
Abstract
Although proteasome inhibitors are effective for treatment of multiple myeloma, resistance eventually develops. The small molecule b-AP15 inhibits proteasomal degradation by inhibiting the USP14/UCHL5 deubiquitinases of the 19S regulatory particle. Here, we show that b-AP15 induces a distinct cellular response compared with a 20S core particle inhibitor. The potent apoptotic activity of b-AP15 is associated with strong increases of chaperone expression and strong induction of oxidative stress. Further, we show that inhibition of PKR-like ER kinase activation during endoplasmic reticulum stress is not essential for the cytotoxicity of proteasome inhibitors. The finding that different modes of blocking proteasome function induce distinct cellular responses has therapeutic implications.
Introduction
T
The efficient degradation of ubiquitinated substrates requires both unfolding and removal of polyubiquitin chains. The translocation of ubiquitin-tagged proteins into the proteolytic core is facilitated by the deubiquitinase (DUB) activity of the 19SRP. In the absence of deubiquitination, bulky ubiquitin moieties would impede entry of the substrate into the 20SCP chamber. DUBs cleave the isopeptide bonds between the C-terminal carboxyl of ubiquitin and the amino group of a lysine residue on an adjacent protein and salvage ubiquitin for reuse by the UPS (23). The 19SRPs of metazoan cells are associated with three different DUBs. USP14 and UCH37/UCHL5 are cysteine isopeptidases that cleave distal polyubiquitin chains and are suggested to promote substrate rescue rather than degradation (33). The third DUB of the 19SRP is the RPN11/POH1 metalloprotease, a Zn2+-dependent protease of the JAMM family that is localized within the lid of the 19SRP. RPN11/POH1 performs ubiquitin chain amputation by cleaving entire chains from substrates in a process that is tightly coupled to degradation (60,64).
Ubiquitinated proteins initially bind to the 19S subunits Rpn10 and Rpn13, but substrate entry requires interactions between polyubiquitin chain and the 19SRP DUBs (21,50). Binding of polyubiquitin chains to USP14 (Ubp6 in yeast) promotes opening of the gate to the 20SCP (50). It was recently shown that substrate deubiquitination is coupled to stimulation of the activities of the AAA+ ATPases in the base of the 19SRP that unwind protein substrates (51). Thus, binding of polyubiquitin conjugates to yeast Ubp6, or to USP14 and UCH37 in mammalian cells, leads to stimulation of 19SRP ATPase activity.
The dipeptidyl boronic acid bortezomib (PS-341, Velcade®) is a selective inhibitor of the proteolytic activity of the 20SCP. Bortezomib has shown activity against several malignant cell types and has been approved by the US Food and Drug Administration as a first line treatment for patients with multiple myeloma (6). The downstream effects of proteasome inhibition that ultimately result in cell death are not completely understood. Proteasome inhibition generates a plethora of cellular responses, including inhibition of NFκB signaling (24), stabilization of Myc (45) and deregulation of Myc signaling (7), induction of endoplasmic reticulum (ER) stress (15), production of reactive oxygen species (ROS) (15,36,49), and amino-acid depletion-induced autophagy (56).
A number of groups have reported that generation of cellular ROS is associated with proteasome inhibitor-induced cell death (15,36,49). The interpretation of some of these studies is complicated by later findings of direct complex formation between proteasome inhibitor scavengers such as N-acetyl cysteine (NAC), vitamin C, edaravone, and Tiron (14,19,37,68). The role of oxidative stress in the induction of apoptosis and cell death is, therefore, somewhat controversial. The mechanism(s) of how ROS is induced in cells exposed to proteasome inhibitors are also unclear.
The ubiquitin system is believed to be highly druggable, but has been underexplored by drug developers (8). We recently identified b-AP15 as an inhibitor of the USP14 and UCHL5 DUBs of the 19SRP (10). Inhibition of both USP14 and UCHL5 leads to a functional block of the proteasome (32) and we, indeed, observed accumulation of various proteasomal substrates in cells exposed to b-AP15 (10). Since b-AP15 blocks the proteasome by a distinct mechanism to that of the clinically used agent bortezomib, it is conceivable that b-AP15 (or an optimized lead) could be used for treatment of multiple myeloma patients who have developed resistance to bortezomib. Our previous studies showed that b-AP15 and bortezomib differ with regard to the apoptotic responses that are elicited. We found that apoptosis by b-AP15, but not by bortezomib, was insensitive to the overexpression of Bcl-2 and genetic disruptions of BAX and TP53 (10). These results were not expected from models in which apoptosis induction by proteasome inhibitors is due to stabilization of various proteasomal substrates such as IkB (24,55), p53 (63), the proapoptotic proteins Bid and Bax (5,35), and Myc (45). To understand and characterize the mechanisms of b-AP15-induced cytotoxicity, we have delineated the various cellular stress pathways affected. We report that disruption of redox homeostasis and induction of high levels of ROS is a major difference between the cellular responses to b-AP15 and bortezomib.
Results
Proteasome inhibition by bortezomib and b-AP15 induces a similar stress response
Proteasome inhibitors show a unique cytotoxic profile in the National Cancer Institute 60 cell line panel (NCI60) when compared with thousands of other compounds (1). We compared the cytotoxic profiles of bortezomib (NSC681239) and b-AP15 (NSC687852) in the NCI60 panel, using data available in the NCI Developmental Therapeutics Program database (
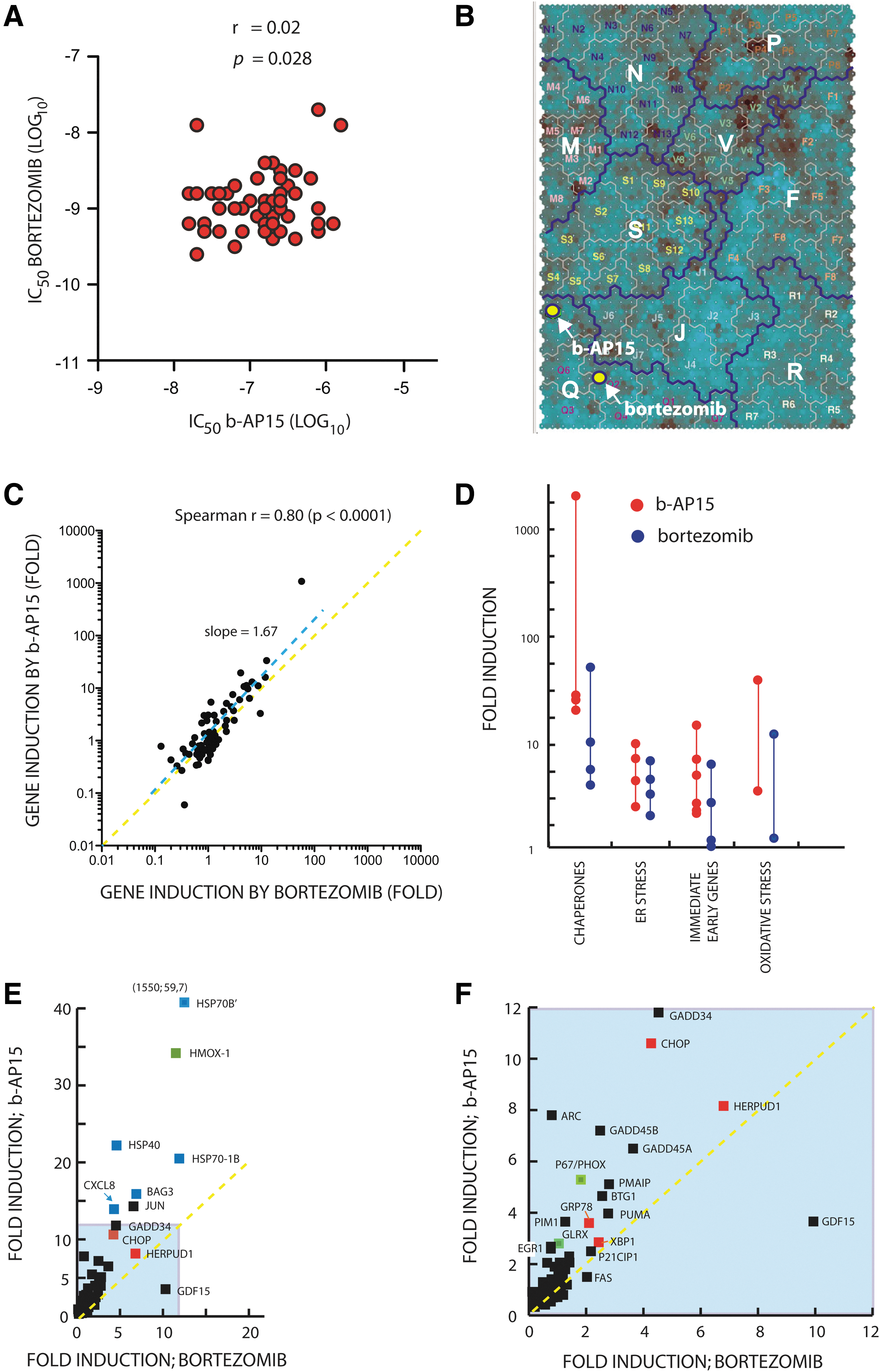
We previously reported that b-AP15 treatment induces a gene expression profile related to that of several characterized proteasome inhibitors (10). We extended this analysis to a subset of stress responsive genes that are commonly affected by chemotherapeutical reagents. Colon carcinoma cells were used, as these are known to be sensitive to b-AP15 (10). HCT116 colon carcinoma cells were exposed to b-AP15 or bortezomib at IC90 concentrations, and gene expression was determined by quantitative polymerase chain reaction (qPCR). Both drugs induced a similar pattern of expression of an 84 gene set (Spearman r=0.80, p<0.0001; Fig. 1C), with genes associated with oxidative stress, ER stress, heat shock, and the immediate early response displaying major alterations in gene expression (Fig. 1D). However, the amplitude of activation of several genes was higher in b-AP15-exposed cells (best-fit slope=1.67). The most conspicuous difference in the response to b-AP15 and bortezomib was the >1000-fold induction of HSPA6 by b-AP15 (Fig. 1E). HSPA6 encodes the Hsp70B’ protein, a heat-shock induced chaperone that acts as the cells' final defense against proteotoxicity (46). Genes regulated by heat shock factor-1, mostly chaperone genes, were in general more strongly induced by b-AP15 when compared with bortezomib (Fig. 1E, blue boxes). Both b-AP15 and bortezomib increased the expression of genes associated with oxidative stress; however, the induction of HMOX1 and PHOX/p67 expression was ∼2.5-fold stronger in b-AP15- compared with bortezomib-exposed cells (Fig. 1E, F; green boxes). The expression of immediate early response genes such as JUN, GADD34, GADD45A, and GADD45B were also more strongly induced by b-AP15 compared with bortezomib (Fig. 1E, F). Finally, similar increases in the expression of genes associated with ER stress were observed after treatment with either drug (Fig. 1E, F; red boxes). We conclude that the two drugs induce similar cellular responses; however, b-AP15 elicited a stronger induction of chaperone genes, genes associated with oxidative stress, and immediate early response genes compared with bortezomib.
Inhibition of proteasome function precedes apoptosis after exposure to the DUB inhibitor b-AP15
Cytotoxicity of b-AP15 to tumor cells is associated with induction of apoptosis (10). Analysis of the kinetics of apoptosis induction showed that b-AP15 elicited a more rapid induction of caspase-cleavage of the endogenous cellular substrate keratin-18 (K18) compared with bortezomib (Fig. 2A), with high levels of cleaved K18 appearing 16 h post drug treatment. We previously reported that b-AP15 induces apoptosis in tumor cells genetically defective in BAX and in cells overexpressing Bcl-2 (10). Bax and Bak are essential for induction of cytochrome c release from mitochondria during apoptosis (62). To extend our previous findings, we used siRNA to down-regulate the expression of Bak in HCT116 colon carcinoma cells. As shown in Figure 2B, down-regulation of Bak did not affect apoptosis induction by b-AP15, but inhibited apoptosis induced by bortezomib. The conformation of Bak is altered during apoptosis, resulting in increased accessibility of antibodies binding to the N-terminal domain (17). Treatment of HCT116 cells with b-AP15 resulted in increased antibody binding to Bak, indicating conformational activation (Fig. 2C). This activation does not, however, appear to be required for b-AP15-induced apoptosis, presumably due to the activation of parallel apoptotic signaling pathways.
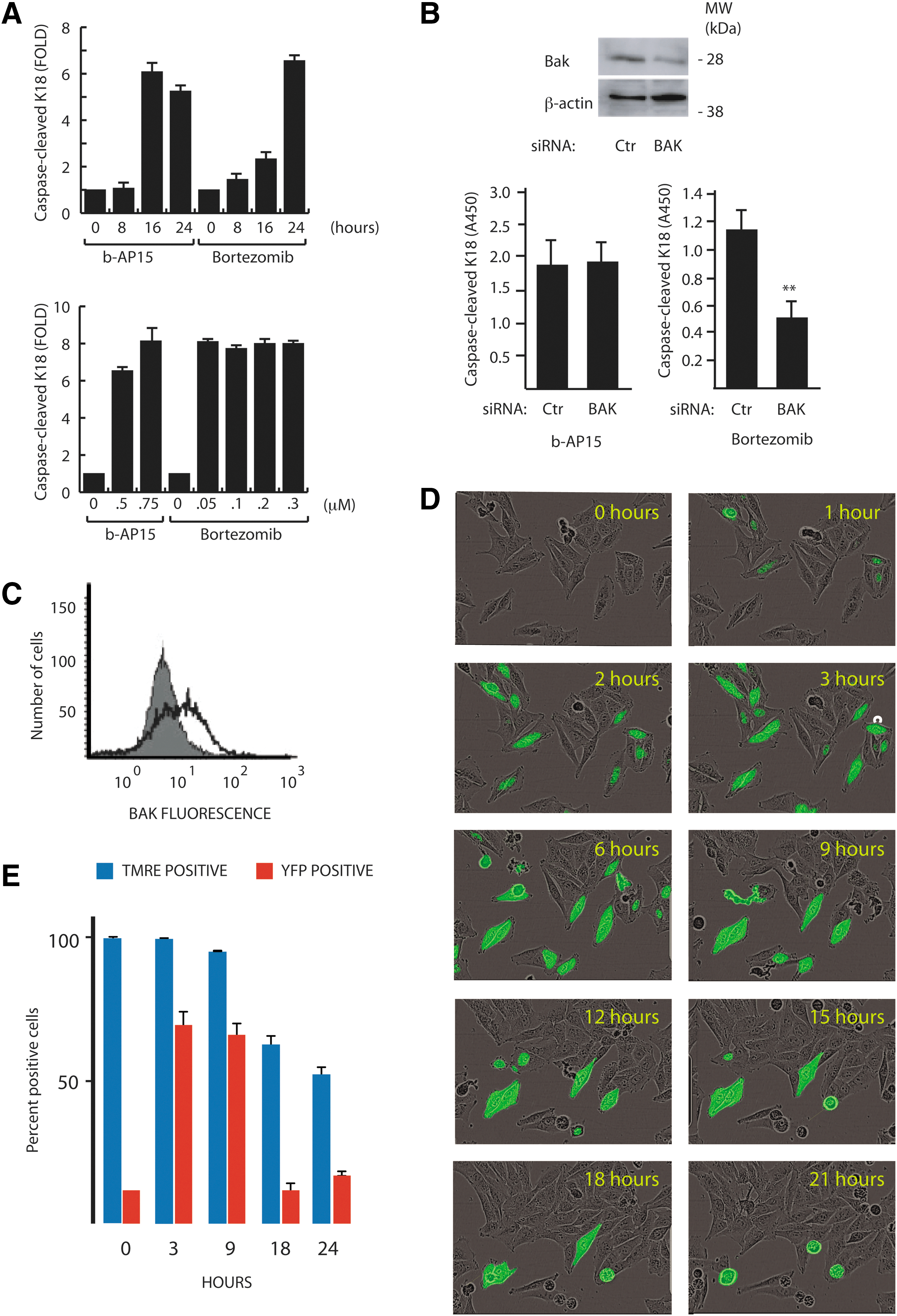
We next examined the effects of b-AP15 on in vivo proteasome function using a reporter cell line, MelJuSo, expressing the proteasome targeted fusion substrate UbG76V–yellow fluorescent protein (YFP) (40). This substrate is intrinsically unstable due to constitutive degradation by the proteasome; thus, impairment of the UPS leads to an increased fluorescent signal. At doses below the IC50 (0.25 μM), a small number of cell nuclei displayed YFP positivity after 1–3 h of b-AP15 drug exposure (Fig. 2D; Supplementary Movie S1 and S2; Supplementary Data are available online at
b-AP15 induces ER stress
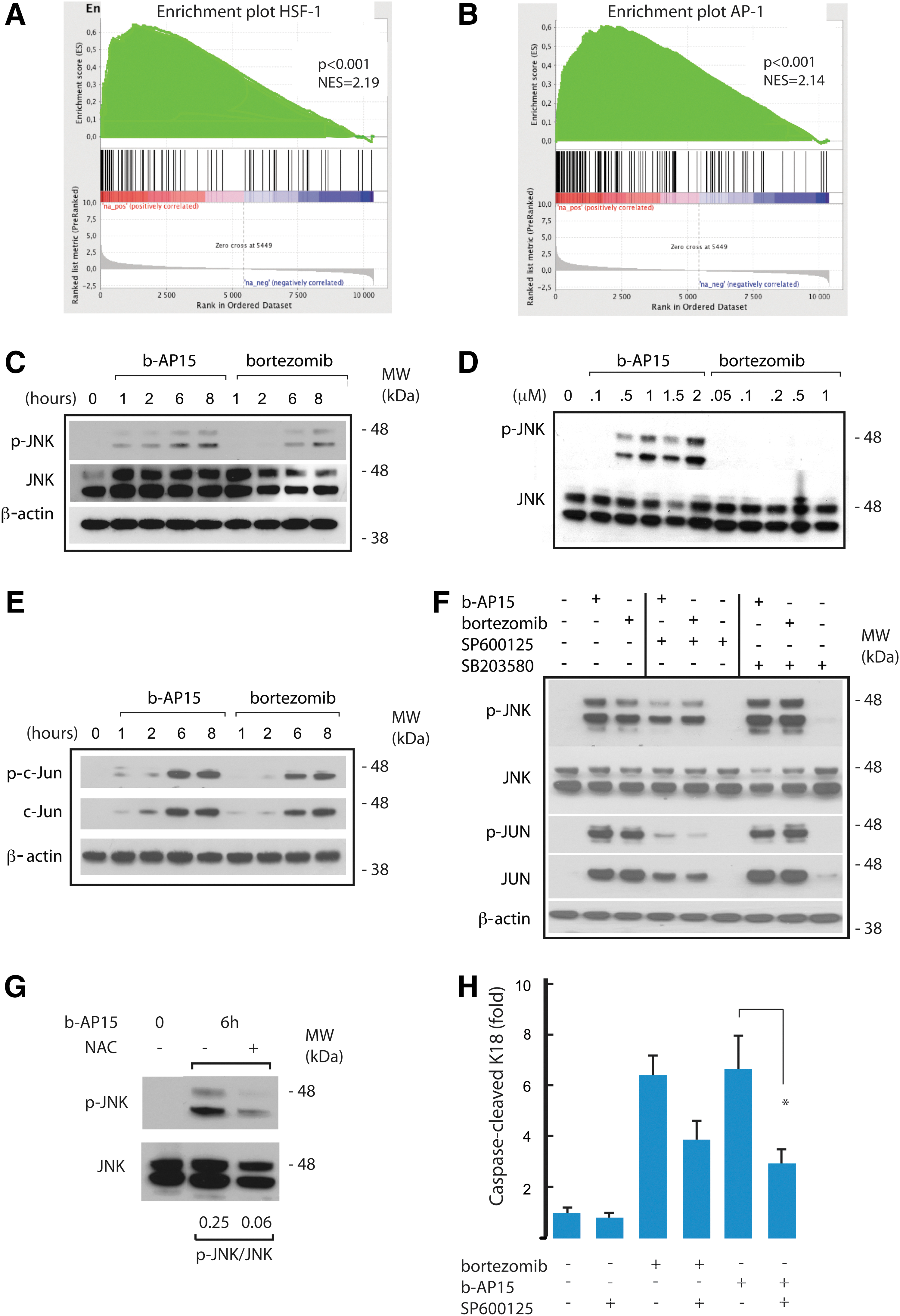
The up-regulation of genes involved in the ER stress response by b-AP15 (Fig. 1D–F) is consistent with previous reports showing induction of ER stress by bortezomib (15,44). As shown in Figure 3A, b-AP15 induced the expression of the ER chaperone Grp78 (BiP) in HCT116 cells. The level of induction was similar to that observed with bortezomib, but weaker than the response to the ER stress-inducer thapsigargin, an inhibitor of sarcoendoplasmic Ca2+ ATPase. b-AP15 induced phosphorylation of α-subunit of eukaryotic initiation factor 2 (eIF2-α) already at 6 h post-drug treatment (Fig. 3A). Phosphorylation of eIF2-α is an integral part of the unfolded protein response, by decreasing the load of misfolded proteins in the ER (22), and decreasing translation of cyclin D1 (20). In accordance with previous findings (44), bortezomib failed to induce phosphorylation of eIF2-α. This is due to inhibition of PKR-like ER kinase (PERK) by bortezomib and leads to a lack of translational suppression, and was suggested to be important for the cytotoxic activity of bortezomib (44). The difference between b-AP15 and bortezomib to induce eIF2-α phosphorylation was verified in three additional colon cancer cell lines, whereas a fourth cell line (OX-CO-1) displayed some induction by bortezomib (Fig. 3B). The underlying mechanism(s) of the variations in the response of the cell lines to b-AP15 and bortezomib was not examined further. We conclude that the rapid apoptotic response of HCT116 cells to b-AP15 occurs despite the induction of phosphorylation of eIF2-α.
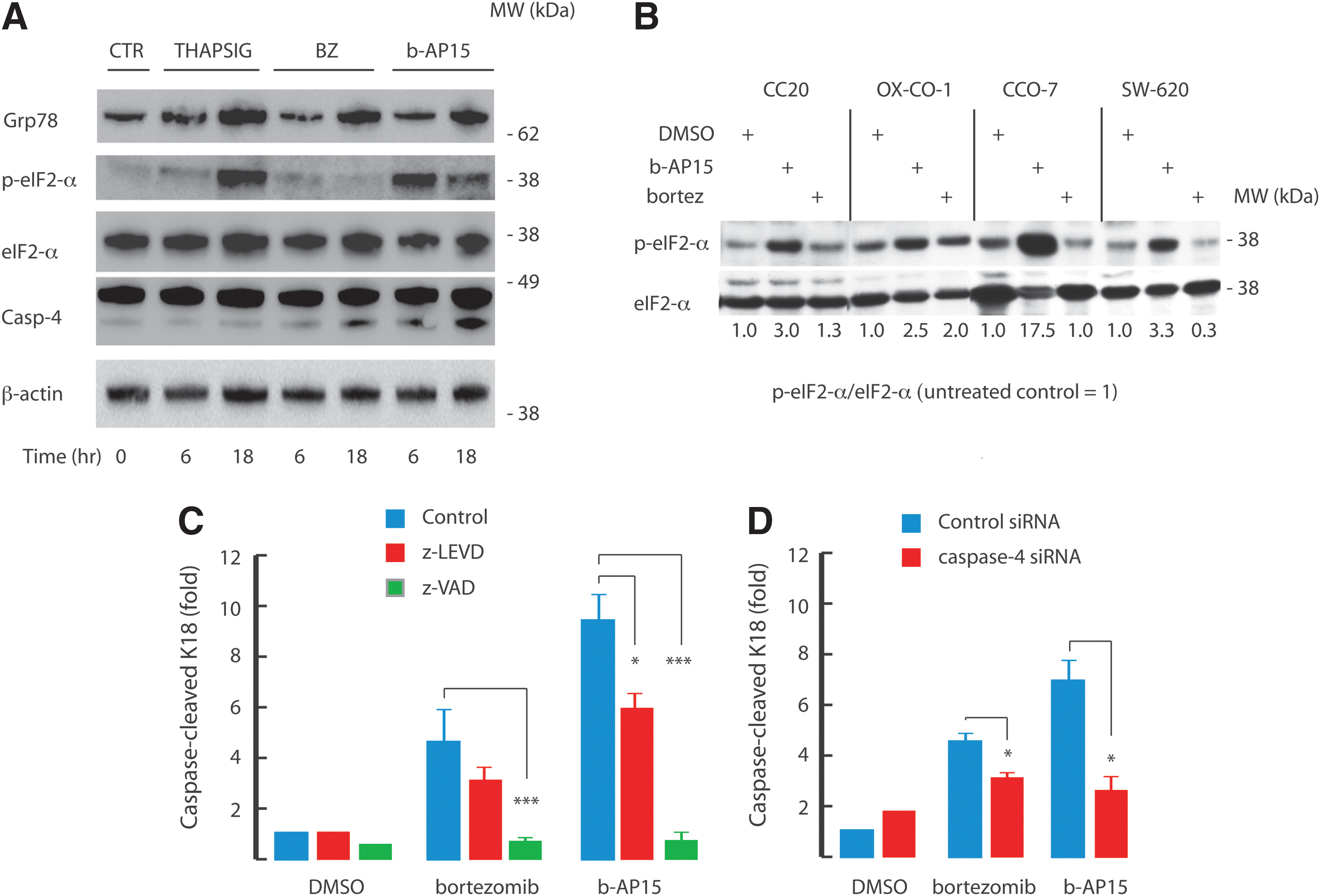
The ER-associated pro-caspase-4 is the human homolog of mouse caspase-12 (43), and is cleaved after acute ER stress (26,44). Caspase-4 cleavage was detected in a time-dependent manner after bortezomib and b-AP15 treatment (Fig. 3A). To investigate the role of caspase-4 in induction of apoptosis, HCT116 cells were exposed to the caspase-4 inhibitor Z-Leu-Glu-Val-Asp-FMK or transfected with caspase-4 siRNA and exposed to either bortezomib or b-AP15. Quantification of the levels of caspase-cleaved K18 showed that inhibition of, or depletion of, caspase-4 reduced both bortezomib- and b-AP15-induced apoptosis (Fig. 3C, D). Taken together, our data suggest that the ER stress induced by b-AP15 contributes to its apoptotic potential.
Role of ROS in b-AP15-induced apoptosis
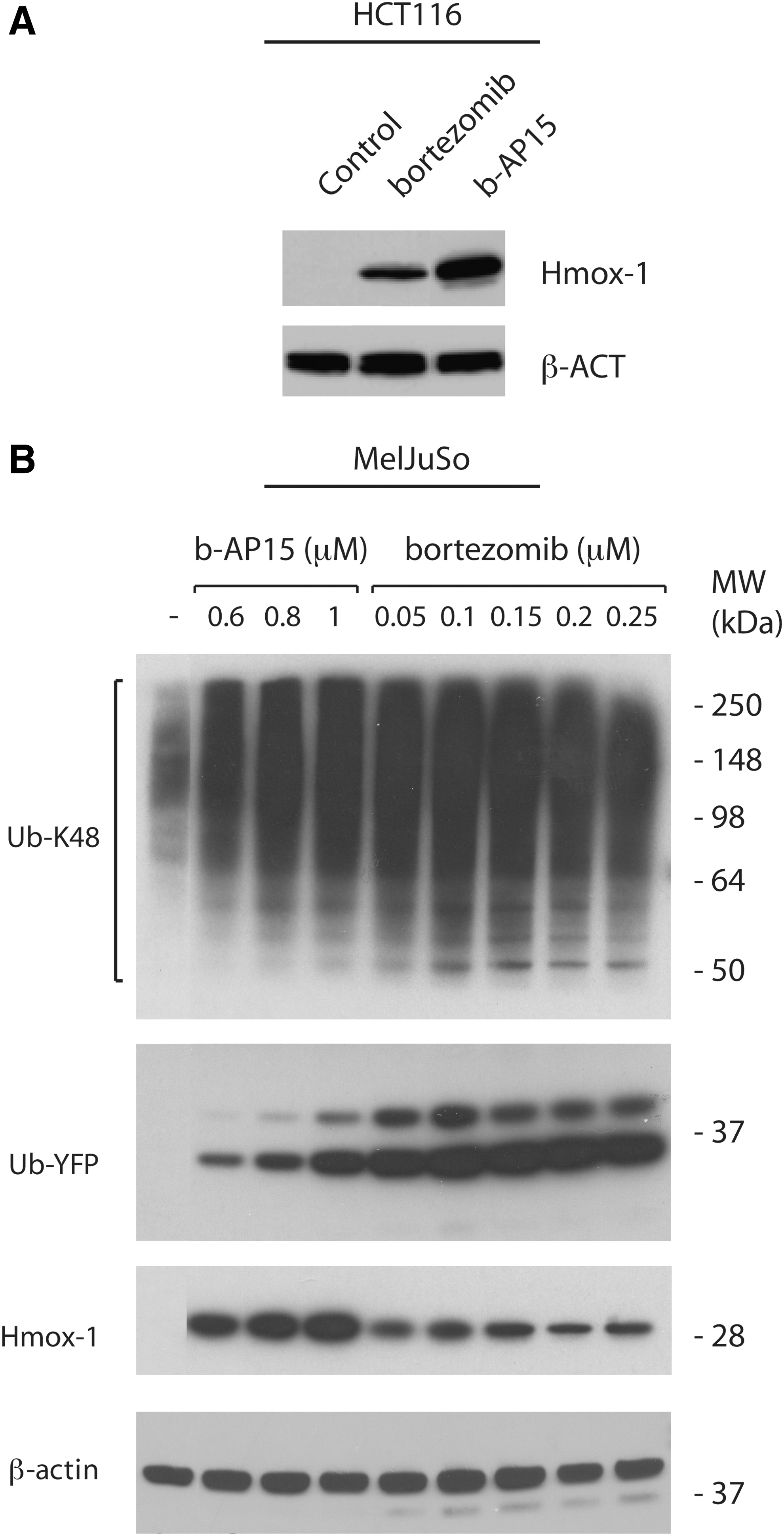
ROS production has been reported to be a critical mediator of bortezomib-induced apoptosis (15,36,49). The strong induction of HMOX1 (Fig. 1E), a target gene of redox-regulated nuclear factor-erythroid-2-related factor 2 (Nrf-2), by b-AP15 is consistent with induction of oxidative stress. Induction of HMOX1 gene expression by b-AP15 in HCT116 cells was confirmed at the protein level by western blotting (Fig. 4A). In MelJuSo UbG76V-YFP cells, Hmox-1 expression was not further increased using higher concentrations (250 nM) of bortezomib (Fig. 4B). The difference in induction of Hmox-1 by bortezomib and b-AP15 was, therefore, not due to the use of insufficient concentrations of bortezomib.
Experiments using the fluorescent ROS probe 2′,7′-dichlorofluorescin diacetate (DCFH-DA) confirmed that b-AP15 induces higher levels of ROS compared with bortezomib (Fig. 5A). To investigate whether ROS was involved in b-AP15-induced apoptosis, we treated HCT116 cells with the water-soluble vitamin E derivative and antioxidant Trolox. Trolox inhibited apoptosis by ∼50% (Fig. 5B). In these experiments, cells were pulsed with b-AP15 for 1 h, after which the drug was removed and fresh media containing Trolox were added. In contrast, no effect on apoptosis was observed when Trolox was present only during the time of drug incubation (Fig. 5B). NAC also inhibited apoptosis induction when added after removal of b-AP15 (Fig. 5C). The inhibitory effects of the ROS scavengers Trolox and NAC on apoptosis are, therefore, not due to a direct chemical reaction with b-AP15 as described for other UPS inhibitors (19), but rather to inhibition of ROS generation downstream from b-AP15-mediated proteasome inhibition. However, when we analyzed the effect of Trolox on rescuing cells from ROS-induced death, no reduction in cytotoxicity was observed (Supplementary Fig. S1). This was in contrast to bortezomib, where Trolox partially inhibited drug-induced cell death (Supplementary Fig. S1). These findings show that apoptosis induction by b-AP15 involves oxidative stress, but that other mechanisms will eventually cause cell death when oxidative stress is alleviated.
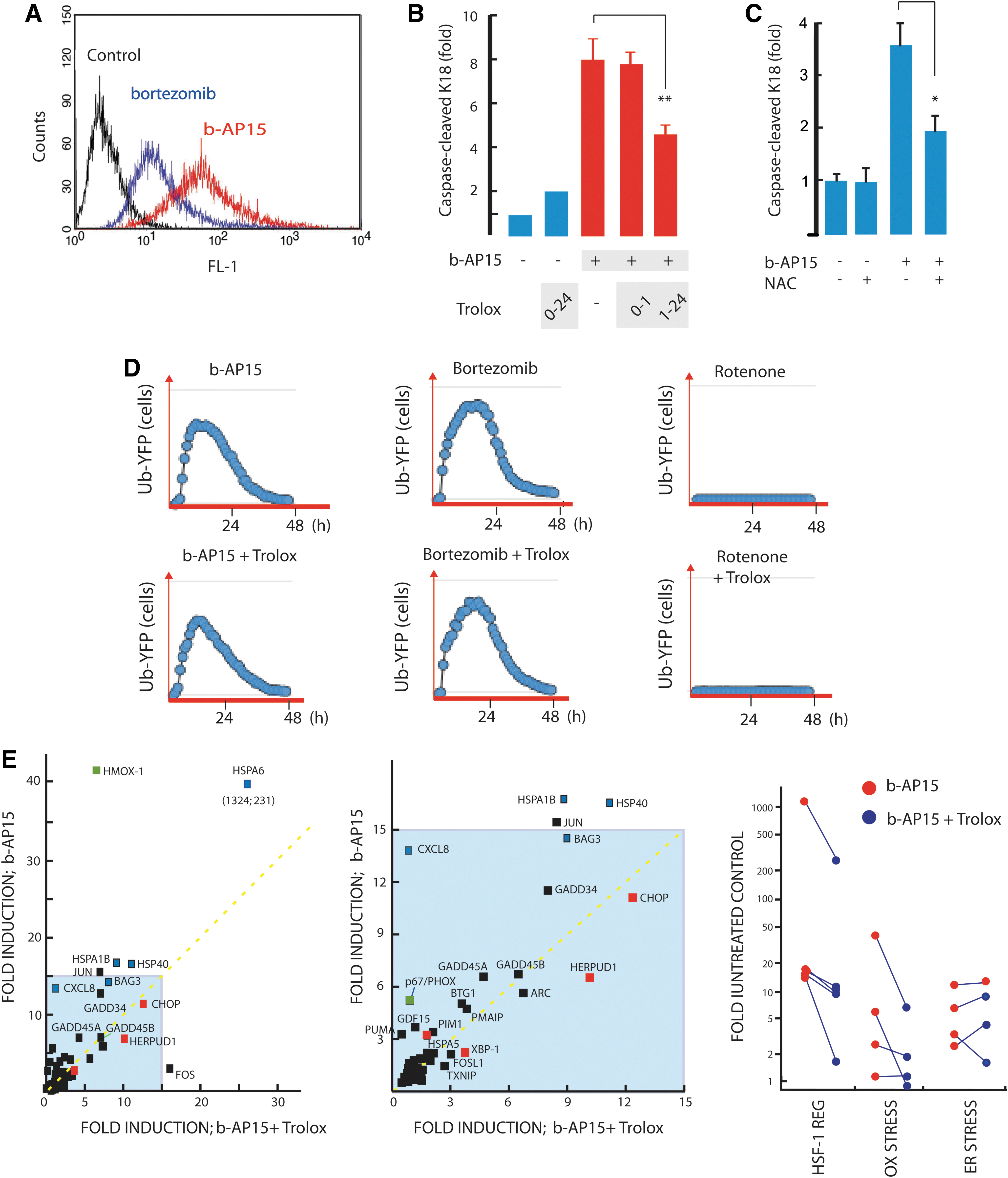
Sustained oxidative stress is known to impair the functionality of the UPS (9,13). The activity of the proteasome appears to be more sensitive to oxidative stress than the DUBs (66). The finding of strong induction of oxidative stress by b-AP15, therefore, raised the possibility that alterations in cellular redox status contribute to inhibition of proteasome function. To examine this question, we exposed MelJuSo UbG76V-YFP reporter cells to bortezomib or b-AP15 in the presence and absence of Trolox as an ROS scavenger (Fig. 5D). The presence of Trolox during the drug treatment had no effect on the accumulation of the UbG76V-YFP reporter, confirming that both drugs inhibit proteasome activity independently of ROS levels (Fig. 5D). We also examined whether proteasome function was affected by rotenone, an agent that interferes with mitochondrial electron transport and induces ROS. No effect on UbG76V-YFP accumulation in cells was observed (Fig. 5D).
To investigate the biological effects of ROS induced by proteasome inhibition, we studied the effect of scavengers on gene expression after treatment with b-AP15. Pretreatment with Trolox resulted in a lower level of expression of a number of stress-induced genes after proteasome inhibition (Fig. 5E). As expected, the expression of the oxidative stress-induced genes HMOX1 and PHOX/p67 was reduced by cotreatment with Trolox. The induction of genes regulated by HSF-1 was also reduced; HSPA6 by approximately fivefold, HSPA1B and DNAJB1 (Hsp40) by approximately twofold. The reduction in chaperone expression after Trolox cotreatment may be due to alleviation of ROS-induced protein oxidation (2). The relationship between chaperone and ROS expression would fit with previous reports showing ROS induction of Hsp70 expression (39). Interestingly, b-AP15 induction of ER stress-regulated genes was not significantly affected by Trolox (Fig. 5E), suggesting that ER stress was independent from oxidative stress.
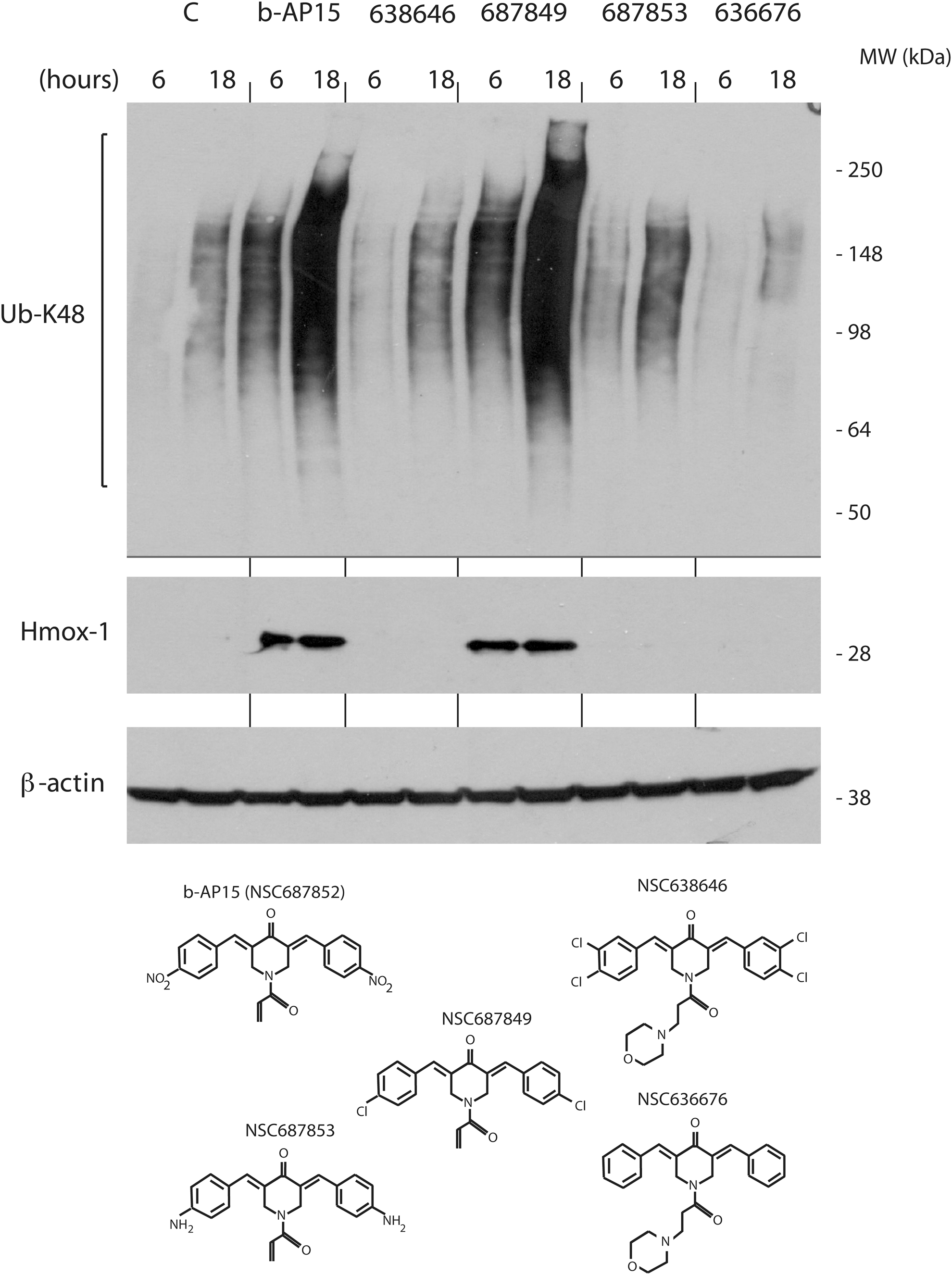
We finally examined the ability of a number of b-AP15 analogues to induce Hmox-1 and to induce the formation of high-molecular-weight poly-ubiquitin conjugates in HCT116 cells. We found that compound NSC687849 generated accumulation of poly-ubiquitin conjugates to a similar extent as b-AP15, whereas three other tested analogues did not show activity at 1 μM (Fig. 6). Interestingly, Hmox-1 levels were increased by b-AP15 and NSC687849, but not by the other compounds, showing an association between proteasomal blocking and induction of oxidative stress.
Induction of stress-activated protein kinases
We examined the gene expression response to b-AP15 in greater detail using Gene Set Enrichment Analysis (GSEA) of microarray data (54). Consistent with the qPCR results shown earlier, we found strong enrichment of genes containing HSF-1-regulated promoters (Fig. 7A). Interestingly, the GSEA also showed a strong enrichment of genes regulated by the activating protein-1 (AP-1) transcription factor (Fig. 7B). Both b-AP15 and bortezomib induced induction of phosphorylation of Jun-N-terminal kinase 1/2 (JNK) in HCT116 cells (Fig. 7C). Phosphorylation of JNK could be detected at 1–2 h after exposure to b-AP15, time points when bortezomib did not induce JNK phosphorylation (Fig. 7C, D). Increasing the concentration of bortezomib to 1 μM did not result in detectable induction of JNK phosphorylation at 2 h (Fig. 7D). At 6 and 8 h, total c-Jun protein levels were increased in both b-AP15- and bortezomib-treated cells (Fig. 7E), as expected from positive autoregulation (3). Increases in the phosphorylation of p38-MAPK were also observed after exposure to b-AP15 and to bortezomib (Supplementary Fig. S2A). Experiments using the JNK inhibitor SP600125 and the p38-MAPK inhibitor SB203580 showed that, as expected, stimulation of phosphorylation of c-Jun at Ser-73 was due to JNK activity (Fig. 7F). NAC was found to reduce JNK activation by b-AP15 in experiments in which the scavenger was added after removal of b-AP15 (Fig. 7G), suggesting that JNK activation was mediated by ROS. We finally examined whether activation of JNK and p38-MAPK was required for apoptosis induction by b-AP15 and bortezomib. Pharmacological inhibition of JNK was found to inhibit apoptosis induced by b-AP15 (Fig. 7H), whereas inhibition of p38-MAPK did not affect apoptosis (Supplementary Fig. S2B).
Discussion
The present study establishes that proteasome inhibition by the USP14/UCHL5 DUB inhibitor b-AP15 induces a distinct cellular response compared with that of the 20S inhibitor bortezomib. The two agents elicit similar, but yet distinct, patterns of cytotoxicity on the NCI60 cancer cell line panel. Further, although both proteasome inhibitors induce apoptosis, the kinetics of b-AP15-induced caspase activation was significantly more rapid (>6 h time difference between treatments). The difference in the timing of apoptosis induction could not be explained by different kinetics of proteasome inhibition (Fig. 5D). Different apoptosis signaling pathways also appear to be induced in response to the two types of proteasome inhibitors, as b-AP15 does not require Bak or Bax for activation of caspase-cleavage activity [Fig. 2B and (10)]. The fact that the response to two agents both of which block the proteasome and that both increase the levels of proteins such as p53, p21, and p27 (10) differs raises questions with regard to the relative importance of stabilization of cellular regulatory proteins for triggering apoptosis. We hypothesize that the distinct cellular response to b-AP15 is related to the strong proteotoxic response induced by this drug, as evidenced by the >1000-fold induction of the HSPA6 chaperone gene by b-AP15. The mechanism underlying this elevated proteotoxicity is not clear but may be due to the accumulation of misfolded proteins with long polyubiquitin chains occurring as a consequence of inhibition of the USP14/UCHL5 DUBs (10). It is possible that misfolded proteins bearing longer ubiquitin chains are processed differently by the cell, resulting in stronger proteotoxic stress. Another possibility is that inhibition of other, non-proteasome-associated, activities of USP14 by b-AP15 will contribute to apoptosis induction [see (41)].
Oxidative stress has been associated with apoptosis induced by proteasome inhibitors (15,36,49). While our results do not contribute to the understanding of why proteasome inhibitors induce oxidative stress, they clearly establish a role of ROS in the induction of apoptosis by b-AP15. Whether ROS is involved in apoptotic signaling in cells exposed to proteasome inhibitors is controversial, as a number of ROS scavengers such as NAC and vitamin C form complexes with bortezomib, piperlongumine, and other UPS inhibitors (14,19,37,68). Here, we found that b-AP15 induced an ∼40-fold induction of HMOX1 expression, a gene known to be regulated by Nrf-2 and induced in response to oxidative stress (42). The amplitude of HMOX1 induction by b-AP15 was significantly higher compared with bortezomib and was concomitant with ROS induction measured by DCFH-DA fluorescence. We found that apoptosis induction by b-AP15 was decreased by Trolox, whereas proteasome blocking was not impaired. By sequential exposure of cells to b-AP15 and scavengers (Trolox or NAC), we ascertained that inhibition of apoptosis by scavengers was not due to an interaction with the drug. Our findings, therefore, establish that ROS is a key mediator of apoptosis induction by b-AP15.
Oxidative stress is known to activate the stress-activated kinases p38-MAPK and JNK through ASK1 (16,29), and these pathways are known to be induced by proteasome inhibitors (25,65). Further, yeast strains carrying mutations in 26S proteasome subunits overexpress Pap1, the Schizosaccharomyces pombe homolog of the human AP-1 transcription factor (48). We indeed found activation of both p38-MAPK and JNK by b-AP15. Similar to previous reports on bortezomib (25,65), a pharmacological inhibitor of JNK impaired b-AP15-induced apoptosis, whereas inhibition of p38-MAPK did not. As expected, phosphorylation of JNK was impaired by treatment with NAC. The relatively rapid induction of JNK implied that early induction of apoptosis is associated with activation of this pathway. These results establish JNK induction as a downstream mediator of b-AP15-induced oxidative stress.
Apoptosis by bortezomib has been attributed to the induction of ER stress (11,15,44,58). b-AP15 was also found to induce ER stress as evidenced by increased expression of CHOP (CCAAT/enhancer-binding protein (C/EBP) homologous protein; GADD153), HERPUD1, GRP78, and XBP1 expression. CHOP is known to be induced by ER stress and by oxidative stress (67). We found that the expression of ER-stress responsive genes after b-AP15 was not affected by Trolox, showing that oxidative stress was not involved in mediating ER stress. A major difference in the ER stress response to b-AP15 and bortezomib was the induction of phosphorylation of the eIF2-α by b-AP15. eIF2-α is usually phosphorylated during ER stress by PERK, leading to attenuation of protein synthesis (31). Such translational attenuation will reduce protein synthesis and, thus, reduce accumulation of misfolded proteins in the ER (22), thereby alleviating ER stress and promoting cell survival. Bortezomib does not induce eIF2-α phosphorylation, due to inhibition of PERK, and the lack of attenuation of protein synthesis during ER stress has been described as being important for the apoptotic activity of this agent (44). It is, therefore, interesting to note that b-AP15, which induces a more rapid induction of caspase cleavage activity compared with bortezomib, elicits eIF2-α phosphorylation. This finding suggests that the ability to induce eIF2-α phosphorylation is not a critical determinant for the apoptotic potential of agents that block proteasome function.
We previously showed that b-AP15 induces lysosomal membrane permeabilization and apoptosis dependent on the lysosomal protease cathepsin-D (4). Interestingly, several apoptotic agents have been described to trigger lysosomal membrane permeabilization and induce cell death by generating oxidative stress (12,47,53,59). Although the precise mechanism is unclear, increased production of ROS may lead to destabilization of the lysosomal membrane via massive peroxidation of membrane lipids. The present observations of high levels of ROS induction by b-AP15 would be consistent with these results.
We conclude that the strong proapoptotic potential of the DUB inhibitor b-AP15 is associated with induction of oxidative stress. This is common territory in the field of cancer therapy, as, in fact, most of our clinically used cancer drugs, as well as ionizing radiation, induce oxidative stress (61). Malignant transformation is known to be associated with enhanced cellular stress, including both oxidative and proteotoxic stress, and the strong oxidative stress associated with b-AP15 may be poorly tolerated by cancer cells, providing a rational for therapy of a wide variety of malignancies (38,57).
Materials and Methods
Materials
b-AP15 was obtained from Oncotargeting AB, bortezomib from ApoEx, SP600125 from Sigma-Aldrich, and SB203580 from Cell Signaling. Compounds were dissolved in dimethyl sulfoxide. Control cultures received the same final concentration of solvent as treated ones.
Cell culture
HCT116 colon carcinoma cells were maintained in McCoy's 5A modified medium/10% fetal calf serum. The proteasome reporter cell line MelJuSo Ub-YFP was generated as described (40) and was cultured in Dulbecco's modified Eagle's medium/10% fetal calf serum. All cells were maintained at 37°C in 5% CO2 and subcultured by trypsinization.
Assessment of apoptosis
HCT116 cells were seeded in 96-well microtiter plates at 10,000 cells per well and incubated overnight. Drugs were then added and cells were incubated further. At the end of the incubation period, NP40 was added to the tissue culture medium to 0.1% and 25 μl of the content of each well was assayed using the M30-CytoDeath® enzyme-linked immunosorbent assay (ELISA) (Peviva AB) (28). This ELISA is based on a specific antibody against a neoepitope of K18 that is generated by the action of caspase-3, −7- and −9 activated in response to apoptosis (34).
Measurement of Bak-associated immunofluorescence
Cells were fixed in paraformaldehyde (0.25%, 5 min) before incubation for 1 h with an antibody recognizing an N-terminal epitope of Bak that is accessible only after conformational activation (AM03; Oncogene Research Products) (17). The primary antibody was diluted in phosphate buffered saline (PBS) containing digitonin (100 μg/ml). Cells were then incubated for 1 h with an Allophycocyanin-conjugated secondary antibody in PBS (550826; BD Biosciences Pharmingen), and fluorescence was measured by flow cytometry.
Measurement of DCFH-DA fluorescence
HCT116 cells were treated with b-AP15, bortezomib, or solvent; stained using DCFH-DA; and subsequently analyzed using an FL1-H wavelength channel of a fluorecescence-activated cell sorting (FACS) Calibur flow cytometer.
siRNA knock-down
HCT116 cells were transfected with siRNA (Qiagen) against human BAK, BAX, or a scrambled control sequence. Two days later, the transfected cells were trypsinized and seeded in 96-well plates. The day after seeding, drugs were added to the cells and drug treatment was allowed to continue for 24 h, after which analysis of apoptosis using the M30-Apoptosense ELISA was conducted.
Western blot analysis
For ubiquitin analysis, cell extract proteins were resolved by Tris-Acetate polyacrylamide gel electrophoresis gels (Invitrogen) and transferred onto a polyvinylidene difluoride membrane for western blotting. Antibodies were obtained from the following sources: c-JUN (BD Transduction Laboratories), p-cJUN (Ser73), eIF2-α, p-eIF2-αp-JNK (T183/Y185) (Cell Signaling), JNK (Santa Cruz), HMOX1, GRP78 (BD Transduction Laboratories), ubiquitin (Chemicon), and β-actin (Sigma-Aldrich).
Live-cell analysis of UPS activity
MelJuSo UbG76V-YFP cells (40) were plated in black optically clear bottom ViewPlates (PerkinElmer) overnight and then treated with b-AP15 or bortezomib. Treatment with compounds that block the UPS leads to accumulation of YFP in these cells, and the generated fluorescence was continuously detected in an IncuCyte FLR instrument (Essen BioScience Inc.).
Transcriptional profiling and GSEA
We examined the pattern of transcripts for 84 genes that were reported to respond to stress. The identity of these genes is given in Supplementary Table S1. Cells were exposed to b-AP15 or bortezomib for 6 h in the presence of absence of Trolox; cDNA was prepared from 1 μg of RNA using the RT2 First-Strand Kit (SA Biosciences). All primers used for qPCR were purchased from SA Biosciences Corp. The array analysis was carried out following the manufacturer's protocol with SYBR Green PCR (ROX) Master Mix (SA Biosciences Corp.). Arrays were assayed for 1 cycle of 10 min at 95°C, then 40 cycles of (15 s at 95°C, followed by 1 min at 60°C) using an ABI7500 system. All conditions were performed in triplicate and normalized to five housekeeping gene mRNA levels. To analyze the PCR-array data, SA Biosciences Web-portal software was used:
The GSEA software and method for microarray result exploration has been described elsewhere (54). Briefly, the pre-ranked gene list from b-AP15 cells versus untreated control (10) was compared with a priori defined and curated gene sets from the MSigDB database (i.e., V$AP1_01 with systematic name M10220 and V$HSF_Q6 with systematic name M16482), using standard GSEA settings. The purpose of GSEA is to find out whether the a priori defined gene sets are significantly enriched toward the upper or lower end of the pre-ranked list. The p-value refers to the nominal p-values after 1000 permutations.
Measurement of mitochondrial membrane potential
Loss of mitochondrial membrane potential was assessed by incubating cells with 25 nM TMRE (Invitrogen) for 30 min. Changes in mitochondrial membrane potential (ΔΨ) were monitored in an FACS Calibur flow cytometer.
Statistical analysis
Statistical analysis (Student's t-test, Pearson correlation coefficient, or Spearman's rank correlation coefficient, as indicated) was performed using Prism software for Apple computers. Statistical significance in t-test was plotted as follows: * (p<0.05), ** (p<0.01), and *** (p<0.001).
Footnotes
Acknowledgments
The authors acknowledge the Developmental Therapeutics Branch at the NCI, Bethesda, for supplying compounds. They are grateful to Maria Rydåker, Lena Lenhammar, and Christina Leek for excellent technical support. They also thank Stephan Feller, University of Oxford, for the gift of colon carcinoma cell lines. This work was supported by Cancerfonden, Radiumhemmets Forskningsfonder, Vetenskapsrådet, and Strategiska Forskningsstiftelsen (SSF).
Author Disclosure Statement
M.F., R.L., and S.L. own shares in Vivolux AB, which develops cancer drugs based on proteasomal inhibition. No competing financial interests exist for the other authors.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.