
Abstract
Introduction
N
Interest in the selenocysteine insertion sequence (SECIS)-binding protein 2 (Secisbp2) is manifold. Since Secisbp2 plays a central role in UGA/Sec recoding and selenoprotein expression, the study of its molecular biology will ultimately lead to a better insight into translation in general and selenoprotein translation in particular. Second, congenital SECISBP2 deficiency causes a syndrome of growth retardation that has been defined as an atypical form of resistance to the thyroid hormone. We provide the first mouse models that allow us to dissect the roles of Secisbp2 in vivo. Finally, mutations in SECISBP2 demonstrate the fundamental effects of selenoproteins on human health, including immunological, metabolic, and neurological processes.
Secisbp2 was purified and cloned based on its ability to bind to SECIS elements in vitro (17). The protein contains an RNA-binding module, the L7Ae domain, which interacts with kink-turn RNA structures such as the one found in the SECIS core that contains a pair of non-Watson–Crick base pairs (30, 54). Secisbp2 facilitates Sec incorporation by binding to the SECIS element, promoting recruitment of EF-Sec and interacting with the ribosome (10, 18). Several other SECIS-binding proteins have since been identified. These include L30 (Rpl30), which is a component of the UGA recoding machinery (14), YB1 (48), NSEP1 (47), eIF4a3 (9), and nucleolin (33, 56), which play roles in regulating UGA recoding. In addition, Secisbp2L binds to SECIS elements, but does not support Sec incorporation (19) [reviewed in Seeher et al. (44)].
Mutations in SECISBP2 were first identified in a family with several children exhibiting a delay in linear growth and bone age. In addition, these patients manifested slightly elevated total thyroxine (T4), low tri-iodothyronine (T3), high reverse T3, and high plasma thyroid-stimulating hormone (TSH) as well as abnormal TSH suppression tests that suggested impaired thyroid hormone metabolism (21). Thyroid hormones are subject to deiodinating reactions catalyzed by deiodinases, a family of selenoenzymes (4). By comparison, deiodinase gene (Dio1) targeted mice show increased reverse T3 (39), while Dio2 deficiency leads to high T4 and TSH (38). Indeed, Dio deficiency, along with reduced expression of selenoprotein P (Sepp) and glutathione peroxidases (Gpx), pointed to a general defect in selenoprotein mRNA translation, since Dio2 mRNA levels were normal in fibroblasts from these patients (21). Similarly, abnormal thyroid function tests led to the identification of new patients carrying several homozygous and compound heterozygous mutations in SECISBP2 (20). Other mutations are apparently more disruptive for selenoprotein biosynthesis and lead to additional symptoms like myopathy [similar to selenoprotein N deficiency (34)], abnormal gait (1), bilateral hearing loss, male infertility, increased photosensitivity, shortened telomere length, and compromised immune function along with abnormal glucose metabolism (40). Mutations in SECISBP2 affect SECIS binding, underlining the importance of SECISBP2 for selenoprotein biosynthesis via SECIS interactions (1, 8).
The many roles of selenoproteins in mammalian health are revealed by phenotypes of mouse models deficient for individual selenoproteins or with inefficient selenoprotein mRNA translation, as well as by human congenital disorders of selenoprotein expression (16, 42). To study the role of Secisbp2 in tissues, organs, and whole organisms, mouse models are needed that allow for biochemical investigations in tissues not accessible in human patients. We describe here the first constitutive and conditional mouse models for Secisbp2 and compare selenoprotein expression in these with our earlier work on similar mouse models lacking tRNA[Ser]Sec.
Results
Constitutive Secisbp2 gene targeting
Secisbp2 gene targeting followed a knockout-first strategy (Fig. 1A) allowing for both classical gene knockout and Cre recombinase-mediated conditional gene ablation. Intercrosses of Secisbp2 +/− mice yielded no live Secisbp2 −/− offspring, but many resorbed conceptuses (Table 1). Dissection of Secisbp2 −/− embryos demonstrated developmental retardation by embryonic day 8 (E8) (Fig. 1B). Whereas control embryos had progressed to TS12 (somites 1–7, first branchial arch develops, beginning of heart formation), Secisbp2 −/− embryos are arrested in TS7, which is characterized by implantation of the conceptus and formation of the egg cylinder. Thus, Secisbp2 −/− embryos failed after implantation, but developed further than tRNA[Ser]Sec-deficient embryos that only reached the morulae or blastocyst stage (E3.5; TS2-3) and failed to implant (6).
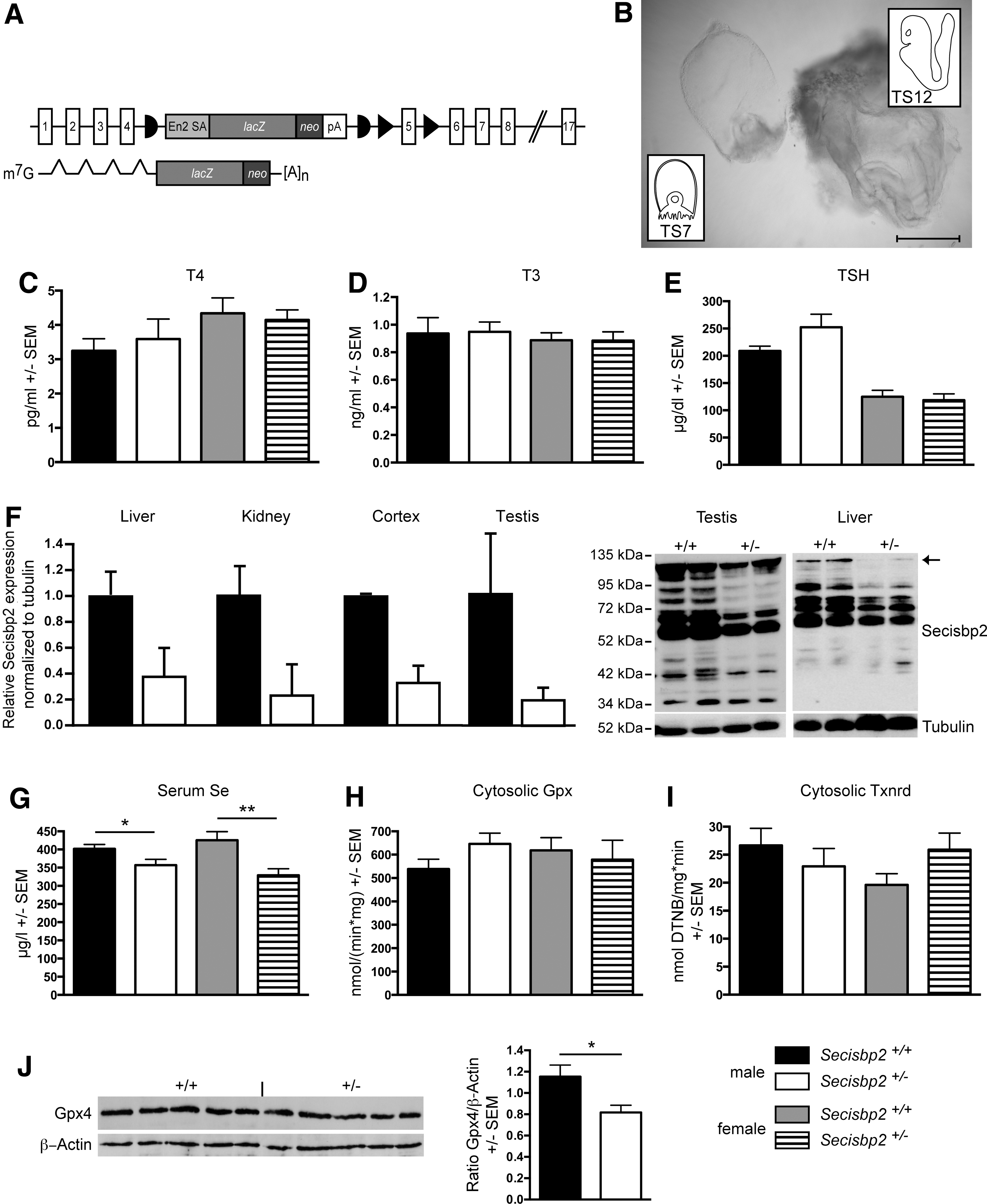
Genotyping of Secisbp2 +/− intercrosses 3 weeks after birth yielded no live Secisbp2 −/− offspring. Analysis of embryos from day 9.5 post coitum (dpc) detected several resorbed structures, which were identified as Secisbp2 −/−. Genotyping resorptions on 13.5 dpc was more complicated because of the more advanced stage of resorption. Most potential Secisbp2 −/− embryos were likely assigned to Secisbp2 +/− due to contamination with maternal tissue.
Genotyping of Secisbp2+/Δ5 intercrosses 3 weeks after birth yielded no live Secisbp2 Δ5/Δ5 offspring. Secisbp2+/fl heterozygous mice were crossed with a germline Cre deleter mouse strain to remove the floxed exon 5. The resulting genotype lacking exon 5 in one allele is denoted Secisbp2+/Δ5 .
N/A, not applicable; Secisbp2, selenocysteine insertion sequence-binding protein 2.
SECISBP2-mutations in humans are associated with delayed bone development, growth defects, and abnormal thyroid hormone constellations. Heterozygous Secisbp2
+/− mice of both sexes exhibit the same body weight and tail length as Secisbp2
+/+ litter mates (Supplementary Fig. S1; Supplementary Data are available online at
Conditional inactivation of Secisbp2 in liver
Embryonic lethality of the knockout mice required the study of selenoprotein biosynthesis in conditional Secisbp2 mice. Germline expression of FLP recombinase removed the gene-trap cassette leaving a conditional (floxed) exon 5 (Fig. 2A). Sequencing of the mutant transcript confirmed that removal of exon 5 causes a frameshift resulting in a premature stop codon in exon 6 (not shown). Homozygous germline deletion of floxed exon 5 in Secisbp2 (Secisbp2 Δ5/Δ5 ) yielded the same embryonic lethal phenotype as the knockout-first allele indicating that the Secisbp2 Δ5 allele is not functional (Table 1). We then targeted hepatocytes using a Cre transgene under control of the albumin promoter (Alb-Cre), which we had previously used to inactivate tRNA[Ser]Sec (gene symbol: Trsp) in hepatocytes. The liver is a good model for the study of selenoprotein expression, since targeted ablation of selenoprotein biosynthesis using the conditional Trsp in our hands does not lead to liver failure or other diseases (43, 46, 52). To test whether liver damage occurs upon Secisbp2 deletion in hepatocytes, we determined liver transaminase activities in serum. No significant changes according to genotype of the animals were noted (n=6–9; alanine-aminotransferase (ALAT, GPT) activity in U/ml±standard error of the mean (S.E.M.): controls 0.23±0.09 versus mutants 0.24±0.12; aspartate-aminotransferase (ASAT, GOT) activity in U/ml: controls 0.36±0.09 versus mutants 0.43±0.13) indicating that no apparent liver damage occurred in Alb-Cre; Secisbp2fl/fl mice. Furthermore, the liver not only expresses many selenoproteins that can be measured reliably, but also provides sufficient material to do so.
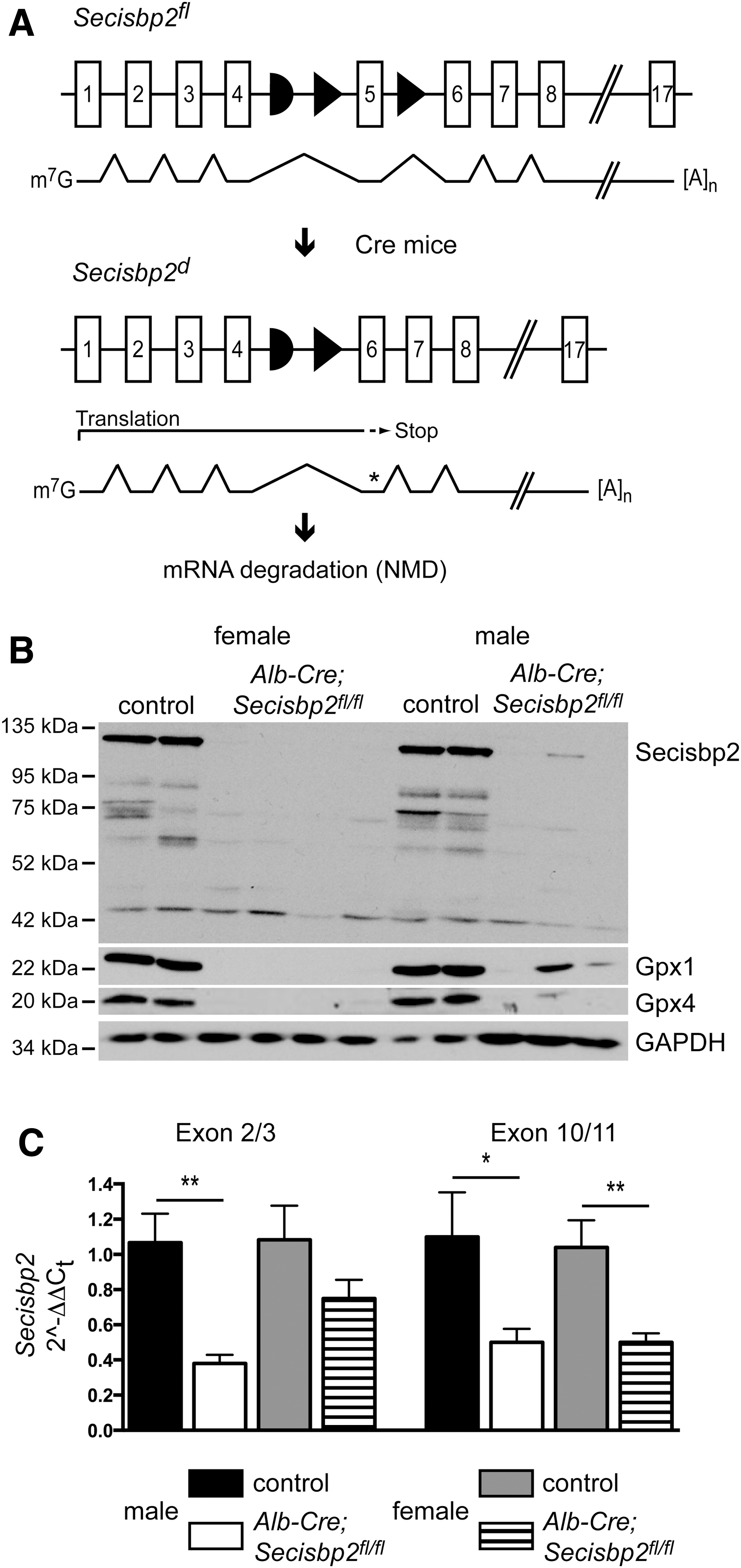
Western blot analysis of control mouse liver extracts using an antibody directed against the C-terminus of Secisbp2 revealed a prominent band of 120 kDa that represents the full-length protein (Fig. 2B, lanes 1, 2, 7, 8). We also observed several minor bands ranging in size from 60 to 90 kDa, which presumably represent degradation products generated during tissue workup, since they varied in pattern and intensity depending on the extraction method. The Secisbp2 protein is quantitatively removed from the livers of Alb-Cre; Secisbp2fl/fl female and male mice (Fig. 2B; lanes 3–6 and 9–11, respectively). Selenoproteins, Gpx1 and Gpx4, were significantly reduced in the Secisbp2-deficient liver samples confirming the dependence of selenoprotein expression on Secisbp2. In only one male mouse, incomplete recombination occurred leaving residual Secisbp2 protein, which accordingly supported partial Gpx1 and Gpx4 expression (Fig. 2B, lane 10). This shows that all phenotypes observed are caused by Secisbp2 inactivation and do not depend on another trait genetically coupled to the transgene. The mouse in which the Cre transgene was apparently nonfunctional was omitted from subsequent analyses except Figure 5C. Quantitative polymerase chain reaction (qPCR) using primers specific for exons 2/3 and exons 10/11 revealed a reduction of Secisbp2 mRNA levels (Fig. 2C). The mutant transcript perhaps is degraded due to the premature stop codon in exon 6. Some of the remaining amounts of Secisbp2 mRNA and protein may arise from endothelial cells and Kupffer cells that are not targeted by Alb-Cre.
Impact of Secisbp2 inactivation on selenoprotein mRNA levels
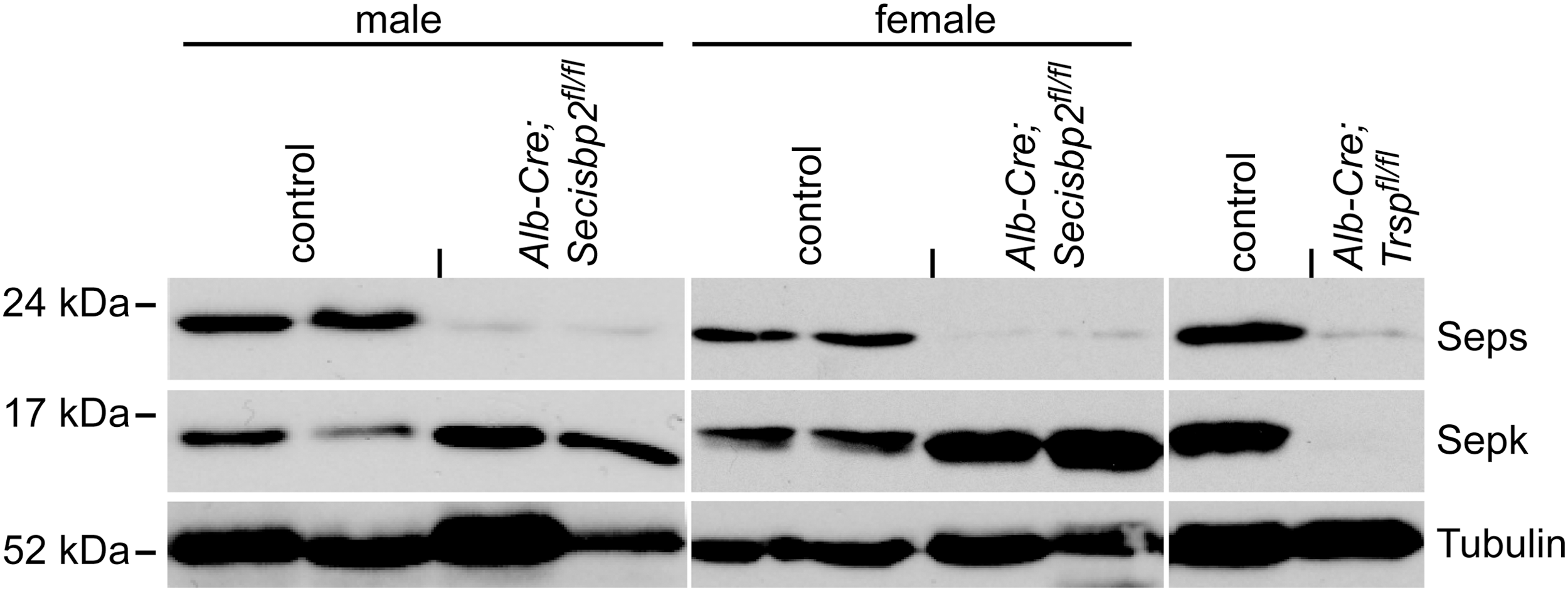
Selenoproteins carry in-frame UGA codons in their mRNAs, which could lead to the transcripts being degraded by nonsense-mediated decay (NMD) when Sec incorporation is impaired. Both Se deficiency and hepatic Trsp deficiency impair selenoprotein biosynthesis and thus reduce the amounts of several selenoprotein mRNAs in mouse livers (13, 53). We therefore tested whether selenoprotein mRNA levels were also impacted in Alb-Cre; Secisbp2fl/fl livers (Fig. 3). We were able to group selenoprotein transcripts into four categories according to their response to Secisbp2 inactivation. Selenoproteins that carry the UGA codon in an upstream exon are in the first group, which potentially fall subject to NMD. Accordingly, many prototypic selenoprotein mRNAs (e.g., Gpx1, Dio1) were significantly reduced in mutant livers (Fig. 3A). The second group comprises selenoprotein mRNAs that also carry the UGA codon in an upstream exon, but in contrast to the first group, the reduction of mRNA is smaller and mostly limited to the male liver (Fig. 3B). In the third group are selenoprotein mRNAs carrying the UGA codon in the last exon. These transcripts are generally not subject to NMD and comprise prototypic examples like Txnrd1 (Fig. 3C). One notable exception, Seps mRNA, falls into the fourth group. This mRNA was reduced in male and female Secisbp2 mutant livers, despite its UGA being located close to the C-terminus in the last exon (Fig. 3D). Accordingly, a clear reduction of Seps protein levels in response to Secisbp2 mutation was apparent (Fig. 4). In contrast, Sepk was expressed at normal levels. Interestingly, in liver homogenate from mice with tRNA[Ser]Sec inactivation, we found undetectable Sepk levels. This finding suggests that lack of Secisbp2 may be mechanistically different from a mere blockade of selenoprotein mRNA translation.
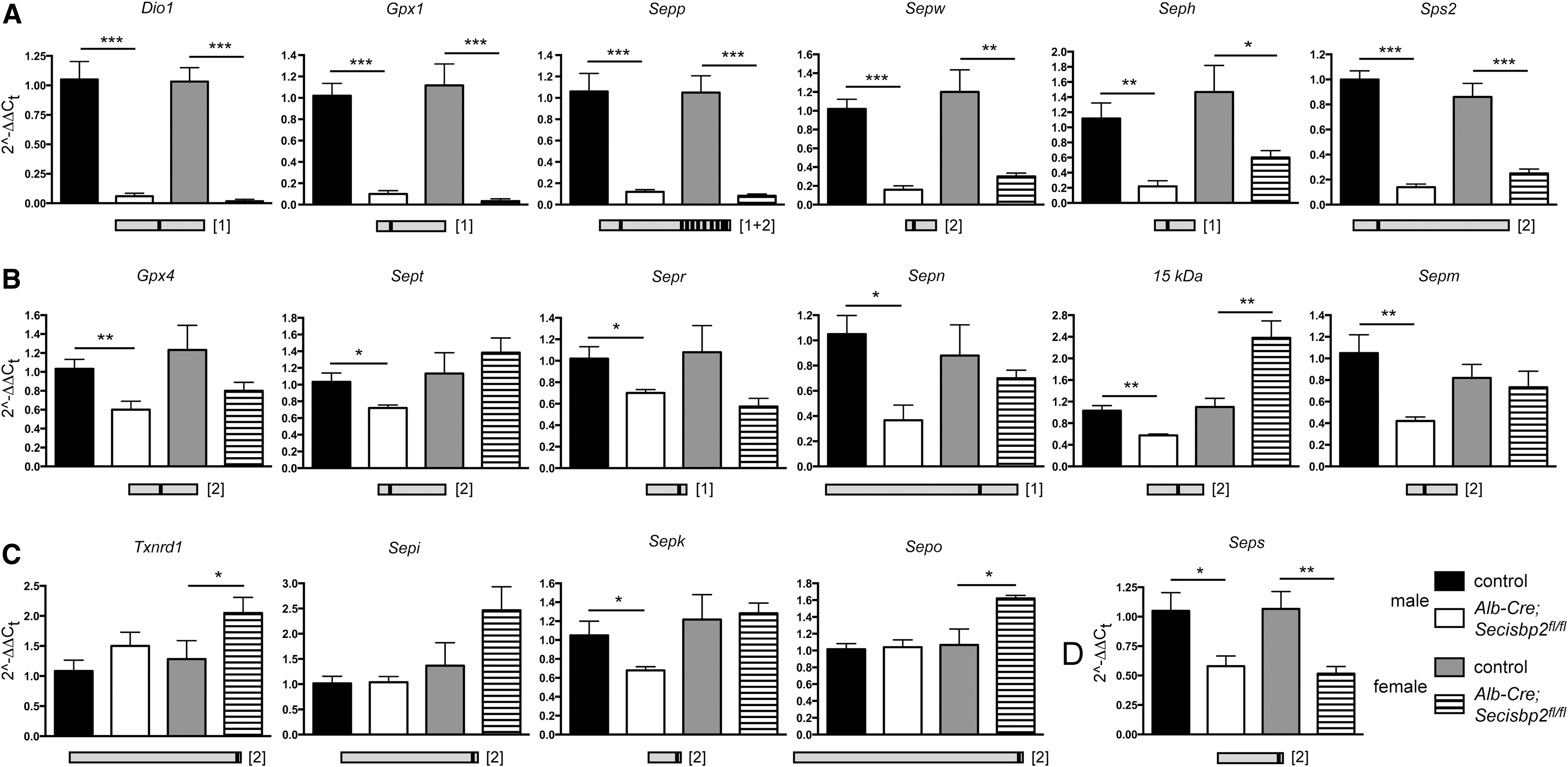
SECIS elements are classified as being type 1 or type 2, which contain either an apical loop or an apical bulge. The differential response of selenoprotein mRNAs did not correlate with the type of SECIS element (Table 2). There was also no correlation between the loss of a particular selenoprotein mRNA and its predicted ability to serve as a substrate for NMD. The current rule for NMD requires that the premature stop codon is more than 50 nucleotides upstream of an exon–exon junction. As shown in Table 2, Sps2 and Seps mRNAs were reduced in spite of the fact that these transcripts have only one exon or contain the UGA in the last exon, respectively. Likewise, several selenoprotein mRNAs are reduced even though the UGA codon is too close to the next downstream exon (e.g., Sepp, Sepw, Sepr, Sep15, and Sepm). Interestingly, all of the selenoprotein mRNAs that are not affected by Secisbp2 deficiency (e.g., Txnrd1, Sepi, Sepk, and Sepo) do not follow the rules for NMD (Table 2). Unexpectedly, several selenoprotein mRNAs that showed a reduction in response to Secisbp2 inactivation were not decreased in the livers of Trsp-deficient or Se-deficient male mice, specifically Dio1, Sps2, Gpx4, Sepr, Sep15, and Seps (11, 53), although Dio1 mRNA was significantly reduced in another study (52). Thus, with respect to hepatic selenoprotein mRNA levels, Trsp deficiency and selenium deficiency more closely mimic each other, whereas Secisbp2 deficiency gives a distinctively different profile.
NMD, nonsense-mediated decay; nt, nucleotides.
Impact of Secisbp2 inactivation on hepatic selenoprotein expression
Sepp is a plasma Se transport protein primarily secreted from the liver by hepatocytes (12, 25, 36, 41, 43). In serum, significant Sepp remained in Alb-Cre; Secisbp2fl/fl mice compared with controls (Fig. 5A), which corresponded to about 30% serum Se remaining in the mutants (Fig. 5B). This is two times higher than in Alb-Cre; Trspfl/fl mice as reported previously (43). In comparison, serum Se in Alb-Cre; Seppfl/fl mice was reduced to 9% (25). We then analyzed liver extracts for remaining Sepp biosynthesis by western blot analysis. We found several bands corresponding to the full-length protein at 41 kDa and a series of increasingly glycosylated forms up to the >55 kDa secreted protein forms. Again, more Sepp proteins are detected in the Alb-Cre; Secisbp2fl/fl liver than in Alb-Cre; Trspfl/fl controls (Fig. 5C). We therefore analyzed again Gpx1 and Gpx4 expression in liver extracts, but this time applied more protein (100 μg per lane). Under these conditions, small amounts of residual Gpx1 and Gpx4 proteins were detectable in the Alb-Cre; Secisbp2fl/fl liver, but not in Alb-Cre; Trspfl/fl (Fig. 5C). Interestingly, the Txnrd1 protein, which carries Sec as the penultimate amino acid, was apparently not reduced (Fig. 5C). However, SDS-PAGE analysis cannot exclude the possibility that a Txnrd1 protein lacking the last two amino acids was synthesized due to premature termination at the UGA/Sec codon.
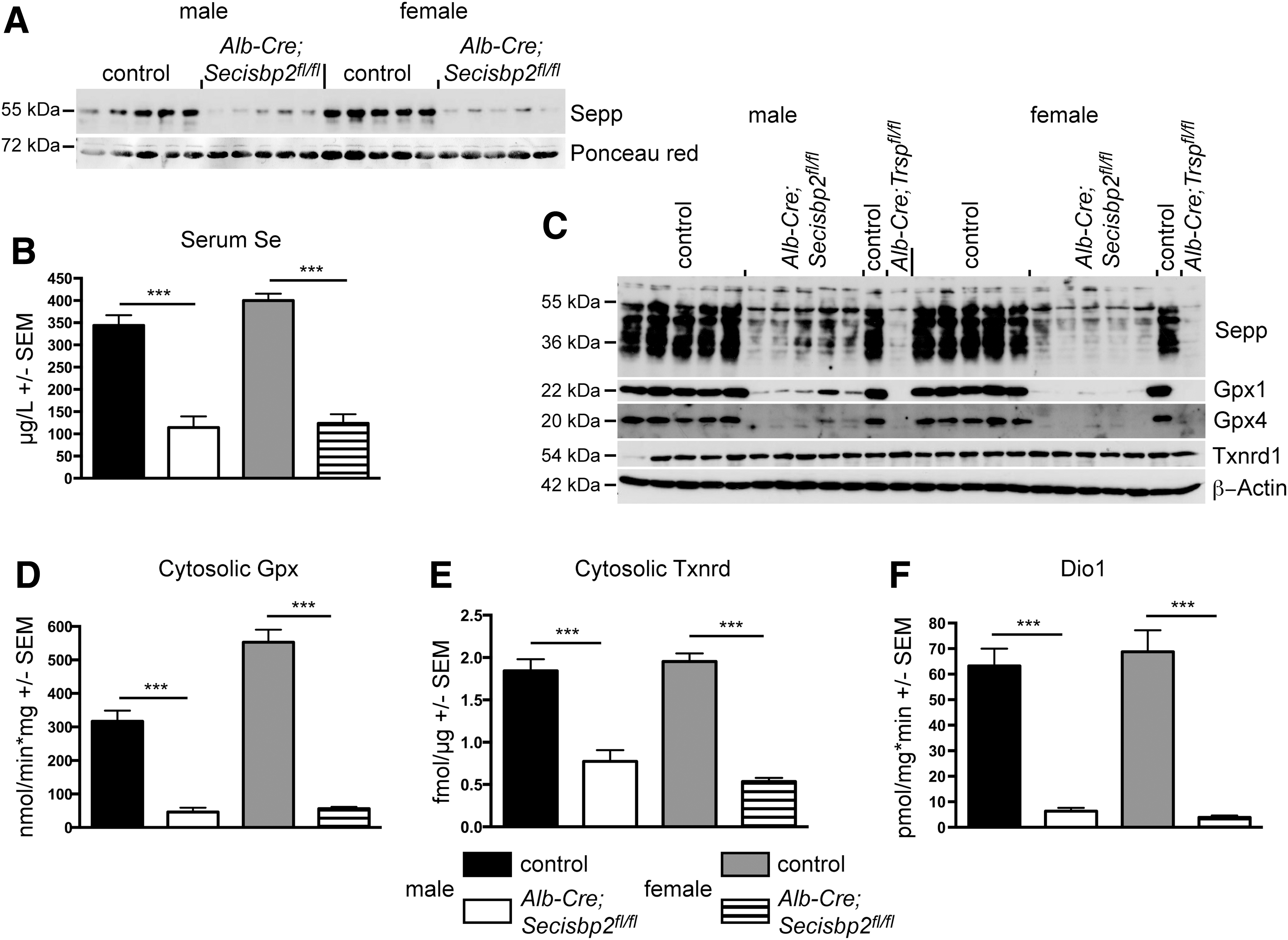
Activity assays are superior to western blots for determination of selenoenzymes, because activity usually critically depends on Sec incorporation. The cellular Gpx activity was reduced by 85% and 90% in male and female Alb-Cre; Secisbp2fl/fl livers, respectively (Fig. 5D). The cytoplasmic Txnrd activity as measured with the 5,5′-Dithiobis(2-nitrobenzoic acid) (DTNB) assay was significantly reduced in the Alb-Cre; Secisbp2fl/fl liver (about 50% not shown) nicely corresponding to our findings in the Alb-Cre; Trspfl/fl liver (52). The insulin-based Txnrd assay is considered more sensitive to the presence of the penultimate Sec residue. Employing this assay, we found an even larger reduction in the Secisbp2 mutants to about 40% of control activity (Fig. 5E).
Since the liver is not only composed of hepatocytes, our analysis may be complicated by proteins from endothelial cells and Kupffer cells. We therefore measured the activity of type I deiodinase (Dio1), which is specific for hepatocytes. Whereas Dio1 mRNA was significantly reduced in the Alb-Cre; Secisbp2fl/fl liver (Fig. 3A), it was obvious that a small fraction of Dio1 activity remained in the Secisbp2-targeted liver (Fig. 5F).
Selenoprotein expression in primary hepatocytes
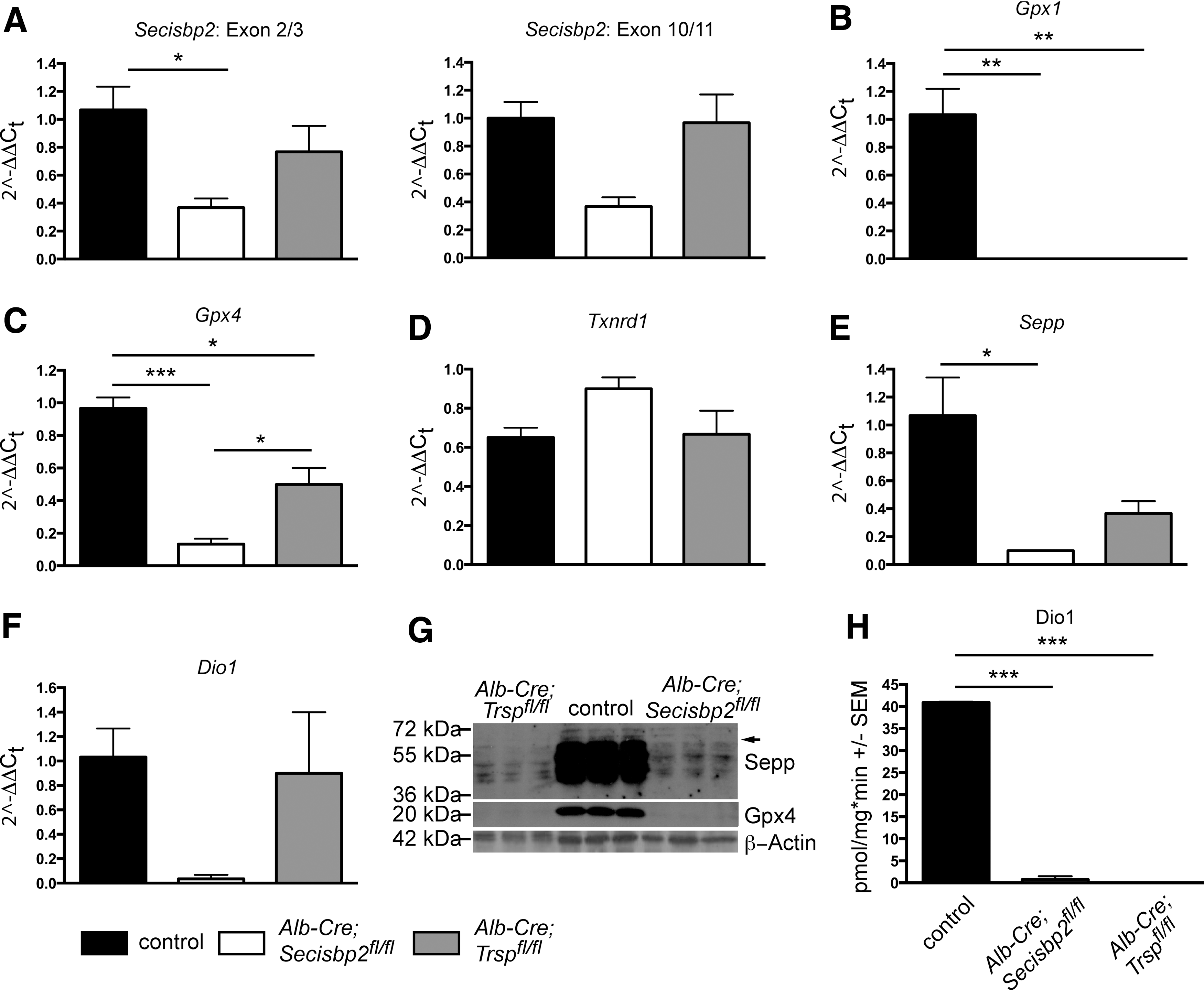
To investigate the possibility further that hepatocytes lacking Secisbp2 can still express small amounts of selenoproteins, we isolated primary hepatocytes from Alb-Cre; Secisbp2fl/fl mice and litter mate controls thus removing background signals derived from endothelium or Kupffer cells. In parallel, we isolated hepatocytes from Alb-Cre; Trspfl/fl mice. Comparing Secisbp2-deficient hepatocytes with Trsp-deficient cells, in the same analyses, allows one to separate effects simply related to impaired selenoprotein mRNA translation from effects specific for Secisbp2 inactivation. Messenger RNA levels for Secisbp2 were only reduced in Alb-Cre; Secisbp2fl/fl cells, but not in Trsp-deficient hepatocytes, consistent with the premature termination codon in exon 6 of the Secisbp2 mutant (Fig. 6A). Gpx1 mRNA levels were almost undetectable in both Secisbp2 and Trsp mutant hepatocytes, consistent with its established sensitivity to NMD (Fig. 6B). Gpx4 was reduced in both mutants, but more pronounced in the Secisbp2 mutant (Fig. 6C). Txnrd1 was not affected by either gene deletion (Fig. 6D). The hepatocyte-specific mRNA for Sepp was reduced in both mutants, but again more severely affected in Secisbp2-deleted cells (Fig. 6E). As in whole liver extracts, we observed Sepp biosynthesis in Secisbp2-deleted cells at a low level, this time however, definitely derived from hepatocytes (Fig. 6G). At close inspection, the Sepp species with the highest molecular weight are not visible in Trsp mutant cells, but it is expressed in Secisbp2 mutant cells (arrow). Low residual expression of Gpx4 was not observed, possibly because of its much lower expression level than Sepp. A remarkable finding was the detection of low Dio1 activity in Secisbp2-deleted cells, but none in Trsp mutant cells (Fig. 6H). This was in stark contrast to higher Dio1 mRNA in Trsp mutants than in Secisbp2 mutants (Fig. 6F). This circumstantial evidence suggests that a very low level of translation of at least some selenoprotein mRNAs may occur in the absence of Secisbp2.
Expression of other known SECIS-binding proteins
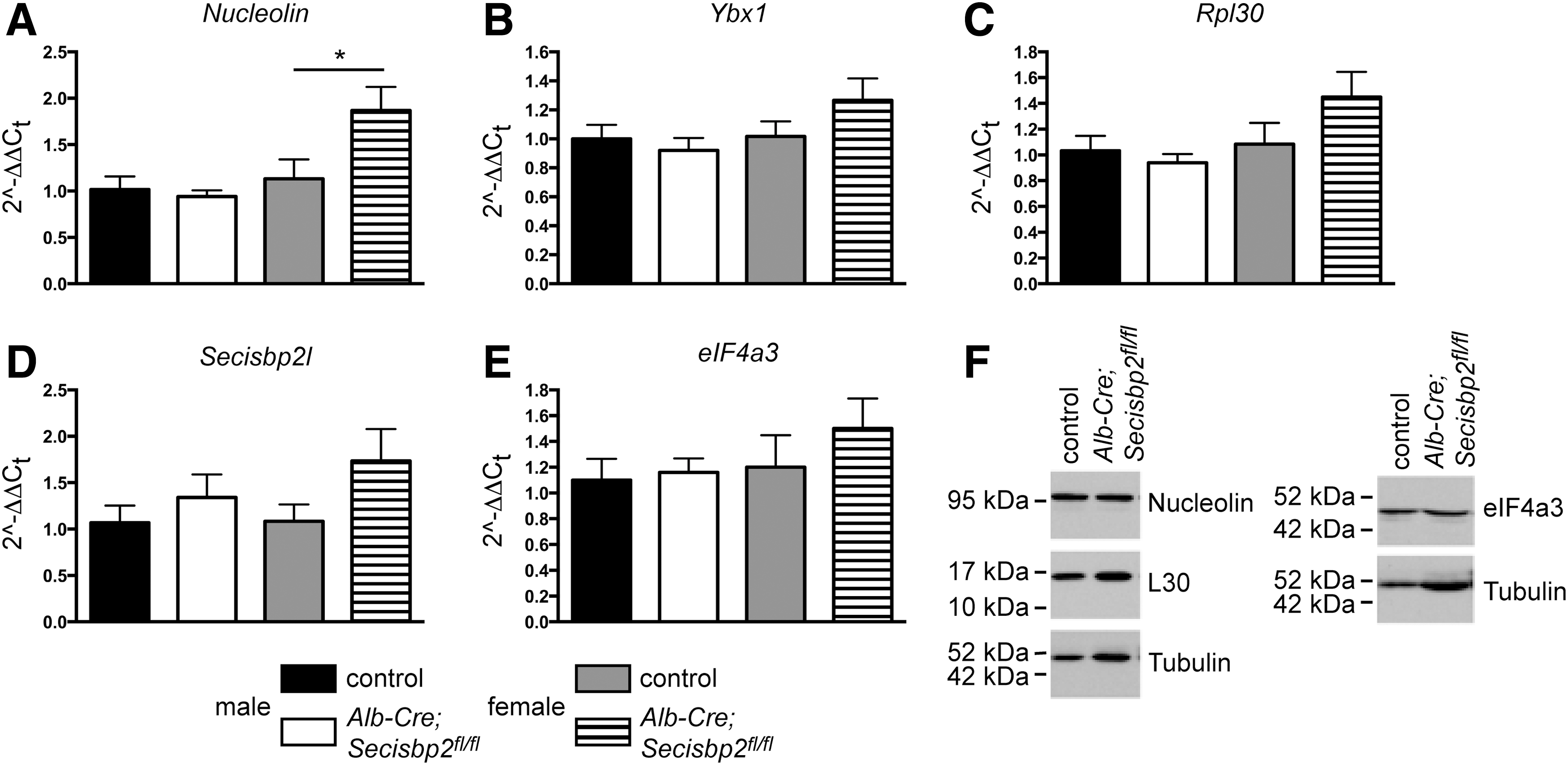
Given the possible persistence of an inefficient mechanism of selenoprotein expression in hepatocytes, we speculated that one of the other SECIS-binding proteins may potentially compensate for the loss of Secisbp2 in hepatocytes. Alternatively, their expression levels may change when Secisbp2 or selenoprotein transcript levels are reduced. We therefore investigated whether Secisbp2 gene targeting changed mRNA expression of any of the other SECIS-binding proteins. Based on qPCR, neither nucleolin (Fig. 7A), Ybx1 (Fig. 7B), Rpl30 (Fig. 7C), or Secisbp2l (Fig. 7D) mRNAs were significantly changed in the livers of male Alb-Cre; Secisbp2 fl/fl mice, although a significant increase in nucleolin mRNA was noted in female mutants (Fig. 7A). The transcript encoding the inhibitory SECIS-binding protein, eIF4a3, which represses Gpx1 mRNA translation in Se deficiency (9), did not change (Fig. 7E). Based on western blotting, the protein levels of L30, nucleolin, and eIF4a3 were not significantly different between the control and Alb-Cre; Secisbp2 fl/fl female mice (Fig. 7F; Supplementary Fig. S2).
Induction of Nrf2 target genes in livers of Secisbp2 mutant mice
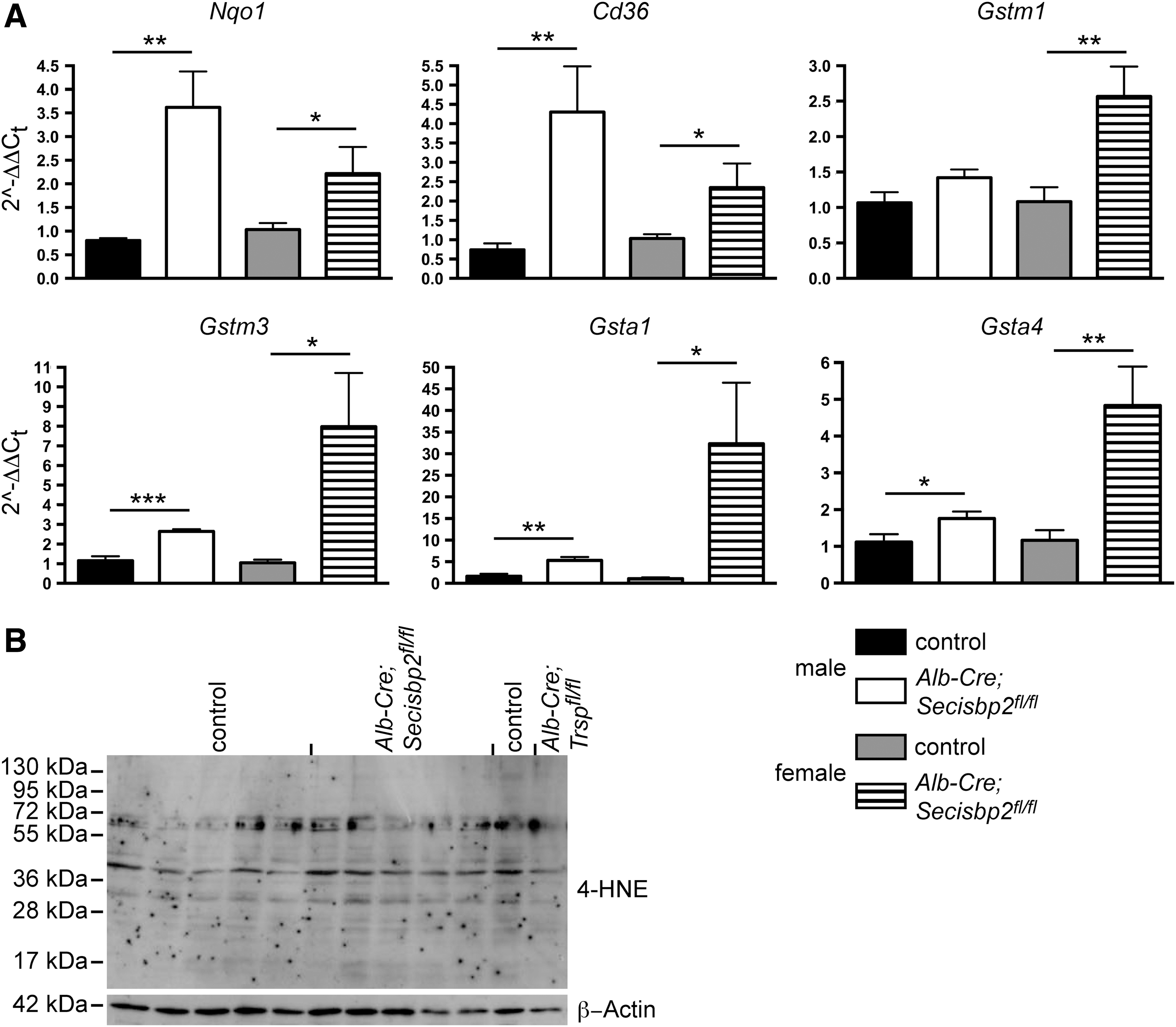
In Alb-Cre; Trsp fl/fl mice, which are globally defective in hepatic selenoprotein expression, nuclear factor, erythroid-derived, like 2 (Nrf2)-dependent gene expression was induced in response to the lack of antioxidant selenoproteins (46). We therefore tested mRNA expression of Nrf2-dependent genes, Nqo1, Cd36, Hmox1, Gstm1, Gstm3, Gsta1, and Gsta4. As expected, we found several-fold induction of most tested genes in male and female mutant mice (Fig. 8A). Interestingly, female Alb-Cre; Secisbp2 fl/fl mice showed higher induction in more genes than males. Staining for proteins modified by the Nrf2-inducing mediator 4-hydroxynonenal (4-HNE) by western blotting did not reveal increased signals in the Secisbp2 mutant liver suggesting efficient detoxification by Nrf2 pathway genes (Fig. 8B).
Discussion
Previous studies in vitro and in cell culture suggested that Secisbp2 is essential for translation of selenoprotein mRNAs (17). Our novel mouse mutants now permit us to investigate Secisbp2 function in embryonic development and in any specific cell type in vivo at any chosen developmental time point, provided a suitable Cre driver is chosen. In summary, our data support that Secisbp2 is required for efficient selenoprotein expression and that it may play a role both in mRNA translation and mRNA stability. Comparison with mice lacking tRNA[Ser]Sec revealed residual selenoprotein expression and a less severe phenotype in Secisbp2-deficient mice, suggesting the existence of a compensatory or Secisbp2-independent molecular mechanism, which needs to be elucidated in future studies.
We chose hepatocytes as a model system, because, unlike mouse embryonic fibroblasts (28, 45), these cells survive abrogation of selenoprotein biosynthesis (43). A reason for this may be efficient transcriptional induction of Nrf2-responsive antioxidant proteins like glutathione-S-transferases (GST) (46). Hepatocytes allowed us to investigate a large number of selenoproteins at the level of protein expression or enzyme activity. We further took advantage of a conditional Trsp allele and compared the effects of Secisbp2 inactivation with tRNA[Ser]Sec inactivation. Throughout, we observed a low level of selenoprotein expression remaining after Secisbp2 inactivation. Similarly, quantification of Nrf2-dependent Gst mRNAs revealed significant induction of these genes in the Secisbp2 mutant liver, but much lower than in mice harboring a conditional deletion of Trsp (46). For example, we found a 1.3-fold induction (normalized to 18S-rRNA) of Gstm1 in our male mutants compared with 3.2-fold in Trsp mutants [normalized to Gusb mRNA in (45)]. Likewise, Gsta1 is induced 3.2-fold in male Secisbp2 mutants compared with 23.4-fold in Trsp mutants. Except for one male (Fig. 2B, lane 10), incomplete gene inactivation is not a likely reason for the milder effect, since we used the same Alb-Cre driver for both Secisbp2 and Trsp. Cre expression starts during embryonic stages, and the animals were analyzed at postnatal day 35 allowing ample time for Secisbp2 recombination, Secisbp2 protein turnover, and loss of Secisbp2-dependent selenoproteins. Cell types not targeted by Alb-Cre-like endothelial cells or Kupffer cells may account for a minor fraction of selenoprotein expression, but should contribute the same background in both Secisbp2 and Trsp mutant liver. Finally, within the liver, Dio1 and Sepp are selenoproteins specific for hepatocytes and residual expression of these proteins thus cannot stem from other cell types. This interpretation is supported by our hepatocyte culture experiments, in which Secisbp2- and Trsp-deficient cells were analyzed in parallel. Again, both the Dio1 activity and Sepp expression were higher in cells lacking Secisbp2 than in cells lacking Trsp. Compared with Trsp mutants, a higher Dio1 activity in Secisbp2-deficient hepatocytes was found despite lower Dio1 mRNA levels. Whereas these considerations are limited by measurements close to the detection limits of the methods, a comparison of embryonic development lends further support to the notion that Secisbp2 inactivation is less detrimental than Trsp inactivation, that is, Secisbp2 −/− embryos fail before gastrulation (clearly after implantation), while Trsp −/− embryos fail before implantation (6). This is consistent with the phenotypes of embryos lacking single essential selenoproteins. Gpx4 −/− embryos die before E7.5 (27, 45, 59). Txnrd1 −/− embryos die around E10.5 (5, 28). Developmental failure of Secisbp2 −/− embryos precedes failure in mice lacking glutathione biosynthesis (Gclc −/−), which do not develop mesoderm during gastrulation (49).
What could be the mechanism involved in the maintenance of a minor fraction of selenoprotein expression? One possibility is the low level expression of a truncated, hypomorphic Secisbp2 protein that initiates from Met302 in exon 6 after the Cre-induced frameshift. It is, however, inherently difficult to exclude the existence of a molecule that remained below the detection limit of our western blot. A second potential mechanism for functional compensation of Secisbp2 inactivation may be the activity of another SECIS-binding protein. The expression of other SECIS-binding proteins does not change at the protein level, although nucleolin mRNA levels were increased in female mutant mice. More importantly, none of the known SECIS-binding proteins have been shown to substitute for Secisbp2 in Sec incorporation, including SECISBP2L (19).
A third possibility is incorporation of another amino acid in place of Sec, ultimately a mechanism of nonsense suppression. Examples of nonsense suppression are already known with regard to selenoproteins. For example, the aminoglycoside G418 can stimulate the incorporation of Arg by a near cognate tRNA into Gpx1 (23). Dietary Se deficiency results in incorporation of Cys at the penultimate UGA codon of Txnrd1 leading to a variant with about 10% activity compared with the Sec-containing enzyme (32, 58). All these mechanisms of nonsense suppression would ultimately lead to full-length, but inactive or minimally active, selenoenzymes. In the case of Txnrd, a significant activity is preserved in male and female Secisbp2 mutants with 41% and 27% of wild-type activity, respectively. Considering that the amount of Txnrd1 mRNA is not reduced, a fourth, although hypothetical, possibility arises: a low-efficiency pathway of Sec incorporation that is independent of Secisbp2 analogous to pyrrolysine insertion (51). It will require future studies in carefully designed systems to uncover such a mechanism.
Finally, an intriguing novel finding of our study is the major role that Secisbp2 plays in selenoprotein mRNA abundance. We did not expect such a massive reduction of selenoprotein mRNAs in the absence of Secisbp2. Clearly, it was expected that failure to translate selenoprotein mRNAs may subject these transcripts to NMD, although to date, only the Gpx1 mRNA has been shown to undergo NMD (35). Our experiments revealed that various selenoprotein mRNAs react differentially to the absence of Secisbp2. Moreover, direct comparison with cells deficient for tRNA[Ser]Sec or cells lacking adequate Se demonstrates that lack of translation and subsequent NMD alone cannot account for reduced selenoprotein mRNA levels. Furthermore, several transcripts that showed a decrease in Secisbp2 deficiency do not follow the canonical rules for NMD (Table 2). Interestingly, some mRNAs were more strongly reduced upon Secisbp2 inactivation than upon Trsp inactivation. Whereas not explored in detail here, these findings point to a potential role of Secisbp2 in the stabilization of a subset of selenoprotein mRNAs in vivo. A previous study showed that stable knockdown of SECISBP2 in a malignant mesothelioma cell line reduced the levels of a different group of selenoprotein mRNAs (50). The differences between the two studies may be due to the cell type, in vitro versus in vivo, or knockdown versus Secisbp2 inactivation.
Differential binding of selenoprotein mRNAs by Secisbp2 has been implicated in establishing the hierarchy among selenoproteins and is sensitive to mutations in the lysine-rich domain of Secisbp2 (8). Patients carrying the R540Q mutation in SECISBP2 exhibit a selective deficiency of selenoprotein expression (20). If patients carry more severe mutations, they suffer from a more global selenoprotein deficiency associated with a more severe clinical syndrome (40). Fibroblasts from these patients show changes of mRNA levels in the same way as in Secisbp2-deficient hepatocytes (40). Nevertheless, phenotypes of patients with mutations in SECISBP2 are still weaker than complete Secisbp2 gene inactivation afforded in our model. This suggests that point mutations observed in patients target specific functions of SECISBP2 or lead to hypomorphic, but not entirely inactive, alleles.
Materials and Methods
Nomenclature
Secisbp2 and SECISBP2 refer to rodent and human proteins, respectively. Names in italics indicate the respective genes.
Mouse models
Transgenic mice harboring the knockout-first allele of Secisbp2 were obtained from the European Conditional Mouse Mutagenesis Consortium (EUCOMM) after the gene was prioritized upon our request. The construct encompasses a FRT-flanked gene-trap cassette in intron 4 leading to an in-frame protein fusion with exon 4 followed by polyadenylation (Secisbp2tm1a(EUCOMM)Wtsi). The gene-trap cassette was removed by intercross with a germline deleter FLPe transgenic mouse resulting in a conditional allele with exon 5 flanked by loxP elements (Secisbp2fl ). Deletion of conditional exon 5 in the germline using a deleter Cre transgenic mouse strain resulted in a Secisbp2 Δ5 allele, a functional knockout. The breeding colony was maintained on breeding diets (Ssniff, Soest, Germany) containing on average 0.2–0.3 ppm Se. Upon weaning, animals that were to be examined by western blotting and used for serum Se analysis were maintained on a low Se diet (Altromin, Lage, Germany; diet C1045 containing 0.08 ppm Se) supplemented with selenite to an adequate Se concentration (0.15 ppm Se, a dose defined as the recommended dietary allowance for mice, designated RDA) as in earlier experiments (36). The Se content of the diets used in the experiments were determined by total reflection X-ray fluorescence (TXRF) analysis that verified the intended Se content. Animal experiments were approved by the local governmental authorities (Landesamt für Gesundheit und Soziales, LAGeSo Berlin, Germany). Genotyping was done by PCR using primers Secisbp2-wt(fwd): GGTTCTGAGTTCCACTTAAAG, Secisbp2-rev2: GGTATGCAAGGGCCACCTTTG and Secisbp2-loxP(rev): GGTATGCAAGGGCCACCTTTG. Alb-Cre and Trsp genotyping was done as described (43). For Secisbp2 Δ5 genotyping, primers Secisbp2-rev2 (described above) and Secisbp2-fwd1: TCTGCTTCTGCCTCCTAAATG were used.
Primary hepatocytes
Primary hepatocytes were isolated as described (55) from female mice. Forty-eight hours after isolation, cells were harvested for analysis.
Hormone measurements
TSH and total serum T4 and T3 were measured by radioimmunoassays adapted to mice as described before (7).
Selenoenzyme analysis
Gpx assays were carried out with tert-butyl hydroperoxide and Txnrd activities were assessed with the DTNB assay in tissue homogenates as described (41). The protein concentration was determined by the Bradford method using IgG as a standard. The Dio1 activity was determined as described with 125I-rT3 as the substrate (52). Fifteen micrograms of membrane fraction protein was used, with 1 μM unlabeled rT3. The reaction time was 60 min and the reaction temperature 37°C. Five to six male and female animals were analyzed for each genotype. The assay was done in triplicate.
Insulin-dependent Txnrd assay
Since DTNB may be reduced by GSTs (which were found increased in the livers of Secisbp2-deficient mice), the insulin-dependent fluorescent Txnrd assay from IMCO (Stockholm, Sweden) was also used to determine Txnrd activities in 35 μg of protein from liver cytosols (26). We followed the manufacturer's recommendations with one minor modification, that is, the Txnrd standards were adjusted to match the range of our samples (in nM): 0.125, 0.25, 0.375, 0.50, 0.625 and results were given in fmol Txnrd per μg cytosolic protein.
Liver transaminase determination
ALAT and ASAT measurements in serum were performed according to standard coupled tests involving 2-ketoglutarate and NADH plus alanine and lactate dehydrogenase (for ALAT) or aspartate and malate dehydrogenase (for ASAT). The decrease in A340 was followed over 3 min and from the slope of the linear part of the curve, the activity was calculated as 1 U=1 μmol/min. The number of animals tested: male controls (6), female controls (9), male mutants (6), and female mutants (9).
Western blot analysis
An antiserum directed against the C-terminal 349 amino acids of mouse Secisbp2 was generated by the Protein Tech Group, Chicago, IL (used at 1:1000). For Gpx1 (1:10,000; rabbit antiserum from Abcam, Cambridge, United Kingdom), 25–100 μg protein from cytosolic fraction was separated on SDS/12% polyacrylamide gels. For Gpx4 (1:10,000; rabbit antiserum against amino acids 32–43 of rat protein made by Biosynthesis, Lewisville), 40 μg of whole testis homogenate and 25 or 100 μg of liver homogenate was used. The antiserum directed toward Sepp (1:400) has been generated in rabbits by immunization with a synthetic C-terminal peptide (ImmunoGlobe, Himmelstadt, Germany), and its specificity was verified using wild-type and Sepp-knockout mice (15, 43). For serum Sepp quantification, 0.2 μl serum was applied per lane. For Txnrd1 (1:1000; mouse monoclonal antibody; Abcam), 100 μg of protein from cytosolic fraction was used for western blot analysis. Antibodies against Sepk, Seps, eIF4a3, and L30 were from Sigma (Munich, Germany; ATLAS Prestige antibodies, rabbit polyclonal) used at 1:1000, 1:20,000, 1:1000, and 1:5000 dilution, respectively. After electrotransfer, nitrocellulose membranes were stained with Ponceau Red, photographed, and blocked with 5% BSA for 1 h at 25°C. As loading controls, the following antibodies were used: rabbit polyclonal β-actin antiserum (Sigma) at 1:2000 dilution, mouse monoclonal against beta I and II tubulin (Abcam) at 1:100,000, or mouse monoclonal 6C5 against GAPDH (Abcam) at 1:300,000.
Selenium measurements
Se was determined by TXRF using a Picofox™ S2 instrument (Bruker nano, Berlin, Germany) (15). Gallium was used as the internal standard for quantification and reference samples for serum (Sero, Billingstad, Norway) were included in all analyses and results were always within the reference range. All samples were measured in duplicate.
qPCR analysis
Total RNA was isolated from powdered mouse tissue according to the TRIzol protocol (Invitrogen, Darmstadt, Germany). Samples were treated with RQ1 RNase-Free DNase (Promega, Madison, WI). Total RNA from primary hepatocytes was isolated with the Aurum Total RNA Minikit (Biorad, München, Germany). cDNA was synthesized using the iScript cDNA synthesis kit (Biorad) according to the manufacturer's protocol. qPCR was performed using SYBR Green from Abgene (Thermo Scientific, Epsom, United Kingdom) on an iCycler (Biorad). Primers used for qPCR detection of Nrf2 target genes were as described previously (46). Primers specific for selenoproteins and Nrf2 target genes are described in Supplementary Table S1. 18S rRNA was used as reference gene for mRNA quantification.
Statistics
For all computations, GraphPad Prism software was used for the tests indicated in the figure legends. Data are expressed as mean±S.E.M. Statistical significance was determined and indicated as *p<0.05, **p<0.01, or ***p<0.001.
Footnotes
Acknowledgments
The authors thank Antje Kretschmer, Anja Fischbach, Vartitér Seher, and Ursula Reuter for excellent technical assistance. Dr. Markus Schupp, Charité-Universitätsmedizin Berlin helped with primary hepatocyte cultures, Dr. Carmen Birchmeier provided FLPe and Cre deleter mice, Dr. Wolfgang Wurst prioritized Secisbp2 in EUCOMM. The study was funded by Deutsche Forschungsgemeinschaft DFG Schw914/2-1 and GK1208 to the U.S. and National Institutes of Health grants R01DK085391 and R01DK07859 to D.M.D.
Author Disclosure Statement
All authors stated that no competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.