
Abstract
Introduction
T
While thioredoxin system (TS) has received a lot of attention as a pharmacological target in cancer, its role is emerging in the cardiovascular field. Our in vitro and in vivo approaches unraveled an extended pathway that links the misfolded and aggregate-prone R120GCRYAB to TS activation through HDAC dependent deacetylation of thioredoxin reductase 1 (TrxR1). This deacetylation activates TrxR1 and subsequently, mitigates protein aggregation due to downstream TS targets, which could include Bcl-2-associated athanogene 3 (BAG3). Ultimately, this pathway underscores the potential pharmacological interferences of therapeutic maneuvers acting on histone deacetylase 3 (HDAC3) and/or TS.
The expression of R120GCRYAB was shown to trigger extensive modifications in the cardiomyocyte transcriptome (45). Since CRYAB is not a transcription factor, its transcriptomic biosignature was likely to be obtained through various mechanisms altering gene regulation such as protein sequestration, which is distinctively relevant to an aggregate-prone disease. For example, Kelch-like ECH-associated protein 1 (Keap-1), the natural negative regulator of a member of the Cap n’ Collar transcription factor family, Nrf2 (nuclear erythroid factor 2-related factor 2 [Nfe2l2]) was relocated into the protein aggregates observed in hR120GTg cardiomyocytes (46). Nrf2, on dissociation and release from Keap1, was stabilized and translocated into the nucleus, where it could bind to and increase the transcription of target genes characterized by DNA binding sequences such as antioxidant responsive elements or electrophile response elements (19, 36). In addition, the oxidative environment of the hR120GTg young cardiomyocyte further stimulated Nrf2 with the expected increase in the expression of its target genes (46).
An important contribution to the Nrf2 antioxidant function relies on the thioredoxin system (TS) and, in particular, on thioredoxin reductase 1 (TrxR1) and thioredoxin 1 (Trx1) (7, 18, 19, 25, 31, 36). TS, first identified in yeast 5 decades ago, broadly consists of thioredoxin (Trx1 and Trx2), thioredoxin reductase (TrxR1 and TrxR2), NADPH, and a natural Trx inhibitor, the thioredoxin-interacting protein (TXNIP) (2, 22, 35, 68). Trx1, substrate of TrxR1, and Trx2, substrate of TrxR2, are located in the nucleo-cytoplasmic and mitochondrial compartment, respectively. Through their redox-sensitive cysteine residues, both protein (e.g., thioredoxin) and nonprotein (e.g., glutathione) thiols play key roles in redox homeostasis in biological systems. The universally conserved active site -Cys-Gly-Pro-Cys in thioredoxin (thioredoxin fold) helps catalyze steps in oxidative protein folding via protein–protein interactions and covalent catalysis acting as chaperones to generate a native fold. TrxR exists as a homodimer, with a head to tail orientation with each monomer consisting of an FAD prosthetic group, an NADPH binding site, and a catalytic site with a redox-active disulfide bond. Due to the significant involvement of TrxR in cell survival and proliferation, numerous investigations have focused on a better understanding of the TrxR structure-function relationship to design efficient inhibitory drugs that could be of benefit to treat diseases such as cancer (6, 60).
In the cardiovascular field, studies using transgenic and knock-out mouse models implicated TS in the protection against cardiac hypertrophy and ischemia/reperfusion (I/R) damages (12),which is a major cardiac stress triggering a complex REDOX response (32, 43, 54, 58). Loss of TrxR1 expression in cardiomyocytes (cTrxR1−/− ) had a tendency to render those cells more sensitive to I/R. (24). In contrast, the same study showed a more drastic implication of the mitochondrial Trx2-TrxR2 system as a key protector against I/R damages. Hence, this work suggested that TrxR1 and TrxR2 are not equally involved in cardiac stress (24).
In addition to cardiac hypertrophy and REDOX dynamic (5, 44), which possibly involve TS, the disease-prone R120GCRYAB is associated with the formation of protein aggregates. Since Trx acts on the REDOX status of cysteine, its activity (coupled to TrxR) can modify protein conformation and, therefore, alter the aggregation property of proteins interacting with Trx. Despite an abundant literature on TS roles (2, 21, 22, 35), a few studies have experimentally examined the impact of TS on the aggregation of misfolded proteins. In yeast, it was shown that TS was critical to prevent the aggregation of ribosomal proteins under REDOX stress (47). In contrast, in vitro experiments using physiological amounts of Trx with reconstituted TS showed the conversion of prion protein into an aggregate-prone form after reduction by Trx (48). In other words, TS activity would be deleterious in promoting the pathogenic form of prion protein. It is important to note that CRYAB does not contain any cysteine, and, thus, it cannot be a direct substrate for TS. However, as a modified chaperone, the mutant R120GCRYAB behaves differently from wild-type (WT) CRYAB and is involved in a distinct complex network of protein–protein interactions with other sHSPs (9, 55) or non HSP proteins [vimentin (1); VDAC/porin (38); Bcl-2 associated athanogene 3 (BAG3) (20)]. It can be speculated that those complexes which are not yet fully identified and characterized could be targeted by TS.
Combined with the previous link between R120GCRYAB cardiomyopathy and Nrf2 activation (46), these data prompted us to test the hypothesis that TS is impacted in the R120GCRYAB-dependent cardiomyopathy. We found that expression of R120GCRYAB alters TS at multiple levels (expression, activity, and regulation) and in a distinct, compartmentalized way (cytosolic vs. mitochondrial). This study combining in vivo and in vitro experiments brings new insights into the involvement of the TS in diseases related to misfolded proteins and chaperone-dependent cardiomyopathy.
Results
Expression and REDOX status of thioredoxin in R120GCRYAB transgenic (hR120GTg) hearts
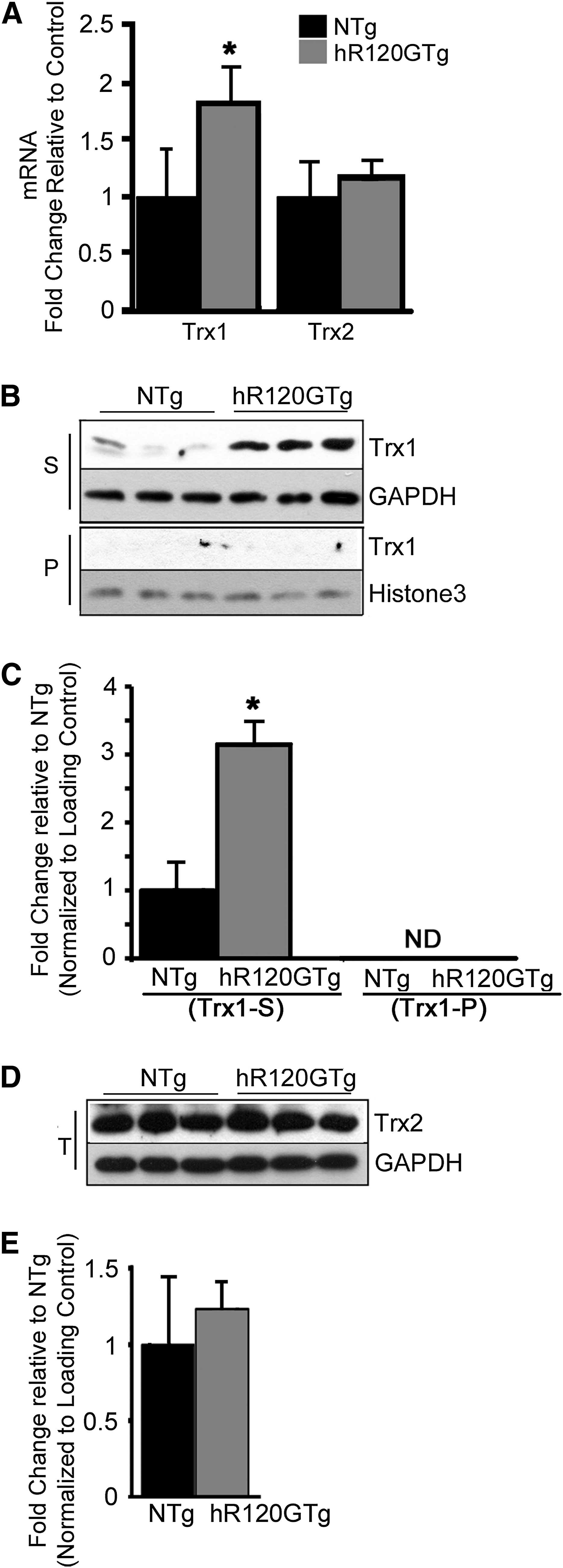
To determine the effects of R120GCRYAB on TS, we first examined the expression of both Trx1 and Trx2 in hearts collected from 6 month-old hR120GTg and nontransgenic (NTg) males. The level for Trx1 transcripts was significantly increased by 1.8-fold in Tg males, but no change was observed for the mitochondrial counterpart, Trx2 (Fig. 1A). The higher Trx1 mRNA level corresponded with a protein level that was significantly augmented by 3.14-fold (p<0.05) (Fig. 1B, C). Trx1 protein was found exclusively in the Triton X100 soluble fraction (S) and not in the insoluble/pellet fraction (P), indicating that Trx1 was not sequestered in the R120GCRYAB aggregates. Trx2 protein level remained unaltered in total extracts (T) (Fig. 1D, E).
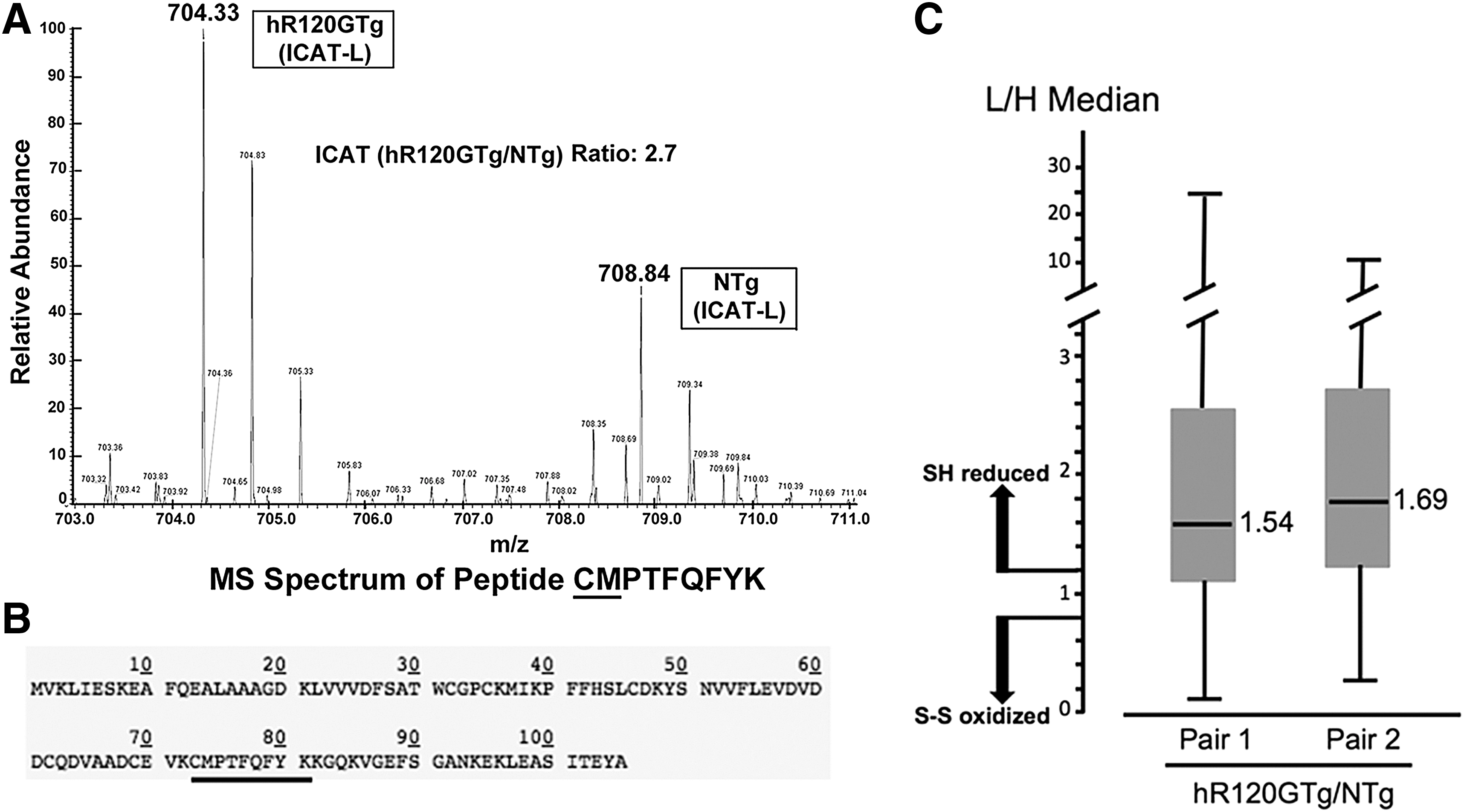
The augmented expression of Trx1, a key actor in reduction-oxidation reaction cycle, prompted us to analyze redox-sensitive cysteines in similar heart samples using isotope coded affinity tag-mass spectrometry (ICAT-MS). ICAT-labeled peptide fragments used for identification and quantifications were performed by searching the MS/MS spectra against international protein index mouse database. Cysteines in Trx1 peptide fragments (73–82) were on average 3.02±0.5 times more reduced in hR120GTg compared with NTg hearts (Fig. 2A, B). This piece of data was consistent with the global median of ICAT ratio for all the identified peptides (on average 1.61±0.11), indicating that redox-sensitive proteins are >60% more reduced in hR120GTg in comparison with NTg hearts (Fig. 2C). Additional information and the complete list of identified peptides are presented in Supplementary Table S1 (Supplementary Data are available online at
TrxR1 activity, expression, and localization
Thioredoxin reductase (TrxR) activity, which is known to reduce Trx1 with NADPH acting as a proton donor, could be involved in the more reduced state of Trx1 found in hR120GTg cardiomyocytes (21). Total extracts from heart samples were used to measure the global TrxR activity (TrxR1+TrxR2), which was 2.35±0.37-fold greater in Tg compared with NTg (p<0.001) (Fig. 3A). To determine the contribution of the mitochondrial TrxR2, mitochondria were isolated and TxR activity was assessed on this fraction (Fig. 3A, Mito Extract). TrxR2 activity was not statistically different between the hR120GTg and NTg samples (Fig. 3A). We concluded from those data that the higher TrxR activity found in the hR120GTg hearts was contributed by TrxR1.
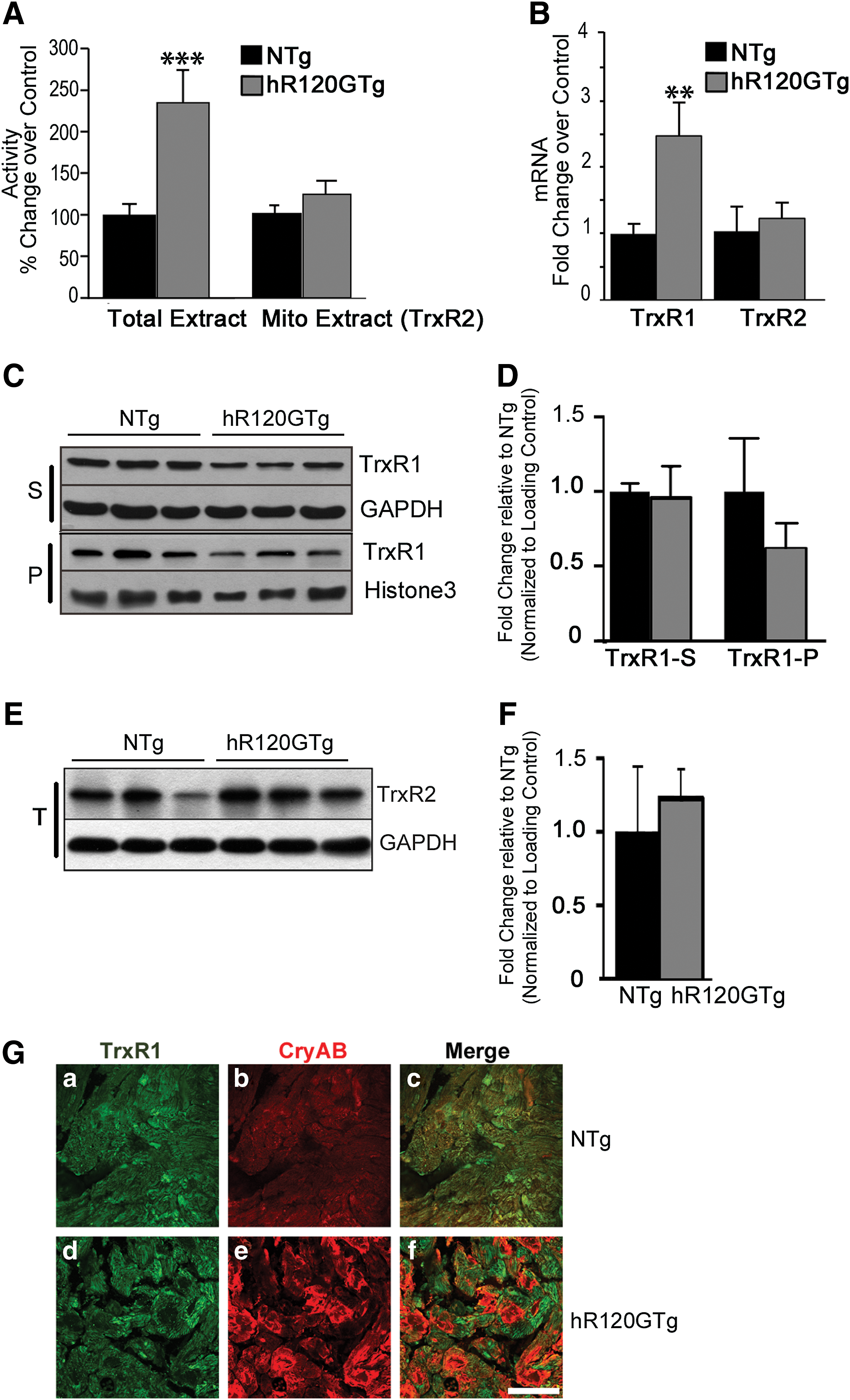
Since an increased TrxR1 activity might be caused by a higher level of TrxR1 expression, the relative amounts of TrxR1 and TrxR2 mRNA were assessed (reverse transcription and quantitative PCR [RTqPCR]). TrxR1 but not TrxR2 transcripts were significantly augmented in the hR120GTg samples (Fig. 3B). However, Western blots showed that the quantity of TrxR1 protein, which could be found in both soluble (S) and pellet (P) fractions of the heart homogenates, was not statistically different between hR120GTg and NTg samples (Fig. 3C, D and Supplementary Fig. S1). The TrxR2 protein level was assessed in parallel and was not different in hR120GTg and NTg samples. This result was consistent with the relative quantification of TrxR2 transcript (Fig. 3E, F).
Next, immunohistochemical detection of TrxR1 protein was performed to determine whether TrxR1 identified in the insoluble/pellet (P) fraction was localized in the R120GCRYAB aggregates (Fig. 3G). TrxR1 signal was similarly visible in the cytoplasm of both NTg (upper panel, Fig. 3G-a) and hR120GTg (lower panel, Fig. 3G-d) cardiomyocytes and did not merge with R120GCRYAB immunodetection that was concentrated in the aggregates (Fig. 3G-f).
All together, these findings revealed compartment-specific changes of TS triggered by hR120GCRYAB expression, with distinct modifications of cytoplasmic Trx1-TrxR1 and not the mitochondrial Trx2-TrxR2 counterpart. Such an observation was also consistent with TrxR1 and TrxR2 being regulated differentially as shown in other studies (13).
Post-translational modifications of TrxR1
Since TrxR1 protein level was not changed in R120GCRYAB samples, the higher level of TrxR1 activity was most likely due to post-translational modifications (PTMs) of the enzyme. Bioinformatic analysis of TrxR1 amino-acid sequence suggested the existence of potential sites for both phosphorylation and acetylation PTMs (Supplementary Fig. S2 and Supplementary Table S2) (23, 62). Therefore, we sought to determine whether TrxR1 PTMs could account for TrxR1 enhanced activity.
First, the global TrxR activity was determined in hR120GTg heart homogenates incubated in vitro with Lambda phosphatase. This treatment did not alter the higher activity observed in hR120GTg samples, leading us to conclude that phosphorylation likely plays a negligible role in TrxR1 activity (Fig. 4A).
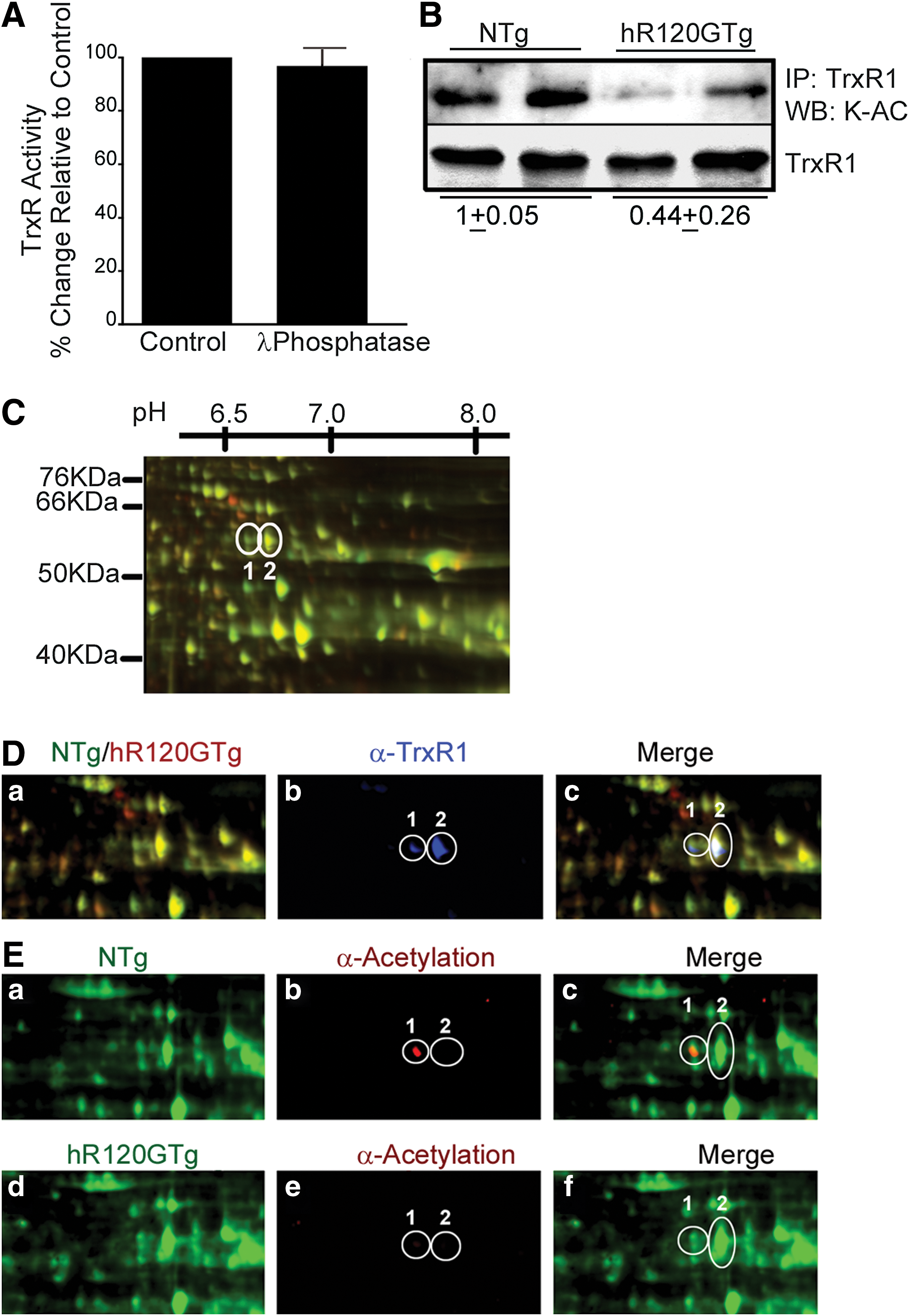
Next, the acetylation of TrxR1 was assessed by two complementary methods. TrxR1 protein from NTg and hR120GTg mice hearts was immunoprecipitated and immunoblotted with antibodies that specifically recognized acetylated lysine (K-Ac) (Fig. 4B). The ratio between NTg and hR120GTg for the acetylated TrxR1 signal was 1/0.44, indicating the acetylation was 2.2±0.7-fold higher in NTg (Fig. 4B). This experimental finding was further supported from data obtained following 2D difference in gel electrophoresis (2D-DIGE) performed with NTg and hR120GTg heart extracts. NTg and hR120GTg samples were labeled with green and red fluorescent reagents, respectively, and Figure 4C displays a part of the gel image where TrxR1 was located, along with an estimated molecular weight (MW) and pI grid (see Supplementary Fig. S3 for the complete gel image). An overlay of 2D-Dige separated protein and TrxR1 immmunoblot (blue) images obtained from NTg and hR120GTg samples is presented in Figure 4D. Anti-TrxR1 antibody (α-TrxR1) recognized two adjacent spots for which MW, and pI matched the calculated values from TrxR1 protein sequence (Fig. 4D-b). Of the two TrxR1 spots, only spot 1 strongly interacted with anti-acetylated lysine (α-Acetylation) antibody (Fig. 4E-b, e). Acetylation level for spot 1 in NTg was at least four-fold higher in comparison to hR120GTg after normalization to protein level (Fig. 4E). Data shown in Figure 4B and E indicate that TrxR1 was predominantly deacetylated in hR120GTg in comparison to NTg mice hearts.
Evidence from animal models that supports HDAC3 as a possible link between hR120GCRYAB and TrxR1 Activity
The augmented TrxR1 activity might be causally linked to the lower acetylation level of TrxR1, which, in turn, could be a consequence of deacetylase activity provided by members of either Class I or Class II Hdacs.
To test this hypothesis, histone deacetylase (HDAC) expression was assessed in hR120GTg hearts. Using RTqPCR, we analyzed mRNA levels for representative members of HDAC Class 1 (Hdac1, Hdac2, and Hdac3) and Class 2 (Hdac4, Hdac5, and Hdac6). Both Hdac1 and Hdac3 were significantly up-regulated in the hR120GTg versus NTg mouse hearts (Fig. 5A). At the protein level, the amount of Hdac3 was augmented by 11.75±6.02-fold (p<0.005) in total extracts from hR120GTg versus NTg hearts (Fig. 5B, C). Western blots probed for Hdac1 and 2 proteins showed that hR120GTg had a less impact on these two other deacetylases (Supplementary Fig. S4). To determine whether the expression of R120GCRYAB was sufficient to provoke the induction of Hdac3 expression, we took advantage of a genetic approach to eliminate the expression of the endogenous WTCRYAB by intercrossing hR120GTg and double knockout (DKO) animals that are deficient in WTCryab and have also lost the neighbor Hspb2 gene (MGI: 2652177) (4). In the absence of an available single knockout for Cryab, Cryab;Hspb2 knockout (DKO) mice represented a valuable model to test the effect of WTCryab deficiency, as we showed that the lack of HspB2 did not exhibit any marked cardiac phenotype (26). Furthermore, hearts from DKO/hR120GTg developed cytoplasmic aggregates as observed in the original hR120GTg line (44) (Supplementary Fig. S5) and second, they did not exhibit a reduced lifespan in comparison with hR120GTg expressing WTCryab (data not shown). Hdac3 protein was similarly increased in heart extracts of hR120GTg mice from DKO or WT background (Fig. 5B–E).
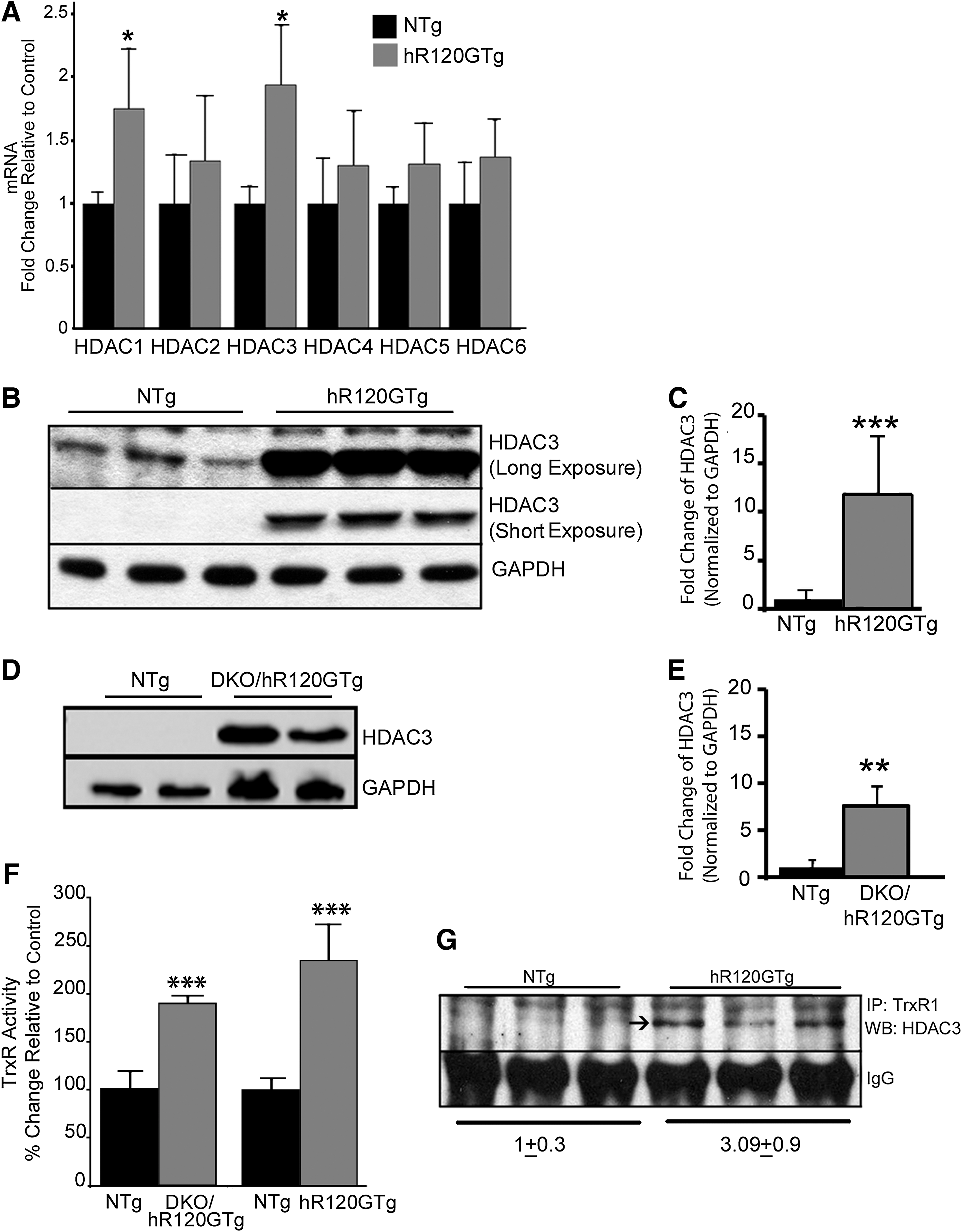
The correlation between Hdac3 and TrxR1 activity was then assessed in hR120GTg animals from either DKO or WT background. A strongly increased TrxR1 activity was observed in both types of animals (NTg vs. hR120GTg, p<0.001) (Fig. 5F). Nevertheless, the lack of WTCryab inflected the induction of TrxR1 activity (hR120Tg versus DKO/hR120GTg, p<0.05). This suggested that, although WTCryab was not required to trigger TS modification, the sHSP could still contribute and modulate those changes.
If Hdac3 is a potential candidate deacetylating TrxR1, those two proteins could be expected to share protein–protein interactions. To test this possibility, immunoprecipitation of TrxR1 was performed and followed by immunoblotting with Hdac3 antibodies. Hdac3 signal was only visible in the TrxR1 immunoprecipitates from Tg samples, suggesting that TrxR1 and Hdac3 were a part of the same protein–protein interacting complex (Fig. 5G). The reciprocal immunoprecipitation using Hdac3 antibodies did not produce exploitable results. To determine whether TrxR1 and Hdac3 were direct interactors, yeast two-hybrid (Y2H) experiments were performed but no interaction was found using this experimental procedure (data not shown).
Functional interactions impacting TrxR1 activity in cardiac cells
To gain more mechanistic insights into the functional interactions modulating TrxR1 activity, we performed a series of gain-of-function experiments. Proof-of-concept experiments were first carried out with the easily transfected HEK 293 cells, and they showed that overexpression of TRXR1 or HDAC3 enhanced TrxR1 activity, which could be subsequently reduced by inhibiting HDAC activity (Supplementary Fig. S6). Then, we used neonatal rat cardiomyocytes (NRCM), a more relevant model for cardiac pathophysiology, to repeat the same experiments. As expected, TrxR1 activity was significantly increased after TRXR1 transfection (close to two-fold, p<0.05 compared with empty vector [EV] transfection, Fig. 6A). HDAC3 transfection also elevated the TrxR1 activity, albeit to a lesser degree (1.5-fold, p<0.05 compared with EV transfection, Fig. 6A). Next we tested the ability of the broad spectrum HDAC inhibitor trichostatin A (TSA) to alter the higher level of TrxR1 activity obtained after TrxR1 transfection. A 6 h treatment (1 μM, TSA) was sufficient to significantly reduce the activity of TrxR1 by 25%, indicating that HDAC activity was critical for modulating TrxR1 function (Fig. 6B). Those experiments combining gain and loss of HDAC3 function were in agreement with the involvement of HDAC activity in regulating TrxR1 activity.
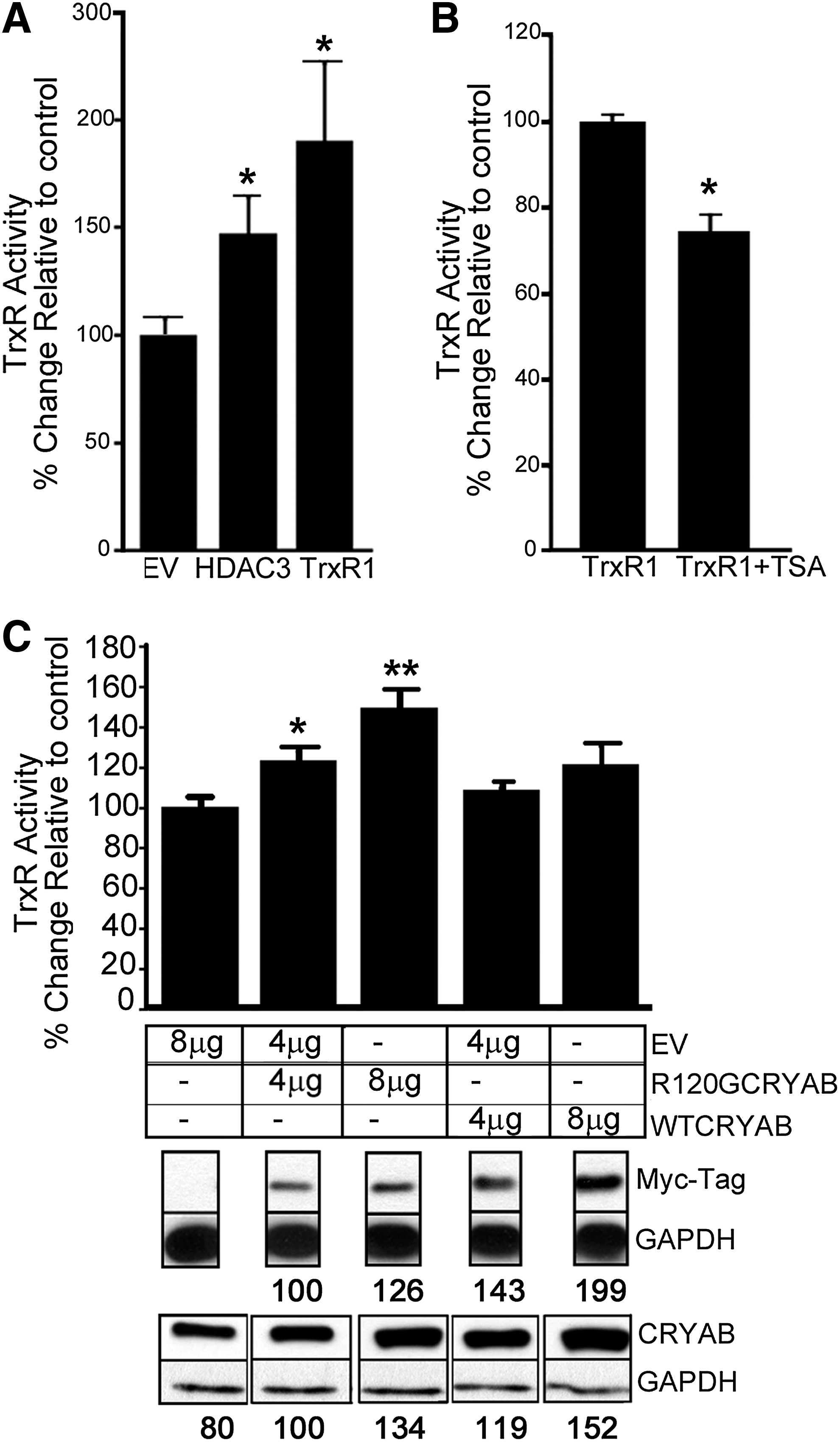
Next, we addressed the question of the respective contribution of WTCRYAB and hR120GCRYAB to the augmented TrxR1 activity as suggested by our in vivo experiments (Fig. 5F). Four or 8 μg of Myc-WTCRYAB and–R120GCRYAB expressing constructs was transfected into H9c2 cardiomyoblasts (Fig. 6C). Western blots were probed with two types of antibodies, either against Myc-Tag detecting only the exogenous proteins or against CRYAB recognizing both the endogenous and exogenous proteins. Relative quantification showed that transfection with WTCRYAB led to a larger amount of synthesized protein than R120GCRYAB (see numbers indicated for each band, Fig. 6C). TrxR1 activity was significantly increased by R120GCRYAB expression in a dose-response manner (4 vs. 8 μg R120GCRYAB vector, p=0.01). In comparison, there was a trend for WTCRYAB expression to cause a modest increase in TrxR1 activity without reaching full significance (EV vs. 8 μg WTCRYAB vector, p=0.057). Therefore, it can be concluded that elevating TrxR1 activity was a part of the gain of function observed with the expression of the mutated hR120GCRYAB.
In vitro and in vivo aggregate growth and TrxR1 activity
R120GCRYAB-related cardiomyopathy is a progressive disease whose major cellular hallmark is the formation of intracytoplasmic aggregates that are resulting from the accumulation of misfolded CRYAB. The detailed mechanisms leading to aggregate development remain overall poorly known. To determine whether TrxR1 activity was functionally linked to aggregate growth, an inhibitor of TrxR1 named aurothioglucose (ATG) was utilized, which could not only be added to cell culture medium but also eventually administered to animals. This drug is approved by the FDA as an anti-inflammatory agent (Solganal®) to treat severe rheumatoid arthritis (40, 56).
First, rat H9c2 myoblasts were transfected with R120GCRYAB expressing vectors and exposed or not to ATG according to the protocol described in Figure 7A. The number of transfected cells presenting CRYAB immuno-positive aggregates was monitored at 24 and 72 h (Fig. 7B). In the presence of ATG, which significantly reduced TrxR1 activity (>90%, data not shown), a higher number of cells contained aggregates. In addition, ATG treated cells contained larger aggregates at 72 h (aggregate area in ATG-treated versus non-treated cells, p=0.05) (Fig. 7C). Nevertheless, some heterogeneity in the morphology (small and dispersed versus more compact) and size of the aggregates was observed in transfected H9c2 as shown in Figure 7D, which presents three microscopic fields for each condition.
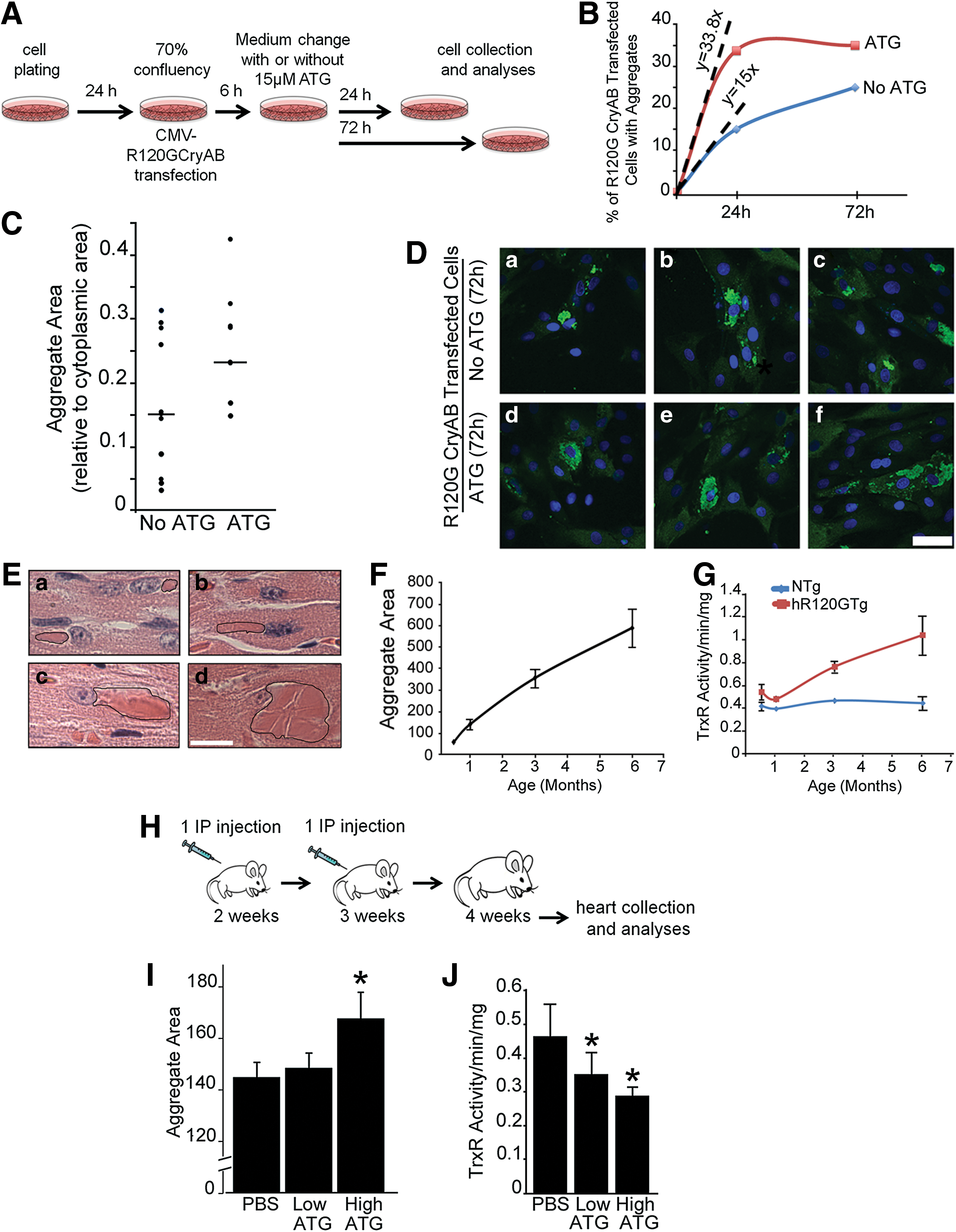
Although the time course of aggregate formation has been briefly reported in transfected cells (9, 53, 55, 59), this process has not been characterized in the hR120GTg mice used in the present study (44). We measured aggregate growth in hR120GTg males from 0.5 to 6 months after birth. Already at 0.5 month, aggregates were located close to the nucleus and then linearly expanded from this position (Fig. 7E, F).
The data presented in Figure 3A revealed that TrxR1 activity was significantly increased in hR120Tg hearts at 6 months, but it was not known whether it was a progressive or an acute change during the life of those cardiomyopathic animals. So, to determine whether a biochemical parameter such as the TrxR1 activity could evolve similarly along time in hR120GTg in comparison to NTg animals, measurements were made at 0.5, 3, and 6 months. In comparison to NTg, TrxR1 activity was already significantly higher in hR120GTg animals at 0.5 month and was further increasing till it reached 6 months (Fig. 7G). Thus, these data indicated that aggregate and TrxR1 activity collinearly progress in hR120GTg transgenic animals.
Next, to set up in vivo experiments using ATG inhibitor, we took into account the data presented in Figure 7F showing a rapid increase in aggregate size from 0.5 to 1 month, delimiting a brief window during which the effect of TrxR1 inhibition could be efficiently revealed. Animals received two injections of the inhibitor by an intraperitoneal injection at 1 week interval following the protocol described in Figure 7H. Hearts were collected 1 week after the second injection when the animals were 1 month old. The same protocol was applied for the administration of the inhibitor at a low (0.025 mg/g body weight) or a high (0.05 mg/g body weight) concentration (Fig. 7H). Aggregate area was measured and compared between ATG and phosphate-buffered saline-treated hR120GTg hearts. ATG at a low dose did not modify aggregate formation, while the administration of a higher dose led to a significant increase in aggregate size by 16% (Fig. 7I). TrxR1 activity was reduced in a nonproportional way when doubling ATG dosage led only to an additional 16.3% reduction (Fig. 7J). Nevertheless, these data suggested that there was a threshold below which lower TrxR1 activity impacted aggregate growth.
Those in vitro and in vivo experiments combined inhibition of TrxR1 and measurement of aggregate development in two different contexts, one being transfected cells continuously exposed to ATG and the other testing ATG in transgenic young animals that had already started displaying distinct cardiac aggregates (Fig. 7E). Despite those experimental differences, H9c2 and transgenic cardiomyocytes expressing R120GCRYAB responded similarly to ATG by enhancing aggregate development. We concluded that drug targeting of TrxR1 activity had direct effects on aggregate formation in vitro and in vivo, suggesting potential therapeutic avenues for modifying disease onset and progression.
R120GCRYAB as a distinct partner orchestrating protein complexes
It is expected and already partially demonstrated that the misfolded R120GCRYAB participates in modified protein complexes (1, 9, 20, 38, 55), although a systematic analysis of this mutated interactome has not yet been described. In the context of this study, TrxR1 was the first candidate interactor to be tested to support the underlying hypothesis that such an interaction could contribute to enhancing TrxR1 activity in hR120GTg heart. TrxR1 immunoprecipitated within a complex recognized by CRYAB antibodies and reciprocally in hR120Tg while NTg heart samples, which expressed only the WT-endogenous CRYAB, failed to provide a visible signal (Fig. 8A). Western blot of TrxR1 after pulling down TrxR1 in both NTg and Tg heart homogenates confirmed the comparable level of immunoprecipitated TrxR1 in both samples. Since antibodies against CRYAB cannot discriminate between the WT-endogenous and the R120GTg forms, it was not technically possible to find out whether WT or R120GCRYAB proteins, or both, were involved in the TrxR1 complexes observed with hR120GTg samples. For this, the animal model DKO/hR120GTg, which is deficient in WTCRYAB, was utilized (44). TrxR1 was immunoprecipitated from DKO/hR120GTg samples and immunoblotted with CRYAB antibodies. As shown in Figure 8B, those antibodies could label TrxR1 immunoprecipitate in the DKO/hR120GTg (but not in NTg) samples, indicating that R120GCRYAB produced in hR120GTg heart was the main and sufficient contributor to this complex.
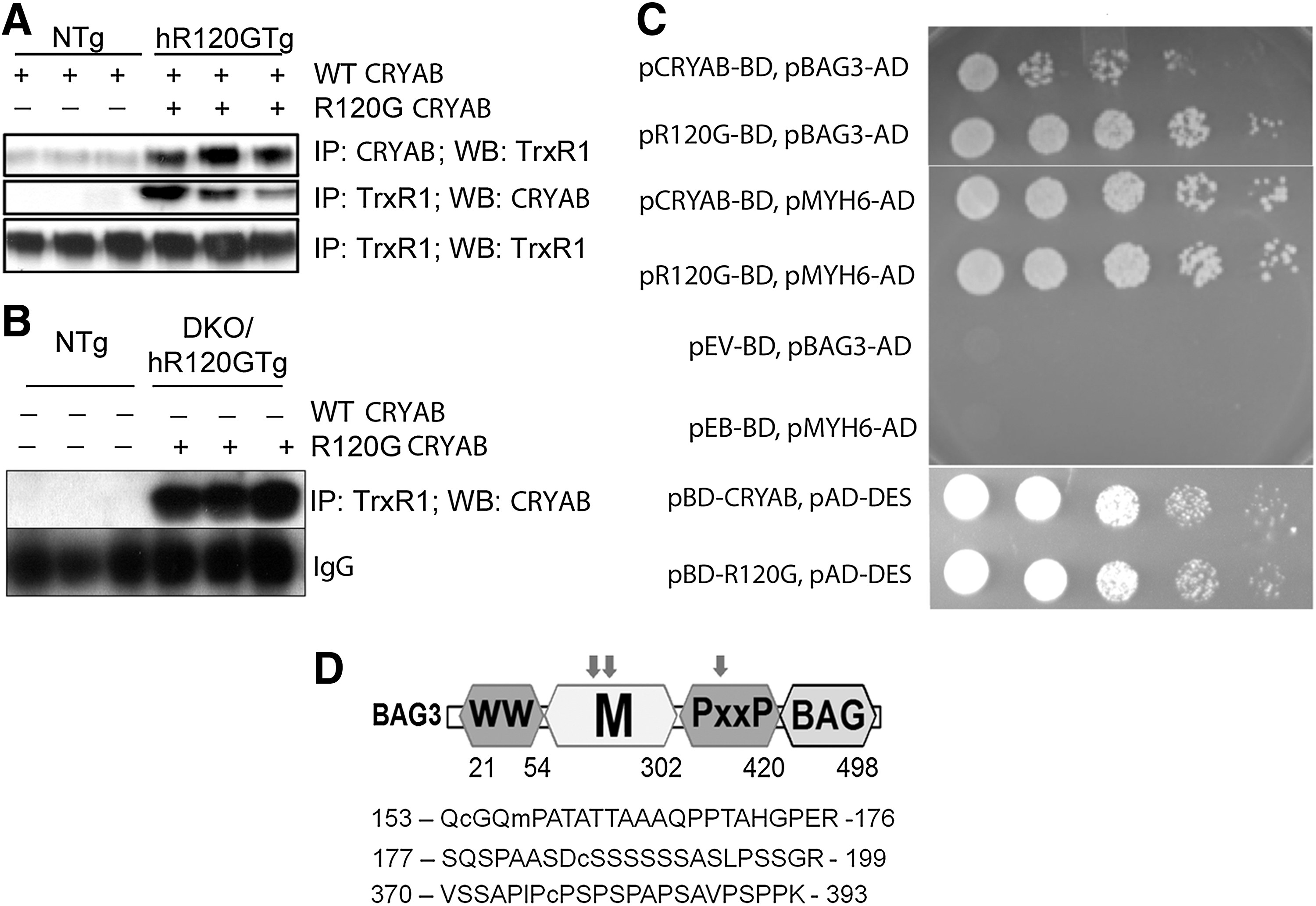
The second candidate we investigated was found through an unbiased Y2H screen that was performed to systematically identify all possible direct binding partners of CRYAB and R120GCRYAB using a cardiac cDNA library (Clontech) (manuscript in preparation). This screen retrieved the BAG3, which was previously shown to play a variety of regulatory roles in cellular adaptation to stress (49). Importantly, BAG3 had been identified as an important protective agent against the R120GCRYAB aggregates and was found, in overexpressing cells and GST pull-down assay, to interact preferentially with the mutated CRYAB (20). We took advantage of the Y2H system to confirm this altered interaction where R120GCRYAB exhibits a much stronger affinity for BAG3 than WTCRYAB (Fig. 8C). Still, this could have been due to a simple and undistinct increase in aggregation of R120GCRYAB with all CRYAB binding partners. To demonstrate the specificity of this interaction, we tested 10 other CRYAB binding partners retrieved from the Y2H screen and found that WT- and R120G-CRYAB equally associated with all of them (e.g., Myh6, Desmin shown in Fig. 8C). Interestingly, three peptides corresponding to BAG3 sequence were listed in the ICAT screen as significantly more reduced (see Supplementary Table S1 and Fig. 8D). These results posit BAG3 as a valuable molecular link between the reducing TS and the limitation of aggregate development (see discussion, Fig. 9).
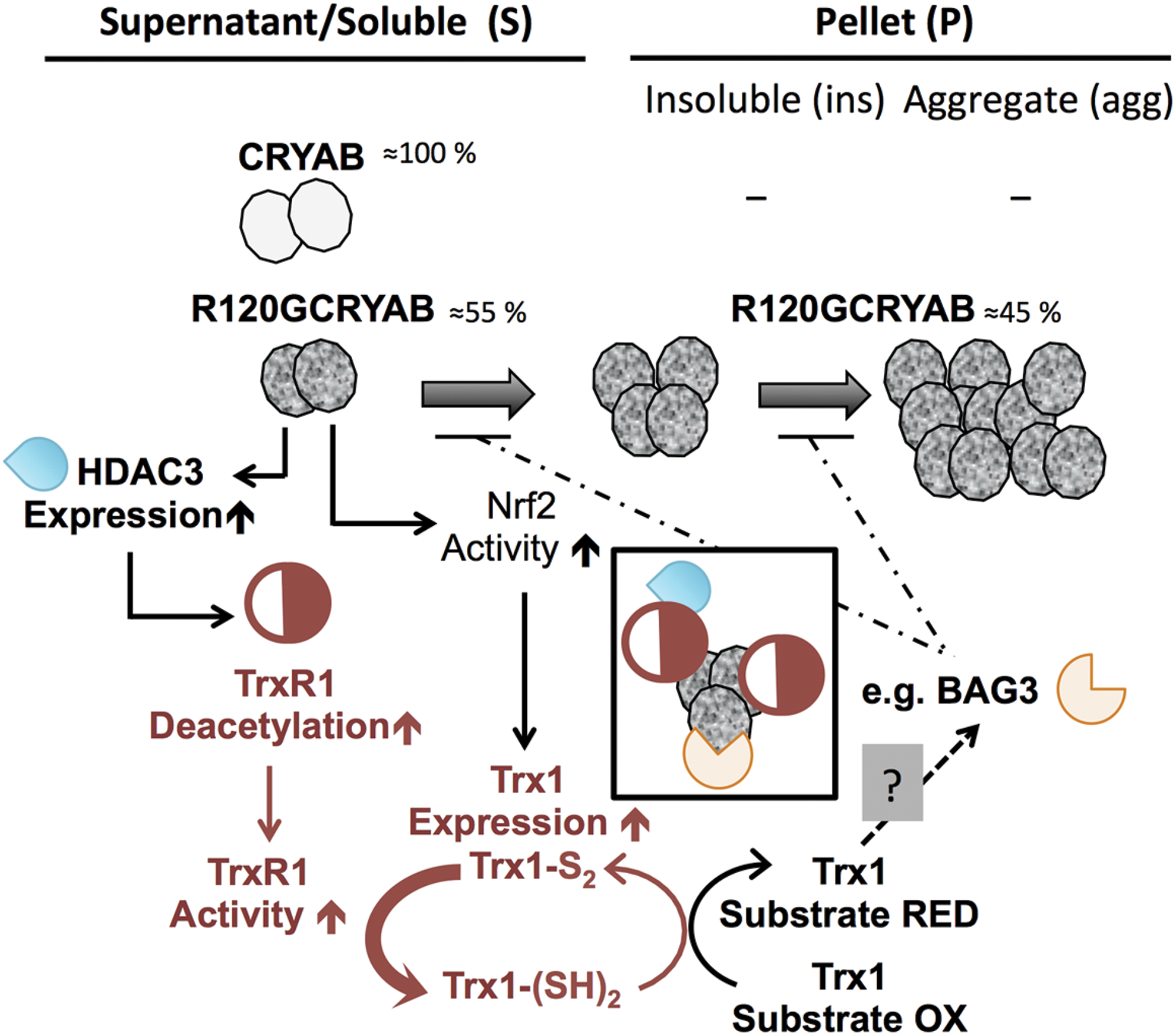
Discussion
The primary finding of our study is the identification of multiple and distinct modifications in the TS, provoked by the cardiac expression of the aggregate-prone R120GCRYAB. This included the discovery of the critical role played by the activity of TrxR1, which mitigated aggregate growth. Molecular dissection of upstream regulators and downstream effectors of TS identified HDAC3 and BAG3, respectively. Figure 9 recapitulates the main findings of our study along with the underlying mechanisms and hypotheses.
Acetylation, a new layer of regulation for the TrxR1 activity
Lysine acetylation is a well-known PTM, which has been the focus of extended proteome-wide analyses to define the so-called acetylome and to determine how acetylome variations could affect (patho)physiological functions (10, 57, 65). Using gain-and-loss-of-function experiments, our study showed that TrxR1 is a member of the cardiac acetylome and acetylation is a molecular mechanism that regulates its activity. This is a new layer of regulation for this selenoenzyme beyond its own protein level, which is first dependent at the transcriptional level, on activation by regulators such as Nrf2 (36), and then, importantly, relies on selenium levels to provide the required selenocysteine for protein synthesis (17). A genetic approach had shown that TrxR1 haploinsufficiency in heterozygous TrxR1 knockout mice correlates with lower TrxR1 activity (30). In fact, TrxR1 expression can be unchanged while its activity is modified. This was observed in the present study, and an analogous situation was reported by Lamoke and collaborators, showing that overexpression of amyloid β in mouse retina was associated to a reduced TrxR1 activity (33). The authors speculated that TrxR1 could be post-translationally modified, but they did not provide any specific data to support their conclusion. Taken together, this study and our work revealed that there might not be a unique or simple correlation between expression of misfolded proteins (i.e., amyloid β or hR120GCRYAB) and a defined change in TrxR1 acetylation/activity (i.e., reduced or augmented, respectively).
The pleiotropic roles of cardiac HDAC3
Human R120GCRYAB transgenic expression was associated with a higher level of HDAC3. However, the regulation of HDAC3 expression in cardiomyocytes is poorly understood and only HDAC3 down-regulation has been reported with isoproterenol treatment while other cardiac stresses (e.g., angiotensin infusion, transaortic banding, and myocardial infarction) had no effect on Hdac3 transcript levels (41). HDAC3 deacetylase activity either modulates gene expression through PTM of histones and/or transcription factors [i.e., aberrant expression of cardiac metabolic genes in Hdac3 cKO (41)] or modifies proteins independently of direct gene expression effects [i.e., cardiac myosin heavy chains (51)]. This dual functionality is particularly relevant for HDAC3, which, among class I HDACs, exhibits several distinct characteristics such as a greater shuttling mobility between the nucleus and the cytoplasm as well as a different molecular organization, suggesting the possibility of HDAC3 specific interactors. Within this context, our study uncovered TrxR1 as a new HDAC3 target protein. This molecular link between HDAC3 and TrxR1 led us to speculate that pharmacological tools inhibiting HDAC might have a synergistic effect with drugs acting on TrxR1. Since both TrxR1 (6, 60) and HDAC inhibitors (15) are a part of the therapeutic agents used in cancer treatment, our data reinforce the importance of evaluating cardiac function during such treatments.
New genes for a molecular pathway modifying R120GCRYAB aggregate formation
Expression of R120GCRYAB causes a progressive disease with adult onset reported in human patients (61). Consistently, we observed a progressive increase in aggregate size as well as a similar evolution of TrxR1 activity in hR120GTg animals from 0.5 to 6 months after birth. A causal link between these observations was supported by the increased development of aggregates when TrxR1 activity was significantly obliterated. The exact mechanisms by which this is executed remain to be further investigated. Nevertheless, it is known that R120GCRYAB protein exists as soluble (s) and insoluble (ins) polydisperse oligomers of a larger size than the WTCRYAB, in addition to supraoligomers organized in aggregates (agg) which are associated to other proteins such as HspB1 or Desmin (3, 44, 61). The TS plays an important role in protein folding by reducing disulfide bond–oxidized cysteines (14) and, while not acting directly on CRYAB, which contains no cysteines, this could affect other proteins interacting with the mutant R120GCRYAB and subsequently, change their aggregation propensity. Our study confirmed BAG3 as preferentially interacting with R120GCRYAB and containing significantly reduced cysteines. Although more biophysical experiments would be needed to understand the conformational consequences of those REDOX modifications, previous work had already shown that BAG3 was involved in stabilizing the soluble counterpart of the expressed R120GCRYAB (20). In other words, BAG3 would slow down the protein flux from soluble to insoluble and aggregate fractions. Besides acting directly on the mechanisms involved in aggregate formation, another nonmutually exclusive possibility would be to alter the degradation processes of those three forms of R120GCRYAB (s, ins, agg). The proteasome would selectively handle the soluble form and insoluble small oligomers (34, 66), while autophagy would be involved in aggregate destruction (42, 59, 64, 67). In support of this later possibility, BAG3 has recently been shown to stimulate autophagy in a complex with the sHSP HspB8 (8).
Mutations in CRYAB gene are responsible for either multisystemic or tissue-restricted diseases, and the induced pathologies present variable expressivity and penetrance (50). This variability can be linked to genetic causes with the intervention of modifier genes (27). Our study provides data supporting a cascade of new genes (R120G→HDAC3→TrxR1→Trx System targets and R120G→Nrf2→Trx System→targets: e.g., BAG3), which could play the role of modifiers in the CRYAB inheritable pathologies (see Fig. 9).
One remaining unresolved issue is the ultimate long-term consequence of the TrxR1 activity on aggregate development. Aggregates are presented as a defensive mechanism that concentrates misfolded proteins and sets them apart from the cytoplasm (67). In particular, preamyloid oligomers, which appeared to exert toxic effects, were found to accumulate in R120GCRYAB aggregates so that preventing aggregate formation could have a deleterious effect by abolishing amyloid sequestration (53). Alternatively, aggregates can sequester various other proteins and, thus, exacerbate the dysfunctional biochemical events in the affected cells. In the particular case of cardiac cells, accumulation of protein aggregates disrupts the sarcomeric organization and consequently, perturbs the contractile apparatus. This was well illustrated in R20GCRYABTg hearts, which exhibited increased myocardial stiffness (37). Based on this mechanical dysfunction associated with aggregate development, it is tempting to speculate that augmentation of TrxR1 activity through a positive feed-back loop could be beneficial for mutant hearts (Fig. 9).
In conclusion, our current study demonstrates that, despite the absence of strong phenotype in TrxR1 cardiac KO (24), TrxR1 is actually a critical target and regulator in hearts. Future research is still needed to fully evaluate the functional consequences of inhibiting either TrxR1 and/or HDAC3 activity at the level of the normal and pathological myocardium.
Materials and Methods
Animals
Mice expressing the human R120G mutated version of CRYAB (hR120G) under the control of the cardiac specific αMHC promoter were previously described (44). NTg, and hR120GTg mice were maintained in the C57BL/6J background.
DKO/hR120GTg mice were obtained by intercrossing hR120GTg mice with CRYAB and HspB2 DKO (Cryab/Hspb2tm1Wawr knockout mice; MGI: 2652177) (4).
All experiments were performed with male mice at the indicated ages. Mice were housed under a controlled environment with 23°C±2°C and 12 h light/dark cycles and had access to water and food (ad libidum). Animals did not receive any specific treatment except for the experiments testing the role of TrxR1, which consisted of intraperitoneal injections of ATG, an inhibitor of TrxR1 (ATG, Fitzgerald (Ireland) Catalog No 51R-U045508) (Fig. 7H).
All experimental protocols followed the US Animal Welfare Acts and NIH guidelines and were approved by the University of Utah Animal Care and Use Committee.
Cell culture, transfection and treatments
H9c2 rat cells obtained from ATCC were grown in DMEM (Invitrogen; Catalog No. 11995-073) that was supplemented with 10% fetal bovine serum (Fisher Scientific, Catalog No SH3007103) and 100 μ penicillin-100 μg streptomycin/ml medium (Invitrogen; Catalog No. 15070-063) in a 5% CO2 humidified atmosphere. One day before transfection, H9c2 cells were trypsinised and plated in growth medium without antibiotics to achieve 80%–90% confluence.
Plasmids used for transfection are described in the “Supplementary Materials and Methods” section. Transfection of both H9c2 and NRCM was performed using Lipofectamine 2000 (Life Technologies; Catalog No. 11668-500) with various amounts of plasmid DNA as required for the experiments. Cells were incubated for 24 to 72 h after transfection depending on the experimental protocol, before testing for transgene expression or performing downstream experiments.
NRCM were transfected with 4 μg TrxR1 and 42 h after transfection, cells were treated with either 1 μM TSA (Cell Signaling; Catalog No. 9950) or the vehicle control for an additional 6 h. The cells were collected and TrxR activity was measured. H9c2 were treated with 15 μm TrxR inhibitor, aurothioglucose (ATG; Catalog No 51R-U045508) as described in the “Results” section (see protocol, Fig. 7A).
RNA and protein analyses
Transcripts were analyzed by RTqPCR as described in the “Supplementary Materials and Methods” section. Proteins were analyzed by Western blots and immunoprecipitation followed by Western blots (Supplementary Materials and Methods). Immunofluorescence was used to detect in situ TrxR1 and CRYAB (Supplementary Materials and Methods).
TrxR assay
TrxR activity was determined by the colorimetric assay using Sigma Thioredoxin Reductase Assay kit (Catalogue No. C-S0170) following the manufacturer's protocol. Activity measurement was performed by NADPH-dependent 5,5′-dithiobis(2-nitrobenzoic) acid (DTNB) reduction to 5-thio-2-nitrobenzoic acid (TNB), which produces a yellow color that is measured at 405 nm (21). Tissue or cells were homogenized in Triton X-100 soluble fraction extraction buffer/mitochondrial isolation buffer or cell lytic buffer, respectively; protein was estimated using BCA method, and homogenates were used for the enzyme activity measurement. One unit of Thioredoxin Activity was defined as an increase in absorbance of 1.0 per minute per ml (when measured in a noncoupled assay containing DTNB alone) at pH 7.0 at 25°C. Activity was normalized to the total protein used for the assay (determined by BCA method) and expressed as activity/min/μg. Phosphatase treatment is described in the “Supplementary Materials and Methods” section.
Isotope coded affinity tag-mass spectroscopy
ICAT-MS was performed at the Center for Advanced Proteomics Research, the New Jersey Medical School Cancer Research Center (14). A detailed description of the procedure is provided in the “Supplementary Materials and Methods” section.
2D difference in gel electrophoresis
2D-DIGE experiments were performed in collaboration with Applied Biomics, Inc. A detailed description of the procedure is provided in the “Supplementary Materials and Methods” section.
Data analysis and statistics
All the experiments were repeated independently twice or thrice. The total number of samples or animals analyzed per genotype, either NTg or hR120Tg, are indicated in the figure legends when appropriate. Transfection experiments were performed by pulling down cells from two to three different independent transfection and or treatment experiments, which were again performed in duplicate. Data are expressed as mean±standard deviation or standard error of the mean, as indicated. Student's t-test was used for statistical analysis. Statistical significance was set at p<0.05, using two-tailed distribution and two-sample equal/unequal variance.
Footnotes
Acknowledgments
The authors are grateful to Brian Foster for his advice and work performed on acetylated TrxR1. They appreciate the contributions of Xiaohui Wang and Shayne Squirres on preliminary studies related to this project. They thank David Coe for mice care and genotyping and Alison Ausman for excellent editorial assistance during the initial preparation of this article.
Sources of Funding
This work was supported by 2 R01 HL063834-06, 5R01HL074370-03, and NIH 5DP1OD006438-03 to I.J.B and Leducq Transatlantic Network “Redox and Nitrosative. Regulation of Cardiac Remodeling: Novel Therapeutic Approaches for Heart Failure.”
Author Disclosure Statement
The authors declare that they have no competing financial interests.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.