
Abstract
Introduction
O
To catalyze oxidative folding, Ero1-flavoproteins yield hydrogen peroxide (H2O2) in stoichiometric amounts to the disulfides formed. We show that glutathione peroxidase 7 (GPx7) can utilize Ero1α-produced H2O2 in vitro and in vivo to oxidize protein disulfide isomerase (PDI) and accelerate oxidative folding. The peroxidase activity of GPx7 reveals novel mechanistic features of the catalytic and resolving cysteines. The Ero1α/GPx7/PDI triad couples H2O2 elimination and disulfide generation to ensure efficient and safe oxidative protein folding.
GPx7 lacks the loop which determines glutathione (GSH) specificity and oxidizes PDI more efficiently than GSH, implying that GPx7 belongs to the family of thioredoxin GPx-like peroxidases (TGPx) (22, 29). In GPx7, the first cysteine residue Cys57 was supposed to be the peroxidatic cysteine (CP), as it is located in the -NVASxC(U)G- reactive (seleno)cysteine-containing motif. The second cysteine residue Cys86 is located in the -FPCNQF- motif that is highly conserved among all GPx homologues, but its function remains to be clarified. Intriguingly, a canonical resolving cysteine (CR) in a ‘Cys block’ region, essential for completing the catalytic cycle of reduction by thioredoxin in typical two-cysteine TGPx, is missing in GPx7 (7, 29). Therefore, GPx7 was defined as an unusual cysteine-based TGPx, and the reaction mechanisms for GPx7 peroxidase activity were, hence, the other main aim of this study.
Our mutagenesis experiments show that Cys57 of GPx7 is oxidized by H2O2 to sulfenic acid, while Cys86 acts as a noncanonical CR, resolving the sulfenylated Cys57 into an intramolecular disulfide bond. Both the disulfide and sulfenic acid forms of GPx7 can oxidize PDI. We demonstrate, moreover, that GPx7 can utilize Ero1α-produced H2O2 to accelerate oxidative folding not only in vitro but also in vivo. In this way, GPx7 allows cells to exploit the potentially harmful peroxides produced by Ero1 to improve oxidative folding, with water being released rather than any reactive oxygen species. Thus, the Ero1α/GPx7/PDI triad represents an efficient and safe oxidative folding system.
Results
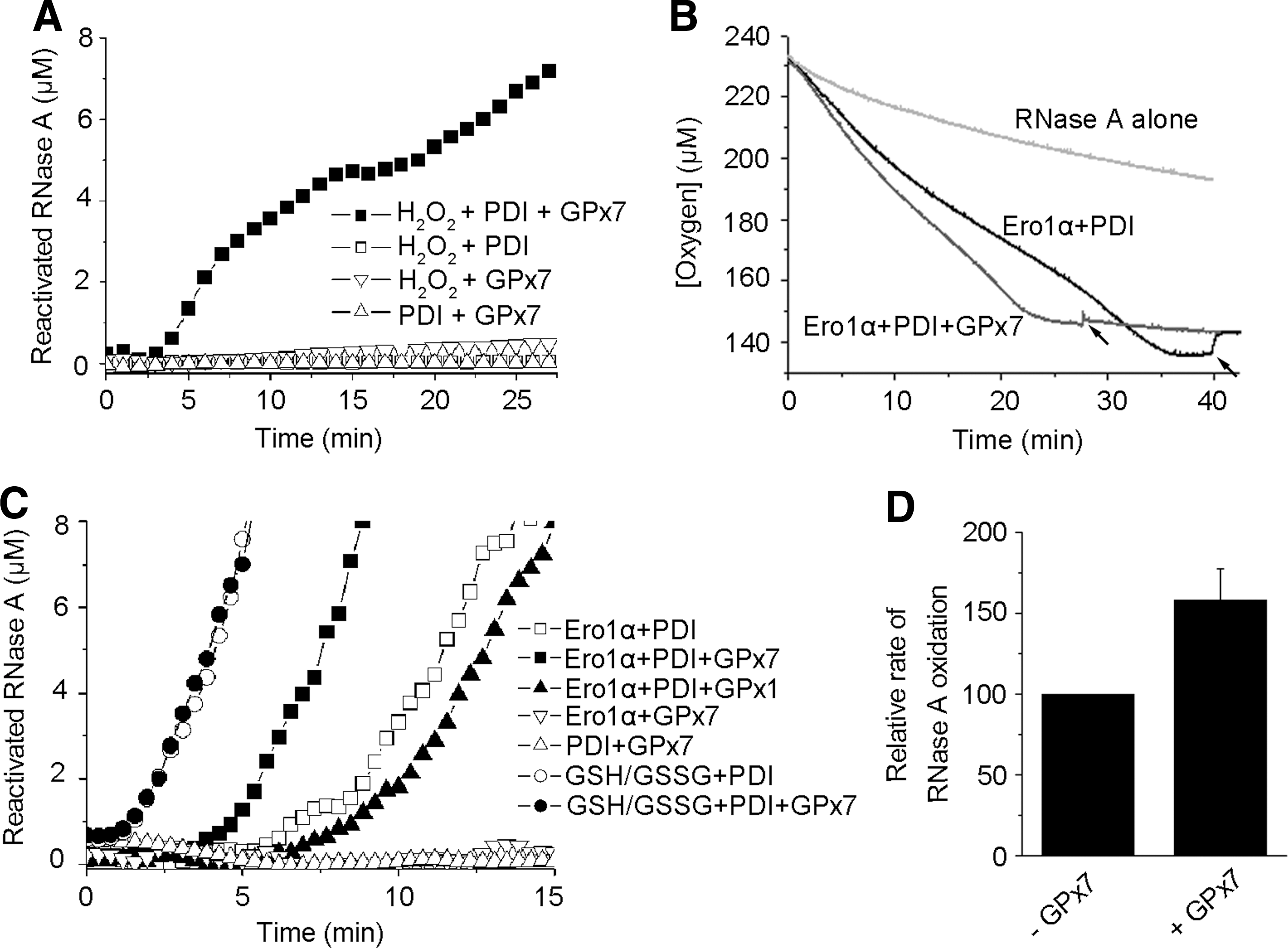
GPx7 accelerates oxidative protein folding in Ero1α/PDI system in vitro
We previously reported that human Ero1 proteins consume one molecule of O2 to produce one disulfide in reduced thioredoxin and equimolar H2O2 (34, 35). Here, we show that GPx7 harbors TGPx activity and can use Ero1α-derived H2O2 to promote the oxidation of thioredoxin. In this system, one O2 was consumed to produce two disulfides in thioredoxin with no H2O2 detected (Supplementary Fig. S1; Supplementary Data are available online at
G
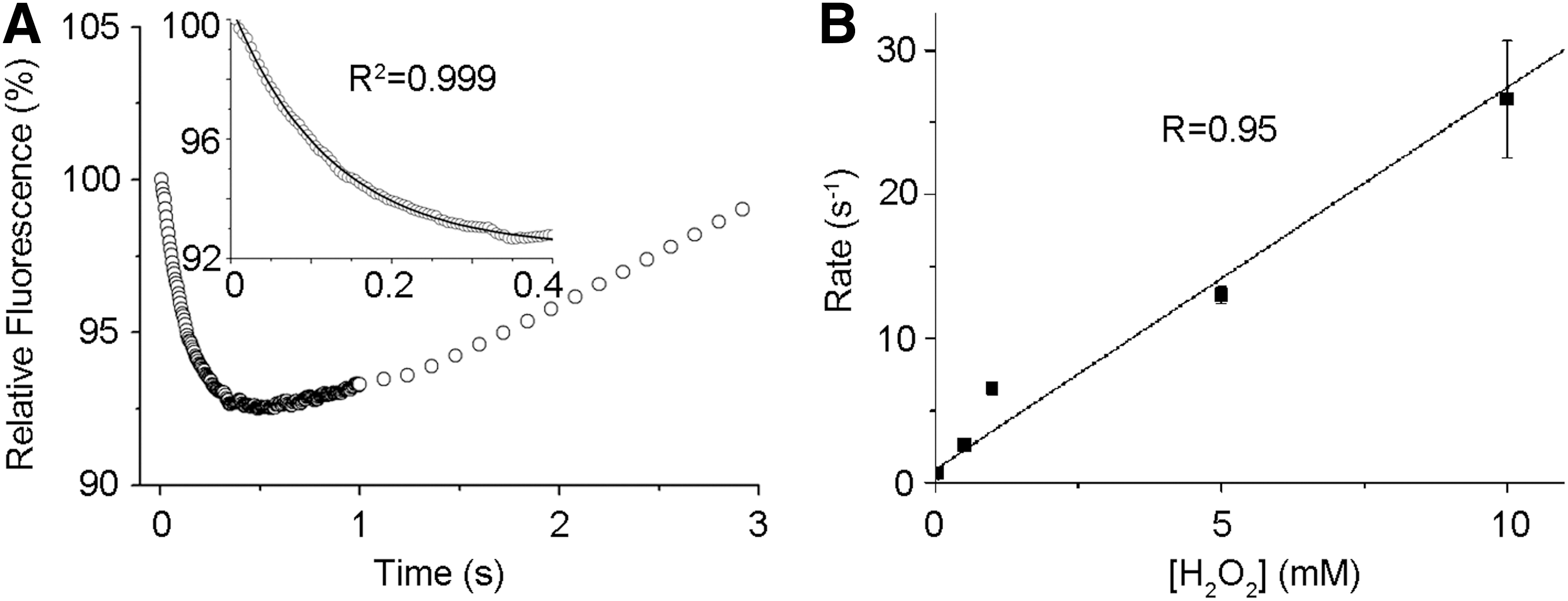
GPx7 is an efficient H2O2 scavenger
Of the three tryptophan residues in GPx7, Trp142 is conserved in the GPx family and located close to the CP active site protein data bank code (PDB: 2P31). On H2O2 addition, a time-dependent decrease of intrinsic GPx7 fluorescence was observed, mainly corresponding to quenching of Trp142 fluorescence by H2O2-oxidized active site. The fluorescence was then increased markedly, probably due to local conformation changes on prolonged H2O2 treatment (Fig. 2A). In the presence of a large excess of H2O2 for kinetics study, the rapid initial decrease in fluorescence was fitted to a single exponential function, representing a pseudo-first-order reaction (see inset in Fig. 2A). The rate constant for this reaction was linearly increasing in a range of 0.5–10 mM H2O2 (Fig. 2B), with the second-order rate constant of 2.6×103 M−1·s−1 calculated, which is comparable to that of a synuclein-fused mouse GPx7 (9.5×103 M−1·s−1) (6), and is about 30- and 300-fold greater than that for the reaction of H2O2 with GPx8 and PDI (22), respectively. These kinetic data indicate that GPx7 is a very efficient H2O2 scavenger.
Cys57 is the CP and Cys86 is a noncanonical CR in GPx7
Mature GPx7 contains two cysteines, Cys57 and Cys86. To identify their roles, we replaced either one (GPx7 C57A and GPx7 C86A) or both (GPx7 DM) with alanine and examined their redox states by using 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS), which reacts with cysteine thiols and adds 500 Da for each modification (Fig. 3A). In the experimental conditions utilized, AMS did not affect the mobility of GPx7 DM, confirming its specific binding to cysteine residues; the single cysteine mutants migrated comparatively slower, indicating that both Cys57 and Cys86 can be modified. Accordingly, GPx7 wild-type (WT) migrated even slower than the single cysteine mutants. After treatment with H2O2, GPx7 C57A still displayed the retarded mobility shift while C86A lost AMS reactivity, indicating that Cys57 but not Cys86 reacts with H2O2. Strikingly, H2O2-treated GPx7 WT migrated even faster than the C86A and DM mutants (compare lane 9 with 11 and 12), which is a characteristic of long-range intramolecular disulfide formation. The redox states of GPx7 and its mutants were further confirmed by methoxy polyethylene glycol maleimide alkylation assays, which also resulted in gel-shift retardation (Supplementary Fig. S4).
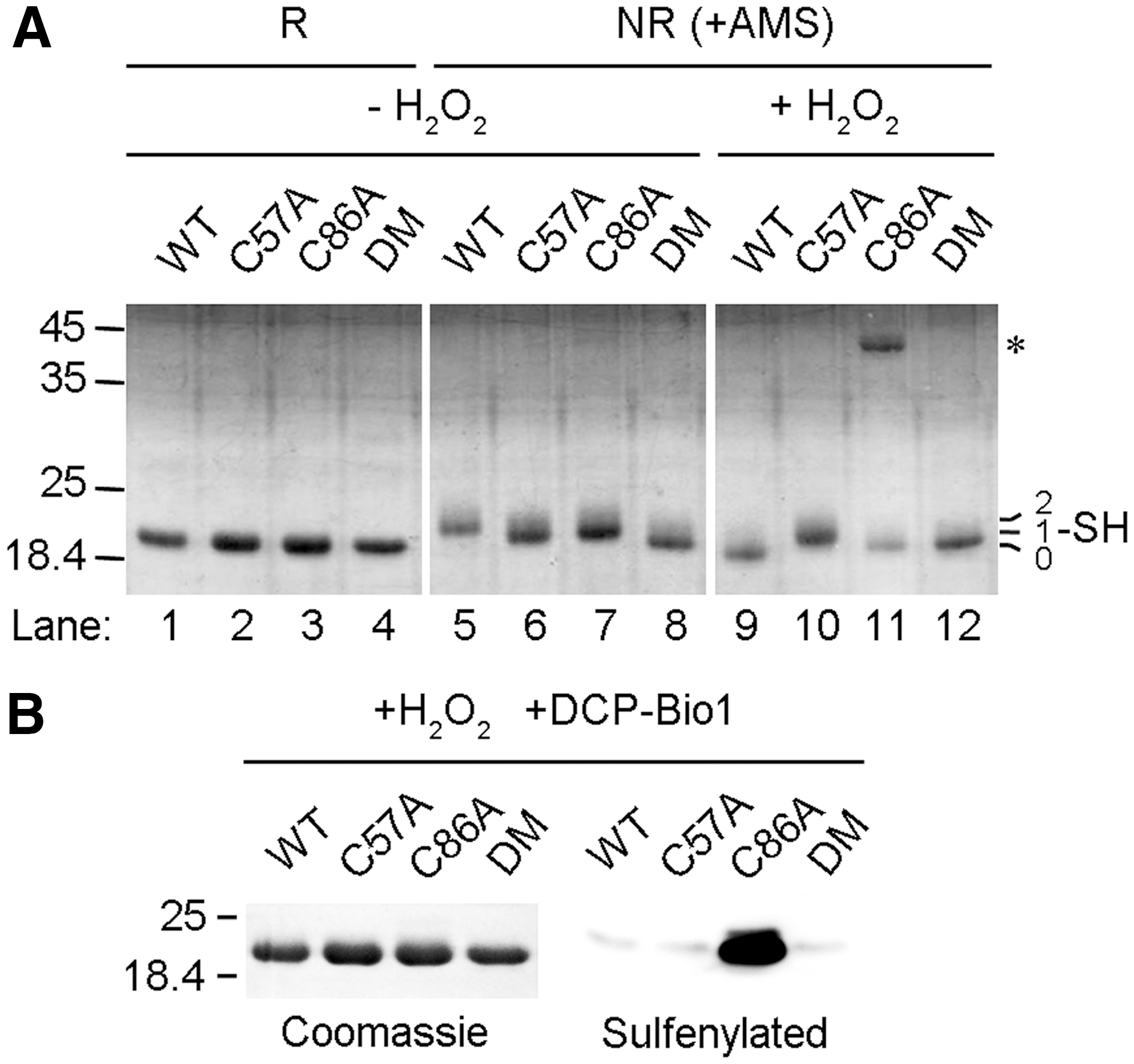
Next, we used the sulfenic acid-specific reagent, dimedone-based probe 3-(2,4-dioxocyclohexyl) propyl with a biotin tag (DCP-Bio1) (18), to characterize the reaction products of GPx7 and H2O2. Consistent with the notion that Cys57 can be sulfenylated by H2O2 and acts as the CP, GPx7 C86A but not C57A was detected by horseradish peroxidise-labeled streptavidin after treating with H2O2 and DCP-Bio1 (Fig. 3B). GPx7 WT was not modified, suggesting fast formation of an intramolecular disulfide between the sulfenylated Cys57 and Cys86, which is consistent with the pronounced mobility shift observed in Figure 3A. Thus, Cys86 plays the resolving function but is a noncanonical CR.
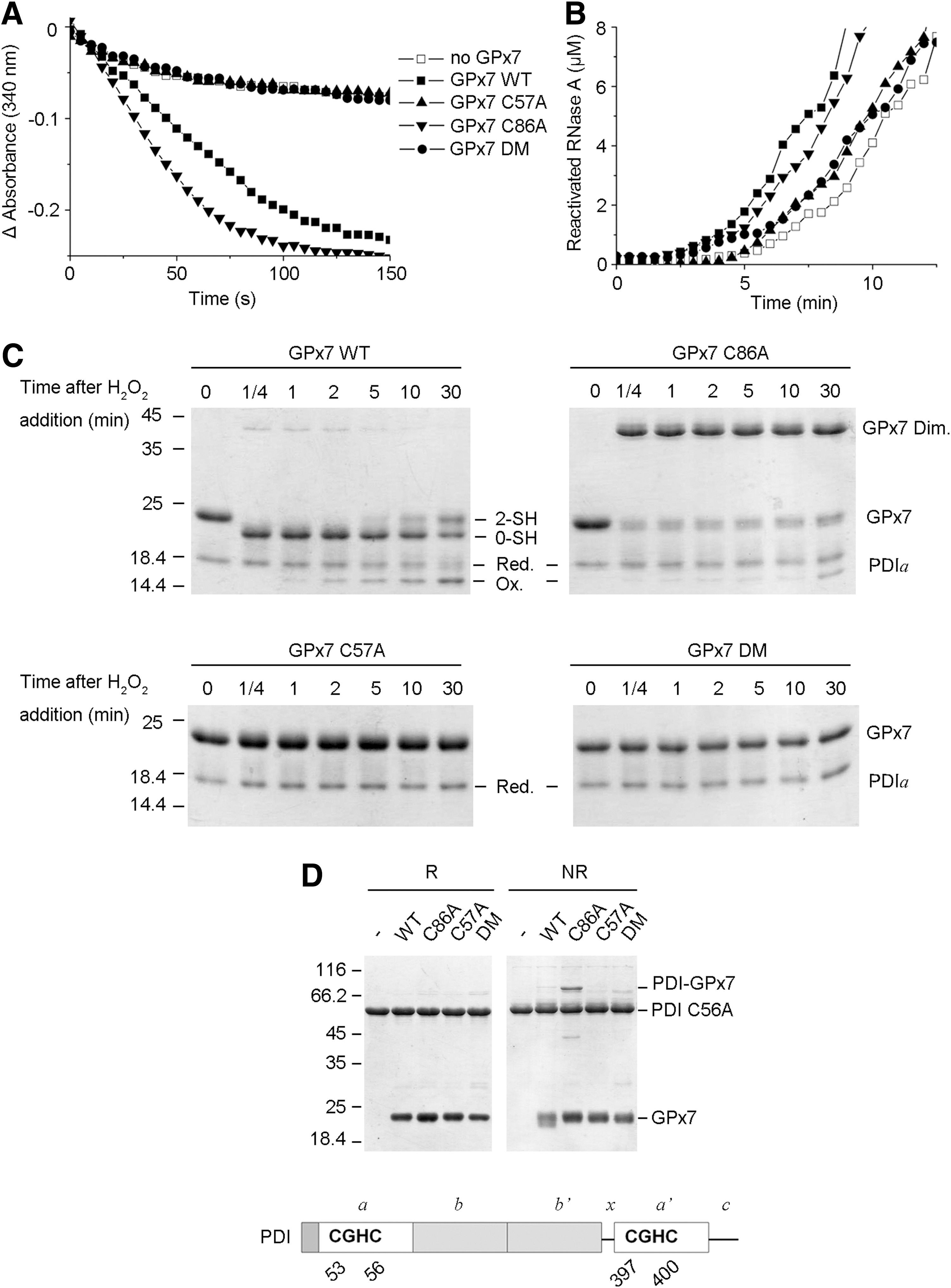
Both the disulfide form and sulfenic acid form of GPx7 are active to oxidize PDI
GPx7 is an efficient PDI peroxidase with extremely weak activity toward GSH (22). To investigate the underlying reaction mechanisms, we determined the enzymatic activities of GPx7 mutants. GPx7 C86A was as active as WT; in contrast, both C57A and DM were inactive (Fig. 4A). C86A also accelerated RNase A refolding when H2O2 was generated by Ero1α, while C57A or DM showed little, if any, effect (Fig. 4B). Thus, Cys57 of GPx7 is critical for PDI peroxidase activity. AMS modification assays were then used to determine changes in the redox state of GPx7 and PDI during the reaction. A single PDI active domain was used instead of full-length PDI in these experiments, because the redox shift is more evident in molecules of lower molecular weight. As shown in Figure 4C, the reduced PDI a domain was gradually oxidized after equimolar H2O2 addition, and was completed after 30 min. Conversely, the intramolecular disulfide form of GPx7 WT due to immediate oxidation by H2O2 gradually shifted to the reduced form. GPx7 C86A with the sulfenylated CP also oxidized PDI a domain, albeit less efficiently than WT, which is probably due to the formation of less-active C86A homodimers in the absence of reducing equivalents in the system. Neither mutant lacking CP (C57A or DM) oxidized PDI a domain. Next, to identify the PDI-reacting cysteine in GPx7, we examined complex formation between GPx7 and PDI in the absence of H2O2. A mutant PDI (C56A) was used, because replacing the C-terminal cysteine in the CGHC active site allows trapping mixed disulfides in interchange reactions (15). PDI C56A formed covalent complexes of ∼70 kDa only with GPx7 C86A (Fig. 4D), indicating that Cys57 forms mixed disulfides with PDI which are resolved by Cys86. Taken together, the results cited earlier suggest that both the disulfide form and sulfenic acid form of GPx7 can directly oxidize PDI and accelerate disulfide formation in substrates, although with different kinetics.
GPx7 accelerates oxidation of reduced J chain in cells depending on Ero1α activity
To investigate whether GPx7 can promote oxidative protein folding in living cells, we monitored disulfide bond formation in myc-tagged Ig J chains (JcM) (21). When HeLa transfectants expressing the reporter were briefly exposed to dithiothreitol (DTT), most JcM migrated as reduced monomers with a few homodimers (Fig. 5A, lane 1). On removal of the reducing agent, oxidized species (monomers, dimers, and high-molecular-weight complexes) progressively appeared (lane 4). As previously described (21), in cells co-expressing Ero1α, oxidized JcM monomers were detected already at the end of the pulse (lane 5), and dimers and high-molecular-weight species formed more rapidly during the chase, confirming that Ero1α functions as an efficient sulfhydryl oxidase. Co-expression of GPx7 resulted in accelerated disappearance of reduced JcM monomers, albeit to a lesser extent compared with Ero1α (compare, for instance, lanes 9 and 5, or 11 and 7). Densitometric quantifications confirmed that GPx7 promotes the oxidation of reduced JcM in living cells (Fig. 5B). Intriguingly, on GPx7 overexpression, fewer JcM-containing covalent complexes accumulated and could be detected only on prolonged exposure (Supplementary Fig. S5). These JcM-containing covalent complexes could still be detected immediately after DTT treatment when Ero1α was overexpressed (Fig. 5A, lane 5), which may represent the “old” molecules that are trapped in partially DTT-resistant complexes in this nonradioactive chase (5). It might be also possible that these covalent complexes were the consequence of indiscriminate oxidation by H2O2, which resulted from robust Ero1α oxidase activity after DTT challenge, and they were inhibited by the peroxidase activity of GPx7.
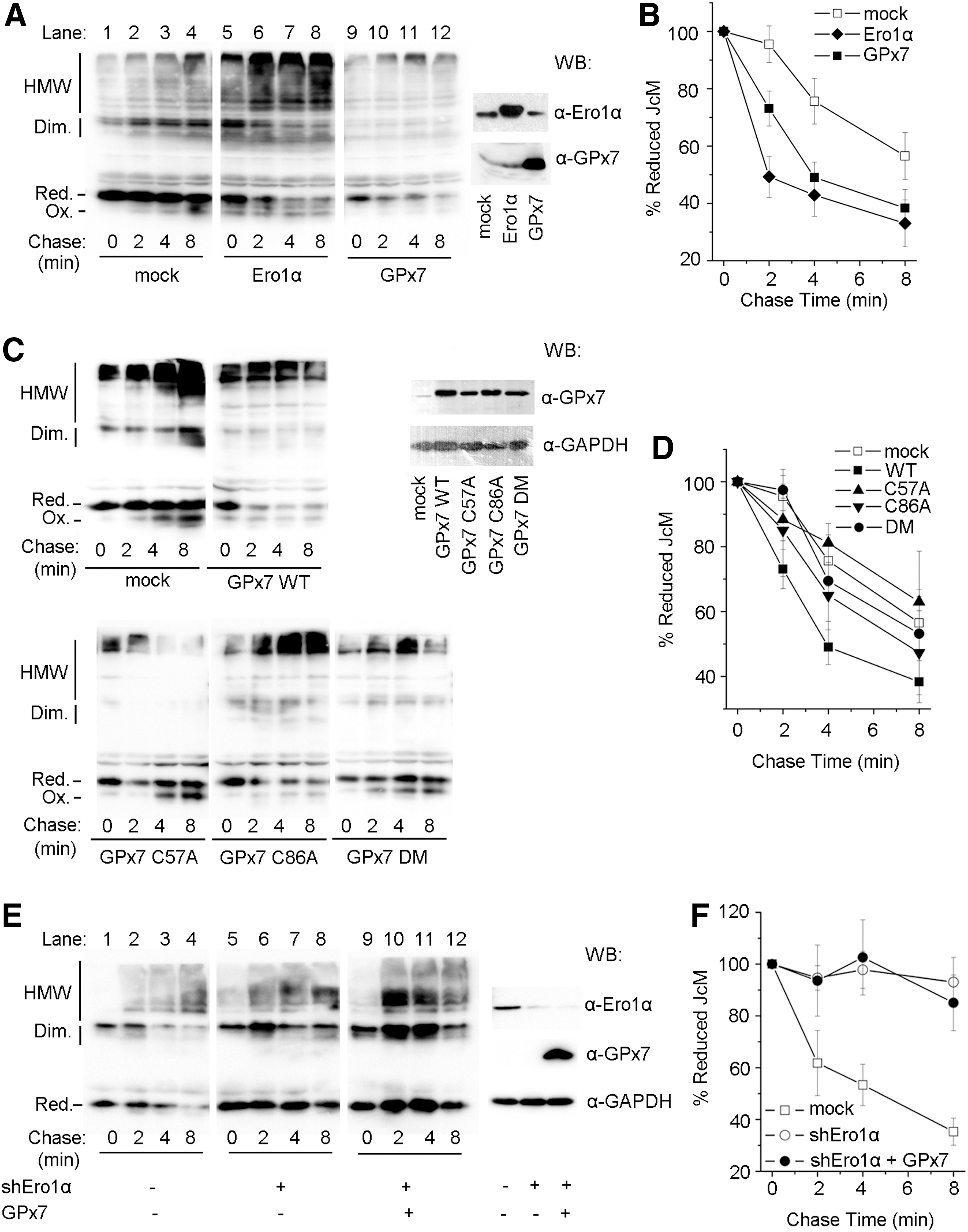
Even if expressed at comparable levels, the cysteine mutants GPx7 C57A and DM were less efficient than WT in inducing the disappearance of reduced JcM monomers (Fig. 5C). GPx7 C86A retained partial activity (Fig. 5D). These observations confirm that Cys57 plays a critical role for peroxidase activity also in vivo, and the presence of Cys86 as a CR is important for catalyzing disulfide formation in cells.
To provide further evidence that the effect of GPx7 on oxidative folding is dependent on Ero1α activity, endogenous Ero1α was knocked down by expressing Ero1α-targeting short hairpin RNA (shEro1α). Reduced JcM monomers in control cells disappeared faster at 30°C than at 20°C (compare lanes 1–4 in Fig. 5E, A). On Ero1α knockdown, the re-oxidation of JcM was significantly delayed (Fig. 5E, lanes 5–8), in line with the observation that the restoration of ER redox homeostasis after DTT challenge is compromised by Ero1α depletion (31). Ectopic expression of GPx7 on the background of Ero1α knockdown did not have a significant effect on JcM re-oxidation (Fig. 5F), strongly suggesting that the acceleration effect of GPx7 on JcM re-oxidation was dependent on Ero1α activity. The results cited earlier implied that in living cells, GPx7 can efficiently utilize Ero1α-derived H2O2, which should be the main source of H2O2 produced after reductive challenge.
GPx7 and Ero1α bind to PDI separately
Having determined that GPx7 potentiates the Ero1α/PDI system in vitro and in vivo, we performed glutathione S-transferase (GST)-pulldown studies to detect possible binary or ternary complexes. Approximately 4% of PDI from HeLa cell lysates specifically bound to GST-GPx7 WT or DM chimeras. Ero1α was not detected in the bound fraction (Fig. 6A). Thus, PDI can bind to GPx7 through noncovalent interactions.
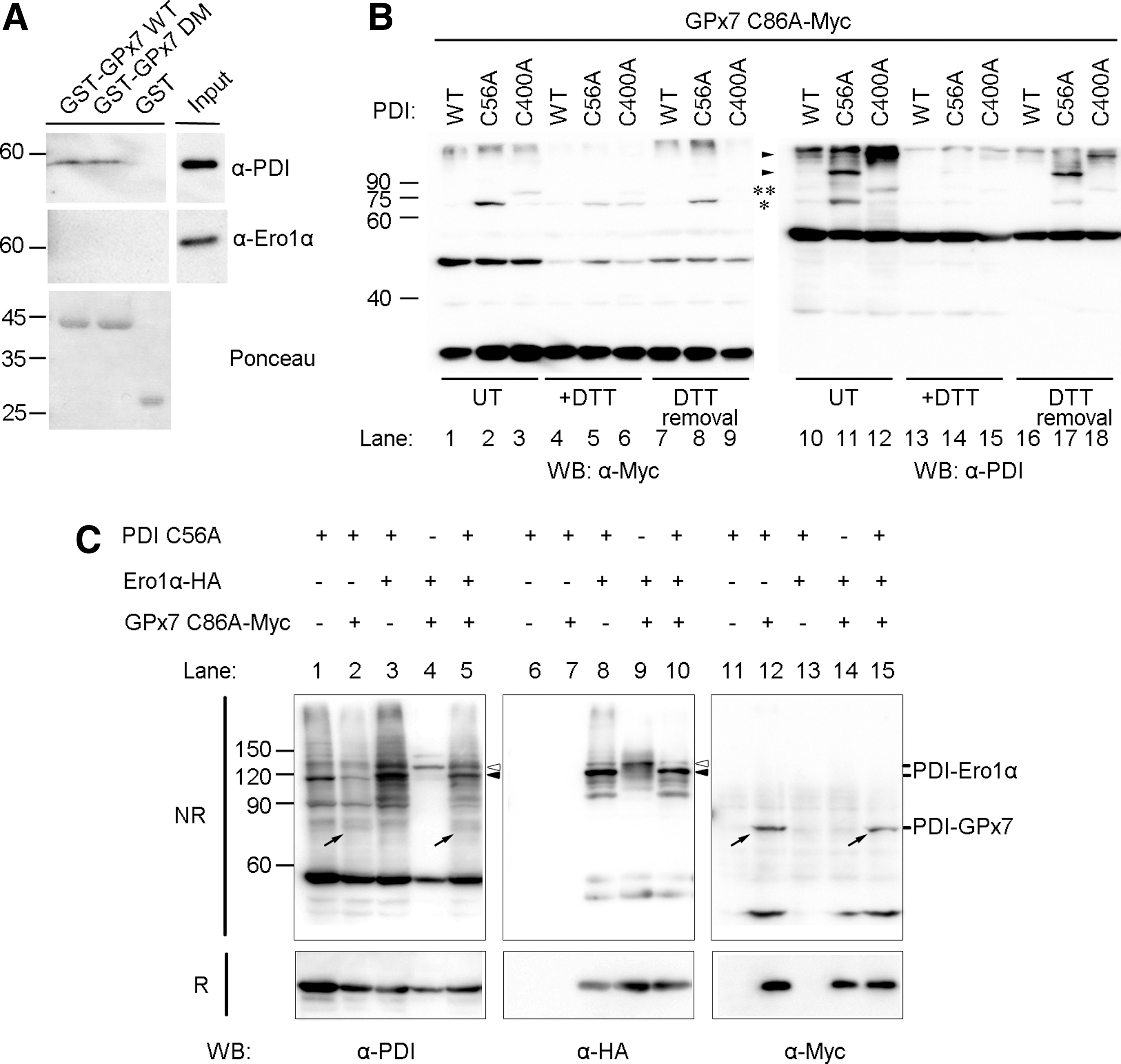
We once again took advantage of the PDI cysteine trapping mutants to analyze the ability of the a and a′ domains to covalently interact with GPx7 in HeLa cells. Clearly, as shown in Figure 6B, the covalent GPx7-PDI complexes were more abundant with PDI C56A (* in lanes 2 and 11) than with PDI C400A (** in lanes 3 and 12), and these bands faded on DTT treatment. After removal of the reducing agent, the complex between GPx7 and PDI a domain re-emerged at 30 min (lanes 8 and 17). These mixed disulfides likely reflect intermediates in the oxidation of PDI by H2O2 catalyzed by GPx7 in cells. Altogether, these results indicate that GPx7 interacts preferentially with the PDI a domain.
Interactions between Ero1α, GPx7, and PDI were further investigated by overexpressing Ero1α, GPx7 C86A, and PDI C56A in combinations (Fig. 6C). Ero1α covalently bound to endogenous PDI (see bands indicated by open arrowheads in lanes 4 and 9), likely via the a′ domain (25), and to both a and a′ domains in PDI C56A mutant (see bands indicated by open and closed arrowheads in lanes 3 and 8). GPx7 C86A was linked to the a domain of PDI C56A (see bands indicated by diagonal arrows in lanes 2 and 12, and also see Fig. 6B). When the three proteins were co-expressed together in cells, the binary complexes (PDI-Ero1α and PDI-GPx7) were clearly visible. In contrast, bands corresponding to ternary complex were not detected (compare, for instance, lanes 10 and 8, or 15 and 12), suggesting that GPx7 and Ero1α covalently bind to PDI in a sequential way rather than simultaneously.
Two active sites of PDI cooperate intramolecularly in the Ero1α/GPx7/PDI system
In order to dissect the Ero1α/GPx7/PDI system in greater detail, we measured the amount of the disulfides generated by monitoring GSH oxidation into GSSG in vitro. Consistent with the notion that PDI metabolizes H2O2 inefficiently (22), very few disulfides were formed in the presence of exogenous H2O2 only (Fig. 7A, white). The presence of GPx7 markedly accelerated disulfide bond formation catalyzed by PDI WT. In the presence of GPx7, PDIC1 and PDIC2 mutants, in which both cysteines were replaced by serines in the a and a′ domain active sites separately, catalyzed GSH oxidation by H2O2, albeit less efficiently than PDI WT. Mixing PDIC1 and PDIC2 restored the peroxidase activity for GPx7, similar with PDI WT (Fig. 7A, black). Thus, GPx7 can readily oxidize either active site of PDI with excess H2O2.
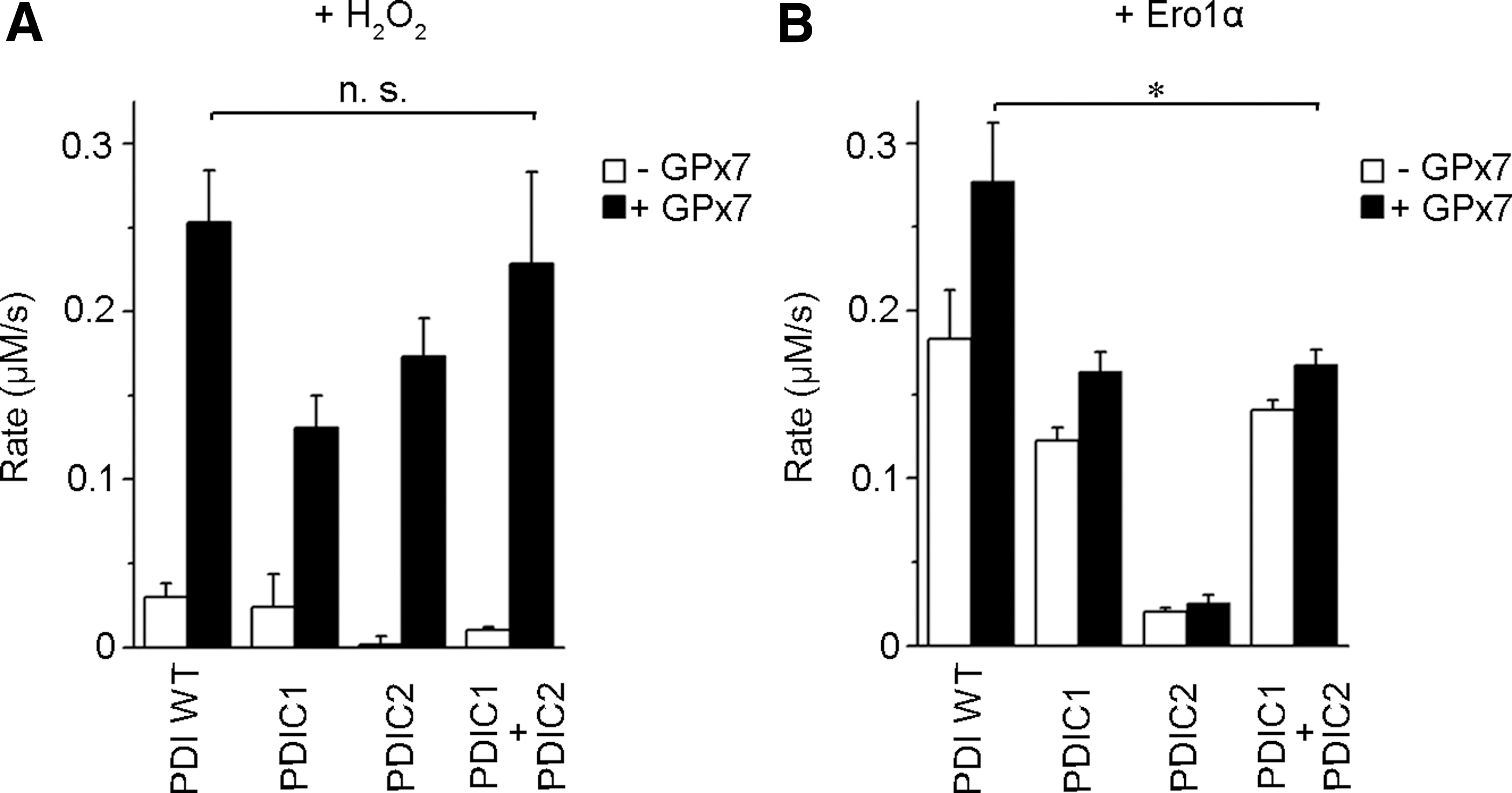
Next, Ero1α was used to supply an oxidizing source instead of exogenous H2O2. In accordance with a previous finding that Ero1α preferentially oxidizes the a′ active site in PDI (3, 34), PDIC1 was able to generate disulfides. In contrast, a few disulfides were produced by PDIC2 (Fig. 7B, white). As expected, GPx7 was active toward PDIC1 but inactive toward PDIC2, as Ero1α does not oxidize the a active site producing little H2O2. Nevertheless, the activity of GPx7 toward PDIC1 was lower than toward PDI WT, suggesting that the active a domains contribute to the interactions with GPx7, although not involved in binding Ero1α. However, adding PDIC2 (with intact a domain) to PDIC1 did not increase the activity in the Ero1α/GPx7/PDI system (Fig. 7B, black), which is different from using exogenous H2O2 as the oxidizing source. The fact that PDIC1 and PDIC2 did not show any synergy for GPx7 in this system suggested that the H2O2 produced on the oxidation of PDI a′ domain by Ero1α can only be efficiently used by GPx7 to oxidize the a domain of the same PDI molecule. Thus, maximal activity of the Ero1α/GPx7/PDI triad requires intramolecular co-operation between the a and a′ sites in PDI.
Discussion
We have reconstructed an efficient, ER-based oxidative folding system composed of a sulfhydryl oxidase (Ero1α), an oxidoreductase/isomerase (PDI), and a peroxidase (GPx7). Our in vitro and in vivo assays enable the depiction of a working model for the Ero1α/GPx7/PDI triad (Fig. 8A). Ero1α preferentially oxidizes the a′ domain of PDI, generating one molecule of H2O2 and a partially oxidized PDI at the expense of one O2 molecule (Step I). GPx7 uses the H2O2 generated by Ero1α in situ for oxidizing the a domain of the same PDI molecule, preventing diffusion of this reactive oxygen species (Step II). Fully oxidized PDI transfers disulfides into substrates for oxidative folding. Via this mechanism, a single O2 molecule can generate two disulfide bonds and two harmless H2O molecules. A bimolecular fluorescence complementation experiment in cells suggested a possible physical association between Ero1α and GPx7 (22), and our model suggests that Ero1α and GPx7 preferentially bind to the a′ and a domain of PDI separately in a sequential way, and maybe transiently close to each other. There are other potential pathways for productive utilization of H2O2 in mammalian ER. GPx8, also localizing in close proximity to Ero1α in cells, shows weaker PDI-peroxidase activity in vitro (22) and has one order of magnitude lower reactivity with H2O2, compared with GPx7 (Fig. 2). Interestingly, GPx8 can prevent accumulation of Ero1α-derived H2O2 and ER stress in living cells (Ramming and Appenzeller-Herzog, personal communication), but whether GPx8 could promote oxidative folding by using Ero1α-derived H2O2 in situ is still an open question. The two-cysteine Prx4 was reported to catalyze disulfide formation in vitro and in vivo (28, 39). However, Prx4 can be inactivated by H2O2, and no enzymes that are capable of reducing over-oxidized Prxs (such as sulfiredoxin) have been found so far in the ER (36). Different from Prx4, GPx7 is hardly inactivated at a sub-millimolar concentration of H2O2 (unpublished data), implying that GPx7 is either resistant to over-oxidation or protected by PDI. A certain degree of redundancy of peroxidase activities in the early secretory compartment may be important to optimize oxidative folding while limiting the risks of oxidative stress.
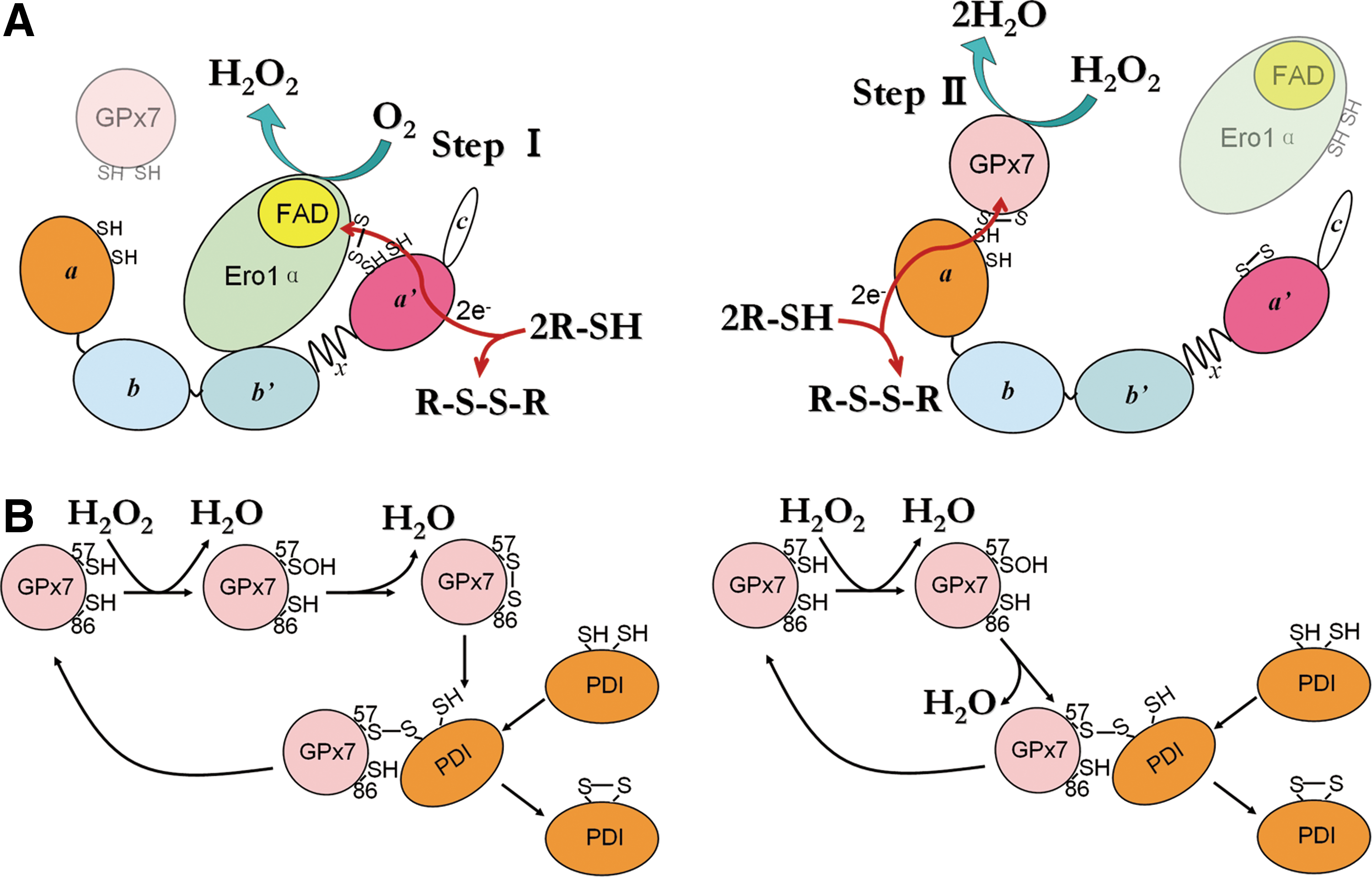
Different from canonical cysteine-based TGPx in invertebrates and plants, GPx7 lacks the corresponding CR known to confer preferential reducibility by thioredoxin (7). Here, we demonstrate the function of Cys86 to resolve the Cys57 sulfenic acid to form an intramolecular disulfide for PDI oxidation (Fig. 8B, left panel). Moreover, the presence of Cys86 also prevents the formation of the less-active GPx7 homodimers bridged by two CP (Fig. 4C). Therefore, Cys86 emerges as a novel noncanonical CR. This cysteine residue is conserved in a similar position among almost all GPx members; however, whether the analogues of Cys86 in other TGPx without canonical CR (e.g., human GPx5) can also play resolving roles is currently unclear. Since the sulfur atoms of the two cysteine residues in reduced GPx7 are located ∼11 Å away protein data bank code (PDB: 2P31), it is expected that the formation of the intramolecular disulfide should result in significant conformational changes. The observation that GPx7 C86A can also oxidize PDI, suggests an alternative pathway (Fig. 8B, right panel), in which the sulfenylated CP directly oxidizes PDI via an intermolecular disulfide. This is similar to the way in which yeast TGPx Orp1 oxidizes the transcription factor Yap1 (9). Therefore, PDI can react with Cys57 of GPx7, either in the sulfenylated form or in the disulfide form. On the other hand, Cys86 was recently reported to be critical for the formation of GPx7-GRP78 covalent complexes in vitro, at an extremely high concentration of H2O2 (37). In this case, the possibility that Cys57 was inactivated to -SO2/SO3 forms by H2O2 cannot be ruled out. Therefore, whether GPx7 could adopt additional ways to interact with its partner proteins still needs further investigation. An interesting feature of GPx7 is that it passes the oxidizing equivalents to PDI rather than directly to GSH (22), and the rate constant for the reduction of GPx7 by PDI is 270-fold faster than that by GSH (6). In this regard, we did not observe any change of oxidized to total glutathione ratio in GPx7-transfected HeLa cells at a steady state (Supplementary Fig. S6). Similarly, Ero1 and Prx4 proteins prefer PDI compared with GSH as substrates (8). In addition, the ER thiol-disulfide homoeostasis is not changed on ectopic expression of WT Ero1α, unless the deregulated hyperactive Ero1α was introduced (1). These results imply that the ER redox homeostasis is tightly controlled, and PDI plays a pivotal role in oxidative folding, preventing futile consumption of GSH and ensuring correct disulfide formation. Moreover, since the chaperone activity and the overall structure of human PDI are redox regulated (32, 33), the oxidation of PDI by GPx7 could result in conformational changes and elevated chaperone activities in cells, which could be another oxidative stress-induced cellular protective pathway.
The observations that GPx7 is down-regulated in breast cancer cells (30) and esophageal adenocarcinoma (23) suggest a tumor suppressor function of GPx7, possibly by preventing cells from high levels of reactive oxygen species and oxidative DNA damage. In accordance with this, GPx7 knockout mice suffered from systemic oxidative stress damage, increased carcinogenesis, and shortened life span (37). Recently, it was revealed that Ero1α is highly expressed in esophageal and gastric cancer cells (4), further emphasizing the importance of ER redox regulation in the pathophysiology of the gastro-intestinal tract. Considering our observations that GPx7 can directly react with Ero1α-derived H2O2, it will be very interesting to investigate whether there exists any coincidence and/or correlation between the decrease of GPx7 and increase of Ero1α in the development of these cancers. In view of the key roles of H2O2 in cell signaling and pathophysiology, further work is also needed to characterize the network of peroxide in terms of sources (38), scavengers (16), and regulation in the secretory compartment. Emerging evidence on the relationship between disulfide formation and diseases associated with redox imbalance also highlight the enzyme molecules involved in ER redox homeostasis as potential targets for both biomarker and drug development in these diseases.
Materials and Methods
Plasmids and protein purification
The pKEHS780 plasmid encoding the mature human GPx7 (Q20-L187) without signal sequence was a kind gift from L. Ruddock. BL21 (DE3) cells (Novagen) expressing GPx7 were grown in Luria-Bertani medium containing 100 μg/ml of ampicillin at 37°C for 4 h, and shaken for 20 h at 25°C after addition of 200 μM isopropyl-β-D-thiogalactoside. Homogenates from cell lysates were applied onto a Ni-Chelating Sepharose Fast Flow column (GE Healthcare). Fractions eluted with 250 mM imidazole were further loaded on a HiPrep 26/10 desalting column (GE Healthcare) that was pre-equilibrated with 50 mM Tris-HCl, pH7.6, 150 mM NaCl, and 1 mM EDTA. The flow-through was concentrated, and aliquots were stored at −80°C. Recombinant mature full-length human Ero1α and PDI proteins were purified as previously described (34).
The cDNA encoding the signal sequence of GPx7 or PDI was added by overlapping PCR using pKEHS780 or pQE30-PDI (34) as a template, and then inserted into pcDNA3.1 vector at XbaI and KpnI sites to generate plasmids suitable for expression in eukaryotic cells. Mutagenesis was carried out using the Fast Mutagenesis Kit (TransGen) according to the manufacturer's instructions. The pcDNA3.1-Ero1α construct was used as previously described (21), and pcDNA3.1-Ero1α with a C-terminal HA tag was constructed by PCR. Each construct was verified by DNA sequencing.
Cell culture, transfection, and antibodies
HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 5% fetal bovine serum (Gibco), 100 units/ml penicillin, and 100 μg/ml streptomycin (Gibco) at 5% CO2. HeLa cells were transfected using Lipofectamine2000 (Invitrogen) according to the manufacturer's instructions.
Antibodies to GPx7 were obtained from rabbits on immunization with purified GPx7 protein as an adjuvant. Mouse monoclonal anti-Ero1α (2G4) and anti-myc (9E10) were used as described (24), while anti-PDI (RL90) and anti-GAPDH (6C5) were purchased from Abcam and Beyotime, respectively.
RNA interference
For transient knockdown of Ero1α, pSUPER-retro-puro retrovirus vector (Oligoengine) expressing shRNA targeting Ero1α sequence 5′-GGGACACAACATTACAGAATTTCAA-3′ (10) was constructed following the manufacturer's instructions. The resulting shEro1α plasmid was transfected into HeLa cells combined with JcM plasmid on day 1, followed by a second-round transfection of shEro1α plasmid with GPx7 plasmid on day 3 and subsequent analysis on day 5.
Enzyme activity assays
Denatured and reduced RNase A was prepared as described (19). For oxygen consumption assays, all components except Ero1α were mixed and added to an Oxygraph Clark-type electrode (Hansatech Instruments), followed by an injection of Ero1α to initiate the reaction. For H2O2 determination, catalase at 20 μg/ml was injected into the reaction vessel when the thiol oxidation reaction was complete.
RNase A reactivation was assayed by monitoring the 296 nm absorbance increase at 25°C due to the hydrolysis of cCMP as described (34).
GPx activity was measured using a coupled assay after the decrease in absorbance at 340 nm due to reduced nicotinamide adenine dinucleotide phosphate (NADPH) (0.15 mM; Roche) consumption by glutathione reductase (0.24 unit; Sigma) (22), with addition of H2O2 or Ero1α to start the reaction. A molar extinction coefficient of 6200 M−1·cm−1 for NADPH was used for calculations. All experiments were performed in 100 mM Tris-HAc (pH 8.0) containing 50 mM NaCl and 1 mM EDTA.
Stopped-flow fluorescence
The oxidation rate of the active site of GPx7 was determined by the decrease of tryptophan fluorescence using a PiStar-180 stopped-flow fluorometer (Applied Photophysics) with an excitation at 280 nm and a band–pass emission >320 nm at 25°C. The reaction was started by an injection of 0–10 mM H2O2 into 10 μM GPx7 in 100 mM citrate (pH 7.0), 200 mM Na2HPO4, and 1 mM EDTA.
In vivo oxidative folding assays
HeLa transfectants expressing JcM were incubated for 5 min at 37°C with 5 mM DTT in DMEM, washed twice with ice-cold phosphate-buffered saline (PBS), and cultured in DMEM without DTT at an indicated temperature (5). Aliquots were taken at different times, quenched with 20 mM N-ethylmaleimide (NEM), lysed in radio immunoprecipitation assay buffer (Beyotime) with NEM and protease inhibitors, and resolved by nonreducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Western blots (WB) were developed by anti-myc and enhanced chemiluminescence (ECL) (Thermo Scientific), followed by using a ChemiScope mini chemiluminescence imaging system (Clinx Science). Band intensities were quantified by densitometry using ImageJ software.
In vitro redox-state measurements, mixed disulfides trapping, and sulfenic acid detection
To measure the redox states of GPx7, 20 μM recombinant GPx7 proteins were incubated with or without 40 μM H2O2 for 15 min. Free thiols were then blocked by adding 2 mM AMS (Invitrogen). For PDI oxidation assays, reactions were initiated by adding 20 μM H2O2 to 20 μM GPx7 and 20 μM reduced PDI a domain, and samples were taken for quenching using 2 mM AMS at the indicated times. To trap the disulfide-linked complex between GPx7 and PDI, 20 μM GPx7 and 10 μM PDI C56A proteins were mixed for 15 min and then blocked by 20 mM NEM. Samples were then analyzed by SDS-PAGE and Coomassie staining. For sulfenic acid detection, 50 μM GPx7 proteins were incubated with 100 μM H2O2 and 1 mM DCP-Bio1 (KeraFAST) for 30 min. Excess DCP-Bio1 was removed by using Amicon Ultra centrifugal filters (Millipore). Samples were then analyzed by SDS-PAGE, developed by either Coomassie staining or WB using horseradish peroxidise-labeled streptavidin (Beyotime) and ECL. All experiments were performed at 25°C in PBS buffer.
Pulldown assays and trapping mixed disulfides in vivo
For pulldown assay, Glutathione Sepharose resins (GE Healthcare) were incubated with 10 μM GST-GPx7 fusion proteins and HeLa lysates (0.5 mg protein/ml) for 4 h at 4°C in PBS, and washed five times with ice-cold PBS. Bound proteins were detected by Ponceau staining and WB, respectively.
To trap mixed disulfides in vivo, HeLa transfectants were incubated for 10 min at 37°C with or without 5 mM DTT in DMEM, quenched with 40 mM NEM immediately or after DTT washout followed by incubation in DTT-free medium for 30 min. Samples were lysed and analyzed by nonreducing or reducing SDS-PAGE and WB.
Footnotes
Acknowledgments
The authors thank Lloyd Ruddock for the kind gift of pKEHS780 plasmid, Jiangyun Wang for generously providing a cell culture room, Si Wu for technical assistance in stopped-flow determination, and Xi Wang, Xi'e Wang, and Claudio Fagioli for invaluable help. This work was supported by grants from the Chinese Ministry of Science and Technology (2011CB910303 and 2012CB911002) to C.C.W., the National Natural Science Foundation of China (31000351 and 31370775) to L.W., and Telethon (GGP11077) and Associazione Italiana Ricerca Cancro (AIRC; IG and 5×1000 program) to R.S.
Author Disclosure Statement
The authors declare that they have no conflicts of interest.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.