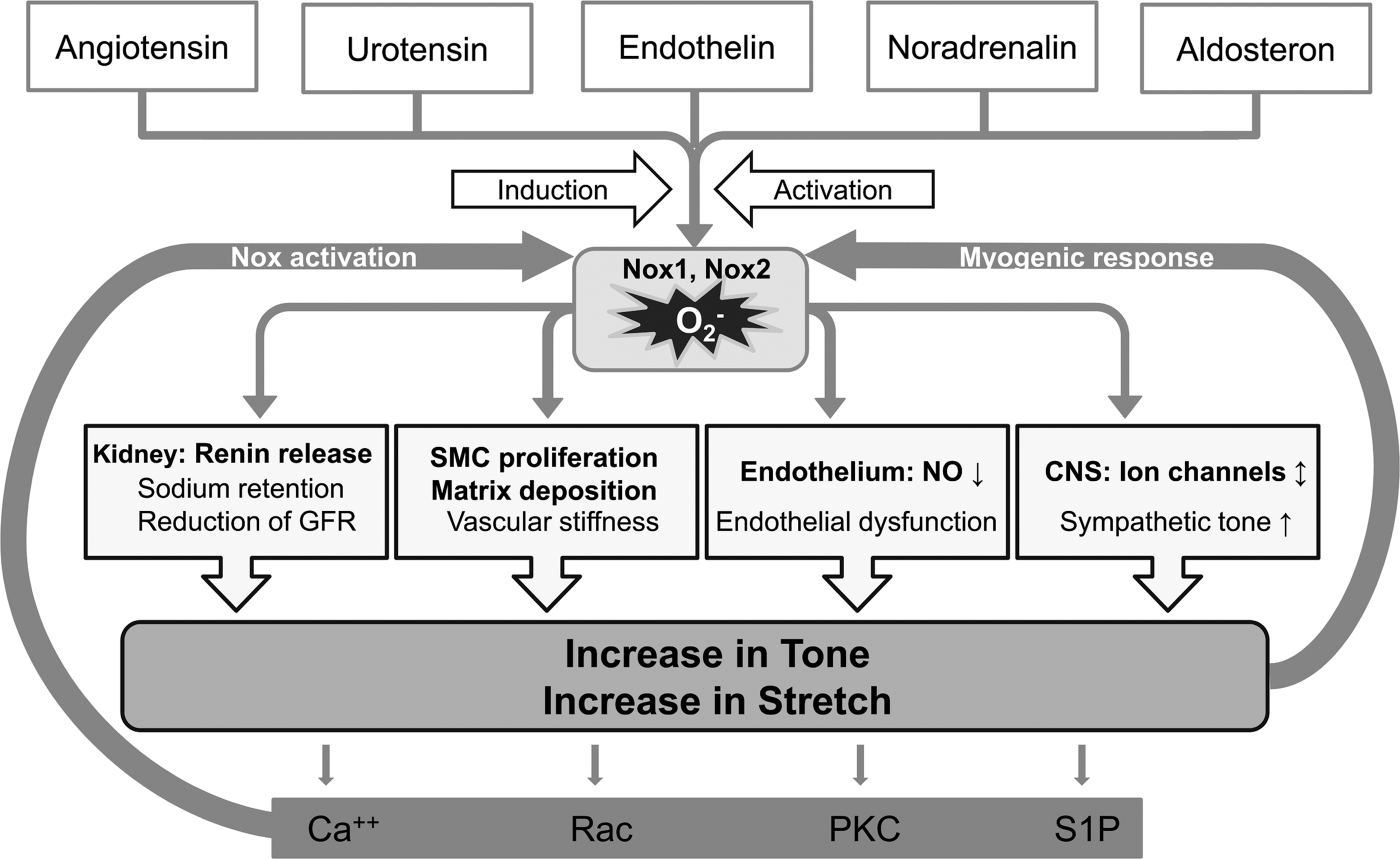
For the myogenic response of arteriolar preparations, it has been demonstrated that increases in stretch result in enhanced ROS production. ROS can activate RhoA (55, 69) and increase intracellular calcium (56) to elevate vascular tone (6, 88). Interestingly, increases in intracellular calcium contribute to ROS formation through the release of arachidonic acid and Nox5 activation (40) so that it is plausible that ROS act as enhancers of myogenic responses. Direct increases in vascular strain in isolated vessels or in transaortic constriction models increase ROS production (116, 120) via increasing intracellular calcium, activation of protein kinase C, sphingosine-1-phosphate production (59) and Rac1 activation (129), which occurs through integrins (121) and an integrin linked kinase-mediated stimulation of the guanine exchange factor β-Pix (120). Similarly, with the Flexcell device, strain was observed to induce NADPH oxidase subunit expression and to activate the enzyme in smooth muscle cells (46) and arteries (89). Obviously, the extent of cellular elongation during stretch determines the magnitude of the cellular reaction. 20% cyclic elongation, for example, induces endothelial cell apoptosis, which was paralleled by increased p22phox protein expression, whereas 5% cyclic elongation may even reduce ROS production by stimulation of AKT-dependent endothelial nitric oxidase synthase (eNOS) activation (44, 45, 63, 72).
As one mechanism involved in this process in rat aortic smooth muscle cells, a localized induction of aldosterone biosynthesis has been suggested. In this model, stretch stimulated and induced NADPH oxidase activity through a mitogen-activated protein kinase (MAP kinase)-mediated induction of CYP11B2 (90). Stretch-induced cell activation and damage was also reported for epithelial cells, also with different patterns. In type II alveolar epithelial cells, cyclic mechanical strain with elongations of 10% and more as well as constant distention inhibited Rac1 activation and thus, blocked migration (27). The mechanism underlying the differential response of epithelial and mesenchymal cells, however, was not explored in that study. Indeed, there is strong heterogeneity in the response to stretch between cells: whereas stretch stimulated Rac1 activation in endothelial cells it increased RhoA activation in smooth muscle cells when 20% elongation and 1 Hz stretch was applied (74). Nevertheless, in the epithelial renal tubular system, increases in stretch stimulate ROS production by NADPH oxidase activation (1, 47) by a mechanism involving protein kinase C alpha. The physiological consequences of this signal, however, remain to be explored (17).
Evidence has been presented that not only increased but also reduced cyclic stretch promotes oxidative stress, although little is known regarding the underlying mechanisms: in an ex vivo system of the perfused porcine carotid artery, reduction of the cyclic stretch from 5% to 1% attenuated endothelium-dependent relaxation and increased ROS production (112). The source of ROS, however, was not identified in this study.
Stretch is also an import stimulus for skeletal muscle (110). Expression of components of Nox complexes are increased in muscles of dystrophic mice and promote an increase in resting intracellular calcium in response to stretch, which leads to an attenuated force development (126). In the heart, myocyte stretch promotes Nox2-dependent ROS formation in close proximity to the ryanodine receptor, which facilitates calcium signaling in the healthy organ. Similar as to skeletal muscle, in dystrophic muscle, the same mechanism leads to calcium overload, which promotes arrhythmias and cardiomyopathy (97). Another proarrhythmogenic effect of stretch is the NADPH oxidase-dependent downregulation of the mRNA of the potassium channel Kv4.3 (131), which leads to action potential prolongation. Also, a link between strain-induced ROS formation and Nox-driven differentiation has been presented: in embryonic stem cells, strain applied by the Flexcell system, induced several components of the Nox system, including Nox1 and Nox4 and increased the cellular ROS production. ROS scavengers blocked the strain-induced differentiation as well as the expression of differentiating transcription factors in these cells (100).
Finally, cellular volume sensing, which is mediated, in part, by stretch-activated ion channels and integrins has been linked to NADPH oxidases (15), and cell swelling increased cellular ROS production (33).
Whether Nox-dependent ROS production facilitates hypertension development or is just a consequence of increased vascular strain as occurs in hypertension is debated. Obviously, both scenarios do not exclude each other. Although it is clear that stretch increases ROS formation and promotes cellular contraction (see above), numerous studies have suggested that development of hypertension is attenuated in NADPH oxidase-deficient mice. This is a consequence of alterations in the central blood pressure control (92) but also of direct vascular effects. Nox-derived ROS promote vascular hypertrophy, fibrosis and the Nox-dependent formation of O2 − limits NO availability (83) and promotes eNOS uncoupling (32). The latter events increase peripheral resistance, which is one aspect of hypertension (114).
Which Nox homologue mediates the response to stretch? Most studies suggest a role of Rac1 for stretch-induced ROS production (71, 124), which would point towards a role of Nox1 or Nox2. Indeed, a selective peptide inhibitor (gp91ds-tat) blocking Nox activity (59, 97) as well as knock-down of p47phox (39) attenuated stretch-induced ROS formation. Importantly, Nox4, different to all other Nox proteins (but not the Duox enzymes), does not contain a Rac-interacting site (57) and is not activated by Rac1 (123), which would argue against a role of Nox4 in stretch-induced ROS production. Indeed, in the heart, stretch-induced ROS production was absent in Nox2 knockout mice (97), but Nox4 knockout mice have not been studied in this context. In human umbilical vein endothelial cells (HUVECs) studied in the Flexcell system applying 12% elongation, Nox4 mRNA expression decreased to 50%, which was, in part, sensitive to NO synthase inhibition (38). Nevertheless, cell swelling-induced ROS production in NIH3T3 mouse fibroblasts was attenuated by Nox4 siRNA, whereas downregulation of p47phox or p67phox did not affect this response (33). Although swelling may activate Rac1 (19) and thus, should stimulate Nox1 and Nox2, this observation may suggest that by a different, so far unidentified pathway, swelling, different to matrix-mediated integrin-dependent strain, may also stimulate Nox4. Indeed, also for smooth muscle cells, alternative pathways appear to exist, which involve transforming growth factor (TGF) β1 signaling [see below, (78)].
Nox-Dependent ROS Production and Oscillatory or Disturbed Flow
Movement of fluid medium, which imposes a certain amount of shear stress on the underlying structure, is another important physical stimulus for cells. Although epithelial cells are often exposed to shear stress, this aspect is best studied in the vascular system, as blood flow is an important determinant of arteriosclerosis development. Whereas continuous laminar flow and uniform shear stress is vasoprotective, turbulent, or oscillatory flow induces endothelial cell activation and leads to endothelial dysfunction and arteriosclerosis (14, 18, 21, 64).
On the cellular level, oscillatory or turbulent flow, due to the perpetually occurring changes in the direction of shear stress, results in continuous movement of the apical cell membrane over the matrix-attached basal parts. This leads to oscillating deformations of the cytoskeleton but also of the bottom membrane and the lateral cell contacts and eventually to the activation of cellular signaling cascades (21).
In endothelial cells, oscillatory flow acutely increases cellular ROS formation (48), which subsequently remains elevated for the duration of shear stress exposure (25, 104). This process is, at least in part, consequence of a continuous activation of Nox2 (79) and of the induction of several components of the NADPH oxidase family (25, 104). The activation of the NADPH oxidases is probably mediated by increases in the intracellular calcium concentration (43) and the prolonged activation of Rac1, which results from the inability of the integrin-related signaling cascades to adapt to the ever changing tensile stretch on the matrix (21, 35, 91, 115).
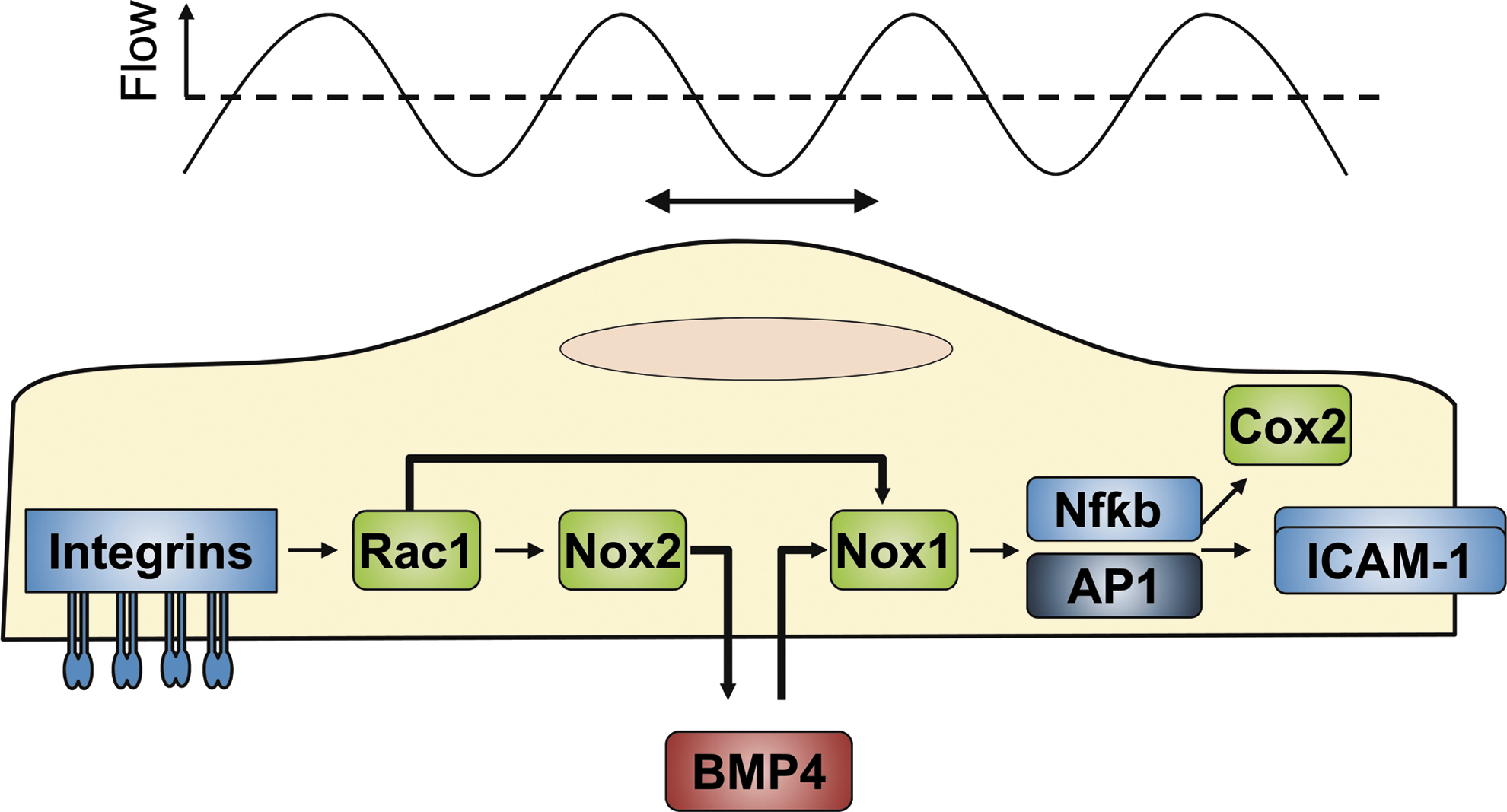
In endothelial cells, cyclic bidirectional shear stress also induces NADPH oxidases, which appears to be related to the bone morphogenic protein (BMP) family. BMP4, which is induced by oscillatory flow (24, 105), increases Nox1 but downregulates Nox4, and also Nox2 mRNA expression (105). Interestingly, blockade of TGF β receptors attenuated the oscillatory flow-mediated induction of BMP4 in the aorta (108), suggesting that activation of latent TGF β is an early step in signaling of oscillatory flow. The subsequently formed BMP4, via the production of ROS induces endothelial dysfunction in the aorta through Cox2 induction. Indeed, inhibition of BMP4 by its receptor antagonist noggin attenuated endothelial dysfunction in mice treated with BMP4 (80) and normalized endothelial function in the aorta of spontaneously hypertensive rats (127). Importantly, the latter effects as well as the inflammatory activation in response to BMP4 are NADPH oxidase-dependent: Blockade of Nox activation by genetic deletion of p47phox prevented the oscillatory shear-induced ROS production and adhesion molecule expression (51, 105) (Fig. 4).
Although these studies provide a link between oscillatory flow and Nox1, it should be mentioned that in bovine aortic endothelial cells different patterns of shear stress-induced Nox expression were observed. Oscillatory shear increased Nox2 and Nox4, whereas pulsatile flow elicited the opposite reaction, Nox1 expression was not studied (50). It is currently not known whether this effect is a cell specific reaction or depends on the experimental model, the shear apparatus used or the intensity or mode of shear stress applied. However, collectively, the observations suggest that oscillatory flow is a situation of particularly high ROS production, which is also underlined by the observation that this type of flow pattern leads to a significant reduction in the intracellular pool of reduced glutathione (84).
Laminar Flow and Nox-Dependent ROS Production
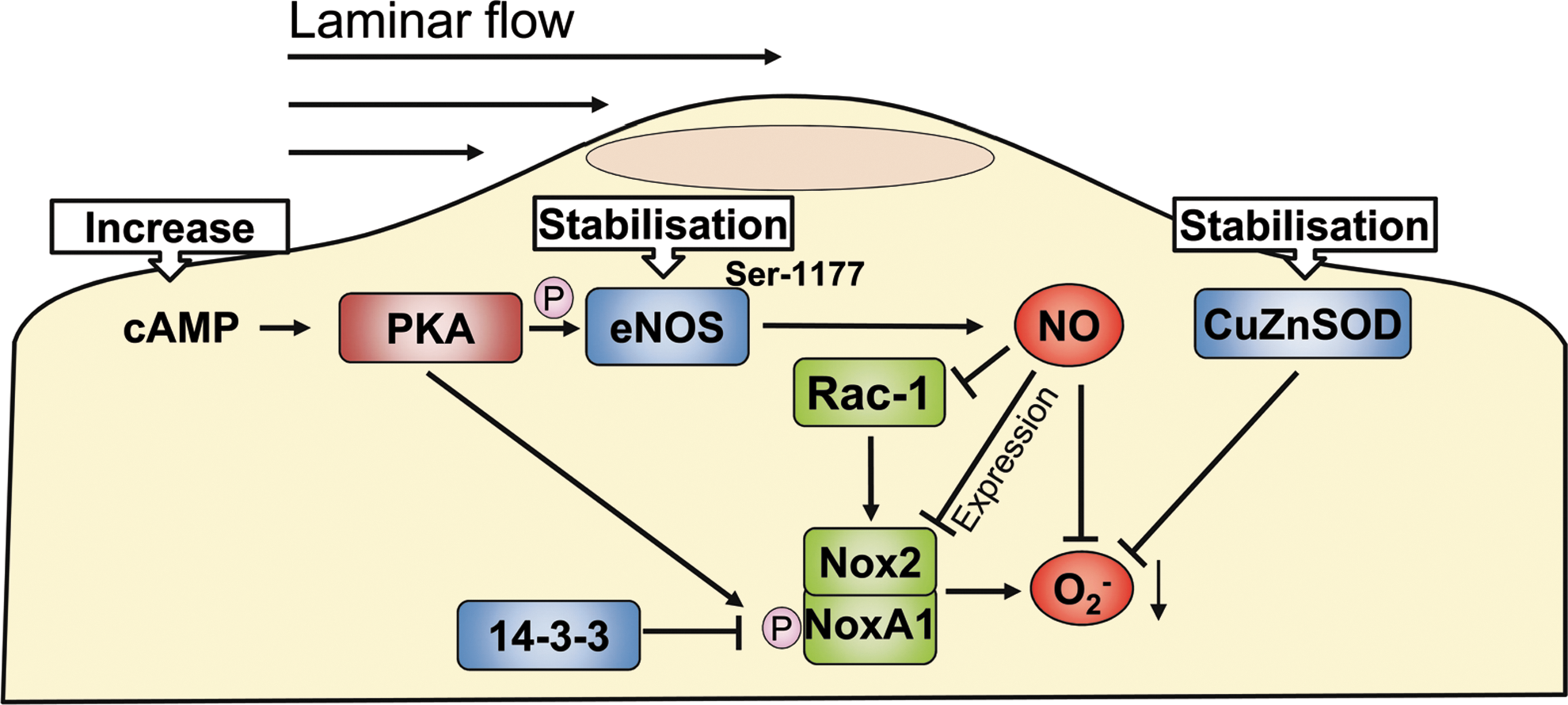
Under steady state conditions, unidirectional shear stress as present during continuous laminar flow is a low ROS condition. The immediate onset of laminar flow, however, activates the same pathways as continuous cellular stimulation during oscillatory flow (49) and likewise the cessation of laminar flow after prolonged exposure, yields a similar signal as oscillatory flow and thus, a transient increase in ROS formation. Both events are mediated by a Rac-1, p47phox-dependent activation of Nox2 (16, 25). Inhibition of Nox2 by the blocking peptide gp91ds-tat prevents the initial increase in ROS formation (30). During prolonged application of continuous unidirectional shear stress, eNOS and CuZnSOD protein expression increase and Rac1 activation, as consequence of adaptation of the cells to the new steady force profile, gradually returns to normal. Moreover, laminar flow decreases the expression of Nox2 (50). This effect is at least, in part, mediated by NO, as NO donors induce a similar downregulation of Nox2 in nonsheared endothelial cells and as the shear stress-induced downregulation of Nox2 is sensitive to NO synthase inhibitors (30). Interestingly, not only the expression of Nox2 but also that of Nox1 is attenuated by NO by a pathway involving cGMP, as demonstrated in rat mesengial cells under static conditions (95).
A significant part of the protective signaling elicited by laminar shear stress is mediated by the Gs-cAMP-protein kinase A-(PKA) axis. PKA phosphorylates the endothelial NO synthase at serine 1177, which increases the activity of the enzyme and thus, the subsequent formation of NO (10). The cAMP-PKA system also limits ROS formation as it reduces Nox1 expression and blocks its interaction with NoxA1, an activator of the enzyme also expressed in vascular cells (5). PKA-mediated phosphorylation of NoxA1 and subsequent recruitment of inhibitory 14-3-3 proteins underlie the acute effects on activity (61). With respect to expression, it was demonstrated that laminar flow downregulates BMP4 (51, 105), a main factor promoting Nox1 expression (105). Also, this cascade is mediated by cAMP as demonstrated in cultured cells and aortic organ culture (24) (Fig. 5).
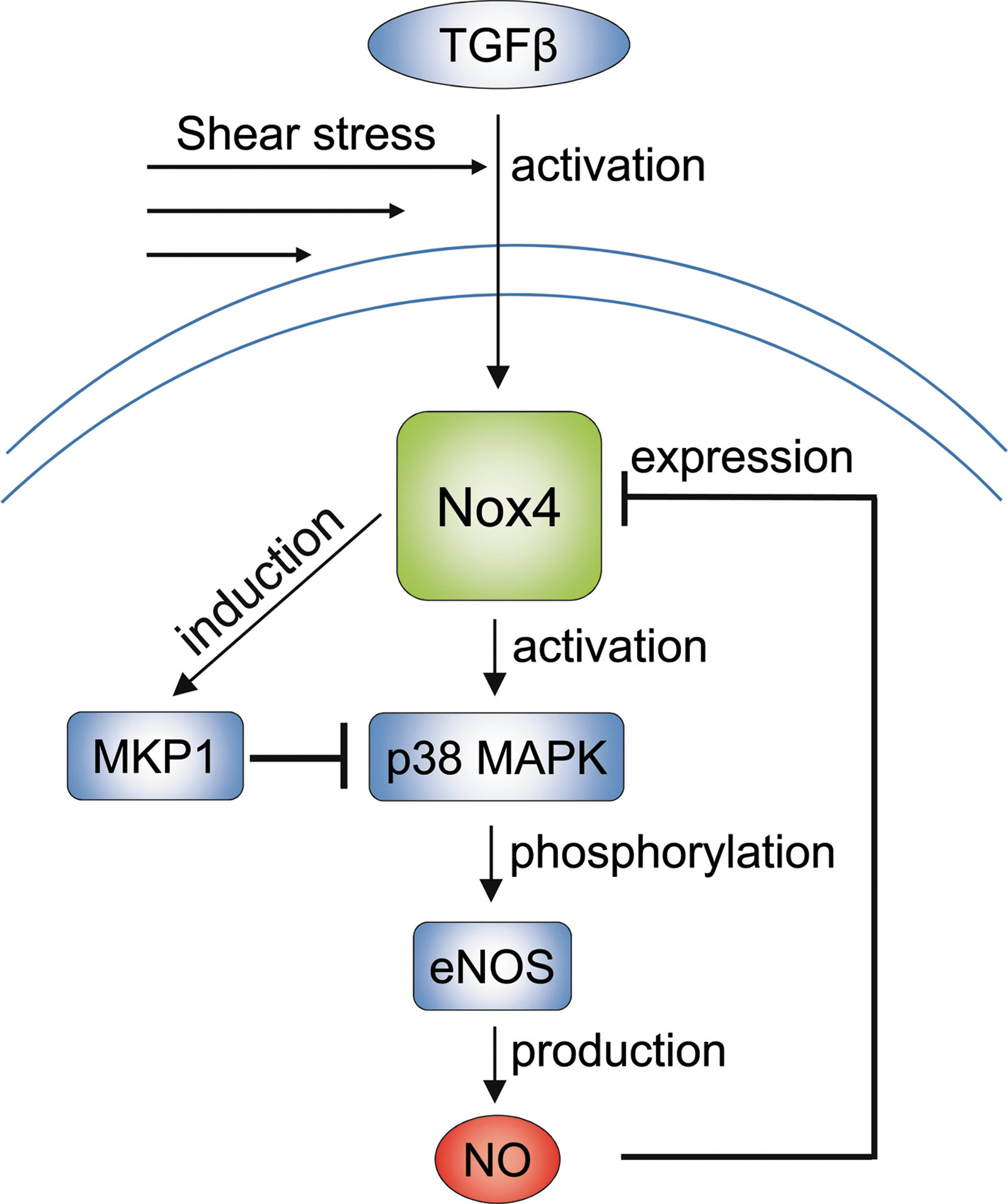
High levels of continuous shear stress induce the expression of the redox-sensitive phosphatase MAP kinase phosphatase (MKP)-1 in the mouse aorta and cultured endothelial cells and increased MKP-1 activity attenuates inflammatory activation (130). Although MKP-1 can be inactivated by oxidative stress, its expression is also induced by Nox4 and thereby limits ERK1/2 activation in response to mitogenic stimuli (101) and p38 MAP kinase activation in endothelial cells in response to laminar flow (130). Laminar flow as generated by a cone and plate device in bovine aortic endothelial cells and HUVECs results in the phosphorylation of p38 MAP kinase and the subsequent activation of eNOS. Importantly, this effect was not observed after siRNA-mediated knockdown of Nox4 (13). It is not completely understood how shear stress could activate Nox4. Shear stress, however, enhances basal TGF-β signaling (111) and TGF-β by a so far undetermined pathway can directly activate Nox4 (41). Latent TGF-β which is present in the blood or cell culture medium in turn is activated in response to shear stress, by a process which displaces the inhibitory complex of latency-associated peptide and latent TGF-β binding protein from the cytokine (4).
Interestingly, these observations may point towards the presence of two closed inhibitory feedback loops to limit shear-stress-induced NO production: Nox4 by means of MKP-1 induction limits p38 MAP kinase and NO itself by downregulation of Nox4 limits p38 MAP kinase activation. These data also support the concept that Nox4 contributes to the maintenance of NO production (23, 102) and this process could be of relevance for the midterm adaptation to shear stress in endothelial cells. (Fig. 6)
Flow Overload
Obviously, beyond a certain rate, laminar flow can be too high to be protective. Very high rates of continuous laminar shear stress activate cells and induce a stress response which is associated with an induction of Nox expression and an accumulation of Nox5 protein in HUVECs (125). The influence of high flow has been studied in animal models: Increases in blood flow in vivo increase vascular ROS production and lead to remodeling. In the carotid artery ligation model in pigs, the flow increase in the nonligated artery induced expression of Nox2, Nox4 as well as of p22phox and p47phox (75). Similarly, in the murine carotid artery shunt model, increases in blood flow were accompanied by an increase in ROS formation and remodeling and both effects were attenuated in p47phox knockout mice (20). Interestingly, genetic deletion of Nox2 had no impact on remodeling and ROS production suggesting that other Nox homologues like Nox1 could be involved in this process.
Exercise, Inactivity and Nox
In a complex, integrated view, also physical exercise imposes mechanical stress on cells. Obviously, the biological effects of exercise are not just force-dependent and among many other factors also involve hormonal regulation, changes in energy state and traumatic affliction of cells. Nevertheless, similarly as for the direct, well defined stimulation in model systems of strain and flow, a U-shaped relation between exercise intensity and ROS level has been documented (96). Importantly, exercise not only affects ROS generation but also alters the expression of antioxidant enzymes. The induction of their genes is mediated by an exercise-induced activation of the nuclear factor erythroid 2-related factor-2 (Nrf2) pathway (85) and Forkhead-box-protein O (Foxo) transcription factors (107). Regular nonexhaustive physical exercise reduces ROS formation and decreases NADPH oxidase expression and activity (62), even in humans (2). Indeed, aging-induced endothelial dysfunction is less frequent in subjects undertaking regular physical exercise and this effect is associated with a reduced vascular expression of Nox subunits (94). Similarly, mild exercise as made possible by providing a running wheel to mice, reduced Nox expression, vascular ROS formation, and improved NO availability (31). Overexercise, exhaustion, and physical inactivity (67) are in contrast situations of high ROS formation. Severe exhaustion leads to trauma, inflammation and apoptosis and increased ROS formation by NADPH oxidases (29, 60, 96, 122). But also physical inactivity (67) is an inflammatory state associated with apoptosis, atrophy and autophagy. Among many mediators, tumor necrosis factor-1 is released, which not only promotes mitochondrial ROS formation but also induces and activates Nox1 and Nox2 (8). A link between Nox4 and autophagy in general has been presented (128) but so far it is unknown whether this NADPH oxidase contributes to ROS formation during physical inactivity.
Consequence of Nox-Mediated Mechano-Transduction
The physiological consequences of mechano-transduction induced ROS production by Nox enzymes, again, are best studied in the vascular system (71). Although general concepts linking ROS formation to vascular responses have been developed, the contribution of individual Nox proteins to specific processes of the vascular system in vivo are less defined. Nox4 has not yet been studied and whether there are differences between the action of Nox1 and Nox2 is unknown. Most studies published to date used either nonselective inhibitors or p47phox knockout mice. Deletion of this subunit, however, most likely affects Nox1 as well as Nox2.
From the whole organ view, ROS are important signaling molecules of many pathways and often rather act as modulators affecting the signal propagation than specifically interfering with a system (12). Consequently, the decision whether Nox proteins are beneficial or harmful (to put this into a more clinical perspective) is dependent on the experimental context. Indeed, positive and negative effects of mechano-transduction may involve increased Nox activity.
Consequences of disturbed flow
This is best illustrated for reactions occurring in response to altered blood flow conditions: disturbed flow clearly has been linked to arteriosclerosis development as consequence of vascular inflammation. Genetic deletion of p47phox, which is required to activate Nox2 or Nox1, prevents inflammation as well as monocyte adhesion. Loss of p47phox reduces arteriosclerosis development (53), attenuates the accelerated arteriosclerosis in the carotid artery ligation model (87), and prevents outward remodeling in the mouse aortic aneurysm model (113). These observations link mechno-transduction by Nox proteins to promotion of vascular diseases. Nevertheless, deletion of p47phox also prevents outward remodeling in a carotid artery shunt model (20) exemplifying a beneficial action of mechano-stimulation. Although proinflammatory, high flow-induced outward remodeling is a very important beneficial physiological response of the vascular system and the basis of collateral artery growth and shunt maturation (42, 99). Mechanistically, high flow promotes endothelial inflammation and activation which results in an accumulation of monocytes. These cells not only themselves contain high levels of Nox proteins, they also secrete proteases, which are needed for the outward remodeling and release factors that stimulate matrix production and smooth muscle cell proliferation and migration (42, 99).
Consequences of low flow
A similar Janus-faced situation is also present for no or low flow conditions. The subsequent high ROS production, on one hand increases neo-intima formation in a Nox-dependent manner as demonstrated by the partial carotid artery ligation model (87). During developmental vasculogenesis, this presumably detrimental process is essential for vascular patterning: no flow leads to vascular obliteration and endothelial cell apoptosis, which is required for the maturation of the vascular network and the resolution of primary capillary plexus.
Consequences of high strain: hypertrophy
A similar situation is present in response to stretch-induced responses: Stretch-induced ROS production takes clearly part in the physiological remodeling process as it mediates cellular hypertrophy and matrix remodeling. H2O2 promotes smooth muscle cell proliferation (98) and Nox-dependent ROS formation contributes to the hypertrophic effects of hypertensive agents like angiotensin II and endothelin (66). Few studies, however, attempted to dissect the direct pressure-induced changes from those indirectly induced by angiotensin II. It is clear that hypertrophic and profibrotic effects of angiotensin II are, in part, mediated by Nox activation (66). In contrast, pressure-induced Nox-induction (119) and activation (120) have been demonstrated but the effect of inhibition or genetic deletion of Nox enzymes on the functional consequence for the vascular wall has not yet been studied. In the heart, however, hypertrophic and profibrotic effects in response to pressure overload are Nox-mediated (86).
Consequences of high strain: matrix remodeling
Not only matrix deposition but also the degradation of matrix contributes to remodeling. Indeed, induction of matrix metalloproteinases in response to stretch in smooth muscle cells is Nox-dependent (39). Cardiac rupture observed in response to excessive concentrations of angiotensin II is Nox1-dependent (36) and angiotensin II-induced aneurysm formation is attenuated by deletion of p47phox (113). Finally, Nox enzymes contribute to cardiac rupture after myocardial infarction (3). Although all these studies provide a robust link between Nox proteins and protease activity, the role of stretch as a driving force in the processes is less clear. Angiotensin II has a massive hyperinflammatory effect which may be sufficient to drive the models in a pressure-independent manner. In the cardiac rupture study, a prominent role of bone marrow-derived NADPH oxidase and thus, leukocyte-driven inflammation was also demonstrated (3). Thus, more studies employing localized increases in pressure, like venous bypass models or responses of the carotid artery in the aortic constriction model are needed, ideally in bone marrow chimeras of Nox proteins. These experiments should help to define the exact role of localized Nox activation in matrix remodeling.
Conclusions
Nox-driven ROS formation plays an integral role in cellular mechano-transduction. The adaptive processes of the cell and the whole body in response to altered mechanical load involves Nox proteins and can lead to beneficial or deleterious remodeling depending on the pattern and the type of stress. Although Nox proteins contribute to physiological signaling, as present in vascular remodeling, mechano-transduction largely links Nox proteins to conditions of vascular pathology. Particularly, Nox1 and Nox2 are induced and activated by mechano-stimuli associated with vascular disease development like oscillatory low or high strain rates. Thus, it is difficult to differentiate the effects of Nox1 and Nox2 in mechano-transduction. Accordingly, protective mechano-stimuli such as laminar flow decrease the expression and activity of both enzymes, Nox1 and Nox2. The calcium-dependency of Nox5 makes this special NADPH oxidase an attractive modulator of myogenic responses but more studies have to be performed on this Nox homologue to define its function in mechano-transduction. Similarly, only few studies linked mechano-transduction to Nox4 and additional studies will be needed to define whether this enzyme is a beneficial or detrimental mediator of mechano-transduction.
Footnotes
Acknowledgment
This work was supported by the DFG Sonderforschungsbereich 815 and 834 and the ECCPS Excellence Cluster Cardio Pulmonary System.