
Abstract
Introduction
W
Nitrite binds to and is reduced at the molybdenum site of mammalian sulfite oxidase (SO) to form nitric oxide (NO), and, thus, contributes to the mammalian nitrate-nitrite-NO signaling pathway.
Such proton and electron transfer reactions can be catalyzed by redox-active hemoproteins, such as hemoglobin, myoglobin, neuroglobin, cytoglobin, and plant hemoglobins (10, 19, 40, 44, 45, 47), and can also be catalyzed by molybdenum cofactor (Moco−)-containing enzymes, such as xanthine oxidase, aldehyde oxidase, and mARC (2, 26, 41). However, despite evidence that these Moco enzymes can contribute to nitrite-dependent signaling in rodents, studies in humans have shown that infusions of allopurinol or oxypurinol, specific xanthine oxidoreductase inhibitors, do not sufficiently block nitrite-mediated vasodilation (12). Therefore, we evaluated sulfite oxidase (SO), a third Moco-containing enzyme, as a potential nitrite reductase and further examined the catalytic mechanism in the purified enzyme system.
In contrast to xanthine oxidase and aldehyde oxidase, eukaryotic nitrate reductase in plants and SO in animals and plants represent a distinct second family of Moco enzymes that catalyze oxygen atom transfer reactions (43). For plant nitrate reductase, nitrite-dependent NO synthesis has been reported (53). Given the high structural similarity between nitrate reductase and SO (9), it is likely that nitrite would react with the reduced molybdenum center of SO. SO is a homodimer (110 kDa) with each monomer containing an N-terminal cytochrome b 5-type heme, a central Moco domain, and a C-terminal dimerization domain. It is located in the mitochondrial intermembrane space of mammalian cells, where it aerobically converts sulfite to sulfate while concomitantly reducing two molecules of cytochrome c, representing the last step in the enzymatic catabolism of cysteine. Mechanistic studies indicate that during the sulfite-SO-cytochrome c catalytic cycle, movement between the molybdenum and heme domain is required to enable efficient single-electron transfer from molybdenum via the heme b 5 cofactor to cytochrome c (21).
Since nitrite-dependent NO synthesis is enhanced under hypoxia, we hypothesized that under hypoxic or ischemic conditions a fully reduced system would serve to switch sulfite-mediated cytochrome c reduction in mitochondria to a nitrite reduction reaction. Here, we report that SO can function as source of bioactive NO by reducing nitrite at the molybdenum domain, coupled to a one-electron oxidation of Mo(IV)-to-Mo(V).
Results
Reduced human SO converts nitrite to NO
To determine whether prereduced SO reduces nitrite to NO at physiological concentrations of nitrite (8, 14), we performed measurements in a purge vessel under anaerobic conditions (helium gas purging through the reaction mixture). Rates were determined by integration of the peak NO generation rate over a 12-second interval (detailed methods are provided in the Supplementary Data; Supplementary Data are available online at
Our experiments used recombinant human SO ([Mo]=0.63 μM), and phenosafranine (one- or two-electron donor) or sulfite (two-electron donor) as reducing substrates. To study the kinetics of SO-dependent nitrite reduction, first the concentration dependence of nitrite was assessed in the presence of a fixed concentration of phenosafranine (40 μM) and human SO. A concentration-dependent increase in the rate of NO formation was observed for nitrite (0.02–100 mM) at pH 7.4 and fitted to the Michaelis–Menten equation (Fig. 1). When concentrations of nitrite were above 4 mM, the NO detected was more than 1000 mV at the high-sensitivity mode, and, thus, the low-sensitivity mode was set to measure NO production when nitrite was in the range of 5–100 mM. Measured vmax, K m, and k cat values are 34 nmol·s−1·mg−1, 80 mM, and 1.9 s−1, respectively. At pH 6.5, the rate of NO formation also increased with the concentration of nitrite, although the kinetic parameters were not determined (Supplementary Fig. S3).
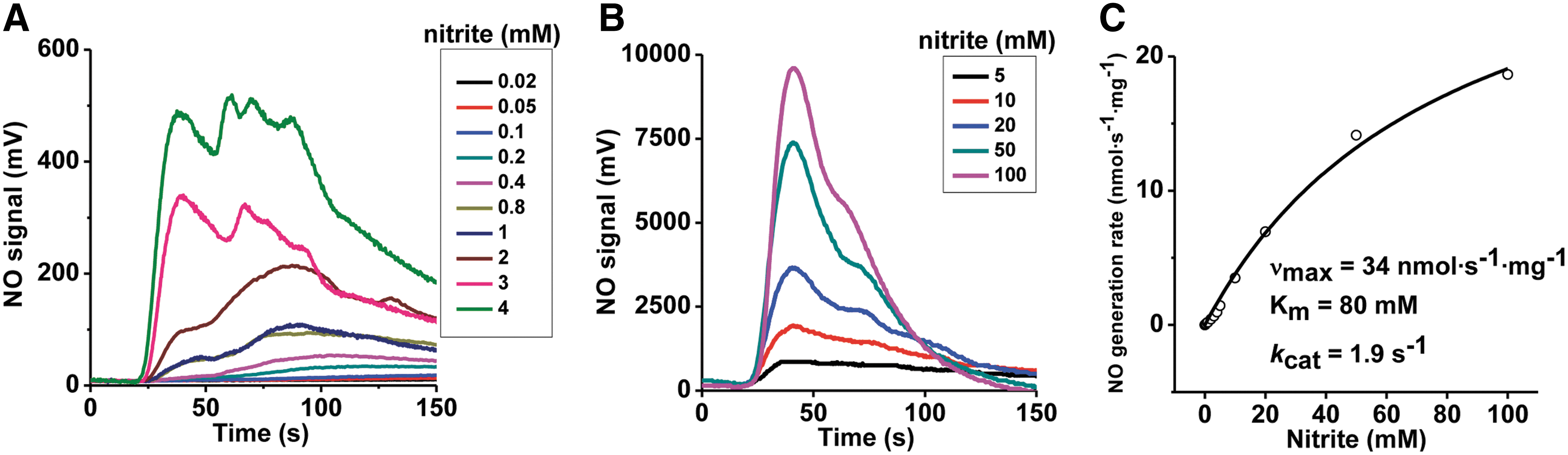
It is notable that SO only catalyzes a single turnover reaction in the presence of sulfite, with no steady-state NO formation observed. However, the relationship between the observed rates and the concentration of nitrite shows a noticeable deviation from a lineal relationship and appears to indicate a possible reaction complex formation. We interpret this fact as the result of a reaction as follows:
where SO(IV) and SO(V) represent SO proteins with their molybdenum atom in the oxidation states IV and V, respectively (25, 27). This kind of single turnover reaction can produce kobs versus [substrate] plots similar to Michaelis–Menten (17). We have renamed the calculated parameters Kd and k et, to indicate that they correspond to an apparent dissociation constant and an electron transfer rate for reactions in the presence of sulfite.
Since sulfite is the only known physiological reducing substrate for SO, NO generation was next studied with sulfite as the reductant. In the absence of SO or in the presence of only sulfite or nitrite, NO formation was not detected; while the combination of nitrite and SO in the presence of either sulfite or phenosafranine resulted in significant NO generation, suggesting redox coupling of substrate oxidation to nitrite reduction (Fig. 2A, B). It is important to note that the addition of nitrate does not lead to NO formation, suggesting that SO is not a nitrate reductase. NO generation with phenosafranine is ∼10-fold greater than that observed with sulfite (Fig. 2B), indicating that phenosafranine is a more efficient electron donor for nitrite reduction. This different reactivity with a strict two-electron donor, sulfite, versus a one- or two-electron donor such as phenosafranine prompted us to study the reaction mechanism in greater detail, as discussed next.
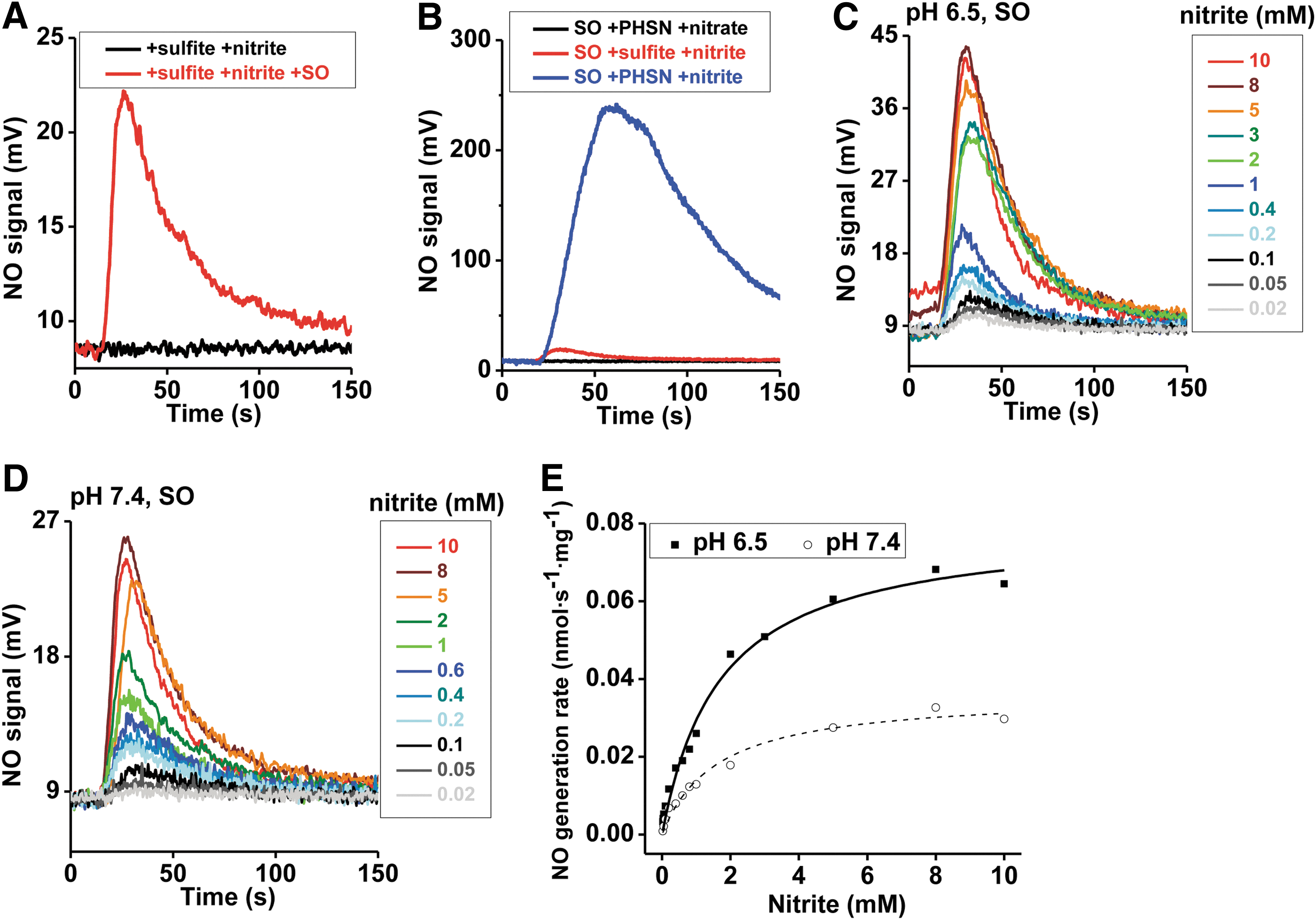
To study the kinetics of SO-dependent nitrite reduction, first the concentration dependence of nitrite was assessed in the presence of a fixed concentration of sulfite (5 μM) and human SO. A concentration-dependent increase in the rate of NO formation was observed for nitrite (0.02–10 mM), and a plot of k obs versus nitrite indicated a hyperbolic fit similar to Michaelis–Menten kinetics for both pH 6.5 and 7.4 (Fig. 2C–E), with vmax, K d, and k et values of 0.0361 nmol·s−1·mg−1, 1.6 mM and 0.002 s−1 at pH 7.4 and 0.0796 nmol·s−1·mg−1, 1.7 mM and 0.0044 s−1 at pH 6.5, respectively. The increases of vmax and k et with the lower pH are not unexpected based on Equation 1 and have been reported for the nitrite reduction reactions catalyzed by hemoglobin, myoglobin, and neuroglobin (10, 40, 46). However, the NO generation rates only increased for about three-fold per unit change of pH, instead of the well-known 10-fold increase for a reaction of heme globins with nitrous acid. The reasons for different influences of pH, which was also observed in other Mo-dependent enzymes (25 –27), are unknown but may involve a direct binding of nitrite or protonation of active site residues at the Mo center. As discussed and evaluated next, the low k et values using sulfite as the reductant suggest that this may not be a catalytic reaction.
When sulfite concentration was examined for effects on NO formation, no significant differences in the rate of nitrite reduction to NO were observed (2 μM–5 mM, pH 6.5) (Supplementary Fig. S4A). However, at pH 7.4, NO formation rates were decreased at sulfite concentrations greater than 1 mM, suggesting a substrate-mediated inhibitory effect on nitrite reduction (Supplementary Fig. S4B). These results suggest that both nitrite and sulfite react at the molybdenum active site. Finally, all results were reproduced with mouse SO (Supplementary Table S1 and Supplementary Fig. S4C–H).
Nitrite reduction to NO at the molybdenum domain of SO
To further assess whether nitrite is reduced at the molybdenum (Mo) center, we next evaluated reactions of nitrite with isolated recombinant Mo- and heme-domains of human SO. Recombinant Mo-domain was used for kinetic studies of NO generation as a function of nitrite or phenosafranine or sulfite concentration, and for quantification of nitrite reduction in the presence of phenosafranine or sulfite.
The concentration dependence of nitrite was first determined at a fixed concentration of 40 μM phenosafranine. On addition of 0.25 μM Mo-domain to nitrite and phenosafranine, NO formation was measureable with the lowest detectable NO concentration at 5 μM nitrite at pH 7.4 (Fig. 3A, B). Michaelis–Menten kinetics were obtained showing vmax, K m, and k cat values with 112 nmol·s−1·mg−1, 31 mM, and 4.7 s−1 at pH 7.4, respectively, with significantly higher vmax and k cat, and lower K m values than observed with the holo-enzyme (Supplementary Table S1). At pH 6.5, when nitrite was in the range of 1–400 μM, NO production by Mo-domain was faster than at pH 7.4 (Supplementary Fig. S5). When sulfite was used as the reducing substrate, the rates showed a similar dependence toward nitrite but were much lower than those observed in the presence of phenosafranine. The rates observed at pH 6.5 were higher than those at pH 7.4, as observed in the presence of phenosafranine (Fig. 4A–C), with vmax, K d, and k et values as 0.179 nmol·s−1·mg−1, 0.973 mM and 0.0075 s−1 at pH 7.4, and 0.323 nmol·s−1·mg−1, 0.496 mM and 0.0136 s−1 at pH 6.5.
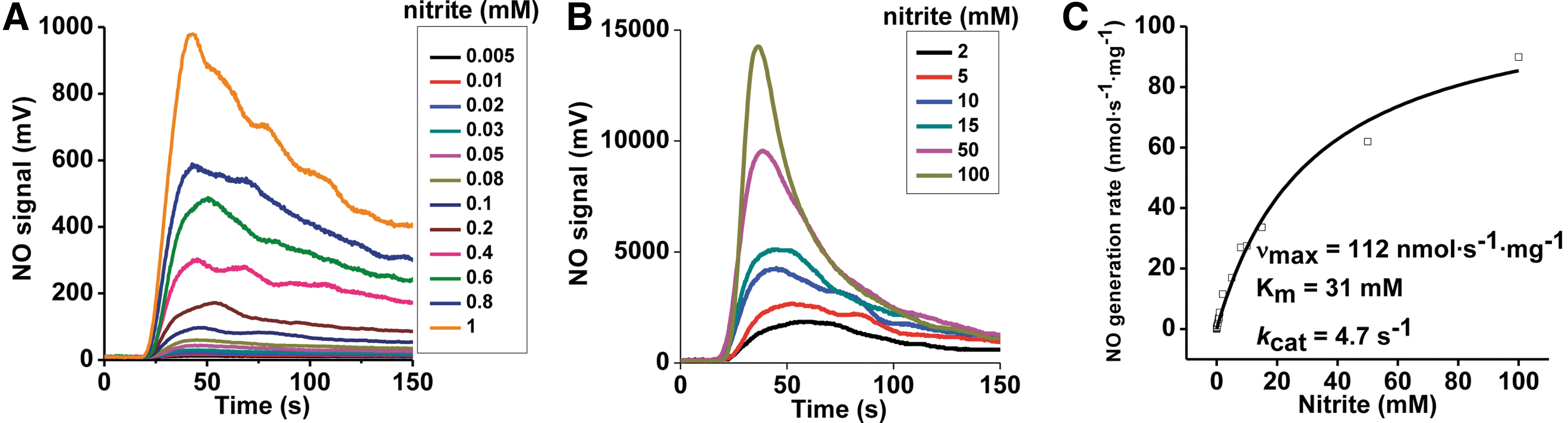
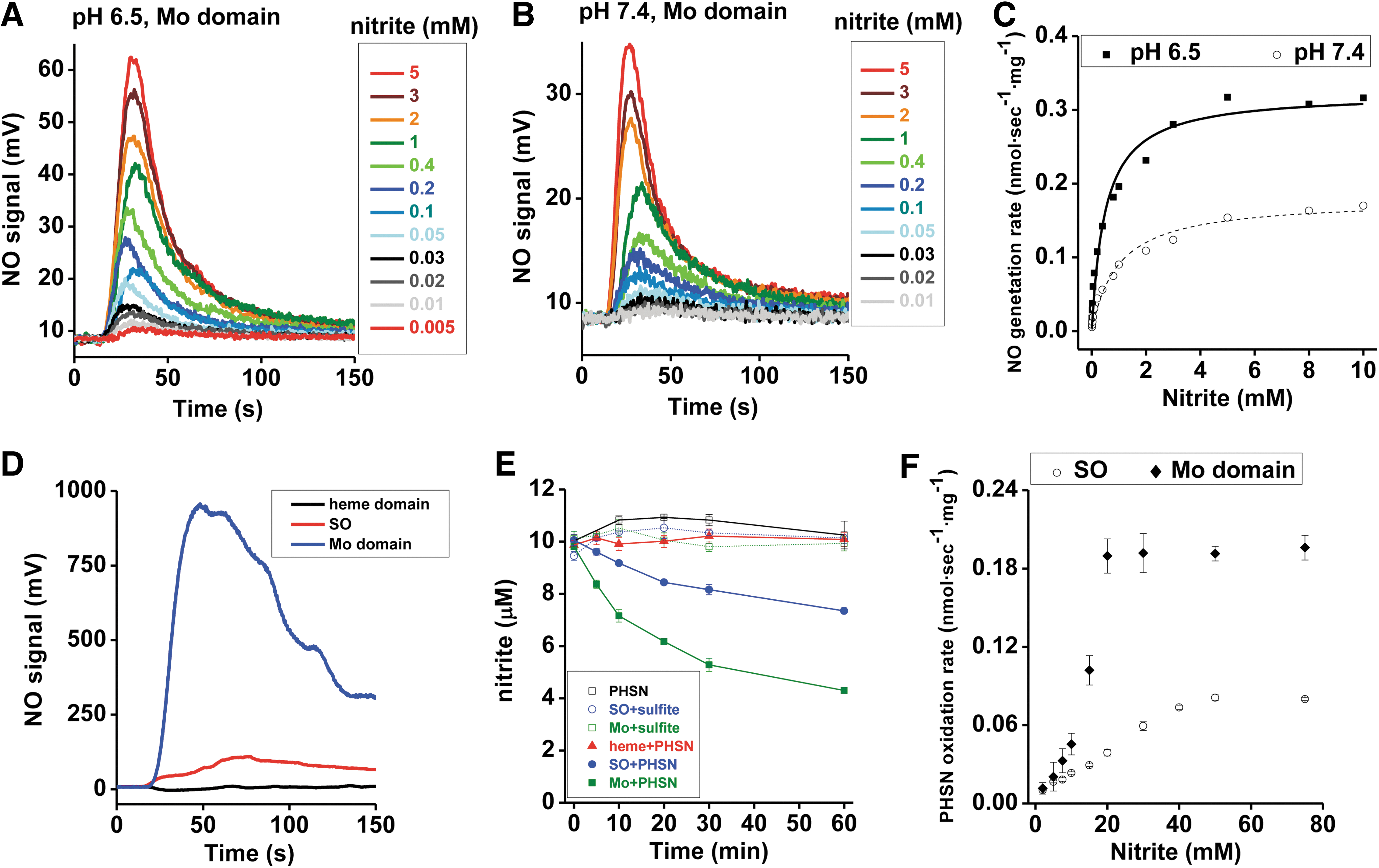
Our studies using phenosafranine or sulfite as reducing substrate show much faster nitrite reduction to NO by the Mo-domain than by holo-SO (Supplementary Table S1). Mechanistically, these data suggest that nitrite reacts with the Mo-domain, and that electron transfer between Mo- and heme domains and/or steric hindrance of the heme domain reduces the reaction rates with nitrite by approximately a factor of three. All results could be reproduced with the mouse Mo-domain using sulfite as the reductant (Supplementary Figs. S6 and S7).
To investigate the inhibitory effect of the heme domain, we evaluated rates of NO formation and nitrite reduction in the presence of the heme domain, the Mo-domain, and holo-SO at the same concentrations (Fig. 4D, E). When 40 μM phenosafranine was mixed with 1 mM nitrite in an anaerobic vessel, addition of the isolated heme domain did not generate NO; whereas addition of the Mo-domain and holo-SO resulted in NO formation (Fig. 4D), suggesting that the heme domain alone does not reduce nitrite to NO. When 10 μM nitrite was mixed with 50 μM sulfite under anoxic conditions, nearly zero nitrite was reduced in 60 min in the presence of holo SO or the Mo-domain (Fig. 4E). However, when 50 μM phenosafranine replaced sulfite, around 25% and 55% nitrite were reduced anaerobically in the presence of holo-SO and the Mo-domain, respectively, while the heme domain did not reduce nitrite (Fig. 4E). Furthermore, nitrite-mediated phenosafranine oxidation by both the Mo-domain and holo-SO confirmed that the catalytic activity of the Mo-domain is ∼2.5-fold greater than observed with the holoenzyme (Fig. 4F). All these results provide further evidence that nitrite reduction occurs at the Mo-domain, with enhanced reaction rates in the presence of phenosafranine compared with sulfite.
Effects of the heme domain on rates of nitrite reduction
To determine whether either the heme-iron center or the heme domain itself negatively modulates NO production within the holoenzyme, we quantified the NO generation rates in the presence of a heme-deficient mouse SO, H119A/H144A variant (Fig. 5 and Supplementary Fig. S8A–C). Loss of His119 and His144, which coordinate heme iron, results in an apo-heme domain, with an overall preservation of the enzyme structure as confirmed by circular dichroism (CD) spectroscopy (Fig. 5A). Functionally, the H119A/H144A SO and holo-SO exhibited ∼30% and 20% of the NO formation activity of the isolated Mo-domain, respectively, suggesting that there is both a steric and an electronic inhibitory effect from the heme domain on Mo-domain-dependent nitrite reduction (Fig. 5B and Supplementary Fig. S8A–C).
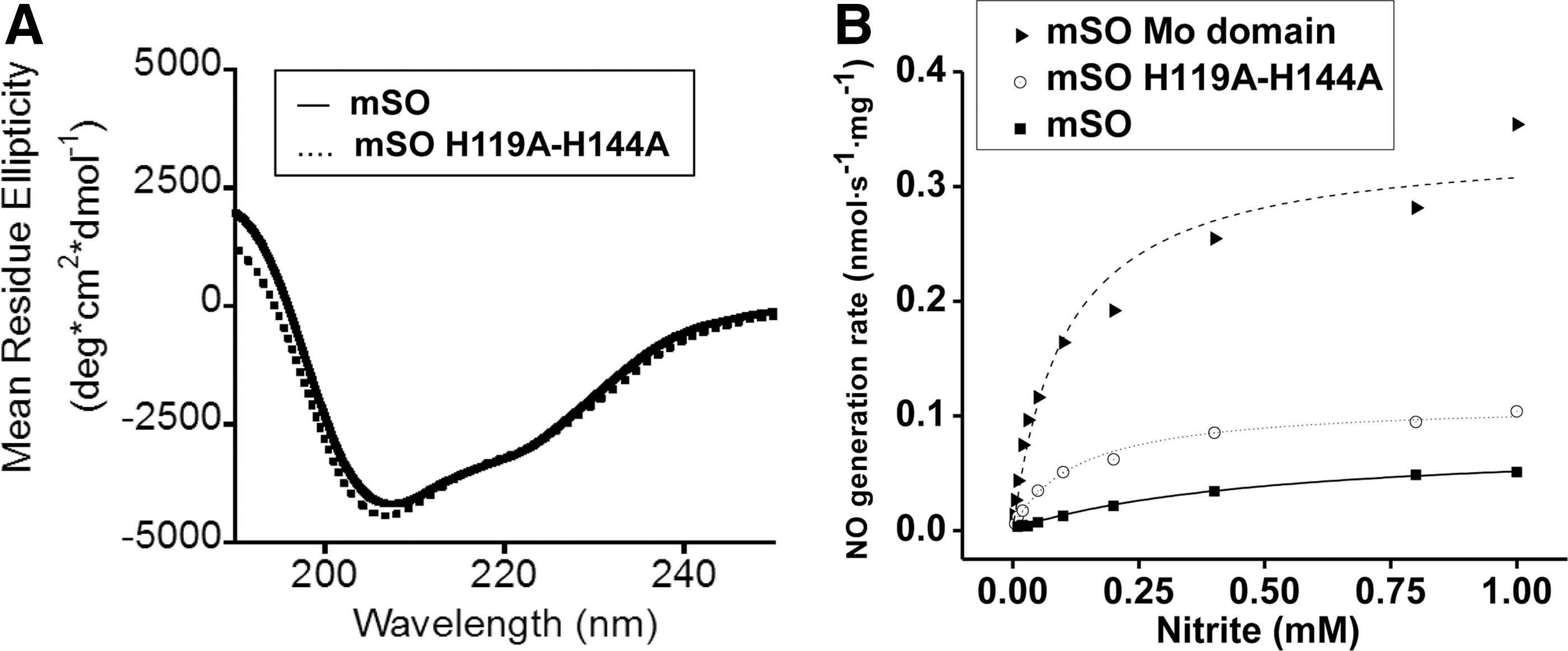
Catalytic Mo(IV) to Mo(V) nitrite reduction cycle
Based on the observation that phenosafranine, a one- or two-electron donor, efficiently catalyzes nitrite reduction by SO, while sulfite (a strict two-electron donor) does not, we studied reactions of prereduced SO with nitrite under anaerobic conditions, in the presence or absence of sulfite. Importantly, addition of sulfite did not promote additional NO generation beyond that of the fully prereduced SO (Fig. 6A). This result suggests that sulfite cannot enter into a catalytic cycle with nitrite reduction, that is, sulfite only modulates one turnover of nitrite reduction. On the other hand, addition of phenosafranine to reduced SO increased NO production, suggesting that a one-electron donor can catalyze nitrite reduction by SO, forming a redox cycle of phenosafranine oxidation and nitrite reduction.
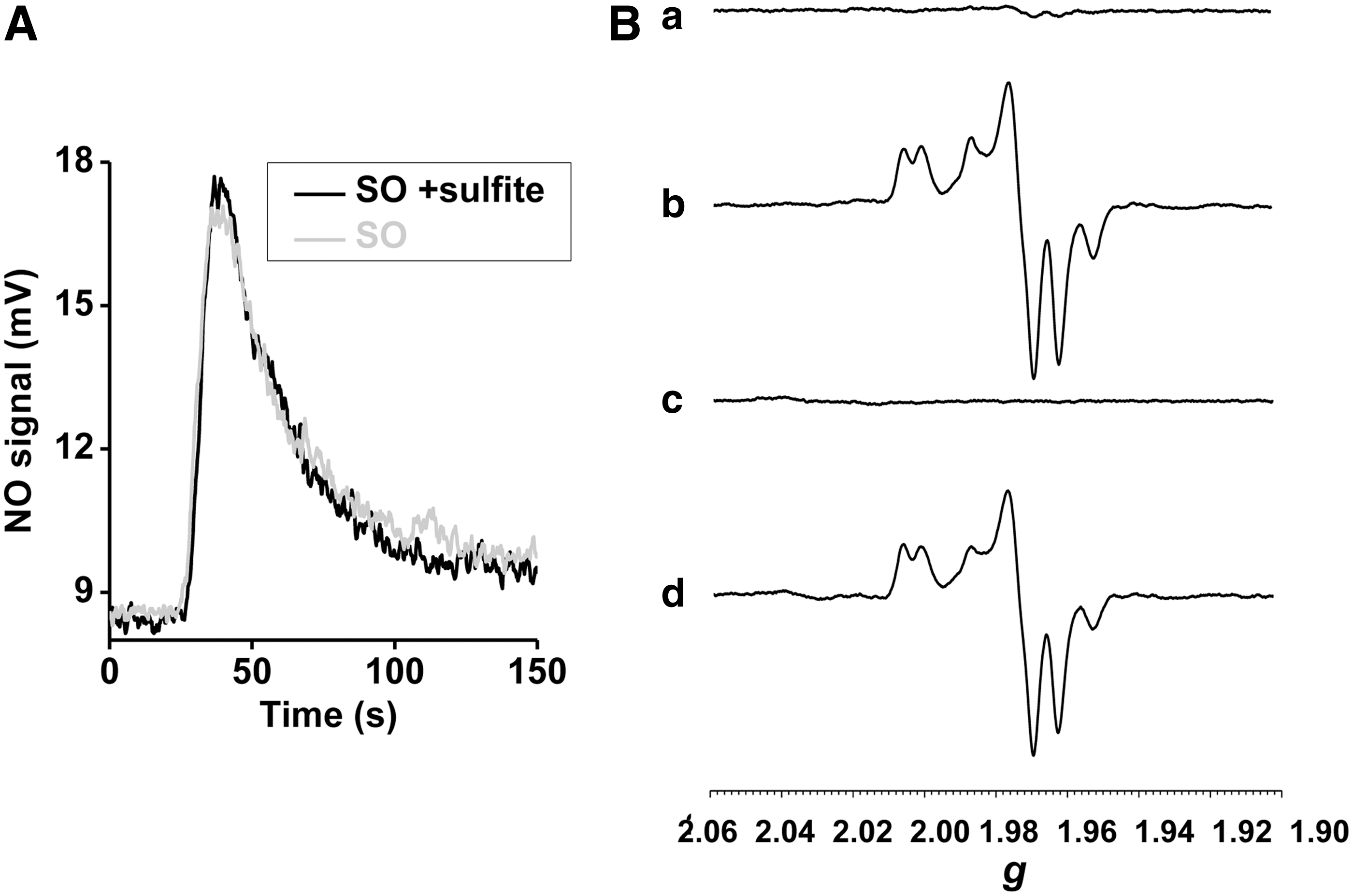
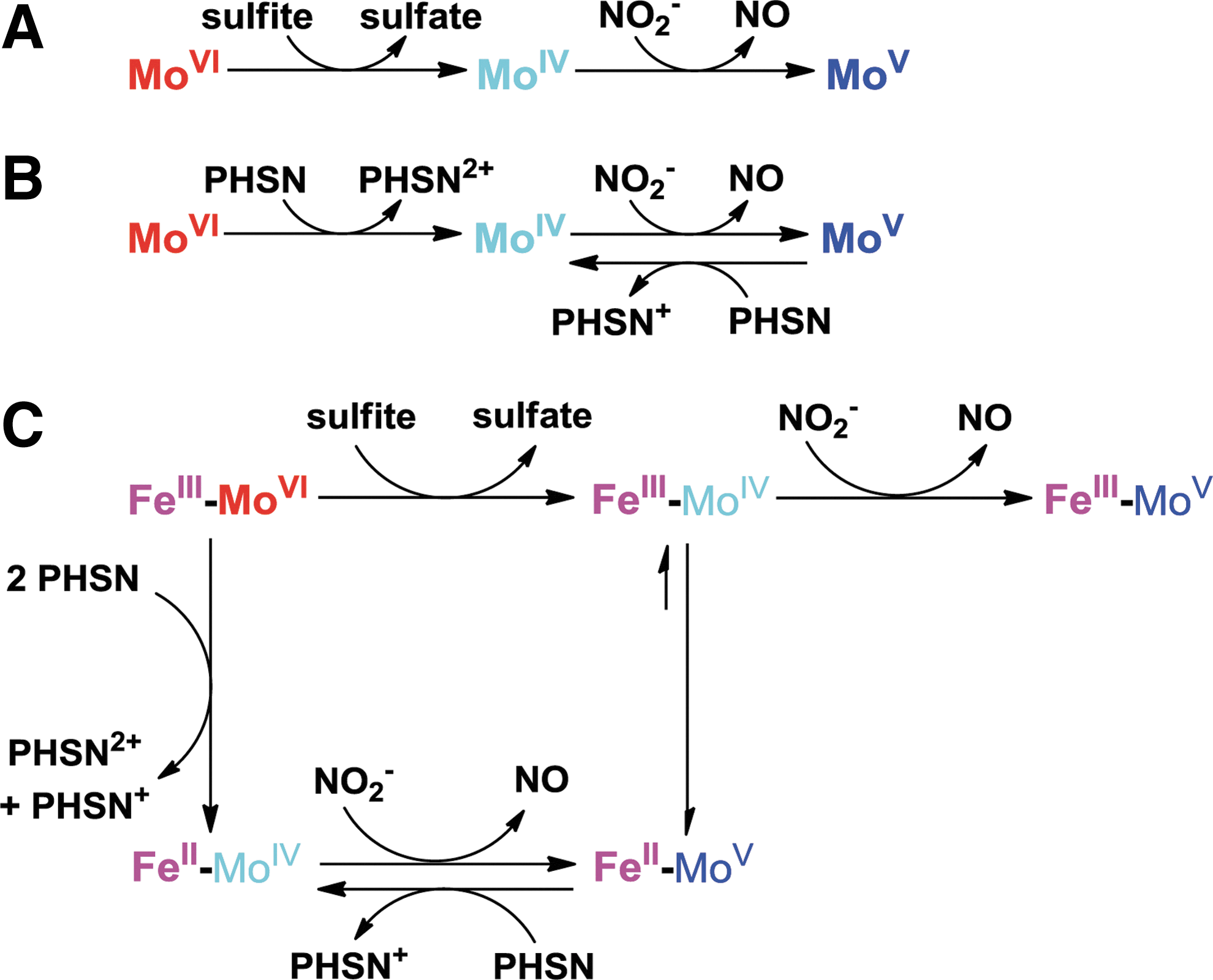
We applied electron paramagnetic resonance (EPR) spectroscopy to monitor changes at the Mo center during its reaction with sulfite, nitrite, and phenosafranine. When 20 μM Mo-domain was mixed with 100 μM sulfite, the Mo center was EPR silent, reflecting the anticipated reduced Mo(IV) state (Fig. 6B-(a)). When excess nitrite (1 mM) was added and incubated for 1.5 h, a stable Mo(V) spectrum appeared (23) and no further change was observed (Fig. 6B-(b)), indicating a one-electron oxidation but inability to convert to the Mo(VI) oxidation state. On addition of 50 μM reduced phenosafranine to the solution and quick freezing afterward (15 s), the Mo(V) spectrum disappeared, suggesting a conversion of Mo(V) back to the EPR-silent Mo(IV) (Fig. 6B-(c)). On thawing and incubation with original added nitrite (1 mM) again for another 1.5 h, more than 80% of the Mo(V) signal was recovered (Fig. 6B-(d)). These data suggest that Mo(IV), formed initially from the two-electron reduction of Mo(VI) by sulfite, is then oxidized to Mo(V) by nitrite (model shown in Fig. 7). Importantly, nitrite cannot further oxidize Mo(V) to EPR-silent Mo(VI). Since sulfite cannot reduce Mo(V) to Mo(IV), the reaction stops at Mo(V). Addition of phenosafranine is able to reduce Mo(V) to Mo(IV), enabling nitrite to be again reduced (Fig. 7). These data, in aggregate, explain the lack of redox cycling of nitrite and sulfite and evident catalytic cycling of nitrite and the one-electron donor phenosafranine (Fig. 7).
SO catalyzes nitrite-NO-cGMP signaling in human fibroblasts
Fibroblasts isolated from patients with Moco deficiency (MOCD), which produces inactive SO, xanthine oxidase, and aldehyde oxidase, as well as fibroblasts from an SO-deficient (SOD) patient were used to study hypoxic nitrite exposure on cGMP generation. Control experiments were performed using fibroblasts from healthy individuals cultured and treated under identical hypoxic conditions (1% O2). SO expression in MOCD or SOD fibroblasts was not detectable when 120 μg protein was loaded for Western analysis assay (Fig. 8A), indicating the deficiency of SO in patients' cells.
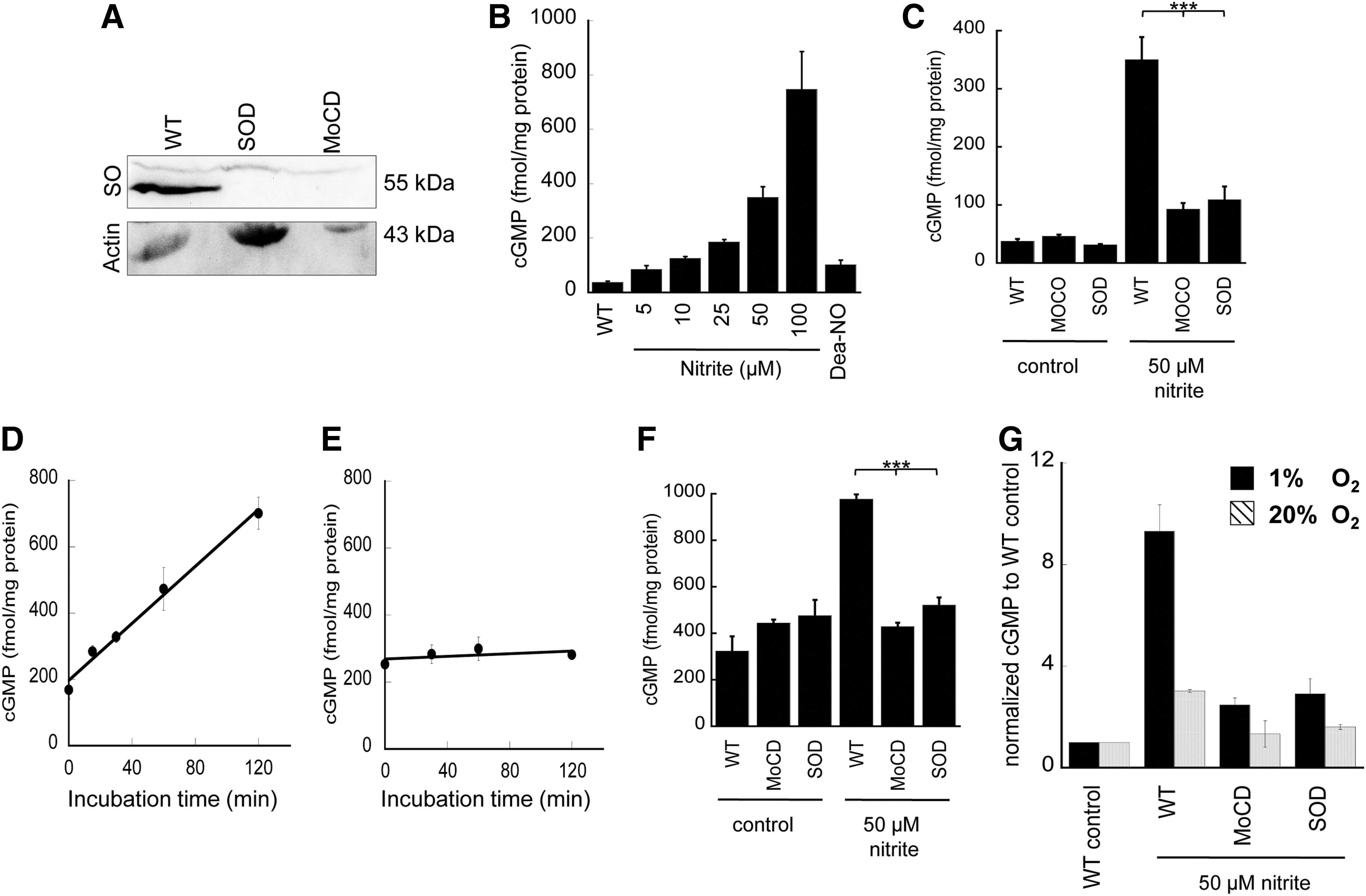
Control experiments were performed using fibroblasts from healthy individuals cultured and treated under identical hypoxic conditions (1% O2). In the presence of an NO donor, DEA-NONOate (DEA-NO), cGMP levels significantly increased (Fig. 8B). Addition of nitrite (5–100 μM) to the medium produced a dose-dependent increase in cellular cGMP (Fig. 8B). For fibroblasts from either Moco- or SO-deficient patients treated with 50 μM nitrite, the cGMP generation was reduced by approximately 80% (Fig. 8C), indicating a classic nitrite-NO-soluble guanylyl cyclase-cGMP response. A time-dependent exposure of wild-type fibroblasts to nitrite showed a linear increase of cGMP levels over time (120 min) with a rate of 4.3 fmol cGMP per mg and minute (Fig. 8D). In contrast, SO-deficient cells showed no significant cGMP production (Fig. 8E) in the same time frame. Similar exploration of cGMP production was performed under normoxia (20% O2). The basal level of cGMP at 20% O2 is higher than at 1% O2 (Fig. 8F), which indicates higher oxygen availability for NO synthase-dependent NO formation in fibroblasts under normoxia. Treatment with nitrite increases cGMP production in fibroblasts even at 20% oxygen, but the fold increase was less under normoxia compared with hypoxia (Fig. 8G), which is also supported by less NO generation under normoxia detected in the NO analyzer (Supplementary Fig. S9). Most importantly, the effect of nitrite on cGMP formation was abrogated in the Moco- or SO-deficient patient's fibroblasts (Fig. 8F). These results demonstrate that SO represent the primary Moco-dependent enzyme required for enzymatic conversion of nitrite to NO in human fibroblasts, given that the observed nitrite-dependent cGMP formation is impaired in SO-deficient cells.
Discussion
Mammals utilize two pathways to produce NO and regulate blood pressure and blood flow: the L-arginine-to-NO pathway and the nitrate-to-nitrite-to-NO pathway (29). These NO biosynthesis pathways respond differently to oxygen tension. The arginine-to-NO pathway requires oxygen as a substrate for NO formation; conversely, the nitrate-to-nitrite-to-NO pathway is potentiated under hypoxic conditions. The upstream pathways for conversion of nitrate to nitrite have been clearly defined, as earlier studies first described the involvement of this pathway in the generation of NO in the stomach, a process shown to be important in regulating stomach mucosal integrity, mucosal host defense, and later mucosal blood flow (5, 6, 28). Since the nitrate-to-nitrite-to-NO pathway exhibits increased potency under lower oxygen tensions, a role for nitrite as an effector of hypoxic signaling and vasodilation has been considered (10, 16, 29). While aerobic NO production has been extensively investigated, enzymes that regulate nitrite-to-NO reduction are less well understood.
Several studies have investigated the possible enzymatic origins for mammalian nitrate and nitrite reduction, and a number of enzyme systems have been reported to control nitrite reduction to NO under certain conditions. As mentioned, xanthine oxidase has been proposed as a mammalian nitrate and nitrite reductase (18). A number of studies have demonstrated that the therapeutic effects of nitrite on pulmonary hypertension require a functional xanthine oxidase system (2, 55); however, the inhibition of xanthine oxidase does not inhibit nitrite-dependent vasodilation in human circulation (12). Several other mammalian nitrite reductase candidates have been proposed, such as the other Mo-containing enzyme aldehyde oxidase and heme enzymes (cytochrome c, eNOS, deoxyhemoglobin, deoxymyoglobin, neuroglobin, and cytoglobin), as well as one nitrite anhydrase (carbonic anhydrase) (1, 4, 19, 38, 40). However, it is likely that additional catalytic human nitrate and nitrite reductases have not yet been identified, such as SO, which has comparable or higher activity than xanthine oxidase and aldehyde oxidase when phenosafranine, a one- or two-electron donor, was used as the reducing substrate. When sulfite, a strict two-electron donor and a known physiological substrate of SO, was used as the reductant, less NO was produced.
These studies suggest that reduced SO can reduce nitrite to form NO, in a reaction analogous to that of xanthine oxidase and aldehyde oxidase. It is now clear that many heme-based and molybdopterin-based proteins have the capacity to reduce nitrite to NO. Enzymes that metabolize nitrite to NO are likely dependent on specific organ location and local oxygen tensions. For example, deoxyhemoglobin reduces nitrite to NO in the blood, and this pathway has been shown to regulate hypoxic vasodilation (10, 11) and, more recently, inhibition of platelet activation (42). Deoxymyoglobin plays a major role in the heart (41, 48) and smooth muscle, where it regulates mitochondrial respiration, myocardial energetics, and hypoxic vasodilation. Consistent with a role for heme globins in vasodilatory and platelet inhibitory effects of nitrite, oxypurinol does not inhibit nitrite-dependent vasodilation.
On the other hand, treatment of rodents with tungsten, which replaces molybdenum in xanthine oxidase, aldehyde oxidase, and SO, inhibits the protective effects of nitrite in vascular injury models, such as carotid wire injury and pulmonary arterial hypertension (2, 54). Allopurinol can also inhibit the effects of nitrite in many preclinical disease models (15). Our present studies using fibroblasts for SO-deficient patients suggest that SO plays a dominant role in nitrite reduction and sGC activation in these cells. These data, in aggregate, suggest that a number of enzymes can reduce nitrite to NO, and the dominant effect of one system likely depends on enzyme concentration and oxygen concentration in specific tissues. Future work with conditional knockout of xanthine oxidase, aldehyde oxidase, and SO will be required to fully explore the relative importance of each in specific tissues and diseases.
In this study, we examined the enzymatic reactions of human and mouse SO in vitro with phenosafranine, sulfite, and nitrite using NO detection, nitrite reduction, and EPR methodologies, revealing that SO can catalyze the reduction of nitrite to NO by the one-electron oxidation of Mo(IV) to Mo(V). We unequivocally demonstrated that only fully reduced Mo(IV) enzyme is capable of nitrite reduction, while Mo(V) does not produce NO. This result indicates that both the electron donor and the redox potential of the enzymatic metal center play an important role in catalytic reactions, which determines the electron flow in SO. SO has two internal cofactors, heme and molybdenum, which have reduction potentials of 68 mV (heme), −239 mV (Mo(V)/Mo(IV)), and 38 mV (Mo(VI)/Mo(V)), respectively (9). Electrons flow from the Mo center to the heme center, and Mo(V) is the favorable oxidation state. In order to efficiently catalyze nitrite reduction in vivo, either an appropriate one-electron donor can reduce Mo(V) to Mo(IV) or an oxidant can oxidize Mo(V) to Mo(VI), enabling sulfite to react with SO again. After the nitrite-dependent formation of Mo(V), the remaining electrons may be transferred to the heme domain, which, in turn, requires one cycle of cytochrome c reduction to complete the catalytic cycling.
Given that reduced SO was not found to react with nitrate, we conclude that SO cannot function as nitrate reductase. A recent study by Qiu et al. has engineered an SO enzyme variant with partial nitrate reductase activity using structure-guided mutagenesis (35). Plant nitrate reductases have three internal cofactors: FAD, heme, and molybdenum cofactor (Moco). Their reduction potentials are −272 to −187 mV, −123 to −174 mV, and −25 to +15 mV, respectively (9). This redox pattern is consistent with a downhill flow of electrons within the enzyme, that is, electrons are readily transferred from heme to Mo for nitrate reduction. In addition, electrons from NAD(P)H, the physiological reductant, are passed to heme and Mo sites one by one via the FAD domain. However, the well-accepted flow of electrons in SO is from the Mo domain to the heme domain, and finally to cytochrome c, the physiological oxidant.
Our studies also suggest that the heme domain reduces the rate of nitrite reduction to NO. The fact that the apo-heme domain also inhibits nitrite reduction, to some extent, suggests an important conformational component in the access of nitrite to sulfite-reduced SO. There is a flexible hinge (14 residues in human SO) that tethers N-terminal heme domain to Mo-domain (21). Previous studies have shown that this hinge controls domain motion during the catalytic cycle of SO, which adopts various conformations during intra-molecular electron transfer (IET) and catalysis (49). Thus, domain motion appears to change domain distance and modulate activity, as supported by molecular dynamic studies that propose a much shorter Mo-heme distance (19 Å) during IET as compared with the distance in the crystal structure of SO (32 Å) (37). Electronically, there are six possible redox states of SO, depending on oxidation states of molybdenum and heme iron. Under hypoxic, ischemic, or reductive conditions, cytochrome c is fully reduced, which, in turn, will slow down re-oxidation of the heme iron and, ultimately, inhibit IET between Mo and heme. This can provide a possible mechanism for hypoxic and/or ischemic nitrite-dependent NO formation catalyzed by SO.
In addition to hypoxia, sulfite accumulation would increase the steady-state levels of Mo(IV)-reduced SO. However, such a condition is only known for Moco- or SO deficiency, genetic disorders leading to rapidly neurodegeneration, mental retardation, and early childhood death. Recently, a first successful experimental treatment for MOCD has been reported (50). Those patients lack Moco and, consequently, sulfite accumulates in large quantities. Due to the reconstitution of Moco biosynthesis in treated patients, SO activity returns and excess sulfite is removed with 72–96 h post-treatment (50). In this study, Moco- or SO deficiency in isolated human fibroblasts caused significant decreases of nitrite-dependent cGMP generation, supporting a role for SO in nitrite-dependent intracellular NO signaling.
This work has identified human SO as a new candidate nitrite reductase that leads to NO production both in vitro and in vivo, which requires a one-electron donor to drive the catalytic cycle between Mo(IV) and Mo(V) in the purified enzyme system. SO contributes to NO-soluble guanylyl cyclase-cGMP signaling in human fibroblasts. Further studies are needed to elucidate the physiological mechanism and significance of how SO function in nitrite-NO signaling in human cells and tissues.
Materials and Methods
Reagents and standard samples preparation
All chemicals were purchased from Sigma-Aldrich. Holo SO, isolated Mo, and heme domains were reduced by sulfite or dithionite; purified by passing through a Sephadex G-25 gel filtration column; and eluted with the indicated buffer anaerobically. Concentrations of the heme cofactor in holo SO and heme domain were determined by measuring absorbance of the heme Soret band using ɛ414=113 mM −1·cm−1 or ɛ423=164 mM −1·cm−1. Concentrations of the Mo cofactor in holo SO and isolated Mo-domain were determined by HPLC Form A analysis (22).
Cloning, expression, and purification of the recombinant proteins
Mature human and mouse SO were polymerase chain reaction cloned into the SacI and SalI sites of pQE-80L (Qiagen). The resulting constructs were used to create the Mo and heme domain expression constructs, as well as the heme-deficient variant (H119A/H144A) using the same restriction sites. All constructs were expressed in Escherichia coli strain TP1000 (33, 46) for 48 h at 24°C. Protein expression from pQE80 was induced after 4 h of incubation at 24°C with 100 μM IPTG. 500 μM sodium molybdate was added to the culture to achieve sufficient levels of Moco production. His-tagged SO was affinity purified using Ni-NTA resin (Qiagen) according to the manufacturer's instructions. Protein purification included further anion exchange with a SourceQ15 column (GE Healthcare) using 50 mM Tris-acetate, pH 8.0, as buffer A. Proteins were eluted with a gradient from 100 to 500 mM NaCl in buffer A.
Measurement of NO formation by ozone-based chemiluminescence
Detailed methods are provided in the Supplementary Data. In summary, NO production was measured by ozone-based chemiluminescence using a Sievers Nitric Oxide Analyzer (NOA 280i) equipped with a liquid sampling purge system (GE Analytical Instruments). Sievers NO Analysis™ Software, Liquid (Version 3.2) was used for data acquisition, at a rate of one data point per 0.25 s.
The standard glass purge vessel (15-ml) was used to mix and deoxygenate reagents. For enzyme reactions, the reaction buffer (20 mM BisTris buffer or 10 mM phosphate buffer pH 6.5 or 7.4) was maintained in the presence of nitrite for 2 min in the sealed purge vessel to equilibrate the temperature to 37°C and to void oxygen from the reagents before initiating the enzyme reaction. Next, reducing substrate (sulfite or phenosafranine) was injected into the purge vessel through the vessel's septa using a Hamilton syringe. Once a stable baseline was established, SO was injected into the mixture and the reaction was monitored for several minutes, until the reaction was completed.
NO production rates were estimated using a calibration curve prepared by the nitrite/I3 − method (see Supplementary Data for details). All the NO generation rates were plotted against the corresponding concentrations of nitrite, and fit to a hyperbolic equation, y=(P1×x)/(P2+x). The values of P1 and P2 correspond to νmax and Km values in the Michaelis–Menten equation, that is, ν=Vmax×[S]/(Km+[S]). In the case of single turnover reactions (nonsteady state), a similar fit was used, where the P1 and P2 terms represent the apparent νmax and Km values, which we refer to as ket (rate of electron transfer) and Kd (apparent dissociation constant) to indicate the single turnover nature of the studied reaction.
Nitrite reduction assay
Time-course nitrite reduction was evaluated by measuring the concentration of nitrite in the reaction mixture with the NOA 280i. 10 μM SO or isolated Mo domain was mixed with 10 μM nitrite, in the presence of sulfite (50 μM) or reduced phenosafranine (50 μM) inside the anaerobic glove box. Afterward, nitrite concentration was measured at 5, 10, 20, 30, and 60 min. Time zero was defined as the same reaction in the absence of protein. Each data point represents the average of three separate measurements.
Steady-state kinetics of nitrite reduction by phenosafranine
Steady-state kinetics were performed anaerobically in a glove box using a plate reader (Biotek). All data recorded are mean values of at least triplicate measurements. Initial velocities were determined by following the oxidation of the artificial electron donor phenosafranine at 520 nm using an extinction coefficient of 29,000 M −1·cm−1. SO was assayed at room temperature in Bis-Tris/acetate buffer, pH 6.5. In the phenosafranine-dependent reduction of nitrite, 12 μM human holo SO or human SO Mo-domain was mixed with 250 μM phenosafranine and 0–75 mM nitrite. The reaction was started with 250 μM sodium dithionite, and control experiments were performed without the addition of enzyme.
CD spectroscopy
Structural similarity between recombinant wild-type mouse SO and the heme-deficient H119A/H144A variant was determined by CD spectroscopy with a JASCO J-715 CD spectropolarimeter. All measurements were performed at 20°C, and secondary structure was measured over a wavelength range of 180–250 nm.
EPR spectroscopy
EPR spectra were recorded at 150 K with the EWWIN 6.0 acquisition and lineshape analysis software (Scientific Software Services) on a Bruker ER 300 spectrometer. Double integration was performed with EWWIN 6.0 using the “very rapid” xanthine oxidase Mo(V) signal as an integration standard (32). All reactions were performed in 50 mM Tris-acetate buffer, pH 8.0 anaerobically. In these experiments, sulfite (100 μM) was added to 20 μM anaerobic Mo domain; the reaction solution was transferred to an argon-flushed EPR tube and frozen; and an EPR spectrum was recorded. The sample was then thawed, 1 mM nitrite was added, and the mixture was incubated for 1.5 h at room temperature (RT) before being re-frozen and a second EPR spectrum was recorded. The sample was then re-thawed, 50 μM dithionite-reduced phenosafranine was added, the sample was re-frozen within 15 s of mixing, and a third EPR spectrum was recorded. Finally, the same solution was once again thawed, and the reaction mixture was incubated for an additional 1.5 h at RT before being frozen for the recording of a final EPR spectrum.
Western blot analysis
Fibroblast cells were washed with ice-cold PBS and homogenized in PBS with sonication. Then, 120 μg total protein of the cell lysate was loaded for electrophoresis. The cell lysates were resolved using 10% Bis-Tris SDS-PAGE gels and transferred to high-quality polyvinylidene difluoride (PVDF) membrane. The membrane was then probed with primary antibodies that were specific to SO (Eurogentec) and beta-actin (Santa Cruz). After incubation with the secondary antibodies coupled to horseradish peroxidase from Santa Cruz, the chemiluminescent signals were detected with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific).
Determination of cGMP production in human fibroblasts
Human fibroblasts derived from SO- and Moco-deficient patients or healthy individuals were grown on 15 cm plates at 37°C with 5% CO2 in RPMI-1640 (Gibco) supplemented with 10% fetal bovine serum (Gibco) and 2 mM L-glutamine (Gibco). Cells were grown to about 90% confluence before treatment with 1 μM sildenafil and 5–100 μM nitrite or 5 μM DEA-NO for 2 h at hypoxic conditions (1% O2) or aerobically (20% O2). Control samples were incubated either without 5 μM DEA-NO or with nitrite addition. Time-dependent cGMP production was measured at 0, 15, 30, 60, and 120 min with 100 μM Nitrite. Cells were harvested and lysed with 100 μl HCl (0.1 M), and total protein concentration was determined by Bradford assay. Triplicates were used for each condition, and cGMP concentration was detected with a cGMP enzyme-linked immunosorbent assay kit (Cayman Chemicals). Results are expressed as the means±standard deviation. Statistical differences between two sets of data were calculated using a Student's t-test. Statistical significance was assumed when p was less than 0.001.
Footnotes
Acknowledgments
The authors thank Dr. Jack Lancaster for helpful discussions on reaction kinetics in NOA purge systems. Dr. Gladwin receives research support from NIH grants R01HL098032, RO1HL096973, and PO1HL103455; the Institute for Transfusion Medicine; and the Hemophilia Center of Western Pennsylvania. Dr. Tejero acknowledges the support from the Competitive Medical Research Fund of the University of Pittsburgh Medical Center (UPMC) Health System. Dr. Guenter Schwarz thanks the German Research Foundation (DFG) and the Fonds der Chemischen Industrie for research support.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.